

Abstract
Peripheral nerve injury causes neuropathic pain, which is characterized by the paradoxical sensations of positive and negative symptoms. Clinically, negative signs are frequently observed; however, their underlying molecular mechanisms are largely unknown. Dysfunction of C-fibers is assumed to underlie negative symptoms and is accompanied by long-lasting downregulation of Nav1.8 sodium channel and μ-opioid receptor (MOP) in the dorsal root ganglion (DRG). In the present study, we found that nerve injury upregulates neuron-restrictive silencer factor (NRSF) expression in the DRG neurons mediated through epigenetic mechanisms. In addition, chromatin immunoprecipitation analysis revealed that nerve injury promotes NRSF binding to the neuron-restrictive silencer element within MOP and Nav1.8 genes, thereby causing epigenetic silencing. Furthermore, NRSF knockdown significantly blocked nerve injury-induced downregulations of MOP and Nav1.8 gene expressions, C-fiber hypoesthesia, and the losses of peripheral morphine analgesia and Nav1.8-selective blocker-induced hypoesthesia. Together, these data suggest that NRSF causes pathological and pharmacological dysfunction of C-fibers, which underlies the negative symptoms in neuropathic pain.
Introduction
Neuropathic pain is characterized by the paradoxical sensations of positive (hyperalgesia, allodynia, and paresthesia) and negative (hypoesthesia, hypoalgesia) symptoms (Baron, 2006), and negative signs are frequently observed during clinical sensory examinations (Devigili et al., 2008; Leffler and Hansson, 2008). The molecular mechanisms underlying positive symptoms have been extensively investigated (Devor, 2006; Costigan et al., 2009); however, those underlying negative symptoms are much less well understood. A possible mechanism for negative symptoms is a dysfunction of small-diameter (C)-fibers (Taylor, 2001; Devigili et al., 2008; Costigan et al., 2009), such as a loss of C-fiber terminals, an impairment of C-fiber-mediated axon-reflex flare responses, or an increase in the threshold against C-fiber-specific stimuli (Fields et al., 1998; Ueda, 2008). Such C-fiber dysfunctions have been implicated in the manifestation of positive symptoms as well as of negative ones, possibly through a synaptic reorganization in the spinal dorsal horn (Taylor, 2001; Ueda, 2008; Costigan et al., 2009). Representative examples for negative symptoms were observed with long-lasting downregulations of Nav1.8 sodium channel and μ-opioid receptor (MOP) in C-fibers (Waxman et al., 1999; Rashid et al., 2004; Kohno et al., 2005), which are essential for C-fiber functions in terms of determining pain thresholds (Akopian et al., 1999) and for the pharmacological actions of μ-opioids (Dickenson and Kieffer, 2006), respectively.
In terms of long-lasting transcriptional regulation, the transcription factor-mediated epigenetic mechanisms have been demonstrated to play a key role (Borrelli et al., 2008). Neuron-restrictive silencer factor (NRSF, also known as REST) functions as a transcriptional repressor of genes that contain neuron-restrictive silencer element (NRSE, also called RE1) (Chong et al., 1995; Schoenherr and Anderson, 1995). NRSF, when it binds to NRSE, recruits histone deacetylase (HDAC) through its corepressors, mSin3 and CoREST, for generating a repressive chromatin environment (Ballas and Mandel, 2005). It has been reported that NRSF represses transcription of MOP gene through HDAC-mediated mechanisms (Kim et al., 2004). Here, we show that nerve injury induces a long-lasting NRSF expression in the dorsal root ganglion (DRG), thereby causing epigenetic silencing of MOP gene and loss of pharmacological target for peripheral morphine analgesia. Furthermore, we also investigated the possible epigenetic silencing of Nav1.8 gene, which has unique forward and reverse NRSE sequences.
Materials and Methods
Animals and surgery.
Male C57BL/6J mice weighing 20–25 g were used. They were kept in a room with a temperature of 21 ± 2°C with ad libitum access to a standard laboratory diet and tap water. All procedures were approved by the Nagasaki University Animal Care Committee (Nagasaki, Japan) and complied with the recommendations of the International Association for the Study of Pain (Zimmermann, 1983). Partial ligation of the sciatic nerve was performed under pentobarbital (50 mg/kg) anesthesia, following the methods of Malmberg and Basbaum (1998).
Oligonucleotide treatments.
The antisense oligodeoxynucleotide (AS-ODN) was designed to target the mouse NRSF sequence and corresponds to the rat sequence targeted for antisense knockdown previously (Calderone et al., 2003). AS-ODN (5′-CGGAAGGGCTTGGCC-3′) and its mismatch scrambled oligodeoxynucleotide (MS-ODN; 5′-GTCGTCGGCGGAGCA-3′) were synthesized and freshly dissolved in artificial CSF (aCSF) containing the following (in mm): 125 NaCl, 3.8 KCl, 2.0 CaCl2, 1.0 MgCl2, 1.2 KH2PO4, 26 NaHCO3, 10 glucose, pH 7.4). AS-ODN or MS-ODN was intrathecally injected at a dose of 10 μg per 5 μl of aCSF on the first, third, and fifth days. Then, nerve injury was performed with subsequent injections of AS-ODN on days 1, 3, 5, and 6 postinjury. The mRNA levels, pain thresholds, peripheral morphine analgesia, and peripheral A-803467 hypoesthesia were assessed at day 7 postinjury.
Nociception test.
In thermal paw withdrawal tests, the nociception threshold was evaluated by the latency of paw withdrawal upon a thermal stimulus (Hargreaves et al., 1988; Inoue et al., 2004). Unanesthetized animals were placed in Plexiglas cages on top of a glass sheet, and an adaptation period of 1 h was allowed. The thermal stimulator (IITC Life Science) was positioned under the glass sheet and the focus of the projection bulb was aimed exactly at the middle of the plantar surface of the animal. A mirror attached to the stimulator permitted visualization of the plantar surface. A cutoff time of 20 s was set to prevent tissue damage. The mechanical paw pressure test was performed as described previously (Rashid et al., 2003; Inoue et al., 2004). Briefly, mice were placed in a Plexiglas chamber on a 6 × 6 mm wire mesh grid floor and allowed to acclimatize for a period of 1 h. A mechanical stimulus was then delivered to the middle of the plantar surface of the right hindpaw using a transducer indicator (model 1601; IITC Life Science). The pressure needed to induce a flexor response was defined as the pain threshold. A cutoff pressure of 20 g was set to avoid tissue damage. In these experiments, using mechanical and thermal tests, the thresholds were determined from three repeated challenges at 10 min intervals, and the averages of responses were evaluated. An electrical stimulation-induced paw withdrawal (EPW) test was performed as described previously (Matsumoto et al., 2008). Briefly, electrodes (Neurotron) were fastened to the plantar surfaces and insteps of mice. Transcutaneous nerve stimuli with each of the three sine-wave pulses (5, 250, and 2000 Hz) were applied using a Neurometer CPT/C system (Neurotron). The minimum intensity (μA) at which each mouse withdrew its paw was defined as the current stimulus threshold. Investigators blind to drug treatments performed all behavioral experiments.
Drug treatments.
Morphine hydrochloride (Takeda Chemical Industries) was dissolved in physiological saline. Saline was used for control injections. Intraplantar injections were given using a Hamilton microsyringe connected to polyethylene tubing with a 30 gauge hypodermic needle. For the time course experiment, we measured the paw-withdrawal latencies at every 10 min interval until 60 min after intraplantar injection of morphine (30 nmol), as reported previously (Rashid et al., 2004). In the area under the curve analysis of peripheral morphine analgesia, we calculated the area under the curve generated by plotting analgesic threshold (after deducting the control threshold from each threshold point) against time, from 10 to 60 min after morphine treatment, using a trapezoidal method. A-803467 (Biomol), a selective blocker for Nav1.8 (Jarvis et al., 2007), was dissolved in dimethyl sulfoxide. Before administration, A-803467 was diluted 15-fold for intraperitoneal injection and 28-fold for intraplantar injection in saline, respectively. The EPW test was performed 30 min after intraperitoneal injection of A-803467 (10 mg/kg), as reported previously (Jarvis et al., 2007).
Quantitative real-time PCR.
Total RNA was extracted from L4-6 DRGs using TRIzol (Invitrogen), and 500 ng of RNA was used for cDNA synthesis. Quantitative real-time PCR was performed with qPCR MasterMix Plus for SYBR Green I (Eurogentec) using the ABI Prism 7000 sequence detection system (Applied Biosystems). The PCR primers used are listed in supplemental Table 1, available at www.jneurosci.org as supplemental material. Some of the primers were published previously (Koenigsberger et al., 2000; Klein et al., 2003; Qiang et al., 2005; Matsumoto et al., 2006; Cheng et al., 2009; Staaf et al., 2009). Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as an internal control for normalization. In all cases, the validity of amplification was confirmed by the presence of a single peak in the melting temperature analysis and by linear amplification with increasing number of PCR cycles.
Western blot analysis.
The L4-6 DRGs from three mice were pooled. DRG samples were homogenized twice in ice-cold cell lysis buffer [10 mm Tris-HCl, pH 8.0, 10 mm NaCl, 0.2% Nonidet P-40, 1 μm (p-amidinophenyl)methanesulfonyl fluoride hydrochloride (p-APMSF)], and then the homogenates were centrifuged to remove contaminating cytosol. Crude nuclear fractions (30 μg) were separated by SDS-PAGE on 7.5% (NRSF) or 15% (histone H3) gels. The primary antibodies were used in the following dilutions: NRSF (1:500; Millipore) and histone H3 (1:500; Millipore). Immunoreactive signals for NRSF (200 kDa) and histone H3 (17 kDa) were detected using enhanced chemiluminescent substrate (SuperSignal West Pico chemiluminescent substrate; Pierce).
Immunohistochemistry.
Mice were deeply anesthetized with pentobarbital (50 mg/kg, i.p.) and perfused transcardially with 20 ml of potassium-free PBS (K+-free PBS, pH 7.4), followed by 50 ml of a 4% paraformaldehyde solution. The L4-6 DRGs were isolated, postfixed for 3 h, and cryoprotected overnight in a 25% sucrose solution. Tissues were fast frozen in cryo-embedding compound in a mixture of ethanol and dry ice and stored at −80°C until use. DRGs were cut on a cryostat at a thickness of 10 μm, thaw mounted on silane-coated glass slides, and air dried overnight at room temperature (RT). Before immunolabeling, antigen unmasking was performed by microwave treatment three times (10 min each) in 10 mm citrate buffer (pH 6.0). The DRG sections were then incubated with 50 and 100% methanol for 5 min, respectively, and washed with PBST (0.1% Triton X-100 in K+-free PBS). The sections were incubated with blocking buffer containing 3% BSA in PBST and subsequently reacted with rabbit polyclonal NRSF antibody (1:200) overnight at 4°C. After washing, the sections were incubated with secondary antibody, Alexa Fluor 594-conjugated anti-rabbit IgG (1:300; Invitrogen), for 2 h at RT. For double immunolabeling, we used the following antibodies: mouse monoclonal antibody against neuron-specific nuclear protein (anti-NeuN; 1:500; Millipore) and Alexa Fluor 488-conjugated anti-mouse IgG (1:300; Invitrogen). After washing, the sections were mounted with Shandon PermaFluor (Thermo Scientific) and analyzed using a confocal laser scanning microscope (PASCAL, Zeiss).
Chromatin immunoprecipitation assay.
Chromatin immunoprecipitation (ChIP) assays were performed using protocols from Millipore and from a previous report (Kubat et al., 2004) with some modifications. For each ChIP assay, the L4-6 DRGs from two mice were pooled. DRG samples were homogenized in ice-cold cell lysis buffer (10 mm Tris-HCl, pH 8.0, 10 mm NaCl, 0.2% Nonidet P-40, 1 μm p-APMSF). Samples were then cross-linked in PBS containing 1% formaldehyde at 37°C for 5 min. The cross-linking reaction was terminated with glycine (0.125 m) and, after repeated washing with PBS, the samples were resuspended in SDS lysis buffer (50 mm Tris-HCl, pH 8.1, 10 mm EDTA, 1% SDS, 1 μm p-APMSF). The chromatin was sheared by sonication into 200–500 bp fragments. Ten percent of each lysate was used as the input control for normalization. The sheared chromatin was diluted 10-fold in ChIP dilution buffer (16.7 mm Tris-HCl, pH 8.1, 1.2 mm EDTA, 167 mm NaCl, 1.1% Triton X-100, 0.01% SDS, 1 μm p-APMSF) and then precleared with protein A-agarose beads (Millipore) for 45 min at 4°C with rotation. The supernatant was incubated overnight at 4°C with anti-NRSF (5 μg), anti-acetyl-H3 (5 μg; Millipore), anti-acetyl-H4 antibodies (5 μl; Millipore), or normal rabbit IgG (5 μg; Santa Cruz Biotechnology). Complexes were collected for 2 h using protein A-agarose beads. Following washing and elution steps, cross-linking was reversed at 65°C for 4 h in the presence of 0.2 m NaCl. After proteinase K treatment for 1 h at 45°C, DNA was purified by phenol/chloroform extraction, dissolved in 50 μl of TE buffer (10 mm Tris-HCl, pH 8.0, 1 mm EDTA), and used for PCR. The PCR products were analyzed on a 2% agarose gel. PCR primers used are listed in supplemental Table 1, available at www.jneurosci.org as supplemental material. The primers for MOP-NRSE were published previously (Kim et al., 2004). Quantitative real-time PCR was performed as described above. In all cases, the validity of amplification was confirmed by the presence of a single peak in the melting temperature analysis and by linear amplification with increasing number of PCR cycles.
Statistical analysis.
The differences between multiple groups were analyzed using a one-way ANOVA with Tukey–Kramer multiple-comparison post hoc analysis (see Figs. 4 B,C, 5, 6 B; also see supplemental Fig. S2, available at www.jneurosci.org as supplemental material). Data were analyzed using Student's t test (see Figs. 1 A, 2, 3, 4 A, 6 A; also see supplemental Fig. S3, available at www.jneurosci.org as supplemental material). The criterion of significance was set at p < 0.05. All results are expressed as means ± SEM.
Figure 4.

Blockade of nerve injury-induced MOP and Nav1.8 downregulations by NRSF knockdown. Mice were intrathecally pretreated with vehicle (Veh), AS-ODN (AS) against NRSF, or the corresponding MS-ODN (MS). A, AS-ODN-induced reduction of NRSF protein expression in the DRG, assessed by Western blot analysis. Data are normalized to histone H3 protein expression levels and then expressed as percentages of the levels in the Veh-treated group. *p < 0.05 versus Veh-treated group. Data represent the means ± SEM from three mice. B, C, The effects of NRSF AS-ODN on nerve injury-induced downregulations of MOP and Nav1.8 (B) and TRPM8, TRPA1, and CGRP (C) in the DRG at day 7 postinjury. The mRNA expression levels were quantified by real-time PCR, and normalized to that of GAPDH mRNA. Results are calculated as percentages of Veh-treated and sham-operated groups and expressed as means ± SEM from at least three mice. *p < 0.05 versus Veh-treated and sham-operated groups and # p < 0.05 versus MS-ODN-treated and nerve-injured groups.
Figure 5.

Blockade of nerve injury-induced C-fiber hypoesthesia and loss of peripheral A-803467 hypoesthesia by NRSF knockdown. A–C, Blockade of nerve injury-induced hypoesthesia of C-fiber (5 Hz) (A), but not hypersensitization of Aδ-fiber (250 Hz) (B) and Aβ-fiber (2000 Hz) (C) by NRSF AS-ODN. Paw withdrawal thresholds to electrical stimulation (μA) were measured using the EPW test. *p < 0.05 versus vehicle (Veh)-treated and sham-operated groups and # p < 0.05 versus Veh-treated and nerve-injured groups. D, Recovery of nerve injury-induced loss of peripheral A-803467 hypoesthesia by AS-ODN. The C-fiber responses were assessed 30 min after intraplantar injection of control (Cont) or A-803467 (30 nmol). *p < 0.05 versus Veh-treated, sham-operated, and Cont-treated groups. # p < 0.05 versus Veh-treated, nerve-injured, and Cont-treated groups. + p < 0.05 versus AS-ODN-treated, nerve-injured, and Cont-treated groups. Data represent the means ± SEM from at least three mice.
Figure 6.

Blockade of nerve injury-induced loss of peripheral morphine analgesia by NRSF knockdown. Thermal pain threshold was assessed at day 7 postinjury by using a thermal paw withdrawal test. A, Time course of thermal paw withdrawal latencies (PWL, in seconds) after morphine (30 nmol, intraplantar) injection. *p, # p, or + p < 0.05 versus corresponding 0 min, respectively. B, Comparison of morphine analgesia by area under the curve (AUC). *p < 0.05 versus vehicle (Veh)-treated and sham-operated groups and # p < 0.05 versus Veh-treated and nerve-injured groups. Data are expressed as the means ± SEM from at least four mice.
Figure 1.

Downregulation of NRSE-containing MOP and Nav1.8 genes after nerve injury. A, Time course of MOP and Nav1.8 mRNA expressions in the DRG after nerve injury. The mRNA expression levels were assessed using quantitative real-time PCR and normalized to that of GAPDH mRNA. Data are calculated as percentages of day 0 and expressed as the means ± SEM from at least three mice. *p < 0.05 versus day 0. B, Schematic diagram indicating the locations of NRSE sequences within MOP and Nav1.8 genes. Coding exons are shown as black boxes, the noncoding exons as open boxes, and the NRSE sequences as gray boxes. The black arrows indicate the translation initiation sites, and the lower open arrows indicate the orientation of the NRSE sequences. C, Deviations of MOP- and Nav1.8-NRSEs from the consensus NRSE. The capital letters are conserved among functional NRSE sequences, and the bold capital letters are important for NRSF binding. The scores were obtained from the TFSEARCH program.
Figure 2.
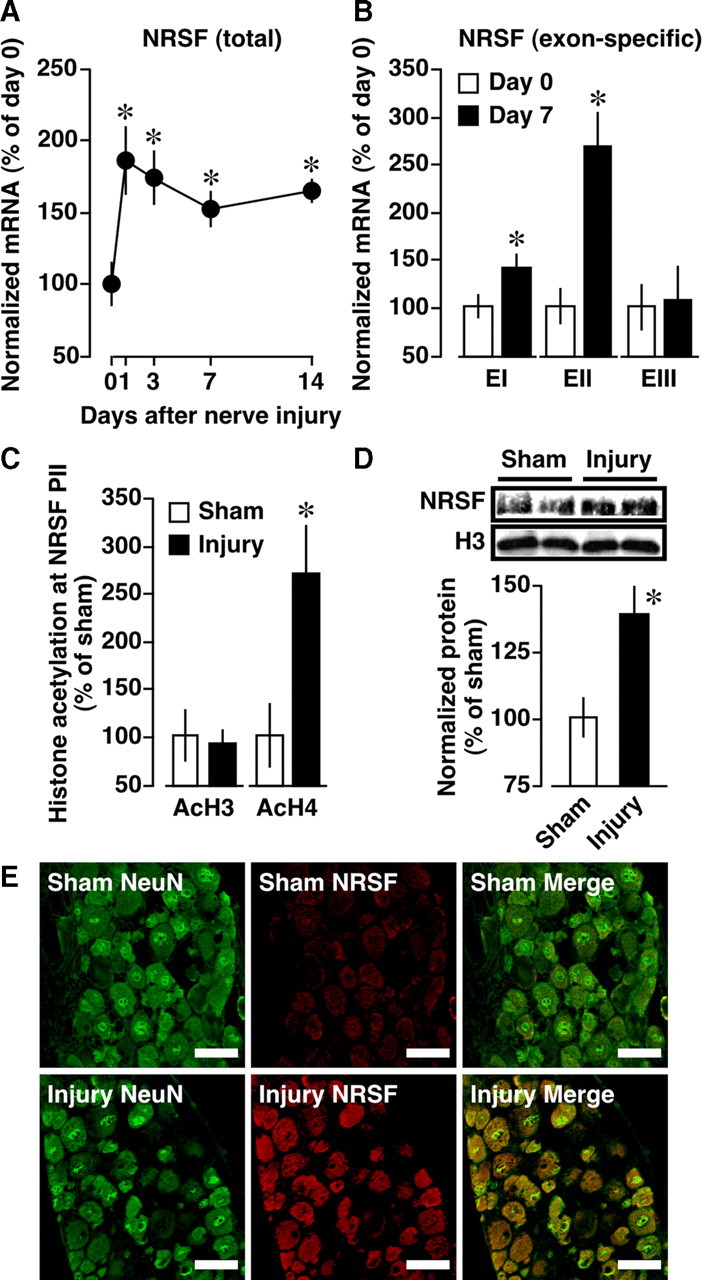
Epigenetic upregulation of NRSF gene expression. A, B, Time course of total (A) and exon-specific (B) NRSF mRNA expressions in the DRG after nerve injury. The mRNA expression levels were assessed using quantitative real-time PCR and normalized to that of GAPDH mRNA. Data are calculated as percentages of day 0. *p < 0.05 versus day 0. EI, EII, EIII, Exons I, II, III, respectively. C, Acetylation of histone H3 (AcH3) and H4 (AcH4) at NRSF promoter II (PII) at day 7 after injury, assessed using ChIP assay. Quantitative analysis was performed using real-time PCR, and the data were normalized to the corresponding input. D, NRSF protein expression at day 7 postinjury, assessed using Western blot analysis. Results are normalized to the histone H3 protein expression level. For C and D, data are calculated as percentages of sham-operated group. *p < 0.05 versus sham-operated group. Data are expressed as the means ± SEM from at least three mice. E, Immunohistochemical double labeling between NRSF (red) and NeuN (green), a neuronal marker, in the DRG of sham-operated and nerve-injured mice. Scale bars, 50 μm.
Figure 3.

Epigenetic silencing of MOP and Nav1.8 genes through NRSF binding. ChIP assay was performed at day 7 postinjury. A, B, ChIP assays using anti-NRSF antibody. A, Gel images show the representative data. B, Quantification of NRSF-binding at MOP-NRSE and at Nav1.8-NRSE-2 (Nav1.8–2). C, D, Scanning ChIP analysis of acetylation levels of histone H3 (AcH3) and H4 (AcH4) in the genomic regions spanning NRSE sequences within MOP (C) and Nav1.8 (D) genes. The schematics at the top show the PCR-targeted regions. Quantitative analysis was performed using real-time PCR, and the data were normalized to the corresponding input. Results are calculated as percentages of sham-operated group and expressed as means ± SEM from at least three mice. *p < 0.05 versus sham-operated group.
Results
Downregulations of NRSE-containing MOP and Nav1.8 gene expressions
To validate that MOP and Nav1.8 are downregulated at the transcriptional level, we isolated L4-6 DRGs at days 1, 3, 7, and 14 postinjury, and mRNA expression levels were quantified by real-time PCR. We found that nerve injury causes a long-lasting reduction in MOP and Nav1.8 mRNA levels in the DRG, starting from days 1 and 3 postinjury, respectively, and these downregulations persisted at least 14 d postinjury (Fig. 1 A).
As reported previously (Kim et al., 2004), analysis using the TFSEARCH program (version 1.3, http://www.cbrc.jp/research/db/TFSEARCHJ.html) with a threshold score of 80.0 revealed that the MOP gene contains a 21 bp NRSE sequence at initiation codon (Fig. 1 B), which is highly conserved among mouse, rat, and human (Fig. 1 C). Based on a previous report indicating the presence of a NRSE in the Nav1.8 gene (Otto et al., 2007), we analyzed its location and sequence. We found that the mouse Nav1.8 gene contains two putative conserved NRSE sites, a forward-oriented sequence within the 5′-untranslated region (NRSE-1) and a reverse-oriented sequence within intron 10 (NRSE-2) (Fig. 1 B,C). Rat and human Nav1.8 genes contain unique NRSEs within the intron 3′ and 5′-untranslated region, respectively (Fig. 1 B,C). Within all NRSE sequences of these genes, the GG nucleotides known to be important for NRSF binding (Mori et al., 1992) were completely conserved (Fig. 1 C).
Epigenetic upregulation of NRSF expression after nerve injury
The expression level of NRSF regulates its silencing activity (Chong et al., 1995; Schoenherr and Anderson, 1995); therefore, we examined NRSF expression in the DRG after nerve injury. As shown in Figure 2 A, NRSF mRNA levels were induced 1–14 d postinjury, which is negatively correlated with the temporal expression patterns of MOP and Nav1.8 (Fig. 1 A). When the transcription of three 5′ noncoding exons (I, II, or III) upstream of a common 3′ coding exon IV (Koenigsberger et al., 2000) was separately quantified, it was revealed that all NRSF transcripts, except those containing exon III, were upregulated at day 7 postinjury (Fig. 2 B). The most prominent induction was observed in the exon II-containing transcript (Fig. 2 B). Then, we assessed the acetylation of histones H3 and H4, which is correlated with transcriptional activation at the NRSF promoter II, upstream of exon II. ChIP analysis revealed that nerve injury causes a robust increase in the acetylation of histone H4, but not of H3, in the NRSF promoter II region (Fig. 2 C), which includes putative transcription start sites and binding sites for AP-1 and Sp1. Furthermore, to assess whether the altered transcription might be reflected in its protein abundance, we performed Western blot analysis using anti-NRSF antibody. We found a significant increase in NRSF protein expression at day 7 postinjury (Fig. 2 D). Using immunohistochemical analysis, we found that almost all NRSF-positive signals are colocalized with NeuN-positive signals in the DRG of sham-operated mice (Fig. 2 E), suggesting that NRSF is extensively expressed in the DRG neurons. Moreover, nerve injury markedly increased NRSF-positive signals in NeuN-positive DRG neurons at day 7 after injury (Fig. 2 E).
Histone hypoacetylation with an increase in NRSF binding at NRSE sequences within MOP and Nav1.8 genes
Next, we used ChIP analysis to examine whether NRSF binds to the NRSE sites of MOP and Nav1.8 genes after nerve injury. Nerve injury caused a drastic increase in NRSF binding to MOP-NRSE, Nav1.8-NRSE-1, and Nav1.8-NRSE-2 sequences (Fig. 3 A), suggesting that these NRSE sequences are capable of serving as NRSF-binding sites. Quantitative real-time PCR analysis showed that there was a threefold increase in NRSF binding to MOP-NRSE and Nav1.8-NRSE-2 (Fig. 3 B), while the fold change could not be calculated in the NRSF binding to Nav1.8-NRSE-1 because no significant signal was detected in sham-operated preparations (supplemental Fig. S1, available at www.jneurosci.org as supplemental material). In contrast, negligible binding was observed following precipitation by normal IgG, confirming the specificity of the immunoprecipitation (Fig. 3 A; supplemental Fig. S1, available at www.jneurosci.org as supplemental material). In addition, we performed scanning ChIP analysis to assess the levels of histone H3 and H4 acetylation in the genomic regions spanning NRSE sequences of MOP and Nav1.8 genes. We found significant reductions of histone H3 and H4 acetylation at MOP-NRSE and Nav1.8-NRSE-2 and H3 acetylation at Nav1.8-NRSE-1 at day 7 after injury (Fig. 3 C,D). Together, these data suggest that nerve injury induces repressive chromatin states around the NRSE sequences of MOP and Nav1.8 genes through NRSF-HDAC-mediated mechanisms.
Blockade of nerve injury-induced reductions in MOP and Nav1.8 gene expressions by NRSF knockdown
To examine whether NRSF could contribute to the downregulation of MOP and Nav1.8 genes after nerve injury, mice were intrathecally pretreated with an AS-ODN against NRSF or a corresponding MS-ODN. Western blot analysis revealed that NRSF protein levels in the DRG were markedly reduced by AS-ODN, but not by MS-ODN (Fig. 4 A). AS-ODN significantly blocked the nerve injury-induced downregulation of MOP and Nav1.8 (Fig. 4 B). However, AS-ODN had no effects on basal MOP and Nav1.8 mRNA levels in sham-operated mice (Fig. 4 B). These findings strongly suggest that NRSF-mediated mechanisms are responsible for the transcriptional suppression of MOP and Nav1.8 genes in the DRG after nerve injury.
On the other hand, it has been reported that nerve injury downregulates transient receptor potential melastatin 8 (TRPM8), TRP ankyrin 1 (TRPA1), and calcitonin gene-related peptide (CGRP) in the DRG (Hökfelt et al., 2006; Caspani et al., 2009; Staaf et al., 2009). Using TFSEARCH program, we found that these genes have putative NRSE sites, which give the TFSEARCH scores below our threshold value, 80.0. However, AS-ODN treatments blocked TRPM8 and TRPA1, but not CGRP, downregulations in the DRG after nerve injury (Fig. 4 C).
Recovery of C-fiber function by NRSF knockdown
We next examined abnormal pain behaviors in AS-ODN-treated mice, including C-fiber hypoesthesia, A-fiber hypersensitization, thermal hyperalgesia, and mechanical allodynia. Using the EPW test to evaluate C-, Aδ-, and Aβ-fiber functions after nerve injury (Koga et al., 2005; Matsumoto et al., 2008; Ueda, 2008), we found that AS-ODN significantly blocks hypoesthesia, seen in response to 5 Hz (C-fiber) stimuli, but does not block hypersensitization, seen in response to 250 Hz (Aδ-fiber) or 2000 Hz (Aβ-fiber) stimuli (Fig. 5 A–C). However, thermal hyperalgesia and mechanical allodynia after nerve injury were normally manifested in AS-ODN-treated mice (supplemental Fig. S2, available at www.jneurosci.org as supplemental material). On the other hand, AS-ODN had no effects on the basal threshold to thermal, mechanical, and electrical stimuli in sham-operated mice (Fig. 5 A–C; supplemental Fig. S2, available at www.jneurosci.org as supplemental material). Consistent with the data for AS-ODN, the treatment with A-803467 (10 mg/kg, i.p.), a selective blocker for Nav1.8 (Jarvis et al., 2007), resulted in a hypoesthesia of C-fiber (control, 93.3 ± 1.7 μA; A-803467, 122.5 ± 7.5 μA; p < 0.05 using Student's t test, n = 3), but not Aδ-fiber (control, 220.0 ± 20.8 μA; A-803467, 210.0 ± 11.5 μA; n = 3) and Aβ-fiber (control, 390.0 ± 10.0 μA; A-803467, 413.3 ± 6.7 μA; n = 3). Moreover, 30 nmol, but not 10 nmol, of intraplantar injection of A-803467 produced C-fiber hypoesthesia in naive mice, and the maximal response was observed at 30 min after injection (supplemental Fig. S3, available at www.jneurosci.org as supplemental material). We found that A-803467 (30 nmol, i.pl.)-induced C-fiber hypoesthesia was diminished after nerve injury, and this loss was significantly recovered by AS-ODN (Fig. 5 D).
Recovery from nerve injury-induced loss of peripheral morphine analgesia by NRSF knockdown
We examined whether AS-ODN ameliorates the loss of peripheral morphine analgesia, which is due to reduced MOP expression in the DRG neurons after nerve injury (Rashid et al., 2004). In this study, we chose a 30 nmol intraplantar morphine injection, which shows a local analgesic effect in the ipsilateral paw but not in the contralateral paw (Rashid et al., 2004). Consistent with the temporal expression pattern of MOP in the DRG after nerve injury (Fig. 1 A), peripheral morphine analgesia was markedly diminished within 3 d after injury (data not shown). The reduced peripheral morphine analgesia at day 7 postinjury was significantly recovered following AS-ODN, but not MS-ODN, administration (Fig. 6 A,B). In contrast, peripheral morphine analgesia in sham-operated mice was not altered by AS-ODN (Fig. 6 A,B), consistent with the mRNA analysis data (Fig. 4 B).
Discussion
Here, we demonstrated the following novel findings: (1) Nerve injury caused an epigenetic induction of NRSF gene expression in the DRG neurons. (2) Nerve injury induces histone hypoacetylation at NRSE sequences within MOP and Nav1.8 genes with an increase in direct NRSF binding. (3) The antisense knockdown of NRSF significantly blocked nerve injury-induced transcriptional repression of MOP, Nav1.8, TRPM8, and TRPA1, but not CGRP, genes in the DRG. (4) All of the nerve injury-induced C-fiber hypoesthesia and the losses of peripheral A-803467 hypoesthesia and peripheral morphine analgesia were markedly recovered by NRSF knockdown.
NRSF is known to repress numerous genes that are essential for neuronal functions, such as ion channels, neurotransmitter receptors, and synaptic vesicle proteins (Schoenherr et al., 1996; Bruce et al., 2004; Otto et al., 2007). It has been reported that NRSF expression is dramatically decreased to negligible levels during the differentiation of cells into mature neurons, this being concomitant with the upregulation of NRSF target genes (Ballas et al., 2005). However, accumulating evidence has revealed that NRSF expression is detected in the adult brain as well as in DRG (Palm et al., 1998; Mori et al., 2002). The present study demonstrated that peripheral nerve injury upregulates NRSF mRNA and protein expression in the DRG, thereby causing epigenetic silencing of MOP and Nav1.8 genes. Considering that NRSF is upregulated after nerve injury in substantially all DRG neurons, it is likely that NRSF-mediated MOP and Nav1.8 downregulations occur in the neuronal populations of DRG. In contrast to the blockade of nerve injury-induced downregulation of MOP and Nav1.8 by NRSF AS-ODN, NRSF knockdown had no effects on basal expression levels of MOP and Nav1.8 in sham-operated mice. Previous reports have suggested that NRSF acts in a concentration-dependent manner, and its silencing activity is insufficient when its expression is below that of a threshold level (Chong et al., 1995; Schoenherr and Anderson, 1995). The present study strongly indicates the presence of such threshold expression levels of NRSF in vivo. On the other hand, a previous report showed that global ischemia induces NRSF expression in the hippocampus, thereby causing repressive chromatin environments around the MOP-NRSE (Calderone et al., 2003; Formisano et al., 2007). However, the authors did not provide direct evidence that NRSF is responsible for MOP downregulation after ischemia, for example by NRSF knockdown.
As multiple promoters regulate NRSF gene expression (Koenigsberger et al., 2000), we analyzed promoter-specific transcription. The exon II-containing NRSF transcript, which is most active in neuronal cells (Koenigsberger et al., 2000), was found to be most responsive to nerve injury. Moreover, this increased transcription was predominantly associated with histone H4, but not H3, hyperacetylation at NRSF promoter II. These results are likely to be consistent with the pioneering report showing that NRSF expression is repressed at transcriptional level by HDAC-mediated mechanisms in mature neuron (Ballas et al., 2005). However, the regulatory mechanisms underlying nerve injury-induced epigenetic upregulation of NRSF in the DRG neurons remain elusive. Considering that NRSF expression is regulated by neuronal activity (Roopra et al., 2001), one possible mechanism underlying aberrant NRSF expression after nerve injury is the nerve injury-induced, long-lasting increase in ectopic activities of both C- and A-fibers (Xie et al., 2005). Neuronal activity regulates gene expression through extracellular signal-regulated kinase (ERK) activation (Ji and Woolf, 2001), a pathway that can lead to the activation of AP-1 and Sp1 (Karin, 1995; Barre et al., 2006), which putatively bind to the NRSF promoter II (Koenigsberger et al., 2000). Future studies should examine whether neuronal activity-induced ERK activation contributes to NRSF induction after nerve injury.
It has been assumed that the long-lasting alterations in pain-related gene expressions underlie the mechanisms of nerve injury-induced neuropathic pain (Devor, 2006; Hökfelt et al., 2006; Ueda, 2006). A unique example is a downregulation of Nav1.8 expression in C-fiber neurons (Waxman et al., 1999; Ueda, 2006). Although Nav1.8 gene is reported to contain putative NRSE (Otto et al., 2007), the transcriptional functions of Nav1.8-NRSE remain unknown. As NRSE function is independent of its position and orientation (Kraner et al., 1992; Mori et al., 1992), it is likely that both the forward- and reverse-oriented Nav1.8-NRSEs could act as a silencer. Indeed, we demonstrated that nerve injury causes NRSF binding and histone hypoacetylation at two putative Nav1.8-NRSEs, and NRSF knockdown blocks nerve injury-induced Nav1.8 downregulation. These results strongly suggest that these putative sites function as a silencer of Nav1.8 gene expression in the DRG after nerve injury. In addition to MOP and Nav1.8 genes, we have recently reported that NRSF plays a role in the nerve injury-induced epigenetic silencing of Kv4.3 gene, which has a NRSE sequence, in the DRG (Uchida et al., 2010), although its functional role remains to be determined. On the other hand, the present study showed that NRSF causes TRPM8 and TRPA1, but not CGRP, downregulations in the DRG after nerve injury, although the putative NRSE sequences in these genes have low TFSEARCH scores. Further studies are required to elucidate the mechanisms for NRSF-directed silencing of TRPM8 and TRPA1 genes after nerve injury.
Epigenetic control, which includes DNA methylation and histone modifications such as acetylation and methylation, alters the accessibility of transcriptional machinery to DNA, thereby regulating gene expression (Abel and Zukin, 2008). Histone acetylation is thought to positively regulate transcription through loosening the DNA–histone interactions (Renthal and Nestler, 2009). In the present study, we successfully demonstrated that long-lasting upregulation of NRSF gene expression following nerve injury is correlated to the histone acetylation located adjacent to NRSF promoter II. Considering that NRSF gene expression is known to be positively regulated by neuronal activity (Roopra et al., 2001), it is suggested that sustained excitation of primary afferent neurons following nerve injury (Xie et al., 2005) may lead to the long-lasting upregulation of NRSF. As NRSF binds to NRSE and represses transcription by recruiting HDAC to induce hypoacetylation of histones (Ballas and Mandel, 2005), its long-lasting upregulation may in turn cause the continued downregulation of target genes in the DRG. Alternatively, the continued downregulation may be related to the fact that histone acetylation links to more stable epigenetic modifications, such as histone methylation and DNA methylation (Abel and Zukin, 2008; Renthal and Nestler, 2009).
Nerve injury abolished the peripheral morphine analgesia, possibly through a downregulation of MOP, whose expression in C-fiber neurons (Rashid et al., 2004) is negatively regulated by NRSF. The NRSF knockdown reversed the loss of peripheral morphine analgesia, suggesting that NRSF is crucial for pharmacological dysfunction of C-fibers in neuropathic pain. The morphine resistance accompanied by MOP downregulation in the DRG is also observed in the case with postherpetic neuralgia and bone cancer pain conditions (Takasaki et al., 2006; Yamamoto et al., 2008), although it remains to be determined whether NRSF-mediated epigenetic mechanisms also occur in these different types of chronic pain. In the present study, we also demonstrated that the peripheral administration of A-803467 produces C-fiber-selective hypoesthesia in naive mice, possibly through a blockade of Nav1.8, whose expression in C-fiber neurons (Waxman et al., 1999) is negatively regulated by NRSF. As seen in the case with morphine analgesia, the NRSF knockdown also reversed the nerve injury-induced loss of peripheral A-803467 hypoesthesia, being consistent with the recovery of Nav1.8 gene expression. Of interest are the findings that NRSF knockdown has no effects on Aδ- and Aβ-hypersensitization, thermal hyperalgesia, and mechanical allodynia after nerve injury. These results are consistent with the report that the ablation of nociceptor neurons expressing Nav1.8 has no effects on neuropathic hyperalgesia, assessed only by thermal and mechanical nociception tests (Abrahamsen et al., 2008). On the other hand, we also demonstrated that NRSF knockdown reverses the nerve injury-induced downregulation of TRPM8 and TRPA1, which have been proposed to function as cold receptors (Levine and Alessandri-Haber, 2007), although their TFSEARCH scores for NRSE are not high enough. It would be an interesting subject to examine whether NRSF-directed TRPM8 and TRPA1 downregulations are crucial for cold hypoesthesia, which is a negative symptom of neuropathic pain as seen in the clinical studies (Devigili et al., 2008; Leffler and Hansson, 2008).
In conclusion, the present study demonstrated that NRSF-directed epigenetic gene silencing of Nav1.8 and MOP genes in the DRG is responsible for pathological and pharmacological dysfunctions of C-fiber after nerve injury. Elucidation of regulatory mechanisms for NRSE-NRSF systems after nerve injury might provide novel therapeutic targets for the unmet negative symptoms in neuropathic pain.
Footnotes
This work was supported by Ministry of Education, Culture, Sports, Science, and Technology Grant-In-Aid for Scientific Research 17109015 (H. Ueda). Health Sciences Research Grants from the Ministry of Health, Labor, and Welfare of Japan (H. Ueda) and Health Labor Sciences Research Grant “Third Term Comprehensive Control Research for Cancer” (398-49) also supported this work. We thank W. Xie, K. Sasaki, and T. Yamasaki for technical help.
References
- Abel T, Zukin RS. Epigenetic targets of HDAC inhibition in neurodegenerative and psychiatric disorders. Curr Opin Pharmacol. 2008;8:57–64. doi: 10.1016/j.coph.2007.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abrahamsen B, Zhao J, Asante CO, Cendan CM, Marsh S, Martinez-Barbera JP, Nassar MA, Dickenson AH, Wood JN. The cell and molecular basis of mechanical, cold, and inflammatory pain. Science. 2008;321:702–705. doi: 10.1126/science.1156916. [DOI] [PubMed] [Google Scholar]
- Akopian AN, Souslova V, England S, Okuse K, Ogata N, Ure J, Smith A, Kerr BJ, McMahon SB, Boyce S, Hill R, Stanfa LC, Dickenson AH, Wood JN. The tetrodotoxin-resistant sodium channel SNS has a specialized function in pain pathways. Nat Neurosci. 1999;2:541–548. doi: 10.1038/9195. [DOI] [PubMed] [Google Scholar]
- Ballas N, Mandel G. The many faces of REST oversee epigenetic programming of neuronal genes. Curr Opin Neurobiol. 2005;15:500–506. doi: 10.1016/j.conb.2005.08.015. [DOI] [PubMed] [Google Scholar]
- Ballas N, Grunseich C, Lu DD, Speh JC, Mandel G. REST and its corepressors mediate plasticity of neuronal gene chromatin throughout neurogenesis. Cell. 2005;121:645–657. doi: 10.1016/j.cell.2005.03.013. [DOI] [PubMed] [Google Scholar]
- Baron R. Mechanisms of disease: neuropathic pain–a clinical perspective. Nat Clin Pract Neurol. 2006;2:95–106. doi: 10.1038/ncpneuro0113. [DOI] [PubMed] [Google Scholar]
- Barre L, Venkatesan N, Magdalou J, Netter P, Fournel-Gigleux S, Ouzzine M. Evidence of calcium-dependent pathway in the regulation of human beta1,3-glucuronosyltransferase-1 (GlcAT-I) gene expression: a key enzyme in proteoglycan synthesis. FASEB J. 2006;20:1692–1694. doi: 10.1096/fj.05-5073fje. [DOI] [PubMed] [Google Scholar]
- Borrelli E, Nestler EJ, Allis CD, Sassone-Corsi P. Decoding the epigenetic language of neuronal plasticity. Neuron. 2008;60:961–974. doi: 10.1016/j.neuron.2008.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruce AW, Donaldson IJ, Wood IC, Yerbury SA, Sadowski MI, Chapman M, Gottgens B, Buckley NJ. Genome-wide analysis of repressor element 1 silencing transcription factor/neuron-restrictive silencing factor (REST/NRSF) target genes. Proc Natl Acad Sci U S A. 2004;101:10458–10463. doi: 10.1073/pnas.0401827101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calderone A, Jover T, Noh KM, Tanaka H, Yokota H, Lin Y, Grooms SY, Regis R, Bennett MV, Zukin RS. Ischemic insults derepress the gene silencer REST in neurons destined to die. J Neurosci. 2003;23:2112–2121. doi: 10.1523/JNEUROSCI.23-06-02112.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caspani O, Zurborg S, Labuz D, Heppenstall PA. The contribution of TRPM8 and TRPA1 channels to cold allodynia and neuropathic pain. PLoS One. 2009;4:e7383. doi: 10.1371/journal.pone.0007383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng HT, Dauch JR, Hayes JM, Hong Y, Feldman EL. Nerve growth factor mediates mechanical allodynia in a mouse model of type 2 diabetes. J Neuropathol Exp Neurol. 2009;68:1229–1243. doi: 10.1097/NEN.0b013e3181bef710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chong JA, Tapia-Ramirez J, Kim S, Toledo-Aral JJ, Zheng Y, Boutros MC, Altshuller YM, Frohman MA, Kraner SD, Mandel G. REST: a mammalian silencer protein that restricts sodium channel gene expression to neurons. Cell. 1995;80:949–957. doi: 10.1016/0092-8674(95)90298-8. [DOI] [PubMed] [Google Scholar]
- Costigan M, Scholz J, Woolf CJ. Neuropathic pain: a maladaptive response of the nervous system to damage. Annu Rev Neurosci. 2009;32:1–32. doi: 10.1146/annurev.neuro.051508.135531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devigili G, Tugnoli V, Penza P, Camozzi F, Lombardi R, Melli G, Broglio L, Granieri E, Lauria G. The diagnostic criteria for small fibre neuropathy: from symptoms to neuropathology. Brain. 2008;131:1912–1925. doi: 10.1093/brain/awn093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Devor M. Response of nerves to injury in relation to neuropathic pain. In: McMahon SB, Koltzenburg M, editors. Wall and Melzack's textbook of pain. Oxford: Churchill Livingstone; 2006. pp. 905–927. [Google Scholar]
- Dickenson A, Kieffer B. Opiates: basic mechanisms. In: McMahon SB, Koltzenburg M, editors. Wall and Melzack's textbook of pain. Oxford: Churchill Livingstone; 2006. pp. 427–442. [Google Scholar]
- Fields HL, Rowbotham M, Baron R. Postherpetic neuralgia: irritable nociceptors and deafferentation. Neurobiol Dis. 1998;5:209–227. doi: 10.1006/nbdi.1998.0204. [DOI] [PubMed] [Google Scholar]
- Formisano L, Noh KM, Miyawaki T, Mashiko T, Bennett MV, Zukin RS. Ischemic insults promote epigenetic reprogramming of mu opioid receptor expression in hippocampal neurons. Proc Natl Acad Sci U S A. 2007;104:4170–4175. doi: 10.1073/pnas.0611704104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hargreaves K, Dubner R, Brown F, Flores C, Joris J. A new and sensitive method for measuring thermal nociception in cutaneous hyperalgesia. Pain. 1988;32:77–88. doi: 10.1016/0304-3959(88)90026-7. [DOI] [PubMed] [Google Scholar]
- Hökfelt T, Zhang X, Xu XJ, Wiesenfeld-Hallin Z. Central consequences of peripheral nerve damage. In: McMahon SB, Koltzenburg M, editors. Wall and Melzack's textbook of pain. Oxford: Churchill Livingstone; 2006. pp. 947–959. [Google Scholar]
- Inoue M, Rashid MH, Fujita R, Contos JJ, Chun J, Ueda H. Initiation of neuropathic pain requires lysophosphatidic acid receptor signaling. Nat Med. 2004;10:712–718. doi: 10.1038/nm1060. [DOI] [PubMed] [Google Scholar]
- Jarvis MF, Honore P, Shieh CC, Chapman M, Joshi S, Zhang XF, Kort M, Carroll W, Marron B, Atkinson R, Thomas J, Liu D, Krambis M, Liu Y, McGaraughty S, Chu K, Roelofts, Zhong C, Mikusa JP, Hernandez G, et al. A-803467, a potent and selective Nav1.8 sodium channel blocker, attenuates neuropathic and inflammatory pain in the rat. Proc Natl Acad Sci U S A. 2007;104:8520–8525. doi: 10.1073/pnas.0611364104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji RR, Woolf CJ. Neuronal plasticity and signal transduction in nociceptive neurons: implications for the initiation and maintenance of pathological pain. Neurobiol Dis. 2001;8:1–10. doi: 10.1006/nbdi.2000.0360. [DOI] [PubMed] [Google Scholar]
- Karin M. The regulation of AP-1 activity by mitogen-activated protein kinases. J Biol Chem. 1995;270:16483–16486. doi: 10.1074/jbc.270.28.16483. [DOI] [PubMed] [Google Scholar]
- Kim CS, Hwang CK, Choi HS, Song KY, Law PY, Wei LN, Loh HH. Neuron-restrictive silencer factor (NRSF) functions as a repressor in neuronal cells to regulate the mu opioid receptor gene. J Biol Chem. 2004;279:46464–46473. doi: 10.1074/jbc.M403633200. [DOI] [PubMed] [Google Scholar]
- Klein JP, Tendi EA, Dib-Hajj SD, Fields RD, Waxman SG. Patterned electrical activity modulates sodium channel expression in sensory neurons. J Neurosci Res. 2003;74:192–198. doi: 10.1002/jnr.10768. [DOI] [PubMed] [Google Scholar]
- Koenigsberger C, Chicca JJ, 2nd, Amoureux MC, Edelman GM, Jones FS. Differential regulation by multiple promoters of the gene encoding the neuron-restrictive silencer factor. Proc Natl Acad Sci U S A. 2000;97:2291–2296. doi: 10.1073/pnas.050578797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koga K, Furue H, Rashid MH, Takaki A, Katafuchi T, Yoshimura M. Selective activation of primary afferent fibers evaluated by sine-wave electrical stimulation. Mol Pain. 2005;1:13. doi: 10.1186/1744-8069-1-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohno T, Ji RR, Ito N, Allchorne AJ, Befort K, Karchewski LA, Woolf CJ. Peripheral axonal injury results in reduced mu opioid receptor pre- and post-synaptic action in the spinal cord. Pain. 2005;117:77–87. doi: 10.1016/j.pain.2005.05.035. [DOI] [PubMed] [Google Scholar]
- Kraner SD, Chong JA, Tsay HJ, Mandel G. Silencing the type II sodium channel gene: a model for neural-specific gene regulation. Neuron. 1992;9:37–44. doi: 10.1016/0896-6273(92)90218-3. [DOI] [PubMed] [Google Scholar]
- Kubat NJ, Amelio AL, Giordani NV, Bloom DC. The herpes simplex virus type 1 latency-associated transcript (LAT) enhancer/rcr is hyperacetylated during latency independently of LAT transcription. J Virol. 2004;78:12508–12518. doi: 10.1128/JVI.78.22.12508-12518.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leffler AS, Hansson P. Painful traumatic peripheral partial nerve injury-sensory dysfunction profiles comparing outcomes of bedside examination and quantitative sensory testing. Eur J Pain. 2008;12:397–402. doi: 10.1016/j.ejpain.2007.08.009. [DOI] [PubMed] [Google Scholar]
- Levine JD, Alessandri-Haber N. TRP channels: targets for the relief of pain. Biochim Biophys Acta. 2007;1772:989–1003. doi: 10.1016/j.bbadis.2007.01.008. [DOI] [PubMed] [Google Scholar]
- Malmberg AB, Basbaum AI. Partial sciatic nerve injury in the mouse as a model of neuropathic pain: behavioral and neuroanatomical correlates. Pain. 1998;76:215–222. doi: 10.1016/s0304-3959(98)00045-1. [DOI] [PubMed] [Google Scholar]
- Matsumoto M, Inoue M, Hald A, Xie W, Ueda H. Inhibition of paclitaxel-induced A-fiber hypersensitization by gabapentin. J Pharmacol Exp Ther. 2006;318:735–740. doi: 10.1124/jpet.106.103614. [DOI] [PubMed] [Google Scholar]
- Matsumoto M, Xie W, Ma L, Ueda H. Pharmacological switch in Abeta-fiber stimulation-induced spinal transmission in mice with partial sciatic nerve injury. Mol Pain. 2008;4:25. doi: 10.1186/1744-8069-4-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mori N, Schoenherr C, Vandenbergh DJ, Anderson DJ. A common silencer element in the SCG10 and type II Na+ channel genes binds a factor present in nonneuronal cells but not in neuronal cells. Neuron. 1992;9:45–54. doi: 10.1016/0896-6273(92)90219-4. [DOI] [PubMed] [Google Scholar]
- Mori N, Mizuno T, Murai K, Nakano I, Yamashita H. Effect of age on the gene expression of neural-restrictive silencing factor NRSF/REST. Neurobiol Aging. 2002;23:255–262. doi: 10.1016/s0197-4580(01)00286-x. [DOI] [PubMed] [Google Scholar]
- Otto SJ, McCorkle SR, Hover J, Conaco C, Han JJ, Impey S, Yochum GS, Dunn JJ, Goodman RH, Mandel G. A new binding motif for the transcriptional repressor REST uncovers large gene networks devoted to neuronal functions. J Neurosci. 2007;27:6729–6739. doi: 10.1523/JNEUROSCI.0091-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palm K, Belluardo N, Metsis M, Timmusk T. Neuronal expression of zinc finger transcription factor REST/NRSF/XBR gene. J Neurosci. 1998;18:1280–1296. doi: 10.1523/JNEUROSCI.18-04-01280.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiang M, Rani CS, Ticku MK. Neuron-restrictive silencer factor regulates the N-methyl-d-aspartate receptor 2B subunit gene in basal and ethanol-induced gene expression in fetal cortical neurons. Mol Pharmacol. 2005;67:2115–2125. doi: 10.1124/mol.104.010751. [DOI] [PubMed] [Google Scholar]
- Rashid MH, Inoue M, Kondo S, Kawashima T, Bakoshi S, Ueda H. Novel expression of vanilloid receptor 1 on capsaicin-insensitive fibers accounts for the analgesic effect of capsaicin cream in neuropathic pain. J Pharmacol Exp Ther. 2003;304:940–948. doi: 10.1124/jpet.102.046250. [DOI] [PubMed] [Google Scholar]
- Rashid MH, Inoue M, Toda K, Ueda H. Loss of peripheral morphine analgesia contributes to the reduced effectiveness of systemic morphine in neuropathic pain. J Pharmacol Exp Ther. 2004;309:380–387. doi: 10.1124/jpet.103.060582. [DOI] [PubMed] [Google Scholar]
- Renthal W, Nestler EJ. Histone acetylation in drug addiction. Semin Cell Dev Biol. 2009;20:387–394. doi: 10.1016/j.semcdb.2009.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roopra A, Huang Y, Dingledine R. Neurological disease: listening to gene silencers. Mol Interv. 2001;1:219–228. [PubMed] [Google Scholar]
- Schoenherr CJ, Anderson DJ. The neuron-restrictive silencer factor (NRSF): a coordinate repressor of multiple neuron-specific genes. Science. 1995;267:1360–1363. doi: 10.1126/science.7871435. [DOI] [PubMed] [Google Scholar]
- Schoenherr CJ, Paquette AJ, Anderson DJ. Identification of potential target genes for the neuron-restrictive silencer factor. Proc Natl Acad Sci U S A. 1996;93:9881–9886. doi: 10.1073/pnas.93.18.9881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staaf S, Oerther S, Lucas G, Mattsson JP, Ernfors P. Differential regulation of TRP channels in a rat model of neuropathic pain. Pain. 2009;144:187–199. doi: 10.1016/j.pain.2009.04.013. [DOI] [PubMed] [Google Scholar]
- Takasaki I, Nojima H, Shiraki K, Kuraishi Y. Specific down-regulation of spinal mu-opioid receptor and reduced analgesic effects of morphine in mice with postherpetic pain. Eur J Pharmacol. 2006;550:62–67. doi: 10.1016/j.ejphar.2006.08.041. [DOI] [PubMed] [Google Scholar]
- Taylor BK. Pathophysiologic mechanisms of neuropathic pain. Curr Pain Headache Rep. 2001;5:151–161. doi: 10.1007/s11916-001-0083-1. [DOI] [PubMed] [Google Scholar]
- Uchida H, Sasaki K, Ma L, Ueda H. Neuron-restrictive silencer factor causes epigenetic silencing of K(v)4.3 gene after peripheral nerve injury. Neuroscience. 2010;166:1–4. doi: 10.1016/j.neuroscience.2009.12.021. [DOI] [PubMed] [Google Scholar]
- Ueda H. Molecular mechanisms of neuropathic pain-phenotypic switch and initiation mechanisms. Pharmacol Ther. 2006;109:57–77. doi: 10.1016/j.pharmthera.2005.06.003. [DOI] [PubMed] [Google Scholar]
- Ueda H. Peripheral mechanisms of neuropathic pain-involvement of lysophosphatidic acid receptor-mediated demyelination. Mol Pain. 2008;4:11. doi: 10.1186/1744-8069-4-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waxman SG, Dib-Hajj S, Cummins TR, Black JA. Sodium channels and pain. Proc Natl Acad Sci U S A. 1999;96:7635–7639. doi: 10.1073/pnas.96.14.7635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie W, Strong JA, Meij JT, Zhang JM, Yu L. Neuropathic pain: early spontaneous afferent activity is the trigger. Pain. 2005;116:243–256. doi: 10.1016/j.pain.2005.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto J, Kawamata T, Niiyama Y, Omote K, Namiki A. Down-regulation of mu opioid receptor expression within distinct subpopulations of dorsal root ganglion neurons in a murine model of bone cancer pain. Neuroscience. 2008;151:843–853. doi: 10.1016/j.neuroscience.2007.11.025. [DOI] [PubMed] [Google Scholar]
- Zimmermann M. Ethical guidelines for investigations of experimental pain in conscious animals. Pain. 1983;16:109–110. doi: 10.1016/0304-3959(83)90201-4. [DOI] [PubMed] [Google Scholar]