

Abstract
GABAergic dysfunction is implicated in a variety of neurodevelopmental and psychiatric disorders. The mechanisms underlying GABAergic differentiation, however, are not well understood. GABA transporter 1 (Gat1; Slc6a1) is an essential component of the GABAergic system, and its ectopic mRNA expression may be responsible for GABAergic malfunction under different pathological conditions. Thus, monitoring the transcriptional regulation of gat1 may help to elucidate the mechanisms that govern the differentiation of GABAergic neurons. In this study, we identified a promoter region that is sufficient to recapitulate endogenous gat1 expression in transgenic mice. A 46 bp cis-regulator in the promoter sequence was responsible for the stimulation of bone morphogenetic protein-2 (BMP2) on gat1 expression in cortical cortex. Furthermore, our study demonstrated that Smad4 and YY1 are physically bound to the element and mediate both the negative and positive regulatory effects in which BMP2 can affect the balance. In summary, we have identified a Smad4/YY1-based bidirectional regulation model for GABAergic gene transcription and demonstrated a molecular cue important for the differentiation of GABAergic neurons.
Introduction
Aberrant development and dysfunction of GABAergic neurons are implicated in a variety of neurodevelopmental and psychiatric disorders, such as epilepsy (DeFelipe, 1999; Cossart et al., 2005), schizophrenia (Woo et al., 1998; Lewis et al., 1999, 2005; Volk and Lewis, 2002; Ruzicka et al., 2007), and anxiety disorders (Baraban, 2002; Heilig and Thorsell, 2002). Signaling molecules that regulate the acquisition and maintenance of GABAergic phenotype include bone morphogenetic proteins (BMPs) (Li et al., 1998; Mabie et al., 1999; Gulacsi and Lillien, 2003), Notch (de la Pompa et al., 1997; Kabos et al., 2002), and glial cell-derived neurotrophic factor (Pozas and Ibáñez, 2005), as well as basic helix-loop-helix (Casarosa et al., 1999; Bae et al., 2000; Fode et al., 2000; Miyoshi et al., 2004; Schuurmans et al., 2004; Nakatani et al., 2007) and homeodomain (Anderson et al., 1997; Sussel et al., 1999; Kroll and O'Leary, 2005) transcription factors. However, the molecular mechanisms regulating gene transcription resulting in GABAergic differentiation are still far from clear.
The GABAergic phenotype requires coordinated activation of glutamate decarboxylases (Gad1-2), the plasma membrane GABA transporters (Gat1-4), and the vesicular inhibitory amino acid transporter (Vgat). In Caenorhabditis elegans, transcription factor unc-30 has been found to determine GABAergic phenotype by regulating the transcriptions of unc-25/gad and unc-47/vgat in a coordinated manner (Eastman et al., 1999; Westmoreland et al., 2001). It enlightens us to elucidate the mechanisms underlying the differentiation of GABAergic neurons by monitoring the transcriptional regulation of functional genes in GABAergic neurons.
GABA transporter 1 (Gat1; Slc6a1) is the major neural GABA transporter, and plays an important role in the termination of GABAergic transmission and the regulation of extracellular GABA concentration (Chiu et al., 2002). Studies of mice overexpressing, or deficient in, gat1 suggest that gat1 is associated with seizures (Ma et al., 2001; Zhao et al., 2003; Chiu et al., 2005) and emotional behaviors such as anxiety (Chiu et al., 2005; Liu et al., 2006). Alterations of gat1 expression on transcription level play a key role in some GABAergic-related pathological circumstances such as epilepsy (Fueta et al., 2003; Sperk et al., 2003; Jiang et al., 2004), schizophrenia (Woo et al., 1998; Lewis et al., 1999; Volk et al., 2001; Volk and Lewis, 2002), and substance abuse (Peng and Simantov, 2003; Zink et al., 2004). Furthermore, polymorphism in the 5′-flanking region of gat1 is highly associated with anxiety disorders (Thoeringer et al., 2009). A recent study demonstrated that 21 bp insertion polymorphism increases gat1 promoter activity (Hirunsatit et al., 2009).
In this study, mouse gat1 gene promoter was identified in transgenic mice and a 46 bp cis-regulatory element was found to regulate gat1 transcription activity in the cerebral cortex. In addition, we found evidence suggesting that functional interaction between Smad4/YY1 and the 46 bp element mediates both the negative and positive regulatory effects in which morphogenetic protein 2 (BMP2) can affect the balance. To the best of our knowledge, this is the first report of bidirectional in vivo transcriptional regulation of gat1.
Materials and Methods
Rapid amplification of 5′ cDNA ends.
Total RNA was isolated from mouse brain and subjected to 5′-race analysis (Invitrogen). The primer used for the first step of amplification was 5′-GCCTTCTTCTGCACCTTGACTACC-3′, located within exon 3. 5′-Race was performed by incubating with an aliquot of first-strand cDNA, a nested PCR primer positioned within exon 3 (5′-CAGGTGGGCGCGAGATGTC-3′), and an abridged anchor primer. The PCR conditions were: initial denaturation at 94°C for 5 min, 35 cycles of 30 s at 94°C, 1 min at 58°C, and 1 min at 72°C. The resulting products were cloned into pMD-18T vector (TaKaRa) and sequenced to determine the transcription initiation site(s).
Constructs and mutagenesis.
A 5.7 kb fragment of mouse gat1 promoter was generated by PCR with mouse genomic DNA as a template. The forward primer was from base 66540243 to 66540265 of the 5′-flanking region of gat1 gene (GI:149255466) with an additional MluI site (italics) (5′-TTAACGCGTGAGAGAGCACAACGCAGGAACAG-3′). The reverse primer was from base 66545546 to 66545570 of the downstream of the exon 1 with an additional XhoI site (underlined) (5′-TTACTCGAGCGAACGAACTAGGACATAGACGGC-3′). The 5.7 kb MluI-XhoI fragment was cloned into pGL3-basic (Promega). This construct is referred to as −5377/luc throughout this manuscript. The construct −3006/luc was assembled by deleting the KpnI-BamHI fragment from the −5377/luc construct. Constructs containing fragments of gat1 gene promoter beginning at −2135, −2085, −1978, −1782, −1493, −1090, −891, −706, −333, −288, −241, −167, −93, or +109 from the transcription initiation site were prepared in a similar manner using forward primers with a MluI site at 5′ terminal (italics) 5′-TTAACGCGTTGCTTTGGTCACGGTGTCTCTTC-3′ (−2135/luc), 5′-TTAACGCGTGAGGTCAAACAGATGCAAAG-3′ (−2085/luc), 5′-TTAACGCGTCCTGGATTTGGTTCCCAGCAC-3′ (−1978/luc), 5′-TTAACGCGTCTTCAGGCACAGCTGGATCAC-3′ (−1782/luc), 5′-TTAACGCGTATGAGACGTGGGGAGAAGACC-3′ (−1493/luc), 5′-TTAACGCGTGTCTGGGCTCTCGAAAGGTTG-3′ (−1090/luc), 5′-TTAACGCGTAGCCTAGATGCTTGTGGGAGG-3′ (−891/luc), 5′-TTAACGCGTTGGGGAACATGGAAAAGGGAGAG-3′ (−706/luc), 5′-TTAACGCGTGTGACAGAGCCAGAGAAACCAAG-3′ (−333/luc), 5′-TTAACGCGTGAGGCCAGGAGACTGAAGGAG-3′ (−288/luc), 5′-TTAACGCGTGGGAGCAGGGCTGGGAGAGAG-3′ (−241/luc), 5′-TTAACGCGTGGCAAGGCGGGCAGGGCCTAG-3′ (−167/luc), 5′-TTAACGCGTGAGGAGGCAGGCAGAGGGAGG-3′ (−93/luc), 5′-TTAACGCGTCTAGAGAGCTGAGAGGTTGCAGG-3′ (+109/luc), respectively. The reverse primer was identical to that used to generate the construct −5377/luc. Restriction analysis and sequencing were used to verify the location of the promoter.
Deletion was introduced to construct −5377m/luc using a two-step PCR method with the −5377/luc DNA as the template. Two overlapping oligonucleotides were synthesized: −5377m/luc (forward) 5′-CTCCTGGCCTCATCTCTCTTGGAAGCATTGTGG-3′ and −5377m/luc (reverse) 5′-GAGAGATGAGGCCAGGAGACTGAAGGAG-3′. The first-step PCR was initiated using one of these primers and an appropriate primer which was used to generate the construct −5377/luc. The products of the first-step PCR were used as a template for the second-step PCR with a set of primers used to generate the construct −5377/luc. The resulting product was digested with MluI and XhoI and cloned into the vector pGL3-basic. The resulting construct lacked fragment −333 to −288, and was verified with sequencing.
The sequence of the mouse gat1 gene was analyzed for potential transcription factor binding sites with P-Match program (BioBase, Wolfenbüttel, Germany) using Transfac 6.0 Public database.
Short hairpin RNA (shRNA) oligonucleotides were designed to specifically target either smad4 or yy1. Two different shRNA oligonucleotides of smad4: shRNA1 5′-CCAGCTACTTACCATCATA-3′ (Thuault et al., 2006) and shRNA2 5′-GCCATAGTGAAGGACTGTT-3′ (Rees et al., 2006). Two different shRNA oligonucleotides of yy1: shRNA1 5′-GAACTCACCTCCTGATTAT-3′ (Allouche et al., 2008) and shRNA2 5′-TGACAGGCAAGAAACTCCC-3′. Two nonspecific control shRNA oligonucleotides with a similar GC content as smad4 shRNA and yy1 shRNA were used. The inhibitory efficiency of each shRNA was determined by Western blot assay.
Generation of transgenic mice.
The pSVβ-galactosidase was obtained from Promega. The 1690 bp HindIII-XbaI fragment was isolated from pGL3-basic. The 3744 bp HindIII-XbaI fragment from pSVβ-galactosidase was inserted to pGL3-basic at the HindIII site. The resulting fusion gene consisted of the Escherichia coli gpt gene fragment containing its translation initiation site, the LacZ gene encoding β-galactosidase from amino acid position 9 and the simian virus 40 (SV40) fragment with the polyadenylation signal. The 5.7 kb fragment, containing the 5′-flanking sequences, exon 1 and part of intron 1, was introduced to the plasmid carrying the LacZ gene at the XhoI and SmaI sites. The final fusion gene was referred to as gat1(5.7)lacz. gat1(5.7m)lacz was prepared in a similar manner to construct −5377m/luc. All clones were verified by sequencing.
gat1(5.7)lacz and gat1(5.7m)lacz constructs were linearized with ScaI and subsequently purified with the Qiaex II Gel Extraction kit (Qiagen). The constructs were microinjected into fertilized eggs of C57BL/6J×DBA/2J hybrid mice. Founders were identified by PCR analysis of tail genomic DNA with primers that amplify a 470 bp region spanning the junction between the mouse gat1 promoter and the lacZ cDNA. Primers for PCR were (forward) 5′-AGCCCCGGCCGCAGGTAGGAA-3′ and (reverse) 5′-GCTGGCGAAAGGGGGATGTGCT-3′.
Quantitative real-time PCR.
Total RNA was extracted from mouse tissues (brain, heart, lung, liver, spleen, and kidney) using Trizol (Invitrogen). RNA samples were treated with RNase-free DNase I (TaKaRa) for 30 min at 37°C to eliminate DNA contamination. Reverse transcription was performed with M-MLV (Promega). Fluorescent signals were generated using SYBR Green PCR Master Mix (Applied Biosystems). PCRs for each gene of interest were run in triplicate on Rotor-Gene 3000 as follows: 10 min at 95°C and 40 cycles of 15 s at 94°C, 15 s at 66°C, and 30 s at 72°C. Primer sequences were: for lacZ (the target gene), (forward): 5′-TCAATCCGCCGTTTGTTCCCAC-3′, and (reverse): 5′-TCCAGATAACTGCCGTACTCCAGC-3′; and for gapdh (the internal control), (forward): 5′-TGATGACATCAAGAAGGTGGTGAAG-3′, and (reverse): 5′-TCCTTGGAGGCCATGTGGGCCAT-3′. A melting curve analysis was performed at the end of the PCR cycle. Electrophoresis with 2% agarose gel was used to verify the amplification a single product. Experimental controls included non-reverse-transcribed RNA samples. Data were analyzed by Rotor-Gene software to determine the threshold cycle (CT) above the background for each reaction. Normalization was performed using the 2–ΔΔCT method (Livak and Schmittgen, 2001). The ▵CT variability calculation revealed a slope value close to zero in a cDNA dilution over a 100-fold range in three independent experiments.
Tissue processing and immunocytochemistry.
Heterozygous transgenic mice (2 months old) were deeply anesthetized and perfused with 4% paraformaldehyde in 0.1 m PB. Whole brains were removed, postfixed in the same fixative for 4 h, and cryoprotected in 20% glycerol/PB overnight at 4°C. Sagittal sections (30 μm) were cut and then stored in an ethylene glycol based cryoprotective solution at −20°C.
Immunohistochemical staining of free-floating sections was performed using an immunoperoxidase kit (VECTA ABC Kit, Vector Laboratories). The sections were incubated in 0.3% H2O2 in PBS for 30 min at room temperature to quench endogenous peroxidase. Sections were then incubated in 10% normal goat serum for 1 h at room temperature to block nonspecific binding. Sections were incubated with rabbit anti-GAT1 (1:100 dilution; Millipore Bioscience Research Reagents) or rabbit anti-LacZ (1: 2000 dilution; Abcam) in 1% normal goat serum overnight at 4°C. After rinsing, the sections were incubated for 30 min each with an appropriate biotinylated goat secondary antibody (ProteinTech Group) and an avidin–biotin complex solution. Staining was developed for 10 min in nickel-DAB solution (0.3%). No staining was observed in the control experiments, in which primary antibodies were omitted. Nontransgenic controls failed to be stained by anti-LacZ antibody under these conditions.
For immunofluorescence staining, free-floating sections were washed twice in PBS and then blocked with 10% normal goat serum in PBS. Double-labeling studies were performed using non-cross-reacting secondary antibodies after primary antibody incubation. The sections were examined under a Nikon Eclipse TE 2000-U fluorescence microscope with filters suitable for selectively detecting the fluorescence of FITC (green) and Cy3 (red) or under a light microscope. For colocalization, images from the same section but showing different antigen signals were overlaid.
The number of positively labeled cells and the intensity of the immunopositive signal were estimated by a blinded observer using Image-Pro Plus program (Media Cybernetics). For an unbiased determination, every 20th serial section at ∼1.2–4.2 mm from the midline were selected. For each brain, five sagittal sections were analyzed, and the average was used to calculate the group means (n = 5 brains).
Cell culture, transfections, and reporter gene assays.
NIH 3T3 fibroblasts and Neuro 2a (mouse neuroblastoma) cell lines were grown in DMEM (Invitrogen) with 10% newborn calf serum supplemented with 100 units/ml penicillin and 100 mg/ml streptomycin (Invitrogen). The mouse embryo teratocarcinoma P19 cells were cultured in DMEM/F12 (Invitrogen) containing 10% fetal bovine serum.
Primary cortical neuron cultures were obtained from embryonic day 18 (E18) mouse embryos, dissociated with 0.125% trypsin (Invitrogen) for 15 min at 37°C, and then dissociated mechanically with a glass Pasteur pipette. Cells were plated in Neurobasal medium (Invitrogen) containing 0.5 mm L-glutamin (Invitrogen), and 2% B-27 supplement (Invitrogen) on poly-l-lysine-coated plates. On day 4 in vitro, one-quarter of the media was replaced with fresh media containing cytosine arabinoside (final concentration, 2 μm) to eliminate non-neuronal cells.
Primary neural stem cell (NSC) cultures were obtained from E14 mouse embryos. Dissected whole brain was transferred to ice-cold Hank's balanced salt solution (Invitrogen) and mechanically dissociated into a single-cell suspension with a fire-polished Pasteur pipette. Cells were seeded in noncoated T-25 culture flask in DMEM/F12 (Invitrogen) containing 2% B-27, 20 ng/ml epidermal growth factor (Peprotech) and 20 ng/ml basic fibroblast growth factor (Peprotech) at a density of 100,000 cells/ml. Primary neurospheres were dissociated by incubation with Accutase (Millipore Bioscience Research Reagents) and reseeded in fresh media at 50,000 cells/ml until secondary spheres were generated. All spheres used for experiments were passaged at least once. All cell lines were kept at 37°C in a humidified atmosphere containing 5% CO2.
Cells were transfected in Opti-MEM (Invitrogen) with Lipofectamine-2000 (Invitrogen). The pRL-SV40 or pRL-TK vector (Promega) was used as internal control. P19 cells and NSCs were treated with bone BMP2 (Peprotech) at 20 and 10 ng/ml, respectively (Lee et al., 2000). Cells were harvested 48–72 h post-transfection and assayed for reporter gene activity with a Dual-Luciferase Reporter Assay System (Promega).
Nuclear extract preparation and electrophoretic mobility shift assay.
Nuclear extracts were prepared according to the method of Jiang et al. (2008). Biotin-labeled double-stranded oligonucleotide from mouse gat1 gene promoter region (probe A 5′-GTGACAGAGCCAGAGAAACCAAGAGACCAATTAAGGTAGACCTTT-3′) or reported smad4-binding site (probe SBE 5′-AGACAGACAATGTCTAGTCTATTTGAAATGCCTGA-3′) was used as a probe. LightShift chemiluminescent EMSA kit (PIERCE) was used for the binding reactions. Before the addition of biotin-labeled probe, 2 μg of nuclear extracts was incubated for 10 min at room temperature in 10 μl of reaction buffer. Biotin-labeled probe was then added, and the incubation was allowed to proceed for 20 min at room temperature. Protein–DNA complexes were separated on nondenaturing polyacrylamide gels. In competition experiments, the nuclear extracts were preincubated with excess unlabeled double-stranded oligonucleotides for 10 min. The sequences of the competitor ASBE3m and SBEm oligonucleotides were: ASBE3m 5′-GTGACAGAGCCAGAGAAACCAATTTTTCAATTAAGGTAGACCTTT-3′, SBEm 5′-AGACAGACAATGTTTATTCTATTTGAAATGCCTGA-3′, and their complementary strands.
Chromatin immunoprecipitation assay.
Chromatin immunoprecipitation (ChIP) assays were performed by using a ChIP assay kit (Millipore Biotechnology). Briefly, P19 cells or primary cultured neurons were cross-linked for 10 min at 37°C by the addition of formaldehyde (final concentration, 1%). After washing with cold PBS, cells were resuspended in SDS lysis buffer supplemented with protease inhibitors and incubated on ice for 10 min. Cell lysate was subsequently sonicated seven times with 3 s bursts at 40 W in a Sonifier (JY92–2D, Ningbo Scientz Biotechnology) to yield input DNA enriched with fragments between 200 and 1000 bp in size. A small proportion of the lysate was immediately heated at 65°C for 4 h in the presence of 5 m NaCl to reverse the cross-links, and was later used for monitoring equal DNA amounts for ChIP (Input). Sonicated lysate obtained from ∼1 × 106 cells was reconstituted in 2 ml of ChIP dilution buffer with protease inhibitors. To reduce nonspecific background, the lysate was treated with salmon sperm DNA/protein A-agarose beads for 30 min at 4°C. The precleared lysate was immunoprecipitated with 5 μg of rabbit anti-YY1 (Santa Cruz Biotechnology), anti-Smad4 (Santa Cruz Biotechnology), or the same amount of normal rabbit IgG at 4°C overnight. Immune complexes were collected with salmon sperm DNA/protein A-agarose. After elution of immune complexes, cross-linking was reversed as described above, and the DNA was then purified by a typical phenol/chloroform procedure and ethanol precipitation. Real-time PCR analysis of ChIP DNA was conducted in three independent experiments, quantified using the standard curve method on ABI 7300 thermocycler, and normalized to Smad4 bound to the region from −333 to −288 of mouse gat1 gene (set at 100%). The primers used were: (forward) 5′-ACACATCCTCCAAGACCAATCCT-3′ and (reverse) 5′-GGCCTCCACCCTCCTTCA-3′.
Statistics.
Results are expressed as the mean ± SD for at least three independent experiments. Differences between two samples were assessed by a two-tailed Student's t test. Differences among multiple means were assessed by one-way ANOVA followed by Bonferroni correction. p values of ≤0.05 were considered statistically significant.
Results
Identification of the mouse Gat1 5′-flanking region
Rapid amplification of 5′ cDNA ends was performed to map the transcription initiation site(s) of mouse gat1 gene. The major PCR product was cloned into pMD-18T vector and then sequenced (Fig. 1A). From the sequences of 12 independent clones, a major transcription initiation site (+1) corresponding to an A residue was identified.
Figure 1.

5′-Race determination of transcription initiation site(s) in mouse gat1 gene. A, Gel analysis of the first PCR and nested PCR products from 5′-race with RNA derived from mouse brain. B, Schematic representation of mouse gat1 genomic structure. The transcription initiation site is indicated by arrow. Open boxes represent the noncoding region and black boxes represent the coding region. The start of translation occurs in exon 3, as indicated by ATG.
Using the sequence information for mouse gat1 gene and flanking sequence (GI:149255466) as a guide for primer design, a 5752 bp genomic fragment was cloned from mouse genomic DNA. The fragment begins inside intron 1 of mouse gat1 gene at the position +375 bp (using transcription initiation site as +1) and extends to −5377 bp upstream of the transcription initiation site. Because the translation initiator ATG sequence is located inside exon 3 of mouse gat1 gene and separated from exon 1 by two introns, the isolated 5′-flanking region of mouse gat1 gene did not contain the translation initiation site (Fig. 1B). The identity of the genomic DNA fragment was established by sequencing.
Gat1 promoter activity in transgenic mice
Transgenic mice with the gat1(5.7)lacz construct harbored a fusion gene consisting of 5.7 kb gat1 5′-flanking region extending 375 bp into the 5′-untranslated leader sequence, a nuclear LacZ expression cassette, and one polyadenylation site derived from the SV40 gene. Five founder lines were obtained, and all expressed the transgene as confirmed by reverse transcriptase PCR assay.
Quantitative reverse transcriptase PCR assay demonstrated almost exclusive distribution of the transgene in the brain, with minimal level of expression in peripheral tissues (Table 1). This pattern is consistent with previous results of endogenous mouse gat1 gene expression (Liu et al., 1993), indicating that the DNA fragment containing the promoter region is sufficient to confer tissue specificity of mouse gat1 gene expression.
Table 1.
Transgene expression in gat1(5.7)lacz transgenic mice
| Transgenic mice tissue |
gat1(5.7)lacz transgenic founder |
|
|---|---|---|
| 88F (n ≥ 3) | 32F (n ≥ 3) | |
| Brain | 100 | 100 |
| Heart | 0 | 11.4 |
| Lung | 2.9 | 5.4 |
| Liver | 5.6 | 4.4 |
| Spleen | 4.4 | 3.0 |
| Kidney | 0.3 | 5.1 |
LacZ mRNA expression in the brain of each line was artificially set at 100. Three replicates of each reaction were performed.
In the two lines analyzed, GAT1 (Fig. 2A–E) and LacZ (Fig. 2A′–E′) immunoreactivity were detected throughout the entire cerebral cortex, hippocampus, cerebellum, and olfactory bulb. These results match the distribution of endogenous mouse GAT1 (Borden, 1996). At higher magnification, GAT1 and LacZ immunoreactivity were strong in layers II and IV and moderate in other layers of the cerebral cortex (Fig. 2B,B′). CA1, CA3, and the dentate gyrus of hippocampus show immunoreactivity for LacZ and GAT1 (Fig. 2C,C′). Immunoreactivity for LacZ and GAT1 was strong in the Purkinje cell layer, moderate in the molecular layer, and very faint or not detected in the granular layer (Fig. 2D,D′). Finally, GAT1 and LacZ immunoreactivity were present in the olfactory bulb (Fig. 2E,E′). LacZ was colocalized with GAT1 in double-staining experiments (Fig. 3). These results further indicate that the 5.7 kb 5′-flanking region of mouse gat1 gene harbors most, if not all, of the cis-regulatory information required for gat1 gene expression in vivo.
Figure 2.

Histological detection of GAT1 and LacZ in the brain of adult gat1(5.7)lacz mice. A–E′, Sagittal brain sections from gat1(5.7)lacz transgenic mice were stained by anti-GAT1 antibodies (A–E) or anti-LacZ antibodies (A′–E′). Shown are images of whole brain (A, A′), cerebral cortex (Cx; B, B′), hippocampus (Hi; C, C′), cerebellum (Cb; D, D′), and olfactory bulb (Ob; E, E′). Abbreviations for this and subsequent figures: CA1, CA3, Fields of the hippocampus; DG, dentate gyrus; GL, ML, and PL are granule, molecular, and Purkinje cell layers, respectively. In all panels, arrows denote immunopositive cells. Scale bars: A, A′, 1 mm; D, D′, 100 μm; B, B′, C, C′, E, E′, 400 μm.
Figure 3.
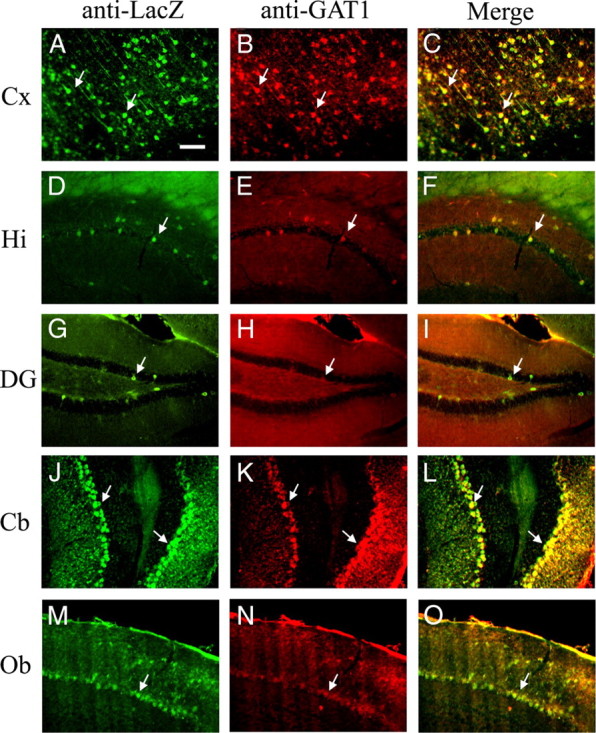
Colocalization of LacZ and GAT1 in the brain of adult gat1(5.7)lacz mice. A–O, Sagittal brain sections from gat1(5.7)lacz transgenic mice were stained by anti-LacZ antibodies (green) and anti-GAT1 antibodies (red). Shown are images of cerebral cortex (Cx; A–C), hippocampus (Hi; D–F), dentate gyrus (DG; G–I), cerebellum (Cb; J–L), and olfactory bulb (Ob; M–O). The third window in each row represents the merging of the red and green channels. Yellow staining indicates colocalization of LacZ and GAT1. A–O, Arrows denote immunopositive cells. See legend of Figure 2 for abbreviations not used in the text. Scale bar, 100 μm.
Definition of regulatory elements within gat1 promoter in vitro
To localize the cis-regulatory elements that negatively regulate gat1 gene expression, reporter gene assays were performed in two cell lines that do not have endogenous gat1 gene expression (NIH 3T3 and Neuro 2a) (Fig. 4A). In both cell lines, a strong gene expression suppression effect was observed in the constructs with 5′-terminal deletion up to nt −333, ∼300 bp upstream from the transcription initiation site. A further deletion of 46 bp (deletion up to nt −288) abolished this effect. Moreover, an internal deletion of the 46 bp element, from −333 to −288, resulted in an increase of full-length promoter activity in both cell types (Fig. 4B). Similar results were achieved both in NSCs and primary cortical neurons (Fig. 4B). Notably, induction of gat1 promoter activity observed in primary cortical neurons was significantly less than that in NSCs, NIH 3T3, and Neuro 2a. These results support the contention that the 46 bp element is an essential requirement for the negative transcriptional regulation of mouse gat1 gene.
Figure 4.

Transient transfection assays define a cis-regulator in mouse gat1 gene promoter. A, Schematic structure of the luciferase constructs used in this study. The boundaries of the promoter constructs are defined relative to the transcription initiation site and are indicated on the left. Independent constructs were transfected into NIH 3T3 or Neuro 2a cells. Luciferase activity was normalized to Renilla luciferase activity encoded by cotransfected control plasmid pRL-SV40 and then normalized to that of −5377/luc construct. B, In the −5377m/luc or −1090∼+52m/luc construct, a 46 bp fragment, from −333 to −288, is indicated by an open rectangle and was deleted. Independent constructs were transfected into NIH 3T3, Neuro 2a cells, primary cortical neurons, or NSCs. Luciferase activity was normalized to Renilla luciferase activity encoded by cotransfected control plasmid, pRL-SV40, or pRL-TK. The promoter activity was normalized to −5377/luc or −1090∼+52m/luc. Induction of promoter activity in different cell lines is indicated as fold increase. Results are shown as the mean ± SD for three independent experiments (n = 3 in each independent experiment).
Identification of the cis-regulatory element in gat1 promoter in vivo
Transgenic mice with the gat1(5.7m)lacz construct, bearing a deletion of the 46 bp element, were established. The overall transgene expression pattern of gat1(5.7m)lacz mice was not significantly different from that of gat1(5.7)lacz mice (Table 2) (two-tailed Student's t test). These findings suggest that silencer elements other than the 46 bp element are required to prevent expression of gat1 in most non-neural tissues. Alternatively, non-neuronal tissues may lack transactivators that are critical for neuronal gene expression.
Table 2.
Transgene expression in GAT1(5.7m)LacZ transgenic mice
| Transgenic mice tissue |
gat1(5.7m)lacZ transgenic founder |
|
|---|---|---|
| 2F (n ≥ 3) | 3F (n ≥ 3) | |
| Brain | 100 | 100 |
| Heart | 7.7 | 15.5 |
| Lung | 5.7 | 18.0 |
| Liver | 12.2 | 7.4 |
| Spleen | 3.8 | 2.3 |
| Kidney | 12.4 | 5.1 |
LacZ mRNA expression in the brain of each line was artificially set at 100. Three replicates of each reaction were performed.
Similar to that of gat1(5.7)lacz mice, a high level of LacZ expression was present in the cerebral cortex, hippocampus, cerebellum, and olfactory bulb in two independent lines of gat1(5.7m)lacz mice (Fig. 5). However, the percentage of GAT1-expressing cells in LacZ-expressing cells was significantly lower in brains of gat1(5.7m)lacz mice than those of gat1(5.7)lacz mice, while the percentages of LacZ-expressing cells in GAT1-expressing cells were not significantly different between two transgenic mice (Fig. 6A–D). These results indicate that deletion of the 46 bp element results in an increased number of LacZ-expressing cells, part of which are GAT1 nonexpressing. It is notable that although the overall LacZ-immunoreactive signal intensity of gat1(5.7m)lacz mice was higher than that of gat1(5.7)lacz mice (Fig. 6E,F), in GAT1-expressing cells the LacZ-immunoreactive signal intensity of gat1(5.7m) lacz mice was lower than that of gat1(5.7) lacz mice (p < 0.05, ANOVA and Bonferroni correction). These results indicate that the 46 bp element has an opposite effect on gat1 gene promoter activity depending on the cellular context.
Figure 5.
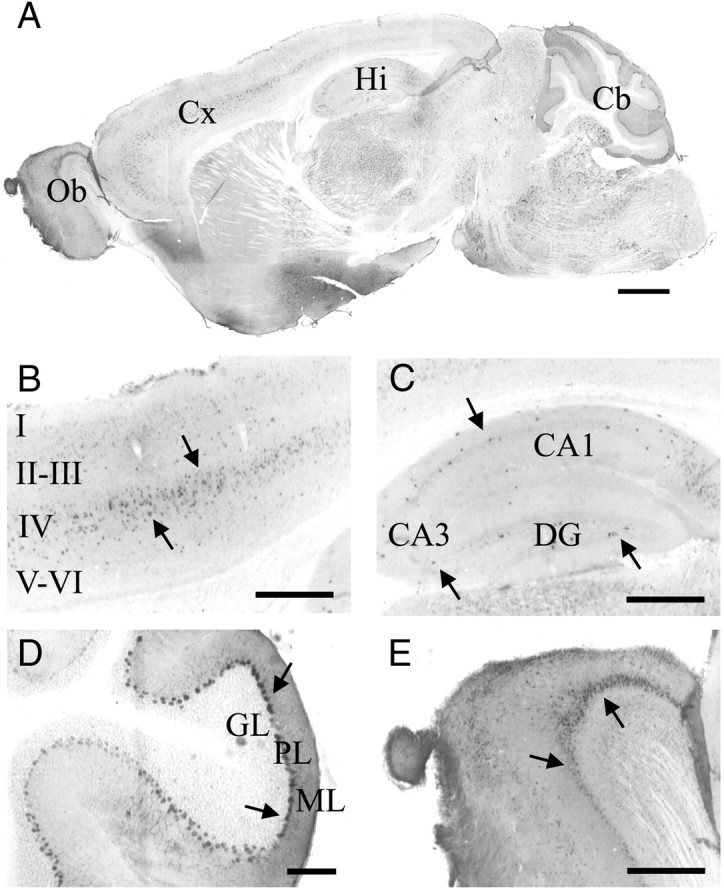
Histological detection of LacZ in the brain of adult gat1(5.7m)lacz mice. A–E, Sagittal brain sections from gat1(5.7m)lacz transgenic mice were stained by anti-LacZ antibodies. Shown are images of whole brain (A), cerebral cortex (Cx; B), hippocampus (Hi; C), cerebellum (Cb; D), and olfactory bulb (Ob; E). A–E, Arrows denote immunopositive cells. Scale bars: A, 1 mm; D, 100 μm; B, C, E, 400 μm.
Figure 6.

Quantitative analysis of LacZ or GAT1 immunopositive cells in cerebral cortex of gat1(5.7)lacz and gat1(5.7m)lacz mice. A, Schematic representation of a sagittal brain section showing image locations. B, Representative images of LacZ and GAT1 immunopositive signal on sagittal brain sections from gat1(5.7)lacz or gat1(5.7m)lacz mice. C, D, The percentage of LacZ-expressing cells in GAT1-expressing cells or the percentage of GAT1-expressing cells in LacZ-expressing cells of region I (C) or region II (D). E, F, Intensity of LacZ-immunoreactive signal in all cells or in GAT1-expressing cells of region I (E) or region II (F). Scale bars: A, 1 mm; B, 100 μm. Data represent mean ± SE (n ≥ 5 mice per line). *p < 0.05 significantly different from gat1(5.7)lacz group (ANOVA followed by Bonferroni correction). DAPI, 4′,6′-Diamidino-2-phenylindole dihydrochloride.
BMP2 induction of gat1 gene expression mediated by Smad4 and YY1
BMP2 is reported to regulate cortical GABAergic neuron differentiation (Mabie et al., 1999; Yung et al., 2002). BMP2 increased the transcriptional activity of wild-type but not mutant gat1 gene promoter construct with the 46 bp element deleted (Fig. 7A,B). These findings indicate that the BMP2 response depends on the region in the mouse gat1 gene promoter between −333 and −288.
Figure 7.

BMP2-induced gat1 promoter activity mediated by Smad4 and YY1. A, B, P19 cells (A) or neural stem cells (B) were transiently transfected with either wild-type or mutant Gat1 gene promoter construct, grown in the presence or absence of BMP2. The activity of wild-type Gat1 promoter construct in the absence of BMP2 was set at 1. Results are expressed as the mean ± SD for three experiments. *p < 0.05, compared with the unstimulated wild-type construct control (Student's t test). C, The inhibitory efficiency of the shRNA directed against smad4 or yy1 evaluated by Western blot analysis. At 48 h after shRNA transfection, total cell lysate was prepared and normalized for protein concentration. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as an internal control. Results shown are representative of three independent experiments. D–G, P19 cells (D, F) or neural stem cells (E, G) were transfected with either wild-type or mutant gat1 gene promoter construct, together with the indicated shRNA expression constructs. Transfected cells were then treated with BMP2 (F, G) or not (D, E), as indicated. The activity of gat1 promoter construct cotransfected with control shRNA obtained in the absence of BMP2 has been set equal to 1. Results are expressed as the mean ± SD for three experiments. *p < 0.05, compared with control shRNA (ANOVA followed by Bonferroni correction).
Sites for Smad4, the major intracellular signaling effector for BMP signals, and YY1, a reported Smad4-interacting protein (Kurisaki et al., 2003), were found in the 46 bp element (P-Match). Each two shRNA oligonucleotides for smad4 or yy1 were designed and their effects were studied (Fig. 7C; supplemental Fig. S1A, available at www.jneurosci.org as supplemental material). Inhibiting the expression of smad4 or yy1 with shRNA in P19 cells or NSCs resulted in an increase of gat1 gene promoter construct transcription activity, and this effect was further accentuated by a combination of Smad4 shRNA and YY1 shRNA (Fig. 7D,E; supplemental Fig. S1B, available at www.jneurosci.org as supplemental material). Neither Smad4 shRNA nor YY1 shRNA had an effect on the transcriptional activity of the mutant gat1 gene promoter construct. However, in the presence of BMP2, knockdown of smad4 or yy1 expression resulted in a decrease of gat1 gene promoter construct transcription activity (Fig. 7F,G; supplemental Fig. S1C, available at www.jneurosci.org as supplemental material). Together, these data strongly suggest that Smad4 and YY1, at least in part, mediate BMP2 regulation of gat1 gene expression.
DNA-binding activity of the 46 bp element on gat1 gene promoter
The biotin-labeled 46 bp oligonucleotide derived from mouse gat1 gene promoter, from −333 to −288, was used as a probe (probe A) in EMSA. Extracts from the primary cortical neurons interacted with probe A and revealed multiple bands on gel electrophoresis (Fig. 8A). The bands were greatly diminished by an excess of unlabeled oligonucleotide probe or unlabeled SBE oligonucleotide containing a canonical Smad4 site. In contrast, a mutant unlabeled probe (ASBE3m) with a mutation in functional Smad4 site (supplemental Fig. S2A,B, available at www.jneurosci.org as supplemental material) failed to inhibit the binding activity. An unlabeled SBEm oligonucleotide containing the mutant Smad4 site also failed to inhibit the binding activity. Similar results also were observed in EMSA studies with extracts of NSCs (supplemental Fig. S3A, available at www.jneurosci.org as supplemental material) and P19 cells (supplemental Fig. S3B, available at www.jneurosci.org as supplemental material). The binding of Smad4 to the 46 bp element was further confirmed by EMSA experiments using a biotinylated SBE as a probe and an unlabeled wild-type or mutant probe A as a competitor (Fig. 8B).
Figure 8.

Traditional EMSA and ChIP localize Smad4 and YY1 bindings to gat1 gene promoter. A, EMSA competition experiments. Nuclear protein was isolated from primary cortical neurons, and EMSA was performed using biotin-labeled oligonucleotide from gat1 gene promoter from −333 to −288 as a probe. Lane 1, free probe; lanes 2 and 5, no competitor; lanes 3 and 4, unlabeled wild-type probe (A) or mutant probe (ASBE3m) as competitor; lanes 6–9, unlabeled wild-type or mutant SBE oligonucleotides as competitor. Indicated are the specific complexes (solid arrow) or nonspecific complexes (open arrows). Results are representative of at least three independent experiments. B, EMSA competition experiments using biotin-labeled SBE oligonucleotides as a probe. Lane 1, free probe; lane 2, no competitor; lanes 3 and 4, wild-type SBE oligonucleotides as competitors; lanes 5 and 6, mutant SBE oligonucleotides as competitors; lanes 7 and 8, wild-type A oligonucleotides as competitors; lanes 9 and 10, mutant ASBE3m (with the functional SBE mutant) oligonucleotides as competitors. Results are representative of at least three independent experiments. C, D, Protein interactions at the region from −333 to −288 of mouse gat1 gene in P19 cells (C) or primary cortical neurons (D) were determined by ChIP assay. IgG was used as control. Chromatin immunoprecipitated DNA was analyzed by real-time PCR.
Unlabeled canonical YY1-binding sites cannot inhibit the binding activity of probe A in competitive EMSAs (data not shown). It is possible that YY1 is tethered to DNA through interaction with another transcription factor (e.g., Smad4) in vivo, instead of binding directly to DNA. This possibility is supported by our observation that YY1 shRNA can induce the gat1 promoter construct activity, but not the mutant lacking Smad4-binding site (supplemental Fig. S2C, available at www.jneurosci.org as supplemental material).
The ChIP assay showed that Smad4 and YY1 are bound to the 46 bp element of gat1 gene promoter (Fig. 8C). Most interestingly, the Smad4-DNA and YY1-DNA interaction were detected in the chromatin from BMP2-treated P19 cells as well as in lysates derived from untreated cells. Similar results were achieved in primary cortical neurons (Fig. 8D).
Discussion
This study identified a promoter region in mouse gat1 gene with sufficient regulatory information to recapitulate endogenous gat1 expression. Deletion of the 46 bp BMP2-responsive element resulted in an increase in the number of LacZ-expressing cells and the overall LacZ-immunoreactive signal intensity. But, in GAT1-expressing cells, deletion of the 46 bp element resulted in a decrease in LacZ-immunoreactive signal intensity. Transcription factors Smad4 and YY1 were found to bind to the 46 bp element and potentiate or inhibit gat1 gene transcription activity depending on the BMP2 signal in the cellular environment.
BMPs and their receptors are abundantly expressed in the brain from early embryogenesis throughout adult life (Mehler et al., 1997; Zhang et al., 1998). Endogenous BMP signaling has been reported to influence the migration of GABAergic neurons (Li et al., 1998; Mabie et al., 1999). BMP2 has been reported to promote the terminal differentiation of striatal (Hattori et al., 1999) and cortical (Mabie et al., 1999; Yung et al., 2002) GABAergic neurons in culture. Loss of bmpr1a, a high-affinity receptor for BMP2, increases the number of calbindin-expressing GABAergic interneurons (Samanta et al., 2007). These observations collectively suggest that BMP2 plays a fundamental role in many aspects of GABAergic neurogenesis and regulation of GABAergic neuron-specific gene expression. Results from this study demonstrated that stimulation of the BMP2 signal could induce gat1 promoter constructs activity via a BMP2 response element.
Smad4 requires partners for regulating transcription of target genes (Attisano and Wrana, 2000). P-Match analysis showed that YY1-binding sites are adjacent to Smad4-binding sites in the 46 bp element of gat1 promoter. YY1 can physically interact with Smad4 both in vitro and in vivo, and functionally cooperate with Smad4 in response to BMP2 signals in epithelial and myoblastic cells (Kurisaki et al., 2003). These previous reports together with our data provide strong evidence that Smad4 and YY1 are synergistic in the regulation of gat1 promoter activity in response to BMP2 stimuli. However, more study is still needed to elucidate whether Smad4 and YY1 are required for BMP2 regulation of endogenous gat1 expression and function.
Smad4–YY1 complex binding to the cis-regulatory element had opposite effects on gat1 promoter constructs transcription activity in the absence versus presence of BMP2, suggesting the involvement of other cofactors in different cellular environment. Both Smad4 and YY1 can recruit histone acetyltransferases (e.g., P300/CBP) to form a transcriptional activation complex or histone deacetylases (HDACs) to form a transcriptional repressor complex (Austen et al., 1997; Thomas and Seto, 1999; Wotton et al., 1999; Yao et al., 2001). YY1 has been reported to repress target promoters by recruiting HDACs during oligodendrocyte differentiation (He et al., 2007). In this context, we hypothesized that the Smad4–YY1 complex recruits HDAC repressing gat1 expression in the absence of BMP2, whereas it recruits P300/CBP activating gat1 expression in the presence of BMP2 (Fig. 9). Support for this hypothesis comes from the observations that inhibition of HDACs by trichostatin A increased the Gat1 promoter constructs activity (supplemental Fig. S4, available at www.jneurosci.org as supplemental material) and that inhibition of endogenous p300 or cbp with shRNA abolished BMP2 induction of gat1 promoter constructs activity (supplemental Fig. S5A,B, available at www.jneurosci.org as supplemental material). The response of CBP–DNA interaction to BMP2 signals was confirmed by ChIP analysis on a 46 bp sequence in P19 cells (supplemental Fig. S5C, available at www.jneurosci.org as supplemental material).
Figure 9.

A model depicting the convergence of Smad4 and YY1 in BMP2-mediated gene transcription. Smad4 and YY1 bind to target gene promoter. In the absence of BMP2, Smad4/YY1 recruits HDACs and represses target gene transcription. In the presence of BMP2, Smad4/YY1 recruits histone acetyltransferases (P300/CBP) and activates target gene transcription.
Proper neuronal function requires orchestrated regulation of many genes that contribute to neurotransmitter synthesis, vesicular packaging, release, and termination. In C. elegans, the unc-25/gad and the unc-47/vgat were regulated by transcription factor unc-30 (Eastman et al., 1999; Westmoreland et al., 2001). So, it is reasonable to think that in mammals several common signaling molecules and transcription factors may also regulate expression of GABAergic neuron-specific genes. The promoters of gad (Makinae et al., 2000; Kobayashi et al., 2003) and vgat (Ebihara et al., 2003; Oh et al., 2005), two essential components of GABAergic neurons, have already been investigated in transgenic and transfection experiments. However, little was known about the upstream regulators, including signaling molecules and transcription factors. It is notable that adjacent Smad4 and YY1 consensus motifs were also found in the promoter regions of other GABAergic neuron-specific genes, such as gat4, gad1-2, and vgat. So, our results also provide a new idea to understand other GABAergic neuron-specific gene expression regulation.
Footnotes
This work was supported by grants from the National Natural Science Foundation of China (30670438), the National Key Basic Research Program (2002CB713803), the National High Technology Research and Development Program (2008AA02Z126), the Science and Technology Commission of Shanghai Municipality (07DZ19503, 06DZ19004), and E-Institutes of Shanghai Municipal Education Commission (E03003). We are grateful to Dr. Fang Huang, Dr. Mei Yu, and Jiajuan Shen for technical assistance, and Dr. Kehong Zhang from Ivy Editing for language editing. We thank Xixia Zhou for animal care.
References
- Allouche A, Nolens G, Tancredi A, Delacroix L, Mardaga J, Fridman V, Winkler R, Boniver J, Delvenne P, Begon DY. The combined immunodetection of AP-2alpha and YY1 transcription factors is associated with ERBB2 gene overexpression in primary breast tumors. Breast Cancer Res. 2008;10:R9. doi: 10.1186/bcr1851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson SA, Eisenstat DD, Shi L, Rubenstein JL. Interneuron migration from basal forebrain to neocortex: dependence on Dlx genes. Science. 1997;278:474–476. doi: 10.1126/science.278.5337.474. [DOI] [PubMed] [Google Scholar]
- Attisano L, Wrana JL. Smads as transcriptional co-modulators. Curr Opin Cell Biol. 2000;12:235–243. doi: 10.1016/s0955-0674(99)00081-2. [DOI] [PubMed] [Google Scholar]
- Austen M, Lüscher B, Lüscher-Firzlaff JM. Characterization of the transcriptional regulator YY1. The bipartite transactivation domain is independent of interaction with the TATA box-binding protein, transcription factor IIB, TAFII55, or cAMP-responsive element-binding protein (CPB)-binding protein. J Biol Chem. 1997;272:1709–1717. doi: 10.1074/jbc.272.3.1709. [DOI] [PubMed] [Google Scholar]
- Bae S, Bessho Y, Hojo M, Kageyama R. The bHLH gene Hes6, an inhibitor of Hes1, promotes neuronal differentiation. Development. 2000;127:2933–2943. doi: 10.1242/dev.127.13.2933. [DOI] [PubMed] [Google Scholar]
- Baraban SC. Antiepileptic actions of neuropeptide Y in the mouse hippocampus require Y5 receptors. Epilepsia. 2002;43([Suppl] 5):9–13. doi: 10.1046/j.1528-1157.43.s.5.13.x. [DOI] [PubMed] [Google Scholar]
- Borden LA. GABA transporter heterogeneity: pharmacology and cellular localization. Neurochem Int. 1996;29:335–356. doi: 10.1016/0197-0186(95)00158-1. [DOI] [PubMed] [Google Scholar]
- Casarosa S, Fode C, Guillemot F. Mash1 regulates neurogenesis in the ventral telencephalon. Development. 1999;126:525–534. doi: 10.1242/dev.126.3.525. [DOI] [PubMed] [Google Scholar]
- Chiu CS, Jensen K, Sokolova I, Wang D, Li M, Deshpande P, Davidson N, Mody I, Quick MW, Quake SR, Lester HA. Number, density, and surface/cytoplasmic distribution of GABA transporters at presynaptic structures of knock-in mice carrying GABA transporter subtype 1-green fluorescent protein fusions. J Neurosci. 2002;22:10251–10266. doi: 10.1523/JNEUROSCI.22-23-10251.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiu CS, Brickley S, Jensen K, Southwell A, Mckinney S, Cull-Candy S, Mody I, Lester HA. GABA transporter deficiency causes tremor, ataxia, nervousness, and increased GABA-induced tonic conductance in cerebellum. J Neurosci. 2005;25:3234–3245. doi: 10.1523/JNEUROSCI.3364-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cossart R, Bernard C, Ben-Ari Y. Multiple facets of GABAergic neurons and synapses: multiple fates of GABA signalling in epilepsies. Trends Neurosci. 2005;28:108–115. doi: 10.1016/j.tins.2004.11.011. [DOI] [PubMed] [Google Scholar]
- DeFelipe J. Chandelier cells and epilepsy. Brain. 1999;122:1807–1822. doi: 10.1093/brain/122.10.1807. [DOI] [PubMed] [Google Scholar]
- de la Pompa JL, Wakeham A, Correia KM, Samper E, Brown S, Aguilera RJ, Nakano T, Honjo T, Mak TW, Rossant J, Conlon RA. Conservation of the Notch signalling pathway in mammalian neurogenesis. Development. 1997;124:1139–1148. doi: 10.1242/dev.124.6.1139. [DOI] [PubMed] [Google Scholar]
- Eastman C, Horvitz HR, Jin Y. Coordinated transcriptional regulation of the unc-25 glutamic acid decarboxylase and the unc-47 GABA vesicular transporter by the Caenorhabditis elegans UNC-30 homeodomain protein. J Neurosci. 1999;19:6225–6234. doi: 10.1523/JNEUROSCI.19-15-06225.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebihara S, Obata K, Yanagawa Y. Mouse vesicular GABA transporter gene: genomic organization, transcriptional regulation and chromosomal localization. Brain Res Mol Brain Res. 2003;110:126–139. doi: 10.1016/s0169-328x(02)00648-4. [DOI] [PubMed] [Google Scholar]
- Fode C, Ma Q, Casarosa S, Ang SL, Anderson DJ, Guillemot F. A role for neural determination genes in specifying the dorsoventral identity of telencephalic neurons. Genes Dev. 2000;14:67–80. [PMC free article] [PubMed] [Google Scholar]
- Fueta Y, Vasilets LA, Takeda K, Kawamura M, Schwarz W. Down-regulation of GABA-transporter function by hippocampal translation products: its possible role in epilepsy. Neuroscience. 2003;118:371–378. doi: 10.1016/s0306-4522(02)00924-7. [DOI] [PubMed] [Google Scholar]
- Gulacsi A, Lillien L. Sonic hedgehog and bone morphogenetic protein regulate interneuron development from dorsal telencephalic progenitors in vitro. J Neurosci. 2003;23:9862–9872. doi: 10.1523/JNEUROSCI.23-30-09862.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hattori A, Katayama M, Iwasaki S, Ishii K, Tsujimoto M, Kohno M. Bone morphogenetic protein-2 promotes survival and differentiation of striatal GABAergic neurons in the absence of glial cell proliferation. J Neurochem. 1999;72:2264–2271. doi: 10.1046/j.1471-4159.1999.0722264.x. [DOI] [PubMed] [Google Scholar]
- He Y, Dupree J, Wang J, Sandoval J, Li J, Liu H, Shi Y, Nave KA, Casaccia-Bonnefil P. The transcription factor Yin Yang 1 is essential for oligodendrocyte progenitor differentiation. Neuron. 2007;55:217–230. doi: 10.1016/j.neuron.2007.06.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heilig M, Thorsell A. Brain neuropeptide Y (NPY) in stress and alcohol dependence. Rev Neurosci. 2002;13:85–94. doi: 10.1515/revneuro.2002.13.1.85. [DOI] [PubMed] [Google Scholar]
- Hirunsatit R, George ED, Lipska BK, Elwafi HM, Sander L, Yrigollen CM, Gelernter J, Grigorenko EL, Lappalainen J, Mane S, Nairn AC, Kleinman JE, Simen AA. Twenty-one-base-pair insertion polymorphism creates an enhancer element and potentiates SLC6A1 GABA transporter promoter activity. Pharmacogenet Genomics. 2009;19:53–65. doi: 10.1097/FPC.0b013e328318b21a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang KW, Gao F, Shui QX, Yu ZS, Xia ZZ. Effect of diazoxide on regulation of vesicular and plasma membrane GABA transporter genes and proteins in hippocampus of rats subjected to picrotoxin-induced kindling. Neurosci Res. 2004;50:319–329. doi: 10.1016/j.neures.2004.08.001. [DOI] [PubMed] [Google Scholar]
- Jiang L, Yao M, Shi J, Shen P, Niu G, Fei J. Yin yang 1 directly regulates the transcription of RE-1 silencing transcription factor. J Neurosci Res. 2008;86:1209–1216. doi: 10.1002/jnr.21595. [DOI] [PubMed] [Google Scholar]
- Kabos P, Kabosova A, Neuman T. Blocking HES1 expression initiates GABAergic differentiation and induces the expression of p21(CIP1/WAF1) in human neural stem cells. J Biol Chem. 2002;277:8763–8766. doi: 10.1074/jbc.C100758200. [DOI] [PubMed] [Google Scholar]
- Kobayashi T, Ebihara S, Ishii K, Kobayashi T, Nishijima M, Endo S, Takaku A, Sakagami H, Kondo H, Tashiro F, Miyazaki J, Obata K, Tamura S, Yanagawa Y. Structural and functional characterization of mouse glutamate decarboxylase 67 gene promoter. Biochim Biophys Acta. 2003;1628:156–168. doi: 10.1016/s0167-4781(03)00138-6. [DOI] [PubMed] [Google Scholar]
- Kroll TT, O'Leary DD. Ventralized dorsal telencephalic progenitors in Pax6 mutant mice generate GABA interneurons of a lateral ganglionic eminence fate. Proc Natl Acad Sci U S A. 2005;102:7374–7379. doi: 10.1073/pnas.0500819102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurisaki K, Kurisaki A, Valcourt U, Terentiev AA, Pardali K, Ten Dijke P, Heldin CH, Ericsson J, Moustakas A. Nuclear factor YY1 inhibits transforming growth factor beta- and bone morphogenetic protein-induced cell differentiation. Mol Cell Biol. 2003;23:4494–4510. doi: 10.1128/MCB.23.13.4494-4510.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JC, Mayer-Proschel M, Rao MS. Gliogenesis in the central nervous system. Glia. 2000;30:105–121. doi: 10.1002/(sici)1098-1136(200004)30:2<105::aid-glia1>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]
- Lewis DA, Pierri JN, Volk DW, Melchitzky DS, Woo TU. Altered GABA neurotransmission and prefrontal cortical dysfunction in schizophrenia. Biol Psychiatry. 1999;46:616–626. doi: 10.1016/s0006-3223(99)00061-x. [DOI] [PubMed] [Google Scholar]
- Lewis DA, Hashimoto T, Volk DW. Cortical inhibitory neurons and schizophrenia. Nat Rev Neurosci. 2005;6:312–324. doi: 10.1038/nrn1648. [DOI] [PubMed] [Google Scholar]
- Liu GX, Cai GQ, Cai YQ, Sheng ZJ, Jiang J, Mei Z, Wang ZG, Guo L, Fei J. Reduced anxiety and depression-like behaviors in mice lacking GABA transporter subtype 1. Neuropsychopharmacology. 2007;32:1531–1539. doi: 10.1038/sj.npp.1301281. [DOI] [PubMed] [Google Scholar]
- Liu QR, López-Corcuera B, Mandiyan S, Nelson H, Nelson N. Molecular characterization of four pharmacologically distinct gamma-aminobutyric acid transporters in mouse brain [corrected] J Biol Chem. 1993;268:2106–2112. [PubMed] [Google Scholar]
- Li W, Cogswell CA, LoTurco JJ. Neuronal differentiation of precursors in the neocortical ventricular zone is triggered by BMP. J Neurosci. 1998;18:8853–8862. doi: 10.1523/JNEUROSCI.18-21-08853.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- Ma Y, Hu JH, Zhao WJ, Fei J, Yu Y, Zhou XG, Mei ZT, Guo LH. Overexpression of gamma-aminobutyric acid transporter subtype I leads to susceptibility to kainic acid-induced seizure in transgenic mice. Cell Res. 2001;11:61–67. doi: 10.1038/sj.cr.7290067. [DOI] [PubMed] [Google Scholar]
- Mabie PC, Mehler MF, Kessler JA. Multiple roles of bone morphogenetic protein signaling in the regulation of cortical cell number and phenotype. J Neurosci. 1999;19:7077–7088. doi: 10.1523/JNEUROSCI.19-16-07077.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makinae K, Kobayashi T, Kobayashi T, Shinkawa H, Sakagami H, Kondo H, Tashiro F, Miyazaki J, Obata K, Tamura S, Yanagawa Y. Structure of the mouse glutamate decarboxylase 65 gene and its promoter: preferential expression of its promoter in the GABAergic neurons of transgenic mice. J Neurochem. 2000;75:1429–1437. doi: 10.1046/j.1471-4159.2000.0751429.x. [DOI] [PubMed] [Google Scholar]
- Mehler MF, Mabie PC, Zhang D, Kessler JA. Bone morphogenetic proteins in the nervous system. Trends Neurosci. 1997;20:309–317. doi: 10.1016/s0166-2236(96)01046-6. [DOI] [PubMed] [Google Scholar]
- Miyoshi G, Bessho Y, Yamada S, Kageyama R. Identification of a novel basic helix-loop-helix gene, Heslike, and its role in GABAergic neurogenesis. J Neurosci. 2004;24:3672–3682. doi: 10.1523/JNEUROSCI.5327-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakatani T, Minaki Y, Kumai M, Ono Y. Helt determines GABAergic over glutamatergic neuronal fate by repressing Ngn genes in the developing mesencephalon. Development. 2007;134:2783–2793. doi: 10.1242/dev.02870. [DOI] [PubMed] [Google Scholar]
- Oh WJ, Noggle SA, Maddox DM, Condie BG. The mouse vesicular inhibitory amino acid transporter gene: expression during embryogenesis, analysis of its core promoter in neural stem cells and a reconsideration of its alternate splicing. Gene. 2005;351:39–49. doi: 10.1016/j.gene.2005.01.009. [DOI] [PubMed] [Google Scholar]
- Peng W, Simantov R. Altered gene expression in frontal cortex and midbrain of 3,4-methylenedioxymethamphetamine (MDMA) treated mice: differential regulation of GABA transporter subtypes. J Neurosci Res. 2003;72:250–258. doi: 10.1002/jnr.10571. [DOI] [PubMed] [Google Scholar]
- Pozas E, Ibáñez CF. GDNF and GFRalpha1 promote differentiation and tangential migration of cortical GABAergic neurons. Neuron. 2005;45:701–713. doi: 10.1016/j.neuron.2005.01.043. [DOI] [PubMed] [Google Scholar]
- Rees JR, Onwuegbusi BA, Save VE, Alderson D, Fitzgerald RC. In vivo and in vitro evidence for transforming growth factor-beta1-mediated epithelial to mesenchymal transition in esophageal adenocarcinoma. Cancer Res. 2006;66:9583–9590. doi: 10.1158/0008-5472.CAN-06-1842. [DOI] [PubMed] [Google Scholar]
- Ruzicka WB, Zhubi A, Veldic M, Grayson DR, Costa E, Guidotti A. Selective epigenetic alteration of layer I GABAergic neurons isolated from prefrontal cortex of schizophrenia patients using laser-assisted microdissection. Mol Psychiatry. 2007;12:385–397. doi: 10.1038/sj.mp.4001954. [DOI] [PubMed] [Google Scholar]
- Samanta J, Burke GM, McGuire T, Pisarek AJ, Mukhopadhyay A, Mishina Y, Kessler JA. BMPR1a signaling determines numbers of oligodendrocytes and calbindin-expressing interneurons in the cortex. J Neurosci. 2007;27:7397–7407. doi: 10.1523/JNEUROSCI.1434-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuurmans C, Armant O, Nieto M, Stenman JM, Britz O, Klenin N, Brown C, Langevin LM, Seibt J, Tang H, Cunningham JM, Dyck R, Walsh C, Campbell K, Polleux F, Guillemot F. Sequential phases of cortical specification involve Neurogenin-dependent and -independent pathways. EMBO J. 2004;23:2892–2902. doi: 10.1038/sj.emboj.7600278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sperk G, Schwarzer C, Heilman J, Furtinger S, Reimer RJ, Edwards RH, Nelson N. Expression of plasma membrane GABA transporters but not of the vesicular GABA transporter in dentate granule cells after kainic acid seizures. Hippocampus. 2003;13:806–815. doi: 10.1002/hipo.10133. [DOI] [PubMed] [Google Scholar]
- Sussel L, Marin O, Kimura S, Rubenstein JL. Loss of Nkx2.1 homeobox gene function results in a ventral to dorsal molecular respecification within the basal telencephalon: evidence for a transformation of the pallidum into the striatum. Development. 1999;126:3359–3370. doi: 10.1242/dev.126.15.3359. [DOI] [PubMed] [Google Scholar]
- Thoeringer CK, Ripke S, Unschuld PG, Lucae S, Ising M, Bettecken T, Uhr M, Keck ME, Mueller-Myhsok B, Holsboer F, Binder EB, Erhardt A. The GABA transporter 1 (SLC6A1): a novel candidate gene for anxiety disorders. J Neural Transm. 2009;116:649–657. doi: 10.1007/s00702-008-0075-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas MJ, Seto E. Unlocking the mechanisms of transcription factor YY1: are chromatin modifying enzymes the key? Gene. 1999;236:197–208. doi: 10.1016/s0378-1119(99)00261-9. [DOI] [PubMed] [Google Scholar]
- Thuault S, Valcourt U, Petersen M, Manfioletti G, Heldin CH, Moustakas A. Transforming growth factor-beta employs HMGA2 to elicit epithelial-mesenchymal transition. J Cell Biol. 2006;174:175–183. doi: 10.1083/jcb.200512110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volk D, Austin M, Pierri J, Sampson A, Lewis D. GABA transporter-1 mRNA in the prefrontal cortex in schizophrenia: decreased expression in a subset of neurons. Am J Psychiatry. 2001;158:256–265. doi: 10.1176/appi.ajp.158.2.256. [DOI] [PubMed] [Google Scholar]
- Volk DW, Lewis DA. Impaired prefrontal inhibition in schizophrenia: relevance for cognitive dysfunction. Physiol Behav. 2002;77:501–505. doi: 10.1016/s0031-9384(02)00936-8. [DOI] [PubMed] [Google Scholar]
- Westmoreland JJ, McEwen J, Moore BA, Jin Y, Condie BG. Conserved function of Caenorhabditis elegans UNC-30 and mouse Pitx2 in controlling GABAergic neuron differentiation. J Neurosci. 2001;21:6810–6819. doi: 10.1523/JNEUROSCI.21-17-06810.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woo TU, Whitehead RE, Melchitzky DS, Lewis DA. A subclass of prefrontal gamma-aminobutyric acid axon terminals are selectively altered in schizophrenia. Proc Natl Acad Sci U S A. 1998;95:5341–5346. doi: 10.1073/pnas.95.9.5341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wotton D, Lo RS, Lee S, Massagué J. A Smad transcriptional corepressor. Cell. 1999;97:29–39. doi: 10.1016/s0092-8674(00)80712-6. [DOI] [PubMed] [Google Scholar]
- Yao YL, Yang WM, Seto E. Regulation of transcription factor YY1 by acetylation and deacetylation. Mol Cell Biol. 2001;21:5979–5991. doi: 10.1128/MCB.21.17.5979-5991.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yung SY, Gokhan S, Jurcsak J, Molero AE, Abrajano JJ, Mehler MF. Differential modulation of BMP signaling promotes the elaboration of cerebral cortical GABAergic neurons or oligodendrocytes from a common sonic hedgehog-responsive ventral forebrain progenitor species. Proc Natl Acad Sci U S A. 2002;99:16273–16278. doi: 10.1073/pnas.232586699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang D, Mehler MF, Song Q, Kessler JA. Development of bone morphogenetic protein receptors in the nervous system and possible roles in regulating trkC expression. J Neurosci. 1998;18:3314–3326. doi: 10.1523/JNEUROSCI.18-09-03314.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao WJ, Ma YH, Fei J, Mei ZT, Guo LH. Increase in drug-induced seizure susceptibility of transgenic mice overexpressing GABA transporter-1. Acta Pharmacol Sin. 2003;24:991–995. [PubMed] [Google Scholar]
- Zink M, Schmitt A, May B, Müller B, Braus DF, Henn FA. Differential effects of long-term treatment with clozapine or haloperidol on GABA transporter expression. Pharmacopsychiatry. 2004;37:171–174. doi: 10.1055/s-2004-827173. [DOI] [PubMed] [Google Scholar]