

Abstract
Mutations in leucine-rich glioma inactivated (LGI1) are a genetic cause of autosomal dominant temporal lobe epilepsy with auditory features. LGI1 is a secreted protein that shares homology with members of the SLIT family, ligands that direct axonal repulsion and growth cone collapse, and we therefore considered the possibility that LGI1 may regulate neuronal process extension or growth cone collapse. Here we report that LGI1 does not affect growth directly but instead enhances neuronal growth on myelin-based inhibitory substrates and antagonizes myelin-induced growth cone collapse. We show that LGI1 mediates this effect by functioning as a specific Nogo receptor 1 (NgR1) ligand that antagonizes the action of myelin-based inhibitory cues. Finally, we demonstrate that NgR1 and ADAM22 physically associate to form a receptor complex in which NgR1 facilitates LGI1 binding to ADAM22.
Introduction
Leucine-rich glioma inactivated 1 (LGI1) is a secreted leucine-rich repeat (LRR) protein expressed in the developing and adult CNS (Head et al., 2007; Ribeiro et al., 2008). LGI1 and its homologs (LGI2-4) each contain four N-terminal LRR domains and a C-terminal epitempin domain and LGI1 has been shown to specifically bind cell surface receptors on PC12 cells, cortical neurons and cerebellar granule neurons (Fukata et al., 2006; Sirerol-Piquer et al., 2006). The gene encoding LGI1 is mutated in patients with autosomal dominant lateral temporal epilepsy (ADLTE) (Kalachikov et al., 2002; Morante-Redolat et al., 2002) and, to date, all of the ADTLE causative mutations identified in LGI1 result in retention of the protein in the endoplasmic reticulum and prevent LGI1 secretion (Senechal et al., 2005; Sirerol-Piquer et al., 2006). Therefore, extracellular LGI1 plays a crucial role regulating neuronal circuits in the intact human brain.
The precise function of LGI1 remains uncertain. An initial proteomic analysis suggested that LGI1 associates with Kvb (Schulte et al., 2006), a cytoplasmic regulatory protein that is a component of the Kv1 (shaker-type)-potassium channel complex but how secreted LGI1 gains access to this cytoplasmic protein remains unknown. Another proteomic study identified the transmembrane proteins ADAM22 and ADAM23 as LGI1 receptors and suggested that LGI1 modulates synaptic function by stabilizing levels of cell surface AMPA receptors (Fukata et al., 2006). More recently, Zhou et al. (2009) reported that LGI1 plays a role in the pruning and maturation of excitatory synapses in the hippocampus.
The LGI proteins are members of a superfamily of secreted LRR family members and within this group, the closest LGI1 homologs are Slit proteins, secreted ligands that bind Robo receptors to mediate axonal repulsion and growth cone collapse (Dolan et al., 2007). The structural relationship between LGIs and Slits raised the possibility that LGI proteins may also regulate neuronal growth or repulsion. We tested this hypothesis and show here that LGI1 enhances growth of neurons plated on growth-inhibitory substrates, such as myelin and Nogo66, and demonstrate that LGI1 blocks growth cone collapse induced by myelin. These data led us to test whether LGI1 binds a component of a myelin-derived growth inhibitor receptor complex and consistent with this, we report that LGI1 is a specific ligand for the NgR1 Nogo receptor and that LGI1 and Nogo66 compete for an overlapping binding site on NgR1.
Since ADAM22 has recently been identified as a LGI1 receptor (Fukata et al., 2006), we considered the possibility that NgR1 and ADAM22 associate to form an LGI1 binding moiety. We show that NgR1 and ADAM22 are physically associated in intact brain and in overexpression paradigms and we demonstrate that NgR1 expression facilitates the interaction of LGI1 with ADAM22. We conclude that LGI1 is an endogenous antagonist of myelin-based inhibitors that binds the NgR1 receptor and show that NgR1 and ADAM22 collaborate to form a functional LGI1 binding moiety.
Materials and Methods
Cell culture, plasmids, and generation of conditioned media.
HEK293T and COS7 cells were maintained as described by Boutilier et al. (2008) and HEK293E cells were maintained as described by Durocher et al. (2002). Secreted alkaline phosphatase (AP), AP-LGI1, and AP-Nogo66 were produced in HEK293E cells and normalized for molar concentrations (Flanagan and Cheng, 2000). To produce biotinylated LGI1, LGI1 cDNA was tagged with a biotin acceptor peptide at the N terminus, carboxy to the signal peptide, and then cotransfected with secreted BirA in HEK293T cells to produce biotinylated LGI1. To deplete LGI1 from conditioned media, the media was dialyzed (Tris, pH 8.0, 150 mm NaCl) to remove free biotin and then biotinylated proteins were captured using streptavidin beads, as directed by the manufacturer (Pierce). For production of Fc-NgR1, the NgR1 open reading frame was amplified by PCR and ligated into a vector containing a secreted N-terminal Fc fusion tag.
Pullouts and immunoblotting.
293T cells overexpressing Fc or Fc-NgR1 were lysed in Nonidet P40 buffer (Boutilier et al., 2008) and incubated overnight with protein A-agarose at 4°C, washed extensively, then incubated with biotinylated-LGI1 and AP media overnight at 4°C. Bound proteins were eluted using sample buffer and analyzed by SDS-PAGE and immunoblotting (Boutilier et al., 2008) using anti-LGI1 (Santa Cruz Biotechnology, #SC-9583), anti-biotin (Jackson Laboratories #200-002-096), anti-ADAM22 (University of California, Davis/National Institutes of Health NeuroMab Facility, #75-083), or anti-NgR1 (R&D Systems, #AF1440).
AP binding assays.
For qualitative assays, COS-7 cells were seeded on poly-l-lysine-coated plates, transfected using Lipofectamine 2000 (Invitrogen) and 2 d later, exposed to AP or AP-LGI1 conditioned medium for 90 min at room temperature. Subsequent steps were performed as described by Boutilier et al. (2008). Qualitative binding assays were performed in a similar manner except that transfected 293T cells were used and cells were plated on poly-l-lysine-coated 96 well plates.
DRG growth cone collapse and neurite outgrowth assays.
For DRG growth cone collapse, E13 DRG explants were plated overnight on laminin-coated chamber slides, then incubated with LGI or control conditioned media for 1 h and then exposed to myelin (Igarashi et al., 1993) for 30 min. Cultures fixed in 4% PFA/20% sucrose were permeabilized and stained with rhodamine-conjugated phalloidin. Growth cones were considered collapsed if they showed no lamellipodial structures or an actin-rich bulbous end. For CGN outgrowth assays, neurons were prepared from P8 Sprague Dawley rats and plated on wells coated with poly-l-lysine and myelin. After 24 h in culture, cells were fixed with 4% PFA, permeabilized, and stained with anti-bIII tubulin antibody (Covance) and Hoechst 33342 (Sigma) to reveal neurites and nuclei, respectively. Neurite outgrowth lengths per cell were assessed using the NeuronJ plug-in as described previously (Fournier et al., 2003). For DRG outgrowth assays, neurons were prepared from P5 Sprague Dawley rats and plated on poly-l-lysine-coated chamber slides coated with GST or GST-Nogo66 (GrandPré et al., 2000). Outgrowth was measured using the neurite outgrowth module of MetaXpress (Molecular Devices).
Results
LGI1 inhibits myelin-induced growth inhibition and growth cone collapse
To determine whether LGI1 affected neuronal growth, we first asked whether LGI1 affected neurite outgrowth from postnatal day 8 (P8) rat cerebellar granule neurons (CGNs) plated on different substrates. LGI1 did not alter outgrowth of CGNs plated on poly-l-lysine but intriguingly, LGI1 consistently potentiated neurite outgrowth of CGNs plated on a range of myelin concentrations (Fig. 1A). We next asked whether LGI1 altered growth cone collapse of embryonic day 13 (E13) chick dorsal root ganglion (DRG) sensory neurons. Collapse was induced using increasing doses of soluble myelin in the absence and presence of control- or LGI1-containing conditioned media. LGI1 did not change the incidence of collapsed growth cones in control conditions but was a potent antagonist of growth cone collapse induced by myelin (Fig. 1B). The LGI1 produced for these assays was engineered such that it was metabolically tagged with biotin (see Materials and Methods) and to confirm that LGI1 mediated these effects, streptavidin-agarose was used to deplete LGI1 from conditioned media. Figure 1, B and C, shows that LGI1-containing media attenuated growth cone collapse whereas LGI1-depleted media did not. We conclude that LGI1 antagonizes myelin-induced neurite outgrowth in CGNs and growth cone collapse in DRG sensory neurons.
Figure 1.
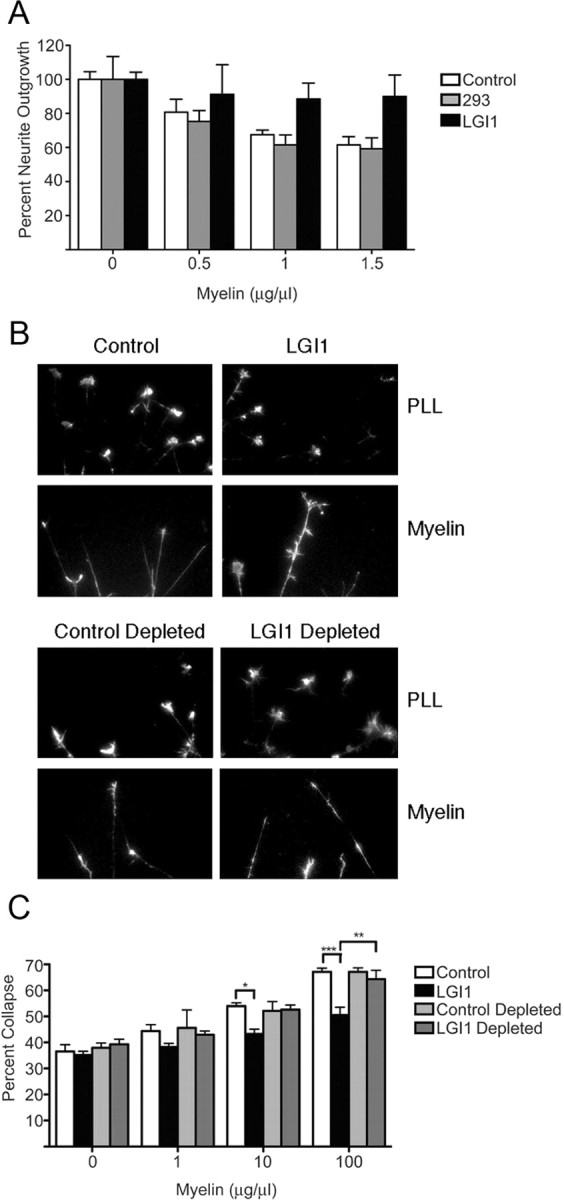
Myelin-induced growth cone collapse and neurite outgrowth inhibition is antagonized by LGI1. A, P8 rat CGNs plated on the indicated concentrations of myelin were maintained in normal media or in media conditioned by 293T cells that had, or had not, been transfected with an LGI1 expression plasmid, as indicated. B, Representative images of E13 chick DRG neurons incubated for 1 h in the presence of the indicated conditioned media then exposed to either PBS or soluble myelin (100 μg/ml) for 30 min. For depletion, media were preincubated with streptavidin agarose beads to remove metabolically biotinylated LGI1 (see Materials and Methods). C, Quantification of DRG neuron growth cone collapse. Neurons were exposed to increasing amounts of myelin in the presence of the indicated conditioned media. For A and C, statistical differences between groups was established using two-way ANOVA followed by Bonferonni post hoc tests (*p < 0.05, **p < 0.01, ***p < 0.001).
AP-LGI1 binds to the surface of COS7 cells overexpressing NgR1
Growth-inhibitory molecules that are present in myelin include Nogo66, myelin-associated glycoprotein (MAG) and oligodendrocyte myelin glycoprotein (OMgp) and we postulated that LGI1 may antagonize these ligands' interaction with the myelin-derived growth inhibitor receptor complex. Components of this receptor complex include NgR1, the p75 neurotrophin receptor (p75NTR) and Lingo1 and we asked whether LGI1 bound any of these cell surface proteins. Figure 2A shows that an alkaline phosphatase fusion of LGI1 (AP-LGI1) showed robust association with COS7 cells that expressed NgR1 but did not bind untransfected cells or those transfected with p75NTR or Lingo1. Cell surface biotinylation assays confirmed that these receptors were expressed on COS7 cell surfaces after transfection (Fig. 2B) and therefore of these, only NgR1 is a specific receptor for LGI1. By performing AP-LGI1 saturation curves on NgR1-transfected COS7 cells, we determined that the Kd of binding of LGI1 was 10.6 nm (Fig. 2C), placing LGI1 in the same range as Nogo66 and MAG, which bind NgR1 with Kd values of 7 and 20 nm, respectively (Fournier et al., 2001; Liu et al., 2002).
Figure 2.
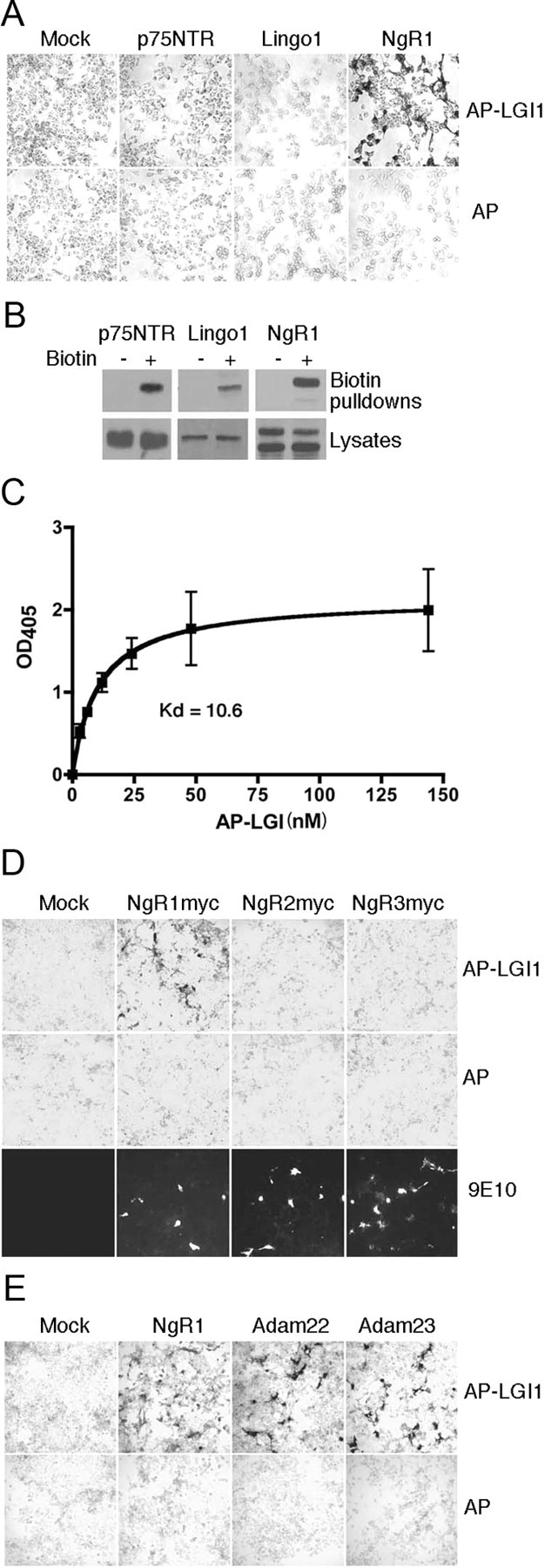
LGI1 binds NgR1 but not NgR2 or NgR3. A, The indicated cDNAs were transfected into COS7 cells and binding of AP-LGI1 to cell surfaces was tested 48 h later, as described in Materials and Methods. AP-LGI1 showed robust binding to NgR1 but did not bind Lingo1 or p75NTR. B, To confirm cell surface expression of receptor components, p75NTR, LINO1 and NgR1 were transfected into COS7 cells and, 48 h later, cell surfaces were biotinylated, subjected to streptavidin pullout and then immunoblotted as indicated. C, Saturation curve of AP-LGI1 binding to NgR1-expressing 293T cells. Data shown are aggregated mean values from three independent experiments; Kd value of 10.6 was calculated from nonlinear best fit regression. D, Interaction between AP-LGI1 and myc-tagged NgR1, NgR2 and NgR3. Upper row shows binding to AP-LGI1, middle row shows binding to AP alone and bottom row confirms NgR expression using anti-myc staining of nonpermeabilized cells. E, Binding of AP-LGI1 to NgR1, ADAM22 and ADAM23 was assessed in transfected COS7 cells, as indicated.
NgR1 has two closely related homologs termed NgR2 and NgR3 (Venkatesh et al., 2005). We expressed myc-tagged forms of each of these in COS7 cells and confirmed that these GPI-linked proteins were abundantly expressed at the cell surface. Binding assays showed that of these, only NgR1 bound LGI1 (Fig. 2D). Recent studies have indicated that ADAM22 and ADAM23, transmembrane proteins that lack the metalloprotease activity typical of the Adams family, may also function as LGI1 receptors (Fukata et al., 2006; Sagane et al., 2008). We therefore compared NgR1, ADAM22, and ADAM23, expressed in COS7 cells, for their ability to bind AP-LGI1. Figure 2E shows that as expected, NgR1, ADAM22, and ADAM23 each show robust binding of AP-LGI1.
LGI1 directly binds NgR1 in a region overlapping the Nogo66 binding site
To determine whether LGI1 directly binds NgR1, we asked whether an Fc-NgR1 fusion protein bound purified biotinylated LGI1 in pulldown assays. LGI1 does not bind control Fc, but an interaction between LGI1 and Fc-NgR1 was readily detected (Fig. 3A). The specificity of this direct interaction was confirmed in competition assays in which AP-LGI1, but not AP alone, antagonized the interaction of biotinylated-LGI1 with Fc-NgR1. Interestingly, the interaction of LGI1 with Fc-NgR1 was reduced, but not abolished, in the presence of AP-Nogo66 (Fig. 3B). Because Nogo66 partially antagonized the interaction of LGI1 with NgR1, we were interested in determining whether LGI1 could antagonize growth inhibition induced by Nogo66. To address this, P5 rat DRG sensory neurons were plated on GST or GST-Nogo66, in the absence and presence of LGI1. Neurons plated on GST-Nogo66 showed a significant inhibition in growth but this effect was completely relieved in the presence of LGI1 (Fig. 3C). Therefore, we conclude that LGI1 and Nogo66 occupy overlapping binding domains on NgR1 and that LGI1 antagonizes Nogo66-induced growth inhibition.
Figure 3.

LGI1 and Nogo66 compete for the same binding site on NgR1. A, Fc or Fc-NgR1 fusion proteins were bound to protein A beads, extensively washed, and then incubated with purified LGI1. After extensive washing, proteins bound to beads were eluted and analyzed by immunoblot, as indicated. Purified LGI1 bound Fc-NgR1 but not Fc alone. The position of Fc-NgR1 is indicated by a single arrowhead, Fc is indicated by a double arrowhead. B, Fc-NgR1 fusion proteins bound to protein A beads were incubated with purified LGI1 in the presence of AP alone, AP-LGI1 or AP-Nogo66. After extensive washing, proteins bound to the protein A beads were eluted and analyzed by immunoblot, as indicated. C, P5 rat DRG neurons were grown on GST or GST-Nogo66 in the absence or presence of purified LGI1 (5 nm). For C, statistical differences between groups was established using Student's t tests (*p < 0.05, **p < 0.001).
NgR1 and ADAM22 associate to form an LGI1 binding complex
LGI1 mutations cause ADTLE in humans and recent studies have shown that the interaction of LGI1 with ADAM22 has profound effects on synaptic transmission and dendritic maturation (Owuor et al., 2009; Zhou et al., 2009). Interestingly, analysis of NgR1 nulls has revealed that NgR1 also functions at central synapses (McGee et al., 2005; Lee et al., 2008). We therefore considered the possibility that NgR1 may form a physiological complex with ADAM22 to mediate synaptic effects of LGI1. To address this, we first compared expression of LGI1, NgR1, and ADAM22 in the developing brain and found that NgR1, LGI1, and ADAM22 are expressed at low levels in embryos and that all three show parallel increases in expression up to P8 (Fig. 4A). Levels of NgR1 and LGI1 remain relatively stable after P8 whereas ADAM22 levels increase from P8 to adulthood. We then asked whether NgR1 and ADAM22 are capable of forming a complex in vivo. Figure 4B shows that NgR1 and ADAM22 robustly associate when overexpressed in HEK293T cells and Figure 4C demonstrates that ADAM22 is present in NgR1 immunoprecipitates prepared from P8 rat brain lysates. We conclude that LGI1, NgR1 and ADAM22 are present in the developing brain and that ADAM22 and NgR1 do indeed associate to form a complex in vivo.
Figure 4.

NgR1 and ADAM22 form a complex that enhances the association of LGI1 with ADAM22. A, Expression of NgR1, LGI1, ADAM22 and actin in lysates prepared from developing rat brain were assessed by immunoblot as indicated. B, NgR1 and Myc-ADAM22 were overexpressed in HEK293T cells and ADAM22-containing immunocomplexes were purified and immunoblotted as indicated. NSG indicates a nonspecific goat Ig used as a negative control for immunoprecipitations. C, NgR1-containing immunocomplexes were isolated from P8 rat brain and then immunoblotted as indicated. Ig indicates a nonspecific mouse monoclonal antibody used as a negative control for immunoprecipitations. D, NgR1 and Myc-ADAM22 were overexpressed in HEK293T cells, pullouts were performed using Fc or Fc-LGI1 and resulting complexes were analyzed by immunoblot, as indicated.
To determine whether coexpression of NgR1 with ADAM22 alters the ability of either receptor to associate with LGI1, we expressed NgR1 and ADAM22 in HEK293T cells and then performed pullouts assays with an Fc-LGI1 fusion protein. Figure 4D shows that when NgR1 and ADAM22 were expressed alone, Fc-LGI1 associated robustly with NgR1 but only modestly with ADAM22. However, when the two receptors were coexpressed, the association of LGI1 with NgR1 was reduced and the association of LGI1 with ADAM22 was dramatically enhanced. These data suggest that NgR1 acts as a coreceptor that enhances the association of LGI1 with ADAM22.
Discussion
Here we identify LGI1 as an antagonist of myelin-induced growth cone collapse and neurite outgrowth inhibition. We show that LGI1 binds directly to NgR1 and show that LGI1 competes with Nogo66 for occupancy of the receptor. The LGI1-NgR1 interaction is highly specific since LGI1 binds to NgR1 but not to NgR2 or NgR3, its closely related structural homologs. Together, these studies establish that LGI1 is a bona fide ligand for the NgR1 receptor that physically and functionally antagonizes myelin-based growth inhibitors. Endogenous NgR1 ligands previously identified function as receptor agonists that promote growth cone collapse and neurite growth inhibition (Ferraro et al., 2004; Zhang et al., 2009). To our knowledge, LGI1 is the first endogenous NgR1 antagonist to be identified and its presence raises the possibility that NgR1 activation in vivo may reflect relative occupancy by endogenous agonists and antagonists. It is noteworthy that one study has shown that NgR1 did not mediate the chronic growth-inhibitory actions of myelin inhibitors but did transduce acute growth cone-collapsing effects of OMgp, MAG and Nogo66 (Chivatakarn et al., 2007). However, our studies show that LGI1, a NgR1 binding protein, not only antagonizes myelin-mediated growth cone collapse but also facilitates neurite extension over myelin and Nogo66. NgR1 is expressed on rat cerebellar granule neurons (supplemental Fig. 1, available at www.jneurosci.org as supplemental material) and it seems likely that LGI1 functions via NgR1 antagonism. However, we cannot rule out the possibility that this ligand may also exert positive effects on neurite extension through alternate mechanisms or through alternate receptors, such as ADAM22 or ADAM23. In this regard, it is noteworthy that Owuor et al. (2009) have recently reported that LGI1 can mediate ADAM22-dependent neurite outgrowth from cortical neurons. Testing the physiological role of LGI1 on neuronal growth in vivo will be an important priority as appropriate genetic resources come available. Since other NgR1 antagonists facilitate growth in vivo (GrandPré et al., 2002; Li et al., 2004; Harvey et al., 2009), we speculate that local application of LGI1 or derivatives will facilitate neuronal growth in vivo after injury or lesion.
ADAM22 had been previously identified as an LGI1 receptor and emerging evidence suggests that an LGI1-ADAM22 complex may regulate dendritic pruning and synaptic function (Fukata et al., 2006; Zhou et al., 2009). Here, we confirm that LGI1 is a ligand for ADAM22 and show that NgR1 and ADAM22 form a stable complex in the developing rat brain. Intriguingly, we found that LGI1 binding to ADAM22 is enhanced in the presence of NgR1, suggesting that a physiological function for NgR1 may be to function as an accessory receptor for ADAM22. NgR1 null mice have defects in dendritic spine morphology and display supranormal experience-driven plasticity. (McGee et al., 2005). Interestingly, a recent study has demonstrated that NgR1 is required for lasting memories (Karlén et al., 2009). Given that LGI1 is critically important for glutamatergic synapse maturation and dendritic pruning (Zhou et al., 2009), determining whether LGI1, NgR1 and ADAM22 function collaboratively to drive synapse maturation and dendritic sculpting is a key priority for future studies. Mice lacking PirB (Atwal et al., 2008), a recently identified receptor for Nogo, MAG, and OMgp, also display supranormal experience-driven plasticity, and it is conceivable that PirB may function in similar pathways.
LGI1 was identified as a causative locus in autosomal dominant partial epilepsy with auditory features, a rare form of human epilepsy (Kalachikov et al., 2002; Morante-Redolat et al., 2002). However, mutations in LGI1 account for only ∼50% of human cases and the loci responsible for the remaining idiopathic cases are not known. Analyses performed to date have ruled out roles for genes encoding ADAM22 and the Kv1 channel subunits KCNA1, KCNA4, and KCNAB1 (Diani et al., 2008). The identification of NgR1 as an LGI1 binding protein suggests that NgR1 is a viable candidate gene for idiopathic cases of ADLTE.
In conclusion, we have identified LGI1 as a novel binding partner for the NgR1 receptor. These findings raise the possibility that LGI1 may function as endogenous antagonists of myelin-based growth inhibitors. We have also found that NgR1 forms a complex with ADAM22 and facilitates LGI1 binding to this receptor and we propose that NgR1 and ADAM22 collaborate to create an LGI1 binding complex in vivo that is important for synapse maturation.
Footnotes
This work was supported by Grant MOP37850 from the Canadian Institute of Health Research (CIHR) (to P.A.B. and A.F.) and by grants from the Ministerio de Ciencia e Innovación (SAF2006-00724) and Generalitat Valenciana (ACOMP09/209; to J.P.-T.). R.T. was supported by CIHR and McGill studentships, and K.F. was supported by a Natural Sciences and Engineering Research Council of Canada studentship. P.A.B. is a McGill Dawson Scholar and a Fond de la Recherche en Santé du Québec Chercheur National.
References
- Atwal JK, Pinkston-Gosse J, Syken J, Stawicki S, Wu Y, Shatz C, Tessier-Lavigne M. PirB is a functional receptor for myelin inhibitors of axonal regeneration. Science. 2008;322:967–970. doi: 10.1126/science.1161151. [DOI] [PubMed] [Google Scholar]
- Boutilier J, Ceni C, Pagdala PC, Forgie A, Neet KE, Barker PA. Proneurotrophins require endocytosis and intracellular proteolysis to induce TrkA activation. J Biol Chem. 2008;283:12709–12716. doi: 10.1074/jbc.M710018200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chivatakarn O, Kaneko S, He Z, Tessier-Lavigne M, Giger RJ. The Nogo-66 receptor NgR1 is required only for the acute growth cone-collapsing but not the chronic growth-inhibitory actions of myelin inhibitors. J Neurosci. 2007;27:7117–7124. doi: 10.1523/JNEUROSCI.1541-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diani E, Di Bonaventura C, Mecarelli O, Gambardella A, Elia M, Bovo G, Bisulli F, Pinardi F, Binelli S, Egeo G, Castellotti B, Striano P, Striano S, Bianchi A, Ferlazzo E, Vianello V, Coppola G, Aguglia U, Tinuper P, Giallonardo AT, et al. Autosomal dominant lateral temporal epilepsy: absence of mutations in ADAM22 and Kv1 channel genes encoding LGI1-associated proteins. Epilepsy Res. 2008;80:1–8. doi: 10.1016/j.eplepsyres.2008.03.001. [DOI] [PubMed] [Google Scholar]
- Dolan J, Walshe K, Alsbury S, Hokamp K, O'Keeffe S, Okafuji T, Miller SF, Tear G, Mitchell KJ. The extracellular leucine-rich repeat superfamily; a comparative survey and analysis of evolutionary relationships and expression patterns. BMC Genomics. 2007;8:320. doi: 10.1186/1471-2164-8-320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durocher Y, Perret S, Kamen A. High-level and high-throughput recombinant protein production by transient transfection of suspension-growing human 293-EBNA1 cells. Nucleic Acids Res. 2002;30:E9. doi: 10.1093/nar/30.2.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferraro GB, Alabed YZ, Fournier AE. Molecular targets to promote central nervous system regeneration. Curr Neurovasc Res. 2004;1:61–75. doi: 10.2174/1567202043480251. [DOI] [PubMed] [Google Scholar]
- Flanagan JG, Cheng HJ. Alkaline phosphatase fusion proteins for molecular characterization and cloning of receptors and their ligands. Methods Enzymol. 2000;327:198–210. doi: 10.1016/s0076-6879(00)27277-7. [DOI] [PubMed] [Google Scholar]
- Fournier AE, GrandPre T, Strittmatter SM. Identification of a receptor mediating Nogo-66 inhibition of axonal regeneration. Nature. 2001;409:341–346. doi: 10.1038/35053072. [DOI] [PubMed] [Google Scholar]
- Fournier AE, Takizawa BT, Strittmatter SM. Rho kinase inhibition enhances axonal regeneration in the injured CNS. J Neurosci. 2003;23:1416–1423. doi: 10.1523/JNEUROSCI.23-04-01416.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukata Y, Adesnik H, Iwanaga T, Bredt DS, Nicoll RA, Fukata M. Epilepsy-related ligand/receptor complex LGI1 and ADAM22 regulate synaptic transmission. Science. 2006;313:1792–1795. doi: 10.1126/science.1129947. [DOI] [PubMed] [Google Scholar]
- GrandPré T, Nakamura F, Vartanian T, Strittmatter SM. Identification of the Nogo inhibitor of axon regeneration as a Reticulon protein. Nature. 2000;403:439–444. doi: 10.1038/35000226. [DOI] [PubMed] [Google Scholar]
- GrandPré T, Li S, Strittmatter SM. Nogo-66 receptor antagonist peptide promotes axonal regeneration. Nature. 2002;417:547–551. doi: 10.1038/417547a. [DOI] [PubMed] [Google Scholar]
- Harvey PA, Lee DH, Qian F, Weinreb PH, Frank E. Blockade of Nogo receptor ligands promotes functional regeneration of sensory axons after dorsal root crush. J Neurosci. 2009;29:6285–6295. doi: 10.1523/JNEUROSCI.5885-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Head K, Gong S, Joseph S, Wang C, Burkhardt T, Rossi MR, LaDuca J, Matsui S, Vaughan M, Hicks DG, Heintz N, Cowell JK. Defining the expression pattern of the LGI1 gene in BAC transgenic mice. Mamm Genome. 2007;18:328–337. doi: 10.1007/s00335-007-9024-6. [DOI] [PubMed] [Google Scholar]
- Igarashi M, Strittmatter SM, Vartanian T, Fishman MC. Mediation by G proteins of signals that cause collapse of growth cones. Science. 1993;259:77–79. doi: 10.1126/science.8418498. [DOI] [PubMed] [Google Scholar]
- Kalachikov S, Evgrafov O, Ross B, Winawer M, Barker-Cummings C, Martinelli Boneschi F, Choi C, Morozov P, Das K, Teplitskaya E, Yu A, Cayanis E, Penchaszadeh G, Kottmann AH, Pedley TA, Hauser WA, Ottman R, Gilliam TC. Mutations in LGI1 cause autosomal-dominant partial epilepsy with auditory features. Nat Genet. 2002;30:335–341. doi: 10.1038/ng832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karlén A, Karlsson TE, Mattsson A, Lundströmer K, Codeluppi S, Pham TM, Bäckman CM, Ogren SO, Aberg E, Hoffman AF, Sherling MA, Lupica CR, Hoffer BJ, Spenger C, Josephson A, Brené S, Olson L. Nogo receptor 1 regulates formation of lasting memories. Proc Natl Acad Sci U S A. 2009;106:20476–20481. doi: 10.1073/pnas.0905390106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee H, Raiker SJ, Venkatesh K, Geary R, Robak LA, Zhang Y, Yeh HH, Shrager P, Giger RJ. Synaptic function for the Nogo-66 receptor NgR1: regulation of dendritic spine morphology and activity-dependent synaptic strength. J Neurosci. 2008;28:2753–2765. doi: 10.1523/JNEUROSCI.5586-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Liu BP, Budel S, Li M, Ji B, Walus L, Li W, Jirik A, Rabacchi S, Choi E, Worley D, Sah DW, Pepinsky B, Lee D, Relton J, Strittmatter SM. Blockade of Nogo-66, myelin-associated glycoprotein, and oligodendrocyte myelin glycoprotein by soluble Nogo-66 receptor promotes axonal sprouting and recovery after spinal injury. J Neurosci. 2004;24:10511–10520. doi: 10.1523/JNEUROSCI.2828-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu BP, Fournier A, GrandPré T, Strittmatter SM. Myelin-associated glycoprotein as a functional ligand for the Nogo-66 receptor. Science. 2002;297:1190–1193. doi: 10.1126/science.1073031. [DOI] [PubMed] [Google Scholar]
- McGee AW, Yang Y, Fischer QS, Daw NW, Strittmatter SM. Experience-driven plasticity of visual cortex limited by myelin and Nogo receptor. Science. 2005;309:2222–2226. doi: 10.1126/science.1114362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morante-Redolat JM, Gorostidi-Pagola A, Piquer-Sirerol S, Sáenz A, Poza JJ, Galán J, Gesk S, Sarafidou T, Mautner VF, Binelli S, Staub E, Hinzmann B, French L, Prud'homme JF, Passarelli D, Scannapieco P, Tassinari CA, Avanzini G, Marti-Masso JF, Kluwe L, et al. Mutations in the LGI1/Epitempin gene on 10q24 cause autosomal dominant lateral temporal epilepsy. Hum Mol Genet. 2002;11:1119–1128. doi: 10.1093/hmg/11.9.1119. [DOI] [PubMed] [Google Scholar]
- Owuor K, Harel NY, Englot DJ, Hisama F, Blumenfeld H, Strittmatter SM. LGI1-associated epilepsy through altered ADAM23-dependent neuronal morphology. Mol Cell Neurosci. 2009;42:448–457. doi: 10.1016/j.mcn.2009.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribeiro PA, Sbragia L, Gilioli R, Langone F, Conte FF, Lopes-Cendes I. Expression profile of Lgi1 gene in mouse brain during development. J Mol Neurosci. 2008;35:323–329. doi: 10.1007/s12031-008-9096-0. [DOI] [PubMed] [Google Scholar]
- Sagane K, Ishihama Y, Sugimoto H. LGI1 and LGI4 bind to ADAM22, ADAM23 and ADAM11. Int J Biol Sci. 2008;4:387–396. doi: 10.7150/ijbs.4.387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulte U, Thumfart JO, Klöcker N, Sailer CA, Bildl W, Biniossek M, Dehn D, Deller T, Eble S, Abbass K, Wangler T, Knaus HG, Fakler B. The epilepsy-linked Lgi1 protein assembles into presynaptic Kv1 channels and inhibits inactivation by Kvbeta1. Neuron. 2006;49:697–706. doi: 10.1016/j.neuron.2006.01.033. [DOI] [PubMed] [Google Scholar]
- Senechal KR, Thaller C, Noebels JL. ADPEAF mutations reduce levels of secreted LGI1, a putative tumor suppressor protein linked to epilepsy. Hum Mol Genet. 2005;14:1613–1620. doi: 10.1093/hmg/ddi169. [DOI] [PubMed] [Google Scholar]
- Sirerol-Piquer MS, Ayerdi-Izquierdo A, Morante-Redolat JM, Herranz-Pérez V, Favell K, Barker PA, Pérez-Tur J. The epilepsy gene LGI1 encodes a secreted glycoprotein that binds to the cell surface. Hum Mol Genet. 2006;15:3436–3445. doi: 10.1093/hmg/ddl421. [DOI] [PubMed] [Google Scholar]
- Venkatesh K, Chivatakarn O, Lee H, Joshi PS, Kantor DB, Newman BA, Mage R, Rader C, Giger RJ. The Nogo-66 receptor homolog NgR2 is a sialic acid-dependent receptor selective for myelin-associated glycoprotein. J Neurosci. 2005;25:808–822. doi: 10.1523/JNEUROSCI.4464-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Zheng S, Wu H, Wu Y, Liu S, Fan M, Zhang J. Identification of BLyS (B lymphocyte stimulator), a non-myelin-associated protein, as a functional ligand for Nogo-66 receptor. J Neurosci. 2009;29:6348–6352. doi: 10.1523/JNEUROSCI.5040-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou YD, Lee S, Jin Z, Wright M, Smith SE, Anderson MP. Arrested maturation of excitatory synapses in autosomal dominant lateral temporal lobe epilepsy. Nat Med. 2009;15:1208–1214. doi: 10.1038/nm.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]