

Abstract
The balance between self-renewal and differentiation of neural progenitor cells is an absolute requirement for the correct formation of the nervous system. Much is known about both the pathways involved in progenitor cell self-renewal, such as Notch signaling, and the expression of genes that initiate progenitor differentiation. However, whether these fundamental processes are mechanistically linked, and specifically how repression of progenitor self-renewal pathways occurs, is poorly understood. Nuclear factor I A (Nfia), a gene known to regulate spinal cord and neocortical development, has recently been implicated as acting downstream of Notch to initiate the expression of astrocyte-specific genes within the cortex. Here we demonstrate that, in addition to activating the expression of astrocyte-specific genes, Nfia also downregulates the activity of the Notch signaling pathway via repression of the key Notch effector Hes1. These data provide a significant conceptual advance in our understanding of neural progenitor differentiation, revealing that a single transcription factor can control both the activation of differentiation genes and the repression of the self-renewal genes, thereby acting as a pivotal regulator of the balance between progenitor and differentiated cell states.
Introduction
Within the developing nervous system, generation of distinct cellular subtypes from radial progenitor cells occurs in a temporally and sequentially distinct fashion, such that neurons are generated first, followed by astrocytes and finally oligodendrocytes. The Notch pathway plays a central role in this process by regulating neural progenitor cell self-renewal (Shimojo et al., 2008). Activation of the Notch receptor results in cleavage and release of the Notch intracellular domain (NICD). The NICD is then translocated to the nucleus, where, in association with the DNA-binding protein RBPj, it induces expression of downstream target genes (Louvi and Artavanis-Tsakonas, 2006). These effector genes include the basic helix-loop-helix transcription factors Hes1 and Hes5, which maintain neural progenitor cells via inhibition of proneural gene expression (Bertrand et al., 2002).
Recent studies have begun to elucidate genes responsible for driving neuronal- or glial-lineage specification (Götz and Huttner, 2005). For instance, the transcription factor nuclear factor I A (Nfia) regulates gliogenesis within the embryonic spinal cord (Deneen et al., 2006). Nfia−/− mice also exhibit aberrant cortical glial development (Shu et al., 2003), and nuclear factor I (NFI) gene family members have been shown to directly bind and regulate the expression of glial-specific genes in vitro (Bisgrove et al., 2000; Cebolla and Vallejo, 2006). Recently it was demonstrated that activation of the Notch pathway in telencephalic neural progenitor cells in vitro culminates in Nfia expression (Namihira et al., 2009), with Nfia overexpression causing demethylation of the STAT3 binding site on the glial fibrillary acidic protein (Gfap) promoter (Namihira et al., 2009). These findings link Notch activity to induction of Nfia expression in neural progenitors, leading to gliogenesis via demethylation of astrocyte-specific genes responsive to JAK-STAT signaling.
Despite these advances, our understanding of the process by which Notch-mediated self-renewal of progenitors is repressed to enable neuronal or glial differentiation to proceed is unclear. Here we investigate progenitor cell differentiation in Nfia−/− mice to address this question. NFIA is expressed within ventricular zone progenitors early in telencephalic development, before astrocyte formation. Moreover, the differentiation of telencephalic ventricular zone progenitors is delayed in Nfia−/− mice. Expression of Hes1 is significantly upregulated in the telencephalic ventricular zone of Nfia mutants, and in silico promoter analysis and chromatin immunoprecipitation assays demonstrate that the promoter of this gene contains a cluster of highly conserved NFI binding sites that is bound by NFIA in vivo. Reporter gene assays further reveal that NFIA is able to directly repress Hes1 promoter activity. Collectively, our data demonstrate that, in addition to its role in activating the expression of astrocyte-specific genes, Nfia plays a crucial role in repressing expression of the key Notch effector Hes1. Thus, Nfia regulates both the repression of the progenitor cell state and the activation of differentiation gene programs that regulate neural development.
Materials and Methods
Animals.
Wild-type C57BL/6J and Nfia−/− mice were used in this study. These animals were bred at the University of Queensland under approval from the institutional Animal Ethics Committee. Embryos were genotyped by PCR (Shu et al., 2003). The Nfia−/− mice were backcrossed for >10 generations onto the C57BL/6J background. No hippocampal defects were detected in wild-type or heterozygote animals. Timed-pregnant females were acquired by placing male and female mice together overnight. The next day, females were inspected for the presence of a vaginal plug and, if present, this day was designated as embryonic day 0. Heterozygous Nfia mutant mice were bred to obtain wild-type, heterozygous, and homozygous progeny.
Reverse transcription and quantitative real-time PCR.
Hippocampi were dissected and snap frozen. Total RNA was extracted using an RNeasy Micro Kit (Qiagen). Reverse transcription was performed using Superscript III (Invitrogen). A 0.5 μg quantity of total RNA was reverse transcribed with random hexamers. Quantitative PCRs (qPCRs) were performed in a Rotor-Gene 3000 (Corbett Life Science) using the SYBR Green PCR Master Mix (Invitrogen). All the samples were diluted 1/100 with RNase/DNase-free water, and 5 μl of these dilutions was used for each SYBR Green PCR containing 10 μl of SYBR Green PCR Master Mix, a 10 μm concentration of each primer, and deionized water. The reactions were incubated for 10 min at 95°C followed by 40 cycles with 15 s denaturation at 95°C, 20 s annealing at 60°C, and 30 s extension at 72°C.
Generation of gene-specific qPCR standards.
The synthesis of these primers was performed by Sigma-Genosys. The following primer sequences were used: Tbr1 forward (GGACACCAATGTGCAAGGAA), Tbr1 reverse (TTTCTTGCCGCATCCAGTGA), Gfap forward (AGTGGTATCGGTCTAAGTTTG), Gfap reverse (CGATAGTCGTTAGCTTCGTG), Glast forward (CACTGCTGTCATTGTGGGTA), Glast reverse (AGCATCCTCATGAGAAGCTC), KA1 forward (GAACTACCTTCGCATGGTAG), KA1 reverse (GCCAATCTGCTGAAACCCAT), nestin forward (GAAGTGGCTACATACAGGAC), nestin reverse (AGCTTCAGCTTGGGGTCAG), Nfia forward (CTTTGTACATGCAGCAGGAC), Nfia reverse (TTCCTGCAGCTATTGGTGTTT), Prox1 forward (TTATTCAGGAAGCGCAATGCA), Prox1 reverse (GAAGTAGGTCTTCAGCATGTT), SCIP forward (TTCGCCAAGCAGTTCAAGCAA), SCIP reverse (TGGTCTGCGAGAACACGTTA), s100β forward (ATTGATGTCTTCCACCAGTAC), s100β reverse (TCCTGCTCCTTGATTTCCTC), Id4 forward (TCACCCTGCTTTGCTGAGACA), Id4 reverse (AGCTCAGCGGCAGAGAATG), Sox8 forward (ATGAATGCCTTCATGGTGTGG), Sox8 reverse (CTCGCTTTCACTCAGCAAGC), Sox10 forward (ACTAGGCAAGCTCTGGAGG), Sox10 reverse (AGGTTGGTACTTGTAGTCCG), Sox3 forward (CAAGAAGGACAAGTACTCGC), Sox3 reverse (TTCACGTGCGTGTACGTGTC), Hey1 forward (GAGAGGCATCATCGAGAAG), Hey1 reverse (TCTAGCTTAGCAGATCCCTG), Ngn2 forward (TTCGCCCACAATTACATCTG), Ngn2 reverse (AGGAGGAAGGTGGAGAAGG), Mash1 forward (ACTGATGCGCTGCAAACGC), Mash1 reverse (GACCAACTTGACCCGGTTG), Notch1 forward (GCTGGACAGAACTGTGAAGA), Notch1 reverse (TCTGTACAGTACTGACCCGT), Hes1 forward (TCTGACCACAGAAAGTCATCA), Hes1 reverse (AGCTATCTTTCTTAAGTGCATC), Hes5 forward (CCAAGGAGAAAAACCGACTG), Hes5 reverse (AACTCCTGCTCCAGCAGCA), Olig1 forward (GCGAAGTTATCCTACCCTACT), Olig1 reverse (AGCGAGCTACCCAGCAGCA), Olig2 forward (AGACAAGAAGCAGATGACTGA), Olig2 reverse (TGGCGATGTTGAGGTCGTG), tenascin C forward (GATAGACTGCTCTGAGGTGT), tenascin C reverse (TCAGTGACCCGCATCTCATT), Dll1 forward (CGCTACTGCGATGAGTGCA), Dll1 reverse (TGAGTACAGTAGTTCAGGTCT), HPRT forward (GCAGTACAGCCCCAAAATGG), and HPRT reverse (AACAAAGTCTGGCCTGTATCCAA).
qPCR data expression and analysis.
After completion of the PCR amplification, the data were analyzed with the Rotor-Gene software as described previously (Piper et al., 2009b). When quantifying the mRNA expression levels, the housekeeping gene HPRT was used as a relative standard. All the samples were tested in triplicate. By means of this strategy, we achieved a relative PCR kinetic of standard and sample. For all qPCR analyses, RNA from three independent replicates for both wild-type and Nfia−/− mice were interrogated. Statistical analyses were performed using a two-tailed unpaired t test. Error bars represent the SEM.
Immunohistochemistry.
Embryos (E14 and below) were drop-fixed in 4% paraformaldehyde (PFA) or transcardially perfused with 0.9% saline, followed by 4% PFA (E15–E18), then postfixed in 4% PFA at 4°C. Brains were removed and sectioned at 50 μm on a vibratome (Leica). Immunohistochemistry using the chromogen 3,3′-diaminobenzidine was performed as described previously (Campbell et al., 2008). For all immunohistochemical analyses, three wild-type and Nfia−/− brains were analyzed. Sections from comparable positions along the rostrocaudal axis were imaged using an upright microscope (Zeiss Z1). Immunofluorescence labeling was performed as described above with minor modifications. The primary antibody was incubated overnight at 4°C, and sections were washed and incubated in secondary antibody, before being mounted. Fluorescence confocal microscopic images revealing expression of hippocampal markers were obtained with a laser scanning microscope (Zeiss 510 Meta). Hippocampal cell culture was performed as described previously (Barry et al., 2008).
Immunohistochemistry on paraffin sections.
Brains were perfused as above, embedded in paraffin, and sectioned coronally at 6 μm. Hematoxylin staining and immunohistochemistry were performed as described previously (Barry et al., 2008).
Antibody parameters.
Primary antibodies used for immunohistochemistry were anti-Tbr1 and anti-Tbr2 (rabbit polyclonal, a gift from Robert Hevner, University of Washington, Seattle, WA, 1/100,000 and 1/30,000 respectively), anti-Prox1 (rabbit polyclonal, 1/100,000; Millipore Bioscience Research Reagents), anti-Pax6 (rabbit polyclonal, 1/25,000; Millipore Bioscience Research Reagents), anti-GFAP (rabbit polyclonal, 1/50,000; DAKO), anti-GLAST (rabbit polyclonal, a gift from Niels Danbolt, University of Oslo, Oslo, Norway, 1/50,000), anti-nestin (mouse monoclonal, 1/1500; Developmental Studies Hybridoma Bank), anti-cleaved caspase 3 (rabbit polyclonal, 1/1000; Cell Signaling Technology), and anti-tenascin C (rabbit polyclonal, 1/2000; Millipore Bioscience Research Reagents). Secondary antibodies used were biotinylated goat-anti-rabbit IgG and biotinylated donkey-anti-mouse IgG (1/500 to 1/2000; Vector Laboratories). The primary antibodies used for immunofluorescent labeling were anti-NFIA (1/1000; rabbit polyclonal, Active Motif), anti β-galactosidase (1/1000; mouse monoclonal, Promega), anti-βIII tubulin (Tuj1 clone; mouse monoclonal, 1/1000; R&D Systems), and anti-GFAP (mouse monoclonal, 1/1000; Millipore Bioscience Research Reagents). The secondary antibodies were goat anti-rabbit IgG AlexaFluor 594 and goat anti-mouse IgG AlexaFluor 488 (each 1/1000; Invitrogen). Primary antibodies used for immunohistochemistry on paraffin sections were anti-NFIA (rabbit polyclonal, 1/1000; Active Motif) and anti-phosphohistone-H3 (rabbit polyclonal, 1/1000; Millipore). A biotinylated goat anti-rabbit IgG secondary antibody (Vector Laboratories) was used at 1/1000.
Quantification of ventricular zone width/hippocampal cell counts.
For measurements of ventricular zone width and phosphohistone-H3 (PH-H3) cell counts, E14, E16, and E18 wild-type and Nfia−/− brains were processed for paraffin sectioning and cut coronally at 6 μm. To measure the ventricular zone width, sections were hematoxylin stained and imaged with an upright microscope (Zeiss ZI) coupled to AxioVision software (Zeiss). The width of the ventricular zone was measured at three points along the hippocampus for each section. Data for both wild-type and knock-out hippocampi were then pooled for comparison of ventricular zone width. For PH-H3 cell counts, the total number of PH-H3-positive cells in the ventricular zone of each hippocampus was counted. For counts of Pax6-positive and Tbr2-positive cells within the hippocampus, the number of immunopositive cells per 100 μm was quantified using confocal images of fluorescently labeled vibratome sections at E16. Images were acquired using a LSM 510 Meta (Zeiss). For the BrdU/Tbr1 hippocampal cell counts, the number of cells in which Tbr1 and BrdU immunoreactivity was colocalized per 300 μm was quantified using confocal images of fluorescently labeled vibratome sections at E16. Images were acquired using a LSM 510 Meta (Zeiss). For all experiments, data represent pooled results from at least 3 wild-type and 3 Nfia−/− brains. Counts were performed blind to the genotype of the sample. Statistical analyses were performed using a two-tailed unpaired t test. Error bars represent the SEM.
In situ hybridization.
Embryos were collected and fixed as described above (n = 3 for both wild-type and knock-out). In situ hybridization was performed using antisense probes as previously described (Piper et al., 2009a) with minor modifications. The hybridization temperatures were 68–70°C. The color reaction solution was BM Purple (Roche). In situ probes were kindly provided by Ryoichiro Kageyama (Kyoto University, Kyoto, Japan) (Hes1 and Hes5), Sally Dunwoodie (Victor Chang Cardiac Research Institute, Sydney, Australia) (Dll1), Jane Johnson (University of Texas, Dallas, TX) (Mash1), and Francois Guillemot (National Institute for Medical Research, London, UK) (Ngn2).
BrdU birth dating.
Pregnant dams were injected with 5-bromo-2′-deoxyuridine (BrdU; 50 mg/g mouse) at E14. Embryos were perfuse-fixed at E16 and processed for immunohistochemistry as described above.
Hippocampal microarrays.
E16 hippocampal tissue was dissected in ice-cold PBS using fine forceps and a dissecting microscope. Tissue was immediately snap frozen in liquid nitrogen. Individual hippocampal samples from Nfia−/− mice and wild-type controls were collected (n = 4 for wild-type and n = 5 for mutant). Total RNA was extracted using the RNeasy Micro Kit (Qiagen). Total RNA concentration was determined using the NanoDrop ND-1000 UV-Vis Spectrophotometer (NanoDrop Technologies) and RNA integrity was confirmed using the Agilent 2100 Bioanalyzer RNA 600 NanoChip (Agilent). Microarray hybridization, washing, and scanning was performed at the ARC (Australian Research Council) Special Research Centre for Functional and Applied Genomics microarray facility (The University of Queensland). Initially, 250 ng of total RNA from each sample was amplified using the Illumina RNA TotalPrep Amplification kit (Ambion) following the manufacturer's instructions. The in vitro transcription reaction was conducted for 14 h and included the labeling of the cRNA by biotinylation.
Labeled and amplified material (1.5 μg/sample) was hybridized to Illumina's Sentrix Mouse-6 Expression BeadChips (Illumina) at 55°C for 18 h according to the Illumina BeadStation 500× protocol. Arrays were washed and then stained with 1 μg/ml cyanine3-streptavidin (GE Healthcare). The Illumina BeadArray reader was used to scan the arrays according to the manufacturer's instructions. Samples were initially evaluated using the BeadStudio software from Illumina. Quality control reports were satisfactory for all samples.
The raw data were then imported into GeneSpring GX v7.3 (Agilent) for data visualization and downstream analysis. Data were initially filtered using GeneSpring normalization algorithms. Quality control data filtering was then performed to remove signals that were not present in at least 7 of 9 of the samples using the bead detection score p value, and with expression values below background, as determined by the cross-gene error model. Differential expression was determined by the one-way ANOVA-Welch's approximate t test without a multiple testing correction. A cutoff p value of 0.05 was used for the mean difference between wild-type and Nfia mutant hippocampal tissue. In addition, a 1.5-fold-change filter was imposed on the genes from the ANOVA dataset.
In silico promoter analysis. A number of genes from the expression analysis were identified as potential direct targets for regulation by NFI. For each gene the University of California Santa Cruz (UCSC) table browser was used to extract the coordinates of 2 kb of sequence upstream of the transcription start site. These genomic coordinates were used to extract a multiple alignment file from the UCSC multiple alignment table multiz30way with mouse as the reference genome. From the multiple alignment sequences were extracted for the mouse, rat, rabbit, and guinea pig genomes. A phylogenetic tree relating these species extracted from the UCSC genome wiki (http://genomewiki.ucsc.edu/index.php/Mm9_multiple_alignment) was used such that phylogenetic relationships in the Newick tree format are taken to be as follows: (((Mouse:0.0763, Rat:0.0844):0.2006, GuineaPig:0.2030):0.0344, Rabbit:0.2085). As our binding site motif, we began with the consensus sequence TTGGC(N5)GCCAA, which has been shown to be the highest-affinity binding site for NFI proteins (Gronostajski, 2000). The consensus sequence was converted directly into a probability matrix. However, to allow for mismatches in the binding site, these probabilities were altered by converting to a frequency matrix under the assumption that we had only five observed binding sites. We then applied a pseudocount of 0.38, shown to be a near-optimal value (Frith et al., 2008), and converted back to a probability matrix. The background frequencies were calculated using only the 2 kb promoter sequences from the mouse genome and used these proportions when applying the pseudocount.
Each of the multiple alignments of promoter sequence was scanned using the Monkey algorithm (Moses et al., 2004). (Note that for some of the promoter regions, there was insufficient sequence in one or more of the genomes, so that the scan was performed over only a subset of the four genomes.) Monkey uses a phylogenetic motif model, which assumes that motif sites are evolving under selective pressure to maintain the affinity to the transcription factor. The algorithm scans a multiple alignment of sequences and produces p values that are estimates of the probability of getting a score as good or better under the assumption that the given section of the multiple alignment evolved from a common ancestor according to a “background” model (HKY) and were not under any selective pressure. To account for multiple testing, we multiplied each predicted site's p value by 2000, the approximate number of positions that could match the motif in a given upstream region. The corrected values were referred to as E values, and represented the expected number of randomly chosen upstream regions that would contain a motif site matching the model as well as the predicted site. As an additional estimate of the reliability of predicted sites, we also estimated the false discovery rate using a technique for estimating the null score distribution (Kheradpour et al., 2007). The columns of the NFI motif were shuffled multiple times to generate candidate null motifs. A number of filters to the candidates (Hawkins et al., 2009) were applied so that we were left with a subset of motifs that did not resemble known motifs. The promoter regions were scanned with these motifs and the average number of hits was taken as an estimate of the number of false positives at a given threshold.
The upstream regions for 11 selected genes were scanned using this method. The genes we selected were Hes1, Hes5, Id4, Hey1, Dll1, Hey2, Gfap, SCIP, Glast, Prox1, and Calretinin. The results are shown in supplemental Table 3 (available at www.jneurosci.org as supplemental material). An extremely probable NFI binding site was found in the Hes1 promoter region (E value = 0.009) and a highly probable site in the SCIP promoter region (E value = 0.07). This strongly suggests that both Hes1 and SCIP are direct NFI targets. Seven of the other eleven selected upstream contain putative, conserved binding sites predicted by Monkey with E values <1.0. Although such large E values are not statistically significant by themselves, the upstream regions of several genes have multiple predicted NFI binding sites. These additional sites and their location relative to the transcription start site lend added support to the prediction that they are direct NFI targets.
The evidence of Hes1 being a direct NFI target provided by the highly conserved site is further increased by the occurrence of two additional potential conserved sites with E values <0.4 in the region we scanned. One would only expect 0.4 matches, so this represents an enrichment of over twofold. Finally, all three of the predicted Hes1 sites are within 200 bp upstream of the transcription start site. Together, these three sites strongly suggest that Hes1 is a direct NFI target.
The Hes5 upstream region contains three possible conserved NFI binding sites with E values <0.5 (representing an approximately sixfold enrichment over what would be expected by chance). Furthermore, the most highly conserved site (E value = 0.3) is located just 169 bp upstream of the transcription start site. We therefore believe Hes5 to be a probable direct regulatory target of NFI.
Support for our prediction that Gfap is a direct NFI target is provided by three sites with E values under 0.5 (sixfold enrichment over chance). Supplemental Table 3 (available at www.jneurosci.org as supplemental material) shows four sites, the most highly conserved of which is only 482 bp upstream of the transcription start site and has an E value of 0.2. There is another, less conserved site 385 bp upstream of the transcription start site (E value 0.49), suggesting that these two putative sites may indicate NFI regulation of Gfap via binding near the proximal promoter. There are two additional putative sites that add additional evidence of direct regulation by NFI, although they should be treated as a single site at ∼1951 bp upstream of the transcription start site (E value 0.2). This site has previously been shown to be bound by NFI proteins in vitro (Miura et al., 1990). The occurrence of (effectively) three conserved putative NFI sites upstream of Gfap suggests that it may also be directly regulated by NFI.
The putative conserved NFI binding sites we predict in the upstream regions of Id4, Hey1, Glast, Prox1, and calretinin are only suggestive, since their E values are not significant, and they each contain only one predicted site. Given the well known difficulty of predicting transcription factor binding sites in silico, however, it may well be that these genes are bona fide NFI targets.
Electrophoretic mobility shift assay.
Total cortex was removed from E18 brains to isolate nuclear extracts. Protease inhibitor tablets (Roche) were added to the extraction buffers (Smith et al., 1998). Electrophoretic mobility shift assay (EMSA) was performed using radiolabeled annealed oligonucleotides containing a control NFI consensus site (Pham et al., 2004) or Hes1 consensus sites, designated Hes1-A, Hes1-B, and Hes1-C. EMSA reactions were performed as described previously using 1 μg of nuclear extract and 1 μg of poly-[dI-dC] as nonspecific competitor per reaction (Smith et al., 1998). Oligonucleotide sequences were as follows: NFI control, 5′-ggTTTTGGATTGAAGCCAATATGATAA-3′ (upper strand), 5′-ggTTATCATATTGGCTTCAATCCAAAA-3′ (lower strand); Hes1-A, 5′-ggCGCCATTGGCCGCCAGACCTTGTG-3′ (upper strand), 5′-ggCACAAGGTCTGGCGGCCAATGGCG-3′ (lower strand); Hes1-B, 5′-ggCCTTGTGCCTAGCGGCCAATGGGG-3′ (upper strand), 5′-ggCCCCATTGGCCGCTAGGCACAAGG-3′ (lower strand); Hes1-C, 5′-ggTCCCATTGGCTGAAAGTTACTGTGG-3′ (upper strand), 5′-ggCCACAGTAACTTTCAGCCAATGGGA-3′ (lower strand). Additional bases used to generate 5′ overhangs for endfill are indicated in lower case.
Chromatin immunoprecipitation.
Total cortex was removed from E18 brains. The chromatin immunoprecipitation (ChIP) assay was performed using ChIP-IT Express kit (Active Motif) as per the manufacturer's instructions. Tissue was homogenized and then fixed in 37% formaldehyde for 10 min followed by 1 ml of glycine buffer for 5 min. Cells were pelleted, washed, then resuspended in lysis buffer for 30 min. Following centrifugation, the pellet was resuspended in shearing buffer with protease inhibitors. Sonication was performed using a Bioruptor (Diagenode), after which the supernatant was used as the input sample for immunoprecipitation experiments. Antibodies used were mouse NFIA (Active Motif), IgG (Active Motif), and RNA polymerase II (Active Motif). The anti-NFIA antibody has previously been shown to be highly specific for NFIA (Plachez et al., 2008). Protein G-coated magnetic beads were used to purify specific antibody/DNA complexes. Following washes in supplied ChIP buffers, DNA was dissociated from the antibody using the supplied elution buffer AM2 and reverse cross-linking buffer. Samples were then incubated at 95°C for 15 min before proteinase K treatment for 1 h at 37°C. DNA fragments bound to NFIA or IgG antibodies were tested by PCR for specific promoter regions predicted to be bound by NFIA.
Primer sequences for chromatin immunoprecipitation.
Sequences for PCR primers used were as follows: Gfap (promoter) forward CTTGACTCTGGGTACAGTG, reverse CCTTTATGGAGGAACGGGTT (149 bp); Gfap negative (exon 6) forward AGTTACCAGGAGGCACTTGC, reverse CGGTTTTCTTCGCCCTCCA (163 bp); Hes1 (promoter) forward GCCAGACCTTGTGCCTAG, reverse ATCTGGGACTGCACGCGAA (142 bp); Hes1 negative (exon 4) forward CCTTTCTCATCCCCAACGG, reverse CCGCCACGGTCTCCACAT (164 bp).
Luciferase reporter assay.
The constructs used in the luciferase assay were a full-length Nfia expression construct driven by the chick β-actin promoter (Nfia pCAGIG) and a construct containing the first 600 bp upstream of the transcription start site of the mouse Hes1 gene containing the predicted NFI binding sites cloned into a luciferase vector (pGL4.23; Promega). DNA was transfected into HEK 293 cells using FuGene (Invitrogen). After 48 h, luciferase activity was assessed using a luciferase substrate (Promega) as per the manufacturer's instructions.
Results
Progenitor cells of the developing hippocampus express NFIA
NFIA is expressed widely in the developing telencephalon (Plachez et al., 2008), including the hippocampus. At E14 and E16, the hippocampal ventricular zone was marked by high levels of NFIA expression (Fig. 1A,B). At E18, expression of NFIA within the ventricular zone remained prominent, and cells within the fimbria, the pyramidal neuronal layer and the emerging dentate gyrus also exhibited NFIA immunoreactivity (Fig. 1C,D). Analysis of dissociated hippocampal cultures in vitro revealed NFIA coexpression with both the neuronal marker TuJ1 and the astrocytic marker GFAP (Fig. 1E,F), demonstrating that NFIA is expressed in both neurons and astrocytes within the hippocampus. However, the expression of NFIA within the ventricular zone suggests that, in addition to regulating astrocyte-specific gene expression (Namihira et al., 2009), Nfia may modulate gene expression within neural progenitors from an early stage in telencephalic development.
Figure 1.
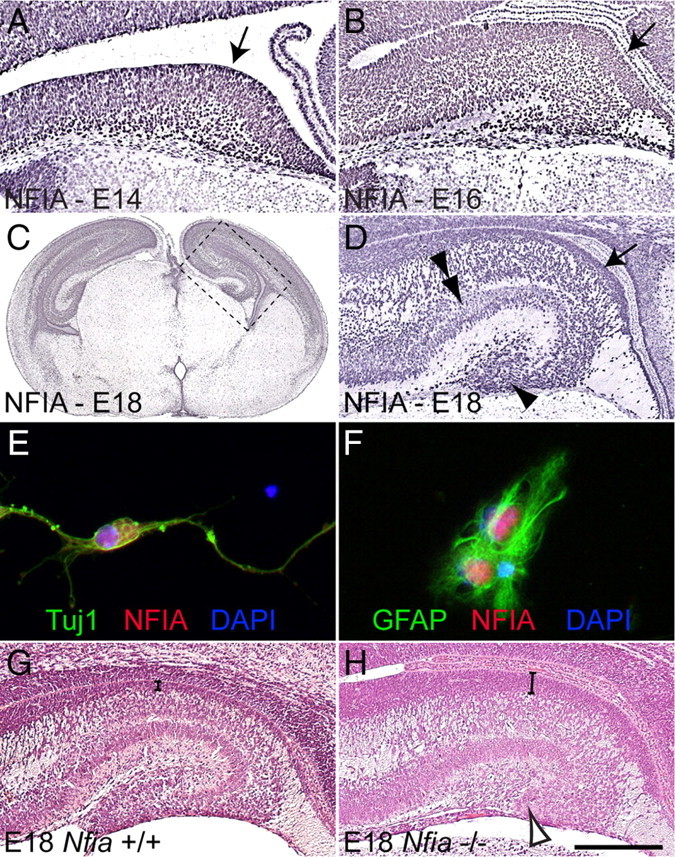
NFIA is expressed broadly within the developing hippocampus. A–D, G, H, Coronal paraffin sections of wild-type (A–D, G) and Nfia−/− (H) brains. At E14 (A) and E16 (B), progenitor cells within the ventricular zone expressed high levels of NFIA (arrows in A, B). C, D, At E18, NFIA expression in the ventricular zone was still prominent (arrow in D), while NFIA expression was also observed in the pyramidal neuronal layer (double arrowhead in D) and dentate gyrus (arrowhead in D). D is a higher-magnification view of the boxed region in C. E, F, In vitro cultures of wild-type hippocampal cells demonstrating that NFIA (red) colocalized with both the neuronal marker Tuj1 (green; E) and the mature glial marker GFAP (green; F). G, H, Blue, DAPI. Hematoxylin staining on wild-type sections at E18 (G) revealed the morphologically folded appearance of the hippocampus. In the mutant (H), however, the dentate gyrus was severely reduced (open arrowhead in H), and the ventricular zone (parentheses in G, H) was markedly thicker than in the control. Scale bar (in H): A, 100 μm; B, 250 μm; C, 1 mm; D, G, H, 300 μm; E, F, 15 μm.
Gross morphological defects are present in the hippocampus of Nfia−/− mice
To address the role played by Nfia during neural progenitor differentiation, we studied hippocampal development in mice lacking this transcription factor. Nfia−/− mice display telencephalic defects, including agenesis of the corpus callosum (Shu et al., 2003). A decrease in the number of glial processes within the dentate gyrus and fimbria has also been reported (Shu et al., 2003), but how this deficit arises is unknown. The absence of hippocampal glia was not the result of excessive apoptosis within the hippocampus of Nfia mutants, as we observed no statistically significant differences in the occurrence of apoptotic cells between mutants and wild-type controls (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). Instead, hematoxylin staining of E18 wild-type and Nfia−/− hippocampi revealed a marked increase in the size of the hippocampal ventricular zone in the mutant at this age (compare brackets in Fig. 1G,H). Furthermore, the dentate gyrus in Nfia−/− mice was morphologically reduced (arrowhead in Fig. 1H) in comparison to that in wild-type controls (Fig. 1G). Mature hippocampal glia and dentate granule neurons, both of which originate in the ammonic ventricular zone, are essential for morphogenesis of the dentate gyrus (Zhou et al., 2004; Barry et al., 2008). Thus, the severe reduction of this structure in the Nfia−/− mice is likely to be the culmination of the delayed differentiation of ventricular zone-derived progenitors.
The neural progenitor population is expanded in the hippocampus of Nfia−/− mice
During cortical development, progenitor cells begin to differentiate from midgestation onwards, and their postmitotic progeny migrate away from the ventricular zone. Consequently, the width of the ventricular zone is reduced over time. The increased ventricular zone width in the Nfia mutant at E18 indicates that differentiation of ventricular zone progenitors may be regulated by this transcription factor. To address this, we initially determined when the enlargement of the ventricular zone first became apparent in Nfia−/− mice, by quantifying the ventricular zone width at E14, E16, and E18 using hematoxylin-stained 6 μm paraffin sections. Whereas at E14 there was no statistically significant difference between the mutant and the control, at both E16 and E18 the width of the ventricular zone was significantly greater in the Nfia mutants (Fig. 2A–C). Furthermore, using immunohistochemistry against the mitotic marker PH-H3, we observed a significant increase in the number of cells undergoing mitosis within the ventricular zone of Nfia−/− hippocampi in comparison to that of littermate controls at these ages (Fig. 2D–F). Together, these data indicate that cells within the hippocampal ventricular zone of Nfia−/− mice remain in a proliferative state for longer than those in littermate controls.
Figure 2.
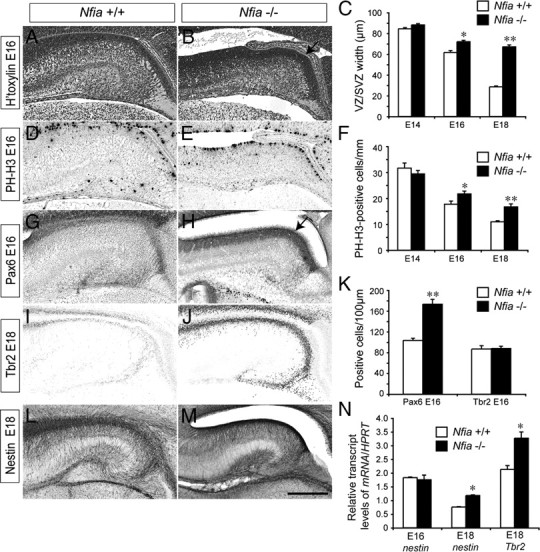
Delayed development of ventricular zone progenitors in Nfia mutants. A, B, Coronal paraffin sections of E16 wild-type and Nfia−/− brains stained with hematoxylin. The ventricular zone was wider in the mutant (arrow in B). C, Quantification of ventricular zone width at E14, E16, and E18 revealed that the ventricular zone was significantly wider in the Nfia mutants compared to controls at both E16 and E18. D, E, PH-H3 immunohistochemistry on coronal paraffin sections of E16 brains. F, Quantification of the number of PH-H3-positive cells revealed significantly more mitotic cells in the mutant ventricular zone at both E16 and E18. G, H, Immunohistochemistry against Pax6 on coronal brain sections at E16. K, Cell counts demonstrated that there were significantly more Pax6-positive cells in the ventricular zone of Nfia mutants (arrow in H) compared to controls. No difference in the number of Tbr2-positive cells within the hippocampus was detected at E16. I, J, Tbr2 immunohistochemistry at E18 revealed higher expression of Tbr2 expression in Nfia−/− hippocampi in late gestation. L, M, Expression of nestin in wild-type (L) and Nfia−/− (M) brains at E18. N, qPCR on wild-type and Nfia−/− hippocampal tissue revealed a significant increase in both nestin and Tbr2 mRNA levels in the mutant at E18. *p < 0.05, **p < 0.001, Student's t test. Scale bar (in M): A–H, 250 μm; I–M, 300 μm.
To confirm this finding, we next investigated expression of the ventricular zone neural stem cell marker Pax6 (Götz et al., 1998; Quinn et al., 2007) at E16. The Pax6-expressing ventricular zone was markedly wider in Nfia−/− sections than in wild-type controls (Fig. 2G,H). Furthermore, quantification of Pax6-positive cells demonstrated that there were significantly more immunopositive cells in the mutant (Fig. 2K). During differentiation, progenitor cells migrate out of the ventricular zone into the subventricular zone to become intermediate progenitor cells that express markers such as Tbr2, while markers such as Pax6 are downregulated (Englund et al., 2005). We hypothesized that the delay in the differentiation of ventricular zone progenitors may culminate in subsequent delays in intermediate progenitor cell formation. There was no significant difference in the number of Tbr2-positive intermediate progenitor cells between controls and Nfia knock-outs at E16 (Fig. 2K). Importantly, however, immunohistochemistry and quantitative real-time PCR (qPCR) 2 d later at E18 revealed significantly higher levels of Tbr2 in the mutant (Fig. 2I,J,N), suggesting that differentiation of intermediate progenitor cells in the subventricular zone is also delayed in the absence of Nfia.
The expansion of the hippocampal ventricular zone and the Pax6-positive progenitor pool in late gestation Nfia mutants led us to speculate that the expression of other neural stem cell markers would also be elevated in these mice. In addition to Pax6, cortical ventricular zone progenitors are characterized by expression of the intermediate filament protein nestin (Lendahl et al., 1990; Yamaguchi et al., 2000). Nestin expression in the Nfia−/− hippocampus from E14 to E18 appeared grossly normal (Fig. 2L,M and data not shown), indicating that ventricular zone progenitors are correctly specified in the absence of Nfia. Crucially, however, we observed a significant increase in the levels of nestin mRNA in the hippocampus of Nfia−/− mice at E18 (Fig. 2N). Collectively, these data indicate that Nfia plays a key role in promoting differentiation of neural stem cells within the embryonic ventricular zone, and that in its absence ventricular zone cells remain in a progenitor state.
Neuronal development is delayed in the embryonic Nfia−/− hippocampus
To address whether the expansion of the ventricular zone in the Nfia−/− hippocampus led to delayed neuronal development, we first analyzed patterning, and the formation of neurons, within the hippocampus. The expression of Wnt3a in the cortical hem of Nfia mutants at E12 was comparable to that in controls, whereas Lhx2 was excluded from the hem (supplemental Fig. 2, available at www.jneurosci.org as supplemental material), indicating that formation of this structure, which is a key signaling center for hippocampal specification (Mangale et al., 2008), occurs normally in Nfia−/− mice. Furthermore, there were no gross defects in the choroid plexus (data not shown), which expresses bone morphogenetic proteins during hippocampal development (Hébert et al., 2002). Cajal–Retzius cells, which originate from multiple sources within the telencephalon, including the cortical hem, were observed at the pial surface of the hippocampus in both the wild-type and the Nfia mutant at E13 (Fig. 3A,B), and expressed reelin at E18 (supplemental Fig. 2, available at www.jneurosci.org as supplemental material). The migration of interneurons into the hippocampus of Nfia−/− mice also appeared normal (supplemental Figs. 3, 4, available at www.jneurosci.org as supplemental material). These data suggest that the phenotypic abnormalities evident in these mice (Fig. 1F) are unlikely to be due to defects in either the function of the cortical hem in patterning the hippocampus, choroid plexus development or the reelin signaling pathway.
Figure 3.
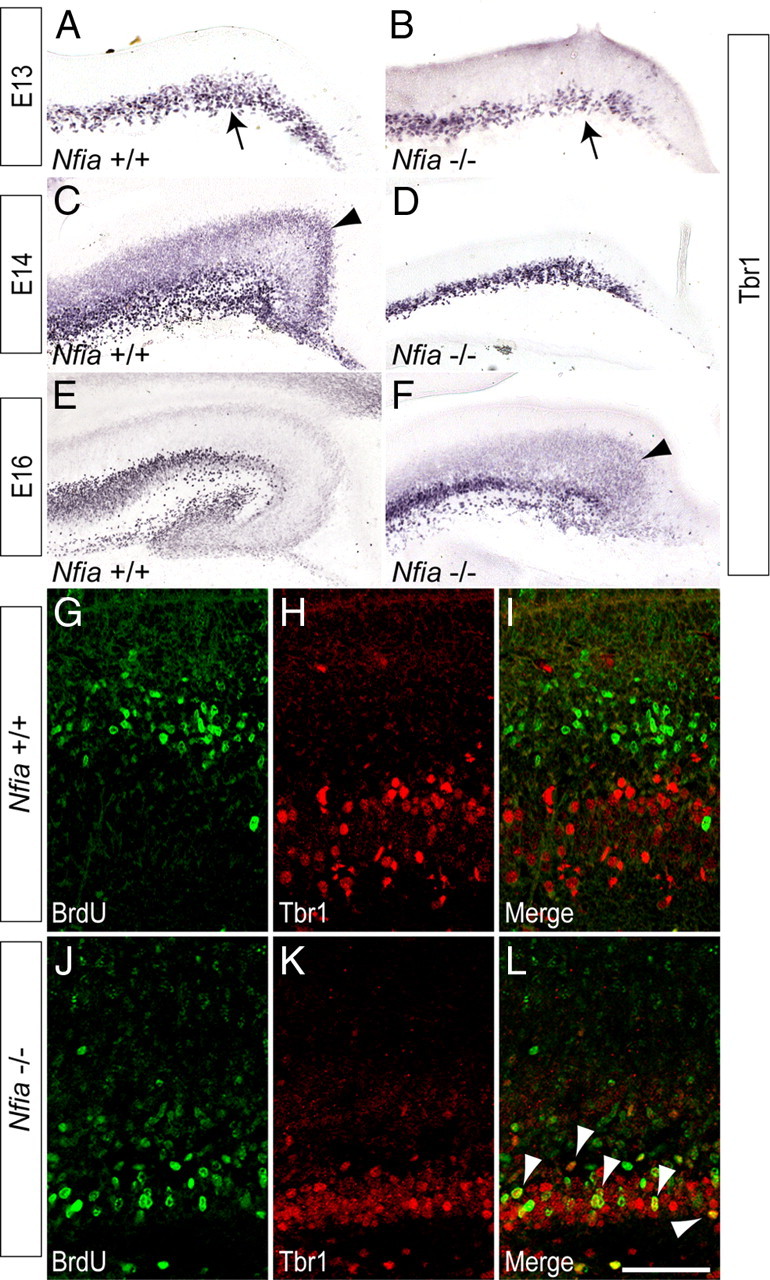
Neuronal development is delayed in the hippocampus of Nfia−/− mice. A–F, Expression of Tbr1 in coronal sections of wild-type (A, C, E) and Nfia−/− (B, D, F) brains. At E13, expression of Tbr1 was seen in the Cajal–Retzius cells at the pial surface of the wild type and mutant (arrows in A, B). At E14 in the wild type, a broad band of ventricular zone-derived Tbr1-positive neurons was observed within the ammonic neuroepithelium (arrowhead in C). In the mutant, however, these cells were not present at E14 (D), and were not detected until E16 (arrowhead in F). G–L, Birth dating of proliferating cells in the hippocampus. At E14, a pulse of BrdU was used to label cells in S-phase. Wild-type and Nfia−/− hippocampi were analyzed 2 d later at E16. In the wild type, little colocalization of BrdU (green) and Tbr1 (red) was evident (G–I). J–L, In Nfia−/− hippocampi, however, there were significantly more cells in which BrdU and Tbr1 were colocalized (L, arrowheads). Scale bar (in L): A, B, 80 μm; C, D, 100 μm; E, F, 250 μm; G–L, 50 μm.
Unlike Cajal–Retzius cells, the postmitotic pyramidal neurons of the cornu ammonis (CA) regions of the hippocampus are derived from the hippocampal ventricular zone. As they differentiate, these cells begin to express neuron-specific genes, including Tbr1 (Hevner et al., 2001). Using Tbr1 expression as a marker, we observed a distinct difference in the differentiation of these neurons in Nfia mutants. In the wild type at E14, a broad band of Tbr1-positive cells was observed, corresponding to postmitotic neurons that had differentiated from the hippocampal ventricular zone and were migrating radially to initiate formation of the pyramidal cell layer (Fig. 3C). In the mutant, however, these cells were not present at E14, most likely indicating that they had yet to differentiate (Fig. 3D). Indeed, such pyramidal neurons were only present in the mutant at E16 (Fig. 3, compare C to F), revealing a delay of ∼2 d in the development of the pyramidal cell layer in Nfia−/− mice. To substantiate this finding, we labeled cells in S-phase with BrdU at E14 and analyzed the proportion of BrdU-positive cells that coexpressed Tbr1 2 d later at E16. In the wild type, BrdU and Tbr1 were colocalized in very few cells (Fig. 3G–I) (2.2 ± 0.9 cells/300 μm), indicating that the majority of Tbr1-positive neurons had already differentiated at the time of the BrdU pulse at E14. Analysis of the mutant, however, revealed a significantly larger number of neurons in which BrdU and Tbr1 were colocalized (Fig. 3J–L) (10.1 ± 4.4 cells/300 μm, p < 0.05, Student's t test), suggestive of a delay in the differentiation of Tbr1-positive neurons in the absence of Nfia. Finally, analysis of the expression of the hippocampal neuronal subfield markers SCIP (CA1) and KA1 (CA3) (Tole et al., 1997) by in situ hybridization (Fig. 4A–F) and qPCR (Fig. 4G) also revealed developmental delays within the Nfia−/− mice. Collectively, these findings indicate that the delay in the differentiation of ventricular zone progenitors in the absence of Nfia culminates in substantial delays in the production of ventricular zone-derived pyramidal neurons.
Figure 4.

Delay in the generation of subfield markers and dentate granule neurons. A–F, In situ hybridization against the hippocampal subfield markers SCIP (A, B) and KA1 (C–F). Expression of SCIP was markedly reduced in the mutant at E18 (open arrowhead in B), whereas expression of KA1 in the mutant was reduced at E16, but appeared comparable to the control at E18. G, qPCR on wild-type and Nfia−/− tissue confirmed these findings, with SCIP mRNA levels at E18 and KA1 mRNA levels at E16 significantly reduced in Nfia−/− mice compared to controls. H–K, Expression of Prox1 in coronal sections of wild-type (H, J) and Nfia−/− (I, K) brains. At E16 in the wild type (H), Prox1-positive dentate granule neurons were observed in the dentate neuroepithelium (arrow in H) and within the incipient dentate gyrus (arrowhead in H). In the mutant, however, expression was only observed in the dentate neuroepithelium (arrow in I). At E18, expression of Prox1 in the mutant had recovered (K), but the dentate granule neurons did not form the distinctive chevron shape of the dentate gyrus that was seen in the control (arrowhead in J). L, qPCR on wild-type and Nfia−/− tissue demonstrated an early delay in the generation of dentate granule neurons, as levels of Prox1 mRNA in the mutant were significantly reduced at E16, but not E18. *p < 0.05, Student's t test. Scale bar (in K): A, B, E, F, J, K, 300 μm; C, D, H, I, 250 μm.
Delay in the development of dentate granule neurons in Nfia−/− mice
Are other ventricular zone-derived hippocampal neuronal populations similarly developmentally delayed? Dentate granule neurons, which can be identified via expression of Prospero-related homeobox 1 (Prox1), are born in the hippocampal dentate neuroepithelium at approximately E14 and migrate subpially into the presumptive dentate gyrus both prenatally and postnatally (Lavado and Oliver, 2007). In wild-type mice, Prox1-positive dentate granule neurons had begun their migration from the dentate neuroepithelium and were populating the presumptive dentate gyrus at E16 (Fig. 4H). However, in Nfia−/− mice at this age, Prox1-positive cells were only apparent in the dentate neuroepithelium, and few were migrating toward the incipient dentate gyrus (Fig. 4I). qPCR analysis also revealed that levels of Prox1 mRNA were significantly lower in the Nfia mutants compared to controls at E16 (Fig. 4L). At E18, the distinctive chevron shape of the dentate gyrus was demarcated by Prox1-positive cells in the wild-type hippocampus (Fig. 4J), but Nfia−/− mice at this age displayed a delayed developmental pattern (Fig. 4K) reminiscent of the wild type at E16 (Fig. 4, compare H to K). The level of Prox1 transcript in the mutant at E18 was, however, comparable to that in the control at this age (Fig. 4L), indicating a recovery of mRNA production in the mutant mice. The recovery in Prox1 mRNA levels may be due to compensation via other members of the Nfi family, such as Nfib, which are also expressed in the hippocampal ventricular zone at this time (Plachez et al., 2008). These findings suggest that Nfia−/− mice display delayed development of multiple neuronal populations, and correlate with our previous data showing a sustained increase in the ventricular zone progenitor population.
Delayed VZ differentiation culminates in deficient glial maturation
During midgestation, ventricular zone progenitors convert from producing neurons to producing glia, a process termed the gliogenic switch. As Nfia has been shown to regulate midline glial formation in the cortex (Shu et al., 2003) and gliogenesis within the spinal cord (Deneen et al., 2006), we next analyzed whether formation of astrocytes within the hippocampus of Nfia−/− mice was impaired. In E14 wild-type mice, expression of the astrocyte-specific glutamate transporter (GLAST), a marker for radial glial cells newly differentiated from neuroepithelial progenitor cells (Shibata et al., 1997; Hartfuss et al., 2001; Götz and Barde, 2005), was apparent throughout the hippocampal formation and fimbria (Fig. 5A). GLAST expression in Nfia−/− mice at E14 was, however, mostly absent from the hippocampal formation, but was still present in the fimbrial region (Fig. 5B). Between E15 and E16, GLAST expression did become evident within the hippocampus of the mutant, but at a significantly lower level than in the control (Fig. 5C–F,I). Expression of another marker for early radial glial cells, the extracellular matrix glycoprotein tenascin C (Götz et al., 1998), was also markedly lower in Nfia−/− mice when compared to controls (supplemental Fig. 5, available at www.jneurosci.org as supplemental material).
Figure 5.
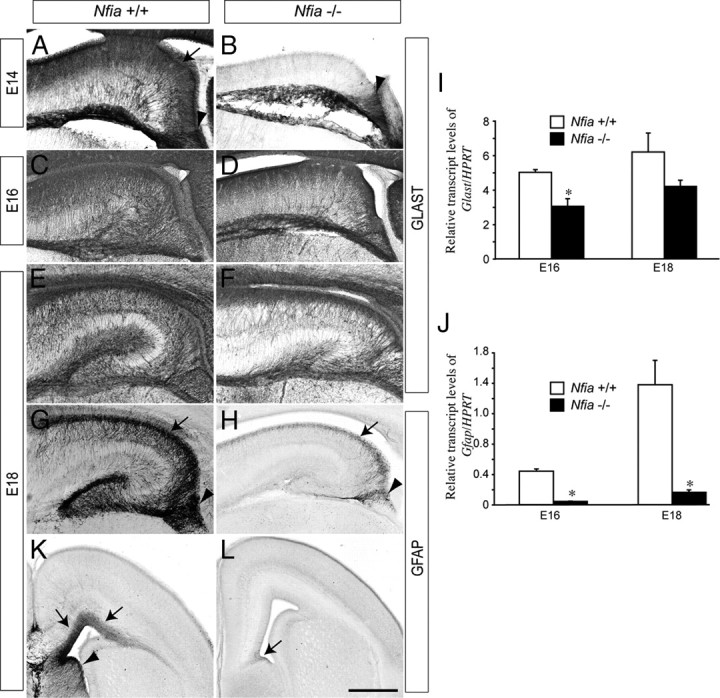
Glial development is curtailed in the telencephalon of Nfia−/− mice. Expression of GLAST (A–F) and GFAP (G, H) in wild-type and Nfia−/− hippocampi. At E14, expression of GLAST in the wild type was detected in the ammonic neuroepithelium (arrow in A) and the fimbrial glioepithelium (arrowhead in A), whereas expression in the mutant was only observed in the fimbrial glioepithelium (arrowhead in B). At both E16 and E18, expression of GLAST in the ammonic epithelium of the mutant became apparent (D, F), but GLAST mRNA levels in the mutant were lower than in the control at both these ages (I). Expression of GFAP within the ammonic epithelium (arrows in G, H) and fimbrial glioepithelium (arrowheads in G, H) was markedly reduced in Nfia−/− hippocampi. This finding was confirmed by qPCR, which demonstrated a significant decrease in Gfap mRNA in the mutant hippocampus at both E16 and E18 (J). At E18, expression of GFAP in the wild type was observed in the dorsal telencephalon (K, arrows) and the septum (arrowhead in K). In the mutant, GFAP expression in the cortex and septum was dramatically reduced, with low levels of GFAP observed only at the cortical midline (arrow in L). *p < 0.05, Student's t test. Scale bar (in L): A, B, 100 μm; C, D, 250 μm; E–H, 300 μm; K, L, 400 μm.
Delays in astrocytic differentiation were even more pronounced in mature hippocampal glial populations. Expression of the astrocytic marker GFAP (Chiu et al., 1981) was initiated in the fimbrial region and neighboring ammonic neuroepithelium of the wild type at E15, whereas GFAP expression was strikingly reduced at this age in the Nfia−/− mice (supplemental Fig. 6, available at www.jneurosci.org as supplemental material). Indeed, from E16 to E18 the marked increase in mature, GFAP-expressing astrocytes within the dentate gyrus, ammonic neuroepithelium and fimbrial glioepithelium seen in the wild-type hippocampus was severely curtailed in the mutant mice (Fig. 5G,H; supplemental Fig. 6, available at www.jneurosci.org as supplemental material). Moreover, expression of GFAP was greatly reduced in the neocortex and septum of Nfia−/− mice (Fig. 5K,L), indicating that the delay in astrocytic development occurred throughout the telencephalon. Transcript levels of Gfap were also significantly reduced in both E16 and E18 mutant hippocampi (Fig. 5J). As Gfap is known to have an NFI binding site in its promoter (Cebolla and Vallejo, 2006), we asked whether the levels of other astrocytic markers were also reduced. Transcript levels of s100β, a calcium-binding protein specifically expressed in astrocytes (Haan et al., 1982), were significantly reduced in E18 Nfia−/− versus wild-type hippocampi (supplemental Fig. 6, available at www.jneurosci.org as supplemental material). Together, these data indicate that the retention of cells within the ventricular zone neural progenitor pool culminates in developmental delays to both neuronal and astrocytic lineages within the hippocampus of Nfia−/− mice.
Increased levels of Notch signaling occur in the absence of Nfia
To identify the molecular pathways regulated by Nfia during morphogenesis of the hippocampus, we next conducted a microarray screen of wild-type and Nfia−/− hippocampal tissue at E16, the age at which the increase in the ventricular zone width first became apparent in the mutant (Fig. 2C). Over 3500 genes were differentially expressed in the mutant hippocampus using both a 1.5-fold cutoff and a significance level of p < 0.05 via ANOVA (supplemental Tables 1, 2, available at www.jneurosci.org as supplemental material). Genes downregulated in the mutant included Prox1, GLAST, and S100α6 (Table 1, Fig. 6S), indicative of the delays in both neuronal and glial differentiation arising from the longer maintenance of the progenitor state in Nfia mutants. Furthermore, expression of many genes in the Notch pathway was significantly upregulated in the mutant hippocampal array (Table 2). These included the Notch ligand Delta-like ligand 1 (Dll1) and the key Notch effectors Hes1 and Hey2, as well as Id4, an inhibitor of stem cell differentiation. Expression of Neurogenin 2 (Ngn2), a proneural gene known to regulate Dll1 (Castro et al., 2006), was also significantly upregulated in the mutant, as was Sox3, which has been implicated in neural stem cell maintenance (Wang et al., 2006). Validation of these array data via both in situ hybridization and qPCR (Fig. 6) confirmed that multiple components of the Notch signaling pathway have elevated levels of expression in the Nfia−/− hippocampus. Furthermore, analysis of cortical ventricular zone progenitors revealed upregulation of Dll1, Ngn2, Hes1, and Hes5 (Fig. 6), suggesting that Nfia regulates ventricular zone differentiation throughout the dorsal telencephalon.
Table 1.
Genes downregulated in the Nfia mutant hippocampus at E16
| Common | Fold change | GenBank |
|---|---|---|
| Sox8 | 0.390 | NM_011447.1 |
| Epha8 | 0.414 | NM_007939.1 |
| Camk2b | 0.472 | NM_007595.2 |
| Prox1 | 0.483 | NM_008937.2 |
| Calretinin | 0.515 | NM_007586.1 |
| Caln1 | 0.551 | NM_021371.1 |
| S100a6 | 0.553 | NM_011313.1 |
| Chl1 | 0.562 | NM_007697.1 |
| Slc1a3 (GLAST) | 0.578 | NM_148938.2 |
| Neurl | 0.581 | NM_021360 |
| Sema5a | 0.603 | NM_009154.1 |
| Syt1 | 0.604 | NM_009306 |
| Fgf9 | 0.610 | NM_013518.1 |
| Sema6b | 0.613 | NM_013662 |
| Sema6c | 0.617 | NM_011351 |
| Slc1a2 (Eaat2) | 0.618 | NM_011393 |
| Sox10 | 0.620 | XM_128139.1 |
| Fgf10 | 0.626 | NM_008002.3 |
| S100a10 | 0.640 | NM_009112.1 |
| Lama2 | 0.651 | NM_008481 |
| Ncam1 | 0.652 | NM_010875.2 |
| Rfng | 0.655 | NM_009053 |
| Gfra1 | 0.661 | NM_010279.2 |
A cutoff p value of 0.05 was used for the mean difference between wild-type and Nfia knock-out hippocampal tissue. In addition, a 1.5-fold change filter was imposed on the genes from the ANOVA dataset.
Figure 6.
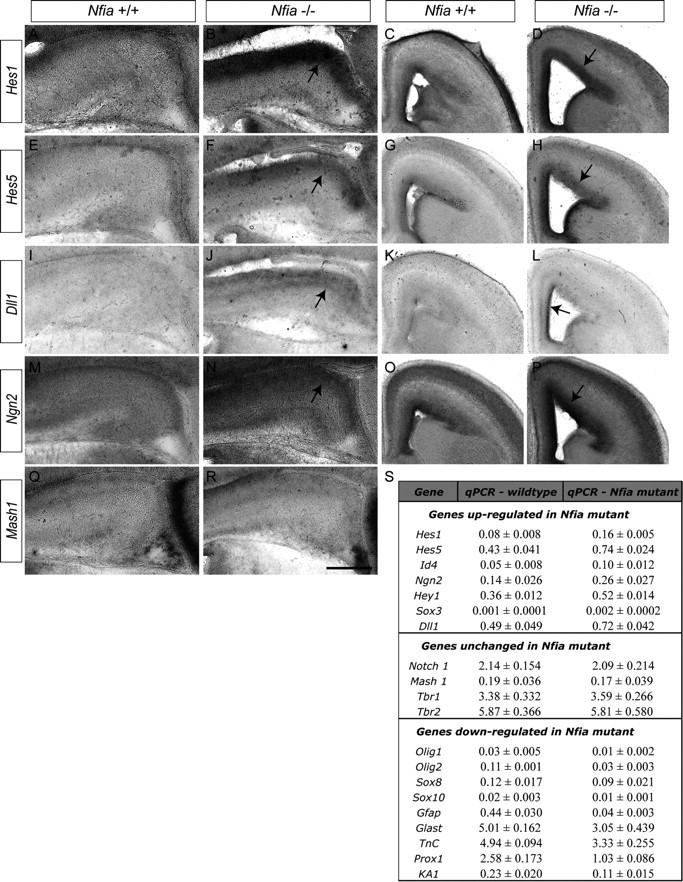
Upregulation of the Notch signaling pathway in Nfia mutants. A–R, In situ hybridization on E16 wild-type and Nfia−/− brains. Expression of the Notch effectors Hes1 (A–D) and Hes5 (E–H) in the ventricular zone of the hippocampus and neocortex was markedly higher in the mutant compared to littermate controls (arrows in B, D, F, H). Dll1 (I–L) was also expressed at higher levels in the hippocampus (arrow in J) and cingulate cortex (arrow in L) of Nfia mutants. The proneural gene Ngn2 (M–P) was also expressed at a higher level in the hippocampus and neocortex of Nfia mutants (arrows in N, P). Expression of Mash1 was comparable between wild-type and mutant hippocampi (Q, R). S, qPCR validation of the microarray results using independent biological replicates from E16 wild-type and Nfia−/− hippocampi. Genes in the top section were significantly upregulated, genes in the middle section were unchanged, and genes in the bottom section were significantly downregulated in Nfia−/− tissue. Statistical significance for upregulated and downregulated genes was determined using a Student's t test with a significance level of p < 0.05. Scale bar (in R): A, B, E, F, I, J, M, N, Q, R, 250 μm; C, D, G, H, K, L, O, P, 400 μm.
Table 2.
Genes upregulated in the Nfia mutant hippocampus at E16
| Common | Fold change | GenBank |
|---|---|---|
| Hey2 (Hesr2) | 2.07 | NM_013904.1 |
| Musashi 2 (Msi2 h) | 1.98 | AK031365 |
| Hes1 | 1.97 | NM_008235.2 |
| Sox6 | 1.97 | AK084290 |
| Gpc6 | 1.97 | AK032866 |
| Id4 (Idb4) | 1.88 | NM_031166.1 |
| Cdon | 1.81 | AK040711 |
| Map2 (Mtap2) | 1.75 | AK086484 |
| Dll1 | 1.72 | NM_007865.2 |
| Sema6a | 1.70 | AK042751 |
| Sox3 | 1.65 | NM_009237.1 |
| Sox4 | 1.63 | NM_009238.1 |
| Gpc3 | 1.58 | NM_016697.2 |
| Camk2d | 1.58 | AK020214 |
| Ephb2 | 1.57 | AK017630 |
| Fn1 | 1.55 | XM_129845.3 |
| Itga6 | 1.54 | NM_008397.2 |
| Elavl4 | 1.53 | NM_010488.1 |
| Gli3 | 1.52 | NM_008130 |
| Sfrp1 | 1.52 | NM_013834.1 |
| Cutl1 (Cux1) | 1.51 | NM_198602.1 |
| Neurog2 (Ngn2) | 1.50 | NM_009718.2 |
A cutoff p value of 0.05 was used for the mean difference between wild-type and Nfia knock-out hippocampal tissue. In addition, a 1.5-fold change filter was imposed on the genes from the ANOVA dataset.
In silico promoter analysis reveals Hes1 as a possible NFI target gene
Nfi genes have been demonstrated to directly regulate both neuronal- and glial-specific genes in vitro (Copertino et al., 1997; Bisgrove et al., 2000; Cebolla and Vallejo, 2006). Is the upregulation of Notch pathway expression observed in the Nfia mutant simply a reflection of ventricular zone progenitors failing to differentiate and so retaining progenitor characteristics, or, alternatively, does Nfia directly repress elements of the Notch pathway, thereby promoting differentiation down either the neuronal or the glial lineage? To address this issue, we first performed in silico promoter analysis to identify potential NFI target genes. Although some potential targets for the Nfi genes have been identified via bioinformatics (Wong et al., 2007), a comprehensive analysis of specific gene promoters, taking into account conservation across species, has yet to be performed. To do this, we used a position weight matrix of the NFI binding motif, TTGGC(N5)GCCAA (Mason et al., 2009), to search for binding sites in the upstream regions of specific genes. This was done using both a simple log-likelihood score, known to be proportional to specific binding constants (Stormo and Fields, 1998), and using a model that included evolutionary conservation (Moses et al., 2004). We first identified four sequences within the Gfap promoter that have previously been identified as NFI binding sites (Miura et al., 1990), thus validating our approach. Using a candidate-based approach, we next found that the promoters of many other neuronal- and glial-specific genes, including Glast, Prox1, and SCIP, possess highly conserved putative NFI binding sites within 2 kilobases of their transcription start site (Fig. 7A). This analysis also indicated that many genes within the Notch signaling pathway contain putative NFI binding sites, including Hey1, Id4, and Hes5, the last of which contained three highly conserved putative NFI binding sites. Hes1, a basic helix-loop-helix transcription factor that is one of the key effectors of the Notch pathway (Kageyama et al., 2008b), was identified as a strong candidate for transcriptional regulation by Nfia, as there were three highly conserved NFI binding sites within 200 bp of the Hes1 transcription start site (Fig. 7A). When considered in combination with our array and qPCR data, this promoter analysis indicates that, in addition to driving expression of genes involved in differentiation, Nfia may also repress expression of key members of the Notch pathway, most notably Hes1.
Figure 7.
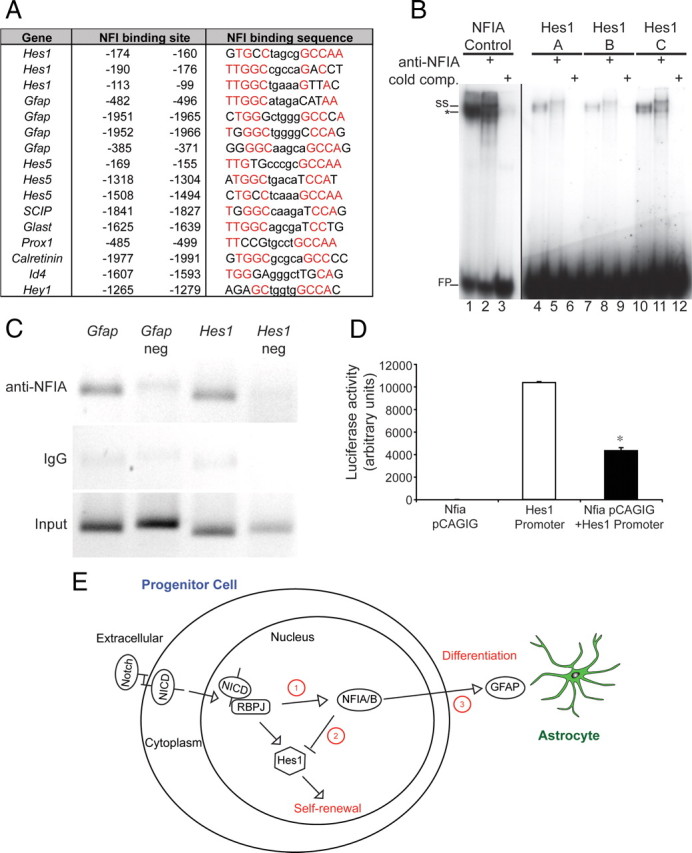
NFIA binds the basal promoter region of Hes1 in vivo and represses Hes1 promoter-driven reporter gene activity. A, Identification of putative conserved NFI binding sites using in silico bioinformatics. The consensus NFI binding site is TTGGC(N5)nnnnnGCCAA (Gronostajski, 2000). Base pairs identified in silico that were identical to the NFI consensus sequence are colored red. The positions of the putative NFI binding sites are given relative to the transcription start site (position 0). The Hes1 promoter has three putative NFI binding sites within 200 bp of the transcription start site. B, EMSA. E18 mouse brain nuclear extracts were incubated with radiolabeled probes for NFI control (lanes 1–3), Hes1-A (lanes 4–6), Hes1-B (lanes 7–9), or Hes1-C (lanes 10–12) consensus sites. Binding reactions included 2× cold (unlabeled) NFI control competitor or specific NFIΑ antibody where indicated. The specific NFIA complex (*) was depleted and produced a supershifted complex (ss) in the presence of the NFIA antibody. FP, Free probe. C, Chromatin immunoprecipitation assays were conducted on E18 wild-type cortices. DNA fragments immunoprecipitated with IgG or anti-NFIA antibodies were analyzed by PCR with primers specifically designed to encompass the predicted NFI binding sites. The NFI binding sites in the Gfap promoter were used as a positive control for NFIA binding (Miura et al., 1990). For both Gfap and Hes1, control primers, designed to an adjacent region of the respective promoters and lacking putative NFI binding sites, were used as negative controls. D, Reporter gene transcriptional assay in HEK 293 cells. Transfection of an Nfia expression vector (Nfia pCAGIG) elicited no luciferase activity, whereas transfection of a luciferase reporter construct under the control of the Hes1 promoter elicited robust induction of the reporter gene. Cotransfection of Nfia with the Hes1 promoter reporter construct resulted in a significantly reduced level of luciferase activity. *p < 0.001, Student's t test. E, Proposed model for the role of Nfia in the regulation of progenitor cell differentiation. Activation of Notch signaling elicits both Nfia transcription (point 1) (Namihira et al., 2009) and Hes1 transcription. NFIA plays a binary role in driving the differentiation of progenitor cells by actively repressing Hes1 expression (point 2) (this study) and activating the transcription of differentiation-specific genes such as GFAP (point 3) (Namihira et al., 2009).
NFIA binds to the promoter of Hes1 in vivo and represses Hes1 promoter-driven reporter gene activity
To determine whether NFIA can bind directly to the putative NFI binding sites identified in the Hes1 promoter, we first performed electrophoretic mobility shift assays using E18 mouse cortex nuclear extracts. E18 mouse cortex nuclear extract bound to radiolabeled oligonucleotide probes containing a control NFI consensus site, as well as to probes containing each of the three potential NFI binding sites within the Hes1 promoter, designated Hes1-A, Hes1-B, and Hes1-C (Fig. 7B). Supershifting of the probes with an anti-NFIA antibody indicated that the protein bound to each oligonucleotide was NFIA. The specificity of binding was further confirmed by the competition of binding with excess unlabeled NFI control probe (Fig. 7B). Binding to the Hes1-A, Hes1-B, and Hes1-C probes was also observed using in vitro translated NFIA (data not shown). These data suggest that NFIA can interact directly with the three NFI binding sites identified within the Hes1 promoter. To determine whether NFIA can bind this region in vivo, we next performed chromatin immunoprecipitation on cortical tissue from E18 wild-type embryos. As a positive control, we amplified a region from the Gfap promoter that has previously been shown to bind NFI protein (Miura et al., 1990). Anti-NFIA antibodies specifically precipitated this promoter region (Fig. 7C), demonstrating in vivo binding of NFIA to the Gfap promoter. Importantly, anti-NFIA antibodies were also able to specifically precipitate the DNA region from the Hes1 promoter containing the three NFI binding sites (Fig. 7C), confirming that NFIA is indeed able to bind the Hes1 promoter in vivo. Finally, using a reporter gene assay, we demonstrated that NFIA was able to directly repress luciferase expression driven by the Hes1 promoter in vitro (Fig. 7D). From these data, we infer that NFIA is a direct transcriptional repressor of Hes1.
Discussion
The self-renewal of progenitor cells is regulated by the Notch pathway during nervous system development. Regulation of Notch signaling is complex, and regulatory control can be exerted at multiple levels in both the extracellular and intracellular milieux (Yoon and Gaiano, 2005; Louvi and Artavanis-Tsakonas, 2006). Here we reveal that, in the absence of Nfia, Hes1 expression is significantly enhanced and progenitor cells remain undifferentiated and accumulate within the telencephalic ventricular zone. Furthermore, using an in silico bioinformatic approach, we have identified three highly conserved putative NFI binding sites within the basal promoter of the Hes1 gene and demonstrated that this region is bound by NFIA using electrophoretic mobility shift assays and chromatin immunoprecipitation, as well as showing that NFIA can repress transcriptional activity under the control of the Hes1 promoter. These data represent a significant advance in our understanding of how the maintenance of progenitor cells is regulated, and provide a mechanistic insight into how the activation of Nfia expression by the Notch pathway (Namihira et al., 2009) culminates not only in the expression of astrocyte-specific genes, but also in the simultaneous abrogation of Notch pathway activity via repression of Hes1 expression (Fig. 7E).
A number of recent findings have highlighted the role played by NFIA in controlling the transition of ventricular zone progenitors from neurogenic to gliogenic division, a process known as the gliogenic switch (Deneen et al., 2006; Namihira et al., 2009). Using the spinal cord as a model system, Deneen et al. (2006) provided evidence that NFIA is required to promote glial-fate specification. Interestingly, expression of Hes5 within the spinal cord ventricular zone was also shown to require NFIA (Deneen et al., 2006), although whether NFIA directly regulated Hes5 was not addressed. The findings that Nfia is required for Hes5 expression (Deneen et al., 2006) and represses Hes1 expression (this study) suggests that the role of Nfia in regulating gene expression within ventricular zone progenitors may be context-dependent. With relation to cortical ventricular zone progenitors, Notch signaling in midgestational telencephalic precursors was recently demonstrated to induce NFIA expression, leading to demethylation of astrocyte-specific genes responsive to JAK-STAT signaling, such as Gfap (Namihira et al., 2009). This study provided a mechanistic insight into the role of NFIA with regard to cortical gliogenesis, placing NFIA downstream of Notch signaling and implicating epigenetic mechanisms in the promotion of the gliogenic switch.
However, a number of studies suggest that the role played by NFI family members is more extensive than this conceptual framework implies. For example, NFIs have been demonstrated to bind to the promoters of neuronal- and glial-specific genes and to regulate their activity in vitro (Copertino et al., 1997; Behrens et al., 2000; Bisgrove et al., 2000; Wang et al., 2004; Cebolla and Vallejo, 2006; Gopalan et al., 2006), implying direct gene regulation by the NFI transcription factors. Moreover, our data clearly indicate that, in addition to regulating astrocytic differentiation within the telencephalon, NFIA also acts to negatively regulate the Notch signaling pathway, in part via direct repression of Hes1. Our working model posits that induction of Nfia expression by the NICD/RBPj complex (Fig. 7E, point 1) (Namihira et al., 2009) results in both repression of the downstream Notch effector Hes1 (Fig. 7E, point 2) and activation of astrocyte-specific genes (Fig. 7E, point 3). As such, in the absence of Nfia, both neuronal and glial differentiation are delayed (Figs. 2–5) as a result of the failure to repress Notch pathway activity. These data imply that inhibition of Notch signaling in Nfia−/− cells would further reduce Gfap expression, as both Notch and NFIA regulate astrocytic development. In vitro culture of E18 Nfia−/− cortical cells with a γ-secretase inhibitor supported this hypothesis, with significantly reduced levels of Gfap mRNA in the γ-secretase inhibitor-treated Nfia−/− cells in comparison to vehicle-treated Nfia−/− cells (data not shown). Thus, these data point to NFIA playing a binary role in telencephalic development and being central to both the expression of genes that drive the differentiation of ventricular zone precursors and the concurrent inhibition of the Notch pathway in the progenitors themselves (Fig. 7E).
The transcriptional repressor Hes1 is central to the process of progenitor self-renewal within the nervous system (Kageyama et al., 2008a). However, the mechanism by which the expression of Hes1 itself is regulated is still unclear. Hes1 is a target for regulation by Notch (Kageyama et al., 2008b), and a recent report has indicated that Gli2 regulates Hes1 activity within retinal progenitors (Wall et al., 2009). Hes1 also negatively autoregulates its own gene expression (Takebayashi et al., 1994). Furthermore, the inhibitor of differentiation (Id) genes were recently shown to release Hes1 from autoregulatory inhibition, thereby promoting the maintenance of the neural stem cell pool during neuronal development (Bai et al., 2007). Our data identify Nfia as a further regulator of Hes1 transcription. Expression of Hes1 is seen very early within neural progenitors in the mouse embryo, from approximately E7.5 (Kageyama et al., 2008b). Expression of Nfia, by contrast, becomes evident only much later, with almost no telencephalic expression of NFIA at E12, and hippocampal expression being initiated at E13 (Plachez et al., 2008). This implies that other factors are required to control the expression of Nfia. Candidates include the transcription factors Pax6 and Emx2, both of which have been implicated upstream of Nfia in cortical development (Gangemi et al., 2006; Holm et al., 2007). The temporal dynamics of NFIA expression may also explain why only relatively subtle defects in neurogenesis occur in Nfia−/− mice, as much neurogenesis has already occurred before induction of Nfia expression, whereas gliogenesis, which occurs from late gestation, is severely affected in these mutants. Negative regulation of Hes1 by Nfia only occurs from approximately E13 onwards at the onset of Nfia expression, providing a mechanism for gliogenic differentiation through regulation of the Notch signaling pathway and activation of astrocyte-specific genes in neural progenitors. Interestingly, Hes1 expression was recently shown to be oscillatory within telencephalic neural progenitors, with instability of Hes1 mRNA and protein, combined with its autoregulatory capacity, contributing to the production of Hes1, Ngn2, and Dll1 oscillatory networks (Shimojo et al., 2008). Whether or not Nfia undergoes a similar oscillatory cycle is unclear at this stage, although the direct regulation of Nfia by Notch signaling (Namihira et al., 2009) indicates this may be a possibility.
One interesting issue raised by our data is that differentiation of ventricular zone progenitors does take place in the absence of Nfia, albeit at a significantly slower pace than in controls (e.g., Fig. 5G,H). This indicates that other factors may also contribute to the repression of Notch signaling within the telencephalon. Another Nfi family member, Nfib, is one potential candidate for mediating the repression of Notch signaling in the Nfia mutant. Like Nfia, Nfib is expressed by progenitor cells within the hippocampal ventricular zone (supplemental Figs. 7, 8, available at www.jneurosci.org as supplemental material), and Nfib−/− mice display delays in hippocampal astrocyte development (Barry et al., 2008). Furthermore, Hes1 mRNA levels are significantly higher in the hippocampus of Nfib−/− mice than that of controls, and chromatin immunoprecipitation experiments demonstrate that the basal Hes1 promoter is bound by NFIB in vivo (supplemental Fig. 8G,H, available at www.jneurosci.org as supplemental material). Finally, reporter gene assays show that NFIB is able to directly repress Hes1 promoter-mediated transcriptional activity (supplemental Fig. 8I, available at www.jneurosci.org as supplemental material), suggesting that both Nfia and Nfib act to repress Notch pathway activity during cortical development.
A further challenge arising from this study is to determine the extent of regulatory control exerted on the Notch signaling pathway by Nfia. In addition to Hes1, our array and qPCR screens identified numerous other genes known to contribute to Notch signaling that were misregulated in Nfia mutants, including Hes5 (Ohtsuka et al., 1999), Dll1 (Grandbarbe et al., 2003), Id4 (Jeon et al., 2008), radical fringe (Mikami et al., 2001), neuralized (Lai et al., 2001), Ngn2 (Shimojo et al., 2008), and Hey1 (Fischer et al., 2004). Although the misregulation of these genes may occur as a secondary consequence of Hes1 overexpression, our bioinformatic promoter screen indicated that many of these genes, including Hes5, Id4, and Hey1 (Fig. 7), also contain conserved putative NFI binding sites within their respective promoters, indicating that they too may be bone fide transcriptional targets of the Nfi family. Furthermore, the overexpression of Ngn2 and Dll1 (Table 2, Fig. 6) suggests that, in addition to regulating Notch signaling in radial progenitors, Nfia may also regulate the expression of Notch ligands on newborn neurons and intermediate progenitors during development. In conclusion, Nfia plays a central role in nervous system development, driving the expression of astrocyte-specific genes but also suppressing the progenitor cell state to allow differentiation to occur.
Footnotes
This work was funded by a National Health and Medical Research Council (NHMRC) project grant (L.J.R.) and a Clive and Vera Ramaciotti grant (M.P.). The following authors were supported by NHMRC fellowships: L.J.R. (Senior Research Fellowship) and M.P. (Howard Florey Centenary Fellowship and Biomedical Career Development Award). S.M. is supported by a University of Queensland F. G. Meade PhD Scholarship. We thank John Baisden, Alex Sykes, Nick Palstra, and Oressia Zalucki for technical assistance, Rowan Tweedale for critical analysis of the manuscript, and Sally Dunwoodie (Victor Chang Cardiac Research Institute), Ryoichiro Kageyama (Kyoto University), Francois Guillemot (National Institute for Medical Research), Jane Johnson (University of Texas), Robert Hevner (University of Washington), Andre Goffinet (University of Louvain Medical School), and Niels Danbolt (University of Oslo) for providing reagents.
References
- Bai G, Sheng N, Xie Z, Bian W, Yokota Y, Benezra R, Kageyama R, Guillemot F, Jing N. Id sustains Hes1 expression to inhibit precocious neurogenesis by releasing negative autoregulation of Hes1. Dev Cell. 2007;13:283–297. doi: 10.1016/j.devcel.2007.05.014. [DOI] [PubMed] [Google Scholar]
- Barry G, Piper M, Lindwall C, Moldrich R, Mason S, Little E, Sarkar A, Tole S, Gronostajski RM, Richards LJ. Specific glial populations regulate hippocampal morphogenesis. J Neurosci. 2008;28:12328–12340. doi: 10.1523/JNEUROSCI.4000-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behrens M, Venkatraman G, Gronostajski RM, Reed RR, Margolis FL. NFI in the development of the olfactory neuroepithelium and the regulation of olfactory marker protein gene expression. Eur J Neurosci. 2000;12:1372–1384. doi: 10.1046/j.1460-9568.2000.00032.x. [DOI] [PubMed] [Google Scholar]
- Bertrand N, Castro DS, Guillemot F. Proneural genes and the specification of neural cell types. Nat Rev Neurosci. 2002;3:517–530. doi: 10.1038/nrn874. [DOI] [PubMed] [Google Scholar]
- Bisgrove DA, Monckton EA, Packer M, Godbout R. Regulation of brain fatty acid-binding protein expression by differential phosphorylation of nuclear factor I in malignant glioma cell lines. J Biol Chem. 2000;275:30668–30676. doi: 10.1074/jbc.M003828200. [DOI] [PubMed] [Google Scholar]
- Campbell CE, Piper M, Plachez C, Yeh YT, Baizer JS, Osinski JM, Litwack ED, Richards LJ, Gronostajski RM. The transcription factor Nfix is essential for normal brain development. BMC Dev Biol. 2008;8:52. doi: 10.1186/1471-213X-8-52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castro DS, Skowronska-Krawczyk D, Armant O, Donaldson IJ, Parras C, Hunt C, Critchley JA, Nguyen L, Gossler A, Göttgens B, Matter JM, Guillemot F. Proneural bHLH and Brn proteins coregulate a neurogenic program through cooperative binding to a conserved DNA motif. Dev Cell. 2006;11:831–844. doi: 10.1016/j.devcel.2006.10.006. [DOI] [PubMed] [Google Scholar]
- Cebolla B, Vallejo M. Nuclear factor-I regulates glial fibrillary acidic protein gene expression in astrocytes differentiated from cortical precursor cells. J Neurochem. 2006;97:1057–1070. doi: 10.1111/j.1471-4159.2006.03804.x. [DOI] [PubMed] [Google Scholar]
- Chiu FC, Norton WT, Fields KL. The cytoskeleton of primary astrocytes in culture contains actin, glial fibrillary acidic protein, and the fibroblast-type filament protein, vimentin. J Neurochem. 1981;37:147–155. doi: 10.1111/j.1471-4159.1981.tb05302.x. [DOI] [PubMed] [Google Scholar]
- Copertino DW, Edelman GM, Jones FS. Multiple promoter elements differentially regulate the expression of the mouse tenascin gene. Proc Natl Acad Sci U S A. 1997;94:1846–1851. doi: 10.1073/pnas.94.5.1846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deneen B, Ho R, Lukaszewicz A, Hochstim CJ, Gronostajski RM, Anderson DJ. The transcription factor NFIA controls the onset of gliogenesis in the developing spinal cord. Neuron. 2006;52:953–968. doi: 10.1016/j.neuron.2006.11.019. [DOI] [PubMed] [Google Scholar]
- Englund C, Fink A, Lau C, Pham D, Daza RA, Bulfone A, Kowalczyk T, Hevner RF. Pax6, Tbr2, and Tbr1 are expressed sequentially by radial glia, intermediate progenitor cells, and postmitotic neurons in developing neocortex. J Neurosci. 2005;25:247–251. doi: 10.1523/JNEUROSCI.2899-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer A, Schumacher N, Maier M, Sendtner M, Gessler M. The Notch target genes Hey1 and Hey2 are required for embryonic vascular development. Genes Dev. 2004;18:901–911. doi: 10.1101/gad.291004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frith MC, Saunders NF, Kobe B, Bailey TL. Discovering sequence motifs with arbitrary insertions and deletions. PLoS Comput Biol. 2008;4:e1000071. doi: 10.1371/journal.pcbi.1000071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gangemi RM, Daga A, Muzio L, Marubbi D, Cocozza S, Perera M, Verardo S, Bordo D, Griffero F, Capra MC, Mallamaci A, Corte G. Effects of Emx2 inactivation on the gene expression profile of neural precursors. Eur J Neurosci. 2006;23:325–334. doi: 10.1111/j.1460-9568.2005.04559.x. [DOI] [PubMed] [Google Scholar]
- Gopalan SM, Wilczynska KM, Konik BS, Bryan L, Kordula T. Nuclear factor-1-X regulates astrocyte-specific expression of the alpha1-antichymotrypsin and glial fibrillary acidic protein genes. J Biol Chem. 2006;281:13126–13133. doi: 10.1074/jbc.M601194200. [DOI] [PubMed] [Google Scholar]
- Götz M, Barde YA. Radial glial cells defined and major intermediates between embryonic stem cells and CNS neurons. Neuron. 2005;46:369–372. doi: 10.1016/j.neuron.2005.04.012. [DOI] [PubMed] [Google Scholar]
- Götz M, Huttner WB. The cell biology of neurogenesis. Nat Rev Mol Cell Biol. 2005;6:777–788. doi: 10.1038/nrm1739. [DOI] [PubMed] [Google Scholar]
- Götz M, Stoykova A, Gruss P. Pax6 controls radial glia differentiation in the cerebral cortex. Neuron. 1998;21:1031–1044. doi: 10.1016/s0896-6273(00)80621-2. [DOI] [PubMed] [Google Scholar]
- Grandbarbe L, Bouissac J, Rand M, Hrabé de Angelis M, Artavanis-Tsakonas S, Mohier E. Delta-Notch signaling controls the generation of neurons/glia from neural stem cells in a stepwise process. Development. 2003;130:1391–1402. doi: 10.1242/dev.00374. [DOI] [PubMed] [Google Scholar]
- Gronostajski RM. Roles of the NFI/CTF gene family in transcription and development. Gene. 2000;249:31–45. doi: 10.1016/s0378-1119(00)00140-2. [DOI] [PubMed] [Google Scholar]
- Haan EA, Boss BD, Cowan WM. Production and characterization of monoclonal antibodies against the “brain-specific” proteins 14-3-2 and S-100. Proc Natl Acad Sci U S A. 1982;79:7585–7589. doi: 10.1073/pnas.79.23.7585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartfuss E, Galli R, Heins N, Götz M. Characterization of CNS precursor subtypes and radial glia. Dev Biol. 2001;229:15–30. doi: 10.1006/dbio.2000.9962. [DOI] [PubMed] [Google Scholar]
- Hawkins J, Grant C, Noble WS, Bailey TL. Assessing phylogenetic motif models for predicting transcription factor binding sites. Bioinformatics. 2009;25:i339–347. doi: 10.1093/bioinformatics/btp201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hébert JM, Mishina Y, McConnell SK. BMP signaling is required locally to pattern the dorsal telencephalic midline. Neuron. 2002;35:1029–1041. doi: 10.1016/s0896-6273(02)00900-5. [DOI] [PubMed] [Google Scholar]
- Hevner RF, Shi L, Justice N, Hsueh Y, Sheng M, Smiga S, Bulfone A, Goffinet AM, Campagnoni AT, Rubenstein JL. Tbr1 regulates differentiation of the preplate and layer 6. Neuron. 2001;29:353–366. doi: 10.1016/s0896-6273(01)00211-2. [DOI] [PubMed] [Google Scholar]
- Holm PC, Mader MT, Haubst N, Wizenmann A, Sigvardsson M, Götz M. Loss- and gain-of-function analyses reveal targets of Pax6 in the developing mouse telencephalon. Mol Cell Neurosci. 2007;34:99–119. doi: 10.1016/j.mcn.2006.10.008. [DOI] [PubMed] [Google Scholar]
- Jeon HM, Jin X, Lee JS, Oh SY, Sohn YW, Park HJ, Joo KM, Park WY, Nam DH, DePinho RA, Chin L, Kim H. Inhibitor of differentiation 4 drives brain tumor-initiating cell genesis through cyclin E and notch signaling. Genes Dev. 2008;22:2028–2033. doi: 10.1101/gad.1668708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kageyama R, Ohtsuka T, Kobayashi T. Roles of Hes genes in neural development. Dev Growth Differ. 2008a;50(Suppl 1):S97–S103. doi: 10.1111/j.1440-169X.2008.00993.x. [DOI] [PubMed] [Google Scholar]
- Kageyama R, Ohtsuka T, Shimojo H, Imayoshi I. Dynamic Notch signaling in neural progenitor cells and a revised view of lateral inhibition. Nat Neurosci. 2008b;11:1247–1251. doi: 10.1038/nn.2208. [DOI] [PubMed] [Google Scholar]
- Kheradpour P, Stark A, Roy S, Kellis M. Reliable prediction of regulator targets using 12 Drosophila genomes. Genome Res. 2007;17:1919–1931. doi: 10.1101/gr.7090407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai EC, Deblandre GA, Kintner C, Rubin GM. Drosophila neuralized is a ubiquitin ligase that promotes the internalization and degradation of delta. Dev Cell. 2001;1:783–794. doi: 10.1016/s1534-5807(01)00092-2. [DOI] [PubMed] [Google Scholar]
- Lavado A, Oliver G. Prox1 expression patterns in the developing and adult murine brain. Dev Dyn. 2007;236:518–524. doi: 10.1002/dvdy.21024. [DOI] [PubMed] [Google Scholar]
- Lendahl U, Zimmerman LB, McKay RD. CNS stem cells express a new class of intermediate filament protein. Cell. 1990;60:585–595. doi: 10.1016/0092-8674(90)90662-x. [DOI] [PubMed] [Google Scholar]
- Louvi A, Artavanis-Tsakonas S. Notch signalling in vertebrate neural development. Nat Rev Neurosci. 2006;7:93–102. doi: 10.1038/nrn1847. [DOI] [PubMed] [Google Scholar]
- Mangale VS, Hirokawa KE, Satyaki PR, Gokulchandran N, Chikbire S, Subramanian L, Shetty AS, Martynoga B, Paul J, Mai MV, Li Y, Flanagan LA, Tole S, Monuki ES. Lhx2 selector activity specifies cortical identity and suppresses hippocampal organizer fate. Science. 2008;319:304–309. doi: 10.1126/science.1151695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mason S, Piper M, Gronostajski RM, Richards LJ. Nuclear factor one transcription factors in CNS development. Mol Neurobiol. 2009;39:10–23. doi: 10.1007/s12035-008-8048-6. [DOI] [PubMed] [Google Scholar]
- Mikami T, Ohnaka Y, Nakamura A, Kurosaka A, Itoh N. Radical fringe negatively modulates Notch signaling in postmitotic neurons of the rat brain. Brain Res Mol Brain Res. 2001;86:138–144. doi: 10.1016/s0169-328x(00)00278-3. [DOI] [PubMed] [Google Scholar]
- Miura M, Tamura T, Mikoshiba K. Cell-specific expression of the mouse glial fibrillary acidic protein gene: identification of the cis- and trans-acting promoter elements for astrocyte-specific expression. J Neurochem. 1990;55:1180–1188. doi: 10.1111/j.1471-4159.1990.tb03123.x. [DOI] [PubMed] [Google Scholar]
- Moses AM, Chiang DY, Pollard DA, Iyer VN, Eisen MB. MONKEY: identifying conserved transcription-factor binding sites in multiple alignments using a binding site-specific evolutionary model. Genome Biol. 2004;5:R98. doi: 10.1186/gb-2004-5-12-r98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Namihira M, Kohyama J, Semi K, Sanosaka T, Deneen B, Taga T, Nakashima K. Committed neuronal precursors confer astrocytic potential on residual neural precursor cells. Dev Cell. 2009;16:245–255. doi: 10.1016/j.devcel.2008.12.014. [DOI] [PubMed] [Google Scholar]
- Ohtsuka T, Ishibashi M, Gradwohl G, Nakanishi S, Guillemot F, Kageyama R. Hes1 and Hes5 as notch effectors in mammalian neuronal differentiation. EMBO J. 1999;18:2196–2207. doi: 10.1093/emboj/18.8.2196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pham TH, Satou K, Ho TB. Mining yeast transcriptional regulatory modules from factor DNA-binding sites and gene expression data. Genome Inform. 2004;15:287–295. [PubMed] [Google Scholar]
- Piper M, Plachez C, Zalucki O, Fothergill T, Goudreau G, Erzurumlu R, Gu C, Richards LJ. Neuropilin 1-Sema signaling regulates crossing of cingulate pioneering axons during development of the corpus callosum. Cereb Cortex. 2009a;19(Suppl 1):i11–i21. doi: 10.1093/cercor/bhp027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piper M, Moldrich RX, Lindwall C, Little E, Barry G, Mason S, Sunn N, Kurniawan ND, Gronostajski RM, Richards LJ. Multiple non-cell-autonomous defects underlie neocortical callosal dysgenesis in Nfib-deficient mice. Neural Dev. 2009b;4:43. doi: 10.1186/1749-8104-4-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plachez C, Lindwall C, Sunn N, Piper M, Moldrich RX, Campbell CE, Osinski JM, Gronostajski RM, Richards LJ. Nuclear factor I gene expression in the developing forebrain. J Comp Neurol. 2008;508:385–401. doi: 10.1002/cne.21645. [DOI] [PubMed] [Google Scholar]
- Quinn JC, Molinek M, Martynoga BS, Zaki PA, Faedo A, Bulfone A, Hevner RF, West JD, Price DJ. Pax6 controls cerebral cortical cell number by regulating exit from the cell cycle and specifies cortical cell identity by a cell autonomous mechanism. Dev Biol. 2007;302:50–65. doi: 10.1016/j.ydbio.2006.08.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibata T, Yamada K, Watanabe M, Ikenaka K, Wada K, Tanaka K, Inoue Y. Glutamate transporter GLAST is expressed in the radial glia-astrocyte lineage of developing mouse spinal cord. J Neurosci. 1997;17:9212–9219. doi: 10.1523/JNEUROSCI.17-23-09212.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimojo H, Ohtsuka T, Kageyama R. Oscillations in notch signaling regulate maintenance of neural progenitors. Neuron. 2008;58:52–64. doi: 10.1016/j.neuron.2008.02.014. [DOI] [PubMed] [Google Scholar]
- Shu T, Butz KG, Plachez C, Gronostajski RM, Richards LJ. Abnormal development of forebrain midline glia and commissural projections in Nfia knock-out mice. J Neurosci. 2003;23:203–212. doi: 10.1523/JNEUROSCI.23-01-00203.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith AG, Brightwell G, Smit SE, Parsons PG, Sturm RA. Redox regulation of Brn-2/N-Oct-3 POU domain DNA binding activity and proteolytic formation of N-Oct-5 during melanoma cell nuclear extraction. Melanoma Res. 1998;8:2–10. doi: 10.1097/00008390-199802000-00002. [DOI] [PubMed] [Google Scholar]
- Stormo GD, Fields DS. Specificity, free energy and information content in protein-DNA interactions. Trends Biochem Sci. 1998;23:109–113. doi: 10.1016/s0968-0004(98)01187-6. [DOI] [PubMed] [Google Scholar]
- Takebayashi K, Sasai Y, Sakai Y, Watanabe T, Nakanishi S, Kageyama R. Structure, chromosomal locus, and promoter analysis of the gene encoding the mouse helix-loop-helix factor HES-1. Negative autoregulation through the multiple N box elements. J Biol Chem. 1994;269:5150–5156. [PubMed] [Google Scholar]
- Tole S, Christian C, Grove EA. Early specification and autonomous development of cortical fields in the mouse hippocampus. Development. 1997;124:4959–4970. doi: 10.1242/dev.124.24.4959. [DOI] [PubMed] [Google Scholar]
- Wall DS, Mears AJ, McNeill B, Mazerolle C, Thurig S, Wang Y, Kageyama R, Wallace VA. Progenitor cell proliferation in the retina is dependent on Notch-independent Sonic hedgehog/Hes1 activity. J Cell Biol. 2009;184:101–112. doi: 10.1083/jcb.200805155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang TW, Stromberg GP, Whitney JT, Brower NW, Klymkowsky MW, Parent JM. Sox3 expression identifies neural progenitors in persistent neonatal and adult mouse forebrain germinative zones. J Comp Neurol. 2006;497:88–100. doi: 10.1002/cne.20984. [DOI] [PubMed] [Google Scholar]
- Wang W, Stock RE, Gronostajski RM, Wong YW, Schachner M, Kilpatrick DL. A role for nuclear factor I in the intrinsic control of cerebellar granule neuron gene expression. J Biol Chem. 2004;279:53491–53497. doi: 10.1074/jbc.M410370200. [DOI] [PubMed] [Google Scholar]
- Wong YW, Schulze C, Streichert T, Gronostajski RM, Schachner M, Tilling T. Gene expression analysis of nuclear factor I-A deficient mice indicates delayed brain maturation. Genome Biol. 2007;8:R72. doi: 10.1186/gb-2007-8-5-r72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi M, Saito H, Suzuki M, Mori K. Visualization of neurogenesis in the central nervous system using nestin promoter-GFP transgenic mice. Neuroreport. 2000;11:1991–1996. doi: 10.1097/00001756-200006260-00037. [DOI] [PubMed] [Google Scholar]
- Yoon K, Gaiano N. Notch signaling in the mammalian central nervous system: insights from mouse mutants. Nat Neurosci. 2005;8:709–715. doi: 10.1038/nn1475. [DOI] [PubMed] [Google Scholar]
- Zhou CJ, Zhao C, Pleasure SJ. Wnt signaling mutants have decreased dentate granule cell production and radial glial scaffolding abnormalities. J Neurosci. 2004;24:121–126. doi: 10.1523/JNEUROSCI.4071-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]