

Abstract
Spinal and bulbar muscular atrophy (SBMA) is a late-onset lower motor neuron disease caused by the expansion of a trinucleotide CAG repeat, which encodes a polyglutamine tract in androgen receptor (AR). Although it is commonly held that the pathogenic polyglutamine proteins accumulate in neurons and thereby induce transcriptional dysregulation, the downstream molecular events have remained elusive. Here, we examined whether TGF-β signaling is dysregulated in SBMA. Nuclear translocation of phosphorylated Smad2/3, a key step in TGF-β signaling, is suppressed in the spinal motor neurons of male transgenic mice carrying the mutant human AR. A similar finding was also observed in the motor neurons, but not in Purkinje cells, of SBMA patients. The pathogenic AR, the causative protein of SBMA, inhibits the transcription of TGF-β receptor type II (TβRII) via abnormal interactions with NF-Y and p300/CBP-associated factor. Furthermore, overexpression of TβRII dampens polyglutamine-induced cytotoxicity in a neuroblastoma cell line expressing the pathogenic AR. The present study thus indicates that disruption of TGF-β due to the transcriptional dysregulation of TβRII is associated with polyglutamine-induced motor neuron damage in SBMA.
Introduction
Polyglutamine disease is a group of hereditary neurodegenerative disorders resulting from the expansion of a genomic trinucleotide CAG repeat, which encodes a polyglutamine tract in causative proteins (Gatchel and Zoghbi, 2005). To date, nine polyglutamine diseases are known: Huntington's disease (HD), spinal and bulbar muscular atrophy (SBMA), dentatorubral-pallidoluysian atrophy, and six forms of spinocerebellar ataxia (SCA1, 2, 3, 6, 7, and 17). SBMA is a lower motor neuron disease caused by an abnormal expansion of a CAG repeat within the androgen receptor (AR) gene (La Spada et al., 1991). Although the causative genes are unrelated except for the expanded CAG repeats, polyglutamine diseases share a common molecular pathogenesis. The expanded polyglutamine tracts alter the conformations of the causative gene products, which then have a strong propensity to accumulate within neurons. It is now commonly held that the accumulation of causative proteins is a pivotal molecular event that triggers the pathogenesis of polyglutamine-mediated neurodegeneration.
Although the molecular mechanism of polyglutamine-induced neuron damage has not been completely elucidated, there is increasing evidence that the accumulation of pathogenic polyglutamine proteins leads to transcriptional dysregulation. This hypothesis has been forwarded by the observation that the polyglutamine aggregates sequester a wide range of transcription factors and coactivators, such as Sp1 and cAMP response element-binding protein-binding protein (CBP) (Nucifora et al., 2001; Dunah et al., 2002). The pathogenic polyglutamine proteins suppress a histone acetyltransferase (HAT) activity of these nuclear proteins, resulting in histone hypoacetylation and eventual downregulation of various gene expressions (Palhan et al., 2005; Sadri-Vakili et al., 2007). In support of this view, pharmacological augmentation of histone acetylation by histone deacetylase (HDAC) inhibitors mitigates the neurodegeneration in cellular and animal models of polyglutamine diseases (Butler and Bates, 2006). Even though it remains unclear which genes are responsible for the pathogenesis of polyglutamine-mediated neurodegeneration, a cDNA microarray study of cultured cells identified a set of genes whose expressions are specifically regulated by HDAC inhibitors (Peart et al., 2005). These genes are components of Myc, transforming growth factor-β (TGF-β), or cycline/cycline-dependent kinase pathways and are relevant to cell survival and proliferation.
Among these HDAC inhibitor-regulated pathways, TGF-β signaling was demonstrated to play a crucial role in the survival and function of adult neurons (Flanders et al., 1998). TGF-β knock-out mice show prominent neuronal cell death together with reduced synaptic density in various parts of the brain (Brionne et al., 2003). TGF-β signaling regulates axon guidance in Caenorhabditis elegans and is required for synaptic growth, neurotransmitter release, and neuron morphogenesis in Drosophila (Colavita et al., 1998; Zheng et al., 2006). These observations suggest that an impaired TGF-β signaling leads to neuron damage. In the present study, we examined whether TGF-β signaling was dysregulated in SBMA, a polyglutamine-induced motor neuron disease, and how this signaling was involved in the molecular underpinnings of polyglutamine-mediated neurodegeneration.
Materials and Methods
Generation, maintenance, and treatment of transgenic mice.
AR-24Q and AR-97Q mice were generated as described previously (Niwa et al., 1991; Katsuno et al., 2002). We used AR-97Q (line 7-8) male mice because they show progressive muscular atrophy and weakness as well as SBMA-like pathology (Katsuno et al., 2003; Waza et al., 2005). In the experiments where it was required, sodium butyrate (a HDAC inhibitor) was administered at a concentration of 4 g/L in distilled water from 5 weeks of age until the end of the analysis, as described previously (Minamiyama et al., 2004). Castration or sham operations were performed on male AR-97Q mice via the abdominal route under pentobarbital anesthesia (40 mg/kg, i.p.) at the age of 5 weeks as described previously (Katsuno et al., 2002). All animal experiments were performed in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and under the approval of the Nagoya University Animal Experiment Committee (Nagoya, Japan).
Immunohistochemistry and immunofluorescence histochemistry.
Mice were deeply anesthetized with pentobarbital and perfused with PBS followed by 4% paraformaldehyde fixative in phosphate buffer, pH 7.4. Whole brains, spinal cords, brainstems, and skeletal muscles were removed and embedded in paraffin. Autopsy specimens of lumbar spinal cord were obtained from genetically diagnosed SBMA patients (52- and 78-year-old males) and from neurologically normal patients (58- and 75-year-old males). The collection of human tissues and their use for this study were approved by the Ethics Committee of Nagoya University Graduate School of Medicine. Six-micrometer-thick sections were prepared from paraffin-embedded tissues, and immunohistochemistry and immunofluorescence were performed as described previously (Katsuno et al., 2006b; Tokui et al., 2009). Sections to be immunostained for TGF-β receptor type II (TβRII) or TGF-β were first microwaved for 20 min in 50 mm citrate buffer, pH 6.0. Sections to be immunostained for polyglutamine (1C2 antibody) were treated with formic acid for 5 min at room temperature. Cells to be immunostained were fixed with 4% paraformaldehyde fixative in phosphate buffer, pH 7.4, and treated with PBS containing 0.1% Triton X-100. All the specimens were treated with TNB blocking buffer (PerkinElmer) before incubation with primary antibodies, and immunoreactivity was detected using EnVision+ System-HRP (DakoCytomation). The immunohistochemical sections were photographed with an optical microscope (BX51, Olympus), and immunofluorescent specimens were photographed with an upright microscope (Axio Imager M1, Zeiss). Fluorescent intensities were measured as described previously (Adachi et al., 2001; Katsuno et al., 2006b). Briefly, fluorescent intensities were measured in >20 neurons within three nonconsecutive sections from each of three AR-97Q mice. For the purposes of counting, a motor neuron was defined by its presence within the anterior horn and the obvious nucleolus in a given 6-μm-thick section. The intensities and cell sizes were quantified with Image Gauge software version 4.22 (Fujifilm). The following primary antibodies were used: anti-pSmad2/3 (3101, Cell Signaling Technology, 1:5000), anti-polyglutamine (1C2) (MAB1574, Millipore, 1:20,000), anti-choline acetyltransferase (AB144, Millipore, 1:1000), anti-pan-specific TGF-β (AB-100-NA, R & D Systems, 1:1000), anti-TβRI (RB-10455, Thermo Scientific, 1:200), anti-TβRII (RB-10345, Thermo Scientific, 1:200), anti-NF-Y (sc-17753, Santa Cruz Biotechnology, 1:500), and anti-p300/CBP-associated factor (P/CAF) (ab12188, Abcam, 1:200). Alexa-conjugated secondary antibodies (Invitrogen) were used for immunofluorescence. Fluorescent Nissl staining was performed using NeuroTrace (Invitrogen, 1:200).
Immunoblotting, immunoprecipitation, and filter trap assay.
Mice were killed under pentobarbital anesthesia. Tissues (whole brains, spinal cord, brainstem, and skeletal muscles) were dissected free, snap frozen with powdered CO2 in acetone, and homogenized in CelLytic lysis buffer (Sigma-Aldrich) containing a phosphatase inhibitor mixture (Sigma-Aldrich) and a protease inhibitor mixture (Thermo Scientific). SH-SY5Y cells were lysed in CelLytic lysis buffer containing a protease inhibitor mixture 36 h after transfection. Nuclear and cytoplasmic fractions were extracted using NE-PER nuclear and cytoplasmic extraction reagents (Thermo Scientific). The homogenates were centrifuged at 2500 × g for 15 min at 4°C. The supernatant fractions were separated on 5–20% SDS-PAGE gels and then transferred to Hybond-P membranes (GE Healthcare) using 25 mm Tris, 192 mm glycine, 0.1% SDS, and 10% methanol as transfer buffer. Primary and secondary antibodies were diluted with Can Get Signal, a signal enhancer solution (NKB-101, Toyobo). The immunoblots were digitalized (LAS-3000 imaging system, Fujifilm), signal intensities of three independent blots were quantified with Image Gauge software version 4.22 (Fujifilm), and the means ± SEM were expressed in arbitrary units. The following primary antibodies were used: anti-pSmad2/3 (3101, Cell Signaling Technology, 1:200), anti-Smad2/3 (3102, Cell Signaling Technology, 1:300), anti-pSmad1/5/8 (9511, Cell Signaling Technology, 1:300), anti-histone H1 (05-457, Millipore, 1:300), anti-α-tubulin (T5168, Sigma-Aldrich, 1:5000), anti-pan-specific TGF-β (AB-100-NA, R & D Systems, 1:200), anti-TβRI (RB-10455, Thermo Scientific, 1:200), anti-TβRII (RB-10345, Thermo Scientific, 1:200), anti-V5 (R960-25, Invitrogen, 1:1000), anti-AR (N-20) (sc-816, Santa Cruz Biotechnology, 1:1000), anti-P/CAF (ab12188, Abcam, 1:500), anti-NF-Y (sc-17753, Santa Cruz Biotechnology 1:500), and anti-acetyl histone H3 (06-599, Millipore, 1:500). Primary antibody binding was probed using horseradish peroxidase-conjugated secondary antibodies (GE Healthcare) at a dilution of 1:5000, and bands were detected using the ECL Plus kit (GE Healthcare). Immunoprecipitation was performed as described previously (Adachi et al., 2003). Briefly, AR-containing immune complexes were precipitated using 300 μg of the total protein lysate in radioimmunoprecipitation assay buffer, 30 μl of protein G-Sepharose (GE Healthcare), and 5 μg of antibody anti-P/CAF (sc-13124, Santa Cruz Biotechnology), anti-NF-Y (sc-17753, Santa Cruz Biotechnology) or normal mouse IgG (Thermo Scientific). Proteins were eluted from beads and loaded on SDS-polyacrylamide gels. Blots were sequentially probed with an anti-AR antibody (sc-816, Santa Cruz Biotechnology, 1:1000) and then with anti-rabbit IgG (TrueBlot, eBioscience, 1:1000). Signal intensities of three independent blots were quantified with Image Gauge software version 4.22 (Fujifilm), and percentage input was calculated. Filter trap assay was performed as described previously (Adachi et al., 2003, 2007). Briefly, SH-SY5Y cells were lysed in 50 mm Tris pH 8.0, 150 mm NaCl, 1% Nonidet P-40, 0.5% deoxycholate, 1 mm 2-mercaptoethanol, and 2% SDS, and samples of protein (100 μg) were filtrated through an upper 0.2 μm cellulose acetate membrane (Sartorius) and a lower 0.45 μm nitrocellulose membrane (Bio-Rad) using a Bio-Dot apparatus (Bio-Rad). Blots were probed as described for immunoblots.
Quantitative real-time PCR.
TGF-β inducible early gene (TIEG) and TβRII1 mRNA levels were determined by real-time PCR as described previously (Katsuno et al., 2006b). Briefly, total RNA was extracted from mouse spinal cord using TRIzol reagent (Invitrogen) and from cells using the RNeasy Mini kit (Qiagen). The extracted RNA was then reverse transcribed into first-strand cDNA using SuperScript III reverse transcriptase (Invitrogen). Real-time PCR was performed in a total volume of 50 μl containing 25 μl of 2× QuantiTect SYBR Green PCR Master Mix and 0.4 μm each primer (Qiagen), and the amplified products were detected by the iCycler system (Bio-Rad). The reaction conditions were 95°C for 15 min and then 45 cycles of 15 s at 95°C followed by 60 s at 55°C. For an internal standard control, the expression level of glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was quantified simultaneously. The following primers were used: 5′-aagctgcttccagtcaggat-3′ and 5′-ttgaactcaggtcgtcttgc-3′ for mouse TβRII, 5′-tcaagaaggtggtgaagcag-3′ and 5′-gttgaagtcgcaggagacaa-3′ for mouse GAPDH, 5′-gaagaacccacggaaatgtt-3′ and 5′-tctcatcaccatcggacact-3′ for mouse TIEG, 5′-taccttcatgggttgcagaa-3′ and 5′-aaggctgggagcagagaata-3′ for human TβRII, and 5′-ctcctcctgttcgacagtca-3′ and 5′-caatacgaccaaatccgttg-3′ for human GAPDH. The threshold cycle of each gene was determined as the number of PCR cycles at which the increase in reporter fluorescence was 10 times the baseline signal. The weight of the gene contained in each sample was equal to the log of the starting quantity, and the standardized expression level in each mouse was equal to the weight ratio of each gene to that of GAPDH. PCRs were repeated four times for each of the indicated numbers of samples.
Plasmid, cell culture, and transfection.
Human truncated AR cDNAs containing 24 or 97 CAGs (1-645 and 1-864 bp, respectively) were isolated from pCR3.1 full-length AR-24Q or AR-97Q vectors (Waza et al., 2005), and subcloned into pcDNA 6.2/V5-GW/D-TOPO (Invitrogen). The fragments were then subcloned into the pcDNA6.2/c-EmGFP vector (Invitrogen). Human TβRII cDNA was also subcloned into pcDNA6/V5-His (Invitrogen). For the luciferase assay, the pTβRII-219/+36 vector encoding firefly luciferase, kindly provided by Dr. Seok Hee Park (Department of Biological Science, Sungkyunkwan University, Korea), and the pRL-TK vector encoding Renilla luciferase (Promega) were transfected into SH-SY5Y cells.
Human neuroblastoma cells (SH-SY5Y, American Type Culture Collection No. CRL-2266) were plated in DMEM/F12 containing 10% fetal bovine serum with penicillin and streptomycin. Each dish was transfected with the indicated vectors using Opti-MEM (Invitrogen) and Lipofectamine 2000 (Invitrogen) and then differentiated in DMEM/F12 supplemented with 2% fetal calf serum, 10 μm retinoic acid, and 2 ng/ml recombinant human TGF-β2 (302-B2, R & D Systems) for 36 h. Sodium butyrate (NaB) dissolved in PBS was added to the differentiation medium at the indicated concentrations. The anti-pan-specific TGF-β antibody (R & D Systems) was applied at a concentration of 5 μg/ml to neutralize TGF-β signaling. The oligonucleotide small interfering RNA (siRNA) duplex against TβRII (Stealth RNAi duplex, HSS 110701) and that against P/CAF (Stealth RNAi duplex, HSS 113059) were synthesized by Invitrogen. SH-SY5Y cells were transfected with either of these siRNAs or with control oligonucleotide (Stealth RNAi negative control duplex, Invitrogen) by using Lipofectamine RNAiMAX (Invitrogen) according to the manufacturer's instructions. Each siRNA was transfected at a concentration of 30 nm.
Luciferase activity and cell viability assay.
For the luciferase assay, cell extracts were prepared 36 h after transfection by a detergent lysis method (Promega). Luciferase activity was measured with a luminometer (Lumi-Counter 2500, Microtec) and the Dual-Luciferase reporter assay system (Promega). The ratio of firefly luciferase activity to Renilla luciferase activity in each sample served as a measure of the relative luciferase activity. Each construct was transfected at least three times, and data for each construct are presented as means ± SEM.
Cell death analysis was performed as described previously (Katsuno et al., 2006b). Briefly, 36 h after transfection, cells were stained with propidium iodide (Invitrogen) and mounted with VECTASHIELD mounting medium (Vector Laboratories). Quantitative analyses were made from triplicate determinations. Duplicate slides were graded blindly in two independent trials as described previously (Katsuno et al., 2005). The cell viability assay was performed using WST-1 (Roche Diagnostics) according to the manufacturer's instructions. Briefly, cells were cultured in 24-well plates. After treatments, cells were incubated with WST-1 substrate for 2–3 h and spectrophotometrically assayed at 440 nm using a plate reader (Powerscan HT, Dainippon Pharmaceutical).
Statistical analyses.
Data were analyzed using the Kaplan–Meier and log-rank tests for survival rate, ANOVA with a post hoc test (Dunnett or Turkey–Kramer) for multiple comparisons, two-way ANOVA with repeated measures for mouse behavior analysis, Pearson's coefficient for correlation, and an unpaired t test from StatView software, version 5 (Hulinks).
Results
Pathogenic AR inhibits nuclear translocation of phosphorylated Smad2/3
The biological properties of TGF-β are mediated by a high-affinity transmembrane receptor serine/threonine kinase complex consisting of TβRI and TβRII. Ligand interaction with a homodimer of TβRII recruits and activates TβRI, which in turn phosphorylates the receptor-regulated Smad proteins (Smad2 and 3). Phosphorylated Smad2/3 (pSmad2/3) then translocate into the nucleus, where the molecules regulate target gene transcription (Shi and Massague 2003). To examine whether TGF-β signaling is altered in SBMA, we first investigated the nuclear translocation of Smad2/3 in a transgenic mouse model of SBMA (Katsuno et al., 2002). In immunohistochemistry, pSmad2/3 was intensely stained in the nuclei of almost all neurons and glial cells within the spinal anterior horn of wild-type mice and AR-24Q transgenic mice bearing normal human AR containing 24 CAGs (Fig. 1A,B). By contrast, immunoreactivity of pSmad2/3 was notably diminished in nuclei of AR-97Q transgenic mice carrying the disease-related mutant AR with 97 CAGs (Fig. 1A). Although the onset of motor impairment in AR-97Q mice is ∼8–10 weeks, decreased pSmad2/3 was evident at 7 weeks, a preonset stage in these mice (Fig. 1A,B). Immunoblotting confirmed a significant reduction in pSmad2/3 protein from the nuclear fractions of the AR-97Q mouse spinal cords (Fig. 1C). Signal intensities of the pSmad2/3-immunoreactive bands normalized to histone H1 were significantly decreased in the spinal cord of AR-97Q mice (AR-24Q, 0.89 ± 0.12; AR-97Q, 0.28 ± 0.05; p < 0.0005; 7 weeks old; n = 3). This profound decrease in pSmad2/3 signal in immunoblot may result from both neuronal and glial accumulation of pathogenic AR due to a wide expression of the transgene driven by a potent promoter in the mouse model (Katsuno et al., 2002). Despite a significant decrease in nuclear pSmad2/3, AR-97Q mice showed no alterations in phosphorylated Smad1/5/8, the downstream signal molecules regulated by bone morphogenetic protein (BMP) (Fig. 1C). To test whether TGF-β signaling is disrupted in the mouse model of SBMA, we measured the levels of TIEG mRNA, the expression of which is controlled by TGF-β. The mRNA levels of TIEG normalized to GAPDH were significantly decreased in the spinal cord of AR-97Q mice (AR-24Q, 3.66 ± 0.59 × 10−3; AR-97Q, 2.22 ± 0.77 × 10−3; p < 0.01; 7 weeks old; n = 4), suggesting that TGF-β signaling is inhibited in the transgenic mouse model of SBMA.
Figure 1.
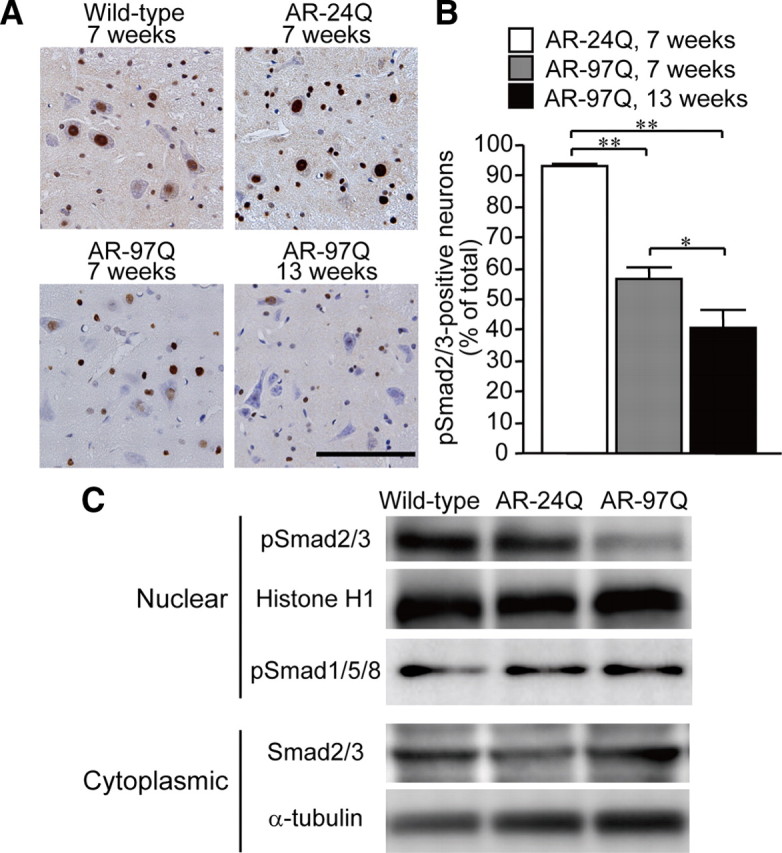
Nuclear translocation of Smad2/3 in SBMA mice. A, Anti-phosphorylated Smad2/3 (pSmad2/3) immunohistochemistry of the spinal cords from wild-type, AR-24Q, and AR-97Q mice. Scale bar, 100 μm. B, The frequency of pSmad2/3 staining in spinal motor neurons of AR-24Q and AR-97Q mice. Error bars indicate SEM; *p < 0.05, **p < 0.01. C, Immunoblots of the nuclear and cytoplasmic fractions from the spinal cords of wild-type, AR-24Q, and AR-97Q mice (7 weeks old).
To confirm that the decreased nuclear translocation of pSmad2/3 is specific to the affected brain regions in SBMA, we compared immunohistochemistry in different areas of the mouse brain. These results showed that nuclear translocation of pSmad2/3 was inhibited in the brainstem, where anti-polyglutamine immunoreactivity is strongly detected, but not in the visual cortex, which is not affected in AR-97Q mice (Fig. 2A). As pathogenic AR also accumulates in the nuclei of non-neural tissues in AR-97Q mice (Katsuno et al., 2002), we also examined TGF-β signaling in skeletal muscle. Phosphorylated Smad2/3 was decreased in the nuclei of skeletal muscle cells from AR-97Q mice, suggesting a noncell type-specific disruption of TGF-β signaling (supplemental Fig. 1A,B). On the contrary, nuclear staining of pSmad2/3 was not diminished in motor neurons within the spinal anterior horn of the mutant superoxide dismutase 1 (SOD1) G93A transgenic mouse, an animal model of amyotrophic lateral sclerosis (ALS), at an early symptomatic stage (supplemental Fig. 2, available at www.jneurosci.org as supplemental material). We further examined whether TGF-β signaling is impaired in patients with SBMA. In autopsied spinal cords from SBMA patients, nuclear immunoreactivity of pSmad2/3 was notably diminished in motor neurons (Fig. 2B). By contrast, nuclear staining of pSmad2/3 was not diminished in the patients' glial cells, in which pathogenic AR accumulation has rarely been observed (Li et al., 1998; Adachi et al., 2005). Furthermore, nuclear translocation of pSmad2/3 was not inhibited in the cerebellar Purkinje cells, which are not affected in SBMA, suggesting that the impairment of TGF-β signaling is specific to pathological lesions of this disease (supplemental Fig. 3, available at www.jneurosci.org as supplemental material).
Figure 2.
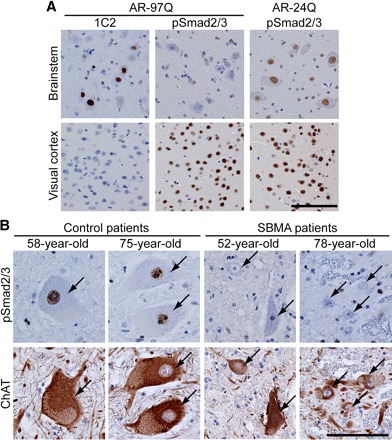
Specificity of Smad2/3 hypophosphorylation in SBMA mice and patients. A, Anti-expanded polyglutamine (1C2) and anti-pSmad2/3 immunohistochemistry of the brainstem and cerebral cortex from AR-24Q, and AR-97Q mice (13 weeks old). Phosphorylation of Smad2/3 is decreased in motor neurons within the motor trigeminal nucleus in the pons, but not in neurons within the visual cortex. B, Anti-pSmad2/3 and anti-choline acetyltransferase (ChAT) immunohistochemistry of serial sections of the spinal cord from control subjects and SBMA patients. Arrows indicate identical neurons in each patient. ChAT-positive motor neurons of the SBMA patient are atrophied and show a decreased immunoreactivity for pSmad2/3 in the nucleus. Scale bars, 100 μm.
The accumulation of pathogenic AR in the nuclei of motor neurons is the pivotal step leading to neurodegeneration in SBMA. To test the relationship between the nuclear accumulation of pathogenic AR and the reduced activation (i.e., phosphorylation and nuclear translocation) of Smad2/3, we performed immunohistochemistry on the spinal cords of AR-97Q mice using antibodies against expanded polyglutamine (1C2) and pSmad2/3. In the spinal anterior horn, there was significantly less pSmad2/3 immunoreactivity in the motor neurons showing nuclear accumulation of pathogenic AR than in those without 1C2 nuclear staining (Fig. 3A). Quantitative analysis showed that the intensity of pSmad2/3 immunoreactivity in 1C2-positive motor neurons from AR-97Q mice was reduced to 36.1 ± 40.1% of that from 1C2-negative neurons (p < 0.05; 8–9 weeks old; n = 3 for each group). The pSmad2/3 immunoreactivity was positively correlated with the size of spinal motor neurons (r = 0.689, p < 0.0001), suggesting a link between TGF-β signal disruption and neuronal dysfunction (Fig. 3B). As observed in the mice, there was less pSmad2/3 immunoreactivity in 1C2-positive motor neurons than in those without nuclear accumulation of pathogenic AR in the autopsied spinal cord of an SBMA patients (Fig. 3C). These observations suggest that TGF-β signaling is interrupted in the neurons bearing nuclear accumulation of pathogenic AR.
Figure 3.
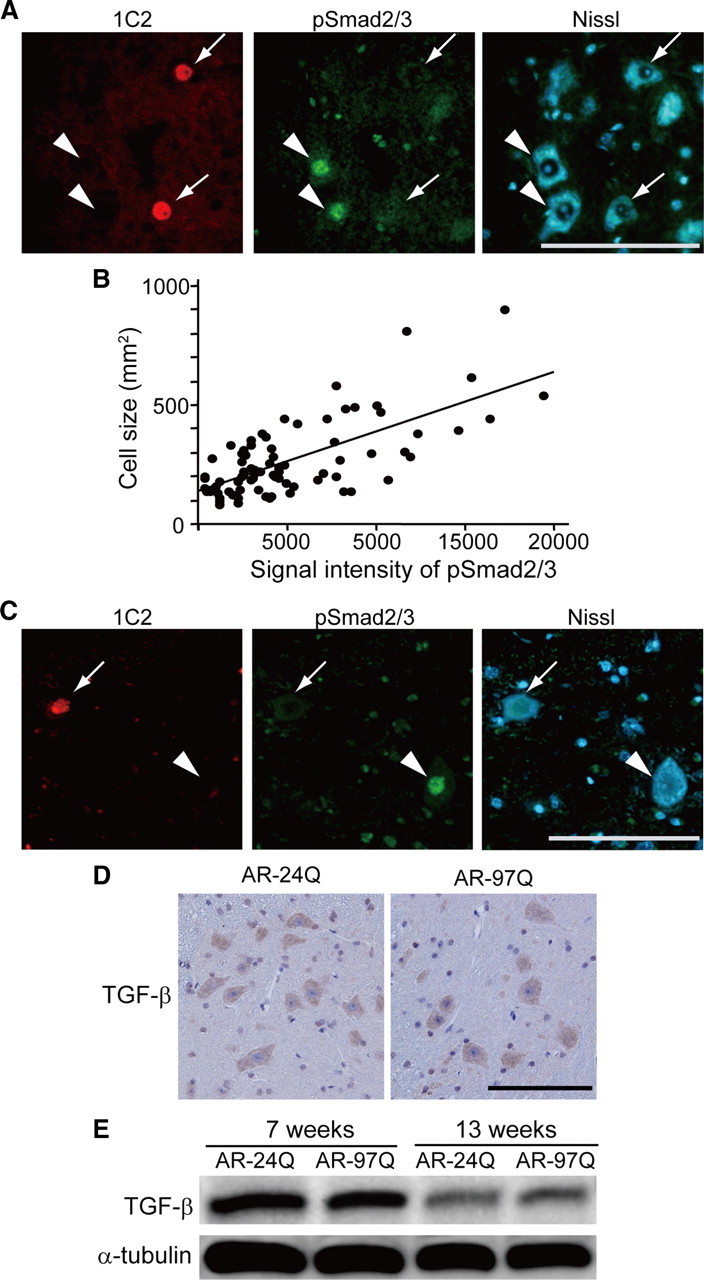
Phosphorylated Smad2/3 and TGF-β expression in SBMA mice. A, Anti-expanded polyglutamine AR (1C2, red) and anti-phosphorylated Smad2/3 (green) double immunofluorescence histochemistry in the spinal cord from an AR-97Q mouse (8 weeks old). Immunoreactivity for pSmad2/3 is detected in 1C2-negative neurons (arrowheads), but not in 1C2-positive cells (arrows). B, Correlation between the intensity of pSmad2/3 immunoreactivity and the size of motor neurons in the spinal cord of AR-97Q mice (8–9 weeks old, n = 4). C, 1C2 and anti-phosphorylated Smad2/3 double immunofluorescence histochemistry in the spinal cord of an SBMA patient. D, Immunohistochemistry in the spinal cords from AR-24Q and AR-97Q mice using a pan-specific antibody against TGF-β isoforms (13 weeks old). E, Immunoblots of the spinal cords of AR-24Q and AR-97Q mice. Scale bars, 100 μm.
To investigate the cause of TGF-β signal disruption, we first examined the levels of the ligand using an antibody against pan TGF-β isoforms. TGF-β immunoreactivity was not decreased in AR-97Q mouse spinal cord (Fig. 3D). This finding was consistent with immunoblotting that demonstrated no reduction in the amount of TGF-β in AR-97Q mice at both preonset and symptomatic stages (Fig. 3E).
Expression of TβRII is decreased in SBMA
The diminished nuclear translocation of pSmad2/3 along with an intact level of TGF-β suggests the involvement of TGF-β receptors. It is known that the TGF-β receptors TβRI and TβRII are widely expressed in the CNS, both during development and in adulthood (Vivien and Ali, 2006). Immunoreactivities for both TβRI and TβRII were detected in the spinal motor neurons of wild-type mice (Fig. 4A); however, spinal motor neuron staining for TβRII, but not for TβRI, was decreased in AR-97Q mice even before the onset of neuromuscular symptoms (Fig. 4A). Immunoblotting confirmed a significant reduction in TβRII protein in the spinal cords of AR-97Q mice (Fig. 4B). Signal intensities of the TβRII-immunoreactive bands normalized to α-tubulin were significantly decreased in the spinal cords of AR-97Q mice (AR-24Q, 0.27 ± 0.03; AR-97Q, 0.16 ± 0.04; p < 0.05; 7 weeks old; n = 3). The mRNA levels of TβRII normalized to GAPDH were significantly decreased in the spinal cords of AR-97Q mice (AR-24Q, 1.38 ± 0.16 × 10−2; AR-97Q, 0.93 ± 0.33 × 10−2; p < 0.001; 7 weeks old; n = 4), suggesting that the expression of this receptor is suppressed.
Figure 4.
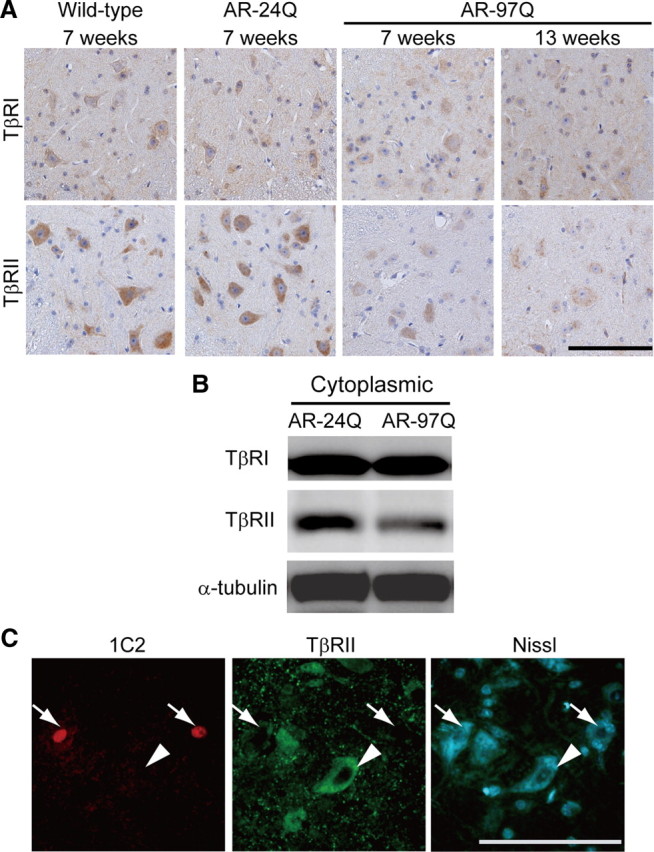
Expression of TGF-β receptors in SBMA mice. A, Immunohistochemistry in the spinal cords from wild-type, AR-24Q, and AR-97Q mice using antibodies against the TGF-β receptors TβRI and TβRII. B, Immunoblots of the cytoplasmic fractions of the spinal cords from AR-24Q and AR-97Q mice (7 weeks old). C, 1C2 and anti-TβRII double immunofluorescence histochemistry in the spinal cord from an AR-97Q mouse (8 weeks old). Immunoreactivity for TβRII is detected in 1C2-negative neurons (arrowheads), but not in 1C2-positive cells (arrows). Scale bars, 100 μm.
To confirm that the decreased TβRII expression is specific to affected neurons, we performed double immunofluorescence histochemistry using 1C2 together with an antibody against TβRII (Fig. 4C). Quantitative analysis showed that the intensity of TβRII immunoreactivity in 1C2-positive motor neurons was reduced to 25.8 ± 37.1% of that from 1C2-negative neurons (p < 0.0001; 8–9 weeks old; n = 3 for each group). Reduced TβRII immunoreactivity was also detected in an autopsied spinal cord from an SBMA patient (Fig. 5A). Furthermore, TβRII immunofluorescence in this SBMA patient was more reduced in 1C2-positive motor neurons than in those without nuclear accumulation of the pathogenic AR (Fig. 5B).
Figure 5.
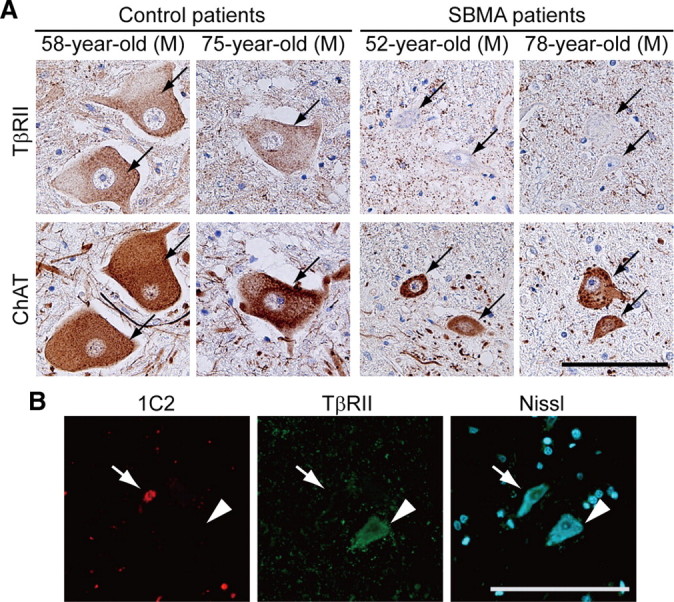
TβRII expression in SBMA patients. A, Anti-TβRII immunohistochemistry in the spinal cords of an SBMA patients and control subjects. Arrows indicate identical neurons in each patient. B, Immunofluorescence of the spinal cord from an SBMA patient using 1C2 in combination with an antibody against TβRII. Immunoreactivity for TβRII is detected in a 1C2-negative neuron (arrowhead) but decreased in a 1C2-positive neuron (arrow). Scale bars, 100 μm.
SBMA only affects males, whereas females carrying the AR mutation scarcely show symptoms (Katsuno et al., 2006a). Previous studies showed that the pathogenic AR containing an expanded polyglutamine tract accumulates in the nuclei of motor neurons in a testosterone-dependent manner, and that inhibition of testosterone production prevents this accumulation (Katsuno et al., 2002; Takeyama et al., 2002). Furthermore, testosterone reduction, by castration or medication, mitigates neurodegeneration in SBMA mice and patients (Katsuno et al., 2003; Chevalier-Larsen et al., 2004; Banno et al., 2009). We thus examined whether castration restores the TGF-β-Smad2/3 signal pathway in male AR-97Q mice. Both immunohistochemistry and immunoblotting demonstrated that castration increased the expression of TβRII and restored the nuclear translocation of pSmad2/3 (supplemental Fig. 4A,B). Together, these findings suggest that the accumulation of the pathogenic AR proteins interferes with TGF-β signaling through downregulation of TβRII expression in SBMA mice and patients.
Pathogenic AR dysregulates transcription of TβRII
To clarify the molecular mechanism by which pathogenic AR suppresses the expression of TβRII, we studied its transcriptional regulation in a cultured cell model of SBMA, SH-SY5Y cells harboring an N-terminal fragment of human AR containing an expanded polyglutamine tract and a part of AF-1 domain [truncated AR-97Q (tAR97Q)]. Phosphorylated Smad2/3 was significantly lower in the nuclear fraction of these cells than in cells bearing tAR24Q (Fig. 6A,B; supplemental Fig. 5, available at www.jneurosci.org as supplemental material), and TβRII protein was reduced in the cytoplasmic fraction of cells carrying tAR97Q (Fig. 6A,C). Quantitative RT-PCR analysis showed that TβRII mRNA normalized to GAPDH was also significantly diminished in tAR97Q-transfected cells compared with those transfected with tAR24Q (tAR24Q, 1.80 ± 0.17 × 10−4; tAR97Q, 1.11 ± 0.02 × 10−4; p < 0.0005; n = 4 per group). To test whether pathogenic AR suppresses the expression of TβRII at the transcriptional level, we investigated TβRII promoter activity using a luciferase reporter assay. The activity of luciferase, controlled by the TβRII promoter, was significantly decreased in a dose-dependent manner in tAR97Q-transfected cells compared with those transfected with tAR24Q (Fig. 6D), suggesting that the pathogenic AR protein bearing an expanded polyglutamine tract downregulates the transcription of TβRII.
Figure 6.
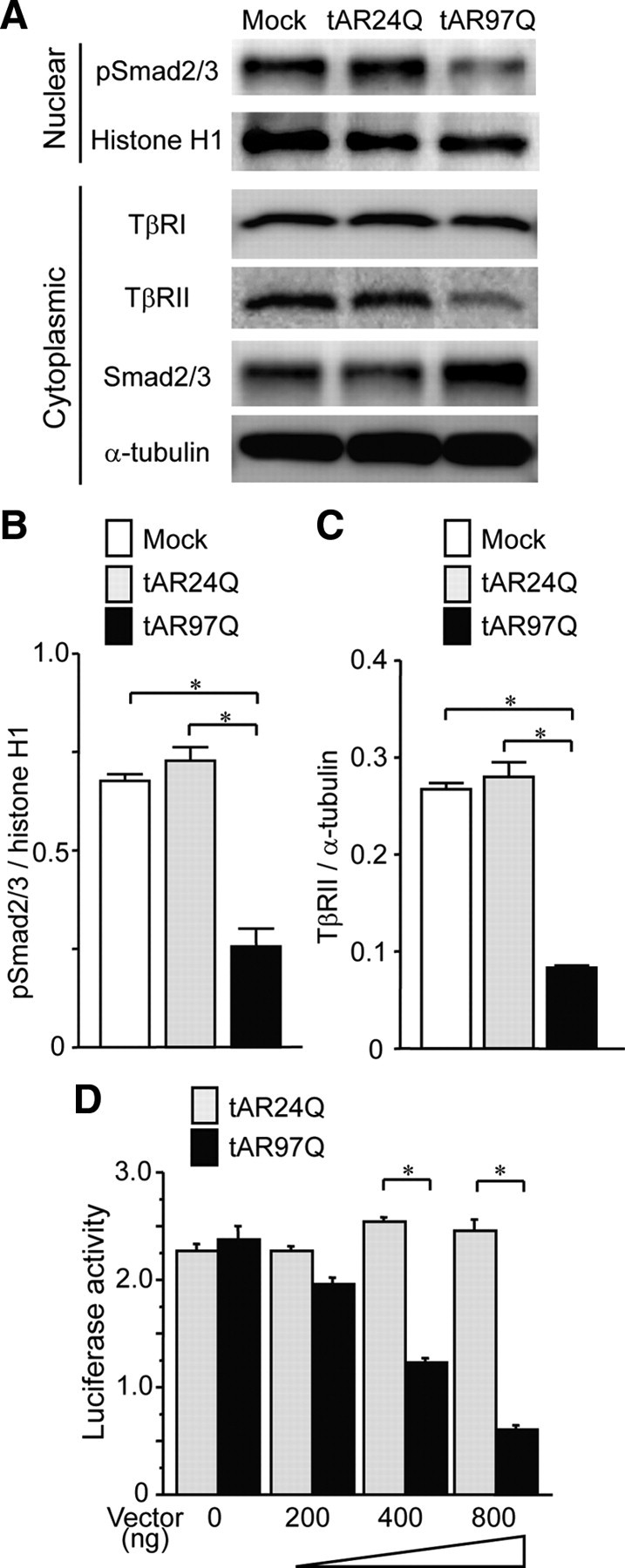
TGF-β-Smad2/3 pathway alteration in cellular model of SBMA. A, Immunoblots of nuclear and cytoplasmic fractions from SH-SY5Y cells transfected with mock, tAR24Q, or tAR97Q vector. B, Signal intensities of the pSmad2/3-immunoreactive bands from the nuclear fractions of SH-SY5Y cells (n = 3 per group). C, Signal intensities of the TβRII-immunoreactive bands from the cytoplasmic fractions of SH-SY5Y cells (n = 3 per group). D, The activity of luciferase, controlled by the TβRII promoter, in SH-SY5Y cells transfected with the indicated amounts of tAR24Q or tAR97Q (n = 3 per group). Error bars indicate SEM; *p < 0.01.
Recent reports indicated that the transcription of TβRII is regulated by the transcription factor NF-Y, which binds to the inverted CCAAT box within the promoter region of TβRII, and by P/CAF, a transcriptional coactivator with HAT activity (Park et al., 2002). Therefore, we next examined whether pathogenic AR interferes with NF-Y and P/CAF in cultured cells. Immunocytochemical analysis demonstrated that green fluorescent protein (GFP)-tagged pathogenic AR was confined to inclusion bodies, to which NF-Y or P/CAF were colocalized (Fig. 7A,B). The AR proteins, tAR24Q and tAR97Q, were coimmunoprecipitated with P/CAF or NF-Y (Fig. 7C). Quantitative analyses showed that the pathogenic AR preferably binds to P/CAF, although this was not the case for NF-Y (Fig. 7D).
Figure 7.
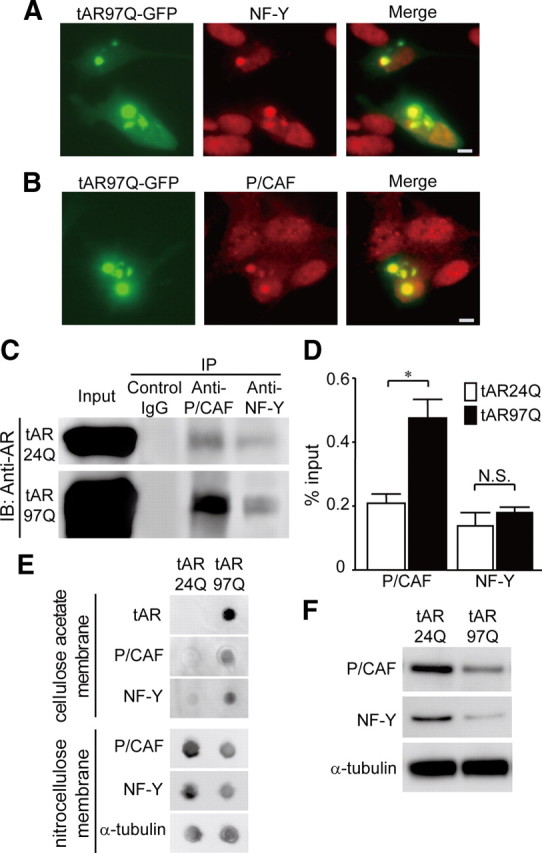
Pathogenic AR interferes with P/CAF and NF-Y. A, Anti-NF-Y immunocytochemistry of SH-SY5Y cells expressing tAR97Q tagged with GFP. B, Anti-P/CAF immunocytochemistry of SH-SY5Y cells expressing tAR97Q tagged with GFP. Scale bars, 10 μm. C, Immunoprecipitation (IP)/immunoblot (IB) of soluble fraction from SH-SY5Y cells transfected with tAR24Q or with tAR97Q. Coimmunoprecipitation of tAR and the nuclear proteins P/CAF and NF-Y was detected. D, Quantification of the amount of tAR proteins that are coimmunoprecipitated with P/CAF or NF-Y. Data are shown as a ratio to input. Error bars indicate SEM *p < 0.01. E, Filter trap assay of the protein lysate from SH-SY5Y cells transfected with tAR24Q or tAR97Q. F, Immunoblots of soluble fraction from SH-SY5Y cells transfected with tAR24Q or tAR97Q.
To confirm the interaction between the pathogenic AR aggregates and the nuclear proteins P/CAF and NF-Y, we performed filter trap assay. In this analysis, the larger-sized protein complex was retained on the cellulose acetate membrane (pore diameter, 0.2 μm), whereas the nitrocellulose membrane captured proteins of all sizes (Adachi et al., 2003). The results showed that both P/CAF and NF-Y were retained on a cellulose acetate membrane together with tAR97Q, but not with tAR24Q, suggesting that these nuclear proteins were confined in a large molecular weight complex of tAR97Q (Fig. 7E). Additionally, the amounts of NF-Y and P/CAF in soluble fraction were decreased (Fig. 7F).
We showed previously that oral administration of NaB, a potent HDAC inhibitor, facilitates histone acetylation and thereby improves motor functions of AR-97Q mice (Minamiyama et al., 2004). Because the induction of histone acetylation was shown to upregulate the TβRII promoter activity (Park et al., 2002), we further tested whether NaB restores TGF-β signaling. In the SH-SY5Y cells expressing tAR97Q, NaB increased the amounts of TβRII and pSmad2/3 (supplemental Fig. 6A, available at www.jneurosci.org as supplemental material). These effects were suppressed by knock-down of P/CAF, suggesting that NaB-mediated facilitation of TGF-β signaling at least partially depends on P/CAF activity (supplemental Fig. 6A, available at www.jneurosci.org as supplemental material). Moreover, the expressions of TβRII and nuclear pSmad2/3 were higher in AR-97Q mice treated with NaB than in untreated mice (supplemental Fig. 6B,C).
TGF-β signaling protects neurons against pathogenic AR toxicity
We next investigated whether the disruption of TGF-β signaling has a causative impact on polyglutamine-mediated neurotoxicity. To this end, we cotransfected SH-SY5Y cells with tAR24Q or tAR97Q and with or without a plasmid vector of TβRII. The cotransfection of TβRII increased the amount of pSmad2/3 in the SH-SY5Y cells expressing tAR97Q (Fig. 8A). Propidium iodide staining demonstrated that overexpression of TβRII inhibited the cell death induced by tAR97Q (Fig. 8B). Inclusion body formation, however, was not suppressed by the coexpression of TβRII (Fig. 8C), suggesting that TGF-β signaling attenuates the pathogenic AR-mediated cell damage without inhibiting the accumulation of abnormal polyglutamine protein.
Figure 8.
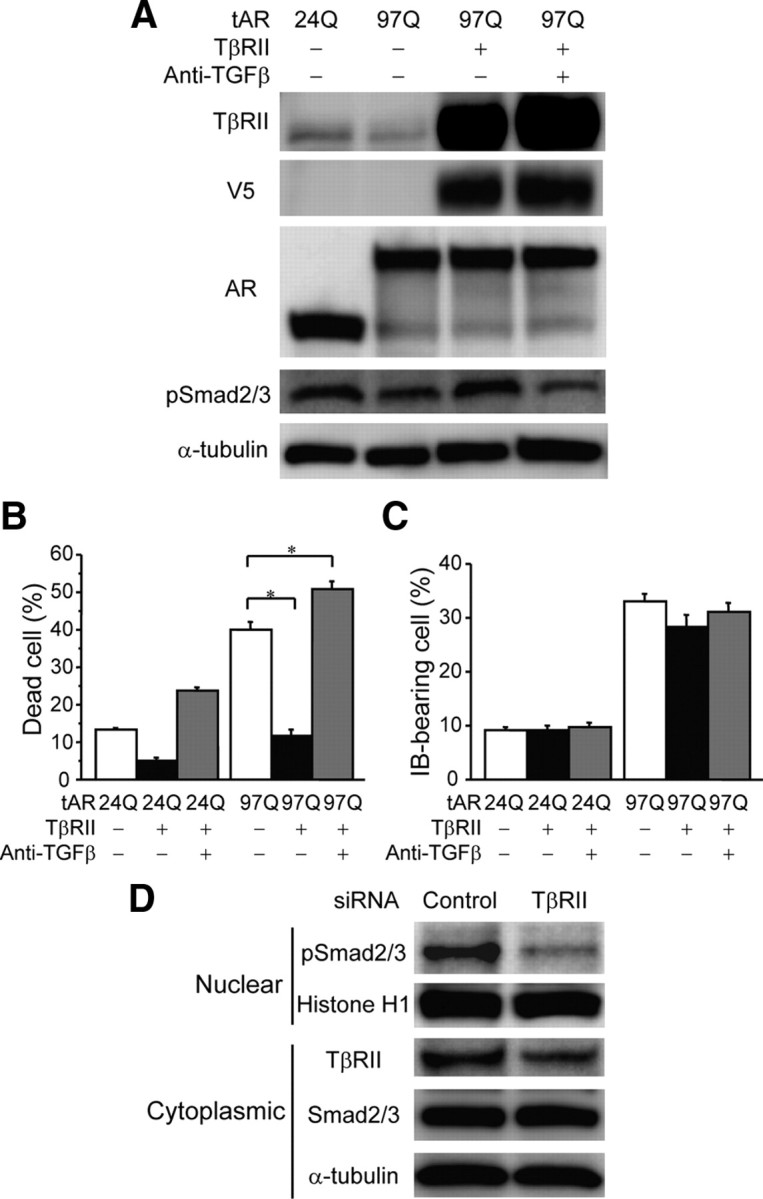
Modulation of TGF-β signaling in SH-SY5Y cells. A, Immunoblots of total homogenates from SH-SY5Y cells transfected with tAR24Q or tAR97Q and with or without the coexpression of V5-tagged TβRII. Anti-TGF-β treatment was used to neutralize TGF-β signaling. B, Frequency of cell death detected by propidium iodide staining in SH-SY5Y cells transfected with tAR24Q or tAR97Q (n = 6 per group) with or without the coexpression of TβRII (n = 6 per group). C, Percentage of SH-SY5Y cells carrying inclusion bodies (IB) containing overexpressed AR protein (n = 6 per group). Error bars indicate SEM; *p < 0.01. D, Immunoblots of the nuclear and cytoplasmic fractions from SH-SY5Y cells transfected with siRNA against TβRII or control oligonucleotide.
To confirm these findings, we evaluated the effects of TβRII knock-down in SH-SY5Y cells. Suppression of TβRII expression using siRNA decreased nuclear pSmad2/3 and cytoplasmic TβRII (Fig. 8D) and significantly increased the number of dead cells detected by propidium iodide staining (control siRNA, 1.19 ± 0.62%; TβRII siRNA, 3.08 ± 0.54%; p < 0.0005; n = 6 per group). In addition, TβRII knock-down decreased cell viability to 77.4 ± 7.7% of control (p < 0.05; n = 6 per group). Given that both pharmacological and genetic suppression of TGF-β signaling was shown to induce degeneration of cultured neuroblastoma cells (Tesseur et al., 2006), our findings suggest that the mutant AR-mediated blockade of TGF-β signaling has cytotoxic effects on neuronal cells and thus is associated with polyglutamine-mediated cellular damage.
Discussion
TGF-β signal perturbation in polyglutamine-mediated neurodegeneration
The present study demonstrated that the accumulation of pathogenic AR proteins containing an expanded polyglutamine tract reduced the expression of TβRII and hampered the nuclear translocation of pSmad2/3 proteins. We also demonstrated that the deficiency in TGF-β signaling had causative effects on neuronal cell damage that was mitigated by the overexpression of TβRII. In support of our findings, the blockade of TGF-β signaling via neuron-specific expression of dominant-negative TβRII was shown to induce an age-dependent neurodegeneration and exacerbate neuropathology in a mouse model of Alzheimer's disease (AD) (Tesseur et al., 2006).
The TGF-β superfamily contains two subfamilies: the TGF-β/Activin/Nodal subfamily and the BMP/growth and differentiation factor/Muellerian inhibiting substance subfamily. TGF-β, a pleiotropic cytokine, regulates a diverse set of cellular responses, including proliferation, differentiation, migration, and apoptosis. TGF-β isoforms TGF-β1, -β2, and -β3 are expressed by both neurons and glial cells, and their receptors are expressed throughout the CNS, including adult human neurons (Flanders et al., 1998). A number of studies have shown that the TGF-β-Smad2/3 pathway has potent neuroprotective effects. The observation that viral vector-mediated overexpression of TGF-β attenuates ischemic brain damage lends support to this view (Zhu et al., 2002). It was postulated that TGF-β signaling plays a fundamental role in neural activity through the regulation of synaptic function (Heupel et al., 2008). TGF-β was also shown to protect neurons from glutamate-mediated excitotoxicity, a putative molecular mechanism underlying the pathogenesis of a variety of neurodegenerative disorders, including polyglutamine diseases (Vivien and Ali, 2006; Fernandes et al., 2007). Furthermore, several studies demonstrated that TGF-β protects neurons from amyloid β-protein, the causative protein of AD (Flanders et al., 1998; Vivien and Ali, 2006). By contrast, genetic reduction of the TGF-β2 isoform was shown to induce a loss of dopaminergic neurons in mice, suggesting a link between TGF-β signaling and Parkinson's disease (Andrews et al., 2006). In the present study, we showed that the expression of TβRII and nuclear translocation of pSmad2/3 are suppressed in the affected neurons of patients with SBMA. A similar decrease in TβRII expression was also reported in AD patients, suggesting that this molecule plays an important role in various pathogeneses of neurodegeneration (Tesseur et al., 2006). Together, these findings indicate that the decreased expression of TβRII and the resulting perturbation of TGF-β signaling appear to underlie polyglutamine-dependent neurodegeneration in SBMA.
Transcriptional dysregulation in polyglutamine diseases
Dysregulation of cellular transcriptional machinery is considered the major molecular mechanism whereby pathogenic polyglutamine proteins induce neuron damage (Riley and Orr, 2006). However, it has yet to be elucidated which molecules are critically implicated in the pathophysiology of polyglutamine-dependent neurodegeneration. In the present study, we demonstrated that the transcription of TβRII, the mediator of TGF-β signaling, is dysregulated by the pathogenic AR protein. This finding was demonstrated in both cellular and animal models of SBMA. Furthermore, a similar finding was also revealed in an SBMA patient, suggesting that transcriptional perturbation of TβRII is a fundamental molecular event in SBMA.
Previous reports demonstrated that transcriptional alteration is an early change in the pathogenesis in mouse models of other polyglutamine diseases as well (Lin et al., 2000; Luthi-Carter et al., 2000). For example, the expression levels of various heat shock proteins were substantially decreased in animal models of SBMA, SCA3, and HD (Hay et al., 2004; Katsuno et al., 2005; Chou et al., 2008). Recent studies also demonstrated that pathogenic polyglutamine proteins repress the transcription of subunits of PGC-1 (peroxisome proliferator-activated receptor gamma coactivator-1), a transcriptional coactivator that regulates the expression of various nuclear-encoded mitochondrial proteins (Cui et al., 2006; Ranganathan et al., 2009). Moreover, we have demonstrated that pathogenic AR impairs retrograde axonal transport via transcriptional dysregulation of dynactin 1 in the mouse model of SBMA (Katsuno et al., 2006b). Given that activated Smads are retrogradely transported in motor axons, pathogenic AR-mediated axonal transport impairment may enhance TGF-β signal disruption in SBMA (Sanyal et al., 2004). Collectively, the pathogenic polyglutamine proteins perturb the transcription of a wide range of genes that play substantial roles in the maintenance of neuronal function, leading to neuronal dysfunction and cell death. Of note is that some of these transcriptional alterations are shared by different diseases, suggesting a common target of therapy development against polyglutamine-mediated neurodegenerative disorders.
Inhibition of histone deacetylase restores TGF-β signaling
The present study showed that the pathogenic AR aggregates sequestrate NF-Y and P/CAF, both of which regulate the transcription of TβRII (Park et al., 2002). This finding should be interpreted cautiously, since our cellular experiments were not performed using the full-length AR or on the gene's endogenous promoter. However, it has been particularly emphasized that a pivotal molecular event in polyglutamine diseases is an abnormal interaction between the respective pathogenic protein and nuclear proteins that regulate transcription. A recent study demonstrated that the components of NF-Y are sequestrated by the aggregates of pathogenic huntingtin in the brains of HD model mice (Yamanaka et al., 2008). P/CAF also interacts with an N-terminal fragment of huntingtin that contains an expanded polyglutamine tract (Steffan et al., 2001). Like other polyglutamine-interacting proteins, P/CAF possesses an intrinsic HAT activity that activates an NF-Y-dependent gene transcription (Jin and Scott, 1998). HDAC inhibition has been shown to upregulate this HAT activity of P/CAF, resulting in mitigation of polyglutamine-dependent neuropathology in a fly model of HD (Steffan et al., 2001). Although augmentation of histone acetylation was shown to attenuate neurodegeneration in several animal models of polyglutamine disease, the genes regulated by HDAC inhibitors in the affected neurons remained elusive (Minamiyama et al., 2004; Hockly et al., 2003; Ying et al., 2006). On the basis of our present findings, it appears reasonable to infer that the therapeutic effects of NaB, a potent HDAC inhibitor, in SBMA mice might stem from the restoration of TβRII expression.
Roles of TGF-β in non-neuronal cells
The present study showed that the pathway whereby Smad2/3 is phosphorylated and translocated to the nucleus is disrupted not only in the CNS but also in skeletal muscle of SBMA mice, suggesting that pathogenic AR disrupts TGF-β signaling in a cell type-independent manner. The TGF-β-Smad2/3 pathway functions in virtually all cell types, including muscle. TGF-β signaling was shown to have negative effects on muscle growth. Myostatin, a member of the TGF-β superfamily, has gained attention as a potential therapeutic target for myopathies, because genetic deletion of this factor results in an increased muscle volume (McPherron et al., 1997). Therefore, disrupted TGF-β signaling in skeletal muscle might play a protective role against muscular damage in SBMA mice. This protection, however, is likely not capable of compensating for the muscle damage due to the polyglutamine-induced degeneration of spinal motor neurons, given the severe muscular atrophy present in the AR-97Q mice (Katsuno et al., 2002). In support of this view, a previous study showed that inhibition of myostatin has little effect on muscle damage in rodent models of ALS (Holzbaur et al., 2006).
The TGF-β-Smad2/3 pathway is also known to regulate the function of glial cells. However, a great deal of debate has been waged on how glial TGF-β signaling modulates the pathogenesis of neurodegeneration. TGF-β was shown to activate astrocytes and to protect against neuron damage caused by brain injury, while the overexpression of TGF-β in astrocytes promotes brain inflammation and AD-like microvascular degeneration (Flanders et al., 1998; Wyss-Coray et al., 2000). In polyglutamine diseases, the pathogenic proteins accumulate exclusively in neurons, suggesting that neuronal involvement is the primary molecular event triggering neurodegeneration. In accordance with this hypothesis, the present study suggests that the neuronal TGF-β-Smad2/3 pathway plays a substantial role in the molecular mechanisms underlying polyglutamine-mediated neurodegeneration, given that TGF-β signaling was disturbed both in transfected cultured neuronal cells and in neurons of diseased patients and mice.
In conclusion, the present study showed that polyglutamine-dependent neuron damage in SBMA is associated with the disruption of TGF-β signaling due to transcriptional dysregulation of TβRII. Our findings further suggest that restoration of the brain TGF-β-Smad2/3 pathway might be a potential therapeutic approach to polyglutamine-induced neurodegenerative diseases.
Footnotes
This study was supported entirely by grants from the Ministry of Education, Culture, Sports, Science, and Technology, Japan, grants from the Ministry of Health, Labor and Welfare, Japan, and the Program for Improvement of Research Environment for Young Researchers from Special Coordination Funds for Promoting Science and Technology commissioned by the Ministry of Education, Culture, Sports, Science and Technology of Japan and Japan Science and Technology Agency, Core Research for Evolutional Science and Technology. We thank Dr. Seok Hee Park for kindly providing the pTβRII-219/+36 vector.
There were no funding sources other than those described above, and the investigators had sole discretion over study design, collection, analysis, and interpretation of data, writing of the report, and the decision to submit it for publication.
References
- Adachi H, Kumiai A, Li M, Nakagomi Y, Niwa H, Do J, Sang C, Kobayashi Y, Doyu M, Sobue G. Transgenic mice with an expanded CAG repeat controlled by the human AR promoter show polyglutamine nuclear inclusions and neuronal dysfunction without neuronal cell death. Hum Mol Genet. 2001;10:1039–1048. doi: 10.1093/hmg/10.10.1039. [DOI] [PubMed] [Google Scholar]
- Adachi H, Katsuno M, Minamiyama M, Sang C, Pagoulatos G, Angelidis C, Kusakabe M, Yoshiki A, Kobayashi Y, Doyu M, Sobue G. Heat shock protein 70 chaperone overexpression ameliorates phenotypes of the spinal and bulbar muscular atrophy transgenic mouse model by reducing nuclear-localized mutant androgen receptor protein. J Neurosci. 2003;23:2203–2211. doi: 10.1523/JNEUROSCI.23-06-02203.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adachi H, Katsuno M, Minamiyama M, Waza M, Sang C, Nakagomi Y, Kobayashi Y, Tanaka F, Doyu M, Inukai A, Yoshida M, Hashizume Y, Sobue G. Widespread nuclear and cytoplasmic accumulation of mutant androgen receptor in SBMA patients. Brain. 2005;128:659–670. doi: 10.1093/brain/awh381. [DOI] [PubMed] [Google Scholar]
- Adachi H, Waza M, Tokui K, Katsuno M, Minamiyama M, Tanaka F, Doyu M, Sobue G. CHIP overexpression reduces mutant androgen receptor protein and ameliorates phenotypes of the spinal and bulbar muscular atrophy transgenic mouse model. J Neurosci. 2007;27:5115–5126. doi: 10.1523/JNEUROSCI.1242-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrews ZB, Zhao H, Frugier T, Meguro R, Grattan DR, Koishi K, McLennan IS. Transforming growth factor beta2 haploinsufficient mice develop age-related nigrostriatal dopamine deficits. Neurobiol Dis. 2006;21:568–575. doi: 10.1016/j.nbd.2005.09.001. [DOI] [PubMed] [Google Scholar]
- Banno H, Katsuno M, Suzuki K, Takeuchi Y, Kawashima M, Suga N, Takamori M, Ito M, Nakamura T, Matsuo K, Yamada S, Oki Y, Adachi H, Minamiyama M, Waza M, Atsuta N, Watanabe H, Fujimoto Y, Nakashima T, Tanaka F, et al. Phase 2 trial of leuprorelin in patients with spinal and bulbar muscular atrophy. Ann Neurol. 2009;65:140–150. doi: 10.1002/ana.21540. [DOI] [PubMed] [Google Scholar]
- Brionne TC, Tesseur I, Masliah E, Wyss-Coray T. Loss of TGF-beta 1 leads to increased neuronal cell death and microgliosis in mouse brain. Neuron. 2003;40:1133–1145. doi: 10.1016/s0896-6273(03)00766-9. [DOI] [PubMed] [Google Scholar]
- Butler R, Bates GP. Histone deacetylase inhibitors as therapeutics for polyglutamine disorders. Nat Rev Neurosci. 2006;7:784–796. doi: 10.1038/nrn1989. [DOI] [PubMed] [Google Scholar]
- Chevalier-Larsen ES, O'Brien CJ, Wang H, Jenkins SC, Holder L, Lieberman AP, Merry DE. Castration restores function and neurofilament alterations of aged symptomatic males in a transgenic mouse model of spinal and bulbar muscular atrophy. J Neurosci. 2004;24:4778–4786. doi: 10.1523/JNEUROSCI.0808-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou AH, Yeh TH, Ouyang P, Chen YL, Chen SY, Wang HL. Polyglutamine-expanded ataxin-3 causes cerebellar dysfunction of SCA3 transgenic mice by inducing transcriptional dysregulation. Neurobiol Dis. 2008;31:89–101. doi: 10.1016/j.nbd.2008.03.011. [DOI] [PubMed] [Google Scholar]
- Colavita A, Krishna S, Zheng H, Padgett RW, Culotti JG. Pioneer axon guidance by UNC-129, a C. elegans TGF-beta. Science. 1998;281:706–709. doi: 10.1126/science.281.5377.706. [DOI] [PubMed] [Google Scholar]
- Cui L, Jeong H, Borovecki F, Parkhurst CN, Tanese N, Krainc D. Transcriptional repression of PGC-1alpha by mutant huntingtin leads to mitochondrial dysfunction and neurodegeneration. Cell. 2006;127:59–69. doi: 10.1016/j.cell.2006.09.015. [DOI] [PubMed] [Google Scholar]
- Dunah AW, Jeong H, Griffin A, Kim YM, Standaert DG, Hersch SM, Mouradian MM, Young AB, Tanese N, Krainc D. Sp1 and TAFII130 transcriptional activity disrupted in early Huntington's disease. Science. 2002;296:2238–2243. doi: 10.1126/science.1072613. [DOI] [PubMed] [Google Scholar]
- Fernandes HB, Baimbridge KG, Church J, Hayden MR, Raymond LA. Mitochondrial sensitivity and altered calcium handling underlie enhanced NMDA-induced apoptosis in YAC128 model of Huntington's disease. J Neurosci. 2007;27:13614–13623. doi: 10.1523/JNEUROSCI.3455-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flanders KC, Ren RF, Lippa CF. Transforming growth factor-betas in neurodegenerative disease. Prog Neurobiol. 1998;54:71–85. doi: 10.1016/s0301-0082(97)00066-x. [DOI] [PubMed] [Google Scholar]
- Gatchel JR, Zoghbi HY. Diseases of unstable repeat expansion: mechanisms and common principles. Nat Rev Genet. 2005;6:743–755. doi: 10.1038/nrg1691. [DOI] [PubMed] [Google Scholar]
- Hay DG, Sathasivam K, Tobaben S, Stahl B, Marber M, Mestril R, Mahal A, Smith DL, Woodman B, Bates GP. Progressive decrease in chaperone protein levels in a mouse model of Huntington's disease and induction of stress proteins as a therapeutic approach. Hum Mol Genet. 2004;13:1389–1405. doi: 10.1093/hmg/ddh144. [DOI] [PubMed] [Google Scholar]
- Heupel K, Sargsyan V, Plomp JJ, Rickmann M, Varoqueaux F, Zhang W, Krieglstein K. Loss of transforming growth factor-beta 2 leads to impairment of central synapse function. Neural Dev. 2008;3:25. doi: 10.1186/1749-8104-3-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hockly E, Richon VM, Woodman B, Smith DL, Zhou X, Rosa E, Sathasivam K, Ghazi-Noori S, Mahal A, Lowden PA, Steffan JS, Marsh JL, Thompson LM, Lewis CM, Marks PA, Bates GP. Suberoylanilide hydroxamic acid, a histone deacetylase inhibitor, ameliorates motor deficits in a mouse model of Huntington's disease. Proc Natl Acad Sci U S A. 2003;100:2041–2046. doi: 10.1073/pnas.0437870100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holzbaur EL, Howland DS, Weber N, Wallace K, She Y, Kwak S, Tchistiakova LA, Murphy E, Hinson J, Karim R, Tan XY, Kelley P, McGill KC, Williams G, Hobbs C, Doherty P, Zaleska MM, Pangalos MN, Walsh FS. Myostatin inhibition slows muscle atrophy in rodent models of amyotrophic lateral sclerosis. Neurobiol Dis. 2006;23:697–707. doi: 10.1016/j.nbd.2006.05.009. [DOI] [PubMed] [Google Scholar]
- Jin S, Scotto KW. Transcriptional regulation of the MDR1 gene by histone acetyltransferase and deacetylase is mediated by NF-Y. Mol Cell Biol. 1998;18:4377–4384. doi: 10.1128/mcb.18.7.4377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katsuno M, Adachi H, Kume A, Li M, Nakagomi Y, Niwa H, Sang C, Kobayashi Y, Doyu M, Sobue G. Testosterone reduction prevents phenotypic expression in a transgenic mouse model of spinal and bulbar muscular atrophy. Neuron. 2002;35:843–854. doi: 10.1016/s0896-6273(02)00834-6. [DOI] [PubMed] [Google Scholar]
- Katsuno M, Adachi H, Doyu M, Minamiyama M, Sang C, Kobayashi Y, Inukai A, Sobue G. Leuprorelin rescues polyglutamine-dependent phenotypes in a transgenic mouse model of spinal and bulbar muscular atrophy. Nat Med. 2003;9:768–773. doi: 10.1038/nm878. [DOI] [PubMed] [Google Scholar]
- Katsuno M, Sang C, Adachi H, Minamiyama M, Waza M, Tanaka F, Doyu M, Sobue G. Pharmacological induction of heat-shock proteins alleviates polyglutamine-mediated motor neuron disease. Proc Natl Acad Sci U S A. 2005;102:16801–16806. doi: 10.1073/pnas.0506249102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katsuno M, Adachi H, Waza M, Banno H, Suzuki K, Tanaka F, Doyu M, Sobue G. Pathogenesis, animal models and therapeutics in spinal and bulbar muscular atrophy (SBMA) Exp Neurol. 2006a;200:8–18. doi: 10.1016/j.expneurol.2006.01.021. [DOI] [PubMed] [Google Scholar]
- Katsuno M, Adachi H, Minamiyama M, Waza M, Tokui K, Banno H, Suzuki K, Onoda Y, Tanaka F, Doyu M, Sobue G. Reversible disruption of dynactin 1-mediated retrograde axonal transport in polyglutamine-induced motor neuron degeneration. J Neurosci. 2006b;26:12106–12117. doi: 10.1523/JNEUROSCI.3032-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- La Spada AR, Wilson EM, Lubahn DB, Harding AE, Fischbeck KH. Androgen receptor gene mutations in X-linked spinal and bulbar muscular atrophy. Nature. 1991;352:77–79. doi: 10.1038/352077a0. [DOI] [PubMed] [Google Scholar]
- Li M, Miwa S, Kobayashi Y, Merry DE, Yamamoto M, Tanaka F, Doyu M, Hashizume Y, Fischbeck KH, Sobue G. Nuclear inclusions of the androgen receptor protein in spinal and bulbar muscular atrophy. Ann Neurol. 1998;44:249–254. doi: 10.1002/ana.410440216. [DOI] [PubMed] [Google Scholar]
- Lin X, Antalffy B, Kang D, Orr HT, Zoghbi HY. Polyglutamine expansion down-regulates specific neuronal genes before pathologic changes in SCA1. Nat Neurosci. 2000;3:157–163. doi: 10.1038/72101. [DOI] [PubMed] [Google Scholar]
- Luthi-Carter R, Strand A, Peters NL, Solano SM, Hollingsworth ZR, Menon AS, Frey AS, Spektor BS, Penney EB, Schilling G, Ross CA, Borchelt DR, Tapscott SJ, Young AB, Cha JH, Olson JM. Decreased expression of striatal signaling genes in a mouse model of Huntington's disease. Hum Mol Genet. 2000;9:1259–1271. doi: 10.1093/hmg/9.9.1259. [DOI] [PubMed] [Google Scholar]
- McPherron AC, Lawler AM, Lee SJ. Regulation of skeletal muscle mass in mice by a new TGF-beta superfamily member. Nature. 1997;387:83–90. doi: 10.1038/387083a0. [DOI] [PubMed] [Google Scholar]
- Minamiyama M, Katsuno M, Adachi H, Waza M, Sang C, Kobayashi Y, Tanaka F, Doyu M, Inukai A, Sobue G. Sodium butyrate ameliorates phenotypic expression in a transgenic mouse model of spinal and bulbar muscular atrophy. Hum Mol Genet. 2004;13:1183–1192. doi: 10.1093/hmg/ddh131. [DOI] [PubMed] [Google Scholar]
- Niwa H, Yamamura K, Miyazaki J. Efficient selection for high-expression transfectants with a novel eukaryotic vector. Gene. 1991;108:193–199. doi: 10.1016/0378-1119(91)90434-d. [DOI] [PubMed] [Google Scholar]
- Nucifora FC, Jr, Sasaki M, Peters MF, Huang H, Cooper JK, Yamada M, Takahashi H, Tsuji S, Troncoso J, Dawson VL, Dawson TM, Ross CA. Interference by huntingtin and atrophin-1 with cbp-mediated transcription leading to cellular toxicity. Science. 2001;291:2423–2428. doi: 10.1126/science.1056784. [DOI] [PubMed] [Google Scholar]
- Palhan VB, Chen S, Peng GH, Tjernberg A, Gamper AM, Fan Y, Chait BT, La Spada AR, Roeder RG. Polyglutamine-expanded ataxin-7 inhibits STAGA histone acetyltransferase activity to produce retinal degeneration. Proc Natl Acad Sci U S A. 2005;102:8472–8477. doi: 10.1073/pnas.0503505102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park SH, Lee SR, Kim BC, Cho EA, Patel SP, Kang HB, Sausville EA, Nakanishi O, Trepel JB, Lee BI, Kim SJ. Transcriptional regulation of the transforming growth factor beta type II receptor gene by histone acetyltransferase and deacetylase is mediated by NF-Y in human breast cancer cells. J Biol Chem. 2002;277:5168–5174. doi: 10.1074/jbc.M106451200. [DOI] [PubMed] [Google Scholar]
- Peart MJ, Smyth GK, van Laar RK, Bowtell DD, Richon VM, Marks PA, Holloway AJ, Johnstone RW. Identification and functional significance of genes regulated by structurally different histone deacetylase inhibitors. Proc Natl Acad Sci U S A. 2005;102:3697–3702. doi: 10.1073/pnas.0500369102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ranganathan S, Harmison GG, Meyertholen K, Pennuto M, Burnett BG, Fischbeck KH. Mitochondrial abnormalities in spinal and bulbar muscular atrophy. Hum Mol Genet. 2009;18:27–42. doi: 10.1093/hmg/ddn310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riley BE, Orr HT. Polyglutamine neurodegenerative diseases and regulation of transcription: assembling the puzzle. Genes Dev. 2006;20:2183–2192. doi: 10.1101/gad.1436506. [DOI] [PubMed] [Google Scholar]
- Sadri-Vakili G, Bouzou B, Benn CL, Kim MO, Chawla P, Overland RP, Glajch KE, Xia E, Qiu Z, Hersch SM, Clark TW, Yohrling GJ, Cha JH. Histones associated with downregulated genes are hypo-acetylated in Huntington's disease models. Hum Mol Genet. 2007;16:1293–1306. doi: 10.1093/hmg/ddm078. [DOI] [PubMed] [Google Scholar]
- Sanyal S, Kim SM, Ramaswami M. Retrograde regulation in the CNS; neuron-specific interpretations of TGF-beta signaling. Neuron. 2004;41:845–848. doi: 10.1016/s0896-6273(04)00152-7. [DOI] [PubMed] [Google Scholar]
- Shi Y, Massague J. Mechanisms of TGF-beta signaling from cell membrane to the nucleus. Cell. 2003;113:685–700. doi: 10.1016/s0092-8674(03)00432-x. [DOI] [PubMed] [Google Scholar]
- Steffan JS, Bodai L, Pallos J, Poelman M, McCampbell A, Apostol BL, Kazantsev A, Schmidt E, Zhu YZ, Greenwald M, Kurokawa R, Housman DE, Jackson GR, Marsh JL, Thompson LM. Histone deacetylase inhibitors arrest polyglutamine-dependent neurodegeneration in Drosophila. Nature. 2001;413:739–743. doi: 10.1038/35099568. [DOI] [PubMed] [Google Scholar]
- Takeyama K, Ito S, Yamamoto A, Tanimoto H, Furutani T, Kanuka H, Miura M, Tabata T, Kato S. Androgen-dependent neurodegeneration by polyglutamine-expanded human androgen receptor in Drosophila. Neuron. 2002;35:855–864. doi: 10.1016/s0896-6273(02)00875-9. [DOI] [PubMed] [Google Scholar]
- Tesseur I, Zou K, Esposito L, Bard F, Berber E, Can JV, Lin AH, Crews L, Tremblay P, Mathews P, Mucke L, Masliah E, Wyss-Coray T. Deficiency in neuronal TGF-beta signaling promotes neurodegeneration and Alzheimer's pathology. J Clin Invest. 2006;116:3060–3069. doi: 10.1172/JCI27341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tokui K, Adachi H, Waza M, Katsuno M, Minamiyama M, Doi H, Tanaka K, Hamazaki J, Murata S, Tanaka F, Sobue G. 17-DMAG ameliorates polyglutamine-mediated motor neuron degeneration through well-preserved proteasome function in an SBMA model mouse. Hum Mol Genet. 2009;18:898–910. doi: 10.1093/hmg/ddn419. [DOI] [PubMed] [Google Scholar]
- Vivien D, Ali C. Transforming growth factor-beta signalling in brain disorders. Cytokine Growth Factor Rev. 2006;17:121–128. doi: 10.1016/j.cytogfr.2005.09.011. [DOI] [PubMed] [Google Scholar]
- Waza M, Adachi H, Katsuno M, Minamiyama M, Sang C, Tanaka F, Inukai A, Doyu M, Sobue G. 17-AAG, an Hsp90 inhibitor, ameliorates polyglutamine-mediated motor neuron degeneration. Nat Med. 2005;11:1088–1095. doi: 10.1038/nm1298. [DOI] [PubMed] [Google Scholar]
- Wyss-Coray T, Lin C, Sanan DA, Mucke L, Masliah E. Chronic overproduction of transforming growth factor-beta1 by astrocytes promotes Alzheimer's disease-like microvascular degeneration in transgenic mice. Am J Pathol. 2000;156:139–150. doi: 10.1016/s0002-9440(10)64713-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamanaka T, Miyazaki H, Oyama F, Kurosawa M, Washizu C, Doi H, Nukina N. Mutant Huntingtin reduces HSP70 expression through the sequestration of NF-Y transcription factor. EMBO J. 2008;27:827–839. doi: 10.1038/emboj.2008.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ying M, Xu R, Wu X, Zhu H, Zhuang Y, Han M, Xu T. Sodium butyrate ameliorates histone hypoacetylation and neurodegenerative phenotypes in a mouse model for DRPLA. J Biol Chem. 2006;281:12580–12586. doi: 10.1074/jbc.M511677200. [DOI] [PubMed] [Google Scholar]
- Zheng X, Zugates CT, Lu Z, Shi L, Bai JM, Lee T. Baboon/dSmad2 TGF-beta signaling is required during late larval stage for development of adult-specific neurons. EMBO J. 2006;25:615–627. doi: 10.1038/sj.emboj.7600962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y, Yang GY, Ahlemeyer B, Pang L, Che XM, Culmsee C, Klumpp S, Krieglstein J. Transforming growth factor-beta 1 increases bad phosphorylation and protects neurons against damage. J Neurosci. 2002;22:3898–3909. doi: 10.1523/JNEUROSCI.22-10-03898.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]