

Abstract
Glucocorticoids are commonly used in treating diseases with white matter lesions, including demyelinating diseases and spinal cord injury (SCI). However, glucocorticoids are ineffective in gray matter injuries, such as head injury and stroke. The differential glucocorticoid effects in white and gray matter injuries are unclear. We report here a novel mechanism of methylprednisolone (MP), a synthetic glucocorticoid widely used for treating multiple sclerosis and SCI, in protecting oligodendrocytes (OLGs) against AMPA-induced excitotoxicity, which has been implicated in the white matter injuries and diseases. The cytoprotective action of MP in OLGs is causally related to its upregulation of a neuroprotective cytokine erythropoietin (Epo). MP transactivation of Epo expression involves dual transcription factors: glucocorticoid receptor (GR) and hypoxia-inducible factor-1α (HIF-1α). Coimmunoprecipitation, chromatin immunoprecipitation analysis, yeast two-hybrid analysis, and structure modeling of three-dimensional protein–protein interactions confirm that MP induces interaction between GR DNA binding domain and HIF-1α PAS domain, with subsequent recruitment of HIF-1β to transactivate Epo expression in OLGs. In contrast, MP activates GR but does not induce GR–HIF-1α interaction, HIF-1α binding to Epo enhancer/promoter, or Epo expression in cultured cortical neurons. The OLG-specific GR–HIF-1α transactivation of Epo provides novel insights into the development of more effective therapies for diseases affecting the white matter.
Introduction
Neuroprotective strategies for treating CNS disorders have been directed mostly at preserving gray matter or neurons (Lipton, 2006). However, white matter injuries, as seen in demyelinating diseases, including multiple sclerosis (MS) and spinal cord injury (SCI), may have an even greater impact on functional outcomes (Bradbury et al., 2002; Stys and Lipton, 2007). For example, disability after SCI is primarily caused by the white matter lesion (Wrathall et al., 1994; Crowe et al., 1997). Gray matter injury, sparing most white matter (e.g., central cord syndrome), results in segmental motor/sensory dysfunction. Disability after SCI can be mostly ascribed to injury to the white matter, which contains the long tracts. Death of oligodendrocytes (OLGs), which form myelin sheath and maintain long tract function in the CNS, is a prominent pathological feature in demyelinating diseases and SCI (Crowe et al., 1997; Miller and Mi, 2007). OLG death in these disorders results, in part, from excitotoxicity involving AMPA receptor (McDonald et al., 1998; Smith et al., 2000; Miller and Mi, 2007). AMPA receptor antagonism is effective in salvaging white matter after traumatic or ischemic SCI (Wrathall et al., 1994; Kanellopoulos et al., 2000).
Glucocorticoids (GCs), primarily methylprednisolone (MP), are the mainstay in treating patients with diseases affecting the white matter, including MS and SCI (Bracken et al., 1990; Zivadinov et al., 2001; Chataway et al., 2006). GCs regulate transcriptional events by activating glucocorticoid receptor (GR), which binds to glucocorticoid response element (GRE) or interacts with other transcription factors to regulate gene expression (Becker et al., 1986). GR modulation of inflammatory/immune or oxidative processes are the proposed mechanisms of MP action in demyelinating diseases and SCI (Schmidt et al., 2000; Xu et al., 2001a; Tsutsui et al., 2008). However, GC or antioxidant therapies were ineffective in gray matter injuries, such as head trauma or stroke (Edwards et al., 2005; Diener et al., 2008). The mechanism underlying preferential effects of GCs in the white, but not gray, matter remains to be defined.
MP upregulates antiapoptotic Bcl-xL gene expression to protect OLGs against AMPA-induced death in vitro and in a rat SCI model in vivo (Lee et al., 2008). Bcl-xL expression is also upregulated by erythropoietin (Epo)/Epo receptor (Silva et al., 1999). Epo is effective in animal models of SCI and MS (Gorio et al., 2002; Li et al., 2004). Epo upregulation involves activation of hypoxia-inducible factor-1α (HIF-1α) (Semenza et al., 1991; Liu et al., 2005), a member of the PAS (PER/ARNT/SIM) family of basic helix–loop–helix (bHLH) transcription factors essential in hypoxia sensing for cell survival (Sharp and Bernaudin, 2004; Semenza, 2009). During activation, HIF-1α translocates into the nucleus and heterodimerizes with aryl hydrocarbon receptor nuclear translocator (ARNT), also known as HIF-1β, to bind to hypoxia-responsive elements (HRE) in the 3′ enhancer of the Epo gene to induce Epo expression (Semenza et al., 1991).
We report here Epo transactivation involving GR and HIF-1α interaction is a causal mechanism of MP protection of OLGs, but not neurons, against AMPA excitotoxicity. These findings offer novel insights into the development of innovative therapies for white matter diseases.
Materials and Methods
Animals.
Sprague Dawley rats were obtained from the National Institute of Experimental Animal Research (Taipei, Taiwan). Animals were killed by an overdose of sevoflurane to minimize pain or discomfort. All animal experimentation procedures were approved by the Experimental Animal Review Committee at Taipei Medical University and are in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals.
Cell cultures.
OLGs in culture were prepared from postnatal day 0–2 neonatal rat brains (Xu et al., 2001b; Lee et al., 2004). OLG purity was >90% based on receptor interacting protein (RIP) immunoreactivity (Xu et al., 2001b; Lee et al., 2004). Cortical neurons were prepared from fetal rat brains at embryonic day 17 as described previously with ∼85% cells in culture expressing the neuronal marker MAP-2 (Lee et al., 2008). Cortical neurons at 10 d in vitro (DIV) that express GR were used in this study.
Quantitative real-time and semiquantitative reverse transcription-PCR.
Total RNA extraction, cDNA preparation, and reverse transcription (RT)-PCR were as described previously (Lee et al., 2008). The real-time PCR reaction was performed using an ABI PRISM 7300 Sequence Detection System. The semiquantitative PCR of Epo, Nr3c1 (GR), Hif1a (HIF-1α), Arnt (HIF-1β), and Bcl-xL cDNAs was performed using the following primers, and cDNA of a reference gene Gapdh (glyceraldehyde-3-phosphate dehydrogenase) was measured in parallel as an internal control: Epo, 5′-TGCGACAGTCGCGTTCTGGAGAGGTAC-3′ and 5′-ATCCGCTGTGAGTGTTCGGAGTGGAGC-3′; Nr3c1 (GR), 5′-AGAATGTCTCTACCCTGCATGTATGAC-3′ and 5′-CTGGAAGCAGTAGGTAAGGAGATTCTC-3′; Hif1a (HIF-1α), 5′-CAAGATCAGCCAGCAAGTCCTTCTGATG-3′ and 5′-AGGTTTCTGTAACTGGGTCTGCTGGAATC-3′; Arnt (HIF-1β) 5′-CTGTTGTCCAGAGGGCTATTAAG-3′ and 5′-CAAGTCTCTCTTTATCGGCAGAG-3′; Bcl-xL, 5′-AGGCTGGCGATGAGTTTGAA-3′ and 5′-TGAAACGCTCCTGGCCTTTC-3′; and Gapdh, 5′- GACCCCTTCATTGACCTCAAC-3′ and 5′- GATGACCTTGCCCACAGCCTT-3′.
SCI animal model.
SCI was conducted using a New York University Impactor (Hashimoto et al., 2007) to injure rat spinal cord at T9–T10 (Lee et al., 2008). Sprague Dawley rats (9 weeks old) were randomly divided into three groups: the sham control, SCI, and SCI with MP treatment groups. Vehicle or MP (30 mg/kg body weight) was administered intravenously 30 min after SCI. Four and 24 h after treatment, animals were anesthetized, and a 1 cm segment of the injured cord centering at the injury epicenter or corresponding sham-operated cord tissue was collected for quantitative real-time RT-PCR analysis.
Western blot analysis.
Western blot analysis was performed as described previously (Xu et al., 2001b; Lee et al., 2008). Primary antibodies used were rabbit anti-Bcl-xL (1:500; Santa Cruz Biotechnology), rabbit anti-GR (1:500; Santa Cruz Biotechnology), mouse anti-HIF-1α (1:500; Novus), mouse anti-HIF-1β (1:1000; Millipore Bioscience Research Reagents), and mouse anti-GAPDH (1:5000; Biogenesis) antibodies. The immune complex was visualized using HRP-reactive ECL reagents (GE Healthcare).
RNA knockdown.
OLGs were transfected with a set of siRNAs specific for GR, HIF-1α, HIF-1β, Epo, or scrambled RNA (Silencer Predesigned siRNA; Ambion) using Lipofectamine reagent (Invitrogen). The siRNA sequences were as follows: GR (Nr3c1; GenBank accession number NM_012576), 5′-GGUUGCUUAAAGAAAGUUtt-3′ (exon 9) and 5′-GCUACAGUCAAGGUUUCUGtt-3′ (exon 1); HIF-1α (Hif1a; GenBank accession number NM_024359), 5′-GCUUGCUCAUCAGUUGCCAtt-3 (exon 2) and 5′-CCAGUUGAAUCUUCAGAUtt-3′ (exon 9); HIF-1β (Arnt; GenBank accession number NM_012780), 5′-GGCUGGAUUUUGAUGAUGAtt-3′ (exon 2) and 5′-CGGCCUACAUUGUCUAACAtt-3′ (exon 15); and Epo (GenBank accession number NM_017001), 5′-GGAGGCAGAAAAUGUCACAtt-3′ (exon 2) and 5′-GGCUGUAGAAGUUUGGCAAtt-3′ (exon 4). Five hours after transfection, the Lipofectamine-containing medium was replaced with OLG culture medium to allow cells to recover for another 67 h.
Luciferase activity assay.
Three copies of human EPO gene enhancer (HREEPO)-containing sequences, 5′-ATAGGTACCGCCCTACGTGCTGTCTCAGCCCTACGTGCTGTCTCAGCCCTACGTGCTGTCTCAGCTAGCTAT-3′, were cloned into promoter of pGL2 promoter vector (Promega) to obtain the luciferase reporter construct pHREEPO–Luc. The cells were transfected with pHREEPO–Luc and pRL–TK (Promega) in Lipofectamine for 36 h, followed by treatments with various agents for 24 h, and then harvested for the luciferase activity assay using the Dual-Luciferase reporter assay system (Promega).
Coimmunoprecipitation.
Nuclear fraction of OLGs was extracted using NE-PER Nuclear and Cytoplasmic Extraction Reagents (Pierce). Nuclear proteins (50 μg) were incubated with mouse anti-GR antibody (2 μg; Abcam) or mouse anti-HIF-1α antibody (2 μg; Novus) for 12 h at 4°C, followed with protein G Sepharose for 1 h at 25°C. The immune complex was then washed and dissociated according to the Protein G Sepharose 4 Fast Flow protocol (GE Healthcare). Dissociated immune complex was then subjected to Western blotting using rabbit anti-GR (1:500; Santa Cruz Biotechnology), mouse anti-HIF-1α (1:500; Novus), and mouse anti-HIF-1β (1:500; Millipore Bioscience Research Reagents) antibodies, respectively.
Yeast two-hybrid assay.
Yeast two-hybrid assay was performed as described previously with minor modifications (Wang et al., 2004). The plasmid pGBKT7–mHIF-1α PAS-B was constructed by cloning a PAS domain (amino acids 238-346) of HIF-1α into pGBKT7 (Clontech). pGADT7–GR–DNA binding domain (DBD) (amino acids 436-516) and pGADT7–GR–ligand binding domain (LBD) (amino acids 601-751) were cloned by inserting DNA fragments of GR derived from pcDNA3.1–rGR (pcDNA3.1–rGR kindly provided by Dr. Trevor K. Archer, LMC, National Institute of Environmental Health Sciences, National Institutes of Health, Research Triangle Park, NC) between the NdeI and XhoI sites of pGADT7 (Clontech). Y190 cells were cotransformed with GAL4AD–GR–DBD (pGADT7–GR–DBD) and GAL4BD–HIF-1α PAS-A (pGBKT7–HIF-1α PAS-A) or with GAL4AD–GR–LBD (pGADT7–GR–LBD) and GAL4BD–HIF-1α PAS-B (pGBKT7–HIF-1α PAS-B) based plasmids. The transformed cells were plated onto a 25 mm 3-amino-1,2,4-triazole (3-AT) containing SD medium (Synthetic Dropout) lacking tryptophan, histidine, and leucine. Five independent colonies were inoculated to examine β-galactosidase activity using o-nitrophenyl-β-d-galactopyranoside as the substrate with A420nm measured in the liquid culture assay. The β-galactosidase unit was calculated by the following equation: β-galactosidase units = 1000 × A420/(t × V × A600), where t is elapsed time (minutes) of incubation, V is 0.1 ml × concentration factor, and A600 the density of yeast culture.
Chromatin immunoprecipitation assay (Lin et al., 2008).
Antibodies used for immunoprecipitation were mouse anti-GR (2 μg; Abcam), mouse anti-HIF-1α (2 μg; Novus), and rabbit anti-CREB (cAMP response element-binding) protein binding protein (CBP) (2 μg; Santa Cruz Biotechnology) antibodies. Immune complex-bound DNA was purified using a PCR purification kit (Qiagen) and amplified by quantitative PCR (Applied Biosystems) with primers flanking an HRE-containing Epo gene 3′ enhancer fragment: (+3497 to +3618; GenBank accession number NM_017001) 5′-TACCTCCCCCCCCCCCCATTCTGGT-3′ and 5′-CAAGCCCAGAGGGGTCAAGAGGTCAGA-3′. Primers flanking an Epo gene promoter fragment (−375 to −221; GenBank accession number NM_017001) were 5′-CAGCCTGCTCTACCCCAGCAAGGA-3′ and 5′-GGGGGTCGGGGATGTTATCAGCA-3′. Primers flanking a GRE-containing Trh (thyrotropin releasing hormone) gene promoter fragment (−267 to −71; GenBank accession number NM_013046) were 5′-CGCGGACTACTCCCGGGACGTCTCT-3′ and 5′-GGGGAGGGGGCGCAGGCCGAAGACA-3′. Normal mouse IgG served as the control.
Computer modeling of protein–protein interaction.
The structures of GR and HIF-1α were obtained as described below for the predictive structural analysis of GR–HIF-1α interaction. For GR, the structure of GR–DBD (amino acids 439-510) is available [Protein Data Bank (PDB) identification number 1GDC], and GR–LBD (amino acids 519-795) was generated by SWISS-MODEL Repository (Schwede et al., 2003) using the protein template PDB identification numbers 1 NHZ, 1M2Z, and 1P93. No crystal structure information of HIF-1α is available. We used conserved domain search of National Center for Biotechnology Information BLASTp (protein–protein basic local alignment search tool) for sequence domain analysis. The PAS-B domain of HIF-1α was predicted by sequence homology (PDB identification numbers 1P97, A24A, A24B, 1XO0, and 1WA9). Furthermore, fold recognition and three-dimensional annotation were conducted to obtain a predicted three-dimensional structure. Prediction of interactions of the GR–DBD and GR–LBD with the PAS-B domain of the HIF-1α was then performed using the program GRAMM (Vakser, 1996). In addition, we predicted the interaction of HIF-1α and HIF-1β using the crystal structures of HIF-2α–ARNT PAS-B domain complex (PDB identification number 2A24) as a template, followed by predicting the interaction of GR–DBD and GR–LBD with the HIF-1α–HIF-1β complex. Finally, the DNA binding capability of GR–DBD in docking into the HIF-1α or HIF-1α–HIF-1β complex was predicted.
Statistic analysis.
Data were expressed as means ± SEM. Statistical analysis was performed by one-way ANOVA, followed by Newman–Keuls multiple comparison post hoc test to compare all groups with the control group or to compare designated pairs of groups. p < 0.05 was considered significant.
Results
MP upregulation of Epo expression in OLGs but not cortical neurons
We noted in a recent study that neuroprotective effect of MP is restricted to OLGs, but not neurons, in the injured spinal cord (Lee et al., 2008). MP or other GCs are effective in treating white (Zivadinov et al., 2001; Chataway et al., 2006), but not gray (Edwards et al., 2005; Diener et al., 2008), matter diseases. Epo has similar effects in SCI (Gorio et al., 2002; Vitellaro-Zuccarello et al., 2007) and the white matter diseases (Li et al., 2004). These findings suggest MP and Epo may share a common neuroprotective mechanism. Figure 1A shows that MP upregulated Epo mRNA expression in OLGs, but not neurons, based on semiquantitative (top panel) and quantitative (bottom panel) RT-PCR analysis. Epo transactivation has been shown to involve HIF-1α, a cytoprotective transcription factor, inducible by hypoxia (Semenza et al., 1991; Sharp and Bernaudin, 2004; Semenza, 2009). Cobalt chloride (CoCl2) activates HIF-1α, mimicking hypoxia. Whereas MP failed to upregulate Epo expression in neurons, chemical hypoxia induced by CoCl2 was effective in neurons as well as OLGs (Fig. 1A, top panel). This finding suggests that neuronal cultures used in the present study maintain a hypoxia-inducible response for upregulating Epo expression but do not share MP responsiveness noted in OLGs. The ELISA results show that MP increased the Epo protein content in OLGs in a dose-dependent manner with an EC50 of 83.11 nm (Fig. 1B).
Figure 1.
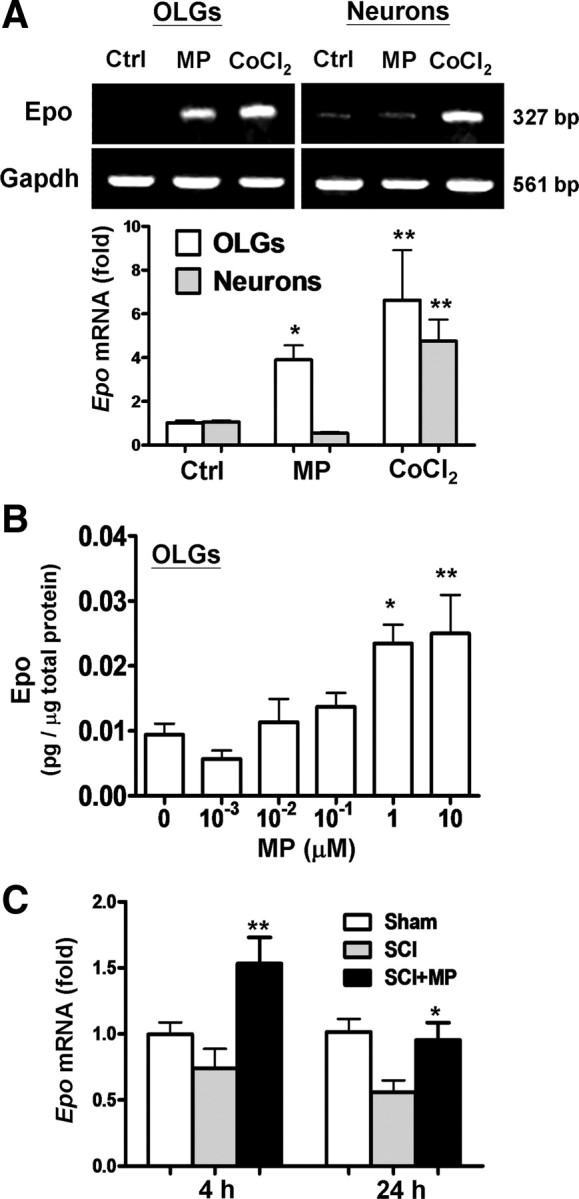
MP upregulation of Epo expression in OLGs and injured spinal cord but not neurons. A, OLGs and 10 DIV cortical neurons were treated with MP (1 μm) or CoCl2 (0.4 mm) for 12 h, followed by RNA extraction. Epo mRNA was analyzed by semiquantitative (top) and quantitative real-time (bottom) RT-PCR, in which Gapdh and β-actin mRNA served as internal controls, respectively. B, Dose–response effect of MP in inducing Epo expression in OLGs was measured by ELISA at 24 h. C, MP (30 mg/kg body weight, i.v.) was given 30 min after SCI. The spinal cord tissues harvested 4 and 24 h after SCI were subjected to quantitative real-time RT-PCR for assessing the extent of Epo mRNA expression. The Ct value was normalized and expressed relative to the sham-operated control (Sham). Data are expressed as means ± SEM (n = 4 in A and B; n = 5 in C). *p < 0.05, **p < 0.01 versus the control (Ctrl) (A), the 0 μm MP group (B), or the SCI group (C) by one-way ANOVA and Newman–Keuls multiple comparison post hoc test.
MP upregulation of Epo expression in the injured spinal cord in vivo
We further examined whether MP treatment affects Epo gene expression in the injured spinal cord in rats. Figure 1C shows MP upregulation of Epo mRNA expression in the spinal cord 4 and 24 h after injury with a dosing schedule (intravenous MP, 30 mg/kg body weight given 30 min after insult) that has been shown to reduce the extent of OLG apoptosis and white matter damage after SCI (Lee et al., 2008). Results suggest that MP-induced Epo expression occurs not only in cultured OLGs in vitro but also in the injured spinal cord in vivo.
Causal role of Epo expression in MP protection of OLGs against excitotoxicity
To establish the causal role of Epo expression in MP protection of OLGs against excitotoxicity, we first studied whether exogenous Epo shares a similar cytoprotective action as MP in OLGs. Both EPO (Silva et al., 1999) and MP (Xu et al., 2009) engage the JAK/STAT5 pathway in upregulating Bcl-xL, an antiapoptotic gene. Both human recombinant EPO (rhEPO, 40 ng/ml) and MP upregulated Bcl-xL expression in AMPA-treated OLGs (Fig. 2A), sharing a common cytoprotective mechanism. Application of rhEPO also protected OLGs against AMPA-induced death (Fig. 2B). To confirm the causal role of Epo expression in MP protection of OLGs, we applied Epo small interfering (si) RNA (siEpo) to knockdown Epo expression (Fig. 2C, inset). The cytoprotective action of MP was abolished in OLGs transfected with siEpo but not scrambled RNA (Fig. 2C). These findings support the contention that Epo expression is causally related to MP protection of OLGs against AMPA excitotoxicity.
Figure 2.

Epo and MP in AMPA-induced OLG death. A, OLGs were treated with AMPA (100 μm) supplemented with cyclothiazide (25 μm) (referred to as AMPA hereafter) with rhEPO (4 U/ml, 40 ng/ml) (left) or MP (right) for 24 h, followed by Western blotting for Bcl-xL. B, rhEPO effect on OLG viability after AMPA treatment based on MTT assay. C, Epo mRNA expression after OLG transfection with scrambled RNA (Scr) or siRNA specific for rat Epo mRNA (siEpo) (top). Effects of Scr or siEpo transfection on MP protection of OLGs against AMPA cytotoxicity (bottom). *p < 0.05, **p < 0.01 versus the control group; #p < 0.05 versus the AMPA-treated group. NS, No significant difference between the two groups (n = 4).
GR-dependent activation of HIF-1α by MP
Transactivation of Epo gene entails the activation of HIF-1α to bind to HRE in the 3′ Epo enhancer region (Semenza et al., 1991). Therefore, we examined whether MP upregulation of Epo expression is mediated by HIF-1α activation. We established a luciferase reporter construct by inserting an HRE-containing DNA fragment of the Epo gene enhancer into a pGL2-promoter vector (pHREEPO–Luc) to study the effect of MP on Epo expression in relation to HIF-1α activity. Treatment with 1 μm MP increased HREEPO-driven luciferase activity in OLGs but not neurons (Fig. 3A), whereas chemical hypoxia (CoCl2) induced a similar degree of HREEPO-driven luciferase activity in both cell types (Fig. 3B), confirming that this expression construct is responsive to an established HIF-1α activation mechanism. Figure 3C shows that dexamethasone, a potent GR agonist, increased HREEPO-driven luciferase activity, comparable with MP. Non-GC steroids, such as aldosterone, a mineralocorticoid receptor (MR) agonist, or 17β-estradiol, an estrogen receptor agonist, were without effects. GR-dependent action of MP was further confirmed by the findings that RU486 [11β-[p-(dimethylamino) phenyl]-17β-hydroxy-17-(1-propynyl)estra-4,9-dien-3-one] (a GR antagonist), but not spironolactone (an MR antagonist), was effective in blocking MP activation of HIF-1α at the basal level (Fig. 3D) or after AMPA treatment (Fig. 3E).
Figure 3.

MP effect on HREEPO-driven luciferase activity in OLGs. OLGs or neurons were cotransfected with pHREEPO–Luc and pRL–TK for 36 h before the respective treatments, followed by the luciferase activity assay. A, OLGs and neurons were with or without MP (1 μm) treatment for 24 h. B, OLGs and neurons were treated with CoCl2 (0.4 mm) for 24 h. Note that both OLGs and neurons are responsive to chemical hypoxia induced by CoCl2. C, OLGs were treated with steroids, including MP, dexamethasone (Dex), aldosterone (Ald), and 17-β-estradiol (E2), 1 μm each for 24 h. D, OLGs were pretreated with 1 μm RU486 (RU; a GR antagonist) or spironolactone (SP; an MR antagonist) for 30 min, followed by MP (1 μm) for 24 h. E, OLGs were pretreated with RU486 (1 μm) for 30 min, followed by AMPA with or without MP. **p < 0.01 versus vehicle-treated control (Ctrl) (n = 4).
Reversal of MP activation of pHREEPO–Luc and upregulation of Epo expression by GR or HIF-1α knockdown
To confirm the causal role of GR and HIF-1α in Epo expression and subsequent Bcl-xL upregulation by MP, we transfected OLGs with specific siRNAs against Nr3c1 (siGR) or Hif1a (siHif1a) to knockdown GR and HIF-1α expression, respectively. Both mRNA and protein levels of GR and HIF-1α were reduced by their specific siRNA (supplemental Fig. 1A,B, available at www.jneurosci.org as supplemental material). To verify that siHif1a indeed reduces HIF-1α at the protein level, we used a strategy reported previously (Siddiq et al., 2009) to confirm the knockdown efficiency of siHIF1a in hypoxia-treated OLGs (supplemental Fig. 1C, available at www.jneurosci.org as supplemental material). GR or HIF-1α knockdown abolished MP activation of pHREEPO–Luc (Fig. 4A), MP upregulation of Epo (Fig. 4B) and Bcl-xL (Fig. 4C), and the protective effect of MP against AMPA-induced cell death in OLGs (Fig. 4D). HIF-1α knockdown even accentuated AMPA excitotoxicity (Fig. 4D). These results support the causal role of GR and HIF-1α in mediating the cytoprotective action of MP via upregulation of Epo expression in OLGs.
Figure 4.

HIF-1α and GR knockdown on MP-induced HREEPO enhancer activity and Epo/Bcl-xL expression. OLGs were transfected with scrambled siRNA (Scr), siNr3c1 (for GR), or siHif1a (for HIF-1α) for 72 h, followed by: A, transfection with pHREEPO–Luc for 24 h and then MP (1 μm) treatment for 24 h, followed by luciferase activity assay; B, quantitative real-time RT-PCR analysis of Epo mRNA; C, semiquantitative RT-PCR analysis of Bcl-xL mRNA; D, AMPA treatment with or without MP (1 μm) for 24 h for cell death analysis by LDH release assay. *p < 0.05, **p < 0.01 versus vehicle-treated control (Ctrl); #p < 0.05 versus the AMPA-treated group (n = 4).
MP enhancement of nuclear GR–HIF-1α interaction in OLGs
GR may regulate gene expression by interacting with other transcription factors (Xu et al., 2009). We explored whether GR interacts with HIF-1α directly in MP transactivation of Epo gene expression. The blotting pattern of HIF-1α, verified by CoCl2 (supplemental Fig. 1C, available at www.jneurosci.org as supplemental material), was used to identify HIF-1α in the coimmunoprecipitation (co-IP) study. MP treatment increased nuclear HIF-1α accumulation in OLGs but not neurons (Fig. 5A). The GR content in the nuclear fraction was increased in OLGs as well as neurons after MP treatment (Fig. 5B). However, an increase in GR–HIF-1α interaction after MP treatment was noted only in OLGs but not neurons when immunoprecipitating the nuclear protein extracted from the MP-treated cells with a GR antibody and blotting with an HIF-1α antibody (Fig. 5B). The MP-increased GR–HIF-1α interaction in OLGs was further verified by using immunoprecipitation with an HIF-1α antibody and blotting with a GR antibody (see Fig. 7B). This result is in agreement with previous findings that MP transactivates Epo expression only in OLGs but not neurons.
Figure 5.

MP activation of GR–HIF-1α interaction in OLGs but not neurons. A, Immunoblotting of nuclear HIF-1α with or without MP treatment in OLGs and neurons. B, Nuclear fractions of OLGs and neurons were immunoprecipitated with an anti-GR antibody (IP-GR) and Western blotted with an anti-HIF-1α or anti-GR antibodies. IgG served as the loading control for the immunoprecipitated complex for quantitative analysis of band intensity. The right shows composite results. **p < 0.01 versus the control (0 min) group, n = 3. C, Domain maps of GR and HIF-1α. Major domains of GR include AF-1 activation domain, DBD and LBD; major domains of HIF-1α include bHLH, PAS-A, PAS-B, and oxygen-dependent degradation (ODD) domain. The shaded domains in gray were used for yeast two-hybrid analysis and computer modeling for protein–protein docking. D, Yeast two-hybrid analysis of GR–HIF-1α interaction. Left, GAL4-activation domain (GAL4AD, amino acids 768-881) hybrid and GAL4–DNA-binding domain (GAL4BD, amino acids 1-147) hybrid. Right, Yeast colony numbers after transformation and β-galactosidase activity (n = 5). N.D., Not detectable.
Figure 7.

HIF-1β interaction with the GR–HIF-1α complex is required for the MP-induced Epo expression in OLGs. A, OLGs were transfected with scrambled siRNA (Scr) and siArnt (for HIF-1β) for 72 h, followed by incubation with MP (1 μm) or CoCl2 (0.4 mm) for 12 h and real-time PCR analysis of Epo mRNA. **p < 0.01, ***p < 0.001 versus vehicle-treated control (Ctrl); #p < 0.05 versus the MP- or CoCl2-treated group (n = 3). B, Nuclear fractions of OLGs were immunoprecipitated with an anti-HIF-1α antibody (IP-HIF-1α) and Western blotted (WB) with an anti-GR or anti-HIF-1β antibody. IgG served as the loading control for the immunoprecipitated complex. The right shows composite results. *p < 0.05 versus control; n = 3.
Co-IP of two proteins can result from either direct or indirect interaction. We conducted yeast two-hybrid analysis to explore possible direct interaction between GR and HIF-1α. Based on the domain maps of GR and HIF-1α (Fig. 5C), we examined the interactions between GR–LBD and GR–DBD, respectively, with the HIF-1α PAS-A (amino acids 85-153) and PAS-B (amino acids 238-346) domains. The GR–DBD, but not the GR–LBD, yielded appreciable number of colonies when hybridized with the HIF-1α PAS-A or PAS-B domain (Fig. 5D). Results suggest that the GR–DBD, but not the GR–LBD, directly interacts with the HIF-1α PAS domains.
Co-recruitment of HIF-1α and GR onto the promoter and 3′ enhancer of the Epo gene in OLGs but not neurons
Because MP induces GR–HIF-1α interaction (Fig. 5), we examined whether the GR–HIF-1α complex is recruited to the promoter or/and 3′ enhancer of the Epo gene during MP treatment. Chromatin immunoprecipitation (ChIP) analysis was performed by immunoprecipitating nuclear HIF-1α or GR, followed by PCR amplification of promoter fragments spanning from −375 to −221 and HRE-containing 3′ enhancer fragments spanning from +3497 to +3618 of the Epo gene, respectively (Fig. 6A). Preferential effects of MP on the GR–HIF-1α association with the Epo promoter and enhancer were observed in OLGs (Fig. 6B) compared with neurons (Fig. 6C). In OLGs but not neurons, the association of HIF-1α and GR to the Epo promoter was increased to threefold at 1 and 3 h after MP treatment (Fig. 6B). Interestingly, GR and HIF-1α binding to the 3′ enhancer was not significant until 3 h after MP treatment, when ∼2.5-fold and 7-fold increase was noticed compared with basal binding activity (0 h), respectively. In neurons, HIF-1α had a slight and transient association with the Epo enhancer after MP treatment. No significant association of HIF-1α with the promoter was observed in neurons. More importantly, no significant GR binding to either the promoter or 3′ enhancer was noted in neurons (Fig. 6C). As a positive control, MP-induced GR binding to a GRE-containing fragment in the promoter of Trh, an established GC-responsive gene, was examined in both OLGs and neurons. As shown in Figure 6D, MP induced GR binding to GRE to a similar extent in both cell types. These findings suggest that co-recruitment of HIF-1α and GR to the Epo promoter/enhancer occurs in OLGs but not in neurons. The association of GR and HIF-1α with the Epo promoter and enhancer in OLGs appears sequential, occurring at 1 and 3 h, respectively, after MP treatment.
Figure 6.

MP recruitment of HIF-1α and GR onto the Epo promoter and 3′ enhancer in OLGs but not neurons. A, Schematic representation of the rat Epo gene promoter and enhancer. The Epo enhancer contains an HIF-1α binding site (HRE). OLGs and neurons were treated with MP (1 μm) for 0, 1, and 3 h, followed by ChIP assay for the GR- and HIF-1α–Epo promoter and enhancer in OLGs (B) and neurons (C). ChIP assay of untreated cells with normal IgG served as a negative control in each set of experiments. D, ChIP assay of the GRE-containing Trh gene promoter fragment (GRETrh) served as a GR activation control. E, OLGs and neurons were treated with MP (1 μm) for 0, 1, and 3 h, followed by ChIP assay for the CBP–Epo promoter. *p < 0.05, **p < 0.01 versus the respective group without MP treatment; n = 4.
The transcriptional activation of Epo gene by HIF-1α requires a DNA looping mechanism that brings the HIF-1α/HIF-1β-bound 3′ enhancer to contact the promoter of Epo gene by interacting with CBP/p300 (Ebert and Bunn, 1998), which serves as a coadaptor to activate an RNA polymerase II-containing general transcription factor complex to initiate gene transcription (von Mikecz et al., 2000). We conducted anti-CBP antibody-based ChIP assay to examine whether MP treatment recruits CBP/p300 to the Epo promoter in OLGs and neurons. Figure 6E shows that CBP association with the Epo promoter was increased by approximately sixfold after MP treatment for 1 h in OLGs but not neurons, when HIF-1α and GR were detected in the same promoter region (Fig. 6B). This finding suggests that MP-induced HIF-1α–GR association with the Epo gene promoter is accompanied by the recruitment of CBP/p300 to the promoter to trigger Epo gene transcription.
HIF-1β association with GR–HIF-1α complex is required for the Epo induction by MP treatment
MP-induced GR–HIF-1α binding to the 3′ enhancer of Epo gene correlated well with its activation of HREEpo–Luc, each requiring binding of both HIF-1α and HIF-1β (Jiang et al., 1996). Knockdown of HIF-1β using a specific siRNA (supplemental Fig. 1A, available at www.jneurosci.org as supplemental material) suppressed Epo expression induced by MP or CoCl2 (Fig. 7A). Furthermore, the co-IP results show that MP significantly increased HIF-1β content to the HIF-1α-immunoprecipitated complex to a similar degree as GR (Fig. 7B). Together, these results suggest that recruitment of HIF-1β to the GR–HIF-1α complex is required for MP transactivation of Epo expression.
Structural analysis of interactions among GR, HIF-1α, HIF-1β, and DNA
To understand the interaction of the HIF-1α PAS domain with the GR–DBD and their relationship with the interaction of HIF-1α and HIF-1β, which is required for HIF-1α binding to HRE, we applied a computational modeling approach to perform a structural analysis of protein–protein interactions among GR, HIF-1α, and HIF-1β. The structures of the GR–DBD and GR–LBD were retrieved from protein data banks (PDB identification numbers 1GDC and 1NHZ, respectively), whereas those of HIF-1α were predicted by homology to obtain a structure for the PAS-B domain at amino acids 238-346. Based on thermal dynamics and intermolecular distances, the modeling results show that the GR–DBD binds HIF-1α on the PAS-B domain while retaining its DNA binding activity (Fig. 8A). Furthermore, the seven residues of the GR–DBD (Gly470, His472, Tyr497, Arg498, Gln502, Asn506, and Glu508) interact with Tyr277, Asp250, Leu249, Glu334, Asn330, Thr297, and Asp294 of the HIF-1α PAS-B domain, respectively. We modeled the structure of the HIF-1α–HIF-1β complex (Fig. 8B) to explore its possible interactions with the GR–DBD. Figure 8C shows that the HIF-1α PAS-B complex interacts with the GR–DBD and HIF-1β simultaneously, preserving the DNA binding activity of the GR–DBD. Results from the protein docking analysis are summarized in Figure 8D.
Figure 8.

Computer modeling of protein–protein interactions among HIF-1α PAS-B domain, GR–DBD, and HIF-1β. A, Predicted structure of HIF-1α PAS-B domain docking with the GR–DBD (PDB identification number 1GDC). B, HIF-1α PAS-B domain docking with the HIF-1β PAS-B domain. C, GR–DBD docking with the (HIF-1α PAS-B)–(HIF-1β PAS-B) complex. Insets in A show predicted amino acid residues for GR–DBD interaction with HIF-1α PAS-B. D, Summary of the modeled protein–protein docking results.
Discussion
Results from the present study are the first to demonstrate that MP protects OLGs against excitotoxicity via the HIF-1α/Epo/Bcl-xL cascade in a cell-type-specific manner under normoxic condition. Several lines of experimental evidence support a causal role of this cascade in MP protection against AMPA-induced excitotoxicity in OLGs. First, exogenous Epo is effective in protecting OLGs against AMPA-induced OLG death. Second, both MP and Epo share a similar cytoprotective mechanism by upregulating Bcl-xL expression in AMPA-treated OLGs. Third, MP transactivates Epo expression in a GR-, HIF-1α-, and HIF-1β-dependent manner. Fourth, the causal roles of GR, HIF-1α, and Epo are substantiated by the findings that knockdown of GR, HIF-1α, or Epo expression abolishes the cytoprotective action of MP in OLGs. Finally, MP activation of HIF-1α occurs in a GR-dependent manner involving stabilization of HIF-1α and complex GR–HIF-1α interaction as supported by pHREEPO–Luc studies, co-IP, ChIP assays, the yeast two-hybrid analysis, and computer modeling of protein–protein interactions.
HIF-1α, as its name implies, is induced by hypoxia. Hypoxia does upregulate Epo expression in both OLGs and neurons. However, MP upregulation of Epo and activation of HIF-1α are selective for OLGs but not neurons under normoxic condition. To the best of our knowledge, this is the first report on activation of HIF-1α under normoxic condition by a therapeutic agent MP. HIF-1α was increased in MP-treated OLGs (30 min) (Fig. 5A) before the increase of GR–HIF-1α interaction (60 min) (Fig. 5B), suggesting that MP-activated GR induces HIF-1α activity via both stabilization of and interaction with HIF-1α. It is noted that, after MP treatment in OLGs for 30 min, both HIF-1α accumulation (Fig. 5A) and GR activation (Fig. 5B, right top panel) occur, but the GR–HIF-1α interaction was not apparent until 60 min (Fig. 5B). Lack of an increase in GR–HIF-1α interaction at the 30 min time point reflects the time lag for activated GR to interact with HIF-1α. The sequence of events leading to interaction of GR and HIF-1α entails the recruitment of GR and HIF-1α to form a protein complex as depicted in Figure 8. More importantly, this mechanism well explains the MP protection of OLGs, but not neurons, against AMPA excitotoxicity as shown in our previous studies (Lee et al., 2008). In the present study, we have established the essential roles of GR, HIF-1α, and Epo in MP upregulation of Bcl-xL as the underlying mechanism of MP protection of OLGs against AMPA toxicity normoxia (Fig. 4C,D). Activation of HIF-1α under normoxia has been demonstrated previously in selected experimental paradigms (Choi et al., 2006; Liu et al., 2007) with RACK1 (the receptor for activated C kinase 1) and FIH-1 (factor inhibiting HIF) playing an important role. It remains to be determined whether these factors are involved in the GR activation of HIF-1α.
In contrast to the GR–HIF-1α crosstalk under hypoxic condition in COS7 cells engaging GR–LBD (Kodama et al., 2003), the present study reveals an essential role of GR–DBD in the normoxic mode of GR–HIF-1α interaction. In addition, the binding activity of the GR–DBD to GRE in the GR–HIF-1α–HIF-1β complex is preserved according to the molecular modeling results (Fig. 8A,C) that are in agreement with the finding that mutation of single amino acid (A458T) essential for the DNA binding activity of GR failed to interrupt GR activation of HIF-1α under hypoxia condition (Kodama et al., 2003). However, we noted another seven amino acid residues of the GR–DBD interacting with seven corresponding amino acid residues in the HIF-1α PAS-B domain. This finding raises the possibility that different binding domains (the GR–LBD under hypoxia vs the GR–DBD under normoxia) may be separately engaged depending on oxygen tension. Additional studies are needed to differentiate GR–HIF-1α interactions under oxygen-independent versus oxygen-dependent paradigms involving the GR–DBD or GR–LBD, respectively.
Cell-type-specific activation of HIF-1α in oligodendrocytes, but not neurons, is another novel finding in the present study. The mechanism of this differential action of MP remains to be fully elucidated. Figures 5 (A and B) and 6 (B, C, and E) suggest that the critical signaling event that differentiates oligodendrocytes from neurons in responding to MP is the association of GR with HIF-1α, which occurs in oligodendrocytes but not neurons. MP activation of GR is a universal event in all the GR-expressing cell types, including OLGs and neurons, as demonstrated in Figure 5B. However, MP does not increase HIF-1α in neurons (Fig. 5A), which matches well with the co-IP results that GR–HIF-1α interaction is not increased in neurons (Fig. 5B, right panel). Figure 6B–D also shows that the differential transcriptional events in response to MP are gene specific with Trh, a typical glucocorticoid-responsive gene, responding to MP in both neurons and oligodendrocytes, in contrast to Epo, responsive to MP only in oligodendrocytes but not neurons. The molecular mechanism leading to the divergence in interaction between two transcription factors should be an important topic in future studies. Furthermore, this cell-type-specific effect of MP is reflected by preferential co-recruitment processes of HIF-1α and GR onto the Epo promoter/enhancer and stabilization of HIF-1α association with the Epo promoter/enhancer in OLGs compared with neurons (Fig. 6). It is noted that both Epo promoter and 3′ enhancer regions contain no sequences with appreciable similarity with GRE (GGTACAnnnTGTTCT), suggesting that the GR–HIF-1α complex is likely to activate the Epo gene via binding to HRE but not GRE. Thus, an apparent increase in GRE binding activity shown by electrophoretic mobility shift assay may reflect GR binding to GRE in the promoter region of other genes, such as Trh expressed in both OLGs and neurons, as shown in Figure 6D, than Epo gene. Importantly, the puzzle of why the GR–HIF-1 complex is recruited to the Epo promoter before its 3′ enhancer remains to be unraveled.
One of the interesting features of MP upregulation of Epo expression is that MP treatment activates the expression of HREEpo-driven reporter construct (Fig. 3), which does not contain GRE. Because activation of HRE requires binding of both HIF-1α and HIF-1β (Jiang et al., 1996), the GR–HIF-1α complex will not bind to HRE unless HIF-1β is recruited to the complex. Our result shows that HIF-1β is recruited to the GR–HIF-1α complex during MP treatment, and its expression is essential for the MP-induced Epo expression (Fig. 7). In addition, ChIP assay results demonstrated that both GR and HIF-1α are recruited to HRE-containing 3′ enhancer of Epo gene at 3 h. Therefore, these findings converge to form a conclusion that GR–HIF-1α–HIF-1β complex binding to HRE is required for the MP activation of HRE–Luc as well as Epo gene expression. A deduced model depicted in supplemental Figure 2 (available at www.jneurosci.org as supplemental material) shows that MP induces interactions among GR, HIF-1α, and HIF-1β, followed by binding to the Epo gene promoter and 3′enhancer along with the CBP/p300 coadaptor to activate Epo gene transcription. GR interaction with HIF-1α is the key step in determining the differential MP effects in OLGs versus neurons. This novel signaling process opens a new avenue to treat white matter diseases by targeting OLGs specifically.
GCs, primarily MP, are the mainstay in treating diseases affecting the white matter, including MS and SCI. Because of the very low blood–brain barrier crossing rate (Chen et al., 1996), large doses of GCs are needed for treating these diseases. High doses of GCs frequently lead to serious side effects, including suppression of immune and adrenal function, loss of connective tissue and bone mass, alteration of cognitive or mental function, hypertension, glucose intolerance, and others (McDonough et al., 2008). These side effects prevent administration of MP or other GCs for extended periods of time to achieve optimal therapeutic effects. Epo, a neuroprotective cytokine, has been reported to improve functional recovery in animal models of demyelinating disorders (Li et al., 2004) and SCI (Gorio et al., 2002). However, systemic administration of Epo is also associated with serious side effects, including cardiovascular, hematological, and other disorders (Maiese et al., 2008). A recent Food and Drug Administration preliminary communication suggests that systemic administration of Epo in patients with acute stroke may be associated with a higher 90 d mortality, with death related to intracerebral hemorrhage fourfold higher in Epo-treated patients than those in the placebo group in a clinical trial of Epo. New therapeutic strategies directed at cell-type-specific upregulation of Epo expression are preferred to systemic Epo or MP administration.
In conclusion, this is the first demonstration that MP protection of OLGs against excitotoxic death is mediated by the HIF-1α/Epo/Bcl-xL cascade. This MP effect is specific in OLGs, but not neurons, mainly determined by the cell-type-specific interaction between GR and HIF-1α. This cell-type-specific signaling process provides an important insight into advancing therapeutic strategies for white matter diseases or SCI by OLG-specific transcriptional regulation of Epo expression to avoid serious adverse side effects associated with currently available therapeutic agents.
Footnotes
This work was supported by National Research Genomic Medicine Projects 93GMP002-1, 93GMP002-2, and 93GMP002-5, NSC96-2628-B-038-010-MY3 of National Science Council, the Ministry of Education (Aim for the Top University Plan), and Taiwan Department of Health Clinical Trial and Research Center of Excellence (DOH99-TD-B-111-004) in Taiwan. We thank C. Y. Kuan, J. Ma, and H. T. Chang for critiques.
References
- Becker PB, Gloss B, Schmid W, Strähle U, Schütz G. In vivo protein-DNA interactions in a glucocorticoid response element require the presence of the hormone. Nature. 1986;324:686–688. doi: 10.1038/324686a0. [DOI] [PubMed] [Google Scholar]
- Bracken MB, Shepard MJ, Collins WF, Holford TR, Young W, Baskin DS, Eisenberg HM, Flamm E, Leo-Summers L, Maroon J, Marshall LF, Perot PL, Piepmeier J, Sonntag VK, Wagner FC, Wilberger JE, Winn HR. A randomized, controlled trial of methylprednisolone or naloxone in the treatment of acute spinal-cord injury. Results of the Second National Acute Spinal Cord Injury Study. N Engl J Med. 1990;322:1405–1411. doi: 10.1056/NEJM199005173222001. [DOI] [PubMed] [Google Scholar]
- Bradbury EJ, Moon LD, Popat RJ, King VR, Bennett GS, Patel PN, Fawcett JW, McMahon SB. Chondroitinase ABC promotes functional recovery after spinal cord injury. Nature. 2002;416:636–640. doi: 10.1038/416636a. [DOI] [PubMed] [Google Scholar]
- Chataway J, Porter B, Riazi A, Heaney D, Watt H, Hobart J, Thompson A. Home versus outpatient administration of intravenous steroids for multiple-sclerosis relapses: a randomised controlled trial. Lancet Neurol. 2006;5:565–571. doi: 10.1016/S1474-4422(06)70450-1. [DOI] [PubMed] [Google Scholar]
- Chen TC, Mackic JB, McComb JG, Giannotta SL, Weiss MH, Zlokovic BV. Cellular uptake and transport of methylprednisolone at the blood-brain barrier. Neurosurgery. 1996;38:348–354. doi: 10.1097/00006123-199602000-00023. [DOI] [PubMed] [Google Scholar]
- Choi SM, Choi KO, Park YK, Cho H, Yang EG, Park H. Clioquinol, a Cu(II)/Zn(II) chelator, inhibits both ubiquitination and asparagine hydroxylation of hypoxia-inducible factor-1alpha, leading to expression of vascular endothelial growth factor and erythropoietin in normoxic cells. J Biol Chem. 2006;281:34056–34063. doi: 10.1074/jbc.M603913200. [DOI] [PubMed] [Google Scholar]
- Crowe MJ, Bresnahan JC, Shuman SL, Masters JN, Beattie MS. Apoptosis and delayed degeneration after spinal cord injury in rats and monkeys. Nat Med. 1997;3:73–76. doi: 10.1038/nm0197-73. [DOI] [PubMed] [Google Scholar]
- Diener HC, Lees KR, Lyden P, Grotta J, Davalos A, Davis SM, Shuaib A, Ashwood T, Wasiewski W, Alderfer V, Hårdemark HG, Rodichok L. NXY-059 for the treatment of acute stroke: pooled analysis of the SAINT I and II trials. Stroke. 2008;39:1751–1758. doi: 10.1161/STROKEAHA.107.503334. [DOI] [PubMed] [Google Scholar]
- Ebert BL, Bunn HF. Regulation of transcription by hypoxia requires a multiprotein complex that includes hypoxia-inducible factor 1, an adjacent transcription factor, and p300/CREB binding protein. Mol Cell Biol. 1998;18:4089–4096. doi: 10.1128/mcb.18.7.4089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards P, Arango M, Balica L, Cottingham R, El-Sayed H, Farrell B, Fernandes J, Gogichaisvili T, Golden N, Hartzenberg B, Husain M, Ulloa MI, Jerbi Z, Khamis H, Komolafe E, Laloë V, Lomas G, Ludwig S, Mazairac G, Muñoz Sanchéz Mde L, et al. Final results of MRC CRASH, a randomised placebo-controlled trial of intravenous corticosteroid in adults with head injury-outcomes at 6 months. Lancet. 2005;365:1957–1959. doi: 10.1016/S0140-6736(05)66552-X. [DOI] [PubMed] [Google Scholar]
- Gorio A, Gokmen N, Erbayraktar S, Yilmaz O, Madaschi L, Cichetti C, Di Giulio AM, Vardar E, Cerami A, Brines M. Recombinant human erythropoietin counteracts secondary injury and markedly enhances neurological recovery from experimental spinal cord trauma. Proc Natl Acad Sci U S A. 2002;99:9450–9455. doi: 10.1073/pnas.142287899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto M, Sun D, Rittling SR, Denhardt DT, Young W. Osteopontin-deficient mice exhibit less inflammation, greater tissue damage, and impaired locomotor recovery from spinal cord injury compared with wild-type controls. J Neurosci. 2007;27:3603–3611. doi: 10.1523/JNEUROSCI.4805-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang BH, Rue E, Wang GL, Roe R, Semenza GL. Dimerization, DNA binding, and transactivation properties of hypoxia-inducible factor 1. J Biol Chem. 1996;271:17771–17778. doi: 10.1074/jbc.271.30.17771. [DOI] [PubMed] [Google Scholar]
- Kanellopoulos GK, Xu XM, Hsu CY, Lu X, Sundt TM, Kouchoukos NT. White matter injury in spinal cord ischemia: protection by AMPA/kainate glutamate receptor antagonism editorial comment: protection by AMPA/kainate glutamate receptor antagonism. Stroke. 2000;31:1945–1952. doi: 10.1161/01.str.31.8.1945. [DOI] [PubMed] [Google Scholar]
- Kodama T, Shimizu N, Yoshikawa N, Makino Y, Ouchida R, Okamoto K, Hisada T, Nakamura H, Morimoto C, Tanaka H. Role of the glucocorticoid receptor for regulation of hypoxia-dependent gene expression. J Biol Chem. 2003;278:33384–33391. doi: 10.1074/jbc.M302581200. [DOI] [PubMed] [Google Scholar]
- Lee JM, Yan P, Xiao Q, Chen S, Lee KY, Hsu CY, Xu J. Methylprednisolone protects oligodendrocytes but not neurons after spinal cord injury. J Neurosci. 2008;28:3141–3149. doi: 10.1523/JNEUROSCI.5547-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JT, Xu J, Lee JM, Ku G, Han X, Yang DI, Chen S, Hsu CY. Amyloid-beta peptide induces oligodendrocyte death by activating the neutral sphingomyelinase-ceramide pathway. J Cell Biol. 2004;164:123–131. doi: 10.1083/jcb.200307017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Maeda Y, Yuan RR, Elkabes S, Cook S, Dowling P. Beneficial effect of erythropoietin on experimental allergic encephalomyelitis. Ann Neurol. 2004;56:767–777. doi: 10.1002/ana.20274. [DOI] [PubMed] [Google Scholar]
- Lin CH, Juan SH, Wang CY, Sun YY, Chou CM, Chang SF, Hu SY, Lee WS, Lee YH. Neuronal activity enhances aryl hydrocarbon receptor-mediated gene expression and dioxin neurotoxicity in cortical neurons. J Neurochem. 2008;104:1415–1429. doi: 10.1111/j.1471-4159.2007.05098.x. [DOI] [PubMed] [Google Scholar]
- Lipton SA. Paradigm shift in neuroprotection by NMDA receptor blockade: memantine and beyond. Nat Rev Drug Discov. 2006;5:160–170. doi: 10.1038/nrd1958. [DOI] [PubMed] [Google Scholar]
- Liu J, Narasimhan P, Yu F, Chan PH. Neuroprotection by hypoxic preconditioning involves oxidative stress-mediated expression of hypoxia-inducible factor and erythropoietin. Stroke. 2005;36:1264–1269. doi: 10.1161/01.STR.0000166180.91042.02. [DOI] [PubMed] [Google Scholar]
- Liu YV, Baek JH, Zhang H, Diez R, Cole RN, Semenza GL. RACK1 competes with HSP90 for binding to HIF-1alpha and is required for O(2)-independent and HSP90 inhibitor-induced degradation of HIF-1alpha. Mol Cell. 2007;25:207–217. doi: 10.1016/j.molcel.2007.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maiese K, Chong ZZ, Shang YC. Raves and risks for erythropoietin. Cytokine Growth Factor Rev. 2008;19:145–155. doi: 10.1016/j.cytogfr.2008.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald JW, Althomsons SP, Hyrc KL, Choi DW, Goldberg MP. Oligodendrocytes from forebrain are highly vulnerable to AMPA/kainate receptor-mediated excitotoxicity. Nat Med. 1998;4:291–297. doi: 10.1038/nm0398-291. [DOI] [PubMed] [Google Scholar]
- McDonough AK, Curtis JR, Saag KG. The epidemiology of glucocorticoid-associated adverse events. Curr Opin Rheumatol. 2008;20:131–137. doi: 10.1097/BOR.0b013e3282f51031. [DOI] [PubMed] [Google Scholar]
- Miller RH, Mi S. Dissecting demyelination. Nat Neurosci. 2007;10:1351–1354. doi: 10.1038/nn1995. [DOI] [PubMed] [Google Scholar]
- Schmidt J, Gold R, Schönrock L, Zettl UK, Hartung HP, Toyka KV. T-cell apoptosis in situ in experimental autoimmune encephalomyelitis following methylprednisolone pulse therapy. Brain. 2000;123:1431–1441. doi: 10.1093/brain/123.7.1431. [DOI] [PubMed] [Google Scholar]
- Schwede T, Kopp J, Guex N, Peitsch MC. SWISS-MODEL: an automated protein homology-modeling server. Nucleic Acids Res. 2003;31:3381–3385. doi: 10.1093/nar/gkg520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semenza GL. Regulation of oxygen homeostasis by hypoxia-inducible factor 1. Physiology. 2009;24:97–106. doi: 10.1152/physiol.00045.2008. [DOI] [PubMed] [Google Scholar]
- Semenza GL, Nejfelt MK, Chi SM, Antonarakis SE. Hypoxia-inducible nuclear factors bind to an enhancer element located 3′ to the human erythropoietin gene. Proc Natl Acad Sci U S A. 1991;88:5680–5684. doi: 10.1073/pnas.88.13.5680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharp FR, Bernaudin M. HIF1 and oxygen sensing in the brain. Nat Rev Neurosci. 2004;5:437–448. doi: 10.1038/nrn1408. [DOI] [PubMed] [Google Scholar]
- Siddiq A, Aminova LR, Troy CM, Suh K, Messer Z, Semenza GL, Ratan RR. Selective inhibition of hypoxia-inducible factor (HIF) prolyl-hydroxylase 1 mediates neuroprotection against normoxic oxidative death via HIF- and CREB-independent pathways. J Neurosci. 2009;29:8828–8838. doi: 10.1523/JNEUROSCI.1779-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva M, Benito A, Sanz C, Prosper F, Ekhterae D, Nuñez G, Fernandez-Luna JL. Erythropoietin can induce the expression of bcl-x(L) through Stat5 in erythropoietin-dependent progenitor cell lines. J Biol Chem. 1999;274:22165–22169. doi: 10.1074/jbc.274.32.22165. [DOI] [PubMed] [Google Scholar]
- Smith T, Groom A, Zhu B, Turski L. Autoimmune encephalomyelitis ameliorated by AMPA antagonists. Nat Med. 2000;6:62–66. doi: 10.1038/71548. [DOI] [PubMed] [Google Scholar]
- Stys PK, Lipton SA. White matter NMDA receptors: an unexpected new therapeutic target? Trends Pharmacol Sci. 2007;28:561–566. doi: 10.1016/j.tips.2007.10.003. [DOI] [PubMed] [Google Scholar]
- Tsutsui S, Vergote D, Shariat N, Warren K, Ferguson SS, Power C. Glucocorticoids regulate innate immunity in a model of multiple sclerosis: reciprocal interactions between the A1 adenosine receptor and beta-arrestin-1 in monocytoid cells. FASEB J. 2008;22:786–796. doi: 10.1096/fj.07-9002com. [DOI] [PubMed] [Google Scholar]
- Vakser IA. Low-resolution docking: prediction of complexes for underdetermined structures. Biopolymers. 1996;39:455–464. doi: 10.1002/(SICI)1097-0282(199609)39:3%3C455::AID-BIP16%3E3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- Vitellaro-Zuccarello L, Mazzetti S, Madaschi L, Bosisio P, Gorio A, De Biasi S. Erythropoietin-mediated preservation of the white matter in rat spinal cord injury. Neuroscience. 2007;144:865–877. doi: 10.1016/j.neuroscience.2006.10.023. [DOI] [PubMed] [Google Scholar]
- von Mikecz A, Zhang S, Montminy M, Tan EM, Hemmerich P. CREB-binding protein (CBP)/p300 and RNA polymerase II colocalize in transcriptionally active domains in the nucleus. J Cell Biol. 2000;150:265–273. doi: 10.1083/jcb.150.1.265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang CY, Liang YJ, Lin YS, Shih HM, Jou YS, Yu WC. YY1AP, a novel co-activator of YY1. J Biol Chem. 2004;279:17750–17755. doi: 10.1074/jbc.M310532200. [DOI] [PubMed] [Google Scholar]
- Wrathall JR, Choiniere D, Teng YD. Dose-dependent reduction of tissue loss and functional impairment after spinal cord trauma with the AMPA/kainate antagonist NBQX. J Neurosci. 1994;14:6598–6607. doi: 10.1523/JNEUROSCI.14-11-06598.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Kim GM, Ahmed SH, Xu J, Yan P, Xu XM, Hsu CY. Glucocorticoid receptor-mediated suppression of activator protein-1 activation and matrix metalloproteinase expression after spinal cord injury. J Neurosci. 2001a;21:92–97. doi: 10.1523/JNEUROSCI.21-01-00092.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Chen S, Ahmed SH, Chen H, Ku G, Goldberg MP, Hsu CY. Amyloid-β peptides are cytotoxic to oligodendrocytes. J Neurosci. 2001b;21:RC118. doi: 10.1523/JNEUROSCI.21-01-j0001.2001. (1–5) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Chen S, Chen H, Xiao Q, Hsu CY, Michael D, Bao J. STAT5 mediates antiapoptotic effects of methylprednisolone on oligodendrocytes. J Neurosci. 2009;29:2022–2026. doi: 10.1523/JNEUROSCI.2621-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zivadinov R, Rudick RA, De Masi R, Nasuelli D, Ukmar M, Pozzi-Mucelli RS, Grop A, Cazzato G, Zorzon M. Effects of IV methylprednisolone on brain atrophy in relapsing-remitting MS. Neurology. 2001;57:1239–1247. doi: 10.1212/wnl.57.7.1239. [DOI] [PubMed] [Google Scholar]