

Abstract
In our previous study, we clearly demonstrated the roles of pro-inflammatory cytokines, including tumor necrosis factor-α, interleukin-1β (IL-1β), and IL-6, and subsequent reactive oxygen species (ROS) generation on the pathogenesis of cisplatin ototoxicity in vitro and in vivo. ROS generation in cisplatin-treated HEI-OC1 auditory cells was also correlated with changing mitochondrial membrane potential. However, the roles of NADPH oxidase in cisplatin-induced ROS generation and ototoxicity have not been fully elucidated. Herein, immunohistochemical studies demonstrated that treatment of cisplatin induced the expression of NADPH oxidase isoforms NOX-1 and NOX-4 in HEI-OC1 auditory cells. Expression of mRNA for NOX-1, NOX-4, NOXO1, NOXA1, p47phox, and p67phox was also increased. Inhibition of NADPH oxidase with diphenyleniodonium chloride or apocynin abolished ROS production and the subsequent apoptotic cell death in cisplatin-treated cells. Furthermore, suppression of NOX1 and NOX4 expression by small interfering RNA transfection markedly abolished the cytotoxicity and ROS generation by cisplatin. Together, our data suggest that ROS generated, in part, through the activation of NADPH oxidase plays an essential role in cisplatin ototoxicity.
Introduction
Cisplatin (cis-diaminedichloroplatinum II; CDDP) is an extensively used chemotherapeutic agent for the treatment of a broad spectrum of tumors (Fram, 1992). However, progressive, irreversible side effects of cisplatin, including nephrotoxicity and ototoxicity, greatly impair the patient's quality of life and frequently result in having to lower dosage during treatment or discontinuation of treatment. In particular, cisplatin ototoxicity occurs primarily in the cochleae. Damage to cochlear structures by apoptosis especially includes outer hair cells (OHCs) (Alam et al., 2000), spiral ganglion cells (Lee et al., 2003), and the marginal cells of the stria vascularis (Lee et al., 2004). In recent years, evidence has accumulated to demonstrate that cisplatin ototoxicity is closely related to the increased production of reactive oxygen species (ROS) (McAlpine and Johnstone, 1990; Rybak et al., 1999). ROS is generated within the cochleae after exposure to noise (Henderson et al., 1999) or ototoxic drugs such as cisplatin (Clerici et al., 1996; Kopke et al., 1997) and aminoglycoside antibiotics (Clerici et al., 1996) as well as within the cultures of rat organ of Corti explants exposed to cisplatin in vitro (Kopke et al., 1997). Although the role of oxidative stress in inner ear damage is well established, its source is poorly elucidated. Potential enzymatic sources of ROS include mitochondrial electron transport chain system (Szeto, 2006), xanthine oxidase (Berry and Hare, 2004), cytochrome p-450 enzymes (Gottlieb, 2003), and NADPH oxidase (Miller et al., 2006). Especially in phagocytic cells such as neutrophils, superoxide is generated by NADPH oxidase enzyme complex (Geiszt, 2006). However, recently, many studies have proven that the expression of superoxide-generating similar NADPH oxidases (NOXs) is not restricted to phagocytic cells, but it is present in a wide variety of nonphagocytic cells and tissues (Quinn et al., 2006). Besides the well known catalytic subunit of the phagocytic NADPH oxidase (gp91phox or NOX2), six other superoxide-producing nonphagocytic NOX enzymes, including NOX1, NOX3, NOX4, NOX5, DUOX1, and DUOX2, have been identified (Quinn et al., 2006). These enzymes are distinct from the phagocyte NOX2 and play essential roles in functions other than host defense. However, structural features of many nonphagocyte oxidase proteins are very similar to or even identical with those of their phagocyte counterparts. Therefore, there is a high level of interest in understanding the physiological functions of these enzymes and their potential roles in pathophysiological events (Quinn et al., 2006). Of note, recently, it has been reported that superoxide-generating NADPH oxidase isoform NOX3 was expressed in the organ of Corti of the cochleae (Bánfi et al., 2004; Mukherjea et al., 2006) and vestibular system (Paffenholz et al., 2004). Bánfi et al. also demonstrated that NOX3-dependent superoxide production was markedly enhanced after cisplatin exposure to NOX3-transfected human embryonic kidney 293 cells (Bánfi et al., 2004). They suggested that NOX3-dependent ROS generation might contribute to the hearing loss and balance problems in response to ototoxic drugs. However, the roles of the other NADPH oxidases in cisplatin-induced ROS generation and ototoxicity have not been fully elucidated. Therefore, we here investigated the expression of NADPH oxidases and their roles on cisplatin ototoxicity in vitro using HEI-OC1 cells and in vivo.
Materials and Methods
Reagents.
Cisplatin, diphenyleniodonium chloride (DPI), apocynin, NADPH, N,NP-dimethyl-9,9P-biacridinium dinitrate (lucigenin), and 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-tetrazolium bromide (MTT) were purchased from Sigma. DMEM, fetal bovine serum (FBS), and other tissue culture reagents were obtained from Invitrogen. Anti-Tumor necrosis factor-α (TNF-α), anti-interleukin-1β (IL-1β), anti-IL-6 antibodies were purchased from R & D Systems. Antibodies specific to NOX1 (catalog #SC-25545), NOX4 (catalog #21860), p22phox (catalog #SC-20781), p47phox (catalog #SC-7660), p67phox (catalog #SC-7663), and caspase-3 (catalog #SC-7148) were obtained from Santa Cruz Biotechnology. Monoclonal anti-phosphotyrosine and anti-phosphoserine/threonine antibodies were obtained from Millipore Corporation. Pharmacological inhibitors of mitogen-activated protein kinase (MAPK), including SB203580 [4-(4-fluorophenyl)-2-(4-methylsulfinylphenyl)-5-(4-pyridyl)-1H-imidazole], SP600125 (anthra[1,9-cd]pyrazol-6 (2H)-one), and U0126 [1,4-diamino-2,3-dicyano-1,4-bis(o-aminophenylmercapto)butadiene], were purchased from Calbiochem.
Cell culture and viability.
The establishment and characterization of the conditionally immortalized HEI-OC1 auditory cells were described previously by Kalinec et al. (2003). Expression of outer hair cell-specific markers, such as Prestin and Math1, were detected in HEI-OC1 cells (data not shown). HEI-OC1 cells were maintained in high-glucose DMEM (Invitrogen) containing 10% FBS. For the experiments described below, HEI-OC1 cells were cultured under permissive conditions: 33°C, 5% CO2 in DMEM supplemented with 10% FBS. Cells (3 × 104 cells per well of 24-well plate) were plated for 16 h and further incubated with 20 μm cisplatin for 24 h. To determine the cell viability, MTT (0.25 mg) was added to 1 ml of cell suspension for 4 h. After washing the cells three times with PBS, pH 7.4, the insoluble formazan product was dissolved in DMSO. Then, the optical density (O.D.) of each culture well was measured using a microplate reader (Titertek Multiskan; Flow Laboratories) at 590 nm. The O.D. in the control cells was taken as 100% of viability.
Determination of ROS production.
The intracellular ROS level was measured using a fluorescent dye 2′,7′-dichlorofluorescein diacetate (DCFH-DA) (Invitrogen). In the presence of an oxidant, DCFH is converted into the highly fluorescent 2′,7′-dichlorofluorescein. Cells were plated in 96-well culture plates overnight and treated with cisplatin in the presence or absence of NOX inhibitors apocynin and DPI for 24 h. After washing with PBS, serum-free DMEM containing 10 μm DCFH-DA was added to each well and incubated at 37°C for 1 h. ROS production was measured using a microplate reader equipped with a spectrofluorometer at an emission wavelength of 538 nm and extinction wavelength of 485 nm. Relative ROS production was expressed as the change in fluorescence of experimental groups compared with that of the appropriate controls (100%).
Flow cytometry analysis.
For measurement of intracellular ROS level by flow cytometry analysis, the oxidation-sensitive probe DCFH-DA was used. Briefly, cells were incubated with 10 μm DCFH-DA for 30 min. For flow cytometry analysis, cells were detached by trypsinization, washed once in PBS buffer, and resuspended in 800 μl of PBS buffer. Flow cytometric analyses (10,000 events per sample) were performed in a FACSCalibur system (BD Biosciences) with extinction and emission at 485 and 538 nm and then evaluated with the CellQuest software.
Transfection of NOX1 and NOX4 small interfering RNA constructs into HEI-OC1 cells.
Inhibition of NOX1 and NOX4 expression was assessed by reverse transcription (RT)-PCR analysis after transfection of HEI-OC1 cells with NOX1 and NOX4 small interfering RNA (siRNA), respectively. The NOX1 siRNA construct is a pool of three sequences of siRNA as follows: duplex 1 sense strand, 5′-GCA ACC GUA CAC UGA GAA ATT-3′; duplex 2 sense strand, 5′-CCU AUC UCA UCC UGA GAA ATT-3′; and duplex 3 sense strand, 5′-GCU UCC AUC UUG AAA UCU ATT-3′ (mRNA GenBank accession number NM_172203.1). The NOX4 siRNAs is also a pool of three sequences of siRNA as follows: duplex 1 sense strand, 5′-CCA UUU GCA UCG AUA CUA ATT-3′; duplex 2 sense strand, 5′-CCA AGA CUC UUC AUA GUU UTT-3′; and duplex 3 sense strand, 5′-CAA GAC CUC UCU CCU UUG ATT-3′ (mRNA GenBank accession number NM_015760.4). All siRNA constructs were purchased from Santa Cruz Biotechnology. Briefly, cells were grown in 24-well plates or 60 mm dishes and transiently transfected with 100 nm NOX1, NOX4, and control siRNA constructs mixed with X-tremeGENE siRNA transfection reagent (Roche Applied Science) according to the protocol of the manufacturer. After incubation at 33°C and 5% CO2 for 36 h, cells were further treated with cisplatin for 24 h. Samples were then prepared and analyzed for viability or Western blot.
Subcellular fractionation and Western blot analysis.
The cells were washed twice with ice-cold PBS and homogenized in a lysis buffer (10 mm HEPES, 10 mm NaCl, 1 mm KH2PO4, 5 mm NaHCO3, 1 mm CaCl2, 0.5 mm MgCl2, 5 mm EDTA, 1 mm PMSF, 0.1% Triton X-100, and protease inhibitor cocktail). The lysates were centrifuged at 13,000 × g for 10 min at 4°C, and supernatant proteins were ultracentrifuged at 100,000 × g for 1 h at 4°C. The supernatant containing cytosolic proteins and pellet containing membrane protein were separated on 12% SDS-polyacrylamide gels for 3 h at 20 mA and then transferred onto nitrocellulose membrane. The membrane was incubated with antibody in 5% (wt/vol) dried milk protein in PBS containing 0.05% (v/v) Tween 20 (PBS-T) for 1 h, washed in PBS-T, and then further reacted with antibodies for p22phox, p47phox, p67phox, and pro-caspase-3 (1:1000) for 1 h. The membrane was extensively washed with PBS-T and then incubated with anti-rabbit IgG antibody conjugated to HRP (1:3000) for 1 h. After extensive washes, protein bands on the membrane were visualized using chemiluminescent reagents according to the instructions of the manufacturer (Supersignal Substrate; Pierce). For the p47phox phosphorylation assay, total lysate (800 μg) was precleared by incubating with protein G-agarose beads (Millipore Corporation) for 1 h at 4°C. After centrifugation, cleared lysates were incubated overnight with 1 μg of p47phox antibody and precipitated with protein G-agarose beads (Millipore Corporation) for 2 h at 4°C. For Western blot analysis, immunoprecipitated proteins were probed with antibodies against phosphotyrosine, phosphoserine/phosphothreonine, and p47phox.
Immunocytochemistry.
Cells were treated with 20 μm cisplatin for 18 h. After removal of the culture medium, cells were washed three times with PBS and fixed with 4% paraformaldehyde in PBS for 30 min at room temperature. Cells were then rinsed twice with PBS, preincubated in the same buffer containing 0.1% Triton X-100, and followed by three washes. Cells were blocked with 1% BSA in PBS for 30 min and then incubated with primary antibodies in PBS containing 1% BSA for 1 h at room temperature. After three washes with PBS, cells were incubated with Alexa Fluor 488 (for anti-NOX4 antibody) or 568 (for anti-NOX1 antibody) secondary antibodies at a dilution of 1:500 in PBS for 1 h at room temperature. Cells were then washed in PBS and further incubated with 0.5 μg/ml 4′,6′-diamidino-2-phenylindole for nuclear image at room temperature for 10 min. Cells were observed under fluorescent microscope equipped with digital camera (IX71; Olympus). The fluorescent images were captured using appropriate filters.
NADPH oxidase activity.
The activity of NADPH oxidase was determined in membrane fractions (50 μg of protein) incubated with 1 mm EGTA and 5 μm lucigenin in phosphate buffer, pH 7.0. The assay was initiated by the addition of 50 μm NADPH to the incubation mixture. Samples were counted immediately using a tabletop luminometer (Berthold Detection Systems FB Luminometer; Zylux) with sampling time every 6 s. Samples were counted over a period of 5 min, and the fluorescence values were recorded for over 2 min of stable readings and averaged for that sample.
Caspase activity assay.
Whole-cell lysate was prepared in a lysis buffer (1% Triton X-100, 0.32 m sucrose, 5 mm EDTA, 1 mm PMSF, 1 μg/ml leupeptin, 1 mm DTT, 10 mm Tris-HCl, pH 8.0) on ice for 30 min and centrifuged at 20,000 × g for 15 min. An equal amount of total protein was quantified by BCA protein quantification kit (Sigma) in each lysate. Catalytic activity of caspase-3 from cell lysate was measured by proteolytic cleavage of 100 μm 7-amino-4-methylcoumarin (AMC)-DEVD, a fluorogeic substate (Calbiochem), for 1 h and AMC as a negative standard in an assay buffer (100 mm HEPES, 10% sucrose, 0.1% 3-[(3-cholamidopropyl)dimethylammonio]-1-propanesulfonate, pH 7.5, 1 mm PMSF, 1 μg/ml aprotinin, 1 μg/ml leupeptin, and 1 mm DTT) at 380 nm of excitation wavelength and 460 nm of emission wavelength.
In vivo experiment of cisplatin ototoxicity.
Male BALB/c mice, weighing between 20 and 23 g at 7 weeks, were used in this study. The animals were fed a standard commercial diet and housed in a room at 20–22°C ambient temperature with a relative humidity of 50 ± 5% and 12 h light/dark cycle. The animals were randomly divided into three groups. Group 1 animals received intraperitoneal injection of PBS. Group 2 animals were intraperitoneally administered with cisplatin alone (4 mg/kg, body weight) for 4 d. To neutralize TNF-α pharmacologically, group 3 animals were intraperitoneally given etanercept (400 μg/kg, body weight; Wyeth Pharmaceuticals) at 12 h before injection of cisplatin. The animals in all groups were killed under anesthesia (CO2 gas) on the next day after final cisplatin injection, and the whole temporal bone was removed. The experimental protocol was approved by the Animal Care and Use Committee at the Wonkwang University School of Medicine.
Immunohistochemical studies and terminal deoxynucleotidyl transferase-mediated dUTP nick end-labeling assay.
The removed temporal bones were fixed and perfused with 4% paraformaldehyde for 16 h, decalcified with 10% EDTA in PBS for 2 weeks, dehydrated, and embedded in paraffin wax. Sections of 5 μm were deparaffinized in xylene and rehydrated through graded concentrations of ethanol. For immunohistochemical study, the kit (LSAB Universal K680; Dako) was used, and all the procedures were performed according to the instructions of the manufacturer. The endogenous peroxidase was blocked with 3% hydrogen peroxide for 5 min at room temperature. After sections were washed in PBS, nonspecific binding was blocked with 1% bovine serum albumin for 1 h. Then, primary antibody (1:200 diluted) was added to the slides and incubation proceeded for 1 h. After repeated washes with PBS, the section was incubated with biotinylated secondary antibody for 1 h and then covered for 15 min with streptavidin–peroxidase. Finally, after repeated washes with PBS, the section was stained in a freshly prepared substrate solution (3 mg of 3-amino-9-ethylcarbazole in 10 ml of sodium acetate buffer, pH 4.9, 500 μl of dimethylformamide, and 0.03% hydrogen peroxide) for 10 min. The nuclei of immunostained cells were counterstained with Mayer's hematoxylin (Sigma-Aldrich). Apoptotic cells were detected in situ using the terminal deoxynucleotidyl transferase-mediated dUTP nick end-labeling (TUNEL) assay (TUNEL POD kit; Roche Molecular Biochemicals). Briefly, a section was deparaffinized and rehydrated. After treatment with proteinase K (20 μg/ml in 10 mm Tris-HCl, pH 7.6) for 30 min, sections were washed in PBS, and the labeling reaction was performed using a labeling solution containing terminal deoxynucleotidyl transferase, its buffer, and fluorescein dUTP at 37°C for 60 min in a humidity chamber. The nuclei were counterstained with propidium iodide (0.5 μg/ml; Invitrogen) for 10 min at room temperature. After washing with PBS, it was examined under a fluorescence microscope.
Culture of the organ of Corti explants.
Sprague Dawley rats were killed on postnatal day 2 (P2), and the cochlea was carefully dissected out. The stria vascularis and spiral ligament were dissected away, leaving the organ of Corti. The middle turn of the cochlea was used for additional analysis. Cochlea explants were treated with high glucose (4.5 g/l) DMEM containing 10% FBS, 20 μm cisplatin, 100 nm DPI, and apocynin and further incubated at 37°C in 5% CO2 for 24 h. Control sample in DMEM containing only serum was run concurrently with the experimental samples. At the end of the experiments, the culture was prepared for histological analysis. The specimen was fixed for 15 min in 2% paraformaldehyde in 0.1 m phosphate buffer, pH 7.4, at room temperature. The specimen was rinsed in 0.1 m PBS, then incubated in 0.25% Triton X-100 for 2 min, and immersed in tetramethylrhodamine isothiocyanate (TRITC)-labeled phalloidin (P1951; 1:100; Sigma) in PBS for 20 min. After three washes with PBS, the specimen was examined under fluorescence microscope with appropriate filters for TRITC (excitation, 510–550 nm; emission, 590 nm).
RT-PCR and quantitative real-time PCR amplification.
Total RNA was extracted from whole cochlea of mice and HEI-OC1 cells with the use of TRIzol (Invitrogen) according to the protocol of the manufacturer. Single-stranded cDNA was synthesized from total RNA. Then, PCR using TaqDNA polymerase (Takara, Takara Shuzo) was performed for 30 cycles using the following protocol: 94°C for 30 s, 60°C for 30 s, and 72°C for 30 s. Then, 10 μl of the PCR products was separated on 1.2% agarose gel and visualized under UV light. The sequences of primers used for PCR amplification are as follows: NOX1 (forward, 5′-TGA ACA ACA GCA CTC ACC AAT GCC-3′; reverse, 5′-TCA TTG TCC CAC ATT GGT CTC CCA-3′); NOX3 (forward, 5′-TTG TGG CAC ACT TGT TCA ACC TGG-3′; reverse, 5′-TCA CAC GCA TAC AAG ACC ACA GGA-3′); NOX4 (forward, 5′-TCA TGG ATC TTT GCC TCG AGG GTT-3′; reverse, 5′-AGT GAC TCC TCA AAT GGG CTT CCA-3′); DUOX1 (forward, 5′-CAC CAT TGG GAC CCT TTG CTG TTT-3′; reverse, 5′-AGC CTT TCA TGA AGA CCA CCA GGA-3′); DUOX2 (forward, 5′-AAC CAC CTA TCT GGG CAT CAT CCT-3′; reverse, 5′-AGC TGC CAT GGA TGA TGA TCT GGA-3′); NOXO1 (forward, 5′-CCA TGC TGT AGC CTT GGT GCA AAT-3′; reverse, 5′-AAA CCA GGC TAC CTG CTG ATC CTT-3′); p22phox (forward, 5′-ATG GGG CAG ATC GAG TGG GCC ATG-3′; reverse, 5′-TCA CAC GAC CTC ATC TGT CAC TGG-3′); NOXA1 (forward, 5′-CAT TCC TGA TGA CCA CAA CG-3′; reverse, 5′-AGT CCA AAT CCT CCG GTC TT-3′); p47phox (forward, 5′-TAT CTG GAG CCC CTT GAC AGT CCC-3′; reverse, 5′-TCA CAC AGC GGA CGT CAG CTT CCG-3′); p67phox (forward, 5′-GTG CTC ATG GAG ACG CGG CTT GGC-3′; reverse, 5′-CTA GAC TTC TCT GGG AAT GCC TTC C-3′); and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (forward, 5′-GGG TGT GAA CCA CGA GAA AT-3′; reverse, 5′-GTC ATG AGC CCT TCC ACA AT-3′). Real-time PCR analysis using the TaqMan fluorescence method was performed for quantitative analysis of mRNAs. The products were amplified using LightCycler (Roche Diagnostics) in a reaction mixture (20 μl) containing 2 μl of LightCycler-FastStart DNA Master Taqman (Roche), 0.5 μm each primer, and 3 mm MgCl2. The forward, reverse, and probe oligonucleotide primers for multiplex real-time TaqMan PCR are as follows: NOX1 (forward, 5′-TGG CTA AAT CCC ATC CAG TC-3′; reverse, 5′-CCC AAG CTC TCC TCT GTT TG-3′); and NOX4 (forward, 5′-ACT TTT CAT TGG GCG TCC TC-3′; reverse, 5′-AGA ACT GGG TCC ACA GCA GA-3′). The copy numbers of mRNA were standardized by using β-actin.
Statistical analysis.
Each experiment was performed at least three times independently, and all values are represented as the means ± SD of triplicates. One-way ANOVA or post hoc test for different interaction within one test was used to analyze a statistical significance of the results. Values of p < 0.05 were considered as statistically significant.
Results
Cisplatin-induced cytotoxicity is associated with an increase in ROS production in HEI-OC1 auditory cells
First, to investigate the effect of cisplatin on intracellular ROS generation and cell viability, HEI-OC1 auditory cells were treated with 20 μm cisplatin for various periods. Cell viability was measured by MTT assay, and the level of intracellular ROS was monitored by DCFH-DA, which is a peroxide-sensitive fluorescent probe. As shown in Figure 1, cisplatin decreased the viability of cells in a time-dependent manner; however, cisplatin significantly increased the production of intracellular ROS in a time-dependent manner also. Therefore, to evaluate whether ROS generation in cisplatin-treated cells was mediated by NADPH oxidase pathway, cells were pretreated with 100 nm DPI or apocynin, which are inhibitors of NADPH oxidases for 30 min, and further incubated with 20 μm cisplatin for 24 h. We did not find any significant cytotoxic effect of DPI or apocynin alone even at 500 nm concentration (data not shown). However, both DPI and apocynin significantly protected HEI-OC1 auditory cells from cisplatin-induced cytotoxicity (Fig. 2A). In addition, both DPI and apocynin also significantly reduced ROS generation in cisplatin-treated cells (Fig. 2B). These inhibitory effects of NOX inhibitors on ROS generation were further confirmed by flow cytometry analysis using DCFH-DA staining. After 24 h of exposure to cisplatin, we observed a rightward shift of fluorescence signals in cisplatin-treated cells compared with control cells, whereas this rightward shift was completely abrogated by DPI or apocynin (Fig. 2C). These results suggest that NADPH oxidase pathways are closely involved in cisplatin-induced ROS generation in HEI-OC1 auditory cells.
Figure 1.

Cisplatin-induced cytotoxicity is associated with an increase in ROS production in HEI-OC1 auditory cells. Cells were treated with 20 μm cisplatin for various time periods. Cell viability was then measured by MTT assay. In addition, the level of intracellular ROS was also monitored at indicated time points using a peroxide-sensitive fluorescent probe, DCFH-DA. The data represent the mean ± SD of triplicate. *,#p < 0.05, **,##p < 0.01, by one-way ANOVA compared with cell viability (*, **) or ROS generation (#, ##) in zero time (0 h) control cells.
Figure 2.

Pharmacological inhibition of NADPH oxidase attenuated the cisplatin cytotoxicity and ROS generation in HEI-OC1 cells. Cells were pretreated with 100 nm DPI or apocynin, an inhibitor of NADPH oxidases, for 30 min and further incubated with 20 μm cisplatin for 24 h. A, Cell viability was then measured by MTT assay. B, After 24 h, the level of intracellular ROS was also monitored using a peroxide-sensitive fluorescent probe, DCFH-DA. The data represent the mean ± SD of three independent experiments. *p < 0.05, by one-way ANOVA compared with cisplatin-only treated cells. C, Intracellular ROS level was also measured by flow cytometry as described in Materials and Methods.
Various NOX isoforms are expressed in the cisplatin-treated HEI-OC1 auditory cells
NADPH oxidases have a number of closely related tissue-specific isoforms (Quinn et al., 2006). NADPH activity is regulated by the specific binding of NOX isoforms and NOX-related regulatory subunits. The unique binding partners of NOX1 are NOXO1 (p47phox homolog) and NOXA1 (p67phox homolog). However, the binding partners of NOX2 are p47phox and p67phox through p22phox. In addition, it has been reported that NOX3 can bind to NOXO1 (or p47phox) and NOXA1 (or p67phox), and NOX3 activity seems dependent on the presence of a specific combination of these organizers and activators. In contrast with NOX1–NOX3, NOX4 seems to function independently of additional cytosolic oxidase cofactors. Therefore, we investigated the mRNA expression of these NOX isoforms and NOX-related regulatory subunits in cisplatin-treated HEI-OC1 auditory cells. The results indicated in Figure 3A showed that treatment with cisplatin could induce an early transcriptional activation of various NOX isoforms and their regulatory subunit mRNAs, including NOX1, NOX4, p47phox, and p67phox at 1 h after treatment, whereas NOXO1, NOXA1, and DUOX1 mRNAs were comparably detectable at 6 h or later. However, p22phox mRNA was not changed by cisplatin exposure. Of note, NOX1, NOX4, NOXO1, and NOXA1 mRNAs were somewhat time dependently increased by cisplatin. Interestingly, NOX3 mRNA was not detected in HEI-OC1 auditory cells (data not shown). Next, to further confirm the expression and cellular localization of the different NOX isoforms in cisplatin-treated HEI-OC1 cells, immunocytochemistry analysis was performed with antibodies against NOX1 (red color; Alexa Fluor 568-labeled) and NOX4 (green color; Alexa Fluor 488-labeled). Especially because these two antibodies were indirectly tagged with two different fluorophores, respectively, we could also elucidate the colocalization of NOX isoforms. As shown in Figure 3B, neither NOX1 nor NOX4 was expressed in control HEI-OC1 auditory cells. However, after exposure to cisplatin, both NOX1 and NOX4 were obviously expressed on the cell membrane. Interesting, the NOX1 isoform displayed a clustered appearance evocative of proteinaceous-like aggregates in lipid raft as for signal transducing platforms, whereas the NOX4 isoform was evenly expressed throughout the cell membrane. Moreover, colocalization of NOX1 and NOX4 was also shown in some areas of cisplatin-treated cells. We also examined the total amount of cellular p22phox, p47phox, and p67phox proteins after cisplatin exposure by Western blotting. As shown in Figure 3C, the total amount of cellular p47phox and p67phox proteins were clearly increased by cisplatin treatment in a time-dependent manner, whereas that of p22phox was not changed by cisplatin exposure. Next, to examine whether the transcriptional activation of various NOX isoforms and their regulatory subunits by cisplatin resulted in the increase of NADPH oxidase activity, HEI-OC1 cells were treated with 20 μm cisplatin for various time periods in the absence or presence of the NOX inhibitors DPI and apocynin. As expected, exposure to cisplatin for 3 h or longer induced a significant increase of NADPH oxidase activity in isolated HEI-OC1 cell membranes, whereas this increased activity of NADPH oxidase induced by cisplatin was markedly abrogated by the NOX inhibitors DPI and apocynin (Fig. 3D).
Figure 3.

Various NOX isoforms are expressed in the cisplatin-treated HEI-OC1 auditory cells. A, Cells were treated with 20 μm cisplatin for the indicated time periods. Total RNA was then isolated by TRIzol, and cDNA was synthesized by reverse transcription. The cDNAs for various NOX isoforms were amplified using specific primer sets as described in Materials and Methods. B, Cells were treated with 20 μm cisplatin for 18 h. After reaction with anti-NOX1 and NOX4 antibodies as described in Materials and Methods, cells were visualized under fluorescent microscope. C, Cells were treated with 20 μm cisplatin for the indicated time periods and subjected to Western blotting with anti-p22phox, anti-p47phox, anti-p67phox, and anti-β-actin, respectively. D, Cells were treated with 20 μm cisplatin for the indicated time periods. Then, the activity of NADPH oxidase was determined using lucigenin as described in Materials and Methods. The data represent the mean ± SD of three independent experiments. *p < 0.05, by one-way ANOVA compared with zero time (0 h) control cells. #p < 0.05, by post hoc test compared with cisplatin-only group at each time point.
Inhibition of cisplatin-induced ROS production by NOX1 and NOX4 siRNAs transfection
Although DPI and apocynin were widely used in many literatures as NOX inhibitors, recently it has been reported that these two compounds are not specific inhibitors for NOXs (Aldieri et al., 2008). Therefore, we further examined the direct involvement of NOXs on cisplatin-induced ROS production by specific knockdown of NOX genes by siRNAs. To determine the roles of NOX1 and NOX4 in cisplatin-mediated ROS production, we transfected HEI-OC1 cells with NOX1/NOX4-specific siRNA constructs or a scrambled control siRNA. We confirmed that transfection of NOX1 or NOX4 siRNAs curtailed the expression of their respective mRNA without influencing GAPDH mRNA levels (Fig. 4A). However, transfection with control siRNA did not affect the cisplatin-induced NOX1 and NOX4 mRNA expression. In addition, protein expression of NOX1 or NOX4 after cisplatin exposure was obviously alleviated by the transfection of NOX1 or NOX4 siRNAs but not by scrambled control siRNAs, respectively (Fig. 4A). HEI-OC1 cells transfected with NOX1 or NOX4 siRNAs showed a significantly lower generation of ROS compared with cisplatin-treated or cisplatin/control siRNA transfected cells (Fig. 4B). Flow cytometry analysis also showed that a rightward shift of fluorescence signals in cisplatin-treated cells was markedly blocked by NOX1 or NOX4 siRNAs but not by control siRNA (Fig. 4C). These results suggest that both NOX1 and NOX4 are directly linked in cisplatin-induced ROS generation in HEI-OC1 auditory cells.
Figure 4.

Knockdown of NOX1 and NOX4 by siRNAs transfection inhibited cisplatin-induced ROS production. Cells were transfected with 100 nm of NOX1, NOX4, and scrambled control siRNAs, incubated for 36 h, and further treated with 20 μm cisplatin for 18 h (for RT-PCR) or 24 h (for ROS generation and flow cytometry analysis), respectively. A, Then, RT-PCR was performed to amplify NOX1, NOX4, and GAPDH genes. In addition, total cell lysates were subjected to 12% SDS-PAGE and immunoblotted with NOX1- and NOX4-specific antibody. W.B., Western blot. B, The level of intracellular ROS was also monitored using a peroxide-sensitive fluorescent probe, DCFH-DA. The data represent the mean ± SD of three independent experiments. *p < 0.05, by one-way ANOVA compared with cisplatin-only treated cells. C, Intracellular ROS level was also measure by flow cytometry as described in Materials and Methods.
Inhibition of cisplatin-induced cytotoxicity and caspase-3 activation by NOX 1 and NOX4 siRNAs transfection
To further evaluate whether ROS generation via the NOX1 and NOX4 pathway was critical for the cisplatin-mediated apoptotic death of HEI-OC1 auditory cells, we examined the effects of NOX1 and NOX4 siRNAs transfection on cisplatin-induced cytotoxicity and caspase-3 activation. As shown in Figure 5A, transfection of either NOX1 or NOX4 siRNAs but not unrelated control siRNAs resulted in a substantial protection against cisplatin cytotoxicity. In addition, both cisplatin-induced caspase-3 enzymatic activation (Fig. 5B) and pro-caspase-3 degradation (Fig. 5C) were markedly decreased by the transfection of either NOX1 or NOX4 siRNAs.
Figure 5.
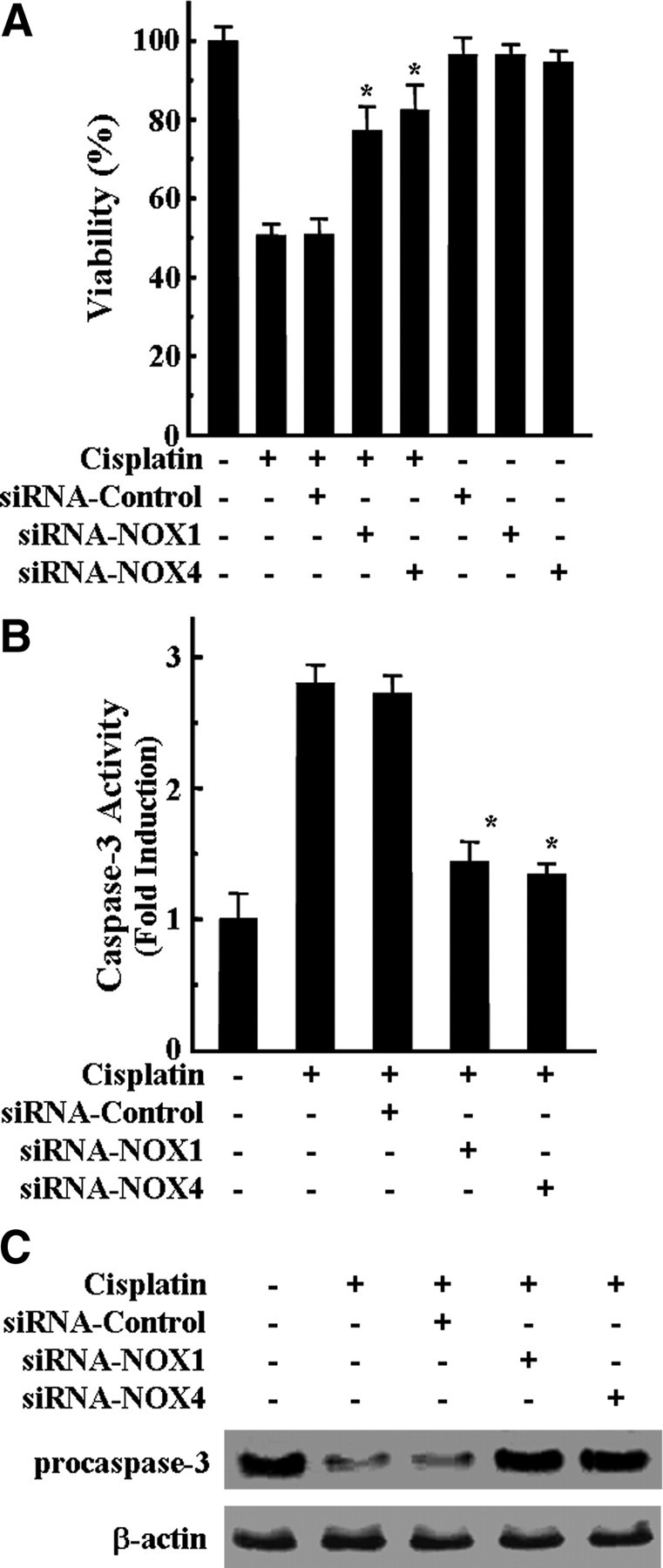
Knockdown of NOX1 and NOX4 by siRNAs transfection inhibited cisplatin-mediated cytotoxicity and caspase-3 activation in HEI-OC1 cells. Cells were transfected with 100 nm NOX1, NOX4, and scrambled control siRNAs, incubated for 36 h, and further treated with 20 μm cisplatin for 24 h. A, Cell viability was measured by MTT assay. The data represent the mean ± SD of three independent experiments. *p < 0.05, by one-way ANOVA compared with cisplatin-only treated cells. B, The activity of caspase-3 was measured by determining the cleaved fluorogenic substrate AMC-DEVD. The data represent the means ± SD of three independent experiments. *p < 0.05, by one-way ANOVA analysis compared with cells treated with cisplatin only. C, Total amount of pro-caspase-3 was determined by Western blotting analysis.
Effect of pro-inflammatory cytokines on cisplatin-induced NADPH oxidase mRNA expression
We have demonstrated previously that pro-inflammatory cytokines play a critical role in cisplatin-induced cochlear injury (So et al., 2007, 2008). TNF-α especially played an important role in this process because the inhibition of TNF-α action significantly attenuated the expression of other pro-inflammatory cytokines after cisplatin injection. Therefore, we assessed the influences of pro-inflammatory cytokine productions by cisplatin on NOX1 and NOX4 mRNA expression. Endogenously produced pro-inflammatory cytokines were neutralized by pretreatment of cell cultures with anti-TNF-α (10 ng/ml), anti-IL-1β (10 ng/ml), and anti-IL-6 (10 ng/ml) antibodies for 30 min before cisplatin exposure. As shown in Figure 6A, each antibody alone successfully inhibited cisplatin-induced NOX1 and NOX4 mRNA expression. Consistent with the attenuation effect of neutralizing antibodies on cisplatin-induced NOX1 and NOX4 mRNA expression, the enzymatic activities of NADPH oxidase were significantly decreased by neutralizing antibodies (Fig. 6B).
Figure 6.
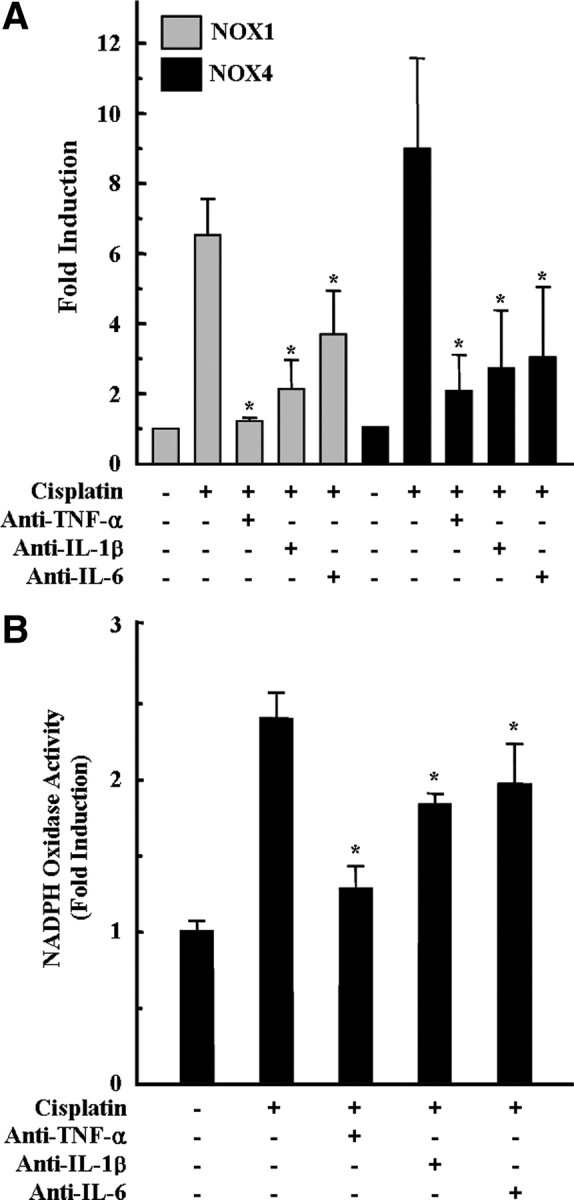
Neutralization of pro-inflammatory cytokines attenuated cisplatin-induced NOX 1 and NOX4 mRNA expressions and NADPH oxidase activity in HEI-OC1 cells. Cells were pretreated with neutralizing antibodies, including anti-TNF-α (10 ng/ml), anti-IL1β (10 ng/ml), and anti-IL-6 (10 ng/ml), for 30 min, and further incubated with 20 μm cisplatin for 6 h (for NADPH oxidase activity) or 18 h (real-time PCR), respectively. A, Then, total RNA was isolated by TRIzol, and cDNA was synthesized by reverse transcription. NOX1, NOX4, and β-actin cDNAs were amplified by quantitative real-time PCR using specific primer sets. The relative copy numbers of mRNA were standardized by those of β-actin. B, The activity of NADPH oxidase was determined using lucigenin as described in Materials and Methods. The data represent the mean ± SD of three independent experiments. *p < 0.05, by one-way ANOVA compared with the cisplatin-only group.
Effect of MAPKs on cisplatin-induced NADPH oxidase mRNA expressions
In our previous report, we demonstrated that cisplatin induced the activations of the MAPKs extracellular signal-regulated kinase (ERK), c-Jun N-terminal protein kinase (JNK), and p38 pathways, whereas only the pharmacologic inhibition of MEK1/ERK activity rescued HEI-OC1 cells from cisplatin cytotoxicity (So et al., 2007). Thus, we examined the effect of MAPK inhibitors on cisplatin-mediated NOX1 and NOX4 mRNA expression. As shown in Figure 7A, pharmacological inhibition of p38 and JNK with SB203580 or SP600125, respectively, did not affect the mRNA expression of NOX4 in cisplatin-treated cells. However, inhibition of JNK by SP600125 did slightly and significantly attenuated cisplatin-mediated NOX1 mRNA expression. On the contrary, pretreatment with the MEK1/ERK inhibitor U0126 blocked cisplatin-induced NOX1 and NOX4 mRNA expression (Fig. 7A). Consistent with the attenuation effect of the MEK1/ERK inhibitor U0126 on cisplatin-induced NOX1 and NOX4 mRNA expression, the enzymatic activity of NADPH oxidase was significantly decreased by MEK1/ERK inhibitor U0126 (Fig. 7B).
Figure 7.
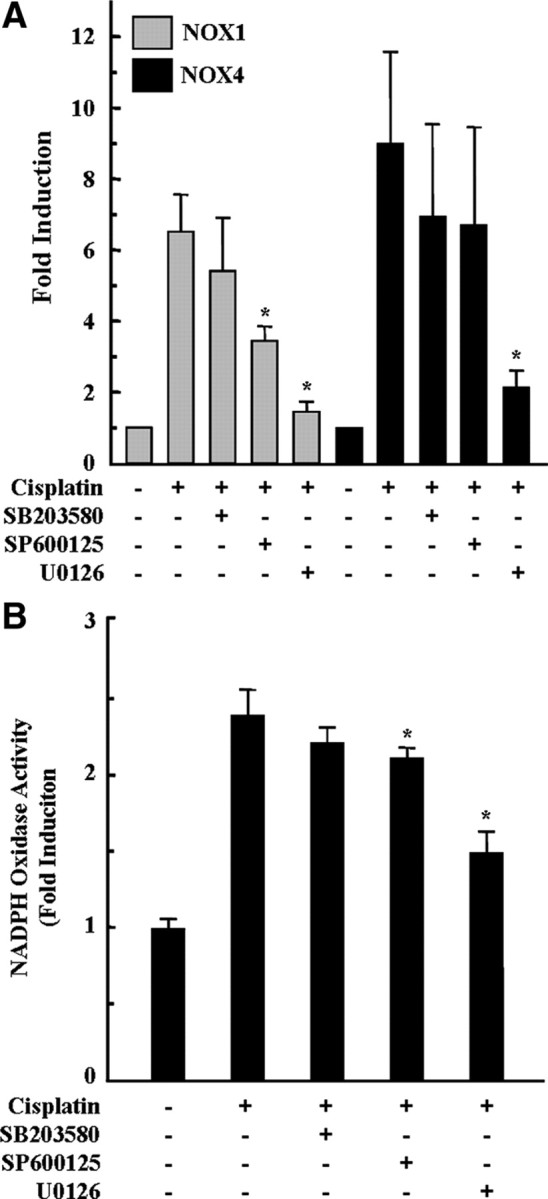
Inhibition of MAPKs, especially ERK, attenuated cisplatin-induced NOX1 and NOX4 mRNA expression and NADPH oxidase activity in HEI-OC1 cells. Cells were pretreated with pharmacological inhibitors of MAPKs for 30 min and then further treated with 20 μm cisplatin for 6 h (for NADPH oxidase activity) or 18 h (real-time PCR), respectively. A, Then, total RNA was isolated by TRIzol, and cDNA was synthesized by reverse transcription. NOX1, NOX4, and β-actin cDNAs were amplified by quantitative real-time PCR using specific primer sets. The relative copy numbers of mRNA were standardized by those of β-actin. B, The activity of NADPH oxidase was determined using lucigenin as described in Materials and Methods. The data represent the mean ± SD of three independent experiments. *p < 0.05, by one-way ANOVA compared with the cisplatin-only group.
Effect of pro-inflammatory cytokines on cisplatin-induced p47phox phosphorylation and membrane translocations of p47phox and p67phox
To characterize the mechanisms by which cisplatin-induced pro-inflammatory cytokines activate NADPH oxidase activity, we examined the phosphorylation of the cytosolic p47phox subunit of NADPH oxidase, which is a requisite step in activation of this complex and thereby induces membrane translocation from cytosol. HEI-OC1 cells were pretreated with anti-TNF-α (10 ng/ml), anti-IL-1β (10 ng/ml), and anti-IL-6 (10 ng/ml) antibodies for 30 min, followed by cisplatin treatment. Phosphorylation of p47phox was determined by immunoprecipitation and Western blotting for phosphoserine and phosphothreonine. Cisplatin obviously increased serine/threonine phosphorylation of p47phox when compared with untreated cells (Fig. 8A,B). However, neutralization of pro-inflammatory cytokine by each specific antibody successfully inhibited cisplatin-induced serine/threonine phosphorylation of p47phox. Next, we investigated whether neutralization of pro-inflammatory cytokines modulated the membrane translocation of p47phox and p67phox in cisplatin-treated HEI-OC1 cells. Cisplatin alone increased the translocations of p47phox and p67phox to the total membrane fraction, whereas it decreased the cytoplasmic amounts of p47phox and p67phox (Fig. 8C). However, neutralization of pro-inflammatory cytokines, especially TNF-α, clearly attenuated the cisplatin-induced membrane translocations of p47phox and p67phox from cytosol. The membrane-associated NADPH oxidase component p22phox and β-actin were used as a loading control for membrane fraction or cytosolic fraction, respectively (Fig. 8C).
Figure 8.
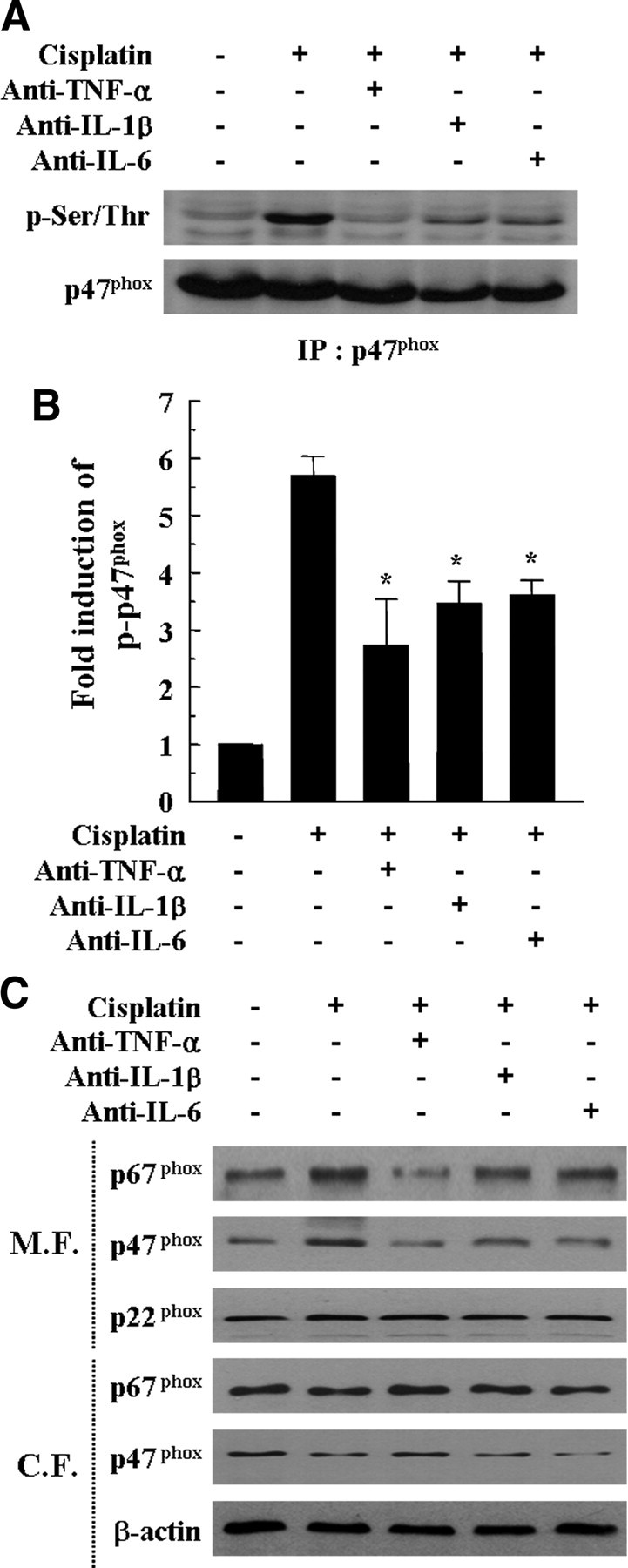
Neutralization of pro-inflammatory cytokines attenuated cisplatin-induced p47phox phosphorylation and membrane translocations of p47phox and p67phox. Cells were pretreated with neutralizing antibodies, including anti-TNF-α (10 ng/ml), anti-IL-1β (10 ng/ml), and anti-IL-6 (10 ng/ml) for 30 min and further incubated with 20 μm cisplatin for 6 h. A, Total cell lysates were subjected to 12% SDS-PAGE and immunoblotted with phosphorylated serine/threonine-specific antibody and p47phox-specific antibody. IP, Immunoprecipitation. B, The band intensities of phosphorylated serine/threonine p47phox and total p47phox were scanned using Scan software (Image Pro Plus 4.5). The relative expression level of the phosphorylated serine/threonine p47phox was quantified by normalization to the level of total p47phox. The data represent the mean ± SD of three independent experiments. *p < 0.05, by one-way ANOVA compared with the cisplatin-only group. C, Cytosolic and membrane fractions were prepared as described in Materials and Methods. These fractions were subjected to 12% SDS-PAGE and immunoblotted with antibodies specific for p22phox, p47phox, p67phox, and β-actin. M.F., Membrane fraction; C.F., cytosolic fraction.
Effect of MAPK inhibitors on cisplatin-induced p47phox phosphorylation and membrane translocations of p47phox and p67phox
Next, we evaluated the effect of MAPK inhibitors on cisplatin-induced p47phox phosphorylation and membrane translocations of p47phox and p67phox. As shown in Figure 9, A and B, pharmacological inhibition of p38 and JNK with SB203580 or SP600125, respectively, did not attenuate the phosphorylation of p47phox in cisplatin-treated cells. However, the MEK1/ERK inhibitor U0126 blocked cisplatin-induced p47phox phosphorylation (Fig. 9A,B). The MEK1/ERK inhibitor U0126 also clearly attenuated the cisplatin-induced membrane translocations of p47phox and p67phox from the cytosol, whereas the p38 inhibitor SB203580 or the JNK inhibitor SP600125 attenuated a small amount or very little cisplatin-induced membrane translocations of p47phox and p67phox from cytosol, respectively (Fig. 9C).
Figure 9.
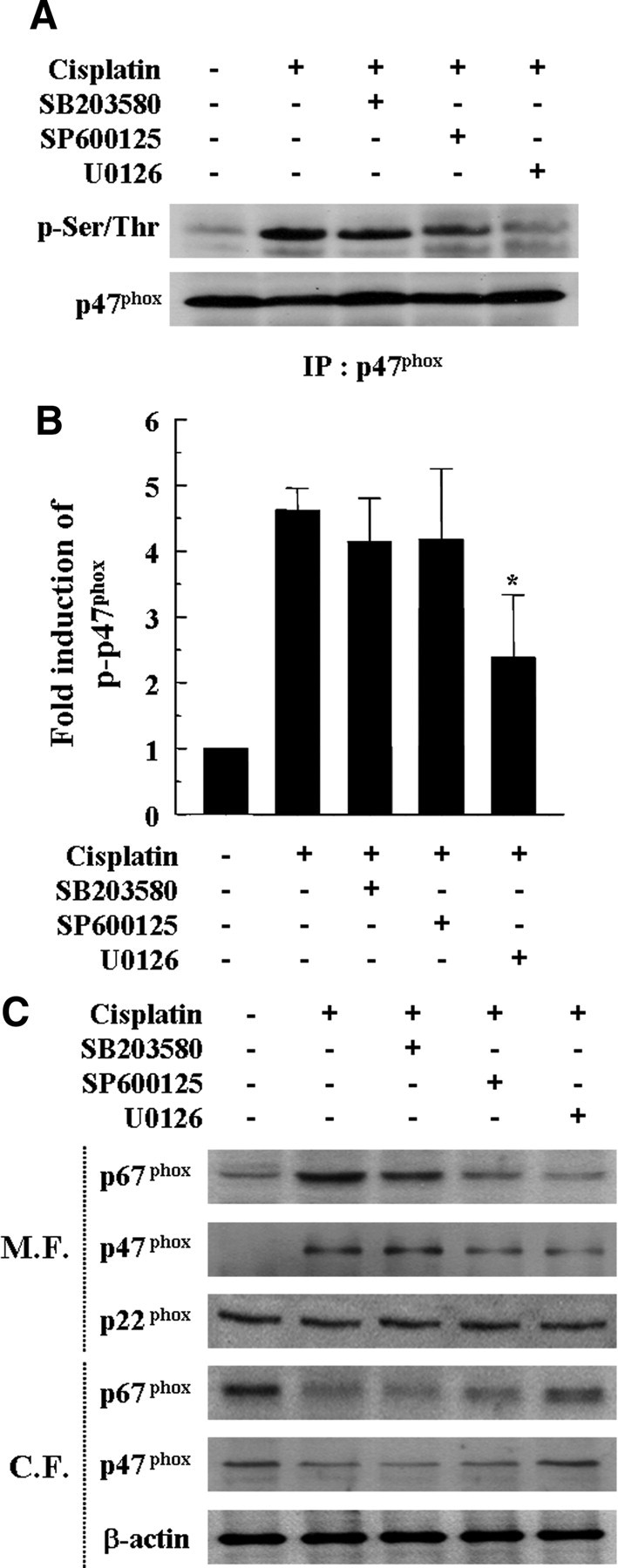
Inhibition of MAPKs, especially ERK, attenuated cisplatin-induced p47phox phosphorylation and membrane translocations of p47phox and p67phox. Cells were pretreated with pharmacological inhibitors of MAPKs for 30 min and then further treated with 20 μm cisplatin for 6 h. A, Total cell lysates were subjected to 12% SDS-PAGE and immunoblotted with phosphorylated serine/threonine-specific antibody and p47phox-specific antibody. IP, Immunoprecipitation. B, The band intensities of phosphorylated serine/threonine p47phox and total p47phox were scanned using Scan software (Image Pro Plus 4.5). The relative expression level of the phosphorylated serine/threonine p47phox was quantified by normalization to the level of total p47phox. The data represent the mean ± SD of three independent experiments. *p < 0.05, by one-way ANOVA compared with the cisplatin-only group. C, Cytosolic and membrane fractions were prepared as described in Materials and Methods. These fractions were subjected to 12% SDS-PAGE and immunoblotted with antibodies specific for p22phox, p47phox, p67phox, and β-actin. M.F., Membrane fraction; C.F., cytosolic fraction.
Protective effect of NADPH oxidase inhibitors on the organ of Corti explant from cisplatin
To examine whether inhibitors of NADPH oxidase, DPI and apocynin, also protect primary organ of Corti explant, the half middle turn of the organ of Corti from P2 Sprague Dawley rat was isolated and treated with 20 μm cisplatin in the presence or absence of 100 nm DPI or apocynin for 24 h and then labeled with TRITC-conjugated phalloidin. As shown in Figure 10A, treatment of media alone did not induce apparent damages on stereocilia bundles. Three rows of OHCs and a single row of inner hair cells (IHCs) were clearly observed in this experiment group. Treatment with cisplatin markedly destroyed the most of stereocilia bundles of hair cells and resulted in the disarray of the stereocilia (Fig. 10B). However, pretreatment of DPI or apocynin apparently provided complete protection against cisplatin-induced loss of stereocilia in P2 rat primary organ of Corti explant (Fig. 10C, D).
Figure 10.

Inhibitors of NADPH oxidase, DPI and apocynin, protected primary organ of Corti explant from cisplatin cytotoxicity. The organ of Corti explant from primary P2 rat was treated with media alone (A), 20 μm cisplatin alone (B), 100 nm DPI plus cisplatin (C), or 100 nm apocynin plus cisplatin (D) for 24 h. The organ of Corti explant was stained with TRITC-conjugated phalloidin and then observed under fluorescent microscope.
Intraperitoneal injection of cisplatin increased the expressions of various NOX isoforms in the cochleae
To investigate the localization and expression profiles of NOX isoforms and their regulatory subunits in the cochleae after cisplatin injection, mice were given intraperitoneal injections of cisplatin for 4 consecutive days (4 mg/kg body weight per injection). We also investigated the effect of the therapeutic neutralization of TNF-α by using etanercept, a TNF-α inhibitor, which has been used successfully to neutralize mouse TNF-α in vivo (Satoh et al., 2002). We first examined mRNA levels of NOX isoforms and their regulatory subunits in whole cochleae using RT-PCR. The total RNAs of whole cochleae from each experiment group were prepared and used for RT-PCR. As shown in Figure 11, the mRNA levels of NOX isoforms and their regulatory subunits were increased after cisplatin injection compared with PBS control mice. Although NOX3 mRNA was not detected in cisplatin-treated HEI-OC1 cells, its expression was obviously detected in control mice and markedly increased in whole cochleae from cisplatin-injected mice. In addition, the simultaneous injection of etanercept and cisplatin markedly blocked the expression of NOX subunit mRNAs. Next, immunohistochemistry was performed to investigate the localization and expression profiles of NOX1 and NOX4 proteins in the cochleae after cisplatin injection. In PBS control mice, the staining of NOX1 proteins was barely detectable except weakly stained spiral prominence regions (Fig. 12A–E). However, after injection with cisplatin, NOX1 staining was prominently observed throughout spiral ligament (Fig. 12F,H), spiral limbus and spiral ganglion neurons (Fig. 12F,J), as well as OHCs and IHCs in the organ of Corti (Fig. 12I). NOX4 was also mostly expressed throughout the spiral ligament (Fig. 13F–H), spiral limbus and spiral ganglion neurons (Fig. 13F,J), as well as OHCs and IHCs in the organ of Corti from cisplatin-injected mice (Fig. 13I). Of note, NOX1 and NOX4 protein expression were barely detected in the stria vascularis of cisplatin-injected mice, respectively (Figs. 12G, 13G). We also examined the in vivo effect of etanercept on cisplatin-induced NOXs expression and cochlear damages. In parallel with the attenuation effect of etanercept on cisplatin-induced NOX mRNAs expression, etanercept also markedly blocked the protein expressions of NOX1 and NOX4 in the cochlea (Fig. 14). In addition, no TUNEL-positive cells were observed in cochleae from PBS control (Fig. 15A) or etanercept/cisplatin-coinjected mice (Fig. 15C). However, histological sections from cisplatin-only treated mice exhibited TUNEL-positive cells in the stria vascularis, spiral ligament, spiral limbus, and the organ of Corti (Fig. 15B).
Figure 11.
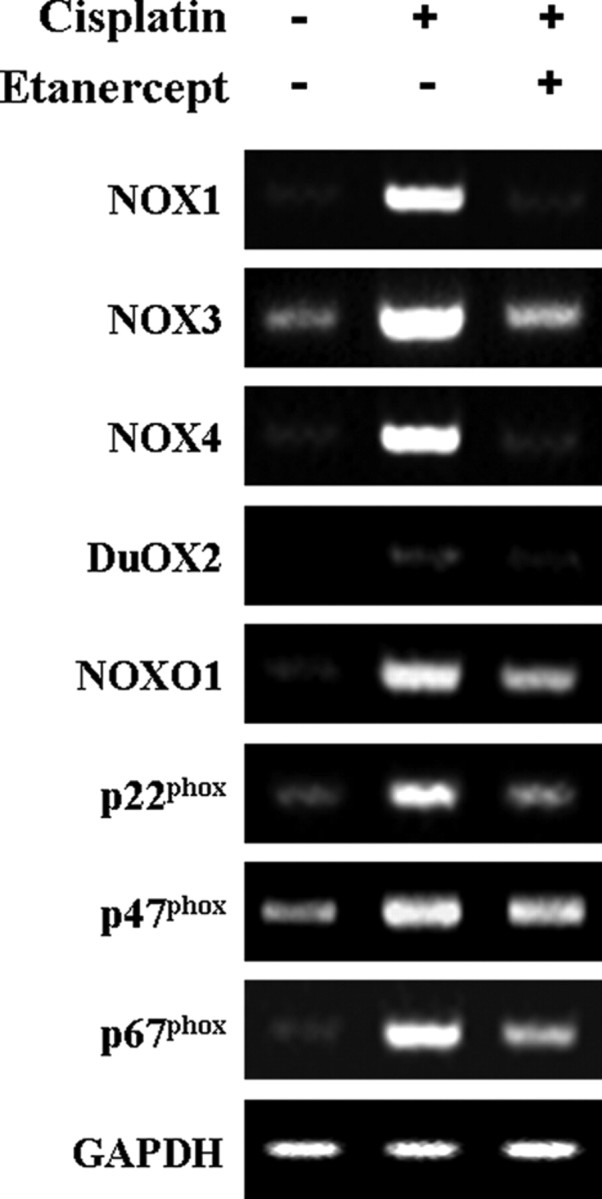
Intraperitoneal injection of cisplatin increased the mRNA expressions of various NOX isoforms and their regulatory subunits in the cochleae of BALB/c mice. Total RNA was then isolated from whole cochleae of each experiment group as described in Materials and Methods, and cDNA was synthesized by reverse transcriptase. NOX1, NOX3, NO4, DuOX2, NOXO1, p22phox, p47phox, p67phox, and GAPDH cDNA sequences were amplified using specific mouse primer sets.
Figure 12.
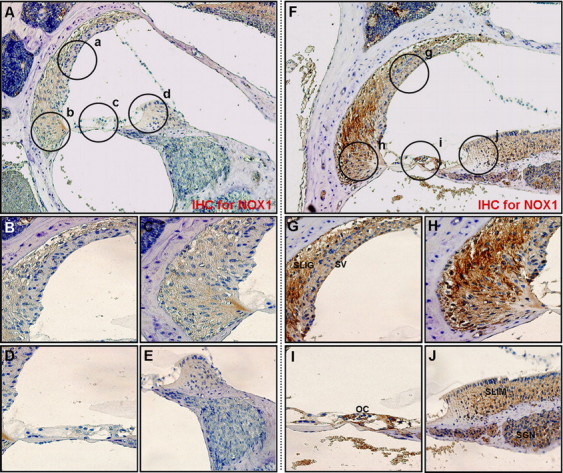
Intraperitoneal injection of cisplatin increased the protein expressions of NOX1 in the cochleae of BALB/c mice. Cochleae from BALB/c mice injected with PBS or cisplatin were removed and embedded in paraffin, and then 5 μm sections were prepared. For immunohistochemistry studies, an immunohistochemistry kit (LSAB Universal K680; Dako) was used, and all the procedures were performed according to the instructions of the manufacturer. The anti-NOX1 was used. A, Whole cochlea from PBS-injected control mice. B–E, Enlarged images of circled part of A. F, Whole cochlea from cisplatin-injected mice. G–J, Enlarged images of circled part of F. SLig, Spiral ligament; SV, stria vascularis; SGN, spiral ganglion neuron; SLim, spiral limbus; OC, organ of Corti.
Figure 13.
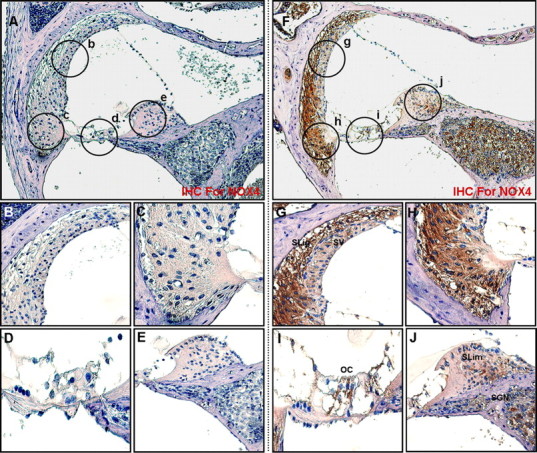
Intraperitoneal injection of cisplatin increased the protein expression of NOX4 in the cochleae of BALB/c mice. Cochleae from BALB/c mice injected with PBS or cisplatin were removed and embedded in paraffin, and then 5 μm sections were prepared. For immunohistochemistry studies, an immunohistochemistry kit (LSAB Universal K680; Dako) was used, and all the procedures were performed according to the instructions of the manufacturer. The anti-NOX4 was used. A, Whole cochlea from PBS-injected control mice. B–E, Enlarged images of circled part of A. F, Whole cochlea from cisplatin-injected mice. G–J, Enlarged images of circled part of F. SLig, Spiral ligament; SV, stria vascularis; SGN, spiral ganglion neuron; SLim, spiral limbus; OC, organ of Corti.
Figure 14.
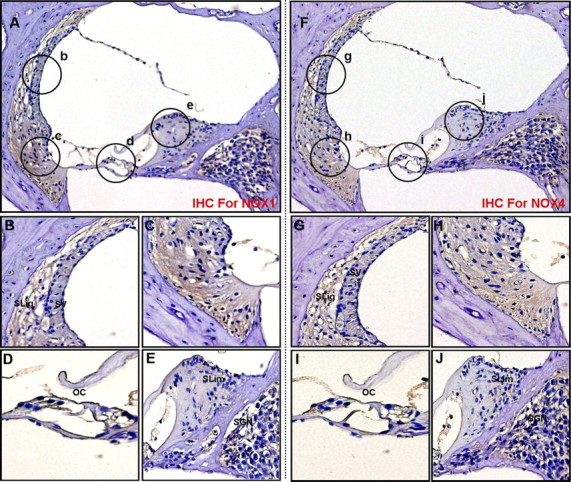
Etanercept obviously attenuated cisplatin-induced protein expressions of NOX1 and NOX4 in the cochleae of BALB/c mice. Cochleae from BALB/c mice injected with cisplatin and etanercept were removed and embedded in paraffin, and then 5 μm sections were prepared. For immunohistochemistry studies, an immunohistochemistry kit (LSAB Universal K680; Dako) was used, and all the procedures were performed according to the instructions of the manufacturer. The anti-NOX1 (A–E) or anti-NOX4 (F–J) was used. A, Whole cochlea; B–E, Enlarged images of circled part of A. F, Whole cochlea. G–J, Enlarged images of circled part of F. SLig, Spiral ligament; SV, stria vascularis; SGN, spiral ganglion neuron; SLim, spiral limbus; OC, organ of Corti.
Figure 15.

Etanercept markedly blocked the cisplatin-induced cochlear damages. Cochleae from BALB/c mice injected with PBS control (A), cisplatin (B), or cisplatin and etanercept (C) were removed and embedded in paraffin. Five micrometer cochlear sections from each experimental group were stained with TUNEL as described in Materials and Methods and then visualized under fluorescent microscope. SLig, Spiral ligament; SV, stria vascularis; SGN, spiral ganglion neuron; SLim, spiral limbus; OC, organ of Corti.
Discussion
Cisplatin is widely used as a chemotherapeutic agent for the treatment of broad spectrums of tumors (Fram, 1992). However, the use of this agent is limited by profound nephrotoxicity and ototoxicity (Humes, 1999). Cisplatin ototoxicity is manifested by severe sensorineural hearing loss after high doses have been administered in cancer chemotherapy (McKeage, 1995). Damage to inner ear structures includes apoptosis of the outer hair cells (Alam et al., 2000), spiral ganglion neuron cells (Lee et al., 2003), and the marginal cells of the stria vascularis (Lee et al., 2004). There are a considerable number of studies that look at the pathological role of excessive ROS production in the inner ear (Henderson et al., 1999; Kopke et al., 1999). Ototoxic drugs such as cisplatin and aminoglycoside antibiotics lead to accumulation of ROS in both the cochlea (Clerici et al., 1996) and the vestibular system (Takumida and Anniko, 2002; Darlington and Smith, 2003; Sergi et al., 2003). Noise trauma has been also demonstrated to be a prominent cause of ROS production in the cochlea (Ohlemiller et al., 1999). A sustained increase of ROS concentration leads primarily to the death of sensory epithelial cells and to a lesser extent the death of innervating neurons (Bánfi et al., 2004). It would be very interesting to elucidate the sources of ROS generated from tissues or cells exposed to a variety of deleterious stimuli. One of the well recognized mechanisms responsible for the generation of oxidative radicals is the disturbance of mitochondrial electron transport chain system, which was accompanied by the mitochondrial membrane potential loss as an indicator of mitochondrial dysfunction. Interestingly, mitochondria themselves are particularly vulnerable to oxidative stress. Oxidative damage to mitochondria causes the impairment of mitochondrial function and subsequently leads to cell death via apoptosis and necrosis (Bai and Cederbaum, 2001). Therefore, ROS-mediated oxidative damage to mitochondria favors more ROS generation, resulting in a vicious cycle. In our previous report, we already demonstrated that cisplatin induced mitochondrial membrane potential loss of HEI-OC1 cells accompanied with cytosolic cytochrome c release from mitochondria (So et al., 2005). These results suggest that mitochondria are the important sources for ROS in cisplatin-mediated auditory cell damages. However, we cannot exclude the non-mitochondrial sources for ROS in cisplatin-mediated auditory cell damages either. Recently, Paffenholz et al. (2004) also suggest a physiological role for ROS in the inner ear by reporting that het (head tilt) mice showed impaired balance and the lack of otoconia formation in the utricle and saccule attributable to the mutations of NOX3 gene, which is one of the non-mitochondrial ROS production pathways (Paffenholz et al., 2004). Interestingly, the het mice did not show any gross hearing deficits, implicating no overt physiological role of NOX3 in hearing. However, a pathological role for NOX3 is possible in the cochlea as well as in the vestibular system. Bánfi et al. (2004) demonstrated that cisplatin treatment is associated with increased NADPH oxidase activity primarily from NOX3 in the cochlea (Bánfi et al., 2004). They suggested that induction of NOX3 could serve as a major source of ROS generation in the cochlea and form part of the pathway leading to cisplatin-mediated damage. However, although they observed the localization of NOX3 mRNA in the inner ear by in situ hybridization, they did not demonstrate ROS generation in the cochlea or in vitro in auditory cells after cisplatin exposure, whereas they only observed ROS generation by cisplatin in NOX3-transfected human embryonic kidney 293 cells. Therefore, the expression and the role of the NADPH oxidase isoforms, including NOX3 in ROS generation and cytotoxicity in cisplatin-exposed cochlea or auditory cells, have yet to still be elucidated.
We report here for the first time that NOX1 and NOX4 protein plays important roles in the overproduction of ROS and subsequent ototoxic damage attributable to cisplatin. In the present study, treatment with cisplatin induced the expression of NADPH oxidase isoforms, including NOX1 and NOX4 in vitro and in vivo (Figs. 4, 11–13). Of note, protein expression of both NOX1 and NOX4 in the cochlear tissues, such as spiral ligament, organ of Corti, spiral limbus, and spiral ganglion neuron, were clearly induced after in vivo injection of cisplatin (Figs. 12, 13). In our in vitro study, inhibition of NADPH oxidase activity with apocynin or DPI abolished ROS production and subsequent apoptotic cell death in cisplatin-treated HEI-OC1 cells (Fig. 2) and organ of Corti explants (Fig. 10), which suggests that the intracellular ROS generation by NADPH oxidase activations was directly involved in the cytotoxic mechanism of cisplatin. However, apocynin or DPI did not further enhance survival or decrease ROS level of cisplatin-treated cells in the absence of NOX1 or NOX4 by siRNA transfection (data not shown). We also confirmed that NOX1 and NOX4 expression played a critically essential role in cisplatin-mediated ROS generation and cytotoxicity by knockdown experiments using specific siRNA transfections (Figs. 4, 5). In our previous study, we demonstrated that cisplatin increased the early immediate release and de novo synthesis of pro-inflammatory cytokines, including TNF-α, IL-1β, and IL-6, through the activation of MEK1/ERK in HEI-OC1 cells. Neutralization of pro-inflammatory cytokines significantly attenuated the death of HEI-OC1 auditory cells caused by cisplatin. Pharmacologic inhibition of MEK1/ERK significantly attenuated the cisplatin-induced cytotoxicity and pro-inflammatory cytokine production. Furthermore, exogenous treatment with these pro-inflammatory cytokines induced intracellular ROS generation in HEI-OC1 cells. Therefore, we suggest that the cisplatin-mediated secretion of pro-inflammatory cytokines is an upstream signal of ROS production. It would also be interesting to elucidate whether pro-inflammatory cytokine and MAPKs are sequentially involved in cisplatin-induced NADPH oxidase activation. In the present study, we demonstrated that cisplatin-induced ERK activation and pro-inflammatory cytokine production were directly involved in the activation of NADPH oxidases (Figs. 6, 7). Of note, TNF-α and ERK activation were involved in the increase of NADPH oxidase activity through NOX1 and NOX4 mRNA expression and NOX subunit p47phox and p67phox protein expression. TNF-α and ERK activation also induced phosphorylation of p47phox and thereby its translocation into the membrane (Fig. 8). Consistent with our results, several studies demonstrated that the phosphorylation of serine 345 in p47phox is mediated by ERK1/2 (Hazan-Halevy et al., 2005; Dang et al., 2006). In the case of p67phox, Serù et al. (2004) also reported that NADPH oxidase activation was induced by the ERK1/2-mediated serine/threonine phosphorylation of p67phox in a human neuroblastoma cells. However, we did not observe the serine/threonine phosphorylation of p67phox in cisplatin-treated HEI-OC1 cells, whereas we observed the tyrosine phosphorylation of p67phox (data not shown). Interestingly, this tyrosine phosphorylation of p67phox has not been reported yet. Considering these results, we suggest that ERKs regulate NADPH oxidase activation through the phosphorylation of their regulatory subunit component as well as their transcriptional activation in cisplatin-mediated auditory cell death.
NADPH oxidase is ubiquitously expressed in a wide variety of cells and organ systems, including kidney, spleen, sperm, mammary glands, and brain (Lambeth, 2004). In our experiments, we demonstrated that the protein expressions of NOX1 and NOX4 were prominently observed throughout spiral ligament, spiral limbus, spiral ganglion neurons, as well as OHCs and IHCs in the organ of Corti. Interestingly, when we compared the localizations of pro-inflammatory cytokine from our previous report (So et al., 2008) and NOX subunit expressions in cisplatin-injected mice, it showed very similar localization patterns of these components except stria vascularis. In stria vascularis, NOX1 and NOX4 protein expression were barely detected in cisplatin-injected mice, whereas pro-inflammatory cytokines were highly expressed (So et al., 2008). However, consistent with our previous report (So et al., 2008), TUNEL-positive cells were obviously observed in the spiral ligament, spiral limbus, the organ of Corti, and even stria vascularis of cisplatin-injected mice (Fig. 15B). Therefore, these results suggest that, although NADPH oxidases, including NOX1 and NOX4, are mainly involved in the cisplatin-mediated auditory cell death of cochlea, other unknown components except NOX 1 or NOX4 are also involved in cisplatin-mediated apoptotic death of stria vascularis. Furthermore, in vivo etanercept injection obviously attenuated the NOX1 and NOX4 protein expressions by cisplatin. Consistent with our results, Pan et al. (2009) recently reported that expression of TNF-α and IL-1β was markedly increased in cisplatin-mediated renal injury, and NOX1 and NOX4 were also critically involved in this injury through ROS production. Considering that cisplatin usually induces nephrotoxicity and ototoxicity, we speculate that a similar damage mechanism through pro-inflammatory cytokines including TNF-α and NOX1 and NOX4 expression may be exploited in the cisplatin toxicity on both kidney and inner ear. Based on the results presented here, the proposed mechanisms of cisplatin-induced auditory cell damages are summarized in Figure 16.
Figure 16.

Proposed mechanisms of cisplatin-induced auditory cell damages. The scheme shows that cisplatin induced pro-inflammatory cytokine production and MAPKs activation and thereby induced ROS through the increase of NOX expression, which caused the oxidative damages of auditory cells. However, NOX inhibitors, DPI and apocynin, attenuated cisplatin cytotoxicity in auditory cells via the inhibition of NOXs activation. HC, Hair cells; SC, supporting cells.
Together, our data demonstrate for the first time that NOX1 and NOX4 may play a critical role in cisplatin-induced cochlear injury. In addition, TNF-α and ERKα played a key role in NADPH oxidase activation because the inhibition of TNF-α or ERK significantly attenuated the expression and activation of NADPH oxidase isoforms after cisplatin injection. These findings are particular interesting because inflammation and ROS generation through NADPH oxidases are one of the mechanisms in the pathogenesis of cisplatin ototoxicity. Furthermore, elucidation of the intracellular mechanisms that participate in cisplatin damage to the inner ear may provide new opportunities for pharmacotherapy with regard to cisplatin and immune-mediated disorders of hearing and balance.
Footnotes
This work was supported by the Ministry of Science and Technology/Korea Science and Engineering Foundation through the Vestibulocochlear Research Center at Wonkwang University in 2008 (Grant R13-2002-055-00000-0).
References
- Alam SA, Ikeda K, Oshima T, Suzuki M, Kawase T, Kikuchi T, Takasaka T. Cisplatin-induced apoptotic cell death in Mongolian gerbil cochlea. Hear Res. 2000;141:28–38. doi: 10.1016/s0378-5955(99)00211-7. [DOI] [PubMed] [Google Scholar]
- Aldieri E, Riganti C, Polimeni M, Gazzano E, Lussiana C, Campia I, Ghigo D. Classical inhibitors of NOX NAD(P)H oxidases are not specific. Curr Drug Metab. 2008;9:686–696. doi: 10.2174/138920008786049285. [DOI] [PubMed] [Google Scholar]
- Bai J, Cederbaum AI. Mitochondrial catalase and oxidative injury. Biol Signals Recept. 2001;10:189–199. doi: 10.1159/000046887. [DOI] [PubMed] [Google Scholar]
- Bánfi B, Malgrange B, Knisz J, Steger K, Dubois-Dauphin M, Krause KH. NOX3, a superoxide-generating NADPH oxidase of the inner ear. J Biol Chem. 2004;279:46065–46072. doi: 10.1074/jbc.M403046200. [DOI] [PubMed] [Google Scholar]
- Berry CE, Hare JM. Xanthine oxidoreductase and cardiovascular disease: molecular mechanisms and pathophysiological implications. J Physiol. 2004;555:589–606. doi: 10.1113/jphysiol.2003.055913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clerici WJ, Hensley K, DiMartino DL, Butterfield DA. Direct detection of ototoxicant-induced reactive oxygen species generation in cochlear explants. Hear Res. 1996;98:116–124. doi: 10.1016/0378-5955(96)00075-5. [DOI] [PubMed] [Google Scholar]
- Dang PM, Elbim C, Marie JC, Chiandotto M, Gougerot-Pocidalo MA, El-Benna J. Anti-inflammatory effect of interleukin-10 on human neutrophil respiratory burst involves inhibition of GM-CSF-induced p47PHOX phosphorylation through a decrease in ERK1/2 activity. FASEB J. 2006;20:1504–1506. doi: 10.1096/fj.05-5395fje. [DOI] [PubMed] [Google Scholar]
- Darlington CL, Smith PF. Vestibulotoxicity following aminoglycoside antibiotics and its prevention. Curr Opin Investig Drugs. 2003;4:841–846. [PubMed] [Google Scholar]
- Fram RJ. Cisplatin and platinum analogues: recent advances. Curr Opin Oncol. 1992;4:1073–1079. doi: 10.1097/00001622-199212000-00012. [DOI] [PubMed] [Google Scholar]
- Geiszt M. NADPH oxidases: new kids on the block. Cardiovasc Res. 2006;71:289–299. doi: 10.1016/j.cardiores.2006.05.004. [DOI] [PubMed] [Google Scholar]
- Gottlieb RA. Cytochrome P450: major player in reperfusion injury. Arch Biochem Biophys. 2003;420:262–267. doi: 10.1016/j.abb.2003.07.004. [DOI] [PubMed] [Google Scholar]
- Hazan-Halevy I, Levy T, Wolak T, Lubarsky I, Levy R, Paran E. Stimulation of NADPH oxidase by angiotensin II in human neutrophils is mediated by ERK, p38 MAP-kinase and cytosolic phospholipase A2. J Hypertens. 2005;23:1183–1190. doi: 10.1097/01.hjh.0000170381.53955.68. [DOI] [PubMed] [Google Scholar]
- Henderson D, McFadden SL, Liu CC, Hight N, Zheng XY. The role of antioxidants in protection from impulse noise. Ann N Y Acad Sci. 1999;884:368–380. doi: 10.1111/j.1749-6632.1999.tb08655.x. [DOI] [PubMed] [Google Scholar]
- Humes HD. Insights into ototoxicity. Analogies to nephrotoxicity. Ann N Y Acad Sci. 1999;884:15–18. doi: 10.1111/j.1749-6632.1999.tb00278.x. [DOI] [PubMed] [Google Scholar]
- Kalinec GM, Webster P, Lim DJ, Kalinec F. A cochlear cell line as an in vitro system for drug ototoxicity screening. Audiol Neurootol. 2003;8:177–189. doi: 10.1159/000071059. [DOI] [PubMed] [Google Scholar]
- Kopke RD, Liu W, Gabaizadeh R, Jacono A, Feghali J, Spray D, Garcia P, Steinman H, Malgrange B, Ruben RJ, Rybak L, Van de Water TR. Use of organotypic cultures of Corti's organ to study the protective effects of antioxidant molecules on cisplatin-induced damage of auditory hair cells. Am J Otol. 1997;18:559–571. [PubMed] [Google Scholar]
- Kopke R, Allen KA, Henderson D, Hoffer M, Frenz D, Van de Water T. A radical demise. Toxins and trauma share common pathways in hair cell death. Ann N Y Acad Sci. 1999;884:171–191. doi: 10.1111/j.1749-6632.1999.tb08641.x. [DOI] [PubMed] [Google Scholar]
- Lambeth JD. NOX enzymes and the biology of reactive oxygen. Nat Rev Immunol. 2004;4:181–189. doi: 10.1038/nri1312. [DOI] [PubMed] [Google Scholar]
- Lee JE, Nakagawa T, Kim TS, Iguchi F, Endo T, Dong Y, Yuki K, Naito Y, Lee SH, Ito J. A novel model for rapid induction of apoptosis in spiral ganglions of mice. Laryngoscope. 2003;113:994–999. doi: 10.1097/00005537-200306000-00015. [DOI] [PubMed] [Google Scholar]
- Lee JE, Nakagawa T, Kita T, Kim TS, Iguchi F, Endo T, Shiga A, Lee SH, Ito J. Mechanisms of apoptosis induced by cisplatin in marginal cells in mouse stria vascularis. ORL J Otorhinolaryngol Relat Spec. 2004;66:111–118. doi: 10.1159/000079329. [DOI] [PubMed] [Google Scholar]
- McAlpine D, Johnstone BM. The ototoxic mechanism of cisplatin. Hear Res. 1990;47:191–203. doi: 10.1016/0378-5955(90)90151-e. [DOI] [PubMed] [Google Scholar]
- McKeage MJ. Comparative adverse effect profiles of platinum drugs. Drug Saf. 1995;13:228–244. doi: 10.2165/00002018-199513040-00003. [DOI] [PubMed] [Google Scholar]
- Miller AA, Drummond GR, Sobey CG. Novel isoforms of NADPH-oxidase in cerebral vascular control. Pharmacol Ther. 2006;111:928–948. doi: 10.1016/j.pharmthera.2006.02.005. [DOI] [PubMed] [Google Scholar]
- Mukherjea D, Whitworth CA, Nandish S, Dunaway GA, Rybak LP, Ramkumar V. Expression of the kidney injury molecule 1 in the rat cochlea and induction by cisplatin. Neuroscience. 2006;139:733–740. doi: 10.1016/j.neuroscience.2005.12.044. [DOI] [PubMed] [Google Scholar]
- Ohlemiller KK, Wright JS, Dugan LL. Early elevation of cochlear reactive oxygen species following noise exposure. Audiol Neurootol. 1999;4:229–236. doi: 10.1159/000013846. [DOI] [PubMed] [Google Scholar]
- Paffenholz R, Bergstrom RA, Pasutto F, Wabnitz P, Munroe RJ, Jagla W, Heinzmann U, Marquardt A, Bareiss A, Laufs J, Russ A, Stumm G, Schimenti JC, Bergstrom DE. Vestibular defects in head-tilt mice result from mutations in Nox3, encoding an NADPH oxidase. Genes Dev. 2004;18:486–491. doi: 10.1101/gad.1172504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan H, Mukhopadhyay P, Rajesh M, Patel V, Mukhopadhyay B, Gao B, Haskó G, Pacher P. Cannabidiol attenuates cisplatin-induced nephrotoxicity by decreasing oxidative/nitrosative stress, inflammation and cell death. J Pharmacol Exp Ther. 2009;328:708–714. doi: 10.1124/jpet.108.147181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinn MT, Ammons MC, Deleo FR. The expanding role of NADPH oxidases in health and disease: no longer just agents of death and destruction. Clin Sci (Lond) 2006;111:1–20. doi: 10.1042/CS20060059. [DOI] [PubMed] [Google Scholar]
- Rybak LP, Whitworth C, Somani S. Application of antioxidants and other agents to prevent cisplatin ototoxicity. Laryngoscope. 1999;109:1740–1744. doi: 10.1097/00005537-199911000-00003. [DOI] [PubMed] [Google Scholar]
- Satoh H, Firestein GS, Billings PB, Harris JP, Keithley EM. Tumor necrosis factor-alpha, an initiator, and etanercept, an inhibitor of cochlear inflammation. Laryngoscope. 2002;112:1627–1634. doi: 10.1097/00005537-200209000-00019. [DOI] [PubMed] [Google Scholar]
- Sergi B, Ferraresi A, Troiani D, Paludetti G, Fetoni AR. Cisplatin ototoxicity in the guinea pig: vestibular and cochlear damage. Hear Res. 2003;182:56–64. doi: 10.1016/s0378-5955(03)00142-4. [DOI] [PubMed] [Google Scholar]
- Serù R, Mondola P, Damiano S, Svegliati S, Agnese S, Avvedimento EV, Santillo M. HaRas activates the NADPH oxidase complex in human neuroblastoma cells via extracellular signal-regulated kinase 1/2 pathway. J Neurochem. 2004;91:613–622. doi: 10.1111/j.1471-4159.2004.02754.x. [DOI] [PubMed] [Google Scholar]
- So H, Kim H, Lee JH, Park C, Kim Y, Kim E, Kim JK, Yun KJ, Lee KM, Lee HY, Moon SK, Lim DJ, Park R. Cisplatin cytotoxicity of auditory cells requires secretions of proinflammatory cytokines via activation of ERK and NF-kappaB. J Assoc Res Otolaryngol. 2007;8:338–355. doi: 10.1007/s10162-007-0084-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- So H, Kim H, Kim Y, Kim E, Pae HO, Chung HT, Kim HJ, Kwon KB, Lee KM, Lee HY, Moon SK, Park R. Evidence that cisplatin-induced auditory damage is attenuated by downregulation of pro-inflammatory cytokines via Nrf2/HO-1. J Assoc Res Otolaryngol. 2008;9:290–306. doi: 10.1007/s10162-008-0126-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- So HS, Park C, Kim HJ, Lee JH, Park SY, Lee ZW, Kim HM, Kalinec F, Lim DJ, Park R. Protective effect of T-type calcium channel blocker flunarizine on cisplatin-induced death of auditory cells. Hear Res. 2005;204:127–139. doi: 10.1016/j.heares.2005.01.011. [DOI] [PubMed] [Google Scholar]
- Szeto HH. Mitochondria-targeted peptide antioxidants: novel neuroprotective agents. AAPS J. 2006;8:E521–E531. doi: 10.1208/aapsj080362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takumida M, Anniko M. Simultaneous detection of both nitric oxide and reactive oxygen species in guinea pig vestibular sensory cells. ORL J Otorhinolaryngol Relat Spec. 2002;64:143–147. doi: 10.1159/000057794. [DOI] [PubMed] [Google Scholar]