

Abstract
In addition to the fundamental role of the extracellular glycoprotein Reelin in neuronal development and adult synaptic plasticity, alterations in Reelin-mediated signaling have been suggested to contribute to neuronal dysfunction associated with Alzheimer's disease (AD). In vitro data revealed a biochemical link between Reelin-mediated signaling, Tau phosphorylation, and amyloid precursor protein (APP) processing. To directly address the role of Reelin in amyloid-β plaque and Tau pathology in vivo, we crossed heterozygous Reelin knock-out mice (reeler) with transgenic AD mice to investigate the temporal and spatial AD-like neuropathology. We demonstrate that a reduction in Reelin expression results in enhanced amyloidogenic APP processing, as indicated by the precocious production of amyloid-β peptides, the significant increase in number and size of amyloid-β plaques, as well as age-related aggravation of plaque pathology in double mutant compared with single AD mutant mice of both sexes. Numerous amyloid-β plaques accumulate in the hippocampal formation and neocortex of double mutants, precisely in layers with strongest Reelin expression and highest accumulation of Reelin plaques in aged wild-type mice. Moreover, concentric accumulations of phosphorylated Tau-positive neurons around amyloid-β plaques were evident in 15-month-old double versus single mutant mice. Silver stainings indicated the presence of neurofibrillary tangles, selectively associated with amyloid-β plaques and dystrophic neurites in the entorhinal cortex and hippocampus. Our findings suggest that age-related Reelin aggregation and concomitant reduction in Reelin-mediated signaling play a proximal role in synaptic dysfunction associated with amyloid-β deposition, sufficient to enhance Tau phosphorylation and tangle formation in the hippocampal formation in aged Reelin-deficient transgenic AD mice.
Introduction
Alzheimer's disease (AD) is characterized by progressive cognitive decline and severe neurodegeneration (Savla and Palmer, 2005). Neuropathological hallmarks include neurofibrillary tangles, consisting of hyperphosphorylated Tau (Grundke-Iqbal et al., 1986) and senile plaques, mainly composed of amyloid-β peptides (Glenner and Wong, 1984), the amyloidogenic cleavage product of amyloid precursor protein (APP) (Estus et al., 1992; Haass et al., 1992). Nonamyloidogenic cleavage produces fragments with neuroprotective and transcriptional functions (Furukawa et al., 1996; Gao and Pimplikar, 2001; von Rotz et al., 2004). Rare early-onset AD is caused by mutations in app or presenilin1/2 genes, all favoring amyloidogenic APP processing (Goate et al., 1991; Levy-Lahad et al., 1995; Rogaev et al., 1995; Sherrington et al., 1995). No apparent familial segregation is evident in the frequent late-onset AD cases. Combinations of genetic, such as apolipoprotein E (ApoE) ε4, and nongenetic risk factors likely determine disease predisposition, age of onset, and progression (Bertram and Tanzi, 2008). The effect of ApoEε4 in late-onset AD neuropathology remains to be elucidated. ApoE isoforms bind to neuronal lipoprotein receptors, involved in a variety of brain functions (Herz, 2009), indirectly implicating ApoE receptors in AD pathophysiology. Two, including very-low-density lipoprotein receptor (VLDLR) and ApoE receptor 2 (ApoER2), bind also the extracellular signaling molecule Reelin, a conserved glycoprotein required for neuronal migration and positioning during development (D'Arcangelo et al., 1995). In adult neurons, Reelin-mediated signaling modulates synaptic NMDA receptor activity and is required for learning and memory (Weeber et al., 2002; Beffert et al., 2005, 2006a; Chen et al., 2005; Qiu et al., 2006; Qiu and Weeber, 2007). In addition to its role in synaptic plasticity and mnemonic functions, Reelin-mediated signaling controls several AD-relevant pathways: it inhibits key mediators of Tau phosphorylation (Hiesberger et al., 1999; Beffert et al., 2002, 2004; Ohkubo et al., 2003) and decreases amyloidogenic APP processing in vitro (Hoe et al., 2006; Hoe and Rebeck, 2008), potentially mediated by direct Reelin–APP interactions (Hoe et al., 2009). In AD patients, increased proteolytic Reelin fragments, Reelin depletion in the entorhinal cortex (Sáez-Valero et al., 2003; Botella-López et al., 2006; Chin et al., 2007), and significant differences in two RELN polymorphisms have been described previously (Seripa et al., 2008).
We have reported in aged rodents and nonhuman primates a pronounced reduction of Reelin-expressing interneurons and concomitant accumulation of Reelin in amyloid-like deposits, colocalizing with proteolytic APP fragments and associated with a reduction in basal forebrain projection neurons that selectively target Reelin-positive hippocampal interneurons (Knuesel et al., 2009; Madhusudan et al., 2009; Doehner et al., 2010). The course and magnitude of the plaque load correlated with cognitive performance and was modulated by exacerbated inflammatory responses and genetic mutations favoring Aβ peptide production (Knuesel et al., 2009). Based on these findings, we hypothesized that reduced Reelin-dependent signaling is a critical upstream factor able to promote amyloidogenic APP processing and Tau hyperphosphorylation. We directly tested this putative association by characterizing AD-like neuropathology in the offspring of transgenic AD mice crossed with heterozygous reeler mice in biochemical, histological, and immunohistochemical experiments across aging.
Materials and Methods
Animals.
All procedures were approved by the local authorities of the Cantonal Veterinary Office in Zurich. All animals were housed in groups of three to four in an optimized in-house hygiene area (Optimierter Hygienebereich, University of Zurich Irchel, Zurich, Switzerland) under 12 h day/night cycle and ad libitum food and water. Heterozygous reeler (B6C3Fe-a/a-Relnrl/+) mice were obtained from The Jackson Laboratory and crossed with transgenic animals expressing the human APP695 gene containing the Swedish (K670N+M671L) and Arctic mutations (E693G) in a C57BL/6 background (Knobloch et al., 2007). The F1 offspring demonstrated Mendelian inheritance, carrying one of the following genotypes at expected frequencies: reln +/+;appwt (wt, 28.6%), reln +/−;appwt (reln, 26.1%), reln +/+;tghappswe,arc (app, 27.6%), and reln +/−;tghappswe,arc (reln/app, 17.7%). Genotype determination was done with postmortem tail biopsies using the following primers to detect the mutant reln gene product (363 bp): forward, 5′-TAATCTGTCCTCACTCTGCC-3′, and reverse, 5′-TGCATTAATGTGCAGTGT-3′. The presence of a human 180 bp app gene product was confirmed with the following primers: 5′-CAGAACTGAACCATTTCAACCGAGC-3′ and 5′-TCAGTGGGTACCTCCAGCGCCCGA-3′. The number of animals used in this study is summarized in Table 1.
Table 1.
Animals used
| Genotype | Abbreviation | Age (months) | No./experiment |
|---|---|---|---|
| reln +/+; appwt | wt | 3 | 4 IHC |
| 6 | 5 IHC | ||
| 9 | 6 IHC | ||
| reln +/−; appwt | reln | 3 | 4 IHC |
| 6 | 4 IHC | ||
| 9 | 9 IHC | ||
| reln +/+; tghappswe,arc | app | 3 | 4 IHC |
| 6 | 4 IHC | ||
| 9 | 8 IHC/8 WB | ||
| 15 | 3 IHC | ||
| reln +/−; tghappswe,arc | reln/app | 3 | 4 IHC |
| 6 | 4 IHC | ||
| 9 | 8 IHC/8 WB | ||
| 15 | 3 IHC |
IHC, Immunohistochemistry, including DAB immunoperoxidase and fluorescence, as well as silver staining; WB, Western blotting and ELISA.
Mouse brain tissue preparation.
Animals were deeply anesthetized with Nembutal (40 mg/kg, i.p.). The tissue was perfusion fixed and processed as described previously (Knuesel et al., 2009; Doehner et al., 2010). Random sampled serial sections were collected throughout the hippocampal formation starting at bregma level −0.85 to −3.5 and stored at −20°C in cryoprotectant solution until immunohistochemical or histological evaluations (Knuesel et al., 2009).
Immunohistochemistry.
A 15 min pepsin pretreatment (0.15 mg/ml in 0.2N HCl at 37°C) was applied to all the free-floating sections for epitope unmasking and reduction of nonspecific staining (Doehner et al., 2010). Sections were then briefly washed with PBS, pH 7.4, and processed for immunohistochemistry. The following antibodies were used: mouse anti-rodent Reelin (clone G10; Millipore; MAB364; 1:1000), rabbit anti-human β-amyloid (1-40/1-42; Millipore; AB5076; 1:2000), mouse anti-human β-amyloid (1-17; clone 6E10; Covance; SIG-39320; 1:2000), mouse anti-mouse NeuN (clone A60; Millipore; MAB377; 1:1000), rabbit anti-human phosphorylated Tau (phospho-T205; Abcam; ab4841; 1:1000), rabbit anti-rat α1-syntrophin [polyclonal affinity purified serum; 1:500 (Haenggi et al., 2005)], and rat anti-mouse CD68 (AbD Serotec; MCA 341R; 1:2000). After completion of the double immunofluorescence stainings, GFAP-positive astrocytes were visualized with direct immunofluorescence using Cy3-conjugated rabbit anti-GFAP IgG (kindly provided by Wolfgang Härtig, University of Leipzig, Leipzig, Germany) (at 10 μg/ml in Tris buffer, pH 7.4, containing 2% BSA and 10% normal rabbit serum). For the nuclear counterstaining, sections were incubated for 5 min in 10 nm SYTOX green nucleic acid stain (Invitrogen) dissolved in PBS. Thioflavin S counterstaining involved 10 min incubation in filtered 1% aqueous Thioflavin S (Sigma-Aldrich) solution at room temperature, followed by two washes for 5 min in 80% EtOH, 5 min wash in 95% EtOH, and three times 5 min washes in distilled water. Brain sections were then air-dried in the dark and mounted with aqueous permanent mounting medium (Dako). Double and triple immunofluorescence labelings were visualized by confocal microscopy (LSM-710; Zeiss) using a 40× [numerical aperture (NA), 1.3] and sequential acquisition of separate channels. Z-stacks of consecutive optical sections (6–12; 512 × 512 pixel; spaced 1 μm in z) were acquired at a magnification of 0.11–0.22 μm/pixel. For visual display, Z-sections of all channels were summed and projected in the z-dimension (maximal intensity) and merged using the image analysis software Imaris (Bitplane). Cropping of images and adjustments of brightness and contrast were identical for each labeling and done using Adobe Photoshop.
Silver staining.
For the detection of dystrophic neurites, extracellular and intracellular proteinaceous aggregates, we used the FD NeuroSilver Kit II (FD NeuroTechnologies). Free-floating perfusion-fixed brain slices (40 μm thick) prepared for normal immunohistochemistry were processed according to the manufacturer's instruction. Control sections obtained from adult wild-type (wt) mice that received unilateral intrahippocampal injections of kainic acid, a procedure that results in rapid degeneration of hippocampal interneurons and CA1 pyramidal cells (Knuesel et al., 2002), served as controls.
Quantification of reactive astrocytes, microglia activation, and colocalization between Thioflavin S, α1-syntrophin, and Aβ.
Quantitative analyses of the immunohistochemical stainings were done on one randomly selected hemisphere and performed blind to the genotype of the animals. Eight single- (GFAP/CD68) or Z-stacks (Aβ/ThioS/α1-syn) of digital images from six to eight brain sections per genotype and age (n = 4–6) were acquired in neocortical areas including LI to LVI, lateral entorhinal cortex, and hippocampus, focusing on the CA1 stratum lacunosum moleculare. Area fractions of the GFAP- or CD68-immunoreactive cells, Thioflavin S, anti-Aβ, and anti-α1-syntrophin immunoreactivity (IR) were quantified using a fixed threshold algorithm of ImageJ software. Colocalization of Thioflavin S- and Aβ-immunoreactive signals in amyloid-β plaques in the same brain areas were measured on projected Z-sections. Red and green channels were first individually segmented and then analyzed with regard to pixel overlap within outlined plaques using a colocalization plug-in of ImageJ. Signals obtained from the green channel were set as 100%. To examine a putative preferential vascular association of amyloid-β plaques in reln/app mice, we measured using the same approach the degree of pixel colocalization between α1-syntrophin and Aβ IR with respect to the Thioflavin S signals, again set as 100%.
Quantification of NeuN-positive neurons and Reelin- and Aβ-positive plaque segregation.
Quantitative analysis of the neuronal density associated with amyloid-β plaques as well as the Reelin/Aβ plaque segregation in 9- and 15-month-old mice were performed on one randomly selected hemisphere in a blinded fashion. Eight single (NeuN density) or Z-stacks (Reelin/Aβ plaque segregation) digital images from four brain sections per genotype and age (n = 6, 9 months; n = 3, 15 months) were acquired in the neocortex (motor and somatosensory areas) as well as in the lateral entorhinal cortex. The numerical density of NeuN-positive neurons around plaques was assessed using a threshold algorithm using ImageJ software to individually outline and quantify the number of cells in 512 × 512 pixel images. Colocalization of Reelin- and Aβ-immunoreactive signals in extracellular plaques were measured on projected Z-sections in LI of the lateral entorhinal cortex. Red and green channels were first individually segmented and then analyzed with regard to pixel overlap and segregation within outlined individual plaques using the colocalization plug-in of ImageJ. Signals obtained from both channels (Reelin and Aβ IR) were calculated as total plaque area and set as 100%.
Volume analysis and quantification of Reelin-positive and amyloid-β plaques.
Volumetric analysis was performed with the Mercator software (Explora Nova). Five to seven sections per series were measured per animal. One hemisphere (for plaque analysis) or individual brain areas included in the volumetric assessments in 15-month-old mice (i.e., neocortex, entorhinal cortex, hippocampus) were delineated with a 2.5× objective (NA, 0.075). The volumes were estimated according to the formula V = ∑A × t nom × 1/ssf, where ∑A is the summed area of one hemisphere or brain area, t nom is the nominal section thickness of 40 μm, and ssf is the section sampling fraction (1/12). Amyloid-β plaques, classified as either granular or fibrillar based on their morphology, were counted exhaustively and their diameter measured within the neocortical layers I, II–VI, entorhinal and piriform cortex, and hippocampus using a graphic tool of Mercator. Reelin-positive neurons and plaques were counted exhaustively within the outlined hippocampus using the 40× objective (NA, 1.3). The total number of neurons (N) and plaques (P) were estimated according to the formula N or P tot = ∑N or P × 1/ssf, where ∑N or P is the sum of neurons or plaques, respectively.
Protein extracts and Western blotting.
Dissected hippocampi and neocortex samples (Table 1) were sonicated on ice in 10 vol of RIPA buffer (20 mm Tris, 150 mm NaCl, 1% Triton X-100, 1% Na-deoxycholate, 0.1% SDS) containing protease (Roche; Mini Complete tablets) and phosphatase inhibitors (Sigma-Aldrich). Soluble and membrane-associated/insoluble fractions were separated by centrifugation at 20,000 × g at 4°C for 20 min. Pellets were resolved in 70% formic acid and neutralized with 8N NaOH. Total protein concentrations were measured with Bradford assays (Bio-Rad). Protein samples (20 μg) were separated by SDS-PAGE using 10–20% Tris-Tricine gels (Invitrogen), blotted onto Protran 0.1 μm nitrocellulose (Sigma-Aldrich), and blocked in TBS containing 5% Western blocking reagent (Roche) for 10 min to 1 h at room temperature. All Western blots were run in duplicate for semiquantitative analysis. Primary antibodies [mouse anti-β-amyloid (clone 6E10; 1:1000), mouse anti-amyloid precursor protein A4 (clone 22C11; Millipore; 1:1000), rabbit anti-human phosphorylated Tau (phospho-T205; 1:1000), rabbit anti-β-actin (A2066; Sigma-Aldrich; 1:15,000)] were incubated overnight at 4°C and visualized by the enhanced chemiluminescence reaction (GE Healthcare). Semiquantitative analyses of the immunoreactive bands were performed on digitalized films using ImageJ software (National Institutes of Health, Bethesda, MD). Sum pixel brightness values (i.e., integrated density) corrected for nonspecific background and equal loading using β-actin as a control were included in the statistical analysis.
ELISA.
Human-specific amyloid-β 1-40 and 1-42 ELISA microtiter plates were obtained from The Genetics Company, and the assays were performed according to the manufacturer's protocol. The 50 μl samples of the brain extracts (supernatant, pellet) of mutant and wild-type mice (Table 1) were used and run in duplicate. After 30 min incubation in the substrate solution [TMB (tetramethylbenzidine)/peroxide mixture], the reaction was stopped and the absorption quantified using the Synergy HT Multi-Mode Microplate Reader (BioTek Instruments) measuring the difference at 450 and 650 nm.
Statistical analyses.
All analyses were performed with the software StatView, version 5.0 (Abacus Concepts). ANOVA with age and genotype as the main between-subjects factors was performed for the immunohistochemical analyses of the GFAP, CD68, NeuN, and Aβ/Reelin plaque stainings. Fisher's least significant difference (LSD) tests were used for post hoc comparisons. Planned comparisons (app vs reln/app) with Mann–Whitney U tests were used for the analysis of the total number of plaques estimated separately for each brain region, the volumetric analysis of the hippocampal formation and neocortex at 15 months of age, the levels of full-length APP, sAPP/N-APP, β-stubs, Aβ40 and Aβ42 (Western blotting and ELISA), full-length Reelin and N-terminal Reelin, phosphoTau (T205), as well as for the size and area fraction of colocalized Thioflavin S and anti-Aβ, and Thioflavin S and α1-syntrophin IR, respectively. The two-sample Kolmogorov–Smirnov test was used for comparing plaques size distribution in app and reln/app subjects. One (of eight) app animal was excluded from the statistical evaluation of the ELISA Aβ42 levels since the measured value was >3 SD above the mean. In addition, one 9-month-old app subject (of six) did not show any amyloid-β plaque staining and was therefore excluded for the statistical analysis of the Reelin/Aβ plaque segregation. Statistical significance was set at p < 0.05.
Results
Early neuropathological changes in the hippocampus of reln mice
We recently reported a significant aging-associated decline in Reelin expression and concomitant accumulation in amyloid-like plaques, which colocalize with murine proteolytic APP fragments (Knuesel et al., 2009; Doehner et al., 2010). We further showed that this pathology was accompanied by a significant reduction in cholinergic and GABAergic basal forebrain projection neurons, the latter known to selectively target Reelin-positive interneurons in the hippocampus (Madhusudan et al., 2009). Here, we tested whether the impact of reduced Reelin levels in heterozygous reeler knock-out mice (reln) correlated with putative accelerated neuropathological changes in the hippocampal formation. The immunohistochemical investigations confirmed the age-dependent reduction in Reelin expression and appearance of Reelin-positive plaques in CA stratum radiatum (sr), lacunosum-moleculare (slm), and dentate gyrus (DG) molecular layer (ml) in wt mice (Fig. 1 A,C). Reelin IR in reln mice was strongly reduced in hippocampal GABAergic interneurons and neuropil compared with wt littermates at all ages investigated (Fig. 1 A–D). Strikingly, already at 3 months of age, Reelin-positive amyloid-like plaques were seen in the slm of reln mice (Fig. 1 B, arrows), whereas the hippocampal formation was devoid of deposits in age-matched littermates (Fig. 1 A,E,F). In agreement with our previous findings (Knuesel et al., 2009), Reelin plaques in wt mice appeared only around 9 months (Fig. 1 C). Statistical evaluation of the stereological estimates of the Reelin plaque density confirmed the significant difference between genotypes at 3 months (supplemental Fig. 1A, available at www.jneurosci.org as supplemental material). At 9 months, however, the plaque density was comparable between wt and reln mice, suggesting a shift in the temporal plaque profile between genotypes and an acceleration of aging-related neuropathological changes in reln versus wt mice (supplemental Fig. 1A,B, available at www.jneurosci.org as supplemental material).
Figure 1.

Early neuropathological changes in the hippocampus of heterozygous reeler (reln) mice. Immunoperoxidase labelings of hippocampal brain sections obtained from 3-month-old (A, B, E–H) and 9-month-old (C, D) wt and heterozygous reeler mice (reln). A, C, Aging-related reduction in Reelin immunoreactivity and concomitant appearance of Reelin-positive plaques (enlarged view in C) in the CA1 stratum radiatum, lacunosum-moleculare (slm), and outer molecular layer (ml) of the DG in 9-month-old compared with 3-month-old wt mice. The two boxes in A are enlarged in E and F to show the dense accumulation of Reelin-positive cells in the stratum oriens and the distinct neuropil staining in the slm and ml, respectively, the latter likely representing the extracellular Reelin secreted from perforant path axonal projections. B, Reelin-positive plaques are already detectable at 3 months of age in heterozygous reeler mice. Note the reduction in Reelin-immunoreactive cells in stratum oriens (box in B is enlarged in G) and selective enrichment in plaques in the slm (arrow; box in B is enlarged in H). D, Prominent accumulation of Reelin in amyloid-like plaques in the subiculum, slm, and outer ml at 9 months of age. Scale bars: D, 500 μm; H, 20 μm.
Early amyloid-β plaque deposition in reln/app double transgenic mice
To assess whether reduced Reelin expression is also associated with altered temporal and spatial amyloid-β plaque pathology, we created a mutant mouse line expressing 50% of the reelin gene product in a transgenic AD background (see Materials and Methods), referred to as reln/app. Littermate controls were either wt, heterozygous for reelin (reln), or transgenic AD mice (app). Immunohistochemical and biochemical experiments were performed across aging, ranging from 3 to 15 months (Table 1). Anti-Aβ immunohistochemistry revealed no fibrillary amyloid-β plaques at 3 months of age independent of the genotype. Likewise, brain sections of 6-month-old wt and reln mice were devoid of fibrillary amyloid-β plaques (data not shown). In line with previous reports (Knobloch et al., 2007), we found no amyloid-β plaques in the hippocampal formation (Fig. 2 A) and only very few in the neocortex (maximum of one to two per section) in app mice at 6 months. This was in contrast to reln/app littermates (Fig. 2 B, arrows), in which amyloid-β plaques of different morphologies including fibrillar and granular types (Fig. 2 C) were evident in the hippocampus and neocortex in all subjects, indicating accelerated amyloid-β plaque formation in Reelin-deficient AD mice. At 9 months, amyloid-β plaques were present in app mice throughout the cortex but mostly missing in the hippocampus (Fig. 2 D, left; supplemental Fig. 1C,D, available at www.jneurosci.org as supplemental material). In reln/app mice, however, the accumulation of granular and fibrillary Aβ IR was strongly increased, strikingly selective in CA1 slm, DG ml, and piriform/entorhinal cortex layer I (Fig. 2 D, right, arrows; supplemental Fig. 1C,D, available at www.jneurosci.org as supplemental material), notably areas with highest Reelin IR in wt mice (Fig. 1 A). Stereological analysis of the plaque load at 9 months revealed a significant increase in plaque size and a corresponding significant increase in the number of large (>75 μm in diameter) granular and fibrillary plaques in hippocampus and cortical layer I of reln/app compared with app subjects (Table 2). In line, the cumulative size distribution revealed a significant shift toward larger plaques (supplemental Fig. 1E–H, available at www.jneurosci.org as supplemental material) (D max ranging from 0.18 to 0.37, Kolmogorov–Smirnov test), despite comparable numerical densities of small- (>25 μm) and medium-sized plaques (25–75 μm) between genotypes (Table 2). We also observed some strikingly large fibrillary and granular plaques in cortical layers II–VI (supplemental Fig. 1C,D, available at www.jneurosci.org as supplemental material); however, the overall number, mean size, as well as the cumulative size distribution of amyloid-β plaques were not different between genotypes in these cortical layers. At 15 months, amyloid-β plaque density further increased and remained significantly higher in reln/app compared with app mice throughout the hippocampal formation and cortex (Fig. 2 E). At this age, app mice also showed amyloid-β plaques in the subiculum and CA1 slm, most prominently seen in the temporal pole of the hippocampus. However, in reln/app mice, these layers were almost completely covered by amyloid-β IR along the entire septal-temporal axis. Moreover, signs of massively enlarged ventricles and a distinct shrinkage in cortical thickness were evident in reln/app mice at 15 months (three of three). Quantitative volumetric analysis of the hippocampus (along its entire septo-temporal axis) and cortex (including piriform, entorhinal, motor and somatosensory areas) revealed a significant volume reduction in 15-month-old reln/app (hip, 10.1 ± 1.5 mm3; ctx, 28.3 ± 1.5 mm3) compared with app mice (hip, 13.8 ± 1.1 mm3; p = 0.029; ctx, 33.3 ± 1.5 mm3; p = 0.017). Volumetric reductions in the whole hippocampal formation of reln/app mice approached a difference of −26% compared with app, whereas neocortical areas were less affected (−9%). The ventricle enlargement in the posterior part was also significantly larger in reln/app compared with app (+11.8%; p = 0.049; Mann–Whitney U test; values are mean ± SD). Together, these results indicate that a reduction in Reelin expression significantly accelerates Aβ deposition and promotes neurodegeneration in aged transgenic AD mice.
Figure 2.

Early and accelerated amyloid-plaque deposition in reln/app double transgenic mice. Representative images of coronal brain sections taken from 6- (A–C), 9- (D), and 15-month-old single mutant (app) and double mutant (reln/app) mice (E) processed for anti-amyloid-β immunoperoxidase staining. A, No amyloid-β plaque deposition was detected in single transgenic AD mice at 6 months. B, In contrast, numerous amyloid-β plaques were evident in both hippocampus and cortex of reln/app mice at this age. C, Typical dense-core fibrillar plaque in the CA1 stratum oriens (so). C′, Granular Aβ plaque, potentially representing precursor amyloid deposits in the DG molecular layer. D, Representative images of half-brain sections of a single (left) and double mutant (right) mouse processed for immunoperoxidase staining using 6E10 monoclonal anti-Aβ antibody. Note the increase in plaque size and the prominent layer-specific localization of amyloid-β plaques in the CA1 lacunosum-moleculare and piriform cortex layer I (arrows) of reln/app mice. E, Representative brain sections of an app (left) and reln/app (right) mouse at 15 months showing the aggravated plaque load in reln/app mice compared with app subjects. Note the ventricular enlargement and reduced cortical thickness in reln/app brains, indicative of progressive neurodegeneration. Scale bars: A, D, E, 500 μm; C', 20 μm.
Table 2.
Stereological estimates of amyloid-β plaques in 9-month-old single and double transgenic AD mice
| app | reln/app | p | |
|---|---|---|---|
| Hippocampus | |||
| Granular | |||
| Small | 175 ± 84 | 224 ± 81 | 0.35 |
| Medium | 125 ± 45 | 249 ± 69 | 0.15 |
| Large | 0 ± 0 | 11 ± 4** | 0.01 |
| Fibrillar | |||
| Small | 434 ± 161 | 1014 ± 313 | 0.15 |
| Medium | 247 ± 82 | 604 ± 175 | 0.20 |
| Large | 3 ± 2 | 38 ± 17* | 0.04 |
| Cortex layer I | |||
| Granular | |||
| Small | 127 ± 57 | 158 ± 49 | 0.35 |
| Medium | 87 ± 31 | 194 ± 53 | 0.15 |
| Large | 0 ± 0 | 6 ± 3 | 0.08 |
| Fibrillar | |||
| Small | 514 ± 183 | 1142 ± 316 | 0.16 |
| Medium | 297 ± 86 | 600 ± 136 | 0.10 |
| Large | 5 ± 4 | 39 ± 17* | 0.04 |
| Cortex layers II–VI | |||
| Granular | |||
| Small | 106 ± 42 | 105 ± 50 | 0.82 |
| Medium | 79 ± 32 | 114 ± 26 | 0.38 |
| Large | 3 ± 2 | 6 ± 5 | 0.99 |
| Fibrillar | |||
| Small | 382 ± 130 | 960 ± 279 | 0.11 |
| Medium | 243 ± 91 | 543 ± 113 | 0.06 |
| Large | 21 ± 13 | 42 ± 11 | 0.12 |
Values are presented as mean (±SEM) for the quantification of the estimated number of granular and fibrillary amyloid-β plaques, classified as either small (<25 μm), medium (25–75 μm), or large (>75 mm in diameter), and expressed as total number in the given brain area of app and reln/app (n = 8) subjects. Plaque counts in the hippocampus were performed along its entire septo-temporal axis; numerical estimates in the cortex included the piriform, lateral entorhinal, as well as the neocortex.
*p < 0.05,
**p ≤ 0.01.
Increased inflammatory responses in double transgenic mice
Neurodegeneration is tightly linked to inflammation, a process that is characterized by the presence of activated microglia and reactive astrocytes around amyloid-β plaques and increased levels of inflammatory mediators (Wyss-Coray, 2006). To test whether the accelerated formation and size increase of amyloid-β plaques in double mutant mice were accompanied by enhanced inflammatory changes, we used markers for activated microglia (CD68) and astrocytes (GFAP) combined with anti-Aβ or -Reelin immunohistochemistry (Fig. 3 A–F). In line with the amyloid-β pathology, GFAP and CD68 IR in hippocampus and cortex at 6 months were only moderately higher in reln and reln/app compared with wt and app mice (Fig. 3 G,H). In contrast, at 9 months of age, both astrocytes and microglia levels were significantly enhanced in reln/app mice compared with app subjects and revealed a pronounced association with amyloid-β as well as Reelin-positive plaques in cortex and hippocampus (Fig. 3 C,F). Interestingly, despite the presence of amyloid-β plaques in app mice, the levels of GFAP and CD68 IR were moderate and not different from wt littermates (Fig. 3 G,H), suggesting the abnormal activation of microglia and astrocytes because of the exacerbated amyloid-β plaque pathology in reln/app mice.
Figure 3.
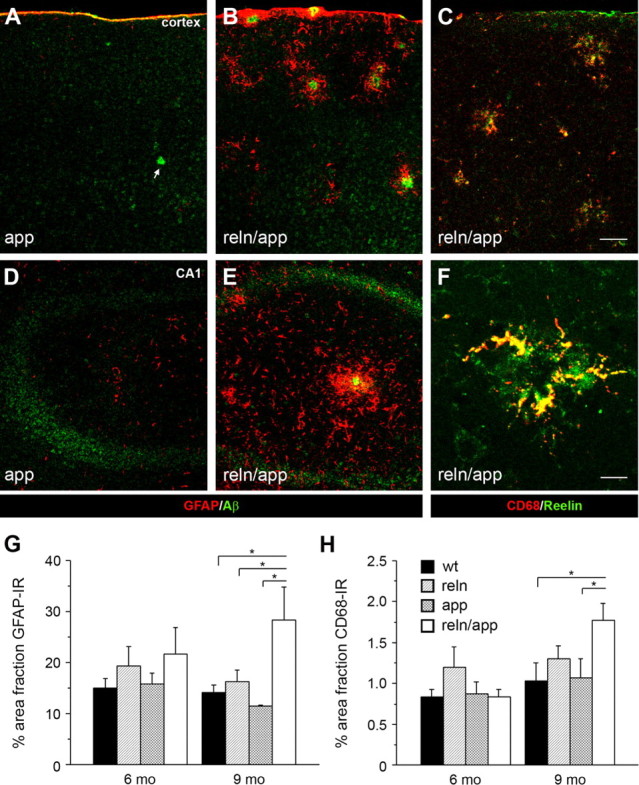
Increased inflammatory responses in reln/app double transgenic mice. Low-magnification images of double immunofluorescence staining of cortical (A–C) and hippocampal (D–F) brain sections obtained from 9-month-old single mutant app and double mutant reln/app mice. A, B, D, E, Pronounced increase in GFAP-positive astrocytes (red) selectively associated with amyloid-β plaques (anti-Aβ1-40/42 antibody; green) was found in reln/app compared with app subjects. C, F, A similar increase in reactive microglia was evident in the neocortex (C) and hippocampus (F) of 9 month reln/app mice. Note the selective accumulation of CD68-positive microglia (red) with Reelin-immunoreactive plaques (green). G, Quantification of the area fraction of GFAP-immunoreactive astrocytes revealed a main effect of genotype (F (3,20) = 4.4; p = 0.016) and significant differences between reln/app versus wt (p = 0.022), reln/app versus reln (p = 0.043), and reln/app versus app (p = 0.010) at 9 months of age. H, Similar differences emerged between reln/app versus wt (p = 0.039) and reln/app versus app (p = 0.046) subjects for the CD68 area fraction. Values are given as mean ± SEM. *p < 0.05 (comparison with reln/app subjects), statistical significance based on Fisher's LSD post hoc analysis. Scale bars: C, 50 μm; F, 10 μm.
Aggravated amyloid-β plaque pathology in reln/app mice
To investigate further the progression of reactive astrocytes, amyloid-β and Reelin IR in reln/app mice, we performed high-magnification confocal imaging of triple-labeled brain sections of 6-, 9-, and 15-month-old mice. We found a prominent elevation in GFAP IR surrounding granular amyloid-β plaques in reln/app mice across aging that was accompanied by a significant enlargement of individual granules and increased colocalization between amyloid-β and Reelin IR in granular plaques (supplemental Fig. 2A,B, available at www.jneurosci.org as supplemental material). In contrast, granular plaque pathology in app subjects was much less pronounced, as indicated by the moderate GFAP, Aβ, and Reelin IR in CA1 area at 9 months of age (supplemental Fig. 2C, available at www.jneurosci.org as supplemental material).
Fibrillary Aβ plaques in 6-month-old reln/app mice were already densely surrounded by astrocytes, particularly prominent in the hippocampus, pointing again to the advanced stage in amyloid-β plaque formation (supplemental Fig. 2E, available at www.jneurosci.org as supplemental material). A similar pattern was seen in the neocortex; however, astrogliosis was much less advanced compared with the hippocampal formation (supplemental Fig. 2D, available at www.jneurosci.org as supplemental material). Interestingly, prominent Reelin IR was associated with fibrillary plaques, correlating with their size and showing partial colocalization with Aβ (supplemental Fig. 2E, arrows, available at www.jneurosci.org as supplemental material), again a feature being much less pronounced in app compared with the reln/app subjects. At 9 months of age, Reelin IR further increased in area and intensity, most prominently in fibrillary plaques in the cortex and CA1 slm (Fig. 4) of reln/app mice, whereas plaque pathology in app littermates resembled the pattern seen in 6 month reln/app mice (Fig. 4 A; supplemental Fig. 2D, available at www.jneurosci.org as supplemental material). Moreover, Reelin-positive plaques appeared to segregate from amyloid-β plaques in hippocampus and cortex of reln/app but not age-matched app mice (Fig. 4 H). Quantitative analysis of the area covered by Reelin and Aβ plaques confirmed the increase in Reelin IR in extracellular deposits (supplemental Fig. 3, available at www.jneurosci.org as supplemental material) and revealed a significantly lower overlap and higher levels of segregation between Reelin and Aβ IR in extracellular plaques in reln/app compared with app mice. These results point to a potential bidirectional facilitation of aggregation between Reelin and amyloid-β peptides. At 15 months, the additional increase in plaque pathology in reln/app mice was mirrored by a selective reduction in neuronal density, accompanied by a massive coverage by activated astrocytes and microglia occupying the neuropil surrounding amyloid-β deposits (Fig. 4 M–O), a phenomenon likely underlying the volumetric reductions of cortical areas measured in reln/app mice at this age (Fig. 2 E). In app littermates, no evidence for a significant reduction in neuronal numbers in the vicinity of amyloid-β plaques was evident (Fig. 4 K), and, correspondingly, glia activation was not as high as in double mutants (Fig. 4 J). Quantitative evaluation of the neuronal density around amyloid-β plaques using NeuN immunohistochemistry supported this observation and revealed a significant reduction in NeuN-positive neurons in reln/app versus app subjects (supplemental Fig. 4A–D, available at www.jneurosci.org as supplemental material), suggesting that the reduction in Reelin-mediated signaling and the elevation in inflammatory markers significantly contribute to progressive neurodegeneration in aged reln/app mice.
Figure 4.

Aggravation and segregation of Reelin and amyloid-β plaques pathology in the hippocampus and cortex of 9- and 15-month-old double mutant reln/app mice. A–I, Triple immunofluorescence stainings using Cy3-GFAP (red), anti-Aβ1-40/42 (green), and anti-Reelin (blue) antibodies on cortical (A–F) and hippocampal (G–I) brain sections of 9-month-old app (A–C) and reln/app mice (D–I). In the double mutants, amyloid-β plaques were densely surrounded and tightly associated with reactive astrocytes, whereas in app subjects only moderate astrogliosis was evident. Note the intense Reelin immunoreactivity associated with fibrillary amyloid-β plaques (B vs E, arrows) and their striking segregation from Aβ deposits (H, arrow) in the CA1 slm in reln/app compared with app subjects. J–O, Aggravation of the microgliosis and astrogliosis in reln/app mice at 15 months of age was particularly prominent in the entorhinal cortex (ECtx). Note the reduction in APP-expressing neurons in the aged double (N) versus single mutants (K). Scale bars: C, F, I, 10 μm; L, O, 20 μm.
Increase in amyloidogenic APP processing and insoluble Aβ species in double mutants
In vitro data revealed reduced amyloidogenic APP processing in the presence of Reelin in the culture medium (Hoe et al., 2006). To test whether the aggravation in amyloid-β plaque pathology was accompanied by enhanced production and increased Aβ peptide levels, we performed Western blot analysis and ELISA of SDS-soluble and -insoluble, formic acid-extracted fractions of hippocampal and cortical brain lysates of app and reln/app mice. In the soluble fraction, enhanced amyloidogenic APP processing in reln/app versus app was evident, as indicated by the presence of Aβ species and the increase in β-secretase-cleaved APP ectodomains (sAPP) and C-terminal fragments (β-stubs) in the hippocampus of reln/app mice compared with app subjects (Fig. 5 A–D). Semiquantitative analysis of the immunoreactive bands in the soluble fractions revealed significantly higher levels of β-stubs and sAPP in the double mutants compared with the single transgenic AD mutants (Fig. 5 B–D). Surprisingly, we also observed elevated levels of N-terminal APP fragments, inversely correlating with the expression levels of full-length Reelin (Fig. 5 C,G), further confirming that reduction in Reelin expression alters proteolytic processing of APP. The insoluble fractions of both hippocampal and cortical brain lysates of reln/app mice contained substantial amounts of Aβ species (Fig. 5 E). Quantitative analysis using ELISA confirmed this observation and revealed significant increases in insoluble Aβ40 and Aβ42 peptides in both hippocampus and cortex in reln/app compared with app littermates (Fig. 5 F). Together, these results suggest that reduction in Reelin increases Aβ levels by favoring amyloidogenic APP processing and by promoting aggregation of Aβ peptides in the neuropil of the hippocampus and cortex.
Figure 5.

Increase in amyloidogenic APP processing and insoluble Aβ levels in double mutant reln/app mice. A, Representative Western blots of the SDS-soluble supernatant (SN) derived from hippocampal brain lysates of 9-month-old mice using anti-Aβ (6E10) antibodies. B, Semiquantitative analysis involving densitometry of the immunoreactive β-cleaved C-terminal APP fragments (β-stubs), run in duplicate, corrected for nonspecific background and equal loading using β-actin as control, revealed a significant increase in reln/app compared with app (p = 0.049; n = 4). Soluble Aβ-levels were very low, but clearly detectable in samples of reln/app subjects. C, Representative Western blots using anti-Reelin (G10) antibody recognizing both full-length (FL-Reln) and the N-terminal 180 kDa fragment (N-Reln). The N-terminal-specific APP antibody (22C11) antibody recognized in addition to full-length APP (FL-APP) and the soluble α- or β-secretase-cleaved APP ectodomains (sAPP) short N-terminal fragments, presumably representing N-APP. D, Statistical analysis of the densitometrical measurements of FL-APP and sAPP-immunoreactive fragments revealed a significant difference between genotypes as indicated by the reduced FL-APP/sAPP ratio in reln/app compared with app subjects (p = 0.033; n = 4). In addition, a marked shift toward higher N-APP-immunoreactive fragments in 9-month-old reln/app mice was evident, as demonstrated by the significant reduction in the FL-Reelin/N-APP ratio (G) (p = 0.034; n = 4). E, Representative Western blots using anti-Aβ (6E10) antibodies of SDS-insoluble fractions (pellet) of hippocampal brain lysates, which was resuspended in formic acid (FA). F, Quantitative analysis using ELISA with Aβ40- and Aβ42-specific antibodies revealed a significant increase in Aβ peptides in the insoluble fractions of reln/app compared with app littermates in the hippocampus (Aβ40, p = 0.021; Aβ42, p = 0.046) and neocortex (Aβ40, p = 0.016; Aβ42, p = 0.016; n = 7–8). Values are expressed as fraction of total protein content and given as mean ± SEM. *p < 0.05 **p < 0.01, statistical significance based on Mann–Whitney U test.
To investigate further the accelerated aggregation state, we compared the density of fibrillary Aβ species between genotypes using Thioflavin S staining in combination with anti-Aβ immunohistochemistry (6E10). In addition, we used an antibody against α1-syntrophin, a marker for astrocytic endfeet, to investigate a putative preferential association of Aβ plaques with cerebral perivasculature. In agreement with our biochemical measurements, we detected a significant increase in the number and size of Thioflavin S-positive fibrillary aggregates in all brain areas investigated, including neocortical and hippocampal formation in reln/app versus app subjects (Fig. 6 A–C). Moreover, the area covered by Aβ- and Thioflavin S-positive signals in these brain regions was significantly larger in reln/app compared with app mice (Fig. 6 D), supporting the view that the reduction in Reelin expression facilitates and accelerates the formation of fibrillary Aβ species. No difference with respect to neuropil versus vascular amyloidosis was found between genotypes (all p > 0.6), indicating that the increase in Aβ plaque deposition in reln/app mice was not associated with a preferential increase in β-amyloid angiopathy.
Figure 6.

Increase in Thioflavin S-positive amyloid-β plaques in reln/app mice in neocortex and hippocampal formation. Double immunofluorescence using anti-Aβ (6E10; red) and anti-α1-syntrophin (αSyn; blue) antibodies combined with Thioflavin S counterstaining (green) were performed to investigate the fibrillary amyloid-β plaque load in both neuropil and cerebral vasculature in 9-month-old app (A) and reln/app mice (B). Note the increase in Thioflavin S signals concomitantly with the anti-Aβ immunoreactivity in the outer layers of the neocortex. C, Densitometric analysis of the brightness and surface area covered by Thioflavin S-positive signals in the neocortical layers I–VI (Ctx), lateral entorhinal cortex (EC), and dorsal and ventral hippocampus (Hip). D, Quantification of the pixel overlap between the green Thioflavin S-positive signals and red anti-Aβ immunoreactive staining, averaged from measurements of eight images (512 × 512 pixel in size) per animal and brain region (n = 4 per genotype) acquired in the areas indicated. Values are given as mean ± SEM. *p < 0.05, statistical significance based on Mann–Whitney U test. Scale bar, 50 μm.
Abundant dystrophic neurites and elevation of phospho-Tau in aged reln/app mice
In addition to direct binding to APP and modulating its proteolytic processing (Hoe et al., 2006, 2009), Reelin-mediated signaling inhibits key mediators of Tau phosphorylation (Hiesberger et al., 1999; Beffert et al., 2002, 2004; Ohkubo et al., 2003). We therefore reasoned that genetic reduction in Reelin expression—in addition to favoring amyloidogenic APP processing—might promote hyperphosphorylation of Tau and facilitate the formation of neurofibrillary tangles in aged subjects. We therefore performed biochemical and immunohistochemical analysis to assess the levels of phospho-Tau using an antibody raised against phosphoT205 across aging. In young and adult brain tissue, highest phospho-Tau levels can be detected in axonal projections throughout the brain, particularly prominent in mossy fibers and terminals in stratum lucidum of the CA3 area (data not shown). At 9 months of age, biochemical analyses revealed a significant elevation of phospho-Tau levels in hippocampal and cortical brain lysates of reln/app compared with app mice (supplemental Fig. 4E–G, available at www.jneurosci.org as supplemental material), confirming that Reelin-mediated signaling in aged subjects remains a major modulator of Tau phosphorylation as described during neurodevelopment (Beffert et al., 2002; Brich et al., 2003). In line with these biochemical data, several neurons with distinct anti-phospho-Tau signals in the soma were detectable, sparsely distributed in plaque-dense areas in the hippocampal formation and neocortex in 9-month-old reln/app but not in app mice (data not shown). At 15 months of age, a striking concentric accumulation of phospho-Tau-positive cells selectively associated with amyloid-β plaques was evident in reln/app. High-magnification confocal microscopy revealed a distinct anti-phospho-Tau IR enriched in neuronal somata and dendrites (Fig. 7 B, arrow, inset). Littermate app mice at the same age showed some anti-phospho-Tau IR; however, this was mostly restricted to extrasomatic sites, likely representing axonal localization (Fig. 7 A, inset). Typically, the area surrounding the plaques also contained pronounced anti-amyloid-β IR in reln/app compared with app mice at 15 months (Fig. 7), suggesting that hyperphosphorylation of Tau in soma and dendrites is selectively promoted around amyloid-β plaques, potentially mediated by reduced Reelin and elevated nonfibrillary Aβ levels, and/or glia cells and their secreted inflammatory cytokines.
Figure 7.

Selective association of anti-phospho-Tau immunoreactivity in plaque-dense areas within CA1 slm. A, B, Double immunofluorescence using anti-phospho-T205 (red) and anti-Aβ (6E10, blue) antibodies combined with Sytox Green nuclear counterstaining to investigate the relationship between amyloid-β plaque and Tau pathology in app (A) and reln/app (B) at 15 months of age. A, Representative amyloid-β plaque in the CA1 slm of app mice with a dense core and some diffuse anti-Aβ immunoreactivity surrounding it (A′). In line with the immunoperoxidase stainings, anti-phospho-Tau immunoreactivity in app subjects was readily detectable in the neuropil, but no evidence for any enrichment in cells surrounding the plaques was found (A″, enlarged view is shown in inset). B–B″, Anti-amyloid-β immunoreactivity covered large areas within the core and surrounding areas of plaques in the CA1 slm of reln/app mice. Note the selective fibrillary enrichment of anti-phospho-Tau signals in the cytoplasm (inset) and dendrites (arrow) of neurons selectively associated with amyloid plaques. The distinct spatial distribution of the amyloid-β and Tau pathology is also evident on the xz and yz view at the bottom and right side of the image. Scale bars: A, B, 30 μm.
A highly similar picture emerged using immunoperoxidase labelings. Both cortical and hippocampal neurons associated with amyloid-β plaques in reln/app but not app mice showed strong somatic anti-phospho-Tau IR (Fig. 8 A,B, inset). Interestingly, some of them stood out by the presence of distinct phospho-Tau aggregates, suggestive of degenerating neurons (Fig. 8 C, inset). This was also evident in the hippocampus, particularly prominent in the CA1 slm and sr, where we detected typical tangle-like structures (Fig. 8 D, inset), indicating the presence of neurofibrillary depositions in aged double mutants. To confirm the presence of this typical AD-like neuropathological hallmark and to assess putative neurodegeneration associated with amyloid-β plaques, we performed also Gallyas silver staining. At 9 months of age, no indication of neurodegenerative processes nor tangle-like aggregates were evident in reln/app and app mice (data not shown). At 15 months, selectively in reln/app subjects, bright-field microscopy revealed the presence of distinct silver precipitates selectively associated with amyloid-β plaques, which likely represent dystrophic neurites (Fig. 8 F,G). This feature was most prominently seen in the entorhinal cortex, accompanied by densely labeled cell bodies and neurites surrounding the plaques in the double mutants but not app mice (Fig. 8 E). In line with the anti-phospho-Tau immunohistochemistry, individual silver-stained intracellular aggregates were detected in the entorhinal cortex of reln/app mice within the dystrophic compartment, suggesting the presence of neurofibrillary tangles selectively associated with amyloid-β plaques in aged Reelin-deficient AD mice.
Figure 8.

Concentric accumulations of phospho-Tau-positive neurons and neurofibrillary tangles around amyloid-β plaques in 15-month-old reln/app mice. Representative images of brain sections obtained from app (A, E) and reln/app mice (B–D, F, G) processed for immunoperoxidase staining using anti-phospho-T205 antibody (A–D) or silver staining (E–G). A, In app subjects, uniform phospho-Tau immunoreactivity was evident in the cortical neuropil, indicative of axonal localization. No particular enrichment was seen in the vicinity of amyloid-β plaques, recognized by their lack of phospho-Tau immunoreactivity (asterisk). B, C, In reln/app mice, strongly stained phospho-Tau-positive neurons were evident around amyloid-β plaques in neocortical areas (insets). D, In the hippocampus of reln/app mice, a similar pattern was evident in close vicinity to plaques; some neurons appeared dystrophic, strongly suggestive of tangle-like intraneuronal accumulations (D, inset). E, Silver staining revealed some dark precipitates (arrowhead) in the vicinity of large plaques (asterisk) in the entorhinal cortex of app mice. F, In contrast, dense silver precipitates surrounding amyloid-β plaques, indicative of dystrophic neurites, were very prominent in reln/app mice. In addition, black-labeled intraneuronal aggregates, likely representing neurofibrillary tangles, were seen in neurons associated with these plaques (arrows; enlarged in the insets). G, Representative image of a large amyloid-β plaque in the entorhinal cortex of a reln/app mouse, showing a concentric pattern of silver-stained cells, likely representing degenerating neurons in the vicinity of plaques (arrowheads). Scale bars, 20 μm.
Together, the findings indicate that a genetic reduction in Reelin expression accelerates the production of amyloidogenic Aβ peptides, which results in earlier plaque formation and an age-related aggravation of the pathology including early signs of neurodegeneration in hippocampal formation and cortex of AD mice that is accompanied by elevated phospho-Tau levels and neurofibrillary tangle formation in aged subjects.
Discussion
The present study provides convergent immunohistochemical and biochemical evidence that a genetic reduction in Reelin expression favors amyloidogenic APP processing in vivo, resulting in precocious formation and age-related aggravation of amyloid-β plaque pathology, accompanied by concentric accumulation of phospho-Tau-positive neurons, neurofibrillary tangles, and neurodegeneration in the hippocampal formation. The animal model thereby unites for the first time amyloid-β plaque and Tau pathology in a temporal and spatial pattern that closely recapitulates the progression of AD neuropathology in humans (Braak and Braak, 1996), importantly in the absence of tau mutations. Furthermore, it complements several previous studies involving genetic studies (Brich et al., 2003), in vitro experiments (Hoe et al., 2006), and in vivo neuronal survival assays (Beffert et al., 2006b) demonstrating that dysfunctional Reelin-mediated signaling through ApoER2 acts as a critical upstream modulator of amyloidogenic APP processing, Tau hyperphosphorylation, and neuronal survival, presumed to underlie AD-associated neuropathology.
Our comprehensive investigations across aging revealed a significantly accelerated extracellular Aβ neuropathology in Reelin-deficient AD mice, as indicated by the presence of amyloid-β plaques in hippocampus and neocortex as early as 6 months. Littermate app mice were virtually devoid of amyloid-β deposits at this age, in line with the original report (Knobloch et al., 2007). At 9 and 15 months, pathology was further aggravated in reln/app mice, resulting in a striking redistribution and selective accumulation of Aβ plaques in entorhinal projection areas, precisely corresponding to areas in which Reelin IR is enriched in extracellular plaques in reln but not app littermates (Figs. 1, 2). Moreover, we detected numerous Aβ plaques in layer I of the entorhinal, piriform, and neocortex, again in areas with strong Reelin expression in young and Reelin plaque depositions in aged subjects (Knuesel et al., 2009). Aβ plaques were also significantly larger in these areas in double compared with single mutants, affecting both granular (presumable nonfibrillary, soluble deposits) as well as fibrillary plaques (Table 2, Figs. 2, 6; supplemental Fig. 1E–H, available at www.jneurosci.org as supplemental material). Biochemical investigations using SDS-soluble fractions of hippocampal and cortical lysates revealed significant decrease in full-length APP and concomitant increases in β-secretase-cleaved APP ectodomains (sAPPβ), C-terminal fragments (β-stubs), and Aβ peptides (Fig. 5), indicating elevated amyloidogenic APP processing, in line with previous in vitro data (Hoe et al., 2006; Hoe and Rebeck, 2008). Western blotting and quantitative ELISA of formic acid-extracted hippocampal and cortical brain lysates revealed also significantly higher levels of insoluble Aβ40/42 peptides in reln/app compared with app littermates (Fig. 5), confirming that reduced Reelin levels, potentially by altering APP trafficking, membrane insertion, and favoring β-secretase cleavage (Hoe et al., 2006), enhances Aβ production. It is further conceivable that reduction in Reelin-signaling promotes APP endocytosis because of reduced binding of the central Reelin domain to the N terminus of APP (Hoe et al., 2009). This scenario fits with previous data showing preferential APP β-secretase cleavage in endocytic vesicles (Rajendran et al., 2006, 2008), in line with the reported endosomal localization and pH optimum of β-secretase activity (Vassar et al., 1999).
In addition to the role of reduced Reelin-mediated signaling on amyloidogenic APP processing, our data also suggest a direct effect on Aβ aggregation. We show that a reduction in Reelin expression favors its own aggregation, as demonstrated by the significantly earlier but highly conserved Reelin plaque pathology (Knuesel et al., 2009) in reln compared with wt mice (Fig. 1). Thus, it is conceivable that both reduced Reelin-dependent signaling promoting Aβ production as well as Reelin aggregation by providing a nucleation site for aggregation-prone peptides, results in aggravation of the amyloid-β plaque phenotype. Indeed, we observe an overlap between Reelin and amyloid-β plaques in LI of the entorhinal cortex and perforant path axonal projection sites where the two selectively associate and partially colocalize in granular and fibrillary plaques (Fig. 4; supplemental Fig. 2, available at www.jneurosci.org as supplemental material). Interestingly, we also detected a segregation of Reelin IR from Aβ-positive fibrillary plaques in aged reln/app mice, indicating that aggregation of Reelin itself aggravates in the vicinity of amyloid-β plaques and reactive glia. This phenomenon was again more prominent in the hippocampal formation compared with neocortical regions, confirming the increased vulnerability of this brain area with respect to AD-like neuropathology. A reciprocal effect is also conceivable, with Aβ peptides promoting Reelin aggregation. Support for this hypothesis has been recently provided by Botella-López et al. (2010), demonstrating that amyloid-β peptides can alter Reelin processing and glycosylation, critical modifications demonstrated to be required for proper Reelin-mediated signaling (Lambert de Rouvroit et al., 1999; Koch et al., 2002; Jossin et al., 2007) and potentially involved in abnormal oligomerization and aggregation of Reelin (Utsunomiya-Tate et al., 2000; Kubo et al., 2002; Yasui et al., 2007).
Difference in plaque pathology between genotypes was also associated with distinct inflammatory responses in reln/app versus app mice. Our investigations of reactive glia revealed a significant aging-related increase in GFAP and CD68 IR, tightly intermingled with amyloid-β plaques in reln/app mice (Figs. 3, 4; supplemental Fig. 2, available at www.jneurosci.org as supplemental material). In contrast, Aβ plaques were detected without or weak GFAP/CD68 IR in app littermates (Fig. 3). In line with the biochemical data, these findings support our interpretation of a significantly more aggravated and advanced fibrillary plaque pathology in aged reln/app compared with app mice. This is in agreement with immunohistochemical studies examining the brains of AD patients (McGeer et al., 1987), as well as with numerous studies using different animal models of AD (for review, see El Khoury and Luster, 2008), demonstrating that microglia accumulation in senile plaques is an integral part of AD pathogenesis. Our data also fit with recent findings showing that the phenotype of accumulating microglia changes as AD-like pathology progresses in tgAPP-PS1 mice (Hickman et al., 2008), from a protective, Aβ-clearing role during early phases to an enhanced production in inflammatory cytokine and loss in Aβ-clearing capabilities during later disease phases. Furthermore, it is conceivable that, in addition to dysfunctional Reelin signaling, proinflammatory cytokines including interferon-γ, TNF-α (tumor necrosis factor-α), and interleukin-1, shown to be secreted from glia (Hickman et al., 2008) and demonstrated to be involved in β-secretase upregulation (Yamamoto et al., 2007) and γ-secretase-mediated APP cleavage (Liao et al., 2004), contribute to Aβ production and accumulation.
The elevated release of proinflammatory cytokines by Aβ-activated glia is also expected to promote neurodegenerative processes (Blurton-Jones and Laferla, 2006; Chiarini et al., 2006). However, neurodegeneration in turn strongly promotes gliosis. Indeed, at 15 months, amyloid-β plaque pathology was accompanied by striking and significant reduction in neuronal density, accompanied by strong astrogliosis and dystrophic neurites surrounding the plaques in reln/app, whereas app littermates showed only very mild signs of plaque-associated neurodegeneration (Fig. 8). Moreover, a significant ventricular enlargement and shrinkage of cortical areas, mostly affecting the hippocampal formation, was evident in double compared with single mutants (Fig. 2), indicating that reduced Reelin levels by promoting Aβ plaque deposition and inflammatory responses, result in neurodegeneration in aged subjects. A potential molecular mechanism has been recently provided by the findings that signaling through a spliced isoform of the Reelin receptor ApoER2 is essential for protection against neuronal cell loss during normal aging (Beffert et al., 2006b), pointing to an exciting mechanistic link between Reelin deficiency and neurodegeneration through dysfunctional ApoER2 signaling. However, we cannot rule out completely that subtle neuroanatomical changes occurring in reln mice may contribute to the observed neuropathology in reln/app mice. However, considering that we did not detect statistical differences in volume and neuronal density between genotypes up to 9 months of age, we consider this putative confounding factor as unlikely.
Another striking effect accompanying these neuropathological changes was the concentric accumulation of phospho-Tau-positive neurons around senile plaques in reln/app mice (Figs. 7, 8), supporting previous genetic interaction studies demonstrating a critical role of Reelin-mediated signaling via Dab1 phosphorylation in inhibiting Tau hyperphosphorylation (Brich et al., 2003). Interestingly, although neocortical neurons contained mainly cytoplasmic anti-phospho-Tau IR, distinct tangle-like structures were found in the hippocampal formation (Fig. 8), in line with the more advanced plaque pathology in this brain region in double versus single mutants. Silver staining revealed the presence of neurofibrillary tangle-like structures, selectively associated with dystrophic neurites surrounding the plaques in the entorhinal cortex, importantly in nontransgenic tau mutants. The temporal and spatial aspects of the neuropathology supports the critical upstream role of dysfunctional Reelin-mediated signaling that is sufficient to accelerate amyloid-β peptide production in the hippocampal formation, which in turn promotes Tau phosphorylation and tangle formation, selectively in plaque- and glia-dense areas. This is in line with the current hypothesis of a upstream role of Aβ species with respect to Tau pathology (Selkoe, 2000; Hardy and Selkoe, 2002; Walsh and Selkoe, 2004) and with the findings from postmortem AD tissue showing a distinct temporal and spatial progression of the neuropathology in humans (Braak and Braak, 1996).
In conclusion, the data revealed for the first time in vivo that reduced expression of Reelin has a significant effect on amyloidogenic APP processing and amyloid-β plaque deposition as well as neurofibrillary tangle formation in the hippocampal formation of aged transgenic AD mice with a genetic reduction in the reeler gene. Considering that Reelin is also an important modulator of adult NMDA receptor-mediated neurotransmission and synaptic plasticity (Weeber et al., 2002; Beffert et al., 2005, 2006a; Chen et al., 2005; Qiu et al., 2006; Qiu and Weeber, 2007), a stronger decline than normally seen during aging is expected to trigger a cascade of downstream events, resulting in the shift from nonamyloidogenic to amyloidogenic pathways, accompanied by immunological changes, Tau hyperphosphorylation, and neurofibrillary tangle formation, which presumably underlie the progressive neurodegeneration and cognitive decline associated with late-onset AD.
Footnotes
This work was supported by the Hartmann Müller Foundation (I.K.), Swiss National Science Foundation Grant 310000-117806 (I.K.), and Swiss National Competence Center in Neural Plasticity and Repair (I.K., R.M.N.). We are extremely grateful to Corinne Sidler and Cornelia Schwerdel for their excellent technical support and to Kofi Kyere for animal husbandry and care.
References
- Beffert U, Morfini G, Bock HH, Reyna H, Brady ST, Herz J. Reelin-mediated signaling locally regulates protein kinase B/Akt and glycogen synthase kinase 3beta. J Biol Chem. 2002;277:49958–49964. doi: 10.1074/jbc.M209205200. [DOI] [PubMed] [Google Scholar]
- Beffert U, Weeber EJ, Morfini G, Ko J, Brady ST, Tsai LH, Sweatt JD, Herz J. Reelin and cyclin-dependent kinase 5-dependent signals cooperate in regulating neuronal migration and synaptic transmission. J Neurosci. 2004;24:1897–1906. doi: 10.1523/JNEUROSCI.4084-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beffert U, Weeber EJ, Durudas A, Qiu S, Masiulis I, Sweatt JD, Li WP, Adelmann G, Frotscher M, Hammer RE, Herz J. Modulation of synaptic plasticity and memory by Reelin involves differential splicing of the lipoprotein receptor Apoer2. Neuron. 2005;47:567–579. doi: 10.1016/j.neuron.2005.07.007. [DOI] [PubMed] [Google Scholar]
- Beffert U, Durudas A, Weeber EJ, Stolt PC, Giehl KM, Sweatt JD, Hammer RE, Herz J. Functional dissection of Reelin signaling by site-directed disruption of Disabled-1 adaptor binding to apolipoprotein E receptor 2: distinct roles in development and synaptic plasticity. J Neurosci. 2006a;26:2041–2052. doi: 10.1523/JNEUROSCI.4566-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beffert U, Nematollah Farsian F, Masiulis I, Hammer RE, Yoon SO, Giehl KM, Herz J. ApoE receptor 2 controls neuronal survival in the adult brain. Curr Biol. 2006b;16:2446–2452. doi: 10.1016/j.cub.2006.10.029. [DOI] [PubMed] [Google Scholar]
- Bertram L, Tanzi RE. Thirty years of Alzheimer's disease genetics: the implications of systematic meta-analyses. Nat Rev Neurosci. 2008;9:768–778. doi: 10.1038/nrn2494. [DOI] [PubMed] [Google Scholar]
- Blurton-Jones M, Laferla FM. Pathways by which Abeta facilitates tau pathology. Curr Alzheimer Res. 2006;3:437–448. doi: 10.2174/156720506779025242. [DOI] [PubMed] [Google Scholar]
- Botella-López A, Burgaya F, Gavín R, García-Ayllón MS, Gómez-Tortosa E, Peña-Casanova J, Ureña JM, Del Río JA, Blesa R, Soriano E, Sáez-Valero J. Reelin expression and glycosylation patterns are altered in Alzheimer's disease. Proc Natl Acad Sci U S A. 2006;103:5573–5578. doi: 10.1073/pnas.0601279103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Botella-López A, Cuchillo-Ibáñez I, Cotrufo T, Mok SS, Li QX, Barquero MS, Dierssen M, Soriano E, Sáez-Valero J. beta-Amyloid controls altered Reelin expression and processing in Alzheimer's disease. Neurobiol Dis. 2010;37:682–691. doi: 10.1016/j.nbd.2009.12.006. [DOI] [PubMed] [Google Scholar]
- Braak H, Braak E. Evolution of the neuropathology of Alzheimer's disease. Acta Neurol Scand Suppl. 1996;165:3–12. doi: 10.1111/j.1600-0404.1996.tb05866.x. [DOI] [PubMed] [Google Scholar]
- Brich J, Shie FS, Howell BW, Li R, Tus K, Wakeland EK, Jin LW, Mumby M, Churchill G, Herz J, Cooper JA. Genetic modulation of tau phosphorylation in the mouse. J Neurosci. 2003;23:187–192. doi: 10.1523/JNEUROSCI.23-01-00187.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Beffert U, Ertunc M, Tang TS, Kavalali ET, Bezprozvanny I, Herz J. Reelin modulates NMDA receptor activity in cortical neurons. J Neurosci. 2005;25:8209–8216. doi: 10.1523/JNEUROSCI.1951-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiarini A, Dal Pra I, Whitfield JF, Armato U. The killing of neurons by beta-amyloid peptides, prions, and pro-inflammatory cytokines. Ital J Anat Embryol. 2006;111:221–246. [PubMed] [Google Scholar]
- Chin J, Massaro CM, Palop JJ, Thwin MT, Yu GQ, Bien-Ly N, Bender A, Mucke L. Reelin depletion in the entorhinal cortex of human amyloid precursor protein transgenic mice and humans with Alzheimer's disease. J Neurosci. 2007;27:2727–2733. doi: 10.1523/JNEUROSCI.3758-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Arcangelo G, Miao GG, Chen SC, Soares HD, Morgan JI, Curran T. A protein related to extracellular matrix proteins deleted in the mouse mutant reeler. Nature. 1995;374:719–723. doi: 10.1038/374719a0. [DOI] [PubMed] [Google Scholar]
- Doehner J, Madhusudan A, Konietzko U, Fritschy JM, Knuesel I. Co-localization of Reelin and proteolytic AbetaPP fragments in hippocampal plaques in aged wild-type mice. J Alzheimers Dis. 2010;19:1339–1357. doi: 10.3233/JAD-2010-1333. [DOI] [PubMed] [Google Scholar]
- El Khoury J, Luster AD. Mechanisms of microglia accumulation in Alzheimer's disease: therapeutic implications. Trends Pharmacol Sci. 2008;29:626–632. doi: 10.1016/j.tips.2008.08.004. [DOI] [PubMed] [Google Scholar]
- Estus S, Golde TE, Kunishita T, Blades D, Lowery D, Eisen M, Usiak M, Qu XM, Tabira T, Greenberg BD. Potentially amyloidogenic, carboxyl-terminal derivatives of the amyloid protein precursor. Science. 1992;255:726–728. doi: 10.1126/science.1738846. [DOI] [PubMed] [Google Scholar]
- Furukawa K, Sopher BL, Rydel RE, Begley JG, Pham DG, Martin GM, Fox M, Mattson MP. Increased activity-regulating and neuroprotective efficacy of alpha-secretase-derived secreted amyloid precursor protein conferred by a C-terminal heparin-binding domain. J Neurochem. 1996;67:1882–1896. doi: 10.1046/j.1471-4159.1996.67051882.x. [DOI] [PubMed] [Google Scholar]
- Gao Y, Pimplikar SW. The gamma-secretase-cleaved C-terminal fragment of amyloid precursor protein mediates signaling to the nucleus. Proc Natl Acad Sci U S A. 2001;98:14979–14984. doi: 10.1073/pnas.261463298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glenner GG, Wong CW. Alzheimer's disease: initial report of the purification and characterization of a novel cerebrovascular amyloid protein. Biochem Biophys Res Commun. 1984;120:885–890. doi: 10.1016/s0006-291x(84)80190-4. [DOI] [PubMed] [Google Scholar]
- Goate A, Chartier-Harlin MC, Mullan M, Brown J, Crawford F, Fidani L, Giuffra L, Haynes A, Irving N, James L. Segregation of a missense mutation in the amyloid precursor protein gene with familial Alzheimer's disease. Nature. 1991;349:704–706. doi: 10.1038/349704a0. [DOI] [PubMed] [Google Scholar]
- Grundke-Iqbal I, Iqbal K, Tung YC, Quinlan M, Wisniewski HM, Binder LI. Abnormal phosphorylation of the microtubule-associated protein tau (tau) in Alzheimer cytoskeletal pathology. Proc Natl Acad Sci U S A. 1986;83:4913–4917. doi: 10.1073/pnas.83.13.4913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haass C, Koo EH, Mellon A, Hung AY, Selkoe DJ. Targeting of cell-surface beta-amyloid precursor protein to lysosomes: alternative processing into amyloid-bearing fragments. Nature. 1992;357:500–503. doi: 10.1038/357500a0. [DOI] [PubMed] [Google Scholar]
- Haenggi T, Schaub MC, Fritschy JM. Molecular heterogeneity of the dystrophin-associated protein complex in the mouse kidney nephron: differential alterations in the absence of utrophin and dystrophin. Cell Tissue Res. 2005;319:299–313. doi: 10.1007/s00441-004-0999-y. [DOI] [PubMed] [Google Scholar]
- Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science. 2002;297:353–356. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- Herz J. Apolipoprotein E receptors in the nervous system. Curr Opin Lipidol. 2009;20:190–196. doi: 10.1097/MOL.0b013e32832d3a10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hickman SE, Allison EK, El Khoury J. Microglial dysfunction and defective β-amyloid clearance pathways in aging Alzheimer's disease mice. J Neurosci. 2008;28:8354–8360. doi: 10.1523/JNEUROSCI.0616-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiesberger T, Trommsdorff M, Howell BW, Goffinet A, Mumby MC, Cooper JA, Herz J. Direct binding of Reelin to VLDL receptor and ApoE receptor 2 induces tyrosine phosphorylation of disabled-1 and modulates tau phosphorylation. Neuron. 1999;24:481–489. doi: 10.1016/s0896-6273(00)80861-2. [DOI] [PubMed] [Google Scholar]
- Hoe HS, Rebeck GW. Regulated proteolysis of APP and ApoE receptors. Mol Neurobiol. 2008;37:64–72. doi: 10.1007/s12035-008-8017-0. [DOI] [PubMed] [Google Scholar]
- Hoe HS, Tran TS, Matsuoka Y, Howell BW, Rebeck GW. Dab1 and Reelin effects on APP and ApoEr2 trafficking and processing. J Biol Chem. 2006;281:35176–35185. doi: 10.1074/jbc.M602162200. [DOI] [PubMed] [Google Scholar]
- Hoe HS, Lee KJ, Carney RS, Lee J, Markova A, Lee JY, Howell BW, Hyman BT, Pak DT, Bu G, Rebeck GW. Interaction of reelin with amyloid precursor protein promotes neurite outgrowth. J Neurosci. 2009;29:7459–7473. doi: 10.1523/JNEUROSCI.4872-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jossin Y, Gui L, Goffinet AM. Processing of Reelin by embryonic neurons is important for function in tissue but not in dissociated cultured neurons. J Neurosci. 2007;27:4243–4252. doi: 10.1523/JNEUROSCI.0023-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knobloch M, Konietzko U, Krebs DC, Nitsch RM. Intracellular Abeta and cognitive deficits precede beta-amyloid deposition in transgenic arcAbeta mice. Neurobiol Aging. 2007;28:1297–1306. doi: 10.1016/j.neurobiolaging.2006.06.019. [DOI] [PubMed] [Google Scholar]
- Knuesel I, Riban V, Zuellig RA, Schaub MC, Grady RM, Sanes JR, Fritschy JM. Increased vulnerability to kainate-induced seizures in utrophin-knockout mice. Eur J Neurosci. 2002;15:1474–1484. doi: 10.1046/j.1460-9568.2002.01980.x. [DOI] [PubMed] [Google Scholar]
- Knuesel I, Nyffeler M, Mormède C, Muhia M, Meyer U, Pietropaolo S, Yee BK, Pryce CR, LaFerla FM, Marighetto A, Feldon J. Age-related accumulation of Reelin in amyloid-like deposits. Neurobiol Aging. 2009;30:697–716. doi: 10.1016/j.neurobiolaging.2007.08.011. [DOI] [PubMed] [Google Scholar]
- Koch S, Strasser V, Hauser C, Fasching D, Brandes C, Bajari TM, Schneider WJ, Nimpf J. A secreted soluble form of ApoE receptor 2 acts as a dominant-negative receptor and inhibits Reelin signaling. EMBO J. 2002;21:5996–6004. doi: 10.1093/emboj/cdf599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubo K, Mikoshiba K, Nakajima K. Secreted Reelin molecules form homodimers. Neurosci Res. 2002;43:381–388. doi: 10.1016/s0168-0102(02)00068-8. [DOI] [PubMed] [Google Scholar]
- Lambert de Rouvroit C, de Bergeyck V, Cortvrindt C, Bar I, Eeckhout Y, Goffinet AM. Reelin, the extracellular matrix protein deficient in reeler mutant mice, is processed by a metalloproteinase. Exp Neurol. 1999;156:214–217. doi: 10.1006/exnr.1998.7007. [DOI] [PubMed] [Google Scholar]
- Levy-Lahad E, Wasco W, Poorkaj P, Romano DM, Oshima J, Pettingell WH, Yu CE, Jondro PD, Schmidt SD, Wang K. Candidate gene for the chromosome 1 familial Alzheimer's disease locus. Science. 1995;269:973–977. doi: 10.1126/science.7638622. [DOI] [PubMed] [Google Scholar]
- Liao YF, Wang BJ, Cheng HT, Kuo LH, Wolfe MS. Tumor necrosis factor-alpha, interleukin-1beta, and interferon-gamma stimulate gamma-secretase-mediated cleavage of amyloid precursor protein through a JNK-dependent MAPK pathway. J Biol Chem. 2004;279:49523–49532. doi: 10.1074/jbc.M402034200. [DOI] [PubMed] [Google Scholar]
- Madhusudan A, Sidler C, Knuesel I. Accumulation of reelin-positive plaques is accompanied by a decline in basal forebrain projection neurons during normal aging. Eur J Neurosci. 2009;30:1064–1076. doi: 10.1111/j.1460-9568.2009.06884.x. [DOI] [PubMed] [Google Scholar]
- McGeer PL, Itagaki S, Tago H, McGeer EG. Reactive microglia in patients with senile dementia of the Alzheimer type are positive for the histocompatibility glycoprotein HLA-DR. Neurosci Lett. 1987;79:195–200. doi: 10.1016/0304-3940(87)90696-3. [DOI] [PubMed] [Google Scholar]
- Ohkubo N, Lee YD, Morishima A, Terashima T, Kikkawa S, Tohyama M, Sakanaka M, Tanaka J, Maeda N, Vitek MP, Mitsuda N. Apolipoprotein E and Reelin ligands modulate tau phosphorylation through an apolipoprotein E receptor/disabled-1/glycogen synthase kinase-3beta cascade. FASEB J. 2003;17:295–297. doi: 10.1096/fj.02-0434fje. [DOI] [PubMed] [Google Scholar]
- Qiu S, Weeber EJ. Reelin signaling facilitates maturation of CA1 glutamatergic synapses. J Neurophysiol. 2007;97:2312–2321. doi: 10.1152/jn.00869.2006. [DOI] [PubMed] [Google Scholar]
- Qiu S, Zhao LF, Korwek KM, Weeber EJ. Differential reelin-induced enhancement of NMDA and AMPA receptor activity in the adult hippocampus. J Neurosci. 2006;26:12943–12955. doi: 10.1523/JNEUROSCI.2561-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajendran L, Honsho M, Zahn TR, Keller P, Geiger KD, Verkade P, Simons K. Alzheimer's disease beta-amyloid peptides are released in association with exosomes. Proc Natl Acad Sci U S A. 2006;103:11172–11177. doi: 10.1073/pnas.0603838103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajendran L, Schneider A, Schlechtingen G, Weidlich S, Ries J, Braxmeier T, Schwille P, Schulz JB, Schroeder C, Simons M, Jennings G, Knölker HJ, Simons K. Efficient inhibition of the Alzheimer's disease beta-secretase by membrane targeting. Science. 2008;320:520–523. doi: 10.1126/science.1156609. [DOI] [PubMed] [Google Scholar]
- Rogaev EI, Sherrington R, Rogaeva EA, Levesque G, Ikeda M, Liang Y, Chi H, Lin C, Holman K, Tsuda T. Familial Alzheimer's disease in kindreds with missense mutations in a gene on chromosome 1 related to the Alzheimer's disease type 3 gene. Nature. 1995;376:775–778. doi: 10.1038/376775a0. [DOI] [PubMed] [Google Scholar]
- Sáez-Valero J, Costell M, Sjögren M, Andreasen N, Blennow K, Luque JM. Altered levels of cerebrospinal fluid reelin in frontotemporal dementia and Alzheimer's disease. J Neurosci Res. 2003;72:132–136. doi: 10.1002/jnr.10554. [DOI] [PubMed] [Google Scholar]
- Savla GN, Palmer BW. Neuropsychology in Alzheimer's disease and other dementia research. Curr Opin Psychiatry. 2005;18:621–627. doi: 10.1097/01.yco.0000184413.36706.8e. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ. Toward a comprehensive theory for Alzheimer's disease. Hypothesis: Alzheimer's disease is caused by the cerebral accumulation and cytotoxicity of amyloid beta-protein. Ann N Y Acad Sci. 2000;924:17–25. doi: 10.1111/j.1749-6632.2000.tb05554.x. [DOI] [PubMed] [Google Scholar]
- Seripa D, Matera MG, Franceschi M, Daniele A, Bizzarro A, Rinaldi M, Panza F, Fazio VM, Gravina C, D'Onofrio G, Solfrizzi V, Masullo C, Pilotto A. The RELN locus in Alzheimer's disease. J Alzheimers Dis. 2008;14:335–344. doi: 10.3233/jad-2008-14308. [DOI] [PubMed] [Google Scholar]
- Sherrington R, Rogaev EI, Liang Y, Rogaeva EA, Levesque G, Ikeda M, Chi H, Lin C, Li G, Holman K. Cloning of a gene bearing missense mutations in early-onset familial Alzheimer's disease. Nature. 1995;375:754–760. doi: 10.1038/375754a0. [DOI] [PubMed] [Google Scholar]
- Utsunomiya-Tate N, Kubo K, Tate S, Kainosho M, Katayama E, Nakajima K, Mikoshiba K. Reelin molecules assemble together to form a large protein complex, which is inhibited by the function-blocking CR-50 antibody. Proc Natl Acad Sci U S A. 2000;97:9729–9734. doi: 10.1073/pnas.160272497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vassar R, Bennett BD, Babu-Khan S, Kahn S, Mendiaz EA, Denis P, Teplow DB, Ross S, Amarante P, Loeloff R, Luo Y, Fisher S, Fuller J, Edenson S, Lile J, Jarosinski MA, Biere AL, Curran E, Burgess T, Louis JC, et al. Beta-secretase cleavage of Alzheimer's amyloid precursor protein by the transmembrane aspartic protease BACE. Science. 1999;286:735–741. doi: 10.1126/science.286.5440.735. [DOI] [PubMed] [Google Scholar]
- von Rotz RC, Kohli BM, Bosset J, Meier M, Suzuki T, Nitsch RM, Konietzko U. The APP intracellular domain forms nuclear multiprotein complexes and regulates the transcription of its own precursor. J Cell Sci. 2004;117:4435–4448. doi: 10.1242/jcs.01323. [DOI] [PubMed] [Google Scholar]
- Walsh DM, Selkoe DJ. Deciphering the molecular basis of memory failure in Alzheimer's disease. Neuron. 2004;44:181–193. doi: 10.1016/j.neuron.2004.09.010. [DOI] [PubMed] [Google Scholar]
- Weeber EJ, Beffert U, Jones C, Christian JM, Forster E, Sweatt JD, Herz J. Reelin and ApoE receptors cooperate to enhance hippocampal synaptic plasticity and learning. J Biol Chem. 2002;277:39944–39952. doi: 10.1074/jbc.M205147200. [DOI] [PubMed] [Google Scholar]
- Wyss-Coray T. Inflammation in Alzheimer disease: driving force, bystander or beneficial response? Nat Med. 2006;12:1005–1015. doi: 10.1038/nm1484. [DOI] [PubMed] [Google Scholar]
- Yamamoto M, Kiyota T, Horiba M, Buescher JL, Walsh SM, Gendelman HE, Ikezu T. Interferon-gamma and tumor necrosis factor-alpha regulate amyloid-beta plaque deposition and beta-secretase expression in Swedish mutant APP transgenic mice. Am J Pathol. 2007;170:680–692. doi: 10.2353/ajpath.2007.060378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasui N, Nogi T, Kitao T, Nakano Y, Hattori M, Takagi J. Structure of a receptor-binding fragment of reelin and mutational analysis reveal a recognition mechanism similar to endocytic receptors. Proc Natl Acad Sci U S A. 2007;104:9988–9993. doi: 10.1073/pnas.0700438104. [DOI] [PMC free article] [PubMed] [Google Scholar]