

Abstract
In the neonatal hippocampus, the activity of interneurons shapes early network bursts that are important for the establishment of neuronal connectivity. However, mechanisms controlling the firing of immature interneurons remain elusive. We now show that the spontaneous firing rate of CA3 stratum lucidum interneurons markedly decreases during early postnatal development because of changes in the properties of GluK1 (formerly known as GluR5) subunit-containing kainate receptors (KARs). In the neonate, activation of KARs by ambient glutamate exerts a tonic inhibition of the medium-duration afterhyperpolarization (mAHP) by a G-protein-dependent mechanism, permitting a high interneuronal firing rate. During development, the amplitude of the apamine-sensitive K+ currents responsible for the mAHP increases dramatically because of decoupling between KAR activation and mAHP modulation, leading to decreased interneuronal firing. The developmental shift in the KAR function and its consequences on interneuronal activity are likely to have a fundamental role in the maturation of the synchronous neuronal oscillations typical for adult hippocampal circuitry.
Introduction
Neuronal excitability and the degree of neuronal synchrony are critical properties in determining the behavior of neuronal networks. Inhibitory interneurons orchestrate network activities by pacing, timing, and synchronizing neural circuits in the adult as well as in the immature brain (McBain and Fisahn, 2001; Ben-Ari et al., 2004). However, despite their vital role in neuronal functioning, surprisingly little is known about the development of intrinsic firing properties of interneurons.
In the adult hippocampus, interneuronal firing is strongly modulated by afterhyperpolarizing K+ currents (Zhang and McBain, 1995). The action potential (AP) firing pattern of stratum radiatum interneurons in CA3 is shaped by a Ca2+-dependent afterhyperpolarization of medium duration (mAHP) (Savic et al., 2001) generated by an apamin-sensitive K+ current mediated by SK channels (IAHP) (Sah, 1996; Savic et al., 2001). Reduction of the mAHP increases the interneuronal firing rate (Savic et al., 2001), and suppression of the apamin-sensitive mAHP can also contribute to hippocampal epileptogenesis (Fernández de Sevilla et al., 2006).
There is increasing evidence showing that kainate receptors (KARs) can modulate slow afterhyperpolarization and neuronal excitability in pyramidal neurons via a metabotropic action (Melyan et al., 2002, 2004; Fisahn et al., 2005; Ruiz et al., 2005). Furthermore, this action can be triggered by synaptic release of glutamate in the CA1 (Melyan et al., 2002) and CA3 pyramidal cells (Ruiz et al., 2005). We now show that in newborn CA3 interneurons, GluK1 subunit-containing KARs markedly increase action potential firing by a tonic G-protein-dependent inhibition of the apamin-sensitive IAHP. We further demonstrate that the KAR-dependent inhibition of the IAHP is age dependent and disappears by the end of the second postnatal week because of a developmental decoupling between KAR activation and mAHP inhibition. Thus, our finding unravels a novel, important physiological mechanism that sets the firing properties of the CA3 hippocampal interneurons during postnatal development.
Materials and Methods
Acute hippocampal slices were prepared from the brain of neonatal [postnatal days 3–5 (P3–P5)] or young (P14–P16) C57/BL/6 and GluK1−/− mice (on a hybrid 129Sv × C57BL/6 background) that have been intercrossed for several generations. In addition, GluK1−/− mice that had recently been backcrossed for 10 generations on a pure C57BL/6 background were also used. In these mice, the difference in AP frequency was similarly impaired as compared with wild-type (WT) controls.
Briefly, the brain was quickly dissected into ice-cold solution containing (in mm): 124 NaCl, 3 KCl, 1.25 NaH2PO4, 10 MgSO4, 26 NaHCO3, 15 d-glucose, 1 CaCl2; saturated with 5% CO2/95% O2. A tissue block containing the hippocampi was dissected and glued into the stage of a vibratome. Three hundred fifty-micrometer-thick slices were cut parasagittally in the above solution and stored at room-temperature in a solution containing (in mm): 124 NaCl, 3 KCl, 1.25 NaH2PO4, 4 MgSO4, 26 NaHCO3, 15 d-glucose, 2 CaCl2; 5% CO2/95% O2. The slices were used 1–4 h after cutting.
For electrophysiological recordings, the slices were placed in a submerged recording chamber and perfused with extracellular solution containing (in mm): 124 NaCl, 3 KCl, 1.25 NaH2PO4, 1 MgSO4, 26 NaHCO3, 15 d-glucose, 2 CaCl2; bubbled with 5% CO2/95% O2 at 32°C. Whole-cell and on-cell recordings were made from CA3 stratum lucidum interneurons using patch electrodes (3–5 MΩ). On-cell recordings were made with pipettes containing extracellular solution. Whole-cell pipettes used for current-clamp experiments contained the following (in mm): 130 K-gluconate, 8 NaCl, 10 HEPES, 0.4 EGTA, 4 Mg-ATP, and 0.3 Na-GTP. Pipettes used to record medium afterhyperpolarizing K+ current (ImAHP) contained the following (in mm): 130 K-gluconate, 10 KCl, 10 HEPES, 0.2 EGTA, 4 Mg-ATP, and 0.3 Na-GTP. For recordings of network activity, the filling solution consisted of the following (in mm): 135 K-gluconate, 10 HEPES, 5 EGTA, 4 Mg-ATP, 0.5 Na-GTP, 2 KCl, and 2 Ca(OH)2. The osmolarity of all intracellular solutions was adjusted to 285 mOsm and the pH to 7.25. For pharmacological blockade of AMPARs, NMDARs, GABAARs, and GABABRs, we used GYKI 53655 (50 μm), AP-5 (50 μm), picrotoxin (100 μm), and CGP 55845 (1 μm), respectively. For the blockade of the metabotropic glutamate receptors mGluR1–mGluR8a, we used 2S-2-amino-2(1S,2S-2-carboxycyclopropyl-1-yl-)3-(xanth-9-yl)propanoic acid (LY 341495; 100 μm). Noradrenergic (NE) receptors were blocked by using propanolol (1 μm), coryanthine (5 μm), and yohimbine (5 μm); serotoninergic (5-HT) receptors were blocked by (2R)-1-(3-hydroxyphenyl)sulfonyl-2–2-(4-methyl-1-piperidinyl)ethylpyrrolidine (SB-269970; 0.1 μm), ketanserin (10 μm), and N-[2-[4-(2-methoxyphenyl)-1-piperazinyl]ethyl]-N-(2-pyridinyl cyclo-hexane carboxamide trihydrochloride (WAY-100635; 1 μm); dopaminergic (DA) receptors were blocked by R(+)-7-chloro-8-hydroxy-3-methyl-1-phenyl-2,3,4,5-tetrahydro-1H-3-benzazepine hydrochloride (SCH-23390; 10 μm) and sulpiride (20 μm); and colinergic (Ach) receptors were blocked by atropine sulfate (1 μm). SR-141716 (5 μm) was used to block CB1 cannabinoid receptors.
Interneurons were visually identified under infrared illumination combined with differential contrast imaging or Dodt gradient optics. In addition, biocytin (Sigma-Aldrich) was routinely added to the filling solution (4 mg/ml), and the cells were visualized by Alexa 568-avidin immunostaining and fluorescence microscopy. The following criteria were used: (1) localization in the CA3 stratum lucidum; and (2) multipolar or bipolar shape of the cell soma (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). Apart from visualization, the following electrophysiological criteria were used to differentiate between the interneurons and pyramidal cells: (1) maximal spiking frequency in response to depolarizing step; (2) instantaneous action potential firing frequency; and (3) spike adaptation rate.
As described previously (Savic et al., 2001), interneurons in CA3 stratum lucidum were mainly multipolar and had variable shapes of soma with diameters of <20 μm. Typically, the dendritic arborizations were limited to stratum radiatum. In terms of firing properties, visually identified interneurons and pyramidal cells could be easily distinguished. The saturating number of action potentials in response to a maximal depolarizing step (500 ms) was higher in interneurons than in pyramidal cells (22.4 ± 2.2 and 8.7 ± 5, respectively; n = 8 and 9; p < 0.005; P3–P5). Furthermore, the instantaneous frequency of action potentials in interneurons was 95 ± 9.2 Hz, whereas in pyramidal cells it was much lower, 43.3 ± 25.2 Hz (p < 0.005). The spike adaptation was more prominent in pyramidal cells than in interneurons. In pyramidal cells the spiking frequency during the 500 ms depolarizing step decreased to 25.5 ± 14.7% of the initial value, and in interneurons it decreased to 46.5 ± 6.8% (p < 0.005).
Action potentials were detected and analyzed using the Mini Analysis Program 5.6.6. (Synaptosoft). The amplitude detection threshold was set between 10 and 15 pA (2–5 times the baseline root mean square noise level), and the detected events were verified visually. The number of spontaneous events was calculated in 60 s bins and normalized to the average baseline value before drug application. Instantaneous frequency (if) was calculated from the interval between the two first action potentials evoked by the maximal current step. Spike adaptation rate was quantified by measuring the if between the last two spikes and normalizing the value against the if measured for the first two spikes. Data are expressed as percentage of control (i.e., 100% = no change). Medium afterhyperpolarizing currents, ImAHP, were evoked by applying a depolarizing 60 mV, 40 ms step. The peak amplitude, duration (from step to 10% of baseline), and decay time were calculated by using pCLAMP software. We also measured the interspike trajectory (time in milliseconds to rise 5 mV from the lowest voltage between the spikes) between the second and the third spike during the 40 pA depolarizing step to quantitate the role of AHP in setting the interspike interval.
For analysis of enzymatic activity, samples of varying glutamate concentrations (0–100 μm, volume 1 ml) were incubated with or without the enzyme glutamic-pyruvic transaminase (GPT; 5 U/ml) (Sigma-Aldrich) and pyruvate (0.3–3 mm), for 10 min at 32°C. The samples were then rapidly frozen for storage before HPLC-based analysis of glutamate concentration.
All the pooled data are given as mean ± SEM for the number of cells indicated. For statistical analysis, ANOVA following Tukey's honestly significant difference (HSD comparison or Student's two-tailed t test was used. p < 0.05 was considered as statistically significant.
All experiments were done in accordance with the guidelines given by the ethics committee for animal research at the University of Helsinki, Helsinki, Finland.
Results
GluK1 subunit-containing KARs regulate spontaneous interneuronal firing at P3–P5 but not at P14–P16
To study the spontaneous action potential firing properties during early development, on-cell recordings were made from CA3 stratum lucidum interneurons from P3–P5 and P14–P16 mice. The firing frequency was analyzed from at least 10 min of stable recording. The mean spontaneous firing frequency of interneurons was found to be strongly downregulated during postnatal development (P3–P5: 1.9 ± 0.4 Hz, n = 16; P14–P16: 0.5 ± 0.0 Hz, n = 8, p < 0.05) (Fig. 1 a).
Figure 1.

GluK1 KARs regulate spontaneous interneuronal firing frequency in CA3 at P3–P5 but not at P14–P16. a, Left, Data from on-cell recordings in interneurons show that the mean AP firing frequency of interneurons in P3–P5 GluK1−/− mice (n = 25), was significantly lower than that of wild types (p < 0.05, n = 16). Sample traces from WT and GluK1 −/− mice and summary statistics for the P3–P5 data are shown. Right, Corresponding data from P14–P16 animals, showing that there was no difference in AP firing rate between WT (n = 8) and GluK1 −/− (n = 10) mice at this age. b, c, Sample traces (b) and summary plot (c) (black, WT; gray, GluK1 −/− ) showing dramatic attenuation of AP firing rate in P3–P5 WT (p = 0.0007, n = 10) but not in GluK1−/− mice (n = 8, p = 0.65) upon application of the GluK1 antagonist, LY 382884. d, Summary statistics for the effect of LY 382884 on interneuronal firing rate in WT (left) and GluK1 −/− mice (right). *p < 0.05; ***p < 0.005.
Interestingly, the mean firing frequency of interneurons in neonatal mice lacking the GluK1 subunit (GluK1−/− mice) (Mulle et al., 2000) was found to be significantly lower compared with WT mice (0.5 ± 0.1 Hz, n = 25, p < 0.05). However, at P14–P16, the spontaneous interneuronal firing rate in GluK1−/− mice was similar to that of WT (0.62 ± 0.18 Hz, n = 10, p = 0.46) (Fig. 1 a) indicating that the developmental regulation of firing rate was absent in GluK1−/− mice.
We next used the specific GluK1 antagonist LY 382884 to further study the role of GluK1 in interneuronal excitability. In line with the results obtained using GluK1−/− mice, in neonatal WT mice LY 382884 (10 μm) reversibly and dramatically decreased the spontaneous firing rate of interneurons (18 ± 3% of control, n = 10, p < 0.005) (Fig. 1 b–d). As expected, LY 382884 had no effect in the GluK1−/− mice (105 ± 12% of control, n = 8, p = 0.65) (Fig. 1 b–d) or WT mice at P14–P16 (97 ± 9% of control, n = 3; data not shown). The excitability of CA3 pyramidal neurons has been previously shown to be regulated by KARs containing the high-affinity GluK5 (formerly known as KA2) subunit (Ruiz et al., 2005). However, we found that the spontaneous firing frequency of interneurons was not different between wild-type and GLUK5−/− mice at P3–P5 (1.9 ± 0.4 Hz, n = 16; 2.1 ± 0.4 Hz, n = 8; data not shown).
These data demonstrate that tonic activation of KARs containing the GluK1 subunit controls the activity of interneurons in the hippocampus in a developmentally restricted manner.
KARs regulate interneuronal excitability and mAHP at P3–P5
The simplest explanation for an effect of tonic GluK1 activation on interneuron firing is a direct depolarization of the neuronal membrane by activation of an inward current. However, recordings from interneurons in the whole-cell current-clamp configuration revealed no significant difference in the resting membrane potential between wild-type (−63 ± 2 mV, n = 13) and GluK1−/− mice (−66 ± 1 mV, n = 17). Furthermore, no tonic inward current was revealed in any of the recorded interneurons upon application of LY 382884 in voltage-clamp mode (data not shown).
We next examined whether the activation of GluK1 KARs changed the excitability of CA3 interneurons as previously demonstrated for CA1 and CA3 pyramidal cells (Melyan et al., 2002, Ruiz et al., 2005). Depolarizing steps of 20–100 pA (500–1000 ms, I = 20 pA) were applied under the current-clamp mode to evoke a train of APs in CA3 interneurons in P3–P5 mice (Fig. 2 a). We found that the firing frequency was significantly lower in GluK1−/− mice compared with WT mice at all depolarizing steps (F = 27,4; Tukey's HSD, p < 0.001; n = 40 and 36 for GluK1−/− and WT, respectively) (Fig. 2 a–c). Interestingly, a distinct afterhyperpolarization was observed after the depolarizing step in GluK1−/− mice but not in wild-type mice (sample recordings and data following a 80 pA depolarizing step; afterhyperpolarization amplitude 1.3 ± 1.6 mV and 6.5 ± 4.3 mV in WT and knock-out mice, respectively; p < 0.005) (Fig. 2 b, arrow). Furthermore, the interspike trajectory, analyzed between the second and third spike (40 pA step), was significantly longer in P3–P5 GluK1 −/− interneurons than in wild-type interneurons (45.8 ± 2.5 ms, n = 40 and 26.1 ± 1.6 ms, n = 36, respectively; p < 0.005) (Fig. 2 c).
Figure 2.

KARs regulate interneuronal excitability and mAHP at P3–P5. a, Pooled data from current-clamp experiments showing the firing of P3–P5 interneurons at depolarizing steps of varying amplitude (500–1000 ms, ΔI = 20 pA). In WT (black circles, n = 36), the firing is attenuated by the GluK1 antagonist LY 382884 (black triangles, n = 8). In GluK1 −/− mice the firing is significantly lower compared with WT (gray squares with solid line, n = 40). b, Representative traces from current-clamp experiments showing firing in WT, GluK1−/−, and WT + LY 382884 interneurons at a 80 pA/500 ms depolarizing step. Note the prominent afterhyperpolarization in GluK1 −/−. c, Summary statistics for the interspike trajectory. d, Pooled data from current-clamp experiments showing the firing of P14–P16 interneurons at depolarizing steps of varying amplitude (500–1000 ms, ΔI = 20 pA). There are no significant differences between the firing rates in WT (black circles, n = 5) and WT + LY 382884 mice (black triangles, n = 7). e, Representative traces from current-clamp experiments showing firing in WT and WT + LY 382884 interneurons at a 80 pA/500 ms depolarizing step. ***p < 0.001.
Consistent with the GluK1−/− data, the GluK1 antagonist LY 382884 decreased the firing evoked by 20–100 pA depolarizing current steps in neonatal WT interneurons (F = 27,4; Tukey's HSD, p < 0.001; n = 8) (Fig. 2 a–c), increased the amplitude of afterhyperpolarization (sample recordings and data following a 80 pA depolarizing step; afterhyperpolarization amplitude, 466 ± 52% of control), and prolonged the interspike trajectory from 25.6 ± 1.8 ms to 42.0 ± 6.6 ms, p < 0.05. At P14–P16, LY 382884 had no effect on depolarization-induced interneuronal firing (Fig. 2 d–e). Together with the data on spontaneous firing rate (see Fig. 1), these data strongly suggest that the developmental attenuation of interneuronal activity is regulated by GluK1-containing KARs.
We then went on to directly test whether KARs regulate the currents responsible for the afterhyperpolarization in interneurons. Intrasomatic depolarizing pulses in the voltage-clamp mode (duration, 40 ms; amplitude, 60 mV; every 20 s) in CA3 interneurons at P3–P5 evoked an outward current with the characteristics of ImAHP (Aoki and Baraban, 2000). Under control conditions, the outward current had an average amplitude of 103.5 ± 30.0 pA and an average duration of 167 ± 29 ms (decay time constant, 16.7 ± 3.1 ms; time to peak, 4.2 ± 0.4 ms; n = 67). The current was partially inhibited by apamin (100 nm, 69.12 ± 6.7% of control, n = 5, p < 0.005) suggesting a contribution by the Ca2+-mediated K+ current previously described in adult CA3 interneurons (Fig. 3 a,d) (Savic et al., 2001).
Figure 3.
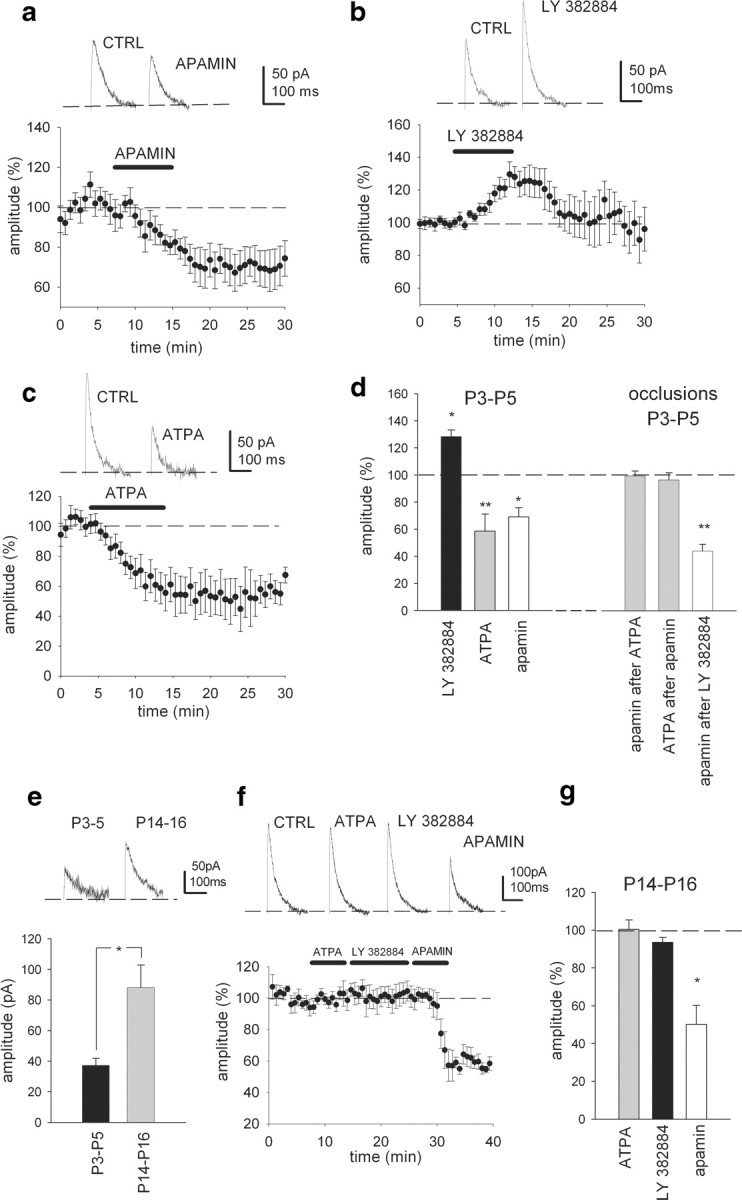
Tonic activity of KAR downregulates apamin-sensitive component of ImAHP at P3–P5 but not at P14–P16. a, Whole-cell voltage-clamp data showing sample ImAHP traces before and after wash-in of apamin and summary plot for the effect of apamin on the afterhyperpolarizing current in P3–P5 interneurons (n = 5, p < 0.005). b, Representative traces and summary plot for the effect of LY 382884 on the afterhyperpolarizing current at P3–P5 (n = 18, p < 0.005). c, Example traces and pooled data showing that ATPA decreases the ImAHP in P3–P5 interneurons (n = 14, p < 0.005). d, Summary data showing the combined effects of LY 382884, ATPA, and apamin on the ImAHP in P3–P5 interneurons. Note that the effect of apamin on the ImAHP is bigger after application of LY 382884 and that the effects of ATPA and apamin are mutually occlusive (ATPA + apamin n, = 5; apamin + ATPA, n = 4; LY 382884 + apamin n = 4). e, Pooled data showing the developmental regulation of the apamin-sensitive component of the ImAHP (P3–P5, n = 8; P14–P16, n = 7; p < 0.05). Data and traces have been obtained by subtracting the ImAHP in the presence of apamin from control current before application of the drug. f, Data showing that in young adult mice (P14–P16) neither GluK1 agonist ATPA nor the antagonist LY 382884 affects the ImAHP amplitude, while apamin still strongly depresses the current (n = 4; p < 0.005). g, Summary data showing the effects of LY 382884, ATPA, and apamin on the ImAHP in P14–P16 interneurons. *p < 0.05; **p < 0.01.
The afterhyperpolarizing K+ current in the neonatal interneurons was sensitive to both GluK1 KAR antagonists and agonists. Upon application of LY 382884 (10 μm), the ImAHP amplitude was significantly and reversibly increased (128 ± 5% of control, p < 0.05, n = 18) (Fig. 3 b,d), while the GluK1 KAR agonist (RS)-2-amino-3-(3-hydroxy-5-tert-butylisoxazol-4-yl) propanoic acid (ATPA; 1 μm) strongly and irreversibly decreased ImAHP amplitude in most of, and, on average, all of the recorded interneurons (58.6 ± 12.6% of control, p < 0.005, n = 14) (Fig. 3 c,d). Moreover, ATPA had no effect on the ImAHP in GluK1−/− mice (94.25 ± 12.96% of control, not significant, n = 7). No apparent correlation between the pharmacological sensitivity and the membrane properties or morphology of the interneurons was detected (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). The effect of apamin on the ImAHP was fully occluded in the presence of ATPA (99.45 ± 3.70%, n = 5) (Fig. 3 d) and, conversely, ATPA had no effect on ImAHP amplitude in the presence of apamin (96.5 ± 5.3%, n = 4). Furthermore, after application of LY 382884, apamin decreased the ImAHP amplitude to 44.1 ± 4.0% (n = 4, p < 0.01), the effect thus being bigger than under control conditions (69.1 ± 6.7%, p < 0.05). These data confirm that the GluK1 selective pharmacological agents indeed modulated the apamin-sensitive current in the neonatal CA3 interneurons (Fig. 3 d). Interestingly, irreversible effects of ATPA on GluR5/GluK1-linked neuronal function have been reported previously (Li et al., 2001; Sallert et al. 2007) (Fig. 3 c). This may be some kind of long-term plasticity induced by strong activation of GluK1-containing KARs (Sallert et al. 2007), but the exact nature of this phenomenon is still unclear.
Developmental regulation of KAR-dependent tonic modulation of ImAHP
The effects of GluK1-containing KARs on interneuron excitability were strongly downregulated during maturation of the circuitry. Therefore, we next tested whether ImAHP and/or its modulation by KAR was also developmentally regulated. Interestingly, the mean amplitude of the apamin-sensitive ImAHP in P14–P16 interneurons was found to be bigger than that at P3–P5 (88.1 ± 14.8 pA, n = 8 and 37.3 ± 4.7 pA, n = 7, respectively; p < 0.05) (Fig. 3 e).
Neither ATPA nor LY 382884 had any effect on the ImAHP amplitude in P14–P16 mice (100 ± 5 and 94 ± 3% of control, respectively, n = 4), while apamin still attenuated the current (50 ± 10% of control, p < 0.005, n = 4) at this developmental stage (Fig. 3 f,g ). These developmental changes might be caused by an overall decrease in GluK1-containing KARs expressed in interneurons. However, a prominent inward current of similar amplitude was seen in response to ATPA application in interneurons at both P3–P5 and P14–P16 (43 ± 5 and 45 ± 5 pA, respectively). Thus, GluK1-containing KARs are expressed and depolarize interneurons at both developmental stages. Since the GluK1-dependent regulation of ImAHP was lost during maturation, albeit with a conserved ionotropic function, it is unlikely that the ionotropic depolarizing action of KARs has a role in the regulation of ImAHP.
Mechanism for the KAR-dependent tonic modulation of ImAHP
The afterhyperpolarizing K+ currents can be modulated by NE, 5-HT, DA, ACh, metabotropic glutamate, and cannabinoid receptors (Vogalis et al., 2001; Sah and Faber, 2002). Thus, KARs could modulate the ImAHP indirectly by activating release of these transmitters in the hippocampus. To test this possibility, we used a cocktail of antagonists to block AMPA, NMDA, GABAA, GABAB, and metabotropic glutamate receptors as well as noradrenalin, serotonin, dopamine, acetylcholine, and cannabinoid receptors (see Materials and Methods).
In the presence of these antagonists ATPA still decreased the ImAHP amplitude in P3–P5 interneurons to 73.8 ± 4.4% (n = 6, p < 0.05) (Fig. 4 a). Finally, in the presence of TTX (1 μm) ATPA still decreased the mAHP amplitude to 59.4 ± 13% of control, p < 0.05, n = 5. These data strongly suggest that KARs do not modulate the function of apamin-sensitive K+ current in the neonatal CA3 interneurons through the release of a neuromodulatory substance, but rather act directly.
Figure 4.
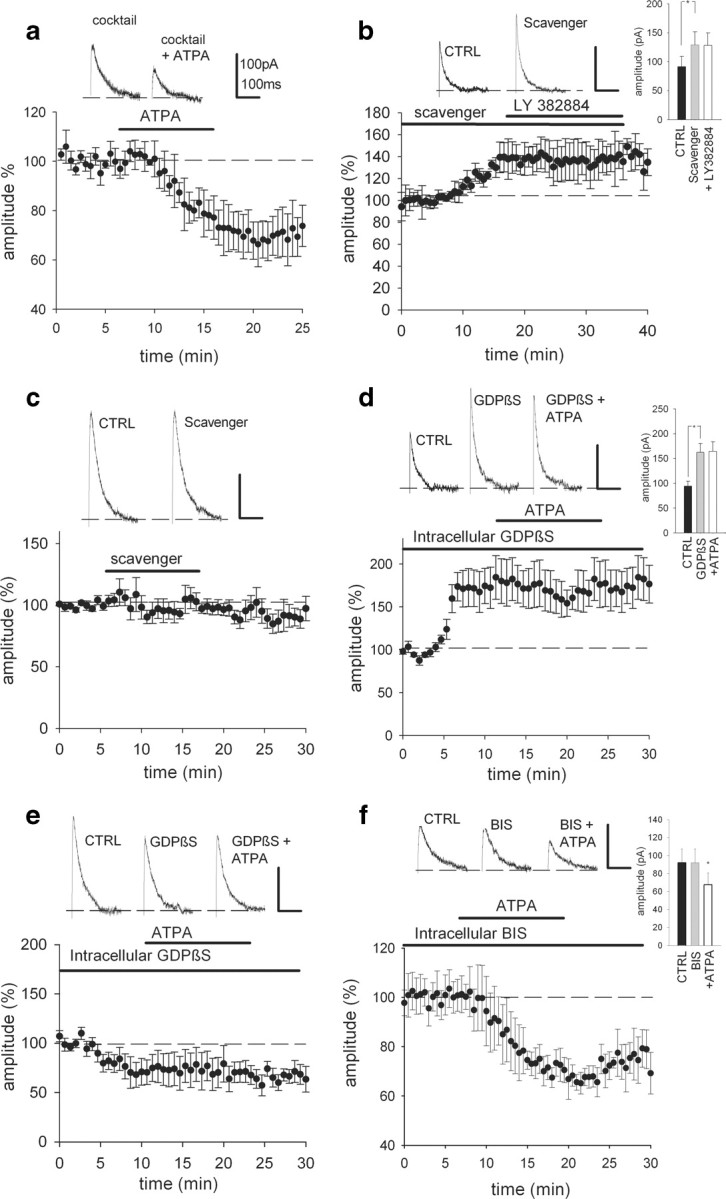
Mechanism for tonic regulation of the mAHP a, Representative traces and summary plot for the effect of ATPA after blockade of AMPAR, NMDAR, GABAA, metabotropic glutamate, 5-HT, NE, DA, ACh, and CB1 receptors (n = 6, p < 0.05). b, Representative traces and summary plot for the effect of glutamate scavenger and LY 382884 on the afterhyperpolarizing current in P3–P5 interneurons. (n = 6, p < 0.01). c, Representative traces and summary plot for the lack of effect of the glutamate scavenger on the afterhyperpolarizing current at P14–P16 (n = 5, p > 0.05). Recordings (a and b) were done in the presence of antagonists for AMPA, NMDA, mGlu, GABAA, and GABAB receptors. d, Example traces and pooled data from P3–P5 mice showing that inhibition of G-protein-mediated signaling by intracellular application of GDPβS accentuates ImAHP (n = 4, p < 0.05). GDPβS occludes the effect of ATPA on the afterhyperpolarizing current. e, Sample traces and summary statistics demonstrating the lack of effect of GDPβS and ATPA on ImAHP in P14–P16 mice (n = 5). f, Representative traces and summary plot for the lack of effect of PKC inhibitor BIS on the afterhyperpolarizing current in P3–P5 interneurons (n = 9) and the effect of ATPA after BIS (p < 0.05, n = 6).
Ambient levels of extracellular glutamate have been previously shown to activate KARs in the immature hippocampus (Lauri et al., 2005). To test whether ambient glutamate could account for the tonic downregulation of ImAHP in the neonatal hippocampal interneurons, we used an enzymatic glutamate scavenger (GPT plus pyruvate) to decrease the extracellular glutamate concentration (Lauri et al., 2005). The efficacy of the scavenger to decrease the extracellular glutamate concentration was verified by HPLC analysis of the artificial CSF (ACSF). The enzymatic treatment reduced the ACSF glutamate concentration to 9.1 ± 2% of control (p < 0.005, n = 12). To isolate the effect from the activity of other glutamate receptors, we also added antagonists for mGluRs and AMPA and NMDA receptors (LY 341495, 100 μm; GYKI 53655, 50 μm; and AP-5, 100 μm, respectively). We found that the ImAHP is highly sensitive to a decrease in the extracellular glutamate concentration at P3–P5. The scavenger caused a significant increase in the ImAHP amplitude (141.7 ± 12.9%, p < 0.01, n = 6) that was very similar to that seen in response to application of a GluK1 antagonist (Fig. 4 b) Also, application of the glutamate scavenger fully occluded the effect of the GluK1 antagonist LY 382884 on the ImAHP (Fig. 4 b) but had no effect on ImAHP in P14–P16 mice (94 ± 8%, n = 5) (Fig. 4 c). Finally, the glutamate scavenger had no effect on the ImAHP amplitude in P3–P5 GluK1−/− mice (current amplitude 102.8 ± 8.8% of control, not significant, n = 9). These results strongly suggest that ambient glutamate at P3–P5 is involved in the tonic regulation of the ImAHP.
To test whether the signaling between GluK1 KARs and K+ channels is G-protein dependent, we used GDPβS (0.3 mm) in the patch-pipette filling solution. We found that intracellular perfusion of GDPβS was associated with a strong increase in the ImAHP amplitude in the neonate (165.3 ± 24.1% of control, p < 0.005, n = 4) (Fig. 4 d). Interestingly, application of ATPA in the presence of GDPβS had no further effect on the ImAHP (166.9 ± 13.4% of control, n = 4) (Fig. 4 d), although the inward current was still present (47 ± 7 pA). The finding thus suggests that KARs regulate ImAHP via a G-protein-dependent mechanism. We found that application of apamin in the presence of GDPβS resulted in an attenuation of the ImAHP, its effect being bigger in the absence of tonic inhibition (37.8 ± 6.7 vs 69.1 ± 6.7%, p < 0.005). However, if the neurons were exposed to apamin before they were perfused with GDPβS, no change in the ImAHP amplitude was seen (99.0 ± 3.7%, n = 4) (data not shown). In contrast to the neonates, in P14–P16 mice GDPβS caused a slight but nonsignificant decrease in the ImAHP amplitude (78.5 ± 12% of control, n = 5) indicating a developmental switch in the mechanisms by which G-proteins regulate the ImAHP in CA3 interneurons (Fig. 4 e). These results indicate that tonically active, GluK1-containing KARs depress apamin-sensitive ImAHP early in development, but the coupling between GluK1-containing KARs and K+ channels is developmentally downregulated.
PKC may be involved in the regulation of the current underlying slow AHP in pyramidal neurons (Melyan et al., 2002). To test for the possible role of PKC activation in the KAR-dependent modulation of ImAHP, we included bisindolylmaleimide VIII acetate (BIS; 1 μm), a selective inhibitor for PKC (Toullec et al., 1991), in the patch-pipette filling solution. However, this treatment had no detectable effect on interneuronal ImAHP (92.1 ± 15.4%, n = 9) (Fig. 4 f). Moreover, in the presence of BIS, ATPA still attenuated the ImAHP to 72.9 ± 4.9% of the control value (p < 0.005, n = 6) (Fig. 4 f). These data thus demonstrate that PKC activation is not involved in the KAR-mediated inhibition of the ImAHP.
ImAHP regulates the physiological activity patterns in the immature hippocampus
Finally, we studied whether the endogenous modulation of ImAHP participates in the regulation of the characteristic synchronous network activity seen in the neonatal but not in the adult hippocampus (Ben-Ari et al., 2004; Lauri et al., 2005). Selective inhibition of the ImAHP by apamin increased the occurrence of spontaneous network bursts recorded in P3–P5 neurons to 202.1 ± 33.9% of the control frequency (p < 0.05, n = 8) (Fig. 5 a). This effect was associated with an increase in the spontaneous firing frequency of interneurons (124.1 ± 16.3% of control, n = 8) (Fig. 5 b). Consistent with the idea of GluK1-mediated tonic inhibition of the ImAHP in the neonatal interneurons, the effect of apamin on the firing frequency was more prominent in the GluK1−/− mice (190.54 ± 10.6%, p < 0.01 compared with WT, n = 6) (Fig. 5 b). In P14–P16, the apamin-induced increase in the firing frequency was more robust (287.6 ± 127.8%, n = 6 and 314.5 ± 94.4%, n = 6 in WT and GluK1−/−, respectively) (Fig. 5 c), but there was no longer a difference between the genotypes. Again, the finding is fully consistent with the idea that the firing activity of neonatal WT but not GluK1−/− interneurons is tonically inhibited by their strong ImAHP, and that the difference between the genotypes in interneuronal firing disappears during the course of development as GluK1-mediated signaling is decoupled from the ImAHP-regulating mechanism.
Figure 5.

The effect of apamin on network activity and interneuronal firing. a, Sample traces and summary plot of whole-cell current-clamp recordings showing that apamin increases the occurrence of synchronous network bursts recorded in P3–P5 interneurons (p < 0.05, n = 8). b, Data from on-cell recordings in P3–P5 interneurons showing the effect of apamin on spontaneous AP firing frequency in WT (n = 8) and GluK1−/− mice (n = 6). Summary graph on the right shows that the effect of apamin on mean firing frequency was significantly (p < 0.01) bigger in GluK1−/− interneurons. c, Data from on-cell recordings in P14–P16 interneurons, showing the effect of apamin on spontaneous AP firing frequency in WT (n = 6) and GluK1 −/− mice (n = 6). Summary graph on the right shows the effect of apamin on mean firing frequency.
Discussion
Our present study provides two fundamental, novel features of interneuronal function. First, we show that the firing activity of CA3 interneurons is dramatically downregulated during early postnatal development. Both spontaneous action potential firing as well as firing in response to a depolarizing step are strongly attenuated during the first postnatal weeks. Second, we show that the developmental shift in the interneuronal output is caused by a decrease in tonic GluK1 and G-protein-dependent inhibition of the apamin-sensitive component of ImAHP. This previously uncharacterized KAR mechanism strongly and tonically depresses the apamin-sensitive K+ current, allowing high firing rates in the neonatal interneurons. The relief of this inhibitory mechanism explains the attenuation of action potential firing during the course of development caused by the emergence of a prominent ImAHP. Moreover, this mechanism likely contributes to the developmental erosion of the spontaneous network bursts involved in the maturation of hippocampal synaptic circuitry (Lauri et al. 2003, 2005; Ben-Ari et al., 2004).
The interneuronal current mediating the mAHP in the newborn interneurons had the electrophysiological and pharmacological characteristics previously reported in the adult CA3 interneurons (Savic et al., 2001). Several neurotransmitter systems, including NE, 5-HT, DA, ACh, and glutamate receptors (Vogalis et al., 2001; Sah and Faber, 2002; Ruiz et al., 2005) have been shown to modulate the afterhyperpolarizing K+ currents in other cell types. However, to the best of our knowledge, endogenous modulation of the apamin-sensitive interneuronal ImAHP has not been described previously. In particular, the results from the experiments using the GluK1 antagonist or the glutamate scavenger strongly argue that interneuronal KARs in the newborn CA3 are continuously activated by endogenous glutamate and that under physiological conditions they exert an inhibitory action on ImAHP. The amplitude of the ImAHP increased from P3 to P14. In parallel, the sensitivity of ImAHP to the KAR antagonist and agonist was lost, although functional GluK1 receptors were expressed in the interneurons also at this developmental stage. These GluRs mediate the conventional, pharmacologically induced ionotropic effect of GluK1 activation at P14. This ionotropic effect is responsible for the GluK1 activation-induced depolarization. We want to emphasize that this nondevelopmentally regulated effect is only seen in response to pharmacological activation, e.g., by ATPA, which is a strong agonist on GluK1-Rs. Thus, during the course of development, the KAR-mediated signaling is decoupled from the ImAHP-regulating mechanism.
Activation of KARs can induce both ionotropic and metabotropic actions in the target neurons (Rodríguez-Moreno and Lerma, 1998; Melyan et al., 2002; Rozas et al., 2003; Lauri et al., 2005). In particular, G-protein-dependent mechanisms have been shown to regulate AHPs and action potential firing in pyramidal cells (Melyan et al., 2002, 2004; Ruiz et al., 2005). In the adult hippocampal pyramidal neurons, slow afterhyperpolarization (sAHP) and mAHP currents (IsAHP and ImAHP, respectively) can be suppressed by activation of GluK2 (formerly known as GluR6), but not by GluK1-containing KARs; this results in a prominent increase in the action potential firing frequency (Fisahn et al., 2005). Furthermore, endogenous activation of KARs by glutamate can inhibit the IsAHP in pyramidal neurons in a G-protein-dependent manner, yet this requires the presence of the high-affinity GluK5 subunit (Ruiz et al., 2005). GluK5 subunits could coassemble with GluK1 in interneurons. However, we found no difference in the firing frequency of interneurons between the WT and GluK5−/− mice. It thus appears that the metabotropic regulation of K+ channels in the neonatal interneurons relies on GluK1, as it does for Ca2+ channels in dorsal root ganglion cells (Rozas et al., 2003). The molecular mechanisms for the metabotropic coupling between GluK1 and K+ channels, as well as its developmental uncoupling, remain to be understood.
Our findings that inhibition of G-protein-dependent signaling postsynaptically reproduces the effect of the GluK1 KAR antagonist on ImAHP, fully occludes the effect of the KAR agonist, and has no effect on ImAHP at P14 reveal a novel type of metabotropic, developmentally regulated, KAR-dependent mechanism. We have described a similar KAR-dependent mechanism that inhibits glutamate release in the newborn CA3-CA1 pyramidal cell synapses (Lauri et al., 2005, 2006). The mechanism is GluK1 KAR dependent, tonically active, G-protein dependent, and erodes by the end of the second postnatal week, thus bearing intriguing phenomenological similarity to the mechanism characterized here. Thus, it appears that metabotropic signaling induced by endogenous activation of GluK1 KARs is intimately involved in the presynaptic, postsynaptic, and interneuronal development of hippocampal circuitry. We have previously shown that the presynaptic GluK1 KAR is involved in the regulation of early network activity in the hippocampus. KARs regulate glutamate release at both pyramidal cell and interneuronal synapses (Lauri et al. 2005). By virtue of this, KARs can set the level of excitation in these neurons, thus modulating the behavior of the hippocampal network. Here, we were particularly interested in how KAR modulation of interneuronal excitability contributes to the spontaneous hippocampal network bursts or giant depolarizing potentials. Direct inhibition of the ImAHP current by apamin provided a clear demonstration for the idea that the mechanism described here can critically contribute to the genesis and synchronization of early hippocampal network activities.
Although not addressed in this study, it is interesting to speculate whether the developmental change in the GluK1-mediated regulation of interneuronal firing pattern is also activity dependent, as is the case for the presynaptic mechanism regulating glutamate release and short-term plasticity in CA3-CA1 (Lauri et al., 2006; Sallert et al. 2009). Interestingly, it has been shown in the developing thalamocortex that long-term potentiation induction results in an increase in the timing precision of evoked action potentials in pyramidal neurons; this was suggested to be an important developmental mechanism for improving cortical information processing (Daw et al., 2006). Since interneurons are the key players in setting the temporal coherence critical for synaptic plasticity and neuronal network oscillations, a mechanism that could switch the interneuronal firing pattern from “newborn” to “adult” type would have a privileged role in the activity-dependent maturation of the brain. There are several reports demonstrating the critical role of early hippocampal network bursts in guiding synaptic maturation (Lauri et al., 2003; Huupponen et al., 2007) and the instrumental role of interneurons in synchronizing this activity (Lamsa et al. 2000; Palva et al., 2000, Ben-Ari et al. 2004). Thus, our findings disclose a critical endogenous mechanism controlling the level of interneuronal output during development that can be of vital importance in the development of functional synaptic circuits in the hippocampus.
Footnotes
This work was supported by the Academy of Finland (to S.E.L. and T.T.), the Sigrid Juselius Foundation (to M.S., S.E.L., and T.T.), and University of Helsinki research funds (to S.E.L.). We thank Eli Lilly and Company for providing LY 382884.
References
- Aoki T, Baraban SC. Properties of a calcium-activated K(+) current on interneurons in the developing rat hippocampus. J Neurophysiol. 2000;83:3453–3461. doi: 10.1152/jn.2000.83.6.3453. [DOI] [PubMed] [Google Scholar]
- Ben-Ari Y, Khalilov I, Represa A, Gozlan H. Interneurons set the tune of developing networks. Trends Neurosci. 2004;27:422–427. doi: 10.1016/j.tins.2004.05.002. [DOI] [PubMed] [Google Scholar]
- Daw MI, Bannister NV, Isaac JT. Rapid, activity-dependent plasticity in timing precision in neonatal barrel cortex. J Neurosci. 2006;26:4178–4187. doi: 10.1523/JNEUROSCI.0150-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernández de Sevilla D, Garduño J, Galván E, Buño W. Calcium-activated afterhyperpolarizations regulate synchronization and timing of epileptiform bursts in hippocampal CA3 pyramidal neurons. J Neurophysiol. 2006;96:3028–3041. doi: 10.1152/jn.00434.2006. [DOI] [PubMed] [Google Scholar]
- Fisahn A, Heinemann SF, McBain CJ. The kainate receptor subunit GluR6 mediates metabotropic regulation of the slow and medium AHP currents in mouse hippocampal neurons. J Physiol. 2005;562:199–203. doi: 10.1113/jphysiol.2004.077412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huupponen J, Molchanova S, Taira T, Lauri SE. Susceptibility for homeostatic plasticity is downregulated in parallel with maturation of the hippocampal synaptic circuitry. J Physiol 581. 2007;2:505–514. doi: 10.1113/jphysiol.2007.130062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamsa K, Palva M, Ruusuvuori E, Kaila K, Taira T. Synaptic GABAA activation inhibits AMPA/kainate receptor-mediated bursting in the newborn (P0–P2) rat hippocampus. J Neurophysiol. 2000;83:359–366. doi: 10.1152/jn.2000.83.1.359. [DOI] [PubMed] [Google Scholar]
- Lauri SE, Lamsa K, Pavlov I, Riekki R, Johnston B, Molnar E, Rauvala H, Taira T. Activity blockade induces formation of functional synapses in the newborn rat hippocampus. Mol Cell Neurosci. 2003;22:107–117. doi: 10.1016/s1044-7431(02)00012-x. [DOI] [PubMed] [Google Scholar]
- Lauri SE, Segerstrale M, Vesikansa A, Maingret F, Mulle C, Collingridge GL, Isaac JTR, Taira T. Endogenous activation of kainate receptors regulate glutamate release and network activity in the developing hippocampus. J Neurosci. 2005;25:4473–4484. doi: 10.1523/JNEUROSCI.4050-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauri SE, Vesikansa A, Segerstrale M, Collingridge GL, Isaac JTR, Taira T. Functional maturation of CA1 synapses involves activity-dependent loss of tonic kainate receptor-mediated inhibition of glutamate release. Neuron. 2006;50:415–429. doi: 10.1016/j.neuron.2006.03.020. [DOI] [PubMed] [Google Scholar]
- Li H, Chen A, Xing G, Wei M-L, Rogawski MA. Kainate receptor-mediated heterosynaptic facilitation in the amygdala. Nat Neurosci. 2001;4:612–620. doi: 10.1038/88432. [DOI] [PubMed] [Google Scholar]
- McBain CJ, Fisahn A. Interneurons unbound. Nat Rev Neurosci. 2001;2:11–23. doi: 10.1038/35049047. [DOI] [PubMed] [Google Scholar]
- Melyan Z, Wheal HV, Lancaster B. Metabotropic-mediated kainate receptor regulation of IsAHP and excitability in pyramidal cells. Neuron. 2002;34:107–114. doi: 10.1016/s0896-6273(02)00624-4. [DOI] [PubMed] [Google Scholar]
- Melyan Z, Wheal HV, Lancaster B. Metabotropic regulation of intrinsic excitability by synaptic activation of kainate receptors. J Neurosci. 2004;24:4530–4534. doi: 10.1523/JNEUROSCI.5356-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulle C, Sailer A, Swanson GT, Brana C, O'Gorman S, Bettler B, Heinemann SF. Subunit composition of kainate receptors in hippocampal interneurons. Neuron. 2000;28:475–484. doi: 10.1016/s0896-6273(00)00126-4. [DOI] [PubMed] [Google Scholar]
- Palva M, Lamsa K, Lauri SE, Rauvala H, Kaila K, Taira T. Fast network oscillations in the newborn rat hippocampus in vitro. J Neurosci. 2000;20:1170–1178. doi: 10.1523/JNEUROSCI.20-03-01170.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodríguez-Moreno A, Lerma J. Kainate receptor modulation of GABA release involves a metabotropic function. Neuron. 1998;20:1211–1218. doi: 10.1016/s0896-6273(00)80501-2. [DOI] [PubMed] [Google Scholar]
- Rozas TL, Paternain AV, Lerma J. Noncanonical signaling by ionotropic kainate receptors. Neuron. 2003;39:543–553. doi: 10.1016/s0896-6273(03)00436-7. [DOI] [PubMed] [Google Scholar]
- Ruiz A, Sachidhanandam S, Utvik JK, Coussen F, Mulle C. Distinct subunits in heteromeric kainate receptors mediate ionotropic and metabotropic function at hippocampal mossy fiber synapses. J Neurosci. 2005;25:11710–11718. doi: 10.1523/JNEUROSCI.4041-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sah P. Ca2+-activated K+ currents in neurones: types, physiological roles and modulation. Trends Neurosci. 1996;19:150–154. doi: 10.1016/s0166-2236(96)80026-9. [DOI] [PubMed] [Google Scholar]
- Sah P, Faber ES. Channels underlying neuronal calcium-activated potassium currents. Prog Neurobiol. 2002;66:345–353. doi: 10.1016/s0301-0082(02)00004-7. [DOI] [PubMed] [Google Scholar]
- Sallert M, Malkki H, Segerstråle M, Taira T, Lauri SE. Effects of the kainate receptor agonist ATPA on glutamatergic synaptic transmission and plasticity during early postnatal development. Neuropharmacology. 2007;52:1354–1365. doi: 10.1016/j.neuropharm.2007.01.015. [DOI] [PubMed] [Google Scholar]
- Sallert M, Rantamäki T, Vesikansa A, Anthoni H, Harju K, Yli-Kauhaluoma J, Taira T, Castren E, Lauri SE. Brain-derived neurotrophic factor controls activity-dependent maturation of CA1 synapses by downregulating tonic activation of presynaptic kainate receptors. J Neurosci. 2009;29:11294–11303. doi: 10.1523/JNEUROSCI.0560-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savic N, Pedarzani P, Sciancalepore M. Medium afterhyperpolarization and firing pattern modulation in interneurons of stratum radiatum in the CA3 hippocampal region. J Neurophysiol. 2001;85:1986–1997. doi: 10.1152/jn.2001.85.5.1986. [DOI] [PubMed] [Google Scholar]
- Toullec D, Pianetti P, Coste H, Bellevergue P, Grand-Perret T, Ajakane M, Baudet V, Boissin P, Boursier E, Loriolle F, Duhamel L, Charon D, Kirilovsky J. The bisindolylmaleimide GF 109203X is a potent and selective inhibitor of protein kinase C. J Biol Chem. 1991;266:15771–15781. [PubMed] [Google Scholar]
- Vogalis F, Furness JB, Kunze WAA. Afterhyperpolarization current in myenteric neurons of the guinea pig duodenum. J Neurophysiol. 2001;85:1941–1951. doi: 10.1152/jn.2001.85.5.1941. [DOI] [PubMed] [Google Scholar]
- Zhang L, McBain CJ. Potassium conductances underlying repolarization and after-hyperpolarization in rat CA1 hippocampal interneurones. J Physiol. 1995;488:661–672. doi: 10.1113/jphysiol.1995.sp020998. [DOI] [PMC free article] [PubMed] [Google Scholar]