

Abstract
Activity-dependent gene expression mediating changes of synaptic efficacy is important for memory storage, but the mechanisms underlying gene transcriptional changes in age-related memory disorders are poorly understood. In this study, we report that gene transcription mediated by the cAMP-response element binding protein (CREB)-regulated transcription coactivator CRTC1 is impaired in neurons and brain from an Alzheimer's disease (AD) transgenic mouse expressing the human β-amyloid precursor protein (APPSw,Ind). Suppression of CRTC1-dependent gene transcription by β-amyloid (Aβ) in response to cAMP and Ca2+ signals is mediated by reduced calcium influx and disruption of PP2B/calcineurin-dependent CRTC1 dephosphorylation at Ser151. Consistently, expression of CRTC1 or active CRTC1 S151A and calcineurin mutants reverse the deficits on CRTC1 transcriptional activity in APPSw,Ind neurons. Inhibition of calcium influx by pharmacological blockade of L-type voltage-gated calcium channels (VGCCs), but not by blocking NMDA or AMPA receptors, mimics the decrease on CRTC1 transcriptional activity observed in APPSw,Ind neurons, whereas agonists of L-type VGCCs reverse efficiently these deficits. Consistent with a role of CRTC1 on Aβ-induced synaptic and memory dysfunction, we demonstrate a selective reduction of CRTC1-dependent genes related to memory (Bdnf, c-fos, and Nr4a2) coinciding with hippocampal-dependent spatial memory deficits in APPSw,Ind mice. These findings suggest that CRTC1 plays a key role in coupling synaptic activity to gene transcription required for hippocampal-dependent memory, and that Aβ could disrupt cognition by affecting CRTC1 function.
Introduction
Gene expression changes in the forebrain occur during normal and pathological aging. Altered gene expression is thought to contribute to the balance between normal aging and age-related memory disorders, including Alzheimer's disease (AD) (Coleman and Yao, 2003; Berchtold et al., 2008). Synaptic dysfunction in AD is apparent before synapse and neuron loss and caused likely by accumulation of β-amyloid (Aβ) peptides (Selkoe, 2002). The cellular mechanisms underlying synaptic and memory dysfunction caused by altered activity-dependent gene transcription in AD are largely unknown. Understanding the molecular pathways regulating gene expression profiles in memory disorders may allow the identification of new signaling pathways for drug discovery (Altar et al., 2009).
Activity-induced gene transcription mediates long-lasting changes of synaptic efficacy essential for neuronal plasticity and memory (Worley et al., 1993; Guzowski et al., 2001; Kandel, 2001). Thus, gene expression mediated by the transcription factor cAMP-response element binding protein (CREB) is essential for synaptic plasticity and long-term memory (Bourtchuladze et al., 1994; Won and Silva, 2008). CREB transcriptional activation depends on calcium- and cAMP-dependent phosphorylation of CREB at Ser133 (Sheng et al., 1991; Mayr and Montminy, 2001), a process mediated by L-type voltage-gated calcium channels (VGCCs) or glutamate ligand-gated receptors (NMDA and AMPA) (Murphy et al., 1991; Cohen and Greenberg, 2008). Interestingly, altered cAMP/PKA-dependent CREB signaling has been postulated to mediate the effect of Aβ on hippocampal synaptic plasticity, memory, and synapse loss (Vitolo et al., 2002; Gong et al., 2006; Smith et al., 2009).
Selective gene transcription by CREB depends on additional events, including other phosphorylation sites and recruitment of specific coactivators. The CREB-regulated transcription coactivators (CRTCs, also known as TORCs) regulate biological events by integrating cellular signals into gene transcriptional responses. Three members of the CRTC family involved in CREB activation (CRTC1, CRTC2, and CRTC3) have been described in mammals (Iourgenko et al., 2003; Ravnskjaer et al., 2007). In non-neuronal cells, selective expression of CREB target genes in response to cAMP and Ca2+ signals, but not by stress stimuli, is achieved by cooperative interaction between CRTC2 and CREB binding protein (CBP)/p300 (Conkright et al., 2003; Ravnskjaer et al., 2007). CRTC1, the most abundant isoform in neurons, mediates the synergistic effect of calcium and cAMP signals on CREB-dependent transcription and long-term potentiation (LTP) (Kovács et al., 2007). CRTC activation by Ca2+ and cAMP signals involves its dephosphorylation by the calcium-dependent phosphatase PP2B/calcineurin and cAMP-mediated inhibition of salt-inducible kinases 1/2 (SIK-1/2) (Bittinger et al., 2004; Screaton et al., 2004; S. Li et al., 2009). Consistent with its role on CREB activation, CRTC1 has been implicated in neuronal dendritic growth, long-term synaptic plasticity, and glucose metabolism (Zhou et al., 2006; Kovács et al., 2007; Altarejos et al., 2008; S. Li et al., 2009).
Whereas the function of CRTC1 on neuronal morphology and plasticity is well established, its role on activity-dependent gene transcription required for memory remains unknown. In this study, we demonstrate that Aβ negatively regulates CRTC1 activation in cultured primary neurons and brain from APPSw,Ind transgenic mice, resulting in a selective and differential disruption of CREB-dependent genes required for hippocampal-dependent memory.
Materials and Methods
Plasmids and antibodies.
Mouse CRTC1-myc, Flag-CRTC2, CREB, CREB R314A, CBP-HA, and p300-HA cloned in pcDNA were previously described (Janknecht et al., 1998; Conkright et al., 2003; Screaton et al., 2004; Kovács et al., 2007). Mouse calcineurin lacking functional CaM-binding and autoinhibitory domains (ΔCnA) was cloned in the CMV-Tag 4A plasmid (O'Keefe et al., 1992). pCRE-luc and TK-Renilla plasmids were purchased from Stratagene and Promega. The CRTC1 S151A mutant was generated from pcDNA3-CRTC1-myc by standard site-directed mutagenesis protocols (Stratagene) with the following forward and reverse primers: 5′-GGAGGAGGACCAACGCTGACTCTGCCCTG-3′ and 5′-CAGGGCAGAGTCAGCGTTGGTCCTCCTCC-3′.
Rabbit phospho-Ser151 CRTC1 antibody was generated by immunizing two rabbits with KLH-conjugated ADTSWRRTN(pS)DSALHQSTMT peptide corresponding to mouse/human CRTC1 (amino acids 142-161). The CRTC1-pSer151 antiserum was sequentially purified by ammonium sulfate precipitation and affinity purification (EZBiolab). The following antibodies were used: APP/Aβ (6E10; 1:2000; Signet), CRTC1 (1:1000; Cell Signaling Technology), CREB and pSer133 CREB (1:1000; Cell Signaling Technology), Egr-1 (1:500; Santa Cruz Biotechnology), BDNF (1:500; Alomone Labs), c-fos (1:500; Santa Cruz Biotechnology), lamin B1 (1:500; Zymed), calcineurin (1:500; BD Transduction Labs), c-myc (9E10; 1:1000; Santa Cruz Biotechnology), β-actin (1:40,000; Abcam), and β-tubulin (1:20,000; Sigma-Aldrich).
APP transgenic mice and behavioral test.
APPSw,Ind (line J9) transgenic mice expressing mutant human APP695 isoform harboring the FAD-linked Swedish (K670N/M671L) and Indiana (V717F) mutations under the neuronal PDGFβ promoter have been previously described (Mucke et al., 2000). Mice were age-matched littermate males obtained by crossing heterozygous APPSw,Ind to nontransgenic (WT) mice (C57BL/6 background). The Morris water maze was performed as previously described (Giménez-Llort et al., 2007; España et al., 2010). Experimenters of the behavioral tests were blind to the genotypes of the mice. Animal procedures were performed in accordance with institutional and national guidelines following approval by the Animal Care and Ethical Committee (CEEAH) of the Universitat Autònoma de Barcelona.
Primary neuronal culture and luciferase reporter assay.
Primary neurons were obtained from mouse embryos (E15.5) of heterozygous APPSw,Ind × nontransgenic crossings. Neurons were dissociated and cultured in Neurobasal medium containing 2% B27, 2 mm glutamine, and 30 mm glucose at a density of 5 · 104 cells/cm2 in 24-well or 35–60 mm dishes. For luciferase assays, 7–15 d in vitro (DIV) neurons in 24-well dishes were transfected for 24 h with pCRE-luc (0.5 μg), TK-Renilla (0.25 μg), and vector or the indicated plasmids (0.5 μg) by using LipofectAMINE 2000 (Invitrogen). For interference assays, neurons at day 0 were infected with shRNA lentiviral vectors (1–2 transducing units per cell). Neurons were treated at 7 DIV with the indicated reagents before stimulation with vehicle, KCl (30 mm), and/or forskolin (20 μm; Sigma) for 4 h. Luciferase activity was measured by triplicate in at least three independent transfections by using the Dual-Luciferase Assay System (Promega) in a Synergy HT luminometer (Bio-Tek).
Lentiviral shRNA and ChIP.
Complementary oligonucleotides for mouse CRTC1 shRNA were as follows: Sh-CRTC1 forward: 5′-gatccccGCAGCGTGACAATCGACCTATttcaagagaATAGGTCGATTGTCACGCTGCttttt-3′; Sh-CRTC1 reverse: 5′-agctaaaaaGCAGCGTGACAATCGACCTATtctcttgaaATAGGTCGATTGTCACGCTGCggg-3′. The scramble control oligonucleotides used were as follows: forward 5′-gatccccGGCTGGGAATGGTAGTCATttcaagagaATGACTACCATTCCCAGCCttttt-3′ and reverse: 5′-agctaaaaaGGCTGGGAATGGTAGTCATtctcttgaaATGACTACCATTCCCAGCCggg-3′. Oligonucleotides were cloned into BglII/HindIII sites of the pSUPER.retro.puro plasmid (OligoEngine). Lentiviral vectors were obtained by digesting EcoR1-ClaI sites from pSUPER-Sh to generate the sequence H1-shRNA that was inserted into pLVTHM vector. Lentiviral particles were generated in HEK293T cells transfected with pLVTHM-Sh, pSPAX2, and pM2G vectors.
For chromatin immunoprecipitation (ChIP) assays, cortical neurons (7 DIV) were treated with vehicle or FSK (20 μm) plus KCl (30 mm) for 2 h. Cells were cross-linked with 1% formaldehyde, lysed in ChIP buffer (25 mm HEPES, pH 8.0, 1.5 mm MgCl2, 10 mm KCl, 0.1% NP-40, 1 mm DTT, and protease and phosphatase inhibitors) and sonicated. Immunoprecipitations of DNA (2.5 μg) were performed overnight with anti-CRTC1 or irrelevant IgG (Cell Signaling Technology). PCR amplification was performed with specific primers for CRE-containing promoters of the genes of interest (supplemental Table 1S, available at www.jneurosci.org as supplemental material).
Biochemical assays.
Primary neurons or mouse hippocampi were lysed in 0.5 ml of cold-lysis buffer (50 mm Tris-HCl, pH 7.4, 150 mm NaCl, 2 mm EDTA, 0.5% Triton X-100, 1% NP-40, 0.1% SDS, 1 mm Na3VO4, 50 mm NaF, and 1 mm PMSF) supplemented with protease and phosphatase inhibitors (Roche). For nuclear fractionation, primary neurons were incubated with the indicated reagents, washed twice in cold-PBS, and lysed using a Dounce homogenizer in buffer A (25 mm HEPES, pH 7.4, 250 mm sucrose, 10 mm KCl, 1.5 mm MgCl, 1 mm Na3VO4, 50 mm NaF, and 1 mm PMSF, supplemented with protease and phosphatase inhibitors). Homogenate was centrifuged (1500 × g) at 4°C for 15 min. The pellet (nuclei) was washed (twice) in buffer A and resuspended and sonicated in lysis buffer (25 mm Tris HCl, pH 7.4, 150 mm NaCl, 1% NP-40, and 10% glycerol) supplemented with protease and phosphatase inhibitors. Proteins were quantified using the BCA protein assay kit (Pierce) and resolved on 8–12.5% SDS-PAGE gels. Proteins were visualized with the enhanced chemiluminescence ECL kit (PerkinElmer) and quantified with the ImageJ software within a linear range of detection for the ECL reagent (España et al., 2010). Soluble Aβ-derived diffusible ligands (ADDLs) were prepared freshly from synthetic Aβ1-42 peptides (Bachem) as previously described (Klein, 2002). The same aggregation protocol was performed on Aβ42-1 peptides. The aggregated Aβ peptides were negative stained and examined in a JEOL JEM-2011 transmission electron microscope. Human Aβ1-40 and Aβ1-40 peptides were measured in conditioned medium using sensitive sandwich ELISA Aβ1-40 and Aβ1-42 kits (Wako) (Guardia-Laguarta et al., 2009).
Calcineurin activity was determined with the calcineurin cellular activity assay kit (Calbiochem). Briefly, mouse brain or cortical neurons were homogenized in lysis buffer (25 mm Tris-HCl, pH 7.5, 0.5 mm dithiothreitol, 50 μm EDTA, 50 μm EGTA, and 0.2% NP-40). Free phosphate was eliminated using a desalting column, and equal amount of protein was incubated with the calcineurin substrate RII phosphopeptide (1.64 mg/ml) for 30 min at 30°C. The reaction was stopped by adding 100 μl of GREEN and fluorescence was measured at 620 nm using a microtiter plate reader.
Immunostaining of primary neurons.
Cortical neurons (10 DIV) were fixed in PBS, pH 7.5, containing 4% paraformaldehyde, 4% sucrose, and 50 mm HEPES for 15 min. Cells were incubated in PBS containing 0.1 m glycine, washed in PBS-Tween 0.1% (PBS-T), and permeabilized with PBS-T plus 0.1% Triton X-100. Cells were blocked with PBS-T containing 0.5% normal goat serum, incubated overnight with mouse anti-PSD-95 (1:50; BD Bioscience) and rabbit anti-synapsin I (1:500; Sigma) antibodies, and detected with the Alexa Fluor 488 or 594 secondary antibodies (1:500) and Hoechst 33258 (Invitrogen). Images from control and APPSw,Ind embryonic neurons (n = 3 per genotype) were analyzed by confocal laser microscopy (Leica TCS SP2 AOBS; Carl Zeiss). For quantitative analyses, acquired neurite images (n ≥ 15 per genotype) were analyzed using LAS AF software (Leica Microsystems). Synaptic boutons were defined as being 0.5–2 μm in length, twofold to threefold more intense than background staining, and stained for synapsin, PSD-95, or both synaptic markers. The number of boutons was divided by the length of the selected dendrite and density presented as boutons per unit length.
Calcium imaging.
Primary cortical neurons grown onto poly-lysine-coated coverslips for 7 d were incubated with the calcium indicator Fura-2/AM (4 μm) for 1 h. Coverslips were washed with Krebs buffer containing (in mm) 119 NaCl, 4.75 KCl, 5 NaHCO3, 1.2 MgSO4, 1.18 KH2PO4, 1.3 CaCl2, 20 HEPES, and 10 glucose, pH 7.4, and mounted in a static chamber at 37°C on an inverted Nikon TE2000U microscope. Cells were excited alternatively at 340 and 380 nm using a monochromator (Cairn Research Limited), and emission light collected at 510 nm every 4 s. Images were acquired by using a 12 bit-CCD ERG ORCA Hamamatsu camera and processed with the Metafluor software (Universal Imaging). When appropriated, cells were treated with KCl (30 mm) and forskolin (20 μm). n ≥ 15 cells/genotype (n = 3 embryos) were analyzed in each experiment.
Real-time RT-PCR.
Total RNA was isolated from mouse primary cortical neurons or hippocampal tissue using the RNeasy Mini Kit (Qiagen). Purified RNA was reverse transcribed using the SuperScript II Reverse Transcriptase Kit (Invitrogen). Briefly, a reaction mix containing 0.25 μg of Oligo(dT) primers, 0.5 mm dNTP, 0.45 mm DTT, RNaseOut (10 U), and SuperScript II Reverse Transcriptase (200 U; Invitrogen) was incubated at 25°C for 10 min, 42°C for 60 min, and 72°C for 10 min. Quantitative RT-PCR of a reaction mix containing cDNA (1 μl), primer pairs (supplemental Table 1S, available at www.jneurosci.org as supplemental material), and the QuantiMix EASY SYG KIT mix (10 μl; Biotools) was performed in an ABI PRISM 7900 Sequence Detector (Applied Biosystems). Data analysis was performed by the comparative ΔΔCt method using the SDS 2.1 software and normalizing to GAPDH.
Statistical analysis.
Statistical analysis was performed using one-way ANOVA and Bonferroni post hoc test. The behavioral data were analyzed using two-way ANOVA with repeated measures and Scheffé's S test for post hoc comparisons. Data were shown as the mean ± SEM. Differences with p < 0.05 were considered significant.
Results
Aβ impairs CRTC1-dependent gene transcription in neurons
To evaluate the possible role of CREB signaling on Aβ-induced transcriptional changes underlying memory dysfunction, we first established primary neurons from an β-amyloid precursor protein (APP) transgenic mouse (APPSw,Ind) that develops age-dependent amyloid pathology and memory deficits (Mucke et al., 2000; España et al., 2010). Cortical neurons from APPSw,Ind embryos expressed human APP (approximately twofold) and released soluble Aβ1-40 and Aβ1-42 peptides without causing gross morphological synaptic changes (supplemental Fig. 1S, available at www.jneurosci.org as supplemental material). Confocal microscopy analysis revealed no significant differences on the number of presynaptic (synapsin), postsynaptic (PSD-95), or active (synapsin/PSD-95) synapses (supplemental Fig. 2S, available at www.jneurosci.org as supplemental material). Since synaptic activity induces efficient expression of immediate-early genes in cortical neurons over the course of 1–3 weeks (Murphy et al., 1991), we performed CREB transcriptional analysis in neurons at 7–15 DIV in conditions mimicking the effects of neuronal activity, such as increasing intracellular Ca2+ by depolarizing concentrations of KCl (30 mm) or cAMP with the adenylate cyclase activator forskolin (FSK) (Greer and Greenberg, 2008). Treatment of control neurons with FSK or KCl resulted in ∼2- and ∼7-fold increase on CRE-luciferase activity, respectively, whereas their combination induced a synergistic effect (∼10-fold) (Fig. 1A). Notably, activation of CRE-dependent transcription was unchanged by FSK but was significantly reduced by KCl (∼25%) or KCl plus FSK (∼50%) in cortical and hippocampal APPSw,Ind neurons (Fig. 1A). Inhibitors of synaptic activity (tetrodotoxin TTX) or calcineurin (FK-506 and cyclosporine) selectively blocked CRE-transcriptional activity induced by Ca2+ and cAMP signals (Fig. 1A).
Figure 1.

Impaired CRTC1-dependent transcription in primary neurons from APPSw,Ind mice. A, Cortical (left) or hippocampal (right) neurons were transfected at 7 DIV with a CRE-luciferase (0.5 μg) and TK-Renilla (0.25 μg) plasmids for 24 h and then treated with vehicle, forskolin (FSK, 20 μm), KCl (30 mm), or FSK/KCl in the presence of cyclosporine (CsA, 5 μm), FK-506 (5 μm), or tetrodotoxin (TTX, 1 μm). B, Cortical neurons were transfected with CRE-luciferase (0.5 μg), TK-Renilla (0.25 μg), and vector, CRTC1, CRTC2, p300, CBP, CREB, or CREB R314A (0.5 μg) for 24 h before FSK/KCl treatment. C, Cortical neurons were untreated (−) or infected at 0 DIV for 7 d with lentiviral CRTC1 or nonsilencing (NS) scramble shRNAs. Western blotting shows that endogenous CRTC1 is efficiently downregulated by CRTC1 shRNA in cortical neurons. CRE-mediated transcription was analyzed as described in A after vehicle or FSK/KCl treatment for 4 h. + and ++ represent 1 and 2 transducing viral units per cell, respectively. D, Top, Real-time RT-PCR analysis performed in control neurons expressing scramble (NS, nonsilencing) or CRTC1 shRNAs reveal that induction of c-fos, Bdnf IV, and Nr4a2 but not Cyr61 are significantly downregulated by CRTC1 shRNA in response to KCl/FSK treatment. Bottom, Quantitative real-time RT-PCR analysis shows differential expression of endogenous CRTC1 target genes in response to FSK/KCl in APPSw,Ind neurons. Values of each gene are normalized to GAPDH and represent percentage of FSK/KCl-treated WT neurons. Bdnf refers to Bdnf IV. E, ChIP assays demonstrate recruitment of CRTC1 to CRE responsive c-fos and Nr4a2 promoters but not to Cyr61 promoter in response to FSK/KCl. IgG indicates immunoprecipitation with an irrelevant antibody. Input lysate is shown as control. Data represent the mean ± SEM of three independent transfections or treatments performed by triplicate. *p < 0.05, **p < 0.01, compared to WT or NS.
Consistent with the idea that CRTC mediates the synergistic effect of cAMP and Ca2+ on CREB-dependent transcription (Screaton et al., 2004), we found that CRTC1, CRTC2, CBP, or p300, but not the CREB R314A mutant lacking the CRTC binding domain (Screaton et al., 2004), potentiated and reversed CRE-transcriptional deficits in APPSw,Ind neurons (Fig. 1B). These results suggested a role of CRTC on altered activity-induced CRE-transcription in APPSw,Ind neurons. We then focused on CRTC1, the most abundant CRTC isoform in neurons and brain (Kovács et al., 2007; Altarejos et al., 2008). We generated lentiviral vectors expressing CRTC1 shRNA that decreased significantly CRTC1 (62–75%) and CRE-mediated transcription induced by cAMP and Ca2+ signals (∼80%) (Fig. 1C). To study the biological significance of CREB transcriptional deficits, we analyzed expression of endogenous genes. Genetic inactivation of CRTC1 by shRNA and gene ChIP analyses demonstrated that CRTC1 is recruited to and activates CRE-containing promoters of several genes, including c-fos, Bdnf IV, and Nr4a2 but not Cyr61 (Fig. 1D,E). Interestingly, induced expression of endogenous CRTC1-dependent genes related to synaptic plasticity and memory such as c-fos, Bdnf IV, and Nr4a2 (∼100- to 1000-fold), but not Cyr61 (∼15-fold), a CREB target gene related to proliferation and activated independently of CRTC1 (Ravnskjaer et al., 2007), was significantly decreased in APPSw,Ind cortical neurons (Fig. 1D).
To test whether Aβ was responsible for mediating the effect of CRTC1 in APPSw,Ind neurons, we used pharmacological and genetic approaches previously shown to reduce Aβ levels (Saura et al., 2005; Oddo et al., 2006). Decreasing Aβ with the γ-secretase inhibitor DAPT or treatment with an anti-Aβ antibody (Ab20.1) reversed significantly CRE-transcriptional deficits in APPSw,Ind neurons (Fig. 2A; supplemental Fig. 1S, available at www.jneurosci.org as supplemental material). Surprisingly, affecting only extracellular Aβ by treatment with Ab20.1 reversed only partially CRE-transcriptional deficits. Furthermore, genetic inactivation of PS1/γ-secretase in APPSw,Ind neurons resulted in normal levels of CRE-transcriptional activity (data not shown). By contrast, media from APPSw,Ind neurons or soluble globular synthetic Aβ1-42 oligomers (ADDLs) containing dimers, trimers, hexamers, and dodecamers at concentrations not affecting neuron morphology or viability (1–20 μm) (Klein, 2002), but not Aβ1-42 monomers or Aβ42-1 peptides submitted to the aggregation protocol, reduced significantly CREB-dependent transcription in a dose-dependent manner (Fig. 2B,C). These results suggested that Aβ negatively affects activity-induced CRTC1-dependent transcription in primary neurons.
Figure 2.

Aβ oligomers impair CRTC1-dependent signaling. A, CRE transcriptional activity in primary cortical neurons treated with vehicle (−), DAPT (125 nm), or anti-Aβ antibody (Ab20.1; 1 μg) for 48 h. B, Biochemical (6E10 antibody) and electron microscopy analysis of Aβ species present in soluble monomeric Aβ1-42 and ADDL preparations. C, CRE-luciferase activity in cortical neurons treated for 48 h with vehicle (−), soluble ADDLs (1-30 μm), aggregated Aβ42-1 (20 μm), Aβ1-42 monomers (1 or 20 μm), or media from control (WT) or APPSw,Ind neurons. Data represent the mean ± SEM of three independent transfections performed by triplicate. *p < 0.05, compared to WT medium; **p < 0.01, compared to FSK/KCI-treated neurons.
Calcineurin-dependent CRTC1 dephosphorylation and activation are impaired in APPSw,Ind mice
The inhibitory effect of calcineurin inhibitors, the CREB R314R mutant, and CRTC1 shRNA on CRTC1 transcription (Fig. 1A,B) prompted us to examine the role of Aβ on calcineurin-mediated CRTC1 activation. Surprisingly, whereas both staurosporine (STS), at doses reported to inhibit SIK and promoting CRTC2 activation [10 nm (Ravnskjaer et al., 2007)], and the active calcineurin mutant ΔCnA (O'Keefe et al., 1992) potentiated CRTC1-dependent transcription, only expression of ΔCnA reversed efficiently transcriptional deficits in APPSw,Ind neurons (Fig. 3A). Biochemical and quantitative analyses revealed that FSK/KCl-induced CRTC1 dephosphorylation was significantly reduced in APPSw,Ind neurons in total and nuclear lysates (p < 0.01). Levels of total CRTC1 (WT: 1.0 ± 0.4 vs APPSw,Ind: 0.86 ± 0.14-fold), CREB (WT: 1.0 ± 0.1 vs APPSw,Ind: 0.9 ± 0.4-fold), or phosphorylated CREB (WT: 1.8 ± 0.6 vs APPSw,Ind: 2.0 ± 0.3-fold) were unchanged in APPSw,Ind neurons (Fig. 3B). To study the biological significance of CRTC1 phosphorylation at Ser151, a phosphorylation site equivalent to CRTC2 Ser 171 (Altarejos et al., 2008), we developed a phosphoSer151-specific CRTC1 antiserum that recognized the endogenous and overexpressed phosphorylated CRTC1 but not a phosphorylation-defective CRTC1 S151A mutant or CRTC2 (Fig. 3C; data not shown). Notably, CRTC1 phosphorylation at Ser151 was significantly increased (∼2-fold) in the hippocampus of APPSw,Ind mice (Fig. 3D). Moreover, both CRTC1 and the active CRTC1 S151A mutant enhanced and reversed CREB transcriptional deficits in APPSwe,Ind neurons (Fig. 3E). These results strongly suggested a deficit on calcineurin-mediated CRTC1 dephosphorylation in neurons and brain of APPSw,Ind mice.
Figure 3.

Aβ impairs CRTC1/CREB-dependent transcription by affecting CRTC1 dephosphorylation. A, CRE transcriptional activity in cortical neurons transfected with CRE-luciferase (0.5 μg), TK-Renilla (0.25 μg), and empty or calcineurin ΔCnA plasmids (0.5 μg). When indicated, neurons were treated with vehicle or STS (10 nm). B, Western blot images and quantitative analyses of CRTC1, CREB, and pCREB (Ser133) in total and nuclear lysates from control (WT) and APPSw,Ind neurons (n ≥ 3). The lower migrating band corresponding to dephosphorylated CRTC1 (top blot, upper graph) and nuclear CRTC1 levels (bottom blot and graph) are significantly decreased in APPSw,Ind neurons stimulated with FSK/KCl for 30 min. In these conditions, pCREB is similarly increased in WT and APPSw,Ind neurons (middle graph). C, Western blot showing that the anti-pSer151 CRTC1 antibody recognizes mouse pCRTC1 but not the CRTC1 S151A mutant expressed in HEK 293T cells. D, Western blot analysis of phosphorylated CRTC1 (Ser151) in hippocampal lysates from WT and APPSw,Ind mice at 6 months of age (n = 6–8 per genotype). E, Expression of CRTC1 and CRTC1 S151A reverses CRE-transcriptional deficits in APPSw,Ind neurons. Data represent the mean ± SEM of three independent transfections performed in duplicate. *p < 0.05, **p < 0.001, ***p < 0.0001.
Aβ reduces calcineurin activity by disrupting Ca2+ influx through L-type calcium channels
Because calcineurin requires Ca2+ for its activation, we next examined the effect of Ca2+ signaling disruption on CRTC1-mediated transcription. Blockers of intracellular Ca2+ (BAPTA), Ca2+ influx (EGTA), and Ca2+ mobilization from endoplasmic reticulum (thapsigargin) significantly reduced CRTC1-dependent transcription in cortical neurons (supplemental Fig. 3S, available at www.jneurosci.org as supplemental material). Indeed, calcineurin activity was significantly reduced in hippocampal (∼ 25%) and cortical (∼40%) neurons and adult brain (∼47%) from APPSw,Ind mice (Fig. 4A). Western blotting analysis revealed unchanged levels of the calcineurin calmodulin-binding catalytic and Ca2+-binding subunits in APPSw,Ind neurons (Fig. 4B). We then measured cytosolic calcium concentration changes elicited by depolarization and cAMP, which are mediated by Ca2+ influx from L-type voltage-gated calcium channels (VGCCs) and Ca2+ mobilization from intracellular stores. Accordingly, Ca2+ imaging experiments showed that the amplitude of Ca2+ changes elicited by FSK/KCl treatment was significantly reduced in APPSw,Ind neurons (Fig. 4C). Similar percentage of control and APPSw,Ind neurons responded to treatment.
Figure 4.

Aβ interferes with calcium-induced CRTC1 activation. A, Decreased calcineurin activity in cultured hippocampal (Hip) or cortical (CX) neurons and brain from APPSw,Ind mice (n = 3 independent cultures and brains). B, Western analysis showing expression of calcineurin in total lysates from WT and APPSw,Ind cortical neurons. C, Intracellular Ca2+ responses in basal and FSK/KCl conditions. Representative calcium images (top) and traces (bottom left) and the mean of peak amplitudes (bottom right) of control and APPSw,Ind neurons are shown. Data represent the mean ± SEM of three independent cultures per genotype (n ≥ 15 cells per culture). D, L-type VGCC blockers nimodipine (5 μm) and verapamil (100 μm) mimic and occlude the effect of Aβ on CRE-transcriptional activity in cortical neurons. E, L-type VGCC agonists Bay K-8644 (10 μm), FPL 64176 (5 μm), and nefiracetam (5 μm) increase and reverse CREB-transcriptional deficits in APPSw,Ind cortical neurons. CRE-luciferase activity was determined as described in Figure 1. *p < 0.05, **p < 0.01.
L-type VGCCs greatly contribute to Ca2+-induced gene expression in hippocampal neurons (Murphy et al., 1991; Mintz et al., 1992; West et al., 2001; Cohen and Greenberg, 2008). Indeed, blockers of postsynaptic L-type (verapamil and nimodipine) or presynaptic N/P/Q-type (ω-conotoxin) VGCCs, but not AMPA (CNQX) or NMDA (MK-801) antagonists, reduced and occluded the effect of cAMP/Ca2+ on CRTC1 transcription in control and APPSw,Ind neurons, respectively (Fig. 4D; supplemental Fig. 3S, available at www.jneurosci.org as supplemental material). Accordingly, the specific L-type Ca2+ channel agonists BayK-8644, FPL 64176, and nefiracetam activated and reversed CREB transcriptional deficits in APPSw,Ind neurons (Fig. 4E). Altogether, these results demonstrated that deficient calcium influx through L-type VGCCs was directly involved in disruption of CRTC1-dependent transcription in APPSw,Ind neurons.
Disruption of CRTC1-dependent gene expression is associated with hippocampal-dependent memory deficits in APPSwe,Ind transgenic mice
Having seen the critical role of Aβ on CRTC1-mediated transcription, we finally analyzed its effect on memory deficits in APPSw,Ind transgenic mice at an age (6 months) coinciding with initial Aβ40/Aβ42 accumulation (Mucke et al., 2000; España et al., 2010) (data not shown). We used the Morris water maze, a spatial memory task that induces expression of immediate early genes 0.5–1 h after training (Guzowski et al., 2001). Six-month-old APPSw,Ind mice required significantly longer latencies and distances to locate the platform during training (two-way ANOVA; latencies: genotype effect, F(1,50) = 19.9; day effect, F(4,50) = 31.6; p < 0.0001). In the probe trial, APPSw,Ind mice spent significantly less time searching and crossing the target platform than controls (genotype effect: F(1,40) = 5.6; quadrant effect: F(3,40) = 6.1, p < 0.002) (Fig. 5A and data not shown). Quantitative real-time RT-PCR analysis revealed a selective reduction of CREB target genes regulated by CRTC1 related to memory (c-fos, Bdnf, and Nr4a2) but not to stress (Rgs2) (Fig. 1) (Ravnskjaer et al., 2007) and unchanged CREB genes regulated independently of CRTC1 involved in memory processing (Egr1), cell proliferation (Cyr61), or stress (Fosb and Junb) in the hippocampus of trained APPSw,Ind mice (Fig. 5B,C). Reduced expression of mRNAs was associated with a significant decreased of c-fos and Bdnf protein levels (Fig. 5D,E). These results revealed specific disruption of CRTC1-dependent genes coinciding with Aβ accumulation and hippocampal-dependent spatial memory deficits in APPSw,Ind mice.
Figure 5.
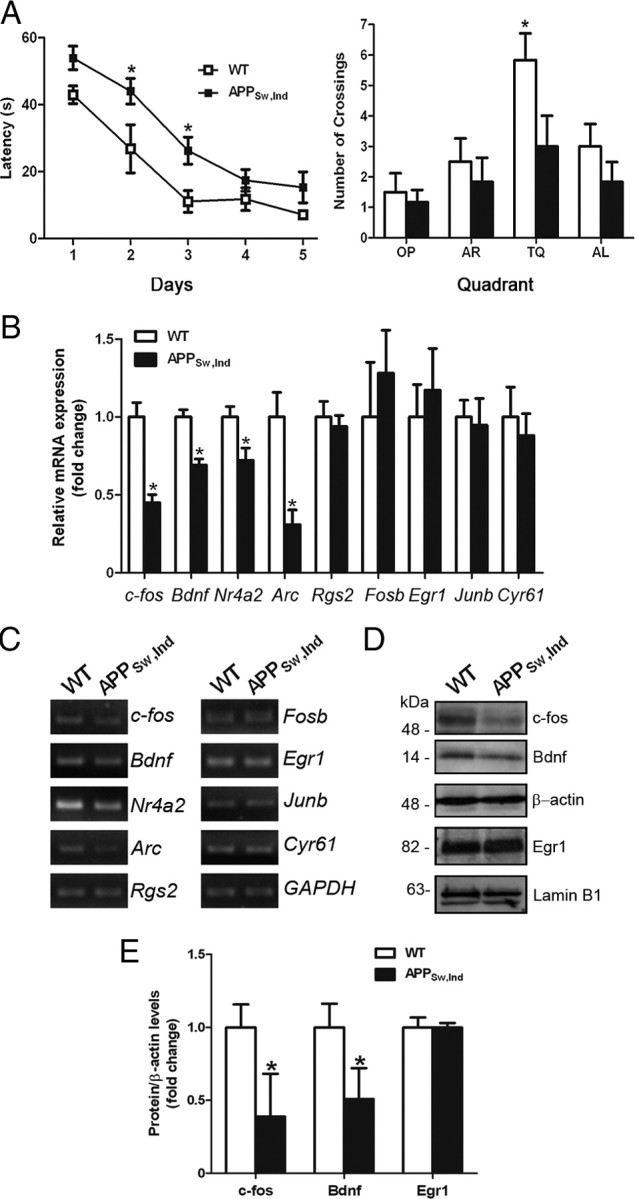
Reduced CRTC1-dependent CREB target genes in the hippocampus of cognitive impaired APPSw,Ind mice. A, APPSw,Ind transgenic mice display learning and spatial memory deficits in the Morris water maze. Six-month-old littermate APPSw,Ind and nontransgenic control mice (n = 6 per genotype) were trained in the MWM for 5 d. APPSw,Ind mice learnt the task but they required significantly longer latencies to locate the platform (two-way ANOVA; latencies: genotype effect, F(1,50) = 19.9; day effect, F(4,50) = 31.6; p < 0.0001). In the probe trial, APPSw,Ind transgenic mice spent significantly less time searching and crossing the target quadrant (TQ) platform location than nontransgenic controls. Data represent the mean ± SEM. *p < 0.05, compared to the same day during training or the rest of quadrants during the probe trial. OP, Opposite platform; AR, adjacent right platform; AL, adjacent left platform. B, Quantitative analysis of hippocampal mRNA of CREB target genes by real-time RT-PCR. Values were normalized to GAPDH. n = 4–5 per genotype. C, PCR analysis showing differential expression of CRTC1 target genes in the hippocampus of APPSw,Ind mice compared to controls. Bdnf refers to Bdnf IV. D, Western blot images showing reduction of c-fos and Bdnf but not Egr1 in the hippocampus of APPSw,Ind mice. E, Quantitative analysis of c-fos, Bdnf, and Egr1 protein levels in hippocampus of 6-month-old WT and APPSw,Ind mice. Data represent the mean ± SEM. *p < 0.05.
Discussion
Gene expression changes in the brain have been suggested to underlie synaptic and cognitive dysfunction during normal and pathological aging (Coleman and Yao, 2003; Berchtold et al., 2008). The molecular mechanisms underlying gene expression changes during memory impairment are largely unknown. In this study, we identified the transcriptional coactivator CRTC1 as mediating the effect of Aβ on disrupting synaptic coupling to activation of genes required for neuronal plasticity and memory. The temporal coincidence of deregulated CRTC1-dependent transcription and cognitive dysfunction in a mouse model of AD strongly argues for a role of CRTC1 on mediating memory processing in normal and pathological conditions.
The transcription factor CREB is a key contributor to cAMP- and calcium-dependent gene transcription during synaptic development and plasticity (Cohen and Greenberg, 2008; Won and Silva, 2008). CREB signaling requires phosphorylation of CREB on Ser133 by cAMP- and Ca2+/calmodulin-dependent kinases (Gonzalez and Montminy, 1989; Dash et al., 1991; Sheng et al., 1991). However, CREB phosphorylation is not sufficient to activate gene transcription (Bito et al., 1996; Zhang et al., 2005), requiring the coactivators CBP, p300, and CRTC (Chrivia et al., 1993; Conkright et al., 2003; Ravnskjaer et al., 2007). Our results showing similar increase of CREB phosphorylation by calcium/cAMP signals in control and APPSw,Ind cortical neurons is consistent with previous reports demonstrating unchanged CREB phosphorylation by Aβ42 in basal or FSK-stimulated mature neurons (Tong et al., 2001; Vitolo et al., 2002; Snyder et al., 2005). By contrast, CREB phosphorylation is decreased in AD brain (Yamamoto-Sasaki et al., 1999) and oligomeric Aβ42 suppresses NMDA- and depolarization-induced CREB phosphorylation in immature neurons (Tong et al., 2001; Ma et al., 2007), an effect that may be due to cAMP/PKA signaling deregulation (Vitolo et al., 2002). Indeed, it was previously shown that Aβ alters hippocampal synaptic plasticity, memory, and synapse morphology through cAMP/PKA-dependent CREB signaling, whereas potentiating this pathway reverses those deficits (Gong et al., 2004, 2006; Smith et al., 2009).
Calcium and cAMP signals do not always cooperate to activate gene expression in response to neuronal activity (Belfield et al., 2006), but they act synergistically on CREB signaling by activating the transcriptional coactivator CRTC (Screaton et al., 2004; Kovács et al., 2007). Consistently, we found deregulation of CRTC1-dependent CREB transcription and reduced Ca2+ responses by naturally secreted Aβ or synthetic Aβ oligomers. These transcriptional deficits are likely attributable to the direct effect of Aβ on CRTC1 because they were prevented by pharmacological or genetic inhibition of Aβ production or by expressing CRTC1 or CRTC1 S151A but not CREB. Surprisingly, treatment of neurons with Ab20.1 antibody reversed only partially CRE-transcriptional deficits, suggesting that both intracellular and extracellular Aβ could contribute to the CRTC1-transcriptional deficits. We therefore propose that deregulation of CRTC1 rather than CREB account for the observed activity-dependent transcriptional deficits in our AD neuronal model.
Our results also provide insights into molecular mechanisms underlying gene expression changes in AD. Activity-dependent gene expression mediated by CREB in excitatory neurons depends on calcium influx through L-type VSCCs or either NMDA or AMPA glutamate receptors (Greer and Greenberg, 2008). Though L-type VGCCs make a minor contribution to synaptic-induced calcium current, they play a critical role in coupling synaptic stimulation to activation of nuclear gene expression (Murphy et al., 1991; Greer and Greenberg, 2008). In agreement with this view, we found that depletion of intracellular calcium and blockers of VGCCs, but not NMDA or AMPA receptor antagonists, reduced significantly CRTC1 transcriptional activity in control neurons, whereas they mimicked the transcriptional defects observed in APPSw,Ind neurons. Our results also agree with previous reports showing that Bdnf, c-fos, Junb, Egr-1, and Fosb are primarily activated by calcium entry through L-type VSCCs rather than NMDA or N-type calcium channels (Murphy et al., 1991; Ghosh et al., 1994). Studies in mice lacking the Cav1.2 channel suggest a role of L-type Ca2+ channels on NMDAR-independent hippocampal LTP and CREB transcription (Moosmang et al., 2005). Because Aβ depresses excitatory synaptic transmission through AMPA and NMDA receptors (Snyder et al., 2005; Hsieh et al., 2006; Dewachter et al., 2009), one possibility is that Aβ modulates differentially glutamatergic signaling depending on its levels (Puzzo et al., 2008).
The finding that agonists of L-type Ca2+ channels reversed the CREB transcriptional deficits in APPSw,Ind cortical neurons strongly implicates altered calcium influx through these channels on the Aβ-induced CRTC1 transcriptional deficits. Indeed, calcium imaging analysis demonstrated decreased intracellular Ca2+ mobilization in response to depolarization and cAMP signals in APPSw,Ind neurons. Although toxic Aβ is known to elevate Ca2+ responses, Aβ reduces P/Q-type calcium currents and spontaneous Ca2+ responses in ∼30% of cortical neurons in APP mice (Busche et al., 2008; Nimmrich et al., 2008). One of the consequences of reduced calcium responses by Aβ may be an impairment of calcineurin activity (Fig. 4) (Lian et al., 2001; Celsi et al., 2007), which in turn may result in decreased L-type Ca2+ channel function (Norris et al., 2002; Tandan et al., 2009). Consistent with an essential role of calcineurin on depolarization-induced CREB-dependent transcription and CRTC1 function (Kingsbury et al., 2007; Kovács et al., 2007), CREB transcriptional activity and CRTC1 dephosphorylation induced by calcium and cAMP signals were fully blocked by calcineurin inhibitors, whereas the active ΔCnA mutant efficiently reversed the transcriptional deficits in APPSw,Ind neurons. In support of a role of altered dephosphorylation of CRTC1 on CRE-transcriptional deficits, we found that SIK inhibition was unable to reverse efficiently CREB transcription or CRTC1 dephosphorylation in APPSw,Ind neurons. Other explanations for reduced calcium responses induced by depolarization and forskolin in APPSw,Ind neurons, including deficient PKA-mediated L-type Ca2+ channel function, cannot be ruled out (Vitolo et al., 2002; Davare and Hell, 2003).
It has been postulated that activity-dependent gene expression plays an essential role in plasticity mechanisms required for memory processing (Guzowski et al., 2005). Neuronal activity and memory training induce expression of Bdnf, c-fos, Junb, Egr-1, and Fosb (Murphy et al., 1991; Worley et al., 1993; Guzowski et al., 2001). Notably, we found that deregulation on CRTC1-dependent genes, including c-fos, Bdnf, and Nr4a2, coincided with the first long-term spatial memory deficits in APPSw,Ind mice (España et al., 2010), suggesting that these events are tightly linked early in the disease process. Notably, reduced c-fos levels were recently associated with learning and memory deficits in APP transgenic mice (Palop et al., 2003; Dewachter et al., 2009), whereas BDNF is decreased in brains of AD patients and transgenic mice (Phillips et al., 1991; Dickey et al., 2003; Palop et al., 2003). In this regard, Bdnf IV, which is induced by calcium influx during neuronal activity and is downregulated by Aβ (Tong et al., 2001; Garzon and Fahnestock, 2007), is particularly important. Our finding that disruption of Bdnf IV and c-fos expression is mediated by deregulation of CRTC1 in APP transgenic mice provides the first reported molecular mechanism underlying deregulation of c-fos and BDNF signaling in AD.
In conclusion, our finding that Aβ disrupts expression of CRTC1 target genes essential for memory processing provides a potential mechanism contributing to cognitive decline in AD. These results may have important therapeutic implications in AD. Indeed, reduced levels of BDNF in CSF were recently associated with age-related cognitive decline (G. Li et al., 2009), whereas BDNF exerts substantial protective effects on neuronal survival and memory circuits in rodent and primate models of AD (Nagahara et al., 2009). Similarly, neural stem cells transplanted in the hippocampus of 3xTg-AD mice enhance synaptic density and improve cognitive function through BDNF (Blurton-Jones et al., 2009). Importantly, agents that activate the PKA/CREB signaling pathway, such as rolipram, ameliorate hippocampal-dependent memory deficits and synapse loss in APP transgenic mice (Gong et al., 2004; Smith et al., 2009). Therefore, understanding the molecular mechanisms regulating CRTC1-dependent signaling and gene responses to therapeutic drugs may provide new targets for memory enhancement in cognitive disorders.
Footnotes
This work was supported by grants from the Ministerio de Ciencia e Innovación (SAF2007-64115 and CIBERNED CB06/05/0042), Ministerio de Sanidad (FIS 04/0937 to C.A.S. and FIS 07/1137 to A.L.) and the 7th Framework Programme of the European Commission (MEMOSAD project, Grant 200611). We thank L. Mucke for providing the APPSw,Ind mice, M. Montminy for CREB and CRTC2 plasmids, J.-R. Cardinaux for mCRTC1 plasmid, R. Janknecht for CBP and p300 plasmids, I. Torres-Alemán for ΔCnA plasmid, and W. Van Nostrand for Ab20.1 antibody. We thank the Servei de Genòmica at the Universitat Autònoma de Barcelona.
References
- Altar CA, Vawter MP, Ginsberg SD. Target identification for CNS diseases by transcriptional profiling. Neuropsychopharmacology. 2009;34:18–54. doi: 10.1038/npp.2008.172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altarejos JY, Goebel N, Conkright MD, Inoue H, Xie J, Arias CM, Sawchenko PE, Montminy M. The Creb1 coactivator Crtc1 is required for energy balance and fertility. Nat Med. 2008;14:1112–1117. doi: 10.1038/nm.1866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belfield JL, Whittaker C, Cader MZ, Chawla S. Differential effects of Ca2+ and cAMP on transcription mediated by MEF2D and cAMP-response element-binding protein in hippocampal neurons. J Biol Chem. 2006;281:27724–27732. doi: 10.1074/jbc.M601485200. [DOI] [PubMed] [Google Scholar]
- Berchtold NC, Cribbs DH, Coleman PD, Rogers J, Head E, Kim R, Beach T, Miller C, Troncoso J, Trojanowski JQ, Zielke HR, Cotman CW. Gene expression changes in the course of normal brain aging are sexually dimorphic. Proc Natl Acad Sci U S A. 2008;105:15605–15610. doi: 10.1073/pnas.0806883105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bito H, Deisseroth K, Tsien RW. CREB phosphorylation and dephosphorylation: a Ca(2+)- and stimulus duration-dependent switch for hippocampal gene expression. Cell. 1996;87:1203–1214. doi: 10.1016/s0092-8674(00)81816-4. [DOI] [PubMed] [Google Scholar]
- Bittinger MA, McWhinnie E, Meltzer J, Iourgenko V, Latario B, Liu X, Chen CH, Song C, Garza D, Labow M. Activation of cAMP response element-mediated gene expression by regulated nuclear transport of TORC proteins. Curr Biol. 2004;14:2156–2161. doi: 10.1016/j.cub.2004.11.002. [DOI] [PubMed] [Google Scholar]
- Blurton-Jones M, Kitazawa M, Martinez-Coria H, Castello NA, Müller FJ, Loring JF, Yamasaki TR, Poon WW, Green KN, LaFerla FM. Neural stem cells improve cognition via BDNF in a transgenic model of Alzheimer disease. Proc Natl Acad Sci U S A. 2009;106:13594–13599. doi: 10.1073/pnas.0901402106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourtchuladze R, Frenguelli B, Blendy J, Cioffi D, Schutz G, Silva AJ. Deficient long-term memory in mice with a targeted mutation of the c-AMP-responsive element binding protein. Cell. 1994;79:59–68. doi: 10.1016/0092-8674(94)90400-6. [DOI] [PubMed] [Google Scholar]
- Busche MA, Eichhoff G, Adelsberger H, Abramowski D, Wiederhold KH, Haass C, Staufenbiel M, Konnerth A, Garaschuk O. Clusters of hyperactive neurons near amyloid plaques in a mouse model of Alzheimer's disease. Science. 2008;321:1686–1689. doi: 10.1126/science.1162844. [DOI] [PubMed] [Google Scholar]
- Celsi F, Svedberg M, Unger C, Cotman CW, Carrì MT, Ottersen OP, Nordberg A, Torp R. β-Amyloid causes downregulation of calcineurin in neurons through induction of oxidative stress. Neurobiol Dis. 2007;26:342–352. doi: 10.1016/j.nbd.2006.12.022. [DOI] [PubMed] [Google Scholar]
- Chrivia JC, Kwok RPS, Lamb N, Hagiwara M, Montminy MR, Goodman RH. Phosphorylated CREB binds specifically to the nuclear protein CBP. Nature. 1993;365:855–859. doi: 10.1038/365855a0. [DOI] [PubMed] [Google Scholar]
- Cohen S, Greenberg ME. Communication between the synapse and the nucleus in neuronal development, plasticity, and disease. Annu Rev Cell Dev Biol. 2008;24:183–209. doi: 10.1146/annurev.cellbio.24.110707.175235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coleman PD, Yao PJ. Synaptic slaughter in Alzheimer's disease. Neurobiol Aging. 2003;24:1023–1027. doi: 10.1016/j.neurobiolaging.2003.09.001. [DOI] [PubMed] [Google Scholar]
- Conkright MD, Canettieri G, Screaton R, Guzman E, Miraglia L, Hogenesch JB, Montminy M. TORCs: transducers of regulated CREB activity. Mol Cell. 2003;12:413–423. doi: 10.1016/j.molcel.2003.08.013. [DOI] [PubMed] [Google Scholar]
- Dash PK, Karl KA, Colicos MA, Prywes R, Kandel ER. cAMP response element-binding protein is activated by Ca2+/calmodulin- as well as cAMP-dependent protein kinase. Proc Natl Acad Sci U S A. 1991;88:5061–5065. doi: 10.1073/pnas.88.11.5061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davare MA, Hell JW. Increased phosphorylation of the neuronal L-type Ca2+ channel Ca(v)1.2 during aging. Proc Natl Acad Sci U S A. 2003;100:16018–16023. doi: 10.1073/pnas.2236970100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dewachter I, Filipkowski RK, Priller C, Ris L, Neyton J, Croes S, Terwel D, Gysemans M, Devijver H, Borghgraef P, Godaux E, Kaczmarek L, Herms J, Van Leuven F. Deregulation of NMDA-receptor function and down-stream signaling in APP[V717I] transgenic mice. Neurobiol Aging. 2009;30:241–256. doi: 10.1016/j.neurobiolaging.2007.06.011. [DOI] [PubMed] [Google Scholar]
- Dickey CA, Loring JF, Montgomery J, Gordon MN, Eastman PS, Morgan D. Selectively reduced expression of synaptic plasticity-related genes in amyloid precursor protein + presenilin-1 transgenic mice. J Neurosci. 2003;23:5219–5226. doi: 10.1523/JNEUROSCI.23-12-05219.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- España J, Giménez-Llort L, Valero J, Miñano A, Rábano A, Rodriguez-Alvarez J, LaFerla FM, Saura CA. Intraneuronal β-amyloid accumulation in the amygdala enhances fear and anxiety in Alzheimer's disease transgenic mice. Biol Psychiatry. 2010;67:513–521. doi: 10.1016/j.biopsych.2009.06.015. [DOI] [PubMed] [Google Scholar]
- Garzon DJ, Fahnestock M. Oligomeric amyloid decreases basal levels of brain-derived neurotrophic factor (BDNF) mRNA via specific downregulation of BDNF transcripts IV and V in differentiated human neuroblastoma cells. J Neurosci. 2007;27:2628–2635. doi: 10.1523/JNEUROSCI.5053-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh A, Carnahan J, Greenberg ME. Requirement for BDNF in activity-dependent survival of cortical neurons. Science. 1994;263:1618–1623. doi: 10.1126/science.7907431. [DOI] [PubMed] [Google Scholar]
- Giménez-Llort L, Blázquez G, Cañete T, Johansson B, Oddo S, Tobeña A, LaFerla FM, Fernández-Teruel A. Modeling behavioral and neuronal symptoms of Alzheimer's disease in mice: a role for intraneuronal amyloid. Neurosci Biobehav Rev. 2007;31:125–147. doi: 10.1016/j.neubiorev.2006.07.007. [DOI] [PubMed] [Google Scholar]
- Gong B, Vitolo OV, Trinchese F, Liu S, Shelanski M, Arancio O. Persistent improvement in synaptic and cognitive functions in an Alzheimer mouse model after rolipram treatment. J Clin Invest. 2004;114:1624–1634. doi: 10.1172/JCI22831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong B, Cao Z, Zheng P, Vitolo OV, Liu S, Staniszewski A, Moolman D, Zhang H, Shelanski M, Arancio O. Ubiquitin hydrolase Uch-L1 rescues β-amyloid-induced decreases in synaptic function and contextual memory. Cell. 2006;126:775–788. doi: 10.1016/j.cell.2006.06.046. [DOI] [PubMed] [Google Scholar]
- Gonzalez GA, Montminy MR. Cyclic AMP stimulates somatostatin gene transcription by phosphorylation of CREB at serine 133. Cell. 1989;59:675–680. doi: 10.1016/0092-8674(89)90013-5. [DOI] [PubMed] [Google Scholar]
- Greer PL, Greenberg ME. From synapse to nucleus: calcium-dependent gene transcription in the control of synapse development and function. Neuron. 2008;59:846–860. doi: 10.1016/j.neuron.2008.09.002. [DOI] [PubMed] [Google Scholar]
- Guardia-Laguarta C, Coma M, Pera M, Clarimón J, Sereno L, Agulló JM, Molina-Porcel L, Gallardo E, Deng A, Berezovska O, Hyman BT, Blesa R, Gómez-Isla T, Lleó A. Mild cholesterol depletion reduces amyloid-β production by impairing APP trafficking to the cell surface. J Neurochem. 2009;110:220–230. doi: 10.1111/j.1471-4159.2009.06126.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guzowski JF, Setlow B, Wagner EK, McGaugh JL. Experience-dependent gene expression in the rat hippocampus after spatial learning: a comparison of the immediate-early genes Arc, c-fos, and zif268. J Neurosci. 2001;21:5089–5098. doi: 10.1523/JNEUROSCI.21-14-05089.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guzowski JF, Timlin JA, Roysam B, McNaughton BL, Worley PF, Barnes CA. Mapping behaviorally relevant neural circuits with immediate-early gene expression. Curr Opin Neurobiol. 2005;15:599–606. doi: 10.1016/j.conb.2005.08.018. [DOI] [PubMed] [Google Scholar]
- Hsieh H, Boehm J, Sato C, Iwatsubo T, Tomita T, Sisodia S, Malinow R. AMPAR removal underlies Aβ-induced synaptic depression and dendritic spine loss. Neuron. 2006;52:831–843. doi: 10.1016/j.neuron.2006.10.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iourgenko V, Zhang W, Mickanin C, Daly I, Jiang C, Hexham JM, Orth AP, Miraglia L, Meltzer J, Garza D, Chirn GW, McWhinnie E, Cohen D, Skelton J, Terry R, Yu Y, Bodian D, Buxton FP, Zhu J, Song C, et al. Identification of a family of cAMP response element-binding protein coactivators by genome-scale functional analysis in mammalian cells. Proc Natl Acad Sci U S A. 2003;100:12147–12152. doi: 10.1073/pnas.1932773100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janknecht R, Wells NJ, Hunter T. TGF-beta-stimulated cooperation of smad proteins with the coactivators CBP/p300. Genes Dev. 1998;12:2114–2119. doi: 10.1101/gad.12.14.2114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kandel ER. The molecular biology of memory storage: a dialogue between genes and synapses. Science. 2001;294:1030–1038. doi: 10.1126/science.1067020. [DOI] [PubMed] [Google Scholar]
- Kingsbury TJ, Bambrick LL, Roby CD, Krueger BK. Calcineurin activity is required for depolarization-induced, CREB-dependent gene transcription in cortical neurons. J Neurochem. 2007;103:761–770. doi: 10.1111/j.1471-4159.2007.04801.x. [DOI] [PubMed] [Google Scholar]
- Klein WL. Aβ toxicity in Alzheimer's disease: globular oligomers (ADDLs) as new vaccine and drug targets. Neurochem Int. 2002;41:345–352. doi: 10.1016/s0197-0186(02)00050-5. [DOI] [PubMed] [Google Scholar]
- Kovács KA, Steullet P, Steinmann M, Do KQ, Magistretti PJ, Halfon O, Cardinaux JR. TORC1 is a calcium- and cAMP-sensitive coincidence detector involved in hippocampal long-term synaptic plasticity. Proc Natl Acad Sci U S A. 2007;104:4700–4705. doi: 10.1073/pnas.0607524104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li G, Peskind ER, Millard SP, Chi P, Sokal I, Yu CE, Bekris LM, Raskind MA, Galasko DR, Montine TJ. Cerebrospinal fluid concentration of brain-derived neurotrophic factor and cognitive function in non-demented subjects. PLoS ONE. 2009;4:e5424. doi: 10.1371/journal.pone.0005424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Zhang C, Takemori H, Zhou Y, Xiong ZQ. TORC1 regulates activity-dependent CREB-target gene transcription and dendritic growth of developing cortical neurons. J Neurosci. 2009;29:2334–2343. doi: 10.1523/JNEUROSCI.2296-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lian Q, Ladner CJ, Magnuson D, Lee JM. Selective changes of calcineurin (protein phosphatase 2B) activity in Alzheimer's disease cerebral cortex. Exp Neurol. 2001;167:158–165. doi: 10.1006/exnr.2000.7534. [DOI] [PubMed] [Google Scholar]
- Ma QL, Harris-White ME, Ubeda OJ, Simmons M, Beech W, Lim GP, Teter B, Frautschy SA, Cole GM. Evidence of Abeta- and transgene-dependent defects in ERK-CREB signaling in Alzheimer's models. J Neurochem. 2007;103:1594–1607. doi: 10.1111/j.1471-4159.2007.04869.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayr B, Montminy M. Transcriptional regulation by the phosphorylation-dependent factor CREB. Nat Rev Mol Cell Biol. 2001;2:599–609. doi: 10.1038/35085068. [DOI] [PubMed] [Google Scholar]
- Mintz IM, Adams ME, Bean BP. P-type calcium channels in rat central and peripheral neurons. Neuron. 1992;9:85–95. doi: 10.1016/0896-6273(92)90223-z. [DOI] [PubMed] [Google Scholar]
- Moosmang S, Haider N, Klugbauer N, Adelsberger H, Langwieser N, Müller J, Stiess M, Marais E, Schulla V, Lacinova L, Goebbels S, Nave KA, Storm DR, Hofmann F, Kleppisch T. Role of hippocampal Cav1.2 Ca2+ channels in NMDA receptor-independent synaptic plasticity and spatial memory. J Neurosci. 2005;25:9883–9892. doi: 10.1523/JNEUROSCI.1531-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mucke L, Masliah E, Yu GQ, Mallory M, Rockenstein EM, Tatsuno G, Hu K, Kholodenko D, Johnson-Wood K, McConlogue L. High-level neuronal expression of Aβ1-42 in wild-type human amyloid protein precursor transgenic mice: synaptotoxicity without plaque formation. J Neurosci. 2000;20:4050–4058. doi: 10.1523/JNEUROSCI.20-11-04050.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy TH, Worley PF, Baraban JM. L-type voltage-sensitive calcium channels mediate synaptic activation of immediate early genes. Neuron. 1991;7:625–635. doi: 10.1016/0896-6273(91)90375-a. [DOI] [PubMed] [Google Scholar]
- Nagahara AH, Merrill DA, Coppola G, Tsukada S, Schroeder BE, Shaked GM, Wang L, Blesch A, Kim A, Conner JM, Rockenstein E, Chao MV, Koo EH, Geschwind D, Masliah E, Chiba AA, Tuszynski MH. Neuroprotective effects of brain-derived neurotrophic factor in rodent and primate models of Alzheimer's disease. Nat Med. 2009;15:331–337. doi: 10.1038/nm.1912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nimmrich V, Grimm C, Draguhn A, Barghorn S, Lehmann A, Schoemaker H, Hillen H, Gross G, Ebert U, Bruehl C. Amyloid β oligomers (Aβ1-42 globulomer) suppress spontaneous synaptic activity by inhibition of P/Q-type calcium currents. J Neurosci. 2008;28:788–797. doi: 10.1523/JNEUROSCI.4771-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norris CM, Blalock EM, Chen KC, Porter NM, Landfield PW. Calcineurin enhances L-type Ca(2+) channel activity in hippocampal neurons: increased effect with age in culture. Neuroscience. 2002;110:213–225. doi: 10.1016/s0306-4522(01)00574-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oddo S, Vasilevko V, Caccamo A, Kitazawa M, Cribbs DH, LaFerla FM. Reduction of soluble abeta and tau, but not soluble abeta alone, ameliorates cognitive decline in transgenic mice with plaques and tangles. J Biol Chem. 2006;281:39413–39423. doi: 10.1074/jbc.M608485200. [DOI] [PubMed] [Google Scholar]
- O'Keefe SJ, Tamura J, Kincaid RL, Tocci MJ, O'Neill EA. FK-506- and CsA-sensitive activation of a interleukin-2 promoter by calcineurin. Nature. 1992;357:692–694. doi: 10.1038/357692a0. [DOI] [PubMed] [Google Scholar]
- Palop JJ, Jones B, Kekonius L, Chin J, Yu GQ, Raber J, Masliah E, Mucke L. Neuronal depletion of calcium-dependent proteins in the dentate gyrus is tightly linked to Alzheimer's disease-related cognitive deficits. Proc Natl Acad Sci U S A. 2003;100:9572–9577. doi: 10.1073/pnas.1133381100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips HS, Hains JM, Armanini M, Laramee GR, Johnson SA, Winslow JW. BDNF mRNA is decreased in the hippocampus of individuals with Alzheimer's disease. Neuron. 1991;7:695–702. doi: 10.1016/0896-6273(91)90273-3. [DOI] [PubMed] [Google Scholar]
- Puzzo D, Privitera L, Leznik E, Fà M, Staniszewski A, Palmeri A, Arancio O. Picomolar amyloid-β positively modulates synaptic plasticity and memory in hippocampus. J Neurosci. 2008;28:14537–14545. doi: 10.1523/JNEUROSCI.2692-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravnskjaer K, Kester H, Liu Y, Zhang X, Lee D, Yates JR, 3rd, Montminy M. Cooperative interactions between CBP and TORC2 confer selectivity to CREB target gene expression. EMBO J. 2007;26:2880–2889. doi: 10.1038/sj.emboj.7601715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saura CA, Chen G, Malkani S, Choi SY, Takahashi RH, Zhang D, Gouras GK, Kirkwood A, Morris RG, Shen J. Conditional inactivation of presenilin-1 prevents amyloid accumulation and temporarily rescues contextual and spatial working memory impairments in amyloid precursor protein transgenic mice. J Neurosci. 2005;25:6755–6764. doi: 10.1523/JNEUROSCI.1247-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Screaton RA, Conkright MD, Katoh Y, Best JL, Canettieri G, Jeffries S, Guzman E, Niessen S, Yates JR, 3rd, Takemori H, Okamoto M, Montminy M. The CREB coactivator TORC2 functions as a calcium- and cAMP sensitive coincidence detector. Cell. 2004;119:61–74. doi: 10.1016/j.cell.2004.09.015. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ. Alzheimer's disease is a synaptic failure. Science. 2002;298:789–791. doi: 10.1126/science.1074069. [DOI] [PubMed] [Google Scholar]
- Sheng M, Thompson MA, Greenberg ME. CREB: a Ca2+-regulated transcription factor phosphorylated by calmodulin-dependent kinases. Science. 1991;252:1427–1430. doi: 10.1126/science.1646483. [DOI] [PubMed] [Google Scholar]
- Smith DL, Pozueta J, Gong B, Arancio O, Shelanski M. Reversal of long-term dendritic spine alterations in Alzheimer disease models. Proc Natl Acad Sci U S A. 2009;106:16877–16882. doi: 10.1073/pnas.0908706106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snyder EM, Nong Y, Almeida CG, Paul S, Moran T, Choi EY, Nairn AC, Salter MW, Lombroso PJ, Gouras GK, Greengard P. Regulation of NMDA receptor trafficking by amyloid-β. Nat Neurosci. 2005;8:1051–1058. doi: 10.1038/nn1503. [DOI] [PubMed] [Google Scholar]
- Tandan S, Wang Y, Wang TT, Jiang N, Hall DD, Hell JW, Luo X, Rothermel BA, Hill JA. Physical and functional interaction between calcineurin and the cardiac L-type Ca2+ channel. Circ Res. 2009;105:51–60. doi: 10.1161/CIRCRESAHA.109.199828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong L, Thornton PL, Balazs R, Cotman CW. β-amyloid-(1-42) impairs activity-dependent cAMP-response element-binding protein signaling in neurons at concentrations in which cell survival is not compromised. J Biol Chem. 2001;276:17301–17306. doi: 10.1074/jbc.M010450200. [DOI] [PubMed] [Google Scholar]
- Vitolo OV, Sant'Angelo A, Costanzo V, Battaglia F, Arancio O, Shelanski M. Amyloid β-peptide inhibition of the PKA/CREB pathway and long-term potentiation: reversibility by drugs that enhance cAMP signaling. Proc Natl Acad Sci U S A. 2002;99:13217–13221. doi: 10.1073/pnas.172504199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West AE, Chen WG, Dalva MB, Dolmetsch RE, Kornhauser JM, Shaywitz AJ, Takasu MA, Tao X, Greenberg ME. Calcium regulation of neuronal gene expression. Proc Natl Acad Sci U S A. 2001;98:11024–11031. doi: 10.1073/pnas.191352298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Won J, Silva AJ. Molecular and cellular mechanisms of memory allocation in neuronetworks. Neurobiol Learn Mem. 2008;89:285–292. doi: 10.1016/j.nlm.2007.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Worley PF, Bhat RV, Baraban JM, Erickson CA, McNaughton BL, Barnes CA. Thresholds for synaptic activation of transcription factors in hippocampus: correlation with long-term enhancement. J Neurosci. 1993;13:4776–4786. doi: 10.1523/JNEUROSCI.13-11-04776.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto-Sasaki M, Ozawa H, Saito T, Rösler M, Riederer P. Impaired phosphorylation of cyclic AMP response element binding protein in the hippocampus of dementia of the Alzheimer type. Brain Res. 1999;824:300–303. doi: 10.1016/s0006-8993(99)01220-2. [DOI] [PubMed] [Google Scholar]
- Zhang X, Odom DT, Koo SH, Conkright MD, Canettieri G, Best J, Chen H, Jenner R, Herbolsheimer E, Jacobsen E, Kadam S, Ecker JR, Emerson B, Hogenesch JB, Unterman T, Young RA, Montminy M. Genome-wide analysis of cAMP-response element binding protein occupancy, phosphorylation, and target gene activation in human tissues. Proc Natl Acad Sci U S A. 2005;102:4459–4464. doi: 10.1073/pnas.0501076102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Wu H, Li S, Chen Q, Cheng XW, Zheng J, Takemori H, Xiong ZQ. Requirement of TORC1 for late-phase long-term potentiation in the hippocampus. PLoS ONE. 2006;1:e16. doi: 10.1371/journal.pone.0000016. [DOI] [PMC free article] [PubMed] [Google Scholar]