

Abstract
Neuropathic pain after peripheral nerve injury, associated with local neuroinflammation in the spinal cord, is a severe incapacitating condition with which clinical treatment remains challenging. Inflammatory molecules signal through various intracellular transduction pathways, activation of which may amplify and cause spreading of the inflammatory response. We showed recently that spinal nerve lesion leads to rapid activation of Janus kinase (JAK)/signal transducer and activator of transcription 3 (STAT3) signal transduction pathway in dorsal spinal cord microglia in relation with enhanced levels of spinal interleukin-6 (IL-6) protein. Here, we selectively inactivated JAK/STAT3 signaling in rat dorsal spinal cord glia through local, lentiviral-mediated production of the suppressor of cytokine signaling SOCS3, a physiologic inhibitory protein of JAK/STAT3, and analyzed its consequences in a preclinical model of neuropathic pain. The targeted blockade of JAK/STAT3 activity prevented the abnormal expression of IL-6, CC chemokine ligand CCL2, and activating transcription factor ATF3 induced in the spinal cord by chronic constriction injury of the sciatic nerve (CCI) and substantially attenuated mechanical hypersensitivity (allodynia) in rats. In naive rats, intrathecal administration of a proalgesic cytokine IL-6 rapidly activated microglial JAK/STAT3 and induced downstream changes closely resembling CCI-evoked alterations. We identified downstream mechanisms through which JAK/STAT3 pathway activation leads to the spreading of neuroinflammation. Our findings reveal that JAK/STAT3 signaling plays a major role in spinal cord plasticity and mechanical allodynia associated with peripheral nerve injury.
Introduction
Neuropathic pain is a highly incapacitating disease that frequently arises after damage to peripheral nerves and is associated with spinal cord plasticity, implicating glial cell activation and inflammatory cytokine signaling (Scholz and Woolf, 2007; Milligan and Watkins, 2009). This neuroinflammatory state involves numerous extracellular signaling molecules acting through complex cascades of intracellular transduction pathways and potentially participating in alteration of neuronal and glial function. We recently reported that the Janus kinase (JAK)/signal transducer and activator of transcription 3 (STAT3) pathway is activated in the dorsal spinal cord as a consequence of peripheral nerve injury. The early activation of JAK/STAT3 is mainly induced by interleukin-6 (IL-6) in spinal microglia and contributes to neuropathic pain development (Dominguez et al., 2008). Adipocytokine leptin was also implicated in neuropathic pain through its interaction with leptin receptor and subsequent activation of JAK/STAT3 in the spinal cord (Lim et al., 2009). STAT3, a member of the JAK/STAT signaling family, is expressed in the CNS. Its active, phosphorylated form is upregulated after CNS damage and participates in the microglial inflammatory response (Kim et al., 2002), reactive astrogliosis, and scar formation (Yamauchi et al., 2006; Herrmann et al., 2008). JAK/STAT pathway is linked to the immune/inflammatory response. Such a peripheral nerve injury-evoked neuroinflammation in the spinal cord plays a crucial role in pathological pain, supporting the idea that rapid activation of JAK/STAT3 signaling may play an important role in the first phase of spinal plasticity and pain induction. The downstream events involved in this early JAK/STAT3-mediated signaling in the spinal cord remain to be understood.
The suppressor of cytokine signaling 3 (SOCS3) protein acts as feedback inhibitor of the JAK/STAT3 pathway, avoiding STAT3 phosphorylation (i.e., activation) (Nicholson et al., 2000). The ability of overproduced SOCS3 to block the JAK/STAT3 pathway and to limit some of the pathophysiological consequences of STAT3-mediated signaling was demonstrated previously at the periphery (Shouda et al., 2001, Jo et al., 2005).
Here, we selectively inhibited JAK/STAT3 function in vivo in rat spinal cord glia by enhancing the local production of SOCS3 using gliotropic lentiviral vectors (LVs) (Meunier et al., 2007, 2008). We assessed the effects of SOCS3 on JAK/STAT3 signaling and explored the consequences of this selective local blockade of JAK/STAT3 on downstream signaling responses and pain hypersensitivity in a preclinical model of neuropathic pain induced by chronic constriction injury of the sciatic nerve (CCI) in rats (Bennett and Xie, 1988). In particular, we analyzed the effects of JAK/STAT3 activity on activating transcription factor 3 (ATF3) production, which is directly related to IL-6 signaling in the spinal cord (Latrémolière et al., 2008), and CC chemokine ligand 2 (CCL2) (also called MCP-1). CCL2 plays an important role in neuroglial interactions and pathological pain (Abbadie, 2005; Thacker et al., 2009), and its expression may also be regulated by the STAT3 transcription factor (Kim et al., 2002).
Our data indicate that early activation of JAK/STAT3 signaling in the spinal cord may play a substantial role in the development of local neuroinflammatory state and mechanical allodynia.
Materials and Methods
Animals.
Animals used in this study (adult male Sprague Dawley rats, 200 g; Centre d'Elevage Janvier) were maintained under controlled conditions (22 ± 1°C, 60 ± 10% relative humidity, 12 h light/dark cycle, food and water ad libitum). Four-day-old Sprague Dawley pups were used for primary glial cell preparations. All experiments were performed in line with institutional guidelines, to comply with national and international law and policies for use of animals in neuroscience research [European Communities Council Directive 87848, October 1987, Ministère de l'Agriculture et de la Forêt, Service Vétérinaire de la Santé et de la Protection Animale; Permission 75-1179 (to M.P.)].
Plasmids.
The expression plasmid pTrip–cytomegalovirus (CMV)–woodchuck posttranscriptional regulatory element (WPRE) (gift from Hammid Mammeri, Centre National de la Recherche Scientifique, Unité Mixte de Recherche 7091, Paris, France) was used to produce LVs. The coding sequence of rat SOCS3, followed by the tag sequence V5 (5′ ggtaagcctatccctaaccctctcctcggtctcgattctacg 3′), or of enhanced green fluorescent protein (EGFP) was inserted (BamHI/XhoI) under the transcriptional control of the CMV promoter in pTrip–CMV–WPRE. The transcomplementation plasmid p8.91 and the plasmid encoding the vesicular stomatitis virus envelope glycoprotein VSV-G (pMD-G) have been described previously (Zufferey et al., 1997).
Lentiviral vector production.
Pseudotyped human immunodeficiency virus vectors were produced as described previously (Zennou et al., 2001; Meunier et al., 2008). Briefly, HEK293T cells were cotransfected with pTrip–SOCS3t–WPRE or pTrip–EGFP–WPRE, p8.91, and pMD-G using calcium phosphate DNA precipitation. Viral particles were obtained by ultracentrifugation (56,000 × g, 1.5 h, 4°C) of the supernatants and resuspended in PBS (D-PBS; Invitrogen). After elimination of remaining cellular debris by successive low-speed centrifugations, the final viral suspension was stored at −80°C until use. Lentiviral suspension was titrated and normalized for the p24 antigen (Beckman Coulter).
Transduction of rat primary mixed glial cells cultures with LV–SOCS3t resulted in expression of transgene-derived SOCS3 transcripts. This was confirmed through the detection of the tag sequence V5 (Fig. 1A). Western blots of total proteins extracted from glial cells transduced with increasing titers of LV–SOCS3t showed titer-dependent production of the transgene-derived SOCS3t (Fig. 1B). Immunohistochemistry (IHC) performed 48 h after transduction of primary glial cells with LV–SOCS3t demonstrated colocalization between SOCS3 immunoreactivity (IR) and V5-IR in transfected glial cells (astrocytes and microglia), confirming expression of the exogenous form of SOCS3 (SOCS3t) (Fig. 1C). Production of the transgene-derived SOCS3t-IR was also detected in transfected HEK293T and BV2 microglial cell lines (data not shown).
Figure 1.
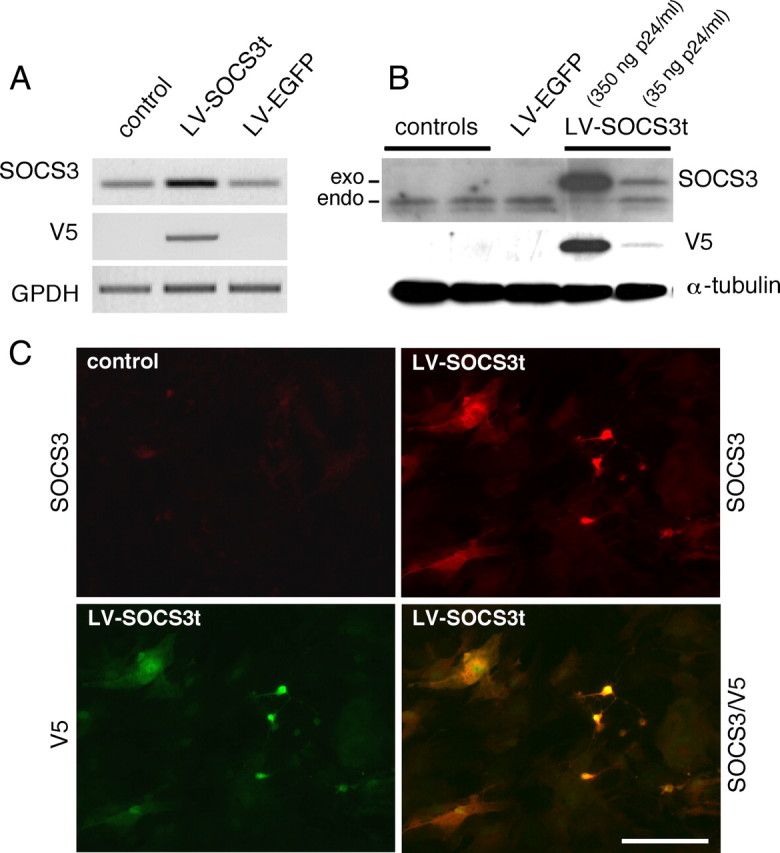
Lentiviral-mediated production of tagged SOCS3 (SOCS3t) in rat primary glial cell cultures. Two days after transduction, the presence of SOCS3 mRNA and of the tag sequence V5 were assessed using conventional RT-PCR with 0.5 μg of total RNA extracted from primary glia transduced with LV–SOCS3t (35 ng of viral envelope protein p24) or LV–EGFP (control lentiviral vectors, 35 ng of p24) or untreated (controls). A, Socs3 and V5 mRNA levels were compared with housekeeping GAPDH mRNA levels for each condition. Western blots were performed with SOCS3- or V5-specific antibodies on total proteins extracted from glial cells, 2 d after transduction. α-Tubulin was used as a loading control. B, Addition of the V5 tag sequence allowed endogenous SOCS3 to be distinguished from transgene-derived SOCS3 protein. C, Whereas in control glia immunohistochemistry could not detect endogenous SOCS3-IR, SOCS3-IR (in red) was present in cells transfected with LV–SOCS3t (35 ng of p24, 2 d before experiments), fully overlapping with V5-immunolabeling (in green), thus confirming the transgene origin of SOCS3-IR in these cells. Scale bar, 50 μm.
Glial cell cultures.
Primary mixed glial cultures were prepared from the cerebral cortex of 4-d-old rat pups (Centre d'Elevage Janvier) following slight modifications of the described procedure (Goslin et al., 1998). Briefly, hemispheres from six to seven pups were dissociated in HBSS containing 50 mm HEPES and centrifuged (400 × g, 30 s). The pellet was resuspended in 10 ml of HBSS containing 50 mm HEPES, 0.25% trypsin, and 0.2 mg/ml Dnase I, incubated 15 min at 37°C. We then added 5% BSA, and the cell suspension was centrifuged (400 × g, 10 min). The resulting pellet was resuspended in 50 ml of DMEM supplemented with 10% heat-inactivated fetal calf serum and 1% (w/v) penicillin/streptomycin (complete culture medium). The suspension was centrifuged as above until a clear supernatant was obtained. The final pellet was resuspended in complete medium and seeded at the density of 104 cells/cm2 in 75 cm2 culture flask (Corning Life Sciences via VWR International).
For immunofluorescence or Western blots and conventional semiquantitative reverse transcription (RT)-PCR experiments, glial cells were plated at a density of 2 × 105 cells/cm2 in four-well plates containing poly-d-lysine-coated coverslips or in six-well plates, respectively. Cells were infected with 35 or 350 ng of p24/ml LV–EGFP or LV–SOCS3t. Cells were incubated for 48 h and then serum starved overnight before being treated with 50 ng/ml recombinant rat IL-6 (Peprotech). Cells were incubated for an additional 15 min, washed thoroughly, and then prepared for Western blot analyses. RT-PCR experiments were performed on total RNA extracted from cells incubated for 6 h in the presence of IL-6.
Delivery of viral vectors into the dorsal horn of the lumbar spinal cord.
Lentiviral vectors were delivered by intraparenchymal injection as described previously (Meunier et al., 2008), with slight modification of the protocol. Briefly, rats were deeply anesthetized with chloral hydrate (400 mg/kg, i.p.). To avoid movements caused by breathing, animal's spine was maintained with two individual bars placed around the L3 vertebra. Under a Carl Zeiss operation microscope (10–25×), the thoracic T13 vertebra was carefully drilled to access the left side of the lumbar spinal cord. After an incision in the intact dura mater and arachnoid mater, 2 μl (∼70 ng of p24) of lentiviral vectors (LV–EGFP or LV–SOCS3t) were delivered using a glass micropipette connected to an automatic microinjection device (KDS 3010; KD Scientific). Muscles and skin were closed with resorbable 4/0 Ethicon stitches (Johnson & Johnson). Rats were then housed in individual cages to recover.
Chronic constriction injury of the sciatic nerve.
Non-injected, sham-injected (LV–EGFP), and LV–SOCS3t-injected (1 week after infection) rats were anesthetized with 3% isoflurane in O2 at 3 L/min and maintained with 1.5% isoflurane in O2. Their left sciatic nerve was exposed at the midthigh region. Four loose ligatures (5-0 chromic catgut, ∼1 mm spacing) were placed around the nerve, taking care to not interrupt the epineural circulation, as originally described by Bennett and Xie (1988). The day on which surgery was performed was referred to as day 0. Sham-operated animals were subjected to the same surgical protocol as CCI animals except that the exposed sciatic nerve was not constricted.
Behavioral testing.
Behavior was analyzed in blind tests. To avoid stress resulting from experimental conditions, all manipulations were performed in quiet conditions in a test room by the same experimenter. For the 7 d preceding the experiments, animals were placed in the test room for 1 h (from 12:00 A.M. to 1:00 P.M.), then gently handled for 5 min, and left to acclimatize in suspended cages with wire-mesh buttons.
Mechanical allodynia was determined as described by Chaplan et al. (1994). The ipsilateral and contralateral hindpaws were probed with calibrated von Frey filaments (Bioseb) applied perpendicularly to the plantar surface and held for ∼5 s. A sharp withdrawal of the paw indicated a positive response. The 50% paw-withdrawal threshold was determined by the nonparametric method of Dixon (1980). The stimulus was incrementally increased until a positive response was obtained and then decreased until a negative result was observed. This protocol was repeated until three changes in behavior had been observed; positive and negative responses were then tabulated. The 50% paw-withdrawal threshold was determined as (10[Xf + kΔ])/10,000, where Xf is the value of the last von Frey filament used, k is Dixon value for the positive/negative pattern, and Δ is the logarithmic difference between stimuli.
Conventional RT-PCR and real-time RT-PCR analysis.
Rats were killed by decapitation, and the lumbar part of the spinal cord was removed in the cold (0–4°C). The lumbar enlargement (L6–L4) was divided into left (injected and lesioned side) and right parts by a sagittal cut and then into dorsal and ventral parts by a horizontal cut passing through the ependymal canal. The median part (∼5 mm, i.e., 2.5 mm rostral and caudal to the injection site) of left dorsal quadrants of the spinal cord were frozen immediately in liquid nitrogen and then stored at −80°C until they were used. Total RNA were extracted from frozen pieces of tissues using NucleoSpin RNA II Purification kit (Macherey-Nagel), and their quality and concentrations were evaluated by optical density using NanoDrop (Thermo Fisher Scientific via Labtech France).
Reverse transcription was immediately followed by PCR using the Access RT-PCR System (Promega) with 0.5 μg of each RNA sample. RT-RNA was amplified with 30 cycles [1 min for each step of denaturing (96°C), annealing (58°C), and extension (72°C)], using 40 pmol of specific primers [the housekeeping gene glyceraldehyde-3-phosphate dehydrogenase (GAPDH) sense, 5′ accacagtccatgccatcac 3′; GAPDH antisense, 5′ tccaccaccctgttgctgta 3′; SOCS3 sense, 5′ cccgctttgactgtgtact 3′; SOCS3 antisense, 5′ tgagtaccagcgggatcttctc 3′; SOCS3–V5 sense, 5′ cccgctttgactgtgtact 3′; SOCS3–V5 antisense, 5′ atggtgatgatgaccggta 3′], following the protocol of the manufacturer. RT-PCR products were separated by electrophoresis on a 1.2% ethidium bromide-stained agarose gel and quantified with the gel analyzer GDS 5000 (Ultra-Violet Products) and NIH ImageJ software (http://rsb.info.nih.gov/ij/). Data were normalized using GAPDH as a reference. For real-time RT-PCR analysis, first-strand cDNA synthesis (0.6 μg of total RNA per 20 μl of reaction) was performed with a High-Capacity cDNA Reverse Transcription kit (Applied Biosystems). Real-time PCR amplification of each sample was performed in triplicate on the ABI Prism 7300 (Applied Biosystems) using ABgene Absolute QPCR ROX Mix. Assay-on-Demand Gene TaqMan PCR probes (Applied Biosystems) were used for target genes: SOCS3 (GenBank accession number Rn00585674_s1), integrin alpha M (ITGAM) (GenBank accession number Rn00709342_m1), GFAP (GenBank accession number Rn01460868_m1), IL-6 (Rn00561420_m1), IL-1β (GenBank accession number Rn00580432_m1), ATF3 (GenBank accession number Rn00563784_m1), CCL2 (GenBank accession number Rn00580555_m1), tumor necrosis factor-α (TNFα) (GenBank accession number Rn99999017_m1), GAPDH (GenBank accession number Rn99999916_s1), and ribosomal subunit 18S (RS 18S) (GenBank accession number Hs 99999901_s1). To perform semiquantitative studies, GAPDH and RS 18S were used as reporter genes. Because the relative expression of GAPDH compared with RS 18S was not significantly different in control versus injured animals at 15 and 21 d after injury, most of the experiments were performed with GAPDH as reporter gene.
Western blotting.
Cell cultures were washed three times in ice-cold 1× PBS and were scraped in 1 ml of this solution. Samples were centrifuged (1500 × g, 3 min, 4°C), and pellets were resuspended in radioimmunoprecipitation assay (RIPA) buffer (20 mm Tris, pH 7.5, 150 mm NaCl, 1% NP-40, 0.5% Na-deoxycholate, 1 mm EDTA, and 0.1% SDS) supplemented with a protease and phosphatase inhibitor cocktail (Sigma-Aldrich). Samples were stored at −80°C until use.
The left dorsal quadrant of the rat lumbar spinal cord was dissected out as described for RT-PCR experiments. Frozen tissue pieces were placed in an ice-chilled Dounce homogenizer and homogenized on ice in 170 μl of RIPA buffer supplemented with a protease and phosphatase inhibitor cocktail. Samples were centrifuged (10,000 × g, 10 min, 4°C), and supernatants were centrifuged once more.
Equal concentrations of proteins, as determined by Bio-Rad protein assay, were mixed with standard Laemli's buffer, sonicated, heated at 95°C for 1 min, and then separated by SDS-PAGE gel (10% acrylamide) and electrotransferred (Trans-Blot SD; Bio-Rad) onto a nitrocellulose membrane (Bio-Rad). Membranes were first saturated in blocking solution (5% nonfat dry milk, 0.1% Tween 20 in 1× PBS) for 1 h at room temperature and then incubated (overnight, 4°C) with mouse anti-V5 (1:250; Invitrogen), rabbit anti-SOCS3 (1:100; IBL), rabbit anti-pSTAT3 (Tyr705, 1:500; Cell Signaling Technology), rabbit anti p-p38 mitogen-activated protein kinase (MAPK) antibody (1:200; Cell Signaling Technology), rabbit anti-phosphorylated extracellular signal-regulated kinase (pERK) antibody (1:500; Cell Signaling Technology), or rabbit anti-GFAP antibody (1:5000; Millipore Bioscience Research Reagents) in the blocking solution. After rinsing with PBS-T (1× PBS, 0.1% Tween 20), blots were incubated (40 min, room temperature) with HRP-linked anti-rabbit Ig (1:5000; Sigma) in the blocking solution. Blots were finally washed in PBS-T and then in PBS. Membranes were treated with ECL Plus kit reagents and exposed to MP-ECL film (GE Healthcare). Membranes were washed in PBS-T and stripped in 32.5 mm Tris-HCl, pH 6.7, 2% SDS, and 100 mm β-mercaptoethanol before incubation with mouse anti-α-tubulin (1:10,000; GE Healthcare) or, in some experiments, with rabbit anti-STAT3 antibody (1:750; Cell Signaling Technology). Relative intensities of the pSTAT3, STAT3, p38, pERK1, and pERK2 immunoreactivity were compared with α-tubulin controls using scanned images of the blots.
Immunochemistry.
The primary antibodies used for this study were mouse anti-V5 (1:200; Invitrogen), goat anti-SOCS3 (1:100; Santa Cruz Biotechnology), goat anti-pSTAT3–Tyr705 (1:150; Santa Cruz Biotechnology), rabbit anti-pSTAT3–Ser727 (1:100; Cell Signaling Technology), mouse anti-GFAP (1:5000; Millipore Bioscience Research Reagents), mouse anti-CD11b (ITGAM) (1:120; Serotec), rabbit anti-Iba1 (1:800; Wako), and mouse anti-neuronal-specific nuclear protein (NeuN) (1:1000; Millipore Bioscience Research Reagents). Secondary antibodies used were Alexa 488- or 594-conjugated donkey anti-rabbit, anti-mouse, and anti-goat Ig (1:500; Invitrogen).
Cells were prepared for fluorescent IHC as described by Meunier et al. (2008). Briefly, after fixation in 4% paraformaldehyde in PBS, coverslips with cells were washed with PBS containing 0.1 mm CaCl2 and 0.1 mm MgCl2 (PBS+) and incubated with blocking buffer (3% donkey serum and 0.3% Triton X-100 in PBS+). Cells were then incubated in the same buffer supplemented with primary antibody (2 h, room temperature), washed with PBS+, and incubated with secondary antibodies in blocking buffer (1 h, room temperature). Coverslips were rinsed (PBS+) and mounted in Fluoromount-G solution (Clinisciences).
Animals were deeply anesthetized with pentobarbital (Nembutal, 50 mg/kg, i.p.) and perfused transcardially with 100 ml of 0.9% NaCl supplemented with 0.1% sodium nitrite, followed by 800 ml of 4% paraformaldehyde in 0.1 m phosphate buffer supplemented with 0.8% picric acid at room temperature. Lumbar spinal cords were dissected out and cryoprotected in 10% sucrose (24 h, 4°C). Cryostat sections (20 μm) were preincubated (1 h, room temperature) in a 1× PBS buffer containing 3% donkey serum (Interchim) and 0.3% Triton X-100. Sections were incubated in the same buffer supplemented with primary antibodies overnight at 4°C. After being washed (1× PBS), sections were incubated for 1 h at room temperature with secondary antibodies, rinsed in 1× PBS, and mounted in Fluoromount-G solution.
Slides were observed and images were generated using a Carl Zeiss microscope (Axio Imager M1, AX10) and AxioVision 4.7 software (Carl Zeiss).
Statistical analysis.
Data are presented as mean ± SEM. Data from behavioral studies were validated with one-way ANOVA, followed by Bonferroni's post hoc test and repeated measures over time. For RT-PCR results, the 2−DDCt method (Livak and Schmittgen, 2001) was used to analyze the relative changes in specific mRNA levels between different groups (RQ Study Software 1.2 version; Applied Biosystems); data were then validated using one-way ANOVA, followed by Scheffé's post hoc test. Western blots and in vitro RT-PCR experiments data were evaluated using one-way ANOVA, followed by Bonferroni's post hoc test. Statistical evaluation was performed with STATVIEW 5.0 software (SAS Institute). When p > 0.05, the corresponding difference was considered to be nonsignificant.
Results
CCI of the rat sciatic nerve results in activation of the JAK/STAT3 pathway in the lesioned side of the dorsal spinal cord
Activation of STAT3 in the spinal cord was assessed by fluorescent IHC using antibodies specific for phospho-STAT3 (Tyr705) or phospho-STAT3 (Ser727) that, in addition to Tyr705 phosphorylation, has been demonstrated in some stimulatory conditions, although its exact role in STAT3 transcriptional activity remains unclear (Ng et al., 2006; Lufei et al., 2007). Our previous data from spinal nerve ligation (SNL) (Dominguez et al., 2008) and sciatic nerve transection (our unpublished observations) models of peripheral nerve injury showed that accumulation of pSTAT3-IR (Western blot, IHC) in spinal cord was maximal 24–48 h after the nerve lesion. We evaluated the accumulation and localization of pSTAT3 in the spinal cord 48 h and 1 week after CCI. Phosphorylated STAT3-IR was not detected in the spinal cord from sham-operated animals (data not shown) or in the side contralateral to the constricted sciatic nerve in CCI rats (Fig. 2A). CCI animals showed a clear accumulation of pSTAT3–Tyr705-IR in the superficial and medial laminae (I–IV) of the dorsal horn, spatially distributed from mid-L6 to L4 spinal cord segments of the ipsilateral lumbar spinal cord (whole lumbar enlargement processed through 20 μm slices) (Fig. 2A,B). In contrast, pSTAT3–Ser727-IR was detected in neither control nor CCI animals (data not shown). We performed double-labeling immunohistofluorescent experiments with markers of microglia (ITGAM and Iba1), astrocytes (GFAP), or neurons (NeuN) to characterize the phenotypes of cell(s) containing pSTAT3-IR. We observed dense ITGAM labeling, indicative of microglial cell activation 48 h after CCI injury in the ipsilateral dorsal horn, which was frequently colocalized with pSTAT3-IR (Fig. 2C). The same labeling pattern, showing that pSTAT3-IR mainly accumulated in microglial cells, was observed using another microglial marker, Iba1. Whereas GFAP-labeled astrocytes did not show any overlap with pSTAT3-immunoreactive signal, some scarce spinal cord neurons were double labeled for NeuN and pSTAT3-IR (Fig. 2C). One week after CCI, pSTAT3-IR remained detectable in the ipsilateral dorsal spinal cord, still mainly colocalizing with Iba1-IR; however, the density of immunolabeling was lower than at 48 h after surgery (Fig. 2D). No GFAP-labeled astrocytes and only a few NeuN-labeled neurons showed positive signal for pSTAT3-IR at this time point (data not shown).
Figure 2.
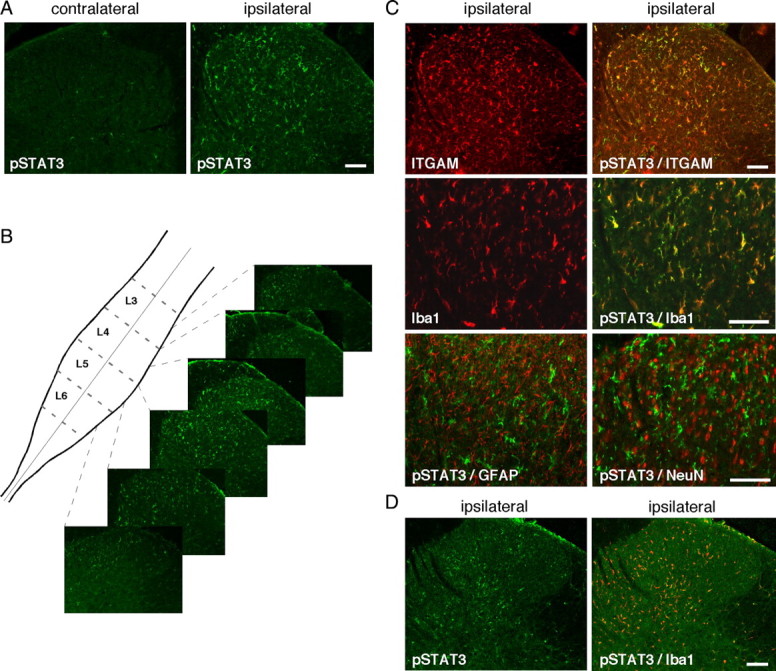
Activation of the JAK/STAT3 transduction pathway. Unilateral CCI of the rat sciatic nerve resulted in the accumulation of the active, phosphorylated form of STAT3 (pSTAT3–Tyr705, in green) 2 d later in numerous cells of the superficial and medial laminae (I–IV) of the dorsal spinal cord, on the side that is ipsilateral to the lesion. A, pSTAT-IR was almost undetectable in the contralateral side of the dorsal spinal cord of CCI rats. B, pSTAT-IR was spatially distributed from approximately the mid-L6 segment to the end of the L4 segment of the spinal cord lumbar enlargement. C, Similarly, CCI injury induced microglial activation mainly in the ipsilateral side of the dorsal spinal cord, as indicated by specific microglial markers ITGAM or Iba1 (in red). Double-labeling experiments for pSTAT3 (in green) with either ITGAM or Iba1 revealed a large colocalization of pSTAT3 with both microglial markers. Colabeling with astrocytes marker GFAP antibodies showed almost no pSTAT-IR (in green) in astrocytes and weak pSTAT3-IR signal in only a very few neurons stained with NeuN antibodies. D, One week after CCI surgery, pSTAT3 labeling (in green) was still detectable but weaker than at 2 d after injury, in the ipsilateral dorsal spinal cord and was mainly present in microglial cells labeled with Iba1-IR (in red). Scale bars, 200 μm.
To further evaluate CCI-induced activation of JAK/STAT3 pathway in the region of the spinal cord ipsilateral to the lesion, we determined the expression profile of the STAT3 target gene socs3 on days 2, 10, 15, and 21 after CCI. Real-time semiquantitative RT-PCR showed that sciatic nerve injury was associated with markedly enhanced levels of SOCS3 mRNA in the ipsilateral dorsal spinal cord (Fig. 3). SOCS3 mRNA concentration was considerably higher 2 d after CCI than in sham animals. It then progressively decreased to reach a level 21 d after nerve injury that was not significantly different from that in sham animals (day 2, ×5.75 ± 0.65, p < 0.001; day 10, ×2.80 ± 0.32, p < 0.01; day 15, ×2.18 ± 0.19, p < 0.05; n = 4–5 for each postinjury time). The ITGAM gene (marker of microglial activation) (Fig. 3) and several other genes associated with spinal cord neuroinflammation (IL-6, CCL2, or ATF3) were also upregulated 2 d after nerve injury, with expression levels remaining higher than those in sham animals over the next 21 d of the experimental procedure (ITGAM day 2, ×2.96 ± 0.13, p < 0.001, day 21, 2.81 ± 0.50, p < 0.001; IL-6 day 2, ×3.08 ± 0.76, p < 0.001; day 21, 4.14 ± 1.01, p < 0.001; CCL2 day 2, ×4.74 ± 1.10, p < 0.001; day 21, 4.26 ± 0.71, p < 0.001; ATF3 day 2, ×18.32 ± 1.64, p < 0.001; day 21, ×15.24 ± 1.15, p < 0.001; n = 4–5 for each postinjury time).
Figure 3.
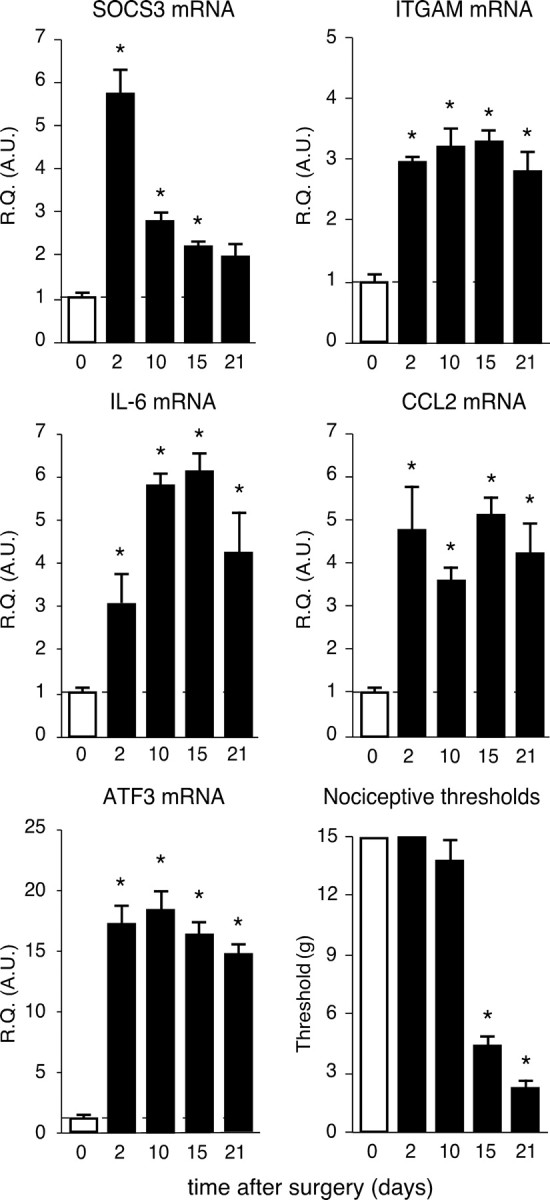
Upregulation of STAT3 target gene SOCS3 and of markers associated with spinal cord inflammatory state. Changes over time of SOCS3, ITGAM, IL-6, CCL2, and ATF3 mRNA levels were determined in the ipsilateral L4–L5 lumbar region of the dorsal spinal cord of CCI rats using semiquantitative real-time RT-PCR. Relative quantification (R.Q.) in arbitrary units (A.U.) corresponds to the ratio of specific mRNA over GAPDH mRNA. In each graph, the dotted line represents the relative quantification of respective mRNA determined in sham animals. Data are representative of different sets of operated rats. Each bar is the mean ± SEM of n = 4–5 rats. Nociceptive threshold of rats to mechanical stimulation was evaluated using von Frey filaments before CCI surgery and then at different postoperative times points. Fifteen grams were chosen as the cutoff threshold to prevent tissue injury. Each bar is the mean ± SEM of n = 8–12 rats. Sham values at each postoperative time point were pooled into one condition referred as 0 (white bars). *p < 0.05, CCI rats versus sham-operated rats at the same respective postoperative time.
We evaluated the mechanical nociceptive threshold of rats using von Frey filaments, at various times after CCI surgery. Rats undergoing CCI surgery exhibited major changes in their response to mechanical stimuli applied to their left hindpaw (the side of sciatic nerve injury) (Fig. 3). Starting postoperative day 15, we observed a markedly lower mechanical threshold in CCI than in sham animals [CCI, 4.53 ± 0.82 g; sham animals, 15 g (maximal force applied); p < 0.001; n = 8–12 for each group]. The allodynia-like behavior of CCI rats persisted on postoperative day 21, consistent with data in the literature (Attal et al., 1990; Field et al., 1999; Latrémolière et al., 2008).
Intrathecal IL-6 administration mimics the rapid activation of JAK/STAT3 and upregulation of inflammatory markers evoked by CCI
We showed previously, by immunoneutralizing IL-6 in the spinal cord of rats suffering from neuropathic pain, that overproduced endogenous IL-6 was directly involved in JAK/STAT3 activation in the spinal cord (Dominguez et al., 2008). Here, to further examine the role of IL-6 in CCI-evoked spinal cord plasticity, IL-6 (1 μg) was injected intrathecally into naive rats and changes of pSTAT3-IR were evaluated by IHC. Whereas pSTAT3 labeling remained almost undetectable in dorsal spinal cord after vehicle intrathecal injection, acute administration of IL-6 (1 μg) led to pSTAT3–Tyr705-IR accumulation bilaterally in the superficial L4–L5 dorsal spinal cord 15 min or 3 h after injection (Fig. 4). pSTAT3–Ser727-IR was not detected in the spinal cord after intrathecal administration of IL-6 (data not shown). Double-labeling immunohistochemistry showed that IL-6-induced pSTAT3-IR accumulation was mainly observed in microglial cells and in only a few spinal cord neurons. Astrocytes were not colabeled for pSTAT3-IR at any of the time points considered (Fig. 4). Moreover, IL-6 intrathecal injection resulted in enhanced expression of several markers that were also consistently upregulated in the spinal cord of CCI rats. Thus, 6 h after IL-6 injection, mRNA concentrations of SOCS3 (×20.9 ± 4.0, n = 4, p < 0.01), IL-6 (×8.8 ± 2.8, n = 4, p < 0.05), TNFα (×12.1 ± 0.8, n = 4, p < 0.001), CCL2 (×50.4 ± 12.3, n = 4, p < 0.02), ATF3 (×5.1 ± 0.4, n = 4, p < 0.005), and ITGAM (×3.2 ± 0.2, n = 4, p < 0.001) were significantly enhanced in the rat dorsal spinal cord.
Figure 4.
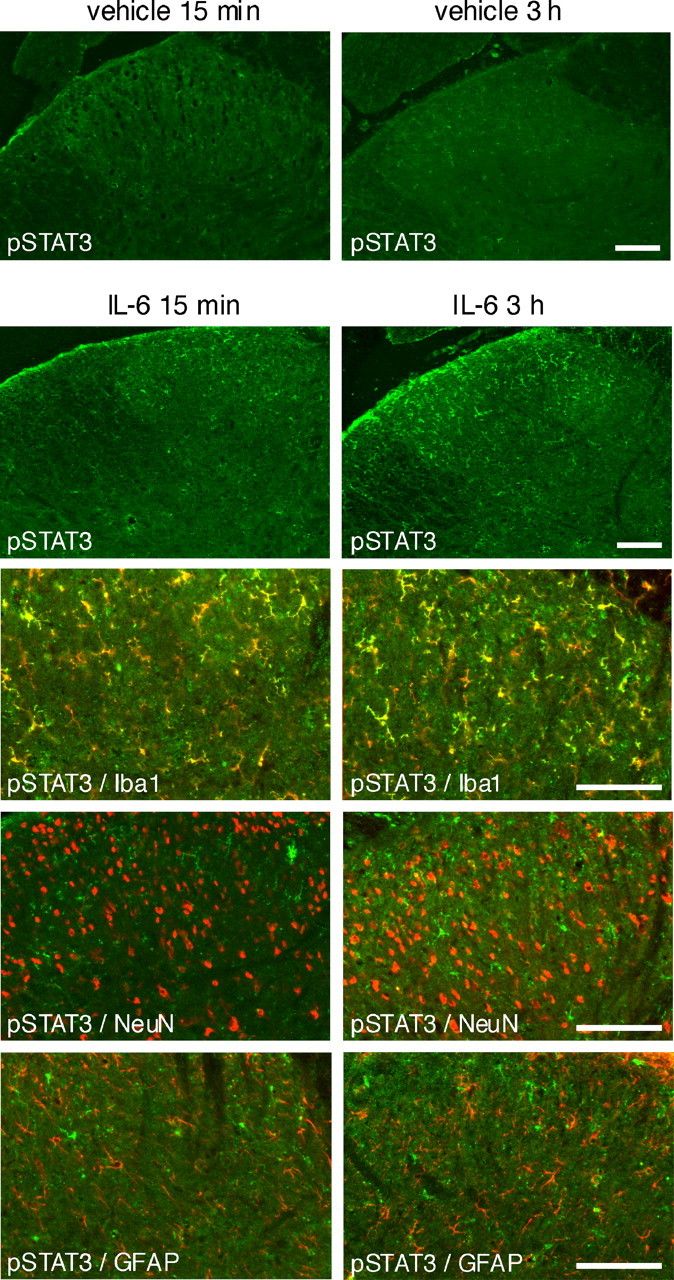
Intrathecal injection of Il-6 resulted in changes reminiscent of CCI-induced alterations in the rat dorsal spinal cord. Vehicle-injected rats showed almost undetectable pSTAT3-IR. In contrast, we observed bilateral accumulation of pSTAT3-IR (pSTAT3–Tyr705, in green) in the superficial layers of the dorsal spinal cord 15 min and 3 h after intrathecal acute injection of IL-6 (1 μg in 25 μl). At both time points, pSTAT3 labeling colocalized mainly with Iba1 microglial marker, showing some colocalization with the NeuN neuronal marker in only a very few cells and almost no colocalization with GFAP-labeled astrocytes. Scale bars, 200 μm.
The induction of rapid JAK/STAT3 activation by IL-6 in the spinal cord and the upregulation of various inflammatory markers led us to further investigate whether IL-6 could also trigger expression of the genes encoding these markers via JAK/STAT3 signaling in primary spinal cord glial cells or in microglial BV2 cells.
IL-6-induced STAT3 phosphorylation and production of inflammatory markers in cultured glial cells can be inhibited by lentivirus-mediated SOCS3 overproduction
Western blot analysis showed that 15 min incubation of primary glial cells with IL-6 (50 ng/ml) resulted in rapid accumulation of pSTAT3 (Fig. 5A). To evaluate the ability of SOCS3 protein to block the IL-6-evoked JAK/STAT3 activation, primary glial cells were transfected with LV–SOCS3t (350 or 35 ng/ml p24) 48 h before their stimulation with IL-6. As shown in Figure 5A, IL-6-induced accumulation of pSTAT3 was completely blocked in LV–SOCS3t-treated cells (350 or 35 ng/ml p24, p < 0.001 when compared with pSTAT3 levels in nontransfected cells incubated with IL-6) (Fig. 5A). Similar results were obtained in BV2 microglial cells line stimulated with IL-6 and infected or not with LV-SOCS3t (data not shown).
Figure 5.

Effects of transducing primary glial cells or BV2 microglia with LV–SOCS3t on JAK/STAT3 pathway activity and inflammatory state markers. A, Stimulation of primary glia with IL-6 (50 ng/ml) resulted in rapid (15 min) pSTAT3 accumulation (i.e., JAK/STAT3 activation, Western blot). This effect was prevented in cells transduced 48 h earlier with LV–SOCS3t (350 or 35 ng/ml p24). Data are shown as mean ± SEM of three independent experiments. #p < 0.001, IL-6-treated versus untreated cell cultures; *p < 0.001, IL-6-treated LV–SOCS3t-transduced cells versus IL-6-treated uninfected cells. B, C, In both primary glial cells (B) and BV2 microglial cell line (C), IL-6-induced production (after 3 h incubation with IL-6) of inflammatory markers (IL-6, CCL2, TNFα) was efficiently inhibited in cells transduced 48 h before with LV–SOCS3t. IL-6 can also induce ATF3 production in BV2 microglia, this effect being significantly prevented in LV–SOCS3t-transduced cells. Each bar is the mean ± SEM (n = 4 for each group). #p < 0.05, IL-6-treated cells versus control cell cultures; *p < 0.05, IL-6-treated LV–SOCS3t-infected cells versus IL-6-treated uninfected cells. R.Q., Relative quantification; A.U., arbitrary unit.
Primary glial cells incubated for 3 h in the presence of IL-6 had higher mRNA levels for several inflammatory markers, including IL-6 (×6.23 ± 0.52, p < 0.001, n = 4), SOCS3 (×12.39 ± 0.43, p < 0.001, n = 4), CCL2 (×5.56 ± 0.28, p < 0.001, n = 4), and TNFα (×16.38 ± 0.50, p < 0.001), than control, untreated glial cells (Fig. 5B). Compared with IL-6 treated cells (infected or not with control LV–EGFP, 35 ng/ml p24), transfection of primary glial cells with LV–SOCS3t (35 ng/ml p24) significantly reduced the IL-6-evoked upregulated expression of IL-6 [×2.02 ± 0.36, p < 0.001, n = 4 (−56%)], CCL2 [×2.65 ± 0.30, p < 0.001, n = 4 (−48%)], and TNFα [×7.57 ± 0.82, p < 0.001, n = 4 (−45%])] (Fig. 5B).
Similarly, compared with controls, stimulation of BV2 microglial cells (infected or not with LV–EGFP) with IL-6 resulted in higher levels of SOCS3 mRNA (×23.21 ± 1.06, p < 0.001) and of several inflammatory markers mRNA (Fig. 5C). Expression of these later markers was significantly inhibited in cell cultures infected with LV–SOCS3t [IL-6, ×155.71 ± 5.09 vs 94.44 ± 2.72, p < 0.001, n = 4 (−39%); CCL2, 13.73 ± 0.49 vs 8.34 ± 0.28, p < 0.005, n = 4 (−40%); TNFα, ×6.91 ± 0.97 vs 3.51 ± 0.25, p < 0.05, n = 4 (−50%)]. The incubation of BV2 cell cultures with IL-6 also resulted in an increased concentration of ATF3 mRNA. This effect was again significantly reduced in cells transfected with LV–SOCS3t [×4.75 ± 0.20 vs 2.76 ± 0.24, p < 0.005, n = 4 (−42%)] (Fig. 5C). We did not observe inhibition of pSTAT3 accumulation or of the increased mRNA levels of various inflammatory markers in either type of glial cell culture infected with control LV–EGFP (35 ng/ml p24) and stimulated with IL-6 (data not shown).
SOCS3 overproduction in the dorsal spinal cord of CCI rats blocks local STAT3 phosphorylation, attenuates the development of CCI-evoked mechanical hypersensitivity, and reduces abnormal expression of inflammatory markers
To evaluate in the spinal cord the role of early activated microglial JAK/STAT3 pathway in the development of biochemical alterations and pain hypersensitivity observed after peripheral nerve lesion, we explored the possibility to locally and selectively block the JAK/STAT3 signaling by the LV-mediated SOCS3 overproduction.
LV–EGFP or LV–SOCS3t vectors were unilaterally microinjected (or not) into the rat dorsal spinal cord of normal animals. These rats were subjected to CCI (or sham) surgery 1 week later. Western blot analysis 2 d after surgery showed increased levels of pSTAT3-IR in the dorsal horn of both non-injected CCI (3.2-fold higher than sham-operated rats, p < 0.001, n = 3) and control vector LV–EGFP-injected CCI (2.9-fold higher than in sham-operated rats, p < 0.001, n = 3) rats (Fig. 6A). pSTAT3-IR levels were markedly reduced (p < 0.001, n = 4), frequently back to basal levels, in the spinal cord of CCI rats treated with LV–SOCS3t. We also examined the status of p38 MAPK and ERK pathways, the activity of which has been reported to play a substantial role in the peripheral nerve injury-evoked neuropathic pain. Three days after CCI (time point for which both p-p38 and pERK were reported to be enhanced in the spinal cord in response to peripheral nerve injury) (Jin et al., 2003; Zhuang et al., 2005; Lee et al., 2009b), Western blots revealed enhanced p-p38-IR and pERK1–2-IR levels in the dorsal spinal cord of non-injected CCI (2.4- or 2.9-fold higher than in sham-operated rats, p < 0.01, n = 3, respectively) and control LV–EGFP vector-injected CCI (2.7-fold higher than in sham-operated rats, p < 0.01, n = 3) rats (Fig. 6B). In LV–SOCS3t-injected CCI rats, p-p38-IR levels tend to decrease (although this effect is not significant), and pERK levels remained elevated and comparable with those measured in control (non-injected and LV–EGFP injected) CCI animals.
Figure 6.
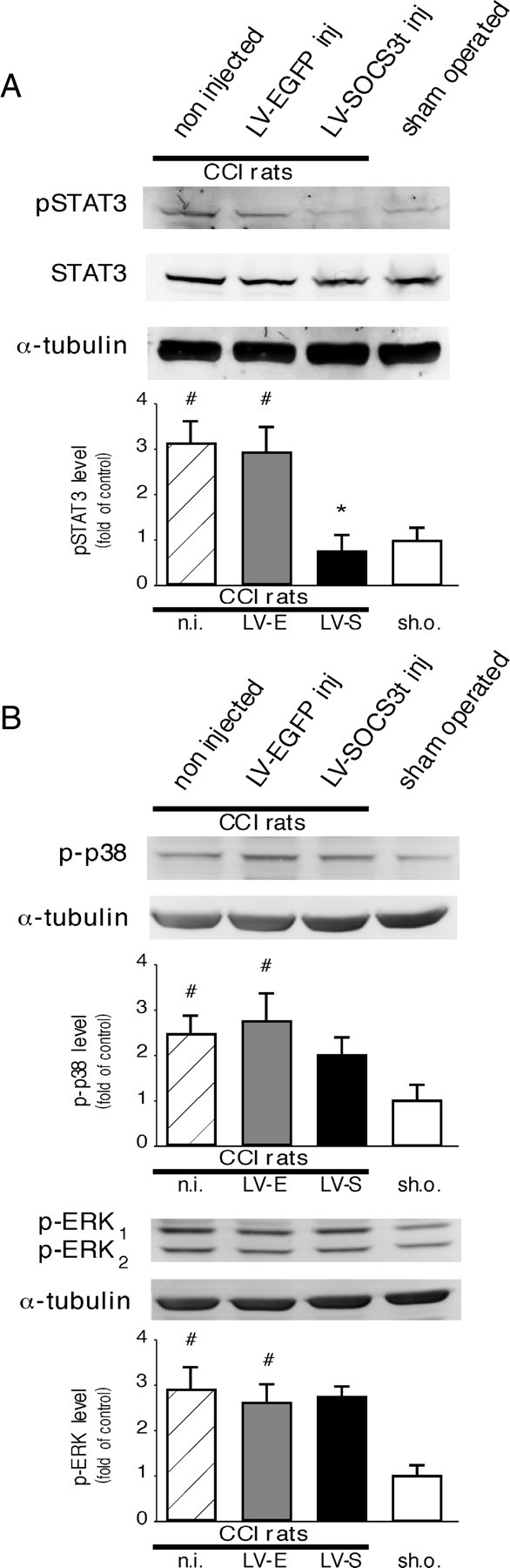
Effects of LV–SOCS3t delivery into the rat dorsal spinal cord on JAK/STAT3, MAPK p38, and ERK pathways activities. A, CCI surgery resulted in pSTAT3 accumulation 2 d later in control or LV–EGFP-infected rats (i.e., JAK/STAT3 activation; Western blots) in the dorsal spinal cord region ipsilateral to the side of the lesion. CCI-evoked pSTAT3 accumulation was completely prevented in rats injected with LV–SOCS3t (2 μl, i.e., 70 ng of p24). Each bar is the mean ± SEM of three independent experiments (n = 3–4 for each group). #p < 0.001, non-injected (n.i.) or LV–EGFP-injected (LV-E) CCI rats versus sham-operated (sh.o.) rats; *p < 0.001, LV–SOCS3t-injected (LV-S) CCI rats versus non-injected or LV–EGFP-injected CCI rats. B, The levels of phosphorylated forms of p38 and ERK MAPK were also increased in the ipsilateral dorsal spinal cord of CCI rats 3 d after surgery (#p < 0.01, #p < 0.001, respectively, vs control). In LV–SOCS3t-injected CCI rats, the level of p38 tend to decrease (although this change was statistically not significant) and pERK levels were unchanged, remaining comparable with those found in control CCI rats (because no difference was observed in pERK1 and pERK2 protein levels under different experimental conditions, only levels of ERK1 protein are represented in quantification graph).
Behavioral experiments were performed 15, 21, and 28 d after CCI. Consistent with our previous data (Meunier et al., 2007, 2008), vector delivery into the spinal cord of rats did not modify their basal pain sensitivity (data not shown). Similarly, the mechanical pain sensitivity of sciatic nerve sham-operated rats assessed with von Frey filaments did not significantly differ from that of naive animals at any time point studied (data not shown). CCI injury resulted in robust mechanical allodynia on the left (operated) side that appeared 2 weeks after nerve constriction and lasted for at least 1 month (Fig. 7A). No significant mechanical hypersensitivity was observed on the right side (contralateral, non-operated) (data not shown). The CCI-induced mechanical hypersensitivity in non-injected rats was indistinguishable from that in LV–EGFP-injected rats. Data from both groups were thus pooled and referred to as CCI sham rats. In contrast, injection of LV–SOCS3t in the dorsal spinal cord resulted in strong attenuation of CCI-induced mechanical hypersensitivity throughout the observation period compared with that developing in CCI sham rats (day 15, p < 0.01; day 21, p < 0.001; day 28, p < 0.01; n = 8–10 for each group) (Fig. 7A).
Figure 7.

Effects of LV–SOCS3t delivery into the rat dorsal spinal cord on pain behavior, local spinal cord inflammation, and glial activity. A, CCI induced mechanical allodynia in control (non-injected) or LV-EGFP-injected rats (2 μl, i.e., 70 ng of p24). This effect was not seen in sham-operated rats and was potently attenuated in LV–SOCS3t-treated rats. Each point is the mean ± SEM of n = 8–10 animals. #p < 0.001, non-injected or LV–EGFP-injected CCI rats versus sham-operated rats; *p < 0.01 LV–SOCS3t-injected CCI rats versus control or LV–EGFP-injected CCI rats. B, LV–SOCS3t-mediated blockade of JAK/STAT3 signaling in the spinal cord effectively prevented the upregulation of IL-6, CCL2, or ATF3 mRNA associated with CCI, 15 and 21 d after surgery. B, However, the high levels of ITGAM mRNA (revealing microglial activation) were unaffected by local STAT3 blockade. C, At these time points, astrocyte activation is also associated with CCI. The upregulated expression of astrocyte activity marker GFAP, observed 21 d after CCI in control rats, was attenuated in LV–SOCS3t-injected CCI animals. IL-1β, whose expression is induced after CCI, particularly in activated astrocytes, was also attenuated in LV–SOCS3t-injected CCI rats. Western blot experiments, showing low levels of GFAP protein in LVSOCS3t-injected CCI rats compared with CCI controls, further confirmed the reduced astrocyte activation in rats with locally inhibited JAK/STAT3 signaling. Data represent the mean ± SEM of n = 4–6 rats for each experimental group. #p < 0.05, non-injected or LV–EGFP-injected CCI rats versus sham operated rats; *p < 0.05, LV–SOCS3t-injected versus LV–EGFP-injected CCI rats. For Western blot experiments, each bar is the mean ± SEM of three independent experiments (n = 3 for each group). R.Q. (A.U.), Relative quantification in arbitrary units; n.i., non-injected CCI rats; LV-E, LV–EGFP-injected CCI rats; LV-S, LV–SOCS3t-injected CCI rats; sh.o., sham-operated rats.
To further investigate the effects of local SOCS3-mediated blockade of JAK/STAT3 signaling, we analyzed the mRNA levels of several markers, the production of which was altered in the spinal cord of CCI rats (ipsilateral side to the lesion). Thus, 15 d (when mechanical hyperalgesia appears) and 21 d (when hyperalgesia is fully developed) after CCI, the expression levels of IL-6 (day 15, −82.4 ± 8.2%, p < 0.01; day 21, −93.2 ± 3.4%, p < 0.01), CCL2 (day 15, −90.1 ± 5.2%, p < 0.01; day 21, −88.1 ± 8.3%, p < 0.05), and the nerve injury marker ATF3 (day 15, −92.4 ± 7.3%, p < 0.001; day 21, −89.0 ± 5.3%, p < 0.01; for each mRNA levels determination, n = 4–6 for each group) were markedly lower in rats injected with LV–SOCS3t than in CCI sham rats (Fig. 7B). However, CCI-evoked enhanced concentrations of the microglia activation marker ITGAM mRNA (p > 0.05, n = 4–6) (Fig. 7B) were not modified by administration of LV–SOCS3t at any time point studied, remaining similar to those measured in CCI sham rats.
Activated astrocytes are also thought to play a role in peripheral nerve injury-evoked neuropathic pain, particularly in its later phase. We thus also assessed a possible effect of local SOCS3 overexpression on astrocyte activity status through the measurement of GFAP astrocyte marker production (both GFAP mRNA and protein levels) and expression of IL-1β, a cytokine also induced in activated astrocytes. As shown in Figure 7C, 21 d after CCI, GFAP protein levels in the spinal cord were higher (2.3-fold higher in non-injected and sham-injected animals, p < 0.01, n = 3) than in naive rats. In contrast, compared with control CCI animals, spinal cord from LV–SOCS3t-injected CCI rats presented significantly decreased GFAP levels (p < 0.05, n = 3). At this time point, GFAP mRNA concentration was also lower in LV–SOCS3t-treated CCI rats than in CCI sham rats (day 21, −68.2 ± 8.8%, p < 0.05). IL-1β mRNA levels in LV–SOCS3t-treated versus sham CCI rats were significantly decreased only 15 d after the nerve injury (day 15, −51.5 ± 9.1%, p < 0.05; for each mRNA levels determination, n = 5 for each group).
The powerful inhibitory action of SOCS3 thus indicated that local activity of JAK/STAT3 transduction pathway, rapidly induced after CCI, is important in spinal cord inflammatory response and development of tactile allodynia after injury of peripheral nerve.
LV–SOCS3t injection into the rat dorsal spinal cord induces local and long-term expression of SOCS3t
To further document that transgene-derived SOCS3 was involved in observed biochemical and behavioral effects, we monitored its production in dorsal spinal cord of injected rats. In line with our previous data (Meunier et al., 2007, 2008), injection of LVs into the rat dorsal horn of the spinal cord resulted in transgene expression preferentially in glial cells, with only a small number of neurons producing low levels of the transgene product (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). LV–SOCS3t or LV–EGFP (control) was microinjected unilaterally into the rat dorsal horn of the spinal cord. Two days after LV–EGFP administration (∼70 ng of p24), SOCS3 mRNA concentration was slightly increased (2.20 ± 0.32 times higher than in naive controls, p < 0.05, n = 3) (Fig. 8A). This increase was transient: 1 week later, SOCS3 mRNA concentration returned to control values (data not shown). However, intraparenchymal microinjection of 2 μl of LV–SOCS3t (70 ng of p24) led to a sustained overexpression of SOCS3 mRNA (real-time semiquantitative RT-PCR) and concomitant expression of the V5 tag sequence (detected using conventional RT-PCR) from 1 week until 6 months after injection (SOCS3 mRNA levels at 1 week, 55.82 ± 10.50 times higher than control naive rats, p < 0.001, n = 4; at 3 months, 76.23 ± 6.03 times higher than control naive rats, p < 0.001, n = 4) (Fig. 8A). In the contralateral dorsal horn of the spinal cord (non-injected), SOCS3 mRNA levels remained similar to those measured in control naive rats, with no specific V5 amplification observed (data not shown). Western blot analysis using anti-V5-specific antibodies 1 week after spinal administration of either LV–EGFP or LV–SOCS3t further confirmed the specific production of transgene-derived SOCS3t protein in spinal cord from rats injected with LV–SOCS3t. V5-IR was not detected in spinal cord extracts from either control naive animals or LV–EGFP-injected rats (Fig. 8B). The spread of viral vector-mediated transgene production through the spinal cord was evaluated using LV–EGFP. In fact, detection of EGFP spontaneous fluorescence was much more sensitive than IHC performed with anti-SOCS3 or anti-V5 antibodies, thus revealing even relatively weakly transfected cells. These experiments confirmed our previous data (Meunier et al., 2007, 2008) showing that transgene-derived production is detectable in a large part of the L4–L5 spinal cord segments (i.e., in the region showing high levels of pSTAT3-IR), spreading rostrocaudally through 4–5 mm but remaining closely restricted to the injected (left) side of the spinal cord (Fig. 8C).
Figure 8.
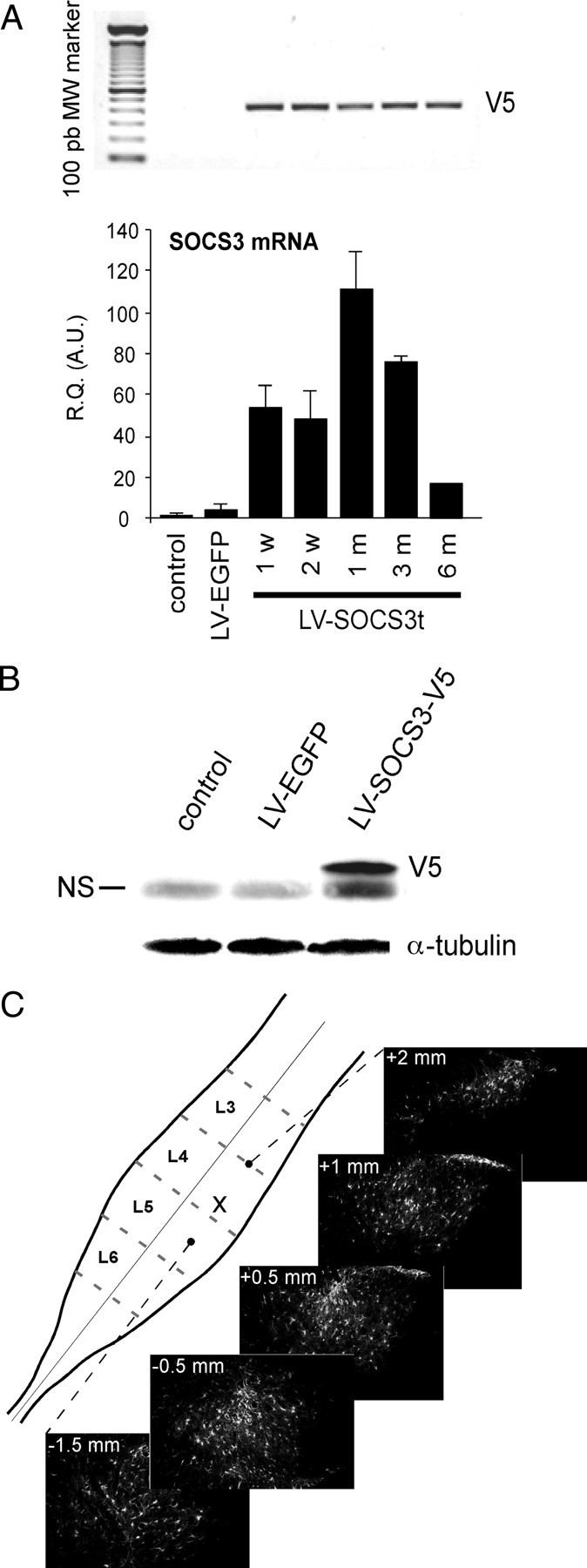
Viral vector-derived transgene production in the dorsal horn of the spinal cord after local intraparenchymal microinjection of LV–SOCS3t or LV–EGFP. A, Local production of transgene-derived mRNA was monitored at different time points after LV–SOCS3t injection using conventional or real-time RT-PCR amplification of the tag (V5) sequence (top) or SOCS3 (bottom), respectively. Data from conventional RT-PCR are representative of different sets of injected rats. Each bar is the mean ± SEM (n = 4, except at the 6 month postinjection time point, n = 2). R.Q. (A.U.), Relative quantification in arbitrary units. Control, Non-injected animals. B, The presence of transgene-derived SOCS3 protein was confirmed 1 week after LV–SOCS3t injection (Western blot for V5 antigen). Blots are representative of three independent experiments with distinct set of animals. NS, Nonspecific immunolabeling. C, Single injection of 2 μl (70 ng of p24) of viral suspension resulted in transgene (EGFP) expression strictly restricted to the injected dorsal horn and spreading rostrocaudally through 4–5 mm (i.e., in the region of the spinal cord in which pSTAT3 was highly accumulating; also see Fig. 2.) x, Microinjection point.
Discussion
We demonstrate that the activity of the JAK/STAT3 signal transduction pathway is rapidly induced in the spinal cord, mainly in microglial cells, in response to peripheral nerve injury (CCI). We show that the physiologic SOCS3 inhibitory protein, locally produced from LVs, may efficiently block JAK/STAT3 function and attenuate spinal cord neuroinflammation. SOCS3 inhibits JAK/STAT3 downstream signaling responses and, in particular, blocks the upregulated expression of inflammatory mediators IL-6, CCL2, and the ATF3 transcription factor observed in the spinal cord of CCI rats. This targeted, mainly glia-oriented blockade of JAK/STAT3 signaling markedly attenuates the development of mechanical allodynia, a hallmark symptom of CCI-induced neuropathic pain.
The inflammatory response that develops in the spinal cord after peripheral nerve injury plays an important role in inducing and maintaining nerve injury-associated pathological pain (Marchand et al., 2005; Scholz and Woolf, 2007; Suter et al., 2007). In this context, we recently demonstrated that ligation of the L6 and L5 spinal nerves (SNL, a neuropathic pain model) leads to activation of the JAK/STAT3 pathway, mostly in the microglia within the ipsilateral dorsal spinal cord (L6–L5 segments) and that IL-6 (the expression of which is ipsilaterally induced first in DRG and then in dorsal spinal cord) is involved in early STAT3 activation (Dominguez et al., 2008). A similar pattern of JAK/STAT3 activation, monitored through the detection of phosphorylated (active) STAT3 accumulation, was shown in this study after CCI (mid-L6 to L4 spinal cord segments). Different types of peripheral nerve injury thus lead to a common response in the spinal cord: pSTAT3 accumulation in regions overlapping the projection zones of the altered peripheral nerves. These observations suggest that JAK/STAT3 activation is triggered principally by signals from the central terminals of primary afferents and that IL-6 may be one of these initial stimuli. The production of leukemia inhibitory factor and ciliary neurotrophic factor, the other members of the IL-6 cytokine family, increases only modestly (or remains unchanged, for CNTF) after peripheral nerve injury, suggesting a minor contribution of these molecules to the early activation of JAK/STAT3 in the spinal cord (Dominguez et al., 2008). Other locally produced and/or released molecules (including IL-6 in the spinal cord, NGF, and BDNF) signaling through this pathway and displaying changes in production levels or activity after peripheral nerve injury may be also involved in the prolonged activation of JAK/STAT3 and its subsequent biological effects. In this context, leptin was recently identified as one of the activators of JAK/STAT3 pathway in the spinal cord and participating in pain after peripheral nerve injury (Lim et al., 2009). However, the cell types in which the JAK/STAT3 pathway was activated by leptin (i.e., contained the phosphorylated form of STAT3) were not identified. In line with our previous data for SNL (Dominguez et al., 2008), we show here that, 2 d or 1 week after CCI, pSTAT3-IR is consistently colocalized with two microglial markers, ITGAM and Iba1, but not with neuronal (NeuN) or astrocyte (GFAP) markers. However, we cannot exclude the possibility that, in the later phases of the spinal cord inflammatory process induced by CCI, the JAK/STAT3 pathway may also be activated in other cell types. Our data showing that acute intrathecal injection of IL-6 results in spinal cord alterations resembling CCI-induced changes (including pSTAT3 accumulation), and the data obtained by Lim et al. (2009) suggest that the JAK/STAT3 pathway, involved in both IL-6 and leptin signaling, plays an important role in the spinal cord plasticity developing after peripheral nerve injury.
Our previous experiments with the intrathecally administered JAK2 tyrosine kinase inhibitor AG490 used to block JAK/STAT3 activity suggested that, in the spinal cord, this pathway is involved in pain development after spinal nerve lesion (Dominguez et al., 2008). The intrathecal codelivery of AG490 and leptin also seems to reduce certain aspects of leptin-induced pain hypersensitivity (Lim et al., 2009), further supporting a role for JAK/STAT3 signaling in pain behavior. However, AG490 is only partially selective for JAK2 kinase, and intrathecal delivery (resulting in extensive spreading of the drug, even to DRG) cannot be used for targeted intervention (Ji et al., 2002; Obata et al., 2004; Zhuang et al., 2006). We thus explored the possibility of blocking JAK/STAT3 activity through the glia-oriented production of the endogenous inhibitory protein SOCS3. The SOCS family of proteins has been identified as a major negative regulator of pathways triggering cytokine signaling (Wang and Campbell, 2002; O'Shea and Murray, 2008). In particular, SOCS3, the production of which is induced directly by IL-6, may specifically inhibit JAK/STAT3 in the periphery, with therapeutic, anti-inflammatory effects in vivo (Jo et al., 2005; Rønn et al., 2008). We recently developed an approach for the preferential expression of transgenes from LVs in glial cells of the rat dorsal spinal cord, to gain further insight into the changes occurring in glia during pathological neuroinflammation in the spinal cord (Meunier et al., 2007, 2008). Here, the microdelivery of LVs encoding a tagged rat SOCS3 led to prolonged local SOCS3 production, resulting in an almost complete inhibition of JAK/STAT3 signaling in the dorsal spinal cord. The activity of the MAPK p38 and ERK pathways, upregulated in the spinal cord in response to peripheral nerve injury and known to play a role in neuropathic pain (Jin et al., 2003; Zhuang et al., 2005; Lee et al., 2009b), was not affected by local SOCS3 production. However, p-p38 protein levels tended to decrease, although not significantly, potentially reflecting the weaker expression of several potential stimulators of this pathway (IL-6, CCL2) in the spinal cord of LV–SOCS3-treated CCI rats. Indeed, the SOCS3-mediated blockade of JAK/STAT3 signaling resulted in markedly lower levels of IL-6 expression, which, as shown in previous studies (Latrémolière et al., 2008; Lee et al., 2009a), is upregulated in the spinal cord 15 and 21 d after CCI, suggesting that, as in immunocompetent cells (O'Shea and Murray, 2008), the JAK/STAT3 pathway plays a major role in regulating IL-6 expression in the spinal cord.
The recently suggested relationship between increases in IL-6 levels and upregulation of the “neuronal injury marker” ATF3 in the dorsal spinal cord of CCI rats (Latrémolière et al., 2008) was further evidenced in this work, showing that a local blockade of JAK/STAT3 signaling prevents the CCI-evoked upregulation of ATF3. ATF3 is classically produced in neurons but may also be strongly induced in macrophages (Gilchrist et al., 2006) and, as shown here, in microglial cell cultures, in which IL-6-induced ATF3 expression is reduced by JAK/STAT3 blockade.
The CCL2 chemokine is another signaling molecule playing a direct role in pain, particularly after peripheral nerve injury (Abbadie, 2005; Thacker et al., 2009). CCL2 expression may be regulated through the nuclear factor-κB (Ping et al., 1999) and STAT3 (Kim et al., 2002) transcription factors. The very weak expression of CCL2 in CCI rats, resulting from a local blockade of JAK/STAT3 activity, suggests that, in the spinal cord, JAK/STAT3 signaling plays an important role in controlling CCL2 expression. A recent study showed that TNFα, by activating the c-Jun N-terminal kinase (JNK) pathway, mediates CCL2 production in spinal astrocytes during SNL-induced neuropathic pain in mice (Gao et al., 2009). Our data for BV2 mouse microglial cells demonstrate that CCL2 production is induced not only in astrocytes but also in microglia, by IL-6, via the JAK/STAT3 pathway. Because IL-6 induced a robust increase in TNFα levels both in vivo, in the spinal cord, and in vitro, in microglial cells, it is possible that CCL2 production may be induced not only in microglia but also in astrocytes, by TNFα acting through JNK.
A blockade of JAK/STAT3 activity and its downstream responses was achieved through the local, glia-oriented production of a physiologically relevant protein, SOCS3. This markedly attenuated mechanical pain hypersensitivity in CCI rats, suggesting that the JAK/STAT3 activity rapidly induced in the spinal cord after peripheral nerve injury is one of the early changes preceding the development of mechanical allodynia (fully expressed in the CCI model 2 weeks after nerve constriction). This attenuation of pain hypersensitivity persisted throughout the 3 week observation period and was accompanied by the continued production of SOCS3 from the transgene and a marked decrease in the levels of mRNA for the proalgesic cytokines IL-6 and CCL2 in the spinal cord. The decrease in the production of these signaling molecules, known for their ability to activate astrocytes (Okada et al., 2004) (for review, see Milligan and Watkins, 2009), may account, at least in part, for the lower levels of GFAP observed in CCI rats in which the JAK/STAT3 pathway was inhibited. In addition, because STAT3 participates in the control of gfap gene transcription (Sriram et al., 2004), the direct LV–SOCS3-mediated blockade of the JAK/STAT3 pathway in astrocytes may be also involved in the downregulation of GFAP production. We therefore cannot rule out the possibility that changes in at least some aspects of astrocyte activity are involved in the attenuation of mechanical allodynia, particularly during the late phase of the response to injury.
As demonstrated previously, LV microinjection results in the production of the transgene-encoded protein strictly within the dorsal horn of the spinal cord (Meunier et al., 2007, 2008). Thus, the biochemical and behavioral effects of the overproduction of SOCS3 in the spinal cord directly reflect the local, targeted blockade of JAK/STAT3 in the spinal cord. In this work, we studied the role of JAK/STAT3 signaling in the development of neuroinflammation in the spinal cord by the preventive blockade of this pathway. In future studies, we will also evaluate the consequences of JAK/STAT3 inhibition in animals with established neuropathic pain.
The treatment of neuropathic pain remains a challenge because of the complex changes and tissue plasticity associated with this pathological state and the diversity of mediators and signaling molecules involved. Intracellular transduction pathways not only mediate the local biological effects of several different molecules but may also amplify their detrimental consequences through the production of other potentially harmful products. Thus, the local control of signal transduction pathways may open up new possibilities for the treatment of neuropathic pain going beyond the blockade of individual signaling molecules or their receptors. As shown here, the local and selective blockade of JAK/STAT3 pathway by an endogenous inhibitory protein clearly has a beneficial effect. Our data demonstrate the key role of this pathway in the development of a neuroinflammatory state in the spinal cord, a major contributory factor in pathological pain development.
Footnotes
This research was supported by grants from the Institut National de la Santé et de la Recherche Médicale, Université Pierre et Marie Curie (Paris 6), and Agence Nationale de la Recherche. E.D. was the recipient of fellowships from the Institut UPSA de la Douleur and Institut de Recherche sur la Moelle et Encéphale. We are grateful to Dr. J. Van Steenwinckel for her help with statistical analyses and Dr. A. Réaux-Le Goazigo for critical reading of this manuscript. Special thanks to Dr. A. Meunier for her initial help with the in vivo microinjection technique.
References
- Abbadie C. Chemokines, chemokine receptors and pain. Trends Immunol. 2005;26:529–534. doi: 10.1016/j.it.2005.08.001. [DOI] [PubMed] [Google Scholar]
- Attal N, Jazat F, Kayser V, Guilbaud G. Further evidence for “pain-related” behaviours in a model of unilateral peripheral mononeuropathy. Pain. 1990;41:235–251. doi: 10.1016/0304-3959(90)90022-6. [DOI] [PubMed] [Google Scholar]
- Bennett GJ, Xie YK. A peripheral mononeuropathy in rat that produces disorders of pain sensation like those seen in man. Pain. 1988;33:87–107. doi: 10.1016/0304-3959(88)90209-6. [DOI] [PubMed] [Google Scholar]
- Chaplan SR, Bach FW, Pogrel JW, Chung JM, Yaksh TL. Quantitative assessment of tactile allodynia in the rat paw. J Neurosci Methods. 1994;53:55–63. doi: 10.1016/0165-0270(94)90144-9. [DOI] [PubMed] [Google Scholar]
- Dixon WJ. Efficient analysis of experimental observations. Annu Rev Pharmacol Toxicol. 1980;20:441–462. doi: 10.1146/annurev.pa.20.040180.002301. [DOI] [PubMed] [Google Scholar]
- Dominguez E, Rivat C, Pommier B, Mauborgne A, Pohl M. JAK/STAT3 pathway is activated in spinal cord microglia after peripheral nerve injury and contributes to neuropathic pain development in rat. J Neurochem. 2008;107:50–60. doi: 10.1111/j.1471-4159.2008.05566.x. [DOI] [PubMed] [Google Scholar]
- Field MJ, Bramwell S, Hughes J, Singh L. Detection of static and dynamic components of mechanical allodynia in rat models of neuropathic pain: are they signaled by distinct primary sensory neurons? Pain. 1999;83:303–311. doi: 10.1016/s0304-3959(99)00111-6. [DOI] [PubMed] [Google Scholar]
- Gao YJ, Zhang L, Samad OA, Suter MR, Yasuhiko K, Xu ZZ, Park JY, Lind AL, Ma Q, Ji RR. JNK-induced MCP-1 production in spinal cord astrocytes contributes to central sensitization and neuropathic pain. J Neurosci. 2009;29:4096–4108. doi: 10.1523/JNEUROSCI.3623-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilchrist M, Thorsson V, Li B, Rust AG, Korb M, Roach JC, Kennedy K, Hai T, Bolouri H, Aderem A. Systems biology approaches identify ATF3 as a negative regulator of Toll-like receptor 4. Nature. 2006;441:173–178. doi: 10.1038/nature04768. [DOI] [PubMed] [Google Scholar]
- Goslin K, Asmussen H, Banker G. Rat hippocampal neurons in low-density culture. In: Banker G, Goslin K, editors. Culturing nerve cells. Cambridge, MA: Massachusetts Institute of Technology; 1998. pp. 339–370. [Google Scholar]
- Herrmann JE, Imura T, Song B, Qi J, Ao Y, Nguyen TK, Korsak RA, Takeda K, Akira S, Sofroniew MV. STAT3 is a critical regulator of astrogliosis and scar formation after spinal cord injury. J Neurosci. 2008;28:7231–7243. doi: 10.1523/JNEUROSCI.1709-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji GC, Zhang YQ, Ma F, Wu GC. Increase of nociceptive threshold induced by intrathecal injection of interleukin-1beta in normal and carrageenan inflammatory rat. Cytokine. 2002;19:31–36. doi: 10.1006/cyto.2002.1949. [DOI] [PubMed] [Google Scholar]
- Jin SX, Zhuang ZY, Woolf CJ, Ji RR. p38 mitogen-activated protein kinase is activated after a spinal nerve ligation in spinal cord microglia and dorsal root ganglion neurons and contributes to the generation of neuropathic pain. J Neurosci. 2003;23:4017–4022. doi: 10.1523/JNEUROSCI.23-10-04017.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jo D, Liu D, Yao S, Collins RD, Hawiger J. Intracellular protein therapy with SOCS3 inhibits inflammation and apoptosis. Nat Med. 2005;11:892–898. doi: 10.1038/nm1269. [DOI] [PubMed] [Google Scholar]
- Kim OS, Park EJ, Joe EH, Jou I. JAK-STAT signaling mediates gangliosides-induced inflammatory responses in brain microglial cells. J Biol Chem. 2002;277:40594–40601. doi: 10.1074/jbc.M203885200. [DOI] [PubMed] [Google Scholar]
- Latrémolière A, Mauborgne A, Masson J, Bourgoin S, Kayser V, Hamon M, Pohl M. Differential implication of proinflammatory cytokine interleukin-6 in the development of cephalic versus extracephalic neuropathic pain in rats. J Neurosci. 2008;28:8489–8501. doi: 10.1523/JNEUROSCI.2552-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee KM, Jeon SM, Cho HJ. Tumor necrosis factor receptor 1 induces interleukin-6 upregulation through NF-kappaB in a rat neuropathic pain model. Eur J Pain. 2009a;13:794–806. doi: 10.1016/j.ejpain.2008.09.009. [DOI] [PubMed] [Google Scholar]
- Lee KM, Jeon SM, Cho HJ Advance online publication. Interleukin-6 induces microglial CX3CR1 expression in the spinal cord after peripheral nerve injury through the activation of p38 MAPK. Eur J Pain. 2009b doi: 10.1016/j.ejpain.2009.10.017. [DOI] [PubMed] [Google Scholar]
- Lim G, Wang S, Zhang Y, Tian Y, Mao J. Spinal leptin contributes to the pathogenesis of neuropathic pain in rodents. J Clin Invest. 2009;119:295–304. doi: 10.1172/JCI36785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- Lufei C, Koh TH, Uchida T, Cao X. Pin1 is required for the Ser727 phosphorylation-dependent Stat3 activity. Oncogene. 2007;26:7656–7664. doi: 10.1038/sj.onc.1210567. [DOI] [PubMed] [Google Scholar]
- Marchand F, Perretti M, McMahon SB. Role of the immune system in chronic pain. Nat Rev Neurosci. 2005;6:521–532. doi: 10.1038/nrn1700. [DOI] [PubMed] [Google Scholar]
- Meunier A, Latrémolière A, Dominguez E, Mauborgne A, Philippe S, Hamon M, Mallet J, Benoliel JJ, Pohl M. Lentiviral-mediated targeted NF-kappaB blockade in dorsal spinal cord glia attenuates sciatic nerve injury-induced neuropathic pain in the rat. Mol Ther. 2007;15:687–697. doi: 10.1038/sj.mt.6300107. [DOI] [PubMed] [Google Scholar]
- Meunier A, Mauborgne A, Masson J, Mallet J, Pohl M. Lentiviral-mediated targeted transgene expression in dorsal spinal cord glia: tool for the study of glial cell implication in mechanisms underlying chronic pain development. J Neurosci Methods. 2008;167:148–159. doi: 10.1016/j.jneumeth.2007.07.022. [DOI] [PubMed] [Google Scholar]
- Milligan ED, Watkins LR. Pathological and protective roles of glia in chronic pain. Nat Rev Neurosci. 2009;10:23–36. doi: 10.1038/nrn2533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng YP, Cheung ZH, Ip NY. STAT3 as a downstream mediator of Trk signalling and functions. J Biol Chem. 2006;281:15636–15644. doi: 10.1074/jbc.M601863200. [DOI] [PubMed] [Google Scholar]
- Nicholson SE, De Souza D, Fabri LJ, Corbin J, Willson TA, Zhang JG, Silva A, Asimakis M, Farley A, Nash AD, Metcalf D, Hilton DJ, Nicola NA, Baca M. Suppressor of cytokine signaling-3 preferentially binds to the SHP-2-binding site on the shared cytokine receptor subunit gp130. Proc Natl Acad Sci U S A. 2000;97:6493–6498. doi: 10.1073/pnas.100135197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obata K, Yamanaka H, Kobayashi K, Dai Y, Mizushima T, Katsura H, Fukuoka T, Tokunaga A, Noguchi K. Role of mitogen-activated protein kinase activation in injured and intact primary afferent neurons for mechanical and heat hypersensitivity after spinal nerve ligation. J Neurosci. 2004;24:10211–10222. doi: 10.1523/JNEUROSCI.3388-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okada S, Nakamura M, Mikami Y, Shimazaki T, Mihara M, Ohsugi Y, Iwamoto Y, Yoshizaki K, Kishimoto T, Toyama Y, Okano H. Blockade of interleukin-6 receptor suppresses reactive astrogliosis and ameliorates functional recovery in experimental spinal cord injury. J Neurosci Res. 2004;76:265–276. doi: 10.1002/jnr.20044. [DOI] [PubMed] [Google Scholar]
- O'Shea JJ, Murray PJ. Cytokine signaling modules in inflammatory responses. Immunity. 2008;28:477–487. doi: 10.1016/j.immuni.2008.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ping D, Boekhoudt GH, Rogers EM, Boss JM. Nuclear factor-kappa B p65 mediates the assembly and activation of the TNF-responsive element of the murine monocyte chemoattractant-1 gene. J Immunol. 1999;162:727–734. [PubMed] [Google Scholar]
- Rønn SG, Börjesson A, Bruun C, Heding PE, Frobøse H, Mandrup-Poulsen T, Karlsen AE, Rasschaert J, Sandler S, Billestrup N. Suppressor of cytokine signalling-3 expression inhibits cytokine-mediated destruction of primary mouse and rat pancreatic islets and delays allograft rejection. Diabetologia. 2008;51:1873–1882. doi: 10.1007/s00125-008-1090-0. [DOI] [PubMed] [Google Scholar]
- Scholz J, Woolf CJ. The neuropathic pain triad: neurons, immune cells and glia. Nat Neurosci. 2007;10:1361–1368. doi: 10.1038/nn1992. [DOI] [PubMed] [Google Scholar]
- Shouda T, Yoshida T, Hanada T, Wakioka T, Oishi M, Miyoshi K, Komiya S, Kosai K, Hanakawa Y, Hashimoto K, Nagata K, Yoshimura A. Induction of the cytokine signal regulator SOCS3/CIS3 as a therapeutic strategy for treating inflammatory arthritis. J Clin Invest. 2001;108:1781–1788. doi: 10.1172/JCI13568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sriram K, Benkovic SA, Hebert MA, Miller DB, O'Callaghan JP. Induction of gp130-related cytokines and activation of JAK2/STAT3 pathway in astrocytes precedes up-regulation of glial fibrillary acidic protein in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine model of neurodegeneration: key signaling pathway for astrogliosis in vivo? J Biol Chem. 2004;279:19936–19947. doi: 10.1074/jbc.M309304200. [DOI] [PubMed] [Google Scholar]
- Suter MR, Wen YR, Decosterd I, Ji RR. Do glial cells control pain? Neuron Glia Biol. 2007;3:255–268. doi: 10.1017/S1740925X08000100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thacker MA, Clark AK, Bishop T, Grist J, Yip PK, Moon LD, Thompson SW, Marchand F, McMahon SB. CCL2 is a key mediator of microglia activation in neuropathic pain states. Eur J Pain. 2009;13:263–272. doi: 10.1016/j.ejpain.2008.04.017. [DOI] [PubMed] [Google Scholar]
- Wang J, Campbell IL. Cytokine signaling in the brain: putting a SOCS in it? J Neurosci Res. 2002;67:423–427. doi: 10.1002/jnr.10145. [DOI] [PubMed] [Google Scholar]
- Yamauchi K, Osuka K, Takayasu M, Usuda N, Nakazawa A, Nakahara N, Yoshida M, Aoshima C, Hara M, Yoshida J. Activation of JAK/STAT signalling in neurons following spinal cord injury in mice. J Neurochem. 2006;96:1060–1070. doi: 10.1111/j.1471-4159.2005.03559.x. [DOI] [PubMed] [Google Scholar]
- Zennou V, Serguera C, Sarkis C, Colin P, Perret E, Mallet J, Charneau P. The HIV-1 DNA flap stimulates HIV vector-mediated cell transduction in the brain. Nat Biotechnol. 2001;19:446–450. doi: 10.1038/88115. [DOI] [PubMed] [Google Scholar]
- Zhuang ZY, Gerner P, Woolf CJ, Ji RR. ERK is sequentially activated in neurons, microglia, and astrocytes by spinal nerve ligation and contributes to mechanical allodynia in this neuropathic pain model. Pain. 2005;114:149–159. doi: 10.1016/j.pain.2004.12.022. [DOI] [PubMed] [Google Scholar]
- Zhuang ZY, Wen YR, Zhang DR, Borsello T, Bonny C, Strichartz GR, Decosterd I, Ji RR. A peptide c-Jun N-terminal kinase (JNK) inhibitor blocks mechanical allodynia after spinal nerve ligation: respective roles of JNK activation in primary sensory neurons and spinal astrocytes for neuropathic pain development and maintenance. J Neurosci. 2006;26:3551–3560. doi: 10.1523/JNEUROSCI.5290-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zufferey R, Nagy D, Mandel RJ, Naldini L, Trono D. Multiply attenuated lentiviral vector achieves efficient gene delivery in vivo. Nat Biotechnol. 1997;15:871–875. doi: 10.1038/nbt0997-871. [DOI] [PubMed] [Google Scholar]