

Abstract
While stressors are known to increase medial prefrontal cortex (PFC) glutamate (GLU) levels, the mechanism(s) subserving this response remain to be elucidated. We used microdialysis and local drug applications to investigate, in male Long–Evans rats, whether the PFC GLU stress response might reflect increased interhemispheric communication by callosal projection neurons. We report here that tail-pinch stress (20 min) elicited comparable increases in GLU in the left and right PFC that were sodium and calcium dependent and insensitive to local glial cystine–GLU exchanger blockade. Unilateral ibotenate-induced PFC lesions abolished the GLU stress response in the opposite hemisphere, as did contralateral mGlu2/3 receptor activation. Local dopamine (DA) D1 receptor blockade in the left PFC potently enhanced the right PFC GLU stress response, whereas the same treatment applied to the right PFC had a much weaker effect on the left PFC GLU response. Finally, the PFC GLU stress response was attenuated and potentiated, respectively, following α1-adrenoreceptor blockade and GABAB receptor activation in the opposite hemisphere. These findings indicate that the PFC GLU stress response reflects, at least in part, activation of callosal neurons located in the opposite hemisphere and that stress-induced activation of these neurons is regulated by GLU-, DA-, norepinephrine-, and GABA-sensitive mechanisms. In the case of DA, this control is asymmetrical, with a marked regulatory bias of the left PFC DA input over the right PFC GLU stress response. Together, these findings suggest that callosal neurons and their afferentation play an important role in the hemispheric specialization of PFC-mediated responses to stressors.
Introduction
Stressors activate several neurochemical systems in the medial prefrontal cortex (PFC), and of these, the dopamine (DA) stress response has been extensively documented (Thierry et al., 1976; Abercrombie et al., 1989). There is evidence now of hemispheric specialization in the various PFC-mediated stress responses in keeping with evidence of inherent asymmetries of cortical DA inputs (Sullivan, 2004; Gratton and Sullivan, 2005). In general, hemispheric biases in PFC DA stress responses depend on the severity, duration, and controllability of the stressor (Carlson et al., 1991, 1993; Sullivan and Szechtman, 1995; Berridge et al., 1999; Stalnaker et al., 2009). These findings suggest that the effects of different stressors and the coping responses they engage are mediated by distinct PFC mechanisms that are inherently lateralized but under DA modulation. They also imply that, in processing stressful events, the left and right PFC must somehow communicate with each other.
This idea led us to examine the role of callosal neurons. Located primarily in layers II, III, and V, these glutamate (GLU)-containing pyramidal neurons send homotopically distributed projections to the opposite PFC, where they synapse on GABA interneurons and pyramidal neurons (Carr and Sesack, 1998; Karayannis et al., 2007). Importantly, DA terminals form synaptic contacts with the dendrites and dendritic spines of callosal neurons (Carr and Sesack, 2000).
Stressors will also increase PFC GLU levels (Moghaddam, 1993; Bagley and Moghaddam, 1997; Steciuk et al., 2000), although it is unclear which of the known GLU inputs to PFC is responsible for this response. The exact nature of the PFC GLU stress response also remains to be fully elucidated; there is some debate as to whether this response is due to increased GLU release from neurons or from glial cells. While evidence that neurons contribute significantly to stress-induced increases in PFC GLU has been reported (Moghaddam, 1993), some have argued otherwise (Timmerman and Westerink, 1997; Timmerman et al., 1999) (but see van der Zeyden et al., 2008).
In the present study, we first documented possible hemispheric differences in the PFC GLU stress responses and, in doing so, established the extent to which this response reflects increased neuronal transmitter release. Next we investigated how the left and right PFC GLU stress responses are altered by excitotoxic lesions to the opposite PFC and, from this, examined whether the PFC GLU stress response requires activation of a GLU input to the opposite hemisphere. We then sought to determine whether the left and right PFC GLU stress responses are differentially regulated by the DA input to the opposite PFC. Finally, we examined the possibility that the PFC GLU stress response is regulated by GABA- and norepinephrine (NE)-sensitive mechanisms in the opposite PFC. In the case of GABA, the rationale here rests on evidence that the PFC DA stress response is modulated locally by GABA (Doherty and Gratton, 1999) and that the activity of pyramidal neurons is regulated both by GABA- and DA-sensitive mechanisms (Seamans et al., 2001; Trantham-Davidson et al., 2004; Tseng and O'Donnell, 2004). The rationale for investigating the role of NE is based on evidence that stressors activate PFC NE transmission (Finlay et al., 1995) and that the nucleus accumbens DA stress response is indirectly modulated by PFC NE (NicNiocaill and Gratton, 2007).
Materials and Methods
Animals
Adult male (300–400 g) Long–Evans rats (Charles River) were used. The animals were singly housed on a reverse 12 h light/dark cycle (lights off at 12:00 P.M.) with food and water available ad libitum. All procedures were approved by McGill's University Animal Care Committee in accordance with the guidelines of the Canadian Council on Animal Care.
Surgery
Animals were pretreated with atropine sulfate (0.1 mg/kg, i.p.), anesthetized with sodium pentobarbital (54.7 mg/kg, i.p.), and stereotaxically implanted with a 22 gauge stainless steel guide cannula (Plastics One) into either the left or right PFC at the following flat skull coordinates (Paxinos and Watson, 1996): anteroposterior (AP): 3.2 mm anterior to bregma, mediolateral (ML): ±0.6 mm off midline, dorsoventral (DV): 2.2 mm below dura. This cannula would later be used to insert the microdialysis probe into the PFC target site. The cannula was secured with acrylic dental cement anchored to four screws threaded into the cranium. An obturator that extended 3.0 mm beyond the tip of the guide cannula was inserted to prevent infection and CSF seepage. The incision was closed with monofilament sutures and antibiotic powder (Cicatrin; neomycin sulfate: 3300 IU) applied to the wound. Carprofen (5 mg/kg, s.c.) was used for postoperative analgesia (once daily for 2 d). Animals were allowed 3–4 d to recover before testing.
In one set of experiments, animals received a unilateral excitotoxic or sham lesion to the left or right PFC 10 d before being implanted with a microdialysis probe guide cannula into the opposite, intact PFC. A 0.5 μl volume of ibotenic acid solution (5 μg/μl; Sigma) or its saline vehicle (sham lesions) was microinjected into each of two adjacent sites within the left or right PFC at the following coordinates: AP: +3.5 mm, ML: ±0.7 mm, DV: −4.7 mm; and AP: +2.5 mm, ML: ±0.7 mm, DV: −3.5 mm. Solutions were slowly injected at a rate of 0.1 μl/min from the tip of a 1 μl Hamilton microsyringe that was stereotaxically lowered into the brain. Following injection, the microsyringe was left in place for 2–3 min to allow diffusion of the solution. Bore holes were sealed with bone wax. These animals were housed singly until cannula implantation.
For those experiments involving central drug microinjections, animals were also implanted with a 27 gauge stainless steel guide cannula (Plastics One) aimed at the opposite infralimbic PFC; that is, contralateral to the microdialysis guide cannula. This cannula was lowered into the PFC at a 30° angle off the anteroposterior plane using the following coordinates: AP entry point: +0.7 mm, ML: ±0.6 mm, DV: −2.2 mm. An obturator was inserted in this cannula as well.
Microdialysis probes
We used I-shaped, microdialysis probes comprised of side-by-side fused silica inlet–outlet lines [internal diameter (ID): 50 μm] that were encased in polyethylene tubing (ID: 0.58–0.38 mm). A length of regenerated, hollow cellulose membrane (Spectrum, molecular weight cutoff: 13 kDa, OD: 216 μm; ID: 200 μm) was secured to the end of a stainless steel cannula (26 gauge) using cyanoacrylate adhesive and was sealed at its tip with epoxy; the active membrane measured 2.5 mm. A stainless steel collar fitted to the probes provided a secure, threaded connection to the animals' indwelling guide cannula. The probe assembly was affixed to a stainless steel spring that was tethered to a liquid swivel (CMA). Probes were calibrated in artificial CSF (aCSF) containing 100 ng/ml aspartate, GLU, and GABA. In vitro probe recovery ranged from 14 to 19% at a flow rate of 2 μl/min. Computer-controlled microinfusion pumps (CMA) were used to deliver perfusate to the probes, and the dialysate was collected from the fused silica outlet line (dead volume: 0.79 μl).
Testing procedures
Microdialysis.
Animals were tested in opaque circular (30 cm diameter) chambers containing 2 cm of bedding. The animals had ad libitum access to food and water and were acclimatized to the chamber for 4 h before the first test day. On the day of testing, a microdialysis probe was inserted into the animals' indwelling guide cannula and perfused with sterile, degassed aCSF (26 mm NaHCO3, 3 mm NaH2PO4, 1.3 mm MgCl2, 2.3 mm CaCl2, 3.0 mm KCl, 126 mm NaCl, 0.2 mm l-ascorbic acid) at a rate of 1.5 μl/min throughout the 4 h stabilization period. One hour before testing, the flow was increased to 2 μl/min. Dialysate samples were collected during this period but were discarded; this served to habituate the animals to the collection process and the presence of the experimenter. Samples were then taken at 10 min intervals for 1 h before, during, and for 2 h after a 20 min exposure to tail-pinch stress, which consisted of securing a plastic-covered metallic clip 2 cm from the base of the animals' tail; this was comprised of an alligator clip the teeth of which were flattened and covered by a length of shrink tubing to prevent injury to the animals' tail. Freezing behavior, vocalization, and defecation was typically observed during the 20 min episode of tail-pinch stress. Most animals also engaged in sporadic bouts of gnawing on the plastic-covered metallic clip; in our previous studies, this relatively mild stressor consistently elicited robust increases in extracellular PFC DA levels (Brake et al., 2000; Stevenson et al., 2003). Each 20 μl dialysate sample was collected in a fraction vial preloaded with 1 μl of 0.25 m perchloric acid to prevent analyte degradation and immediately stored at 4°C for subsequent analysis.
Reverse dialysis.
Reverse dialysis was used to study the effects of locally perfused treatments on basal and stress-induced increases in extracellular GLU levels. The aim of these experiments was to confirm that the PFC GLU stress responses are due to increased synaptic release of the transmitter. The sodium channel blocker—tetrodotoxin (TTX with citrate, 10 μm, Alomone Labs)—was used to determine whether stress-induced increases in PFC GLU were derived from an impulse-dependent neuronal pool. The calcium dependency of PFC GLU stress responses was also tested by replacing aCSF CaCl2 with an equimolar concentration MgCl2 (final concentration: 3.6 mm). A possible contribution of non-neuronal GLU to the stress response was examined by locally perfusing (S)-4-carboxyphenylglycine (CPG, 50 μm, Tocris Bioscience), a glial cystine–glutamate exchanger blocker (Baker et al., 2002; Melendez et al., 2005). At 30 min before onset of tail-pinch stress, the aCSF perfusate was changed to aCSF containing one of the above three treatments using a liquid switch (CMA) and was maintained throughout the 20 min stress episode and subsequent recovery period. The three perfusate conditions were tested in separate groups of animals. Animals were each tested on separate days under both the control (aCSF) and one of the three perfusate conditions (TTX, Ca2+-free aCSF, or CPG). The treatment order was counterbalanced across animals, and at least 5 d separated the two tests; a fresh dialysis probe was used for each test.
Local drug microinjections.
Each animal received a drug and vehicle control treatment. All drugs were dissolved in aCSF on the test day, and the treatment order was counterbalanced across animals with at least 5 d separating each treatment; a fresh dialysis probe was used on each treatment day. A 0.5 μl volume of solution was slowly ejected over 5 min from the tip of a 30 gauge cannula that was connected to a Hamilton microsyringe by a length of polyethylene tubing such that the tip extended 2 mm beyond the animals' indwelling guide cannula and into the PFC target site. A sample was collected 10 min after injection just before the onset of the stress episode and stored as described above.
Given evidence of hemispheric asymmetries in the PFC DA stress response, stress-induced increases in GLU levels were sampled in either the left or right PFC following local D1 receptor blockade in the opposite PFC with SCH23390 hydrochloride (6 or 0.06 nmol; Sigma). For all subsequent experiments stress-induced increases in GLU levels were monitored in the right PFC following drug microinjections into the left PFC. The following drugs were tested: (1) LY379268, a group II metabotropic glutamate receptor (mGluR2/3) agonist (10 nmol; Tocris Bioscience), (2) benoxathian (BENOX), an α1-adrenoreceptor antagonist (10 nmol; Sigma), and (3) R(+)-baclofen HCl, a GABAB receptor agonist (10 nmol; Sigma).
HPLC
Glutamate levels were determined by precolumn derivatization using HPLC with electrochemical detection (HPLC-EC). The chromatographic system consisted of an ESA pump (model 582) and an ESA injector (model 542) coupled to a Waters Xterra MS C18 3.0 × 50 mm 5 μm analytical column. The mobile phase was prepared as needed and consisted of 3.5% acetonitrile, 20% methanol, and 100 mm sodium phosphate dibasic (Na2HPO4) adjusted to pH 6.7 with 85% phosphoric acid. The flow rate was set at 0.5 ml/min, and the electrochemical detector (ESA Coularray, model no. 5600A) was set at potentials of +150 mV and +550 mV.
Working standards (100 ng/ml) and derivatization reagents were prepared fresh daily from stock solutions and loaded with samples into a refrigerated (10°C) ESA autosampler (model 542). Before injection onto the analytical column, each fraction was sequentially mixed with 20 μl of o-phthaldehyde (0.0143 mol/L) diluted with 0.1 m sodium tetraborate and 20 μl of 3-mercaptopropionic acid (0.071 mol/L) diluted with H2O and allowed to react for 5 min. Following each injection, the injection loop was flushed with 20% methanol to prevent contamination of subsequent samples. Under these conditions, the retention time for GLU was ∼2.4 min with a total run time of 30 min/sample. Chromatographic peak analysis was accomplished by identification of unknown peaks in a sample matched according to retention times from known standards using ESA's CoulArray software.
Histology
Animals were deeply anesthetized with sodium pentobarbital (70 mg/kg, i.p.) and perfused transcardially with 0.9% heparinized saline, followed by a 4% paraformaldehyde solution. The brains were extracted and stored in 4% paraformaldehyde and subsequently cryoprotected in a 30% sucrose solution before being sliced. Lesion sites and probe and microinjector tip placements were confirmed from 30 μm thionin-stained coronal sections.
Data format and statistical analyses
Unless stated otherwise, the data are expressed as the mean (±SEM) percentage change in GLU levels relative to the pooled average of the five baseline samples collected immediately before central drug injection and/or stress; the data were not corrected for the probes' in vitro recovery. Treatment effects were tested for statistical significance using a repeated measures ANOVA using the appropriate two- or three-factorial design with time as a within-group factor and treatment and, in some cases, hemisphere, as between-group factors. When indicated, post hoc comparisons were performed using Tukey's honestly significant difference test (α = 0.05). Analyses were performed using Datasim software (version 1.2, Drake R. Bradley, Bates College, Lewiston, ME).
Results
A total of 86 animals with histologically confirmed PFC cannula and microdialysis probe placements were included in the study (ibotenic acid lesion experiment: n = 28; TTX experiment: n = 6; Ca2+-free aCSF experiment: n = 6; CPG experiment: n = 7; LY379268 experiment: n = 6; SCH23390 experiment: n = 20; BENOX experiment: n = 7; baclofen experiment: n = 6). As can be seen in Figure 1, the active (2.5 mm) segment of the dialysis probes spanned the ventral–dorsal extent of the infralimbic PFC (IL-PFC) and prelimbic PFC (PL-PFC), and in all cases, damage produced by the tips of the injection cannulae was found to extend into the IL-PFC target site.
Figure 1.
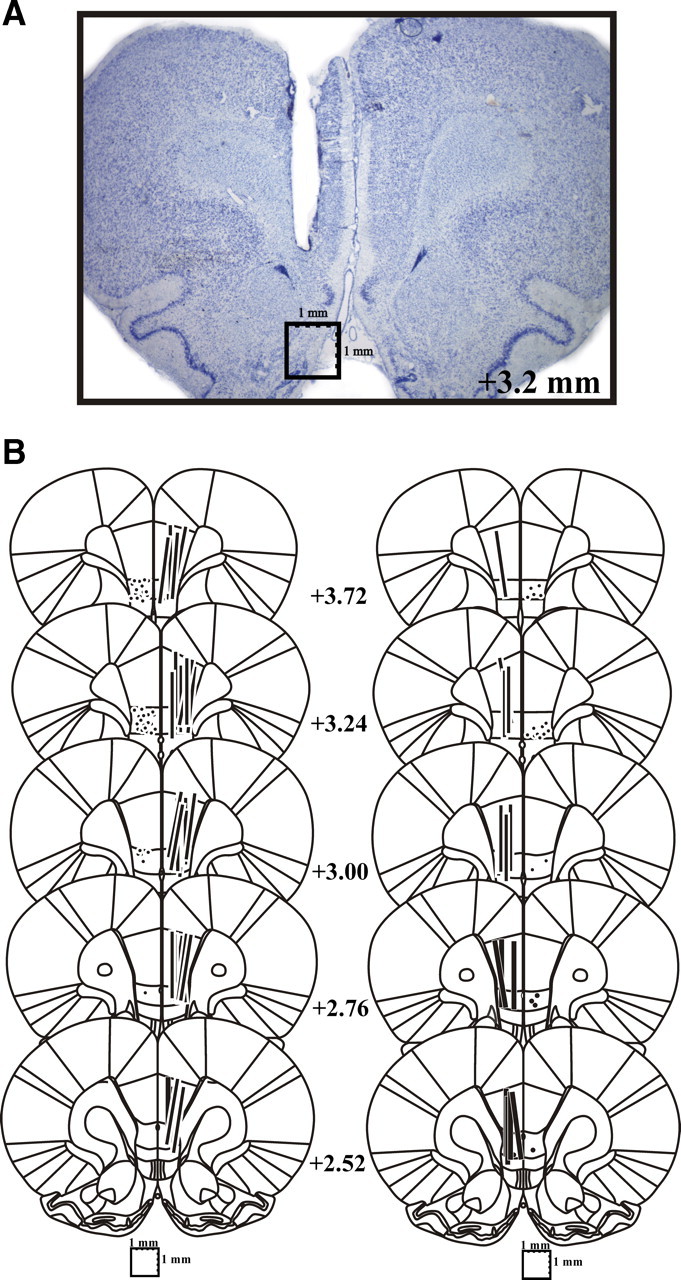
A, B, Histological verification of PFC microdialysis probe and injection cannula placements. A, Photomicrograph of typical damage produced by PFC microdialysis probe. B, Schematic reconstruction of microdialysis probe (vertical bars) and injection cannula (filled circles) placements within the IL-PFC. Length of vertical bars corresponds to the length of the active portion of the dialysis membrane (2.5 mm).
Left versus right PFC GLU stress responses
Figure 2 is a comparison of the stress-induced increases in GLU levels observed in left and right PFC. The data shown in Figure 2B are the averaged percentage changes in GLU levels observed in the left (n = 10) and right (n = 28) PFC of those animals that received control vehicle microinjections into the opposite PFC before being stressed. Two findings emerged from this analysis. First, there were no significant hemispheric differences in the magnitude of stress-induced increases in PFC GLU levels. Second, GLU levels in both the left and right PFC increased significantly above basal levels not only during the 20 min tail-pinch stress episode (10 and 20 min time points) but also immediately before stress onset (0 min time point). This initial rise in GLU levels was associated with the (5 min) microinjection procedure performed shortly after collecting the last baseline sample (−10 min time point). Although repeatedly handled and acclimatized before each experiment, animals had to be briefly immobilized to insert the microinjector into their guide cannula, and this, apparently, was sufficiently stressful to stimulate GLU efflux. This explanation is supported by the fact that no such early increase in GLU levels was seen when animals were not in any way manipulated before tail-pinch stress as was the case with the sham-lesioned animals that served as controls for the ibotenate-lesioned animals. As can be seen in Figure 2A, increases in GLU levels were seen only during the 20 min tail-pinch stress episode, but here too, there were no significant hemispheric differences in the magnitude of the GLU stress response. We also compared basal dialysate levels of GLU in the left and right PFC. This was done by pooling and averaging the absolute GLU levels in the first five prestress dialysate samples. The analysis revealed that basal PFC GLU levels in the right (mean ± SEM = 0.452 ± 0.036 pmol/μl) and left (0.372 ± 0.027 pmol/μl) hemispheres did not differ significantly (t(359) = 1.68, p = 0.0942).
Figure 2.
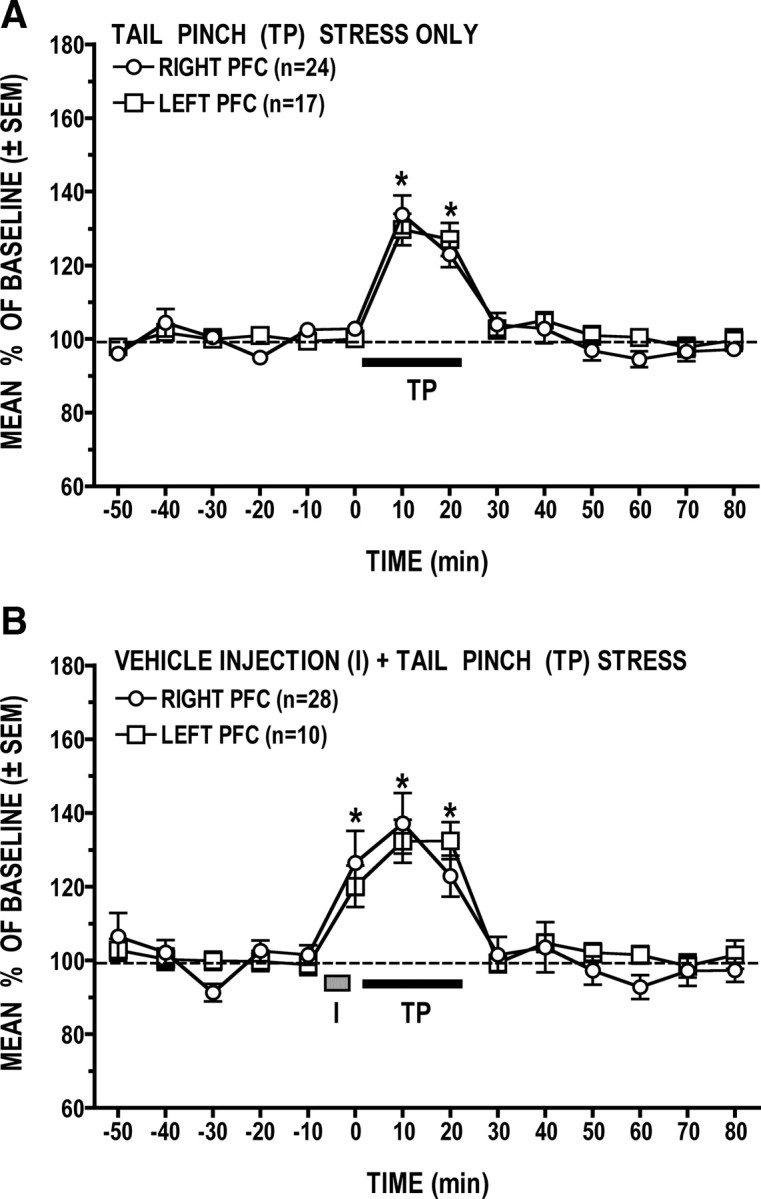
A, B, Comparison of stress-induced increases in left versus right PFC dialysate levels of GLU. A, Comparable increases in GLU levels were elicited in the left and right PFC during tail-pinch (TP) stress. B, Significant increases in GLU levels were also elicited in both hemispheres as animals were handled during the vehicle microinjection procedure (I) that preceded tail-pinch stress. In this and subsequent figures, the length of the black bar and of the shaded rectangle correspond to the duration, respectively, of the tail-pinch stress episode (20 min) and of the microinjection procedure. *p < 0.05 versus prestress baseline.
Effects of TTX, Ca2+-free aCSF, and CPG
Figure 3A shows that the PFC GLU response to tail-pinch stress is significantly attenuated by sodium channel blockade with TTX (treatment × time interaction: F(13,65) = 2.86, p = 0.0026). Post hoc analysis revealed that the peak GLU stress response seen (at 20 min) during local perfusion of the aCSF vehicle was all but abolished by TTX (p < 0.05), indicating that the stress-induced increases in extracellular PFC GLU reflects primarily transmitter release from an impulse-dependent neuronal pool. Interestingly, TTX had no effect on basal (prestress) dialysate levels of GLU (Fig. 3A, inset). The PFC GLU stress response was similarly attenuated when samples were collected using a Ca2+-free aCSF perfusate (Fig. 3B) (treatment × time interaction: F(13,65) = 3.34, p < 0.001, n = 6), and as was the case with TTX, there was no effect of this condition on basal (prestress) levels of GLU (inset). Finally, as can be seen in Figure 3C, locally applied CPG had no effect on the GLU stress response (p > 0.05), indicating that it does not require activation of the glial cystine–GLU antiporter. It is noteworthy, however, that CPG did cause a transient increase in basal (prestress) GLU at ∼20 min following the start of drug application. In the PFC, CPG has been reported to act also as a competitive mGlu1 receptor antagonist (Baker et al., 2002; Melendez and Kalivas, 2003; Melendez et al., 2005) but also displays agonist (Hayashi et al., 1994; Baker et al., 2002) and antagonist (Thomsen et al., 1994) properties at mGlu2/3 receptors in other preparations. The transient CPG effect seen before stress is perhaps best explained by its action at mGlu2/3 receptors. Specifically, decreased cystine–GLU antiporter activity would result in reduced mGlu2/3 stimulation that would in turn lead to a transient increase in GLU release. A direct, antagonist action of CPG at mGlu2/3 receptors could also account for the transient increase in GLU levels. Additionally, activation of mGlu2/3 receptors on GABA interneurons has been reported to inhibit GABA release in the cerebellum (Mitchell and Silver, 2000) and nucleus tractus solitarii (Chen and Bonham, 2005). Thus, it is also conceivable that CPG could transiently stimulate GLU release as an indirect consequence of its inhibitory action on GABA transmission.
Figure 3.
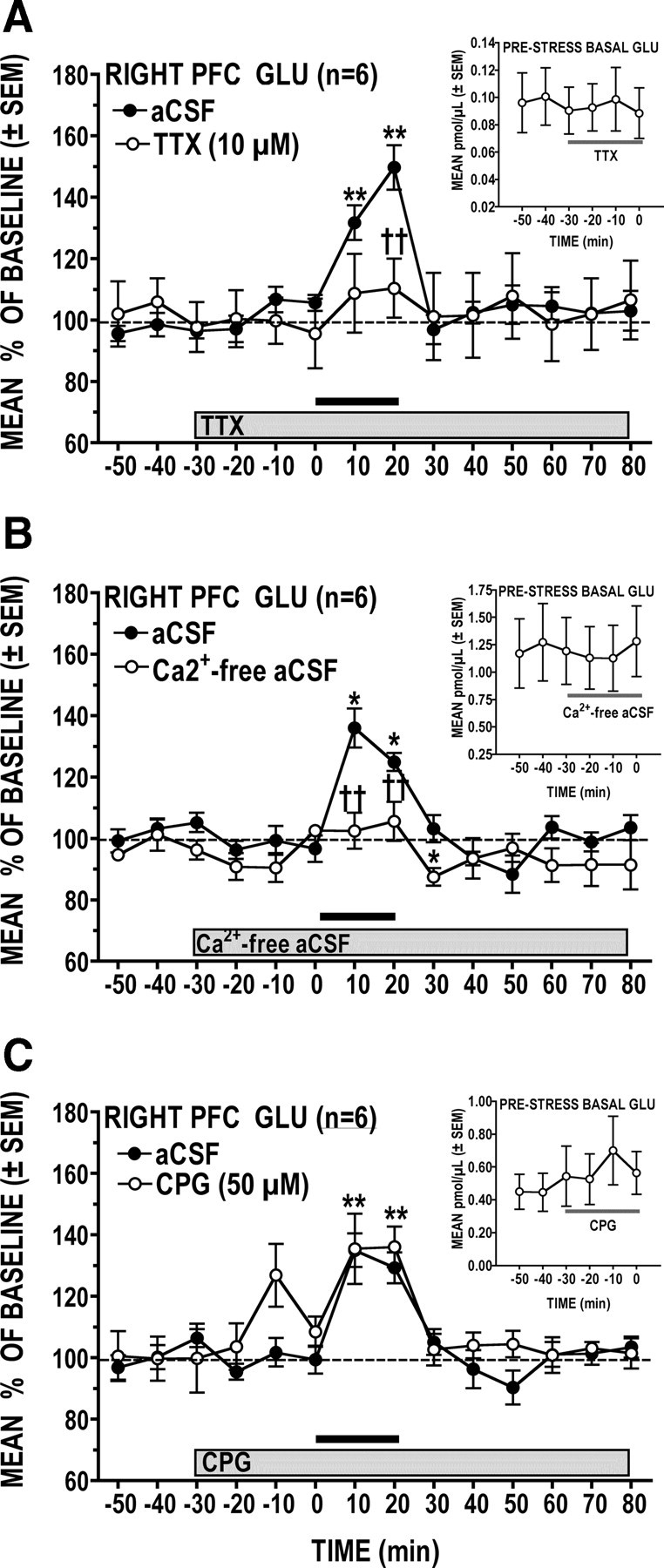
A–C, Effects of locally perfused TTX (A), Ca2+-free aCSF (B), and CPG (C) on stress-induced increases in PFC GLU levels. Insets show absolute levels of basal (prestress) GLU before and during locally perfused treatment. A, Sodium channel blockade with TTX significantly attenuated the GLU response to tail-pinch stress but did not affect basal GLU levels. B, A significantly attenuated GLU stress response was also observed when the perfusate was switched to Ca2+-free aCSF; basal GLU levels were unaffected. C, Glial cystine–GLU antiporter blockade with CPG had no effect on the GLU response to stress but did produce a transient increase in basal GLU levels. *p < 0.05, **p < 0.01 versus prestress baseline; ††p < 0.01 versus time-matched vehicle control.
Effects of left versus right PFC excitotoxic lesions on the contralateral GLU stress response
The histological analysis (Fig. 4A,B) revealed damage characterized by neuronal loss, atrophy, and cavitation; no evidence of such damage was seen in sham-lesioned animals. While damage was clearly confined to either the left or right PFC, there was some between-animal variability in the extent of the lesions; in many animals, tissue damage was restricted mostly to the IL-PFC and the ventral portion of the PL-PFC, but in several cases, neuronal loss was also observed in the dorsal PL-PFC, which occasionally extended into the cingulate cortex. When compared to sham-lesioned animals, the PFC GLU response to stress was significantly attenuated in animals with ibotenic acid lesions to the opposite PFC (Fig. 4C, group × time interaction: F(13,156) = 2.89, p < 0.001; and Fig. 4D, group × time interaction: F(13,156) = 7.10, p < 0.0001); poststress GLU levels in lesioned animals tended to remain slightly (at times significantly) elevated over those of sham controls. Although the left and right PFC GLU stress responses were similarly attenuated by lesions to the opposite hemisphere, post hoc analysis revealed subtle yet significant hemispheric differences. Thus, whereas lesions to the left PFC significantly attenuated the right PFC GLU stress response throughout the 20 min tail-pinch episode (p < 0.01, p < 0.05), right PFC lesions significantly attenuated the left PFC GLU response only during the first 10 min of the stress period (p < 0.01). On close inspection, however, this apparent hemispheric difference in lesion effects is likely due to the greater variability of the left PFC GLU stress response in the sham-lesioned animals. In addition, when compared to the respective sham controls, prestress PFC GLU levels were found to be higher in lesioned animals (Fig. 4D, inset). The magnitude of this effect was hemisphere dependent; that is, in left-lesioned animals, prestress GLU levels in right PFC were elevated by <10% over those of sham controls (sham: 0.572 pmol/μl ± 0.0167 vs lesioned: 0.627 pmol/μl ± 0.0277), but in right-lesioned animals, prestress GLU levels in left PFC were significantly elevated (p < 0.05) by almost 30% over those of sham controls (sham: 0.553 pmol/μl ± 0.0285 vs lesioned: 0.705 pmol/μl ± 0.0580). Although intriguing, the functional significance of this finding is unclear, particularly in view of the fact that the basal GLU levels in the left versus right PFC of lesioned animals did not differ.
Figure 4.
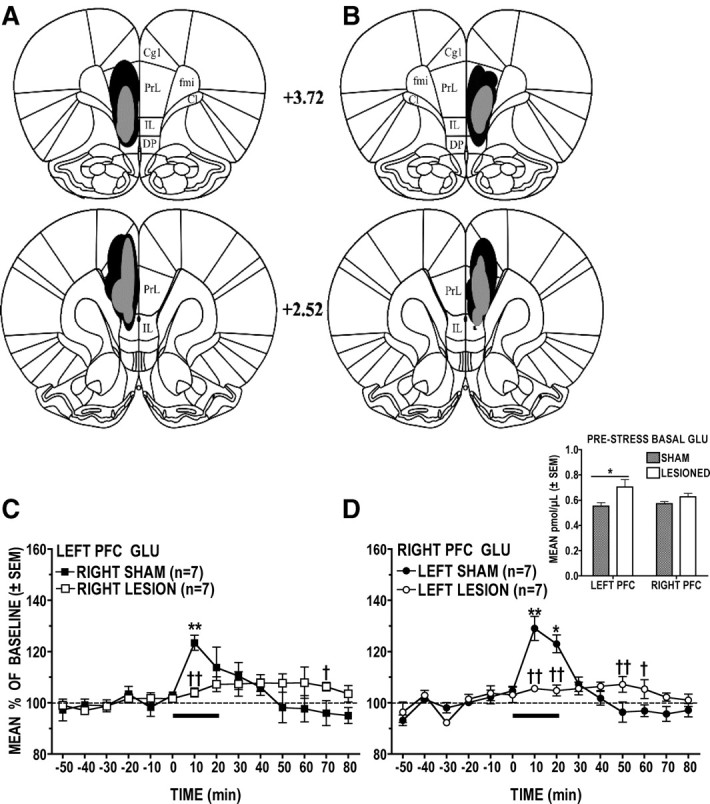
A–D, Effects of unilateral PFC sham and ibotenic acid lesions on the contralateral GLU stress response. A, B, Schematic reconstruction of maximum (black) and minimum (shaded) extent of excitotoxic damage to the left (A) and right (B) PFC. C, Right PFC lesions resulted in significantly attenuated left PFC GLU stress responses. D, Right PFC GLU stress responses were significantly attenuated in animals with left PFC lesions. The inset shows that right PFC-lesioned animals had significantly elevated basal GLU levels in the left PFC. *p < 0.05, **p < 0.01 versus prestress baseline; †p < 0.05, ††p < 0.01 versus time-matched sham control. Cg1, Cingulate cortex; DP, dorsal peduncular cortex; fmi, forceps minor of the corpus callosum; Cl, claustrum.
Effects of left PFC mGlu2/3 receptor activation on the right PFC GLU stress response
When compared to vehicle, LY379268 (10 nmol) significantly attenuated the right PFC GLU stress response (Fig. 5) (treatment × time interaction: F(14,70) = 5.48, p < 0.0001). Post hoc analysis confirmed that, following LY379268, GLU dialysate levels were significantly lower at times 0, 10, and 20 min than those seen following the vehicle treatment (p < 0.05); although not statistically significant, GLU levels during tail-pinch stress following LY379268 were noticeably lower than those seen before injection and following stress.
Figure 5.

Local left PFC application of the mGlu2/3 receptor agonist LY379268 produced a stress-dependent decrease in right PFC GLU levels. **p < 0.01 versus prestress baseline; ††p < 0.01 versus time-matched vehicle control.
Effects of left versus right PFC D1 receptor blockade on the contralateral GLU stress response
Figure 6, A and B, shows that the PFC GLU stress response was significantly potentiated following local application of SCH23390 (6 nmol) in the opposite PFC and that the magnitude of this effect was hemisphere dependent (hemisphere × treatment × time interaction: F(14,140) = 2.18, p = 0.0114). Under the control (vehicle) condition, tail-pinch stress significantly increased (p < 0.05 at 10 and 20 min) GLU to comparable levels in the left and right PFC. Local D1 receptor blockade in the left PFC resulted in a significant (p < 0.05) long-lasting potentiation of the right PFC GLU stress response (Fig. 6B). However, the same treatment applied to the right PFC produced a relatively modest and short-lasting potentiation of the left PFC GLU stress response (Fig. 6A).
Figure 6.

A–D, Effects of unilateral PFC D1 receptor blockade on the contralateral PFC GLU response to stress. A, B, At the higher dose, SCH23390 injected into the left PFC produced a potent, long-lasting enhancement of the right PFC GLU stress response (B), but when applied to the right PFC, the same treatment produced a comparatively weaker effect on the left PFC GLU response (A). C, D, When injected into the left PFC, a lower dose of SCH23390 still produced a noticeable but shorter-lasting (nonsignificant) potentiation of the right PFC GLU stress response (D). Right PFC application of the low dose of SCH23390 had a negligible effect on the left PFC GLU response (C). *p < 0.05 versus prestress baseline; †p < 0.05, ††p < 0.01 versus time-matched vehicle control.
The prolonged potentiation of the right PFC GLU stress response produced by SCH23390 administration into the left PFC raised the possibility that the main effect of this treatment was to elevate basal levels of GLU, independently of the stressor. To rule out this possibility, a separate group of four animals was prepared to examine the right PFC GLU stress response following left PFC microinjections of a 100-fold lower dose of SCH23390 (0.06 nmol). As can be seen in Figure 6D, this low dose of SCH23390 resulted in a similar, albeit nonsignificant, potentiation of the right PFC GLU stress response. Importantly, though, this effect was transient, with GLU levels returning to basal values within 20 min after stress. When applied to the right PFC of a separate group of animals (n = 4), the same low dose of SCH23390 resulted in a slight nonsignificant potentiation of left PFC GLU stress response (Fig. 6C).
Effects of left PFC α1-adrenoreceptor blockade on the right PFC GLU stress response
Local application of BENOX (10 nmol) into the left PFC significantly attenuated the GLU stress response in the right PFC (Fig. 7) (treatment × time interaction: F(14,84) = 3.46, p = 0.002); post hoc analysis revealed that this effect was significant at 10 min into the stress period (p < 0.01) Under the vehicle condition, tail-pinch stress significantly elevated right PFC GLU above baseline at 10 (p < 0.01) and 20 (p < 0.05) min. Following BENOX, however, GLU levels during the 20 min stress period did not differ significantly (p > 0.05) from prestress levels, and while poststress GLU levels appeared to be elevated, this effect was not significant (p > 0.05).
Figure 7.

Local left PFC application of the α1-adrenoreceptor antagonist BENOX significantly attenuated the right PFC GLU stress response. *p < 0.05, **p < 0.01 versus prestress baseline; ††p < 0.01 versus time-matched vehicle control.
Effects of left PFC GABAB receptor activation on the right PFC GLU stress response
Local baclofen (10 nmol) application into the left PFC significantly potentiated the GLU stress response in the right PFC (Fig. 8) (treatment × time interaction: F(14,70) = 2.20 p = 0.016); post hoc analysis revealed that this effect was significant after 20 min of stress (p < 0.05). Poststress GLU levels remained significantly elevated above basal levels for 30 min following the cessation of tail pinch stress (p < 0.05).
Figure 8.

Local left PFC application of the GABAB receptor agonist baclofen significantly potentiated the right PFC GLU stress response. *p < 0.05 versus prestress baseline; †p < 0.01 versus time-matched vehicle control.
Discussion
Origin of the PFC GLU stress response
Our findings indicate that the PFC GLU stress response reflects, at least in part, activation of GLU-containing neurons located in the opposite hemisphere and, thus, implicate callosal neurons in PFC-mediated responses to stressors. A perennial question is whether this GLU response originates from neurons or glial cells; basal and stressed-induced increases in PFC GLU levels are reported to be insensitive (Hashimoto et al., 1995; Timmerman and Westerink, 1997) or partially sensitive (Moghaddam, 1993) to sodium channel blockade. In the present study, TTX had little effect on basal GLU levels but practically abolished the GLU stress response. Stress-induced GLU increases were abolished also when perfusate Ca2+ was omitted but were unaffected by glial cystine–GLU transporter blockade. Thus, although a glial contribution cannot be ruled out entirely, based on these criteria and the fact that the PFC GLU stress response was attenuated following excitotoxic lesions to the opposite hemisphere and was sensitive to contralateral drug injections, we conclude that stress-induced increases in PFC GLU are due mostly to increased neuronal release of the transmitter.
Effects of excitotoxic lesions
That the GLU response to stress was profoundly diminished by excitotoxic lesions to the opposite hemisphere is surprising because it suggests that other known GLU inputs to PFC do not contribute significantly to the GLU stress response. Such a possibility is difficult to envisage, particularly in the case of the basolateral amygdala (BLA). In addition to receiving a stress-responsive DA input (Inglis and Moghaddam, 1999), the BLA shares reciprocal GLU-containing connections with the PFC (McDonald, 1991, 1996; Pinto and Sesack, 2008) and has been implicated in processing fear responses (Goldstein et al., 1996; Anglada-Figueroa and Quirk, 2005; Likhtik et al., 2005; Malin and McGaugh, 2006). Furthermore, it remains that stress did reliably elicit, in lesioned animals, small yet prolonged increases in GLU. It is therefore possible that activation of other PFC GLU inputs is responsible for this residual increase in transmitter levels. In considering this possibility, it is important to keep in mind that the poor temporal (and spatial) resolution of microdialysis precludes drawing any conclusions on the functional significance of this activation. Such protracted increases in GLU might just as well reflect the integrated sum of multiple bursts of transmitter release as it would a small sustained elevation of transmitter levels.
Role of mGlu2/3 receptors
Unilateral PFC mGlu2/3 receptor activation blocked the opposite PFC GLU stress response but also caused GLU to decrease below basal levels during the stress episode. This suggests that stress-induced activation of interhemispheric GLU transmission is itself regulated by GLU, although the mechanism by which this occurs is unclear. Activation of presynaptic mGlu2/3 receptors inhibits GLU release (Kilbride et al., 1998; Cartmell and Schoepp, 2000; Schoepp, 2001), and this should result in less activation of callosal neurons projecting to the opposite PFC. However, the fact that LY379268 caused a stress-dependent reduction in GLU suggests a more complex mechanism. While LY379268 might directly influence the excitability of callosal neurons via postsynaptic mGlu2/3 receptors (Sekizawa et al., 2009), there is evidence that GLU can inhibit GABA release by acting at presynaptic mGlu2/3 receptors (Mitchell and Silver, 2000; Chen and Bonham, 2005). Moreover, local mGlu2/3 receptor activation inhibits stress-induced increases in PFC NE but has no effect on the PFC DA stress response (Swanson et al., 2004). Elucidating how activation of heterosynaptic mGlu2/3 receptors might result in such stress-dependent decreases in PFC GLU will require additional experimentation. Still, evidence of mGlu2/3 receptors on GABA and NE terminals is significant; given that the GLU stress response was sensitive to GABAB receptor activation and α1-adrenoreceptor blockade, it suggests that mGlu2/3 receptor-mediated modulation of PFC GABA and NE release plays a role in regulating stress-induced activation of callosal neurons.
Involvement of NE and GABA
Unilateral α1-adrenoreceptor blockade attenuated the opposite PFC GLU stress response; this effect was followed by a small, sustained increase in GLU suggesting that the drug acted at multiple sites. While α1-adrenoreceptors are mostly postsynaptic, some might also be located presynaptically (Nakadate et al., 2006), and although α1-adrenoreceptor activation was initially reported to increase EPSCs in PFC pyramidal neurons (Marek and Aghajanian,1999), recent evidence suggests instead that it suppresses GLU-, AMPA-, and NMDA-induced EPSCs (Kobayashi et al., 2009). Furthermore, locally applied NE increases PFC DA levels, an effect that is inhibited by local α1-adrenoreceptor blockade (Pan et al., 2004). Thus it is presently unclear whether the effects of benoxathian were mediated directly by α1-adrenoreceptors on callosal neurons or indirectly via a DA-dependent mechanism.
The PFC GLU stress response was facilitated by contralateral GABAB receptor activation, suggesting an indirect action of GABA on the activity of callosal neurons; the direct action of GABA on pyramidal neurons is mediated primarily by GABAA receptors. Given that baclofen and D1 receptor blockade (with SCH23390) similarly enhanced the GLU stress response and that local GABAB receptor activation inhibits PFC DA release (Santiago et al., 1993; Balla et al., 2009) and the PFC DA stress response (Doherty and Gratton, 1999), it is conceivable that baclofen facilitated the PFC GLU stress response as a consequence of inhibiting stress-induced DA release in the opposite hemisphere. As discussed below, an interaction between GABA- and DA-sensitive mechanisms is likely to account also for the enhanced GLU stress response produced by SCH23390.
Role of D1 receptors
Our data suggest that DA exerts a D1 receptor-mediated inhibitory influence on PFC callosal neurons. This control is asymmetrical in that the left PFC DA input exerts a stronger influence on the contralateral GLU stress response than does the right PFC DA input. D1 and D2 receptors are located on PFC pyramidal neurons, including callosal neurons, most of which also express NMDA receptors (Kruse et al., 2009; Santana et al., 2009). Electrophysiological evidence indicates that D1 receptor activation potentiates NMDA receptor-mediated increases in cell excitability, while activation of D2 receptors has the opposite effect (Yang and Seamans, 1996; Trantham-Davidson et al., 2004; Tseng and O'Donnell, 2004; Tseng et al., 2006). In light of this, SCH23390 would presumably have prevented the synergistic effect of concurrent D1 and NMDA receptor activation on the excitability of callosal neurons. If this was indeed the case, then blocking D1 receptors on callosal neurons should not have potentiated, as it did, the contralateral PFC GLU stress response; a dampened GLU stress response would be expected instead, assuming that DA released during stress activated only D2 receptors. Another possibility is that the effect of SCH23390 was mediated indirectly via D1 receptors located on GABA interneurons. Activation of these receptors causes a prolonged increase in IPSCs in PFC pyramidal neurons, ostensibly as a result of increasing the excitability of GABA interneurons; in contrast, activation of D2 receptors on GABA interneurons appears to diminish PFC pyramidal cell IPSCs, presumably as a result of inhibiting GABA transmission (Seamans et al., 2001; Trantham-Davidson et al., 2004). Thus, one possible consequence of GABA interneuron D1 receptor blockade might have been to unmask the indirect disinhibitory effect of GABA interneuron D2 receptor activation (by DA released during stress) on pyramidal cell activity.
Asymmetric modulation of the GLU stress response by DA
That interhemispheric modulation of the PFC GLU stress response by DA is asymmetric is consistent with evidence that the left and right PFC DA inputs are activated to different degrees depending on the stressor and play functionally distinct but complementary roles in the various responses (i.e., behavioral, endocrine, autonomic) engaged to counter the immediate and long-term effects of stressors. The PFC DA response to short-lasting stressors is typically biased toward the left hemisphere. However, activation of PFC DA transmission will shift from this initial left-brain bias to a right-brain bias in response to severe stressors or when exposure to such stressors becomes prolonged or is perceived as uncontrollable (Carlson et al., 1991, 1993; Sullivan and Szechtman, 1995; Berridge et al., 1999; Stalnaker et al., 2009). As proposed by Denenberg et al. (1986), the initial left-biased PFC DA response might mediate preemptive responses to stressors before they become unmanageable and start engaging the right PFC mechanisms involved in activating physiological stress responses (Sullivan and Szechtman, 1995; Sullivan and Gratton, 1998, 1999, 2002b). However, activation of the right PFC would eventually come to predominate when, in the face of a prolonged and inescapable stressor, the early left PFC-mediated responses prove to be ineffective. Under these conditions, the right-biased increase in PFC DA transmission might serve to prevent the negative consequences of sustained right PFC activity. Indeed, impaired right PFC DA function is typically associated with maladaptive responses to stressors and increased vulnerability to stress-related disorders (Kalin et al., 1998; Stefanatos and Wasserstein, 2001; Sullivan and Gratton, 2002a; Gratton and Sullivan, 2005). We can only speculate as to mechanism(s) underlying the asymmetric effects of SCH23390 on stress-induced activation of interhemispheric GLU transmission. It is reasonable to assume, however, that these reflect left versus right hemispheric differences in the cellular and subcellular distributions of D1 and D2 receptors and of other receptor mechanisms known to regulate the excitability of PFC callosal neurons.
Conclusions
The present findings indicate that stressors stimulate PFC GLU transmission in one hemisphere as a consequence of activating or disinhibiting pyramidal neurons located in the opposite hemisphere. While our data do not preclude a role for other GLU inputs to PFC, they do indicate that an important component of the PFC GLU stress response represents increased interhemispheric communication by callosal neurons. Together, these findings implicate callosal neurons in the circuitry regulating the noted hemispheric specialization of PFC-mediated responses to stressors.
Footnotes
This study was made possible by a Canadian Institutes for Health Research grant to A.G. and a Fonds de la recherche en santé du Québec doctoral fellowship to D.L.
References
- Abercrombie ED, Keefe KA, DiFrischia DS, Zigmond MJ. Differential effect of stress on in vivo dopamine release in striatum, nucleus accumbens, and medial frontal cortex. J Neurochem. 1989;52:1655–1658. doi: 10.1111/j.1471-4159.1989.tb09224.x. [DOI] [PubMed] [Google Scholar]
- Anglada-Figueroa D, Quirk GJ. Lesions of the basal amygdala block expression of conditioned fear but not extinction. J Neurosci. 2005;25:9680–9685. doi: 10.1523/JNEUROSCI.2600-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagley J, Moghaddam B. Temporal dynamics of glutamate efflux in the prefrontal cortex and in the hippocampus following repeated stress: effects of pretreatment with saline or diazepam. Neuroscience. 1997;77:65–73. doi: 10.1016/s0306-4522(96)00435-6. [DOI] [PubMed] [Google Scholar]
- Baker DA, Xi ZX, Shen H, Swanson CJ, Kalivas PW. The origin and neuronal function of in vivo nonsynaptic glutamate. J Neurosci. 2002;22:9134–9141. doi: 10.1523/JNEUROSCI.22-20-09134.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balla A, Nattini ME, Sershen H, Lajtha A, Dunlop DS, Javitt DC. GABAB/NMDA receptor interaction in the regulation of extracellular dopamine levels in rodent prefrontal cortex and striatum. Neuropharmacology. 2009;56:915–921. doi: 10.1016/j.neuropharm.2009.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berridge CW, Mitton E, Clark W, Roth RH. Engagement in a non-escape (displacement) behavior elicits a selective and lateralized suppression of frontal cortical dopaminergic utilization in stress. Synapse. 1999;32:187–197. doi: 10.1002/(SICI)1098-2396(19990601)32:3<187::AID-SYN5>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- Brake WG, Sullivan RM, Gratton A. Perinatal distress leads to lateralized medial prefrontal cortical dopamine hypofunction in adult rats. J Neurosci. 2000;20:5538–5543. doi: 10.1523/JNEUROSCI.20-14-05538.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlson JN, Fitzgerald LW, Keller RW, Jr, Glick SD. Side and region dependent changes in dopamine activation with various durations of restraint stress. Brain Res. 1991;550:313–318. doi: 10.1016/0006-8993(91)91333-v. [DOI] [PubMed] [Google Scholar]
- Carlson JN, Fitzgerald LW, Keller RW, Jr, Glick SD. Lateralized changes in prefrontal cortical dopamine activity induced by controllable and uncontrollable stress in the rat. Brain Res. 1993;630:178–187. doi: 10.1016/0006-8993(93)90655-7. [DOI] [PubMed] [Google Scholar]
- Carr DB, Sesack SR. Callosal terminals in the rat prefrontal cortex: synaptic targets and association with GABA-immunoreactive structures. Synapse. 1998;29:193–205. doi: 10.1002/(SICI)1098-2396(199807)29:3<193::AID-SYN1>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- Carr DB, Sesack SR. Dopamine terminals synapse on callosal projection neurons in the rat prefrontal cortex. J Comp Neurol. 2000;425:275–283. doi: 10.1002/1096-9861(20000918)425:2<275::aid-cne9>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- Cartmell J, Schoepp DD. Regulation of neurotransmitter release by metabotropic glutamate receptors. J Neurochem. 2000;75:889–907. doi: 10.1046/j.1471-4159.2000.0750889.x. [DOI] [PubMed] [Google Scholar]
- Chen CY, Bonham AC. Glutamate suppresses GABA release via presynaptic metabotropic glutamate receptors at baroreceptor neurones in rats. J Physiol. 2005;562:535–551. doi: 10.1113/jphysiol.2004.076885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denenberg VH, Gall JS, Berrebi A, Yutzey DA. Callosal mediation of cortical inhibition in the lateralized rat brain. Brain Res. 1986;397:327–332. doi: 10.1016/0006-8993(86)90634-7. [DOI] [PubMed] [Google Scholar]
- Doherty MD, Gratton A. Effects of medial prefrontal cortical injections of GABA receptor agonists and antagonists on the local and nucleus accumbens dopamine responses to stress. Synapse. 1999;32:288–300. doi: 10.1002/(SICI)1098-2396(19990615)32:4<288::AID-SYN5>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- Finlay JM, Zigmond MJ, Abercrombie ED. Increased dopamine and norepinephrine release in medial prefrontal cortex induced by acute and chronic stress: effects of diazepam. Neuroscience. 1995;64:619–928. doi: 10.1016/0306-4522(94)00331-x. [DOI] [PubMed] [Google Scholar]
- Goldstein LE, Rasmusson AM, Bunney BS, Roth RH. Role of the amygdala in the coordination of behavioral, neuroendocrine, and prefrontal cortical monoamine responses to psychological stress in the rat. J Neurosci. 1996;16:4787–4798. doi: 10.1523/JNEUROSCI.16-15-04787.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gratton A, Sullivan RM. Role of the prefrontal cortex in stress responsivity. In: Steckler T, Kalin N, Reul JMHM, editors. Handbook of stress and the brain, Pt 1, The neurobiology of stress. Vol 15. Amsterdam: Elsevier Science; 2005. pp. 807–818. [Google Scholar]
- Hashimoto A, Oka T, Nishikawa T. Extracellular concentration of endogenous free D-serine in the rat brain as revealed by in vivo microdialysis. Neuroscience. 1995;66:635–643. doi: 10.1016/0306-4522(94)00597-x. [DOI] [PubMed] [Google Scholar]
- Hayashi Y, Sekiyama N, Nakanishi S, Jane DE, Sunter DC, Birse EF, Udvarhelyi PM, Watkins JC. Analysis of agonist and antagonist activities of phenylglycine derivatives for different cloned metabotropic glutamate receptor subtypes. J Neurosci. 1994;14:3370–3377. doi: 10.1523/JNEUROSCI.14-05-03370.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inglis FM, Moghaddam B. Dopaminergic innervation of the amygdala is highly responsive to stress. J Neurochem. 1999;72:1088–1094. doi: 10.1046/j.1471-4159.1999.0721088.x. [DOI] [PubMed] [Google Scholar]
- Kalin NH, Larson C, Shelton SE, Davidson RJ. Asymmetric frontal brain activity, cortisol, and behavior associated with fearful temperament in rhesus monkeys. Behav Neurosci. 1998;112:286–292. doi: 10.1037//0735-7044.112.2.286. [DOI] [PubMed] [Google Scholar]
- Karayannis T, Huerta-Ocampo I, Capogna M. GABAergic and pyramidal neurons of deep cortical layers directly receive and differently integrate callosal input. Cereb Cortex. 2007;17:1213–1226. doi: 10.1093/cercor/bhl035. [DOI] [PubMed] [Google Scholar]
- Kilbride J, Huang LQ, Rowan MJ, Anwyl R. Presynaptic inhibitory action of the group II metabotropic glutamate receptor agonists, LY354740 and DCG-IV. Eur J Pharmacol. 1998;356:149–157. doi: 10.1016/s0014-2999(98)00526-3. [DOI] [PubMed] [Google Scholar]
- Kobayashi M, Kojima M, Koyanagi Y, Adachi K, Imamura K, Koshikawa N. Presynaptic and postsynaptic modulation of glutamatergic synaptic transmission by activation of a1- and b-adrenoceptors in layer v pyramidal neurons of rat cerebral cortex. Synapse. 2009;63:269–281. doi: 10.1002/syn.20604. [DOI] [PubMed] [Google Scholar]
- Kruse MS, Prémont J, Krebs M-O, Jay TM. Interaction of dopamine D1 and NMDA NR1 receptors in rat prefrontal cortex. Eur Neuropsychopharmacol. 2009;19:296–304. doi: 10.1016/j.euroneuro.2008.12.006. [DOI] [PubMed] [Google Scholar]
- Likhtik E, Pelletier JG, Paz R, Paré D. Prefrontal control of the amygdala. J Neurosci. 2005;25:7429–7437. doi: 10.1523/JNEUROSCI.2314-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malin EL, McGaugh JL. Differential involvement of the hippocampus, anterior cingulate cortex, and basolateral amygdala in memory for context and footshock. Proc Natl Acad Sci U S A. 2006;103:1959–1963. doi: 10.1073/pnas.0510890103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marek GJ, Aghajanian GK. 5-HT2A receptor or alpha1-adrenoceptor activation induces excitatory postsynaptic currents in layer V pyramidal cells of the medial prefrontal cortex. Eur J Pharmacol. 1999;367:197–206. doi: 10.1016/s0014-2999(98)00945-5. [DOI] [PubMed] [Google Scholar]
- McDonald AJ. Organization of amygdaloid projections to the prefrontal cortex and associated striatum in the rat. Neuroscience. 1991;44:1–14. doi: 10.1016/0306-4522(91)90247-l. [DOI] [PubMed] [Google Scholar]
- McDonald AJ. Glutamate and aspartate immunoreactive neurons of the rat basolateral amygdala: colocalization of excitatory amino acids and projections to the limbic circuit. J Comp Neurol. 1996;365:367–379. doi: 10.1002/(SICI)1096-9861(19960212)365:3<367::AID-CNE3>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- Melendez RI, Kalivas PW. Metabotropic glutamate receptor regulation of extracellular glutamate levels in the prefrontal cortex. Ann N Y Acad Sci. 2003;1003:443–444. doi: 10.1196/annals.1300.047. [DOI] [PubMed] [Google Scholar]
- Melendez RI, Vuthiganon J, Kalivas PW. Regulation of extracellular glutamate in the prefrontal cortex: focus on the cystine glutamate exchanger and group I metabotropic glutamate receptors. J Pharmacol Exp Ther. 2005;314:139–147. doi: 10.1124/jpet.104.081521. [DOI] [PubMed] [Google Scholar]
- Mitchell SJ, Silver RA. Glutamate spillover suppresses inhibition by activating presynaptic mGluRs. Nature. 2000;404:498–502. doi: 10.1038/35006649. [DOI] [PubMed] [Google Scholar]
- Moghaddam B. Stress preferentially increases extraneuronal levels of excitatory amino acids in the prefrontal cortex: comparison to hippocampus and basal ganglia. J Neurochem. 1993;60:1650–1657. doi: 10.1111/j.1471-4159.1993.tb13387.x. [DOI] [PubMed] [Google Scholar]
- Nakadate K, Imamura K, Watanabe Y. Cellular and subcellular localization of a1-adrenoceptors in the rat visual cortex. Neuroscience. 2006;141:1783–1792. doi: 10.1016/j.neuroscience.2006.05.031. [DOI] [PubMed] [Google Scholar]
- NicNiocaill B, Gratton A. Medial prefrontal cortical alpha1 adrenoreceptor modulation of the nucleus accumbens dopamine response to stress in Long-Evans rats. Psychopharmacology. 2007;191:835–842. doi: 10.1007/s00213-007-0723-1. [DOI] [PubMed] [Google Scholar]
- Pan WHT, Yang S-Y, Lin S-K. Neurochemical interaction between dopaminergic and noradrenergic neurons in the medial prefrontal cortex. Synapse. 2004;53:44–52. doi: 10.1002/syn.20034. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Watson C. The rat brain in stereotaxic coordinates. New York: Academic; 1996. [DOI] [PubMed] [Google Scholar]
- Pinto A, Sesack SR. Ultrastructural analysis of prefrontal cortical inputs to the rat amygdala: spatial relationships to presumed dopamine axons and D1 and D2 receptors. Brain Struct Funct. 2008;213:159–175. doi: 10.1007/s00429-008-0180-6. [DOI] [PubMed] [Google Scholar]
- Santana N, Mengod G, Artigas F. Quantitative analysis of the expression of dopamine D1 and D2 receptors in pyramidal and GABAergic neurons of the rat prefrontal cortex. Cerebral Cortex. 2009;19:849–860. doi: 10.1093/cercor/bhn134. [DOI] [PubMed] [Google Scholar]
- Santiago M, Machado A, Cano J. Regulation of the prefrontal cortical dopamine release by GABAA and GABAB receptor agonists and antagonists. Brain Res. 1993;630:28–31. doi: 10.1016/0006-8993(93)90638-4. [DOI] [PubMed] [Google Scholar]
- Schoepp DD. Unveiling the functions of presynaptic metabotropic glutamate receptors in the central nervous system. J Pharmacol Exp Ther. 2001;299:12–20. [PubMed] [Google Scholar]
- Seamans JK, Gorelova N, Durstewitz D, Yang CR. Bidirectional dopamine modulation of GABAergic inhibition in prefrontal cortical pyramidal neurons. J Neurosci. 2001;21:3628–3638. doi: 10.1523/JNEUROSCI.21-10-03628.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sekizawa S, Bechtold AG, Tham RC, Bonham AC. A novel postsynaptic group II metabotropic glutamate receptor role in modulating baroreceptor signal transmission. J Neurosci. 2009;29:11807–11816. doi: 10.1523/JNEUROSCI.2617-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stalnaker TA, España RA, Berridge CW. Coping behavior causes asymmetric changes in neuronal activation in the prefrontal cortex and amygdale. Synapse. 2009;63:82–85. doi: 10.1002/syn.20583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steciuk M, Kram M, Kramer GL, Petty F. Immobilization-induced glutamate efflux in medial prefrontal cortex: blockade by (+)-Mk-801, a selective NMDA receptor antagonist. Stress. 2000;3:195–199. doi: 10.3109/10253890009001123. [DOI] [PubMed] [Google Scholar]
- Stefanatos GA, Wasserstein J. Attention deficit/hyperactivity disorder as a right hemisphere syndrome. Selective literature review and detailed neuropsychological case studies. Ann N Y Acad Sci. 2001;931:172–195. [PubMed] [Google Scholar]
- Stevenson CW, Sullivan RM, Gratton A. Effects of basolateral amygdala dopamine depletion on the nucleus accumbens and medial prefrontal cortical dopamine responses to stress. Neuroscience. 2003;116:285–293. doi: 10.1016/s0306-4522(02)00553-5. [DOI] [PubMed] [Google Scholar]
- Sullivan RM. Hemispheric asymmetry in stress processing in rat prefrontal cortex and the role of mesocortical dopamine. Stress. 2004;7:131–143. doi: 10.1080/102538900410001679310. [DOI] [PubMed] [Google Scholar]
- Sullivan RM, Gratton A. Relationships between stress-induced increases in medial prefrontal cortical dopamine and plasma corticosterone levels in rats: role of cerebral laterality. Neuroscience. 1998;83:81–91. doi: 10.1016/s0306-4522(97)00370-9. [DOI] [PubMed] [Google Scholar]
- Sullivan RM, Gratton A. Lateralized effects of medial prefrontal cortex lesions on neuroendocrine and autonomic stress responses in rats. J Neurosci. 1999;19:2834–2840. doi: 10.1523/JNEUROSCI.19-07-02834.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan RM, Gratton A. Behavioral effects of excitotoxic lesions of ventral medial prefrontal cortex in the rat are hemisphere-dependent. Brain Res. 2002a;927:69–79. doi: 10.1016/s0006-8993(01)03328-5. [DOI] [PubMed] [Google Scholar]
- Sullivan RM, Gratton A. Prefrontal cortical regulation of hypothalamic-pituitary-adrenal function in the rat and implications for psychopathology: side matters. Psychoneuroendocrinology. 2002b;27:99–114. doi: 10.1016/s0306-4530(01)00038-5. [DOI] [PubMed] [Google Scholar]
- Sullivan RM, Szechtman H. Asymmetrical influence of mesocortical dopamine depletion on stress ulcer development and subcortical dopamine systems in rats: implications for psychopathology. Neuroscience. 1995;65:757–766. doi: 10.1016/0306-4522(94)00531-9. [DOI] [PubMed] [Google Scholar]
- Swanson CJ, Perry KW, Schoepp DD. The mGlu2/3 receptor agonist, LY354740, blocks immobilization-induced increases in noradrenaline and dopamine release in the rat medial prefrontal cortex. J Neurochem. 2004;88:194–202. doi: 10.1046/j.1471-4159.2003.02125.x. [DOI] [PubMed] [Google Scholar]
- Thierry AM, Tassin JP, Blanc G, Glowinski J. Selective activation of mesocortical DA system by stress. Nature. 1976;263:242–244. doi: 10.1038/263242a0. [DOI] [PubMed] [Google Scholar]
- Thomsen C, Boel E, Suzdak PD. Actions of phenylglycine analogs at subtypes of the metabotropic glutamate receptor family. Eur J Pharmacol. 1994;267:77–84. doi: 10.1016/0922-4106(94)90227-5. [DOI] [PubMed] [Google Scholar]
- Timmerman W, Westerink BHC. Brain microdialysis of GABA and glutamate: what does it signify? Synapse. 1997;27:242–261. doi: 10.1002/(SICI)1098-2396(199711)27:3<242::AID-SYN9>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- Timmerman W, Cisci G, Nap A, de Vries JB, Westerink BH. Effects of handling on extracellular levels of glutamate and other amino acids in various areas of the brain measured by microdialysis. Brain Res. 1999;833:150–160. doi: 10.1016/s0006-8993(99)01538-3. [DOI] [PubMed] [Google Scholar]
- Trantham-Davidson H, Neely LC, Lavin A, Seamans JK. Mechanisms underlying differential D1 versus D2 dopamine receptor regulation of inhibition in prefrontal cortex. J Neurosci. 2004;24:10652–10659. doi: 10.1523/JNEUROSCI.3179-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tseng KY, O'Donnell P. Dopamine–glutamate interactions controlling prefrontal cortical pyramidal cell excitability involves multiple signaling mechanisms. J Neurosci. 2004;24:5131–5139. doi: 10.1523/JNEUROSCI.1021-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tseng KY, Mallet N, Toreson KL, Le Moine C, Gonon F, O'Donnell P. Excitatory response of prefrontal cortical fast-spiking interneurons to ventral tegmental area stimulation in vivo. Synapse. 2006;59:412–417. doi: 10.1002/syn.20255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Zeyden M, Oldenziel WH, Rea K, Cremers TI, Westerink BH. Microdialysis of GABA and glutamate: analysis, interpretation and comparison with microsensors. Pharmacol Biochem Behav. 2008;90:135–147. doi: 10.1016/j.pbb.2007.09.004. [DOI] [PubMed] [Google Scholar]
- Yang CR, Seamans JK. Dopamine D1 receptor actions in layers V–VI rat prefrontal cortex neurons in vitro. Modulation of dendritic–somatic signal integration. J Neurosci. 1996;16:1922–1935. doi: 10.1523/JNEUROSCI.16-05-01922.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]