

Abstract
Dicer is responsible for the generation of mature micro-RNAs (miRNAs) and loading them into RNA-induced silencing complex (RISC). RISC functions as a probe that targets mRNAs leading to translational suppression and mRNA degradation. Schwann cells (SCs) in the peripheral nervous system undergo remarkable differentiation both in morphology and gene expression patterns throughout lineage progression to myelinating and nonmyelinating phenotypes. Gene expression in SCs is particularly tightly regulated and critical for the organism, as highlighted by the fact that a 50% decrease or an increase to 150% of normal gene expression of some myelin proteins, like PMP22, results in peripheral neuropathies. Here, we selectively deleted Dicer and consequently gene expression regulation by mature miRNAs from Mus musculus SCs. Our results show that in the absence of Dicer, most SCs arrest at the promyelinating stage and fail to start forming myelin. At the molecular level, the promyelinating transcription factor Krox20 and several myelin proteins [including myelin associated glycoprotein (MAG) and PMP22] were strongly reduced in mutant sciatic nerves. In contrast, the myelination inhibitors SOX2, Notch1, and Hes1 were increased, providing an additional potential basis for impaired myelination. A minor fraction of SCs, with some peculiar differences between sensory and motor fibers, overcame the myelination block and formed unusually thin myelin, in line with observed impaired neuregulin and AKT signaling. Surprisingly, we also found signs of axonal degeneration in Dicer mutant mice. Thus, our data indicate that miRNAs critically regulate Schwann cell gene expression that is required for myelination and to maintain axons via axon–glia interactions.
Introduction
Micro-RNAs (miRNAs) are small endogenous RNA-duplex transcripts of ∼22 nt that play a pivotal role in the regulation of mRNAs in eukaryotic cells (Rana, 2007; Amaral et al., 2008). Transcribed as long precursors that can contain multiple mature miRNA segments (Kim et al., 2009), they undergo a processing step within the nucleus, mediated by the microprocessor complex, generating ∼60 nt pre-miRNAs (Lee et al., 2003). Pre-miRNAs are exported to the cytoplasm, where the final maturation step is mediated by a complex containing Dicer, a type-III ribonuclease that cleaves the ∼60 nt precursor into ∼22 nt duplexes (Kim et al., 2009). The miRNA is then loaded into RNA-induced silencing complex (RISC) and functions as a probe that targets the 3′-untranslated regions of particular mRNAs (Hannon et al., 2006). This results in RISC-mediated translational modulation or, sometimes, mRNA degradation (Hannon et al., 2006). Because of its crucial role in miRNA maturation, Dicer deletion results in loss of miRNA activity (Bernstein et al., 2003). Several studies have applied this strategy to underscore the importance of miRNAs in morphogenesis of multiple tissues and organs, such as the liver (Sekine et al., 2009), heart (Chen et al., 2008), kidney (Ho et al., 2008), retina (Damiani et al., 2008), hair follicles (Lei et al.), inner ear (Soukup et al., 2009), female reproductive system (Gonzalez and Behringer, 2009), pancreas (Lynn et al., 2007), cortex, and hippocampus (Davis et al., 2008).
Schwann cells (SCs) ensheath and myelinate large-caliber axons in the peripheral nervous system (Bunge, 1993), which greatly increases conduction velocity of action potentials (Peters and Vaughn, 1970). The importance of myelin is highlighted in demyelinating neuropathies, which result in loss of motor and sensory abilities (Scherer and Wrabetz, 2008). SCs arise from neural crest cells (NCCs) (Jessen and Mirsky, 2005), a migratory population that delaminates from the neural tube early in embryonic development (Woodhoo and Sommer, 2008). During differentiation, SCs surround groups of axons and progressively single them out in a process known as “radial sorting” (Pereira et al., 2009). When the promyelinating SC is attached to the selected axon, it substantially alters gene expression and activates Krox20 (Jessen and Mirsky, 2005), a transcription factor with a central role in PNS myelin formation (Topilko et al., 1994). For the onset of myelination, SCs must upregulate expression of promyelinating factors, such as Krox20, as well as myelin structural components (Topilko et al., 1994; Jessen and Mirsky, 2005), and downregulate myelination inhibitors, including Jun, Notch, and SOX2 (Le et al., 2005; Parkinson et al., 2008; Woodhoo et al., 2009).
Here, we used a conditional gene disruption approach to investigate the importance of Dicer-dependent small RNAs during SC maturation. We show that loss of Dicer impairs SC differentiation with a major impact on the onset of myelination by disrupting the balance between stimulating and inhibiting pathways for the myelination process. Furthermore, we observed signs of axonal degeneration, suggesting that SC–axon interactions are crucially affected by SC-specific loss of miRNA-based gene regulation.
Materials and Methods
Generation of conditional knock-out mice.
The generation of conditional Dicer mutant has been described (Bernstein et al., 2003). Mice homozygous for the Dicer floxed allele (Dicerlox/lox) were crossed with mice heterozygous for the floxed allele and additionally expressing the Cre-recombinase under the control of the Dhh promoter (Dhh-cre+ Dicerlox/wt) (Lindeboom et al., 2003), to obtain Dhh-cre+ Dicerlox/lox mice (hereafter called Dicer mutant mice) and Dhh-cre− Dicerlox/wt (hereafter called Dicer control mice). Genotypes were determined by PCR using genomic DNA derived from tail biopsies.
Electron microscopy.
Mice were deeply anesthetized with pentobarbital (Nembutal, Abbott Laboratories; 150 mg/kg, i.p.) and then perfused with 0.1 m phosphate buffer (pH, 7.4) followed by buffer containing 3% glutaraldehyde and 4% paraformaldehyde (PFA). Fixed tissues were post-fixed in 2% osmium tetroxide, dehydrated through a graded acetone series, and embedded in Spurrs resin (Electron Microscopy Sciences). Semithin sections were stained with 1% toluidine blue for analysis at the light microscope, and ultrathin sections were contrasted with 3% uranyl acetate and 1% lead citrate before observation in an FEI Morgagni 268 transmission electron microscope.
Scanning electron microscopy after in situ focused ion beam milling of resin-embedded mouse sciatic nerves.
The resin was trimmed to position the nerve for longitudinal sectioning by focused ion beam milling (FIB). FIB milling and automatic recording of the images was performed with a Zeiss NVision 40 scanning electron microscope combined with an FIB gun system at a 6 nA milling rate, where Ga+ ions were accelerated by a voltage of 30 kV.
Scanning electron microscopy (SEM) images were obtained every 75 nm by an in-lens energy selective backscattered detector at 1.5 kV imaging voltage. Image processing was performed using IMARIS software, version 6.0.0 (Bitplane AG) for cross-sectional stacks and for inverting the images. The raw data block consists of longitudinal images of 75 nm. The raw data covered a volume of 76.5 μm in the x-axis, 25 μm in z-axis, and 48 μm in y-axis. The data were treated with IMARIS software to obtain a transversal plan seriated over 400 individual images. The x-/y-axis voxel resolution of each individual frame was 37.5 nm, and that for the z-axis was 75 nm, resulting in a distance of 140 nm between adjacent images in the movies.
Terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling staining.
For terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling (TUNEL) staining, sections were fixed in 4% PFA for 10 min and blocked for 1 h with 10% goat serum, 1% Triton X-100, and 0.1% BSA in PBS. TUNEL assays were performed using indirect immunofluorescence to visualize nicked DNA according to the manufacturer's instructions (Roche).
Bromodeoxyuridine incorporation and staining.
For proliferation analysis, control and mutant mice were injected with bromodeoxyuridine (BrdU) (Sigma, 10 mg/ml in H2O) at a ratio of 0.1 mg/g body weight 1 h before being killed. After killing, sciatic nerves (SNs) were fixed with 4% PFA in PBS for 10 min and frozen in OCT (Tissue Tek). Ten micrometer frozen sections were submitted to antigen retrieval in 10 mm citrate buffer for 15 min at 90°C. After cooling, sections were incubated for 1 h in PBS with 0.2% Triton X-100 (Sigma), and the DNA was denatured with 2N HCl for 15 min at room temperature (RT). The pH was neutralized with 0.1 m Na-tetraborate for 15 min at RT, and sections were blocked for 1 h with 5% goat serum (Invitrogen), 1% BSA (Sigma), and 0.1% Triton X-100 dissolved in PBS. Primary antibody against BrdU produced in mouse (Sigma, 1:250) was used overnight at 4°C. The following day, sections were incubated with secondary antibody against mouse coupled to Cy3 (Jackson ImmunoResearch, 1:300) for 1 h at RT. The sections were washed, incubated for 5 min in DAPI, washed again, and mounted with IMMU-MOUNT (Thermo Scientific).
Immunofluorescence.
For enhanced yellow fluorescence protein (eYFP) immunohistochemistry, frozen sections were treated for 20 min with prechilled methanol (−20°C) and then fixed in 4% PFA for 5 min. Sections were blocked [10% goat serum (GS) in PBS with 0.3% Triton X-100] for 1 h at RT and incubated with mouse antibodies against green fluorescent protein (abcam, 1:200) and rabbit anti-S100β (DAKO, 1:200) overnight at 4°C in staining buffer (10% GS in PBS with 0.05% Triton X-100). After washing with PBS, sections were incubated with antibody against mouse coupled to Cy3 (Jackson ImmunoResearch, 1:300), and rabbit coupled to Alexa Fluor 488 (Invitrogen, 1:300) for 1 h at RT. Sections were washed with PBS, incubated for 5 min with DAPI, and mounted with IMMU-MOUNT (Thermo Scientific). For SMI32 staining, frozen sections were treated for 20 min with prechilled methanol (−20°C) and then fixed in 4% PFA for 5 min. Sections were blocked (10% GS in PBS with 0.1% Triton X-100) for 1 h at RT and incubated with mouse antibodies against a nonphosphorylated epitope in neurofilament H (clone SMI32, Sternberger Monoclonals, 1:200) and rabbit anti-Neurofilament-M (Millipore Bioscience Research Reagents, 1:200) overnight at 4°C in blocking buffer. After washing with PBS, sections were incubated with antibody against mouse coupled to Cy3 (Jackson ImmunoResearch, 1:300) and rabbit coupled to Alexa Fluor 488 (Invitrogen, 1:300) for 1 h at RT. Sections were washed with PBS, incubated for 5 min with DAPI, and mounted with IMMU-MOUNT (Thermo Scientific).
All images were acquired at RT using Carl Zeiss microscopes (Axioplan 2, with objectives 20×/0.5; and Imager.A1 equipped with objectives 20×/0.5) equipped with Carl Zeiss AxioCam CCD camera (AxioCam MRm). Acquisition software was AxioVision 4.6 (Carl Zeiss). Images were further processed (levels adjusted) using Photoshop CS3 software (Adobe).
Quantitative real-time-PCR of mRNA.
SNs were taken from P5 control and mutant mice, separated from the epineurium and perineurium, and total RNA was extracted using the miRNAeasy Mini Kit (Qiagen) according to the manufacturer's recommendations. cDNA was produced using SuperScript III Reverse Transcriptase (Invitrogen). Quantitative real-time PCR (qRT-PCR) analyses were performed on an ABI 7000 Sequence Detection System (Applied Biosystems) using the 2X SYBR Green PCR Mastermix (Applied Biosystems) according to manufacturer's recommendations. The amplification program was as follows: 5 min at 96°C; and 40 steps of 30 s at 96°C, 30 s at 57°C, 30 s at 72°C, and 5 min at 72°C. The primer sequences were as follows:
Hes1 Forward, TCTGGAAATGACTGTGAAGCA; Hes1 Reverse, GTCACCTCGTTCATGCACTC; Notch1 Forward, GAATGGAGGTAGGTGCGAAG; Notch1 Reverse, CTGAGGCAAGGATTGGAGTC; Notch2 Forward, TGCTGTGGCTCTGGCTGT; Notch2 Reverse, TGTGCCGTTGTGGTAGGTAA; Hes5 Forward, GTA GTC CTG GTG CAG GCT CT; Hes5 Reverse, AAC TCC AAG CTG GAG AAG GC; C-Jun Forward, ACC CCC ACT CAG TTC TTG TG; C-Jun Reverse, AGT TGC TGA GGT TGG CGT AG; SOX2 Forward, ATG GGC TCT GTG GTC AAG TC; SOX2 Reverse, ACC CCT CCC AAT TCT CTT GT; Id2 Forward, CAG CCA TTT CAC CAG GAG AAC A; Id2 Reverse, CAG CAT TCA GTA GGC TCG TGT CA; Dicer Forward, ACC AGC GCT TAG AAT TCC TGG GAG; Dicer Reverse, GCA GCA GAC TTG GCG ATC CTG TAG; ErbB2 Forward, GATCATCATGGAGCTGGC; ErbB2 Reverse, GTCGCAACTTCATGTCGGTA; ErbB3 Forward, AGGTGCTGGGTTTCCTTCTC; ErbB3 Reverse, TCACACTCAGCCCGTTTAGA; Sox10 Forward, GAG GAG GTG GGC GTT GGG CTC TTC; Sox10 Reverse, AGC TCT GTC TTT GGG GTG GTT GGA; MAG Forward, CTGCCTTCAACCTGTCTGTG; MAG Reverse, CGGGTTGGATTTTACCACAC; myelin protein zero (MPZ) Forward, CCCTGGCCATTGTGGTTTAC; MPZ Reverse, CCATTCACTGGACCAGAAGGAG; PMP22 Forward, GGGATCCTGTTCCTGCACAT; PMP22 Reverse, TGCCAGAGATCAGTCGTGTGT; Krox-20 Forward, TTGACCAGATGAACGGAGTG; Krox-20 Reverse, CAGAGATGGGAGCGAAGCTA; Oct-6 Forward, GTTCTCGCAGACCACCATCT; Oct-6 Reverse, GTCTCCTCCAGCCACTTGTT; HMGCR Forward, GGCCTCCATTGAGATCCG; HMGCR Reverse, CACAATAACTTCCCAGGGGT; GAPDH Forward, CGTCCCGTAGACAAAATGGT; and GAPDH Reverse. TTGATGGCAACAATCTCCAC.
qRT-PCR of miRNA.
SNs were taken from P5 control and mutant mice, separated from the epineurium and perineurium, and total RNA was extracted using the miRNAeasy Mini Kit (Qiagen) according to the manufacturer's recommendations. Using the Taqman MicroRNA reverse transcription kit (Applied Biosystems) and the microRNA assays kit (Applied Biosystems), we generated cDNA specific for mature miRNA20a and miRNA9, according to manufacturer's instructions. qRT-PCR analyses were performed on a light cycler 480 (Roche) using the 2× Taqman PCR Mastermix (Applied Biosystems) according to manufacturer's recommendations. The amplification program was as follows: 10 min at 95°C; and 40 cycles of 15 s at 95°C and 60 s at 60°C.
Gene chip hybridization of mRNA.
SNs were collected from four control and four mutant P5 animals, separated from the epineurium and perineurium, and total RNA was extracted using the miRNAeasy Mini Kit (Qiagen) according to the manufacturer's recommendations. The RNA was quality tested and hybridized on gene chips (Agilent), using one-color labeling, by the Functional Genomics Center of Zürich (FGCZ). The data scanned from the chips was submitted to “robust multiarray normalization” by the FGCZ to solidify the statistical comparison between control and mutant animals.
Western blot.
Mice at different developmental stages were killed, and SNs were isolated and separated from the epineurium and perineurium. The nerves were homogenized with a chilled mortar and pestle in lysis buffer (0.1% SDS, 10 mm TrisHCl, 150 mm NaCl, 50 mm NaF, 1 mm NaVO4, 1 mm EDTA, 0.5% sodium-deoxycholate, protease inhibitor mixture; Sigma). Extracts were processed using standard SDS-PAGE and Western blotting procedures. The following antibodies were used: AKT-P Serine 473 (Cell Signaling Technology, 1:1000); AKT (Cell Signaling Technology, 1:1000); Krox20 (Covance, 1:1000); Oct6 (courtesy of Prof. Dies Meijer, Erasmus University, Rotterdam, The Netherlands, 1:200); MAG (Zymed, 1:1000); PMP22 (our laboratory, 1:1000); SOX2 (courtesy of Prof. Michael Wegner, Erlangen-N̈urnberg University, Germany, 1:5000), Notch1 intracellular domain (NICD) (NOTCH1, Millipore, 1:1000), HES1 (Abcam, 1:500) ErbB2-P Tyrosine 1248 (Abcam, 1:1000), ErbB2 (Abcam, 1:1000). Secondary antibodies were obtained from Promega and Southern Biotech. Bands were quantified using “Quantity one” software (Bio-Rad).
Reagents were obtained from Sigma unless otherwise stated.
Morphometric analysis.
The morphometric measurements were performed in toluidine-blue-stained semithin sections (0.5 μm). For this purpose, a cross section per animal of the entire nerve area (quadriceps, and saphenous, dorsal and ventral roots) was reconstructed by merging several high-magnification photographs. The quantification of the myelinated fibers was performed by counting all the figures present in the entire cross section. The ratio of the axon diameter/fiber diameter (g-ratio) was measured in pixels using Adobe Photoshop CS3 software and then converted to micrometers. The number of myelinated fibers was determined manually using the count plugin from Adobe photoshop CS3.
Statistical analysis.
The data show the mean ± SEM in graphic representations, and the mean ± SD in text representations. Statistical significance was determined using a two-tailed Student's t test. Significance was set at *p < 0.05, **p < 0.01, and ***p < 0.001. n represents the number of independent experiments.
Results
Conditional deletion of Dicer in Schwann cells
To delete Dicer specifically from SCs, we used a transgenic mouse that expressed the cre recombinase (Cre) under the control of the desert hedgehog (dhh) gene regulatory sequences (Fig. 1A). Using this construct, Cre becomes active in SCs but not in neurons from embryonic day 12 (Joseph et al., 2004). The Cre-expressing mice were bred with animals containing loxP sequences flanking the Dicer exons 22 and 23. A Rosa26 reporter driving the expression of eYFP (Srinivas et al., 2001) was also bred into the Dicer floxed animals. On Cre expression, the floxed stop cassette is excised and allows the expression of eYFP (Fig. 1A). Genotypes were determined by PCR amplification of genomic DNA (Fig. 1B). qRT-PCR, performed on SN lysates from postnatal day 5 (P5) animals, revealed a dramatic decrease of Dicer mRNA in mutant animals, with some residual levels most likely mainly because of non-recombined endoneurial fibroblasts (Fig. 1C, top). qRT-PCR analysis, designed to target specifically the mature form of miRNAs, revealed a strong reduction of miRNA20a (Fig. 1C, middle) and miRNA9 (Fig. 1C, bottom) in mutant SNs.
Figure 1.
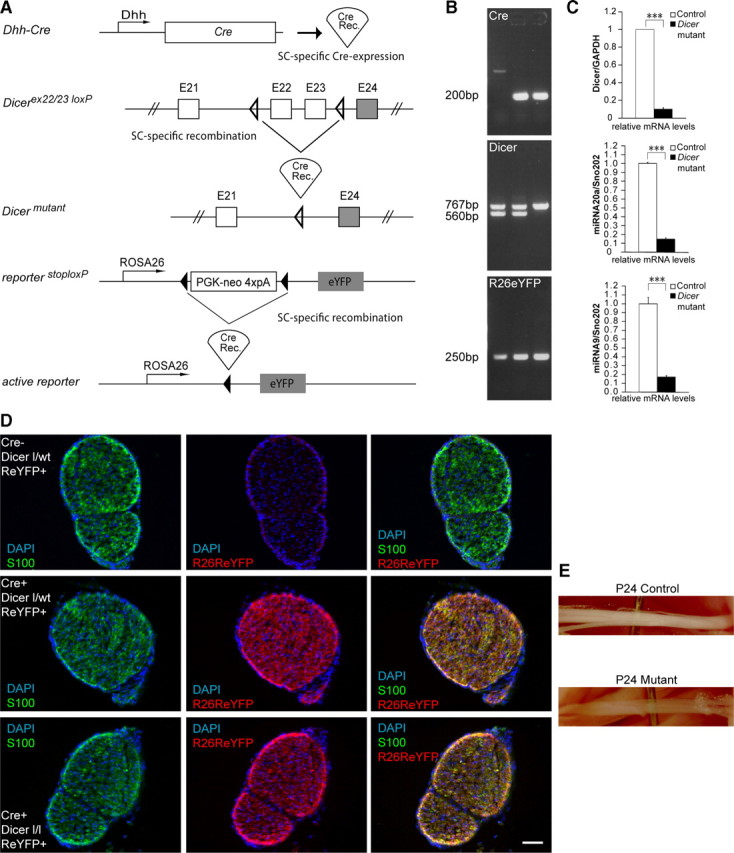
Recombination of the conditional Dicer allele in Schwann cells of mutant mice. A, Regulatory elements of the Dhh promoter drive the expression of Cre in SCs. Schematic map of the Dicer allele depicts the location of the loxP sequences. On Cre-mediated recombination, the genomic region located between the two loxP sites, which includes exon 22 and 23, is excised, thereby inactivating the conditional Dicer allele. The Rosa26 stop/flox locus annexed to an eYFP reporter construct was bred into the floxed animals. When cre recombinase is expressed in SCs, it excises the stop cassette and allows expression of eYFP. B, PCR performed on genomic DNA using primers against Cre (top; 200 bp), Dicer (middle; floxed 767 bp, wt 560 bp), and R26R locus (bottom; 250 bp) showed a cre-negative Dicer flox/wt animal in lane 1 (control), cre-positive Dicer flox/wt animal in lane 2 (heterozygous), and a cre-positive Dicer flox/flox animal in lane 3 (mutant). All three animals were carrying the R26R eYFP construct. C, qRT-PCR performed on RNA extracted from three independent P5 animals showed a dramatic reduction of Dicer (top) mRNA in mutant animals (n = 3, p < 0.0001). The low residual Dicer mRNA levels detected in mutant lysates are likely the result of the presence of endoneurial fibroblasts. qRT-PCR targeting mature miRNAs 20a (middle) (n = 3, p = 0.00031) and 9 (bottom) (n = 3, p = 0.00032) revealed a dramatic decrease of mature miRNAs in Dicer mutant SNs at P5. D, Immunohistochemistry performed on SNs from P3 animals using antibodies against S100 (green channel) and Rosa26 eYFP (red channel). Animals not expressing Cre showed no signal for Rosa26 eYFP (top), whereas control (middle) or mutant mice (bottom) carrying the Cre and Rosa26 reporter displayed an abundant signal for the eYFP antibody, colocalizing with S100, indicating that recombination successfully occurred in the SCs of these animals. E, Control and mutant SNs from P24 littermates. Note that SNs of mutant mice were thinner and more transparent than controls. Error bars display ± SEM. Scale bars, 50 μm.
To show that SCs were effectively recombined, we performed immunohistochemistry, using an antibody against eYFP, on SN frozen sections extracted from P3 animals (Fig. 1D). SNs that did not express Cre showed no eYFP staining and were positive for the SC marker S100 (Fig. 1D, top). Control heterozygous (Fig. 1D, middle) or mutant (Fig. 1D, bottom) animals, which expressed both Cre and Rosa26 reporter, presented abundant and overlapping eYFP and S100 signals, showing that SCs were efficiently recombined. Morphologically, P24 mutant animals had thinner and more transparent SNs than their control littermates (Fig. 1E).
Overall, mutant animals displayed discoordination, tremor, and ataxia pronounced at P24 (supplemental Movies 1 and 2, available at www.jneurosci.org as supplemental material), characteristics that tended to aggravate as the animals grew older. At the age of 5 weeks, mutant animals were barely able to move their hindlimbs (supplemental Movie 3, available at www.jneurosci.org as supplemental material). Because of the severity of the phenotype, and in line with legal requirements, all subsequent experiments were performed at P24 or earlier.
Myelination defects in Dicer mutant SCs
During postnatal development, immature SCs initially engage and surround groups of axons (Fig. 2a,c, asterisks) and then select a single large-caliber axon from the bundle to form a 1:1 relation, a process referred to as “radial sorting” (Jessen and Mirsky, 2005; Pereira et al., 2009). SCs elongate along a segment of the axon to structure an internode, and this segment was subsequently myelinated (Fig. 2c,e, white arrowheads). By analyzing semithin sections (0.5 μm), we observed that Dicer mutant SCs successfully performed radial sorting to a large extent, although some small bundles of unsorted axons remained in P24 mutant animals (Fig. 2b,d,f, asterisks). However, only a few of the axons sorted from the bundles were successfully myelinated by Dicer mutant SCs (Fig. 2d,f, white arrowheads).
Figure 2.
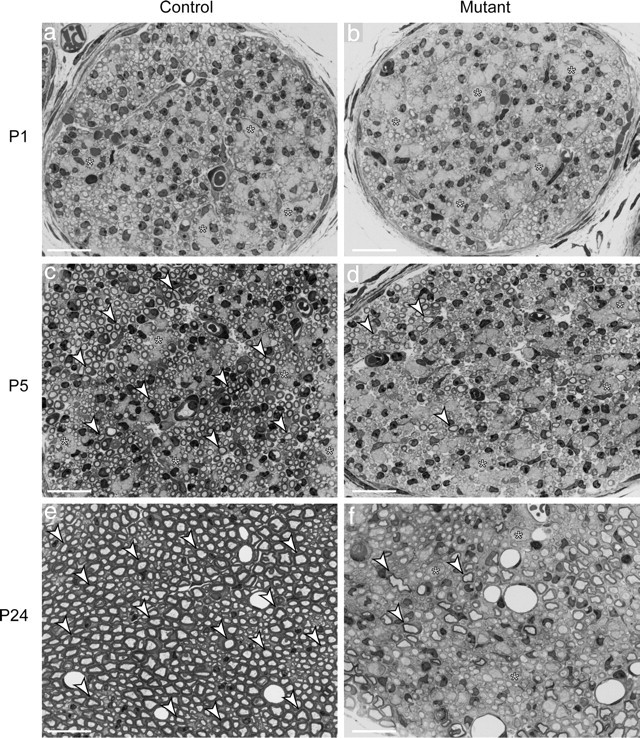
Myelination is impaired on loss of Dicer in sciatic nerves. a–f, SN cross sections (0.5 μm) stained with toluidine blue. a–d, Early in development, SCs are in close association with bundles of axons (asterisks). Progressively, individual axons are sorted from these bundles in a process referred to as radial sorting. E, In control nerves, individual large-caliber axons were wrapped and myelinated. f, In the mutant nerves, myelination was impaired, and only a few myelinated axons were visible at P24 (arrowheads), together with a few small bundles of unsorted axons (asterisks) and many 1:1 relations between axons and SCs, which did not present myelin. Scale bars, 20 μm.
Ultrastructural analysis of control SNs at P5 (Fig. 3Aa) showed SCs at multiple stages of differentiation, some still engaging axon bundles (asterisks), others engaging a single axon but not yet myelinating (white arrowheads), and more advanced ones already myelinating (black arrowheads). In the mutant SNs (Fig. 3Ab), we could also find axonal bundles (asterisks) and many relations not myelinated at 1:1 (white arrowheads), but only a minor number of SCs were myelinating axons in these nerves (black arrowheads).
Figure 3.
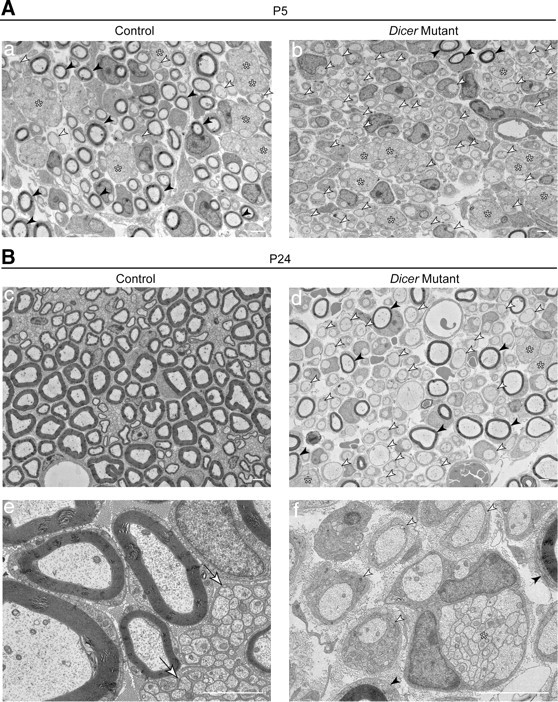
Schwann cells were mainly arrested at the promyelinating stage in the absence of mature miRNAs. A, B, EM micrographs of SNs ultrathin sections at P5 (A) and P24 (B). A, At P5, a panoramic merge of several high-magnification photographs showed in controls (a) a mixture of unsorted axonal bundles (asterisks), 1:1 relations that were not yet myelinated (white arrowheads), and numerous myelinated figures (black arrowheads). Mutant sciatic nerves (b) also displayed several bundles (asterisks), but very few myelinated fibers (black arrowheads) and many 1:1 not-myelinated profiles (white arrowheads). B, At P24, control SNs were extensively myelinated (c, e) and nonmyelinating SCs had established close relations with very small-caliber axons (e, white arrows). In Dicer mutants, a panoramic photomerge (d) showed abundant 1:1 not-myelinated relations (d, f, white arrowheads), some remaining unsorted bundles of naked axons (d, f, asterisks) and a few fibers that had been successfully myelinated (d, f, black arrowheads). Scale bars, 2 μm.
At P24 (Fig. 3B), SNs obtained from control animals were thoroughly myelinated (Fig. 3Bc), and the very small-caliber axons were engaged by nonmyelinating SCs as Remak bundles (Fig. 3Be, white arrows), a structure that differs from bundles of unsorted axons because the SC extends processes that contact every single axon (Griffin and Thompson, 2008) instead of simply surrounding the group. Dicer mutant SNs still presented a large number of 1:1 not-myelinated profiles (Fig. 3Bd,f, white arrowheads), some myelinating SCs (Fig. 3Bd,f, black arrowheads), and sporadically some bundles of unsorted axons (Fig. 3Bd,f, asterisks).
The impairment seen in mutant SNs was not a result of the lack of SCs, since virtually every sorted axon was engaged by an SC (Fig. 3Ab,Bd). Quantification of the cell number in the distal SN (at the proximal part of the tibialis nerve) showed no significant differences between controls and mutants at P5 (256 ± 24 for controls and 218 ± 14 for mutants, n = 3, p = 0.078). Proliferation was also not altered in Dicer mutant SNs at P1 (11.2 ± 1.1% BrdU-positive cells for controls and 10.9 ± 1.9% for mutants, n = 3, p = 0.813) or P5 (8.6 ± 2% BrdU-positive cells for controls and 11.6 ± 2.8% for mutants, n = 3, p = 0.190). Cell death was within normal levels at P1 (0.27 ± 0.13% TUNEL-positive cells for controls and 0.28 ± 0.13% for mutants, n = 3, p = 0.941) but was significantly increased at P5 compared with control animals (0.11 ± 0.03% TUNEL-positive cells for controls and 0.68 ± 0.12% for mutants, n = 3, p = 0.002). In line with increased TUNEL-positive labeling, the expression of several caspases was significantly increased in mutant animals (supplemental Fig. 1, available at www.jneurosci.org as supplemental material).
Together, these data indicated that Dicer mutant SCs were largely successful in performing radial sorting but failed to proceed past the promyelinating stage and to produce a myelin sheet.
Different peripheral nerves were affected on loss of Dicer in SCs, although with phenotypic regional differences
Nerve roots are the PNS region most proximal to the spinal cord. The dorsal roots are composed of sensory axons and the ventral roots of motor axons. At P24, control dorsal and ventral lumbar 5 (L5) roots were thoroughly myelinated (Fig. 4A,Da,g), whereas Dicer mutant roots were not. Dorsal mutant roots showed many SCs arrested at the promyelinating stage (Fig. 4A,Dc,d, white arrowheads), some remaining bundles of axons (Fig. 4A,Db,c, asterisks), and several SCs that successfully produced myelin (Fig. 4A,Dd, black arrowhead). Dorsal roots displayed only a small fraction of myelinated fibers compared with control counterparts (Fig. 4C, left). Myelinating SCs in Dicer mutants produced thinner myelin compared with controls (Fig. 4B, top).
Figure 4.
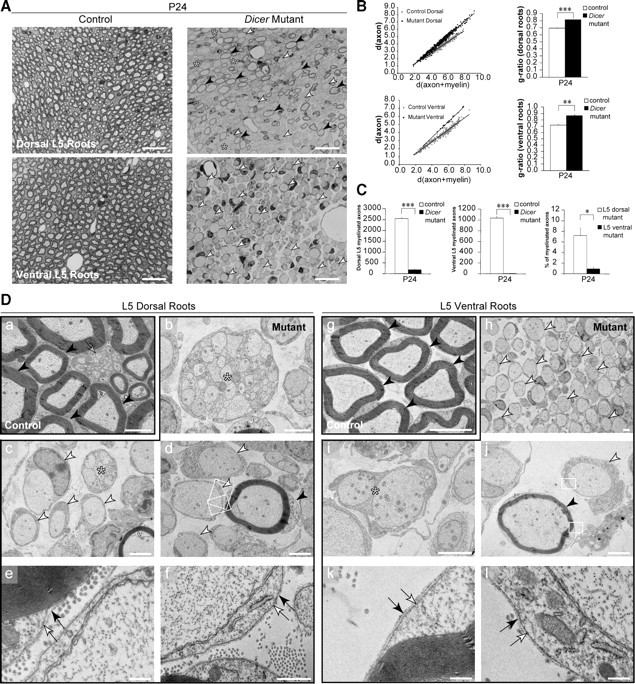
Dorsal and ventral roots were distinctively affected on loss of Dicer in Schwann cells. A, Toluidine blue-stained sections (0.5 μm) of P24 dorsal (sensory fiber-rich) and ventral (motor fiber-rich) roots showed widespread myelination in control animals, opposed to impaired myelination in the mutants. Mutant dorsal roots presented few myelinated fibers (black arrowheads) and small bundles of unsorted axons (asterisks). Dorsal and ventral mutant roots displayed many 1:1 not-myelinated profiles (white arrowheads). Scale bars, 20 μm. B, Scatterplot graphics of measurements performed in control and mutant animals showed thinner myelin sheaths on fibers that managed to get myelinated in mutant animals, both in dorsal and ventral roots. Thinner myelin sheaths were translated into higher values for g-ratio in every animal analyzed (n = 3, p < 0.0001 for dorsal roots, and n = 3, p = 0.0013 for ventral roots). C, The total number of myelinated fibers was quantified in the L5 roots of control and mutant animals. Both dorsal (n = 3, p < 0.0001) and ventral (n = 3, p < 0.0001) roots displayed a small fraction of myelinated fibers when compared with control counterparts. Interestingly, when comparing mutant dorsal to mutant ventral roots we observed that the percentage of myelinated fibers in dorsal roots was about eightfold higher (n = 3, p = 0.011). Da–l, EM micrographs of dorsal roots (a–f) and ventral roots (g–l). Control roots (a, g) displayed thoroughly myelinated fibers (black arrowheads), and in control dorsal roots we could also find nonmyelinating SCs (gray arrow). Mutant dorsal roots displayed bundles of naked axons (b, c, asterisks), 1:1 not-myelinated profiles (c, d, white arrowheads), and some myelinated fibers (d, black arrowhead). Mutant ventral roots were filled with 1:1 not-myelinated profiles (h, white arrowheads), and sporadically SCs were engaging more than a single axon (i, asterisk), although these structures were different from the bundles observed in the dorsal roots. The myelinating cells and the 1:1 profiles in the roots displayed a basal lamina (e, f, k, l, black arrows) apposed to the plasmalemma (e, f, k, l, white arrows), indicating that these were SCs and not invading oligodendrocytes. Scale bars: g, 5 μm; a–d, h–j, 2 μm; e, 0.5 μm; f, k, l, 0.2 μm. Error bars display ± SEM.
Ventral mutant roots showed almost exclusively 1:1 not-myelinated fibers (Fig. 4A,Dh, white arrowheads), with a few cells engaging more than one axon (Fig. 4Di, asterisk), and very few myelinated fibers (Fig. 4Dj, black arrowhead). The number of myelinated fibers in ventral L5 roots was ∼1% of a control littermate (Fig. 4C, middle). The few myelinating SCs also displayed a thinner myelin sheath, which translated into a higher g-ratio compared with controls (Fig. 4B, bottom).
When analyzed at high magnification, the myelinating and the 1:1 profiles in the roots presented a basal lamina (Fig. 4De,f,k,l, black arrow) adjacent to the plasmalemma (Fig. 4De,f,k,l, white arrow) in the external (abaxonal) domain, indicating that the cells were SCs rather than oligodendrocytes invading from the spinal cord.
Interestingly, when we compared the percentage of myelinated fibers in mutant dorsal and ventral roots, we observed that dorsal roots had about eightfold more myelinated axons than ventral roots (Fig. 4C, right).
The differences between the sensory and motor nerve fibers in the roots prompted us to analyze distal nerves with a distinct population of sensory and motor axons. Since in rodents, SNs are composed of ∼85% sensory and only 15% myelinated motor fibers (Schmalbruch, 1986), we decided to examine an additional nerve with a more convenient separation between sensory and motor axons. In rodents, the femoralis nerve branches into the saphenous nerve with purely sensory myelinated fibers and the quadriceps nerve that contains ∼40–50% of myelinated motor axons (R. Martini, unpublished data) (Peyronnard et al., 1986). At P24, control saphenous and quadriceps nerves were thoroughly myelinated (Fig. 5A,Da,g, black arrowheads) and presented mature nonmyelinating SCs engaging small-caliber axons (Fig. 5Da,g, gray arrows). The purely sensory mutant saphenous nerve displayed several not-myelinated 1:1 profiles (Fig. 5A,Db, white arrowheads) and axonal bundles (Fig. 5Dc,d, asterisks). Approximately 2% of Dicer mutant SCs (Fig. 5C, left) successfully proceeded to myelinate axons in the saphenous nerve (Fig. 5A,Dd, black arrowhead).
Figure 5.
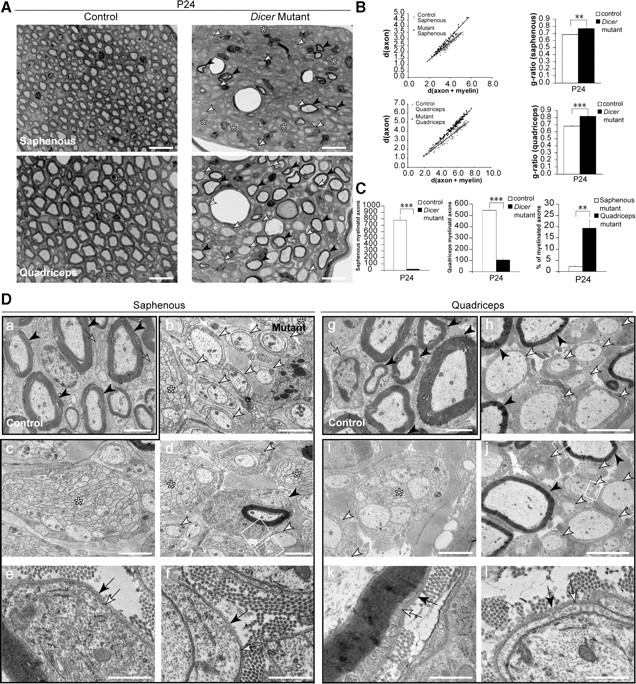
Sensory- and motor-enriched femoralis branches were distinctively affected on loss of Dicer in Schwann cells. A, Toluidine blue-stained sections (0.5 μm) of P24 saphenous nerve, exclusively composed of sensory axons, and quadriceps nerve, composed of 40–50% myelinated motor axons. Control nerves were widely myelinated, but mutant nerves displayed very few myelinated fibers (black arrowheads), several promyelinating profiles (white arrowheads), and some small axonal bundles (asterisks). Scale bars, 20 μm. B, Scatterplot graphics of measurements performed in control and mutant animals showed thinner myelin sheaths on fibers that were myelinated in mutant animals. Every mutant animal analyzed displayed a higher average g-ratio than control littermates (n = 3, p = 0.0014 for dorsal roots; n = 3, p < 0.0001 for ventral roots). C, The total number of myelinated fibers was quantified in the entire nerve cross section of control and mutant animals. Both saphenous (n = 3, p = 0.00011) and quadriceps (n = 3, p < 0.0001) nerves displayed a small fraction of myelinated fibers when compared with control counterparts. Surprisingly, when comparing myelinated fibers in the mutant saphenous nerves to the mutant quadriceps nerves, the saphenous nerve showed an approximately ninefold lower percentage of myelinated axons than the quadriceps (n = 3, p = 0.0077). Da–l, EM micrographs of saphenous (a–f) and quadriceps (g–l) nerves. Control nerves (a, g) displayed thoroughly myelinated fibers (black arrowheads) and mature nonmyelinating SCs (gray arrows). Mutant saphenous nerves displayed bundles of naked axons (b–d, asterisks), 1:1 not-myelinated profiles (b, d, white arrowheads), and very rare myelinated fibers (d, black arrowhead). Quadriceps nerves from Dicer mutants presented 1:1 not-myelinated profiles (h–j, white arrowheads), bundles of unsorted axons (i, j, asterisks), and some myelinating SCs (h, j, black arrowheads). The myelinating and promyelinating SCs presented a basal lamina (e, f, k, l, black arrow) tightly apposed to the SC abaxonal plasmalemma (e, f, k, l, white arrow). Both mutant nerves displayed very high levels of collagen when compared with controls. Scale bars: h, j, 5 μm; a–d, g, i, 2 μm; e, f, k, l, 0.5 μm. Error bars display ± SEM.
In the quadriceps mutant nerves, we found promyelinating relations (Fig. 5A,Dh, white arrowheads) and some unsorted bundles of axons (Fig. 5Di, asterisk). A significant fraction of mutant SCs (Fig. 5C, middle) successfully assembled myelin (Fig. 5A,Dh,j, black arrowheads). Both myelinating and promyelinating SCs in the saphenous and quadriceps nerves displayed a well defined basal lamina (Fig. 5De,f,k,l, black arrow) apposed to the plasma membrane (Fig. 5De,f,k,l, white arrow).
The g-ratio of Dicer mutant myelinated structures in both the saphenous and the quadriceps nerve was significantly increased compared with controls, indicating that mutant cells produced a thinner myelin sheath than controls for axons of the same diameter (Fig. 5B). When comparing the percentage of myelinated fibers among mutant nerves, we observed that the mutant saphenous nerve possessed ∼2% myelinated fibers, whereas the mutant quadriceps had 18%, a difference of approximately ninefold (Fig. 5C, right).
We interpret these results that SCs devoid of Dicer could not normally activate their final differentiation program to myelinate axons. Those SCs that managed to myelinate anyhow displayed impairments in the amount of myelin produced. The phenotypic differences between sensory and motor axon-enriched mutant nerves suggest that SCs associated with motor axons or sensory axons react differently to loss of Dicer-mediated gene regulation. Similar regional phenotypic variations have been previously described in mouse models for human neuropathies (Sancho et al., 1999; Feltri and Wrabetz, 2005). How many of these effects are the result of intrinsic SC differences or deficits involving transcellular communication between SCs and the different types of axons, also taking potential distal–proximal nerve differences into account, remains to be elucidated.
Lack of Dicer and miRNAs prevented the downregulation of myelination inhibitors and led to reduced neuregulin signaling
Our data suggested that the myelination program was suppressed in SCs lacking Dicer. Therefore, we performed an mRNA gene chip array on P5 SNs to screen for transcripts deregulated in mutant animals. As expected, the master transcription activator of myelin genes, Krox20 (also known as Egr2), was significantly reduced in mutants when compared with control SNs (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). The other transcriptional activators SOX10 and Oct6 (also known as Pou3f1) were not significantly altered (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). Several myelin structural proteins (MAG, MPZ, myelin basic protein, periaxin) were also strongly reduced in mutant animals. The enzyme 3-hydroxy-3-methylglutaryl coenzyme A reductase (HMGCR) involved in the synthesis of cholesterol (supplemental Fig. 1, available at www.jneurosci.org as supplemental material) was also significantly decreased in Dicer mutants. We performed qRT-PCR in P5 SNs of control and mutant animals, and confirmed the decrease in mRNA levels of Krox20 (mutant at 17 ± 5% of control levels, n = 3, p < 0.0001) and also of MAG (mutant at 16 ± 6% of control levels, n = 3, p < 0.0001), MPZ (mutant at 15 ± 10% of control levels, n = 3, p = 0.0001), PMP22 (mutant at 15 ± 3% of control levels, n = 3, p < 0.0001), and HMGCR (mutant at 13 ± 4% of control levels, n = 3, p < 0.0001). As in the array screen, Oct6 (mutant levels at 67 ± 22% of controls, n = 3, p = 0.0633) and SOX10 (mutant levels at 113 ± 18% of controls, n = 3, p = 0.274) were not significantly changed. At the protein level, Krox20 was dramatically reduced at P5 and even as early as P1 (Fig. 6A). Oct6 was only slightly reduced in the mutants (Fig. 6A), and SOX10 was not significantly altered (data not shown). The protein amounts of MAG and PMP22 were strongly reduced in mutant SNs (Fig. 6B).
Figure 6.
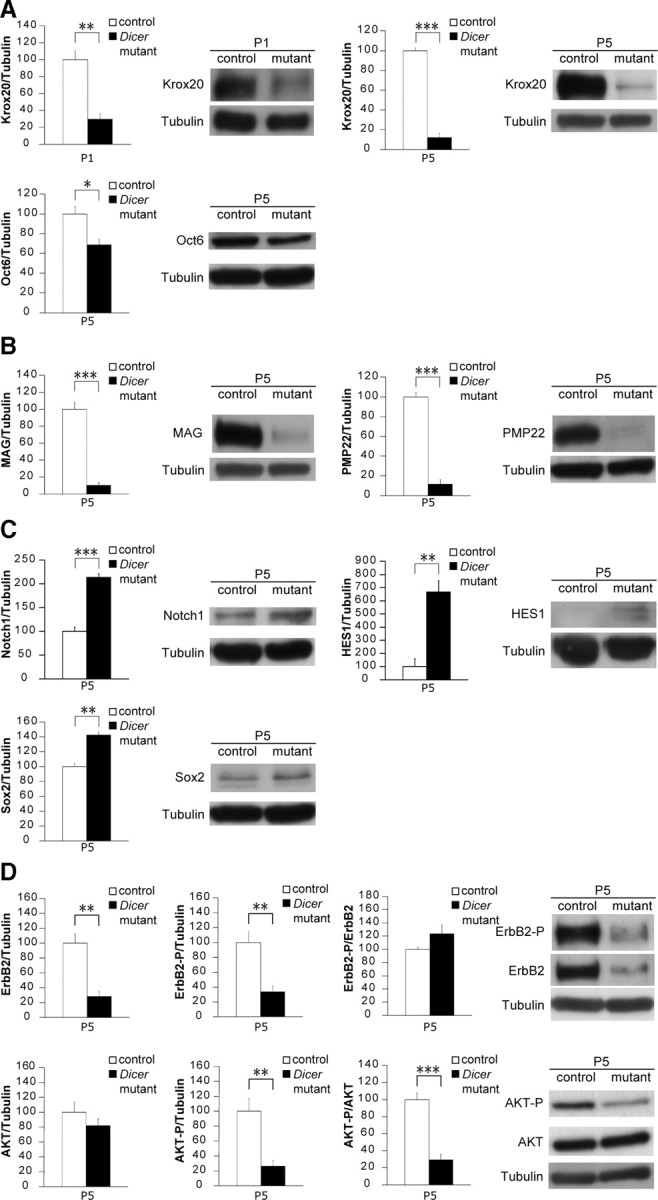
Loss of Dicer resulted in failure to downregulate myelination inhibitors and impaired myelin-stimulating pathways in mouse sciatic nerves. A, Krox20 protein levels were heavily reduced at P5 (n = 5, p < 0.0001) and even as early as P1 (n = 3, p = 0.0045) in mutant Dicer SN lysates. Oct6 levels were also slightly reduced in mutants at P5 (n = 3, p = 0.031). B, The myelin proteins MAG (n = 5, p < 0.0001) and PMP22 (n = 3, p = 0.00017) were also reduced in Dicer mutant mice. C, The transcription factor SOX2, an inhibitor of myelination, was increased in mutant SNs at P5 compared with controls (n = 3, p = 0.0022). NICD was present at higher levels in mutants (n = 3, p = 0.00068), as was its downstream effector HES1 (n = 3, p = 0.0049), which can also act as myelination inhibitor. D, The neuregulin receptor ErbB2 in SCs was decreased at the total protein level (n = 7, p = 0.0018), which is reflected in a reduction of the phosphorylated form (tyrosine 1248) when normalized to tubulin (n = 7, p = 0.0021). AKT phosphorylation on serine 473 was also reduced in P5 mutant nerves when compared with total AKT (n = 6, p < 0.0001) or when compared with tubulin (n = 6, p = 0.0029). AKT total levels were unchanged between control and mutant SNs. Error bars display ± SEM.
Further analysis of the gene array screen revealed that well established myelination suppressors, such as SOX2, Notch1, and HES1, were present at higher levels in mutant SCs (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). However, Jun, Notch2, HES5, and Id2 were not altered (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). We validated these results using qRT-PCR in P5 SNs and verified a pronounced increase of SOX2 (mutant levels at 258 ± 24% of controls, n = 3, p = 0.0004), Notch1 (mutant levels at 195 ± 52% of controls, n = 3, p = 0.033), and HES1 (mutant levels at 554 ± 218% of controls, n = 3, p = 0.022), as well as a slight variation of Notch2 (mutant levels at 115 ± 9% of controls, n = 3, p = 0.037). The increases of SOX2, Notch1, and HES1 were also detectable at the protein level (Fig. 6C), strongly suggesting that these molecules are actively inhibiting the SC differentiation program.
Axonal neuregulin activates the heterodimer ErbB2/ErbB3 at the SC surface and is crucial to stimulate myelination (Nave and Salzer, 2006), prompting us to analyze this particular pathway. We found that total protein levels of ErbB2 and P-ErbB2 (tyrosine 1248) were reduced in Dicer mutant SNs compared with controls (Fig. 6D). In contrast, the mRNA levels of ErbB2 and ErbB3 were not altered as initially seen in the gene array screen (supplemental Fig. 1, available at www.jneurosci.org as supplemental material) and were verified by qRT-PCR (for ErbB2, mutant levels at 93 ± 24% of controls, n = 3, p = 0.646; for ErbB3, mutant levels at 102 ± 35% of controls, n = 3, p = 0.927). Erb2/Erb3 signaling is thought to activate the downstream regulator AKT in myelination, and indeed, in line with our previous findings, we observed a reduction in the phosphorylation of AKT (serine 473) in Dicer mutant SN lysates (Fig. 6D).
In summary, our results indicate that myelination inhibitors are not downregulated in the absence of Dicer. This failure does not allow the start of the myelination program in the majority of SCs. In the SCs that managed to overcome this barrier and myelinate, impaired neuregulin and AKT signaling is likely to result in hypomyelination in mutant peripheral nerves.
Loss of Dicer specifically in Schwann cells resulted in signs of axonal degeneration
While analyzing SN samples obtained from mutant animals under the electron microscope, we observed several axons that presented an unusual accumulation of vesicular structures (Fig. 7Bd–f, black arrowheads) compared with control samples (Fig. 7Ba–c). The figures indicated that these axons might be degenerating and were present in all mutant animals analyzed. Thus, we performed immunohistochemistry using antibodies that specifically recognize a nonphosphorylated epitope in neurofilament H, clone SMI32, in combination with an antibody that generally recognizes neurofilament. SMI32-positive staining is increased in degenerating axons (Meller et al., 1994; Roussarie et al., 2007). In all mutants, we observed strong SMI32 labeling in every section, a signal colocalizing with the neurofilament staining (Fig. 7A, bottom). We never found SMI32 labeling in sections obtained from control animals (Fig. 7A, top). We conclude that some axons become susceptible to degeneration when engaged by Dicer mutant SCs.
Figure 7.
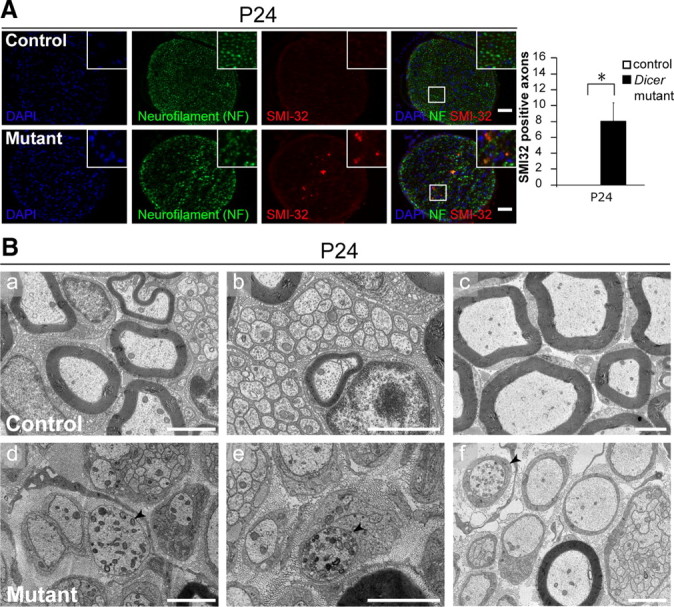
Axonal degeneration on conditional ablation of Dicer in Schwann cells. A, Immunohistochemistry in P24 control and mutant SNs using antibodies against neurofilament (green channel) and SMI32, which specifically recognizes nonphosphorylated neurofilament H (red channel). Zoom boxes highlight the colocalization of the SMI32 and the NF signals. Nerves obtained from Dicer mutant animals showed an SMI32-positive signal in every cross section analyzed, whereas control animals did not show a single positive signal (three different control and mutant animals were analyzed, four full cross sections per animal). Quantification depicts SMI32-positive signal count per section (n = 3, p = 0.026). Scale bars, 50 μm. Error bars display ± SEM. B, Electron micrographs of P24 control (a–c) and mutant (d–f) SNs. In controls, axons that were either myelinated or engaged by a nonmyelinating SC displayed sporadic organelles at the cross-section level (a–c); however, in the mutant animals there were several axons that presented an unusually high accumulation of vesicular structures (d–f, black arrowheads), an indication that the axon might be affected. Scale bars, 2 μm.
Serial electron microscopy imaging revealed in-depth ultrastructural details of Dicer mutant sciatic nerves including degeneration of axon–Schwann cell units
Our morphological analysis of Dicer mutant SNs to this point was mainly based on static electron microscopy (EM) photographs that represent small sections of the whole nerve. Although powerful, this technical approach limited our understanding of the fate of the same nerve fiber and axons in different portions of the nerve. Thus, we performed SEM coupled with in situ FIB (FIB-SEM) (Knott et al., 2008). Through this fascinating technique, we verified that the same axon could be myelinated at several levels and not myelinated at others (supplemental Movie 4 and supplemental Fig. 2a,b, available at www.jneurosci.org as supplemental material, examples are marked with white arrowheads). We also found several SCs loaded with myelin debris and axonal remains (supplemental Movie 4 and supplemental Fig. 2a,b, available at www.jneurosci.org as supplemental material, examples are marked with white arrows), indicating that axonal degeneration is likely coupled to myelin degeneration on loss of Dicer in SCs. Furthermore, it was striking to observe that axons that are integrated in a bundle (supplemental Movie 5 and supplemental Fig. 2c, available at www.jneurosci.org as supplemental material, examples are marked by asterisks) could be found to be sorted by an SC at different depths along the axon (supplemental Movie 5 and supplemental Fig. 2d, available at www.jneurosci.org as supplemental material, black arrowheads). Based on the substantial gain of knowledge in our particular analysis shown here, FIB-SEM appears to have a high potential for future detailed examinations of developing and diseased peripheral nerves.
Discussion
In this study, we have demonstrated that Dicer-dependent miRNAs are critical regulators of SC differentiation. In the absence of Dicer, the vast majority of SCs are blocked at the promyelinating stage although some delayed progression or blocking at the radial sorting stage is also evident. We provide evidence that the defect in myelination progression is likely the result of a combination of failed downregulation of myelination inhibitors and reduced expression of positive myelination regulators and myelin components. Some SCs can escape the blockage and myelinate. However, this myelination is incomplete and is associated with decreased neuregulin signaling in the mutant SNs. Using immunohistochemistry and, in particular FIB-SEM, we show that SC-specific deletion of Dicer leads also to signs of axonal degeneration including entire myelinating axon–SC units.
Loss of Dicer dysregulates the balance between myelination-suppressing and -inducing pathways
The maturation steps of myelinating SCs are a remarkable process of differentiation, requiring pronounced morphological and gene expression specifications (Jessen and Mirsky, 2005). In particular, the onset of myelination depends on the interplay between stimulating and suppressing pathways of SC differentiation (Mirsky et al., 2008). Krox20 is one of the most prominent transcription factors that induces myelination (Topilko et al., 1994) and was strongly reduced in Dicer mutant SNs. Similar to observations in Krox20-deficient animals (Topilko et al., 1994; Le et al., 2005), Dicer mutant SCs were frequently found arrested at the promyelinating stage and showed decreased expression of several myelin proteins, a finding consistent with Krox20-enforced overexpression resulting in activation of multiple myelin-related genes (Nagarajan et al., 2001). Interestingly, we found that MAG was heavily decreased in Dicer mutant SNs but not in Krox20-deficient SNs (Topilko et al., 1994), indicating miRNA-dependent regulation of myelination beyond Krox20. At the promyelinating stage, SOX10 and Oct6 synergize to induce Krox20 expression (Ghislain and Charnay, 2006). In Dicer mutant SNs, we found normal levels of SOX10, whereas Oct6 was slightly reduced. Since Oct6-deficient mice show delayed Krox20 expression but are still able to myelinate (Jaegle and Meijer, 1998; Svaren and Meijer, 2008), the mild Oct6 reduction is unlikely to explain the long-term block of Krox20 expression on Dicer deletion. Additional factors must contribute.
Myelination inhibitors that counteract promyelinating factors need to be downregulated at myelination onset, and their enforced expression in myelinating SCs will induce dedifferentiation and demyelination (Mirsky et al., 2008). SOX2 is such a negative regulator of myelination (Le et al., 2005) and enforced SOX2 expression results in impaired SC differentiation by preventing transcription of several myelin genes (Le et al., 2005). In Dicer mutant nerves, we detected a significant increase in SOX2 compared with controls that is likely to contribute to the observed phenotype. We also found a paired increase of Notch1 and HES1 at RNA and protein levels. During normal SC development, the expression of Krox20 is necessary and sufficient to downregulate Notch, whereas enforced Notch expression actively suppresses Krox20 (Woodhoo et al., 2009). The molecular mechanism by which Notch acts as a myelination inhibitor involves activation of HES genes (Woodhoo et al., 2009). Thus, active suppression of Krox20 expression via Notch is likely to be part of the barrier that prevents most Dicer mutant SCs from myelination.
Loss of Dicer impairs neuregulin and AKT signaling
Neuregulin I type III (NRGI-III) is expressed at the surface of axons and functions as a crucial signal to instruct SCs to start myelination and to define how much myelin should be produced (Nave and Salzer, 2006). NRGI-III acts on SCs through epidermal growth factor-like receptor dimers of ErbB2 and ErbB3 (Britsch et al., 1998). NRGI-III induces a dose-dependent response in SCs since reduced expression results in thinner or no myelin, whereas overexpression induces hypermyelination (Michailov et al., 2004). We found that Dicer mutant SNs contain reduced levels of phosphorylated ErbB2, and total ErbB2 levels were reduced at the protein but not the mRNA levels. Although it remains to be elucidated how loss of Dicer leads to a reduction in ErbB2 receptors, the resulting decrease in neuregulin signaling is likely to further impair the maturation of mutant SCs toward a myelinating fate, in particular by affecting axonal sorting. Furthermore, it is likely to contribute to the observed thinner myelin sheath of mutant SCs that escaped the myelination blockage.
The amount of neuregulin can also influence the degree of AKT phosphorylation in SCs (Hu et al., 2006; Nave and Salzer, 2006), a process thought to be important for myelination (Campana et al., 1999; Maurel and Salzer, 2000; Ogata et al., 2004; Nave and Salzer, 2006). In Dicer mutant SNs, AKT levels were normal but phosphorylation of Ser-473 was impaired. Thus, this defect is likely to be a contributing part of the impaired progression from immature to myelinating Dicer mutant SCs.
Dicer deletion in Schwann cells leads to heterogeneous phenotypes in different PNS regions
The nerve roots are composed of distinct axon populations, exclusively sensory on the dorsal side and motor on the ventral side (Coulpier et al., 2009). The SCs that colonize the roots are ontologically distinct from those in the distal peripheral nerves (Coulpier et al., 2009). The early NCC ventrolateral migratory stream generates SCs in distal nerves. In contrast, the late NCC migration gives rise to the boundary cap (BC) cells. BC cells will differentiate into dorsal root ganglia (DRG) neurons, satellite cells within the DRG, and SCs that myelinate the dorsal and the ventral roots (Maro et al., 2004). Dicer mutant SCs are more successful in myelinating dorsal roots compared with ventral roots. This intriguing result could derive from the type of axon engaged or from differences inherent to SCs that populate both roots (Coulpier et al., 2009). In Dicer mutant distal nerves, the purely sensory saphenous nerve showed a lower percentage of myelinated fibers than the quadriceps nerve that, in controls, is composed of 40–50% myelinated motor axons. The identity of myelinated axons in mutant quadriceps nerves is unclear, and it is possible that sensory axons were preferentially myelinated, as seen in the dorsal roots. However, the observations in mutant saphenous nerves make this an unlikely scenario. Differences in SCs between distal nerves and roots (Maro et al., 2004) may also account for some of the phenotypic variations observed. Other features in mutants, such as thinner myelin sheaths, widespread arrest at the 1:1 stage, and some remaining axonal bundles, were present in all regions analyzed. The mild radial-sorting defects may indicate altered ECM components, receptors, or downstream effectors (Feltri and Wrabetz, 2005). Future studies aiming to dissect SC population differences and axonal type-specific features are necessary for proper interpretation of these tantalizing observations.
Schwann cell survival is compromised in the absence of Dicer
Dicer inactivation results in programmed cell death in various cell types, including Purkinje cells and cortical neurons (Davis et al., 2008), retinal and lens cells (Damiani et al., 2008), inner-ear hair cells and vestibular neurons (Soukup et al., 2009), and cardiomyocytes (Chen et al., 2008). At P1, Dicer mutant SNs showed similar apoptosis rates when compared with controls. However, at P5 the number of TUNEL-positive SCs was significantly increased in mutants. We do not think that SC apoptosis in mutant nerve maturation was a major contributing factor. In P5 mutants, apoptosis remained low, and at P24 virtually every sorted, not-myelinated axon was engaged by a promyelinating SC, despite there being no detectable changes in the proliferation of SCs.
Selective Dicer inactivation in Schwann cells results in axonal damage
In the progression of demyelinating neuropathies, axons eventually become affected and atrophic, and some neurons may even die (Suter and Scherer, 2003). This factor is most likely the major contributor to long-term disability in human patients and animal models (Scherer and Wrabetz, 2008). It is thought that the damage inflicted on axons on an SC-specific mutation more likely derives from demyelination, disruption of SC trophic support toward axons, or the production of toxic influences than from a primary lack of myelin (Nave and Trapp, 2008). EM serial sectioning revealed SCs filled with myelin debris and axonal remains in Dicer mutants at P24. When the animals reached 5 weeks, they were more affected than at P24, and it is feasible that demyelination and axonal loss potentiated the aggravation of their condition, similar to patients with demyelinating neuropathies (Suter and Scherer, 2003; Nave and Trapp, 2008). It will be an exciting task to find the factors that are affected by the Schwann cell-specific loss of Dicer that lead to axonal degeneration.
In summary, we demonstrate that Dicer plays multiple functional roles in SC biology, directly or indirectly regulating Krox20-dependent and -independent myelin components, diverse inhibitors of myelination, neuregulin receptors, and AKT signaling. The cellular processes affected on SC-specific Dicer deletion include myelination (at least axonal sorting, initiation of myelination from the promyelinating state, and myelin growth), SC survival, demyelination, and ultimately axonal integrity. Identifying and validating thoroughly the clusters of miRNAs that regulate the pathways affected remain important tasks for the future. The information gained will be fundamental for in-depth understanding of SC biology and Schwann cell–axon interactions, and might also carry clinical relevance for optimizing therapies toward patients suffering from hypomyelinating and demyelinating disorders.
Footnotes
This work was supported by Eidgenössische Technische Hochschule (ETH) Independent Investigators' Research Awards from the ETH Zurich, by the Swiss National Science Foundation, and the National Centre for Competence in Research grant “Neural Plasticity and Repair.” We thank the Suter laboratory members for all fruitful discussions during this work, and Prof. Rudolf Martini, Janos Groh, and Dennis Klein (University of Würzburg, Würzburg, Germany) for their excellent assistance with femoral nerve and spinal root preparations, and for sharing unpublished data. We also thank Dr. Gregory Hannon (Cold Spring Harbor Laboratory, Watson School of Biological Sciences, and Howard Hughes Medical Institute, Cold Spring Harbor, NY) for providing the Dicer floxed mice. All of the electron microscopy data presented in this work were collected in the Electron Microscopy Center of the ETH Zurich. The Agilent gene chip hybridization and data analysis were performed at the Functional Genomics Center Zürich.
References
- Amaral PP, Dinger ME, Mercer TR, Mattick JS. The eukaryotic genome as an RNA machine. Science. 2008;319:1787–1789. doi: 10.1126/science.1155472. [DOI] [PubMed] [Google Scholar]
- Bernstein E, Kim SY, Carmell MA, Murchison EP, Alcorn H, Li MZ, Mills AA, Elledge SJ, Anderson KV, Hannon GJ. Dicer is essential for mouse development. Nat Genet. 2003;35:215–217. doi: 10.1038/ng1253. [DOI] [PubMed] [Google Scholar]
- Britsch S, Li L, Kirchhoff S, Theuring F, Brinkmann V, Birchmeier C, Riethmacher D. The ErbB2 and ErbB3 receptors and their ligand, neuregulin-1, are essential for development of the sympathetic nervous system. Genes Dev. 1998;12:1825–1836. doi: 10.1101/gad.12.12.1825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bunge RP. Expanding roles for the Schwann cell: ensheathment, myelination, trophism and regeneration. Curr Opin Neurobiol. 1993;3:805–809. doi: 10.1016/0959-4388(93)90157-t. [DOI] [PubMed] [Google Scholar]
- Campana WM, Darin SJ, O'Brien JS. Phosphatidylinositol 3-kinase and Akt protein kinase mediate IGF-I- and prosaptide-induced survival in Schwann cells. J Neurosci Res. 1999;57:332–341. [PubMed] [Google Scholar]
- Chen JF, Murchison EP, Tang R, Callis TE, Tatsuguchi M, Deng Z, Rojas M, Hammond SM, Schneider MD, Selzman CH, Meissner G, Patterson C, Hannon GJ, Wang DZ. Targeted deletion of Dicer in the heart leads to dilated cardiomyopathy and heart failure. Proc Natl Acad Sci U S A. 2008;105:2111–2116. doi: 10.1073/pnas.0710228105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coulpier F, Le Crom S, Maro GS, Manent J, Giovannini M, Maciorowski Z, Fischer A, Gessler M, Charnay P, Topilko P. Novel features of boundary cap cells revealed by the analysis of newly identified molecular markers. Glia. 2009;57:1450–1457. doi: 10.1002/glia.20862. [DOI] [PubMed] [Google Scholar]
- Damiani D, Alexander JJ, O'Rourke JR, McManus M, Jadhav AP, Cepko CL, Hauswirth WW, Harfe BD, Strettoi E. Dicer inactivation leads to progressive functional and structural degeneration of the mouse retina. J Neurosci. 2008;28:4878–4887. doi: 10.1523/JNEUROSCI.0828-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis TH, Cuellar TL, Koch SM, Barker AJ, Harfe BD, McManus MT, Ullian EM. Conditional loss of Dicer disrupts cellular and tissue morphogenesis in the cortex and hippocampus. J Neurosci. 2008;28:4322–4330. doi: 10.1523/JNEUROSCI.4815-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feltri ML, Wrabetz L. Laminins and their receptors in Schwann cells and hereditary neuropathies. J Peripher Nerv Syst. 2005;10:128–143. doi: 10.1111/j.1085-9489.2005.0010204.x. [DOI] [PubMed] [Google Scholar]
- Ghislain J, Charnay P. Control of myelination in Schwann cells: a Krox20 cis-regulatory element integrates Oct6, Brn2 and Sox10 activities. EMBO Rep. 2006;7:52–58. doi: 10.1038/sj.embor.7400573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez G, Behringer RR. Dicer is required for female reproductive tract development and fertility in the mouse. Mol Reprod Dev. 2009;76:678–688. doi: 10.1002/mrd.21010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffin JW, Thompson WJ. Biology and pathology of nonmyelinating Schwann cells. Glia. 2008;56:1518–1531. doi: 10.1002/glia.20778. [DOI] [PubMed] [Google Scholar]
- Hannon GJ, Rivas FV, Murchison EP, Steitz JA. The expanding universe of noncoding RNAs. Cold Spring Harb Symp Quant Biol. 2006;71:551–564. doi: 10.1101/sqb.2006.71.064. [DOI] [PubMed] [Google Scholar]
- Ho J, Ng KH, Rosen S, Dostal A, Gregory RI, Kreidberg JA. Podocyte-specific loss of functional microRNAs leads to rapid glomerular and tubular injury. J Am Soc Nephrol. 2008;19:2069–2075. doi: 10.1681/ASN.2008020162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu X, Hicks CW, He W, Wong P, Macklin WB, Trapp BD, Yan R. Bace1 modulates myelination in the central and peripheral nervous system. Nat Neurosci. 2006;9:1520–1525. doi: 10.1038/nn1797. [DOI] [PubMed] [Google Scholar]
- Jaegle M, Meijer D. Role of Oct-6 in Schwann cell differentiation. Microsc Res Tech. 1998;41:372–378. doi: 10.1002/(SICI)1097-0029(19980601)41:5<372::AID-JEMT4>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- Jessen KR, Mirsky R. The origin and development of glial cells in peripheral nerves. Nat Rev Neurosci. 2005;6:671–682. doi: 10.1038/nrn1746. [DOI] [PubMed] [Google Scholar]
- Joseph NM, Mukouyama YS, Mosher JT, Jaegle M, Crone SA, Dormand EL, Lee KF, Meijer D, Anderson DJ, Morrison SJ. Neural crest stem cells undergo multilineage differentiation in developing peripheral nerves to generate endoneurial fibroblasts in addition to Schwann cells. Development. 2004;131:5599–5612. doi: 10.1242/dev.01429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim VN, Han J, Siomi MC. Biogenesis of small RNAs in animals. Nat Rev Mol Cell Biol. 2009;10:126–139. doi: 10.1038/nrm2632. [DOI] [PubMed] [Google Scholar]
- Knott G, Marchman H, Wall D, Lich B. Serial section scanning electron microscopy of adult brain tissue using focused ion beam milling. J Neurosci. 2008;28:2959–2964. doi: 10.1523/JNEUROSCI.3189-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le N, Nagarajan R, Wang JY, Araki T, Schmidt RE, Milbrandt J. Analysis of congenital hypomyelinating Egr2Lo/Lo nerves identifies Sox2 as an inhibitor of Schwann cell differentiation and myelination. Proc Natl Acad Sci U S A. 2005;102:2596–2601. doi: 10.1073/pnas.0407836102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee Y, Ahn C, Han J, Choi H, Kim J, Yim J, Lee J, Provost P, Rådmark O, Kim S, Kim VN. The nuclear RNase III Drosha initiates microRNA processing. Nature. 2003;425:415–419. doi: 10.1038/nature01957. [DOI] [PubMed] [Google Scholar]
- Lei L, Jin S, Gonzalez G, Behringer RR, Woodruff TK. The regulatory role of Dicer in folliculogenesis in mice. Mol Cell Endocrinol. 315:63–73. doi: 10.1016/j.mce.2009.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindeboom F, Gillemans N, Karis A, Jaegle M, Meijer D, Grosveld F, Philipsen S. A tissue-specific knockout reveals that Gata1 is not essential for Sertoli cell function in the mouse. Nucleic Acids Res. 2003;31:5405–5412. doi: 10.1093/nar/gkg723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynn FC, Skewes-Cox P, Kosaka Y, McManus MT, Harfe BD, German MS. MicroRNA expression is required for pancreatic islet cell genesis in the mouse. Diabetes. 2007;56:2938–2945. doi: 10.2337/db07-0175. [DOI] [PubMed] [Google Scholar]
- Maro GS, Vermeren M, Voiculescu O, Melton L, Cohen J, Charnay P, Topilko P. Neural crest boundary cap cells constitute a source of neuronal and glial cells of the PNS. Nat Neurosci. 2004;7:930–938. doi: 10.1038/nn1299. [DOI] [PubMed] [Google Scholar]
- Maurel P, Salzer JL. Axonal regulation of Schwann cell proliferation and survival and the initial events of myelination requires PI 3-kinase activity. J Neurosci. 2000;20:4635–4645. doi: 10.1523/JNEUROSCI.20-12-04635.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meller D, Eysel UT, Schmidt-Kastner R. Transient immunohistochemical labelling of rat retinal axons during Wallerian degeneration by a monoclonal antibody to neurofilaments. Brain Res. 1994;648:162–166. doi: 10.1016/0006-8993(94)91917-8. [DOI] [PubMed] [Google Scholar]
- Michailov GV, Sereda MW, Brinkmann BG, Fischer TM, Haug B, Birchmeier C, Role L, Lai C, Schwab MH, Nave KA. Axonal neuregulin-1 regulates myelin sheath thickness. Science. 2004;304:700–703. doi: 10.1126/science.1095862. [DOI] [PubMed] [Google Scholar]
- Mirsky R, Woodhoo A, Parkinson DB, Arthur-Farraj P, Bhaskaran A, Jessen KR. Novel signals controlling embryonic Schwann cell development, myelination and dedifferentiation. J Peripher Nerv Syst. 2008;13:122–135. doi: 10.1111/j.1529-8027.2008.00168.x. [DOI] [PubMed] [Google Scholar]
- Nagarajan R, Svaren J, Le N, Araki T, Watson M, Milbrandt J. EGR2 mutations in inherited neuropathies dominant-negatively inhibit myelin gene expression. Neuron. 2001;30:355–368. doi: 10.1016/s0896-6273(01)00282-3. [DOI] [PubMed] [Google Scholar]
- Nave KA, Salzer JL. Axonal regulation of myelination by neuregulin 1. Curr Opin Neurobiol. 2006;16:492–500. doi: 10.1016/j.conb.2006.08.008. [DOI] [PubMed] [Google Scholar]
- Nave KA, Trapp BD. Axon-glial signaling and the glial support of axon function. Annu Rev Neurosci. 2008;31:535–561. doi: 10.1146/annurev.neuro.30.051606.094309. [DOI] [PubMed] [Google Scholar]
- Ogata T, Iijima S, Hoshikawa S, Miura T, Yamamoto S, Oda H, Nakamura K, Tanaka S. Opposing extracellular signal-regulated kinase and Akt pathways control Schwann cell myelination. J Neurosci. 2004;24:6724–6732. doi: 10.1523/JNEUROSCI.5520-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parkinson DB, Bhaskaran A, Arthur-Farraj P, Noon LA, Woodhoo A, Lloyd AC, Feltri ML, Wrabetz L, Behrens A, Mirsky R, Jessen KR. c-Jun is a negative regulator of myelination. J Cell Biol. 2008;181:625–637. doi: 10.1083/jcb.200803013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pereira JA, Benninger Y, Baumann R, Gonçalves AF, Ozçelik M, Thurnherr T, Tricaud N, Meijer D, Fässler R, Suter U, Relvas JB. Integrin-linked kinase is required for radial sorting of axons and Schwann cell remyelination in the peripheral nervous system. J Cell Biol. 2009;185:147–161. doi: 10.1083/jcb.200809008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters A, Vaughn JE. Morphology and development of myelin sheath. In: Davison A, Peters A, editors. Myelination. Springfield, IL: Charles C. Thomas; 1970. [Google Scholar]
- Peyronnard JM, Charron LF, Lavoie J, Messier JP. Motor, sympathetic and sensory innervation of rat skeletal muscles. Brain Res. 1986;373:288–302. doi: 10.1016/0006-8993(86)90343-4. [DOI] [PubMed] [Google Scholar]
- Rana TM. Illuminating the silence: understanding the structure and function of small RNAs. Nat Rev Mol Cell Biol. 2007;8:23–36. doi: 10.1038/nrm2085. [DOI] [PubMed] [Google Scholar]
- Roussarie JP, Ruffié C, Edgar JM, Griffiths I, Brahic M. Axon myelin transfer of a non-enveloped virus. PLoS One. 2007;2:e1331. doi: 10.1371/journal.pone.0001331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sancho S, Magyar JP, Aguzzi A, Suter U. Distal axonopathy in peripheral nerves of PMP22-mutant mice. Brain. 1999;122:1563–1577. doi: 10.1093/brain/122.8.1563. [DOI] [PubMed] [Google Scholar]
- Scherer SS, Wrabetz L. Molecular mechanisms of inherited demyelinating neuropathies. Glia. 2008;56:1578–1589. doi: 10.1002/glia.20751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmalbruch H. Fiber composition of the rat sciatic nerve. Anat Rec. 1986;215:71–81. doi: 10.1002/ar.1092150111. [DOI] [PubMed] [Google Scholar]
- Sekine S, Ogawa R, Mcmanus MT, Kanai Y, Hebrok M. Dicer is required for proper liver zonation. J Pathol. 2009;219:365–372. doi: 10.1002/path.2606. [DOI] [PubMed] [Google Scholar]
- Soukup GA, Fritzsch B, Pierce ML, Weston MD, Jahan I, McManus MT, Harfe BD. Residual microRNA expression dictates the extent of inner ear development in conditional Dicer knockout mice. Dev Biol. 2009;328:328–341. doi: 10.1016/j.ydbio.2009.01.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srinivas S, Watanabe T, Lin CS, William CM, Tanabe Y, Jessell TM, Costantini F. Cre reporter strains produced by targeted insertion of EYFP and ECFP into the ROSA26 locus. BMC Dev Biol. 2001;1:4. doi: 10.1186/1471-213X-1-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suter U, Scherer SS. Disease mechanisms in inherited neuropathies. Nat Rev Neurosci. 2003;4:714–726. doi: 10.1038/nrn1196. [DOI] [PubMed] [Google Scholar]
- Svaren J, Meijer D. The molecular machinery of myelin gene transcription in Schwann cells. Glia. 2008;56:1541–1551. doi: 10.1002/glia.20767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Topilko P, Schneider-Maunoury S, Levi G, Baron-Van Evercooren A, Chennoufi AB, Seitanidou T, Babinet C, Charnay P. Krox-20 controls myelination in the peripheral nervous system. Nature. 1994;371:796–799. doi: 10.1038/371796a0. [DOI] [PubMed] [Google Scholar]
- Woodhoo A, Sommer L. Development of the Schwann cell lineage: from the neural crest to the myelinated nerve. Glia. 2008;56:1481–1490. doi: 10.1002/glia.20723. [DOI] [PubMed] [Google Scholar]
- Woodhoo A, Alonso MB, Droggiti A, Turmaine M, D'Antonio M, Parkinson DB, Wilton DK, Al-Shawi R, Simons P, Shen J, Guillemot F, Radtke F, Meijer D, Feltri ML, Wrabetz L, Mirsky R, Jessen KR. Notch controls embryonic Schwann cell differentiation, postnatal myelination and adult plasticity. Nat Neurosci. 2009;12:839–847. doi: 10.1038/nn.2323. [DOI] [PMC free article] [PubMed] [Google Scholar]