

Abstract
Erbin, a binding partner of ErbB2, was identified as the first member of the LAP family of proteins. Erbin was shown at postsynaptic membranes of the neuromuscular junction (NMJ) or in cultured C2C12 myotubes (1) to be concentrated, (2) to regulate the Ras–Raf–Mek pathway, and (3) to inhibit TGF-β signaling. In the CNS, Erbin interacts with PSD-95. Furthermore, agrin–MuSK signaling initiates formation of AChR aggregates at the postsynaptic membrane. In search of proteins interacting with MuSK, we identified Erbin as a MuSK binding protein. We verified the interaction of MuSK with Erbin, or both concomitantly with ErbB2 by coimmunoprecipitation, and we mapped the interacting epitopes between Erbin and MuSK. We demonstrated elevated mRNA levels of Erbin at synaptic nuclei and colocalized Erbin and MuSK at postsynaptic membranes. We identified several Erbin isoforms at the NMJ, all of which contained the MuSK binding domain. By knocking down Erbin, we observed agrin-dependent AChR aggregates on murine primary skeletal myotubes and C2C12 cells, and in the absence of agrin, microclusters, both of significantly lower density. Complementary, AChR-ε-reporter expression was reduced in myotubes overexpressing Erbin. We show that myotubes also express other LAP protein family members, namely Scribble and Lano, and that both affect physical dimensions of agrin-dependent AChR aggregates and density of microclusters formed in the absence of agrin. Moreover, MuSK–Erbin–ErbB2 signaling influences TGF-β signaling. Our data define the requirement of Erbin on the cross talk between agrin and neuregulin signaling pathways at the NMJ.
Introduction
The neuromuscular junction (NMJ) transmits signals from motor neurons to multinucleated muscle fibers. A hallmark of the postsynaptic apparatus is the aggregation of acetylcholine receptors (AChRs) (Sanes and Lichtman, 1999). Preferentially, muscle nuclei below the synapse transcribe genes encoding AChR subunits (Merlie and Sanes, 1985). Classically, the aggregation of AChRs is induced by motor neuron-derived agrin and neuregulins. The latter are believed to increase transcription of AChR genes (Falls et al., 1993). However, neuregulins might not be essential for synapse-specific transcription of AChR subunit genes because modest or no phenotypes were detected in mice lacking ErbB2/4 in skeletal muscle or neuregulin in motor neurons and muscle fibers (Escher et al., 2005; Jaworski and Burden, 2006). Other data suggest that the primary role of neuregulins on the postsynaptic side is involved in regulating clustering and trafficking of AChRs to the surface of cells (Trinidad and Cohen, 2004; Ponomareva et al., 2006).
Neuregulins activate receptor tyrosine kinases of the EGF receptor (ErbB) family. The ErbB2/4 heterodimer is the most likely functional receptor at the postsynaptic apparatus (Trinidad et al., 2000). ErbB proteins are enriched at postsynaptic sites at the NMJ (Altiok et al., 1995; Moscoso et al., 1995; Zhu et al., 1995; Trinidad et al., 2000) and in the CNS (Garcia et al., 2000; Huang et al., 2000). The synaptic localization of ErbB4 in the CNS is mediated by the interaction of its C terminus with the PDZ domain of the postsynaptic density protein (PSD)-95 (Garcia et al., 2000; Huang et al., 2000). In contrast, PSD-95 interacts weakly or not at all with ErbB2/3. Another PDZ-domain-containing adapter protein, Erbin, interacts specifically with ErbB2 (Borg et al., 2000; Huang et al., 2001). Erbin is the founding member of a leucine-rich repeat (LRR) and PDZ domain (LAP) family of proteins, characterized by 16 leucine-rich repeats at the N terminus and one to four PDZ domains at the C terminus (Borg et al., 2000). Recently, the region between LRR and PDZ of Erbin was shown to interact with EBP50 (Rangwala et al., 2005) and Smad3 (Dai et al., 2007).
At the NMJ, the role and molecular interactions of Erbin have not been investigated. Here, we show that (1) Erbin is localized to the NMJ; (2) muscle cells express three Erbin isoforms; (3) Erbin interacts with ErbB2 and the receptor tyrosine kinase MuSK, which mediates the actions of agrin; (4) the domain of Erbin interacting with MuSK overlaps with the domain interacting with Smad3; and (5) Erbin, MuSK, or constitutively active mutants of both attenuated Smad3-mediated transcription. Knockdown experiments emphasize the role of Erbin in ensuring the appropriate density of AChR aggregates and microclusters. Interestingly, in the absence of Erbin, expression of constitutively active mutants of ErbB2 and MuSK end up in AChR aggregates of higher density than aggregates formed in the presence of Erbin. Next to Erbin, other LAP protein family members, Lano and Scribble, but not Densin-180, were detected in myotubes.
Materials and Methods
Plasmid constructs and yeast two-hybrid experiments.
Two DNA fragments, each encoding the intracellular epitope of rat MuSK [GenBank accession number (Acc. No.) U34985], were linked (named MuSK2xwt) as described before (Cheusova et al., 2006b). The same strategy was applied for the generation of two intracellular domains from a MuSK kinase defective mutant (named MuSK2xkd). MuSK2xwt and MuSK2xkd were subcloned either in pGBKT7 (BD Biosciences Clontech) to generate yeast two-hybrid bait plasmids, or in pCMV5 containing a T7- or a myc-tag to allow expression in mammalian cells. A prey plasmid (pGAD-GH) containing a C-terminal part of human Erbin (GenBank Acc. No. NM_018695.2; starting at position 3017 of the open reading frame and coding for 365 aa) fused in frame to the Gal4 activation domain was identified by yeast two-hybrid. A human HeLa S3 Matchmaker cDNA library (BD Biosciences Clontech) was used for the screen. In total 8.5 × 106 independent yeast clones were screened. Yeast two-hybrid analysis was performed as described previously (Cheusova et al., 2006b).
For epitope-mapping studies, different intracellular epitopes of mouse MuSK (GenBank Acc. No. MMU37709) were used as described before (Cheusova et al., 2006b).
Expression plasmids containing different MuSK mutants were generated by cloning a T7-tagged intracellular domain of mouse MuSK (GenBank Acc. No. MMU37709) or 3′ truncations thereof into pCMX-PL1 using EcoRI and XhoI. PCR fragments were generated using the following primers: 5′-GTA TTG CTG CCG AAG GAG GAA A-3′ in combination with different 3′ primers: (1) MuSK-515-563: 5′-TTA CTT AGG ATT CAG AAG GAG-3′, (2) MuSK-515-682: 5′-TTA CAG GTC ACT GTG GCT GAG-3′, (3) MuSK-857: 5′-TTA GCG CTG CAG GAT CCT GTG-3′, (4) MuSK-515-868: 5′-TTA GAC ACC CAC CGT TCC CTC-3′.
A CMV-driven expression plasmid containing the human myc-tagged Erbin (pRK5 myc-Erbin) was kindly provided by Jean-Paul Borg (INSERM, Marseille, France) and will be referred to as hErbin-v2. The PDZ domain of human Erbin (amino acid residues 1280–1371, Acc. No. NM_018695.2) was cloned into pCMV5-myc using KpnI and XbaI.
PCR-amplified human Erbin-encoding epitopes were fused either to the Gal4 activation domain by using EcoRI and XhoI of pGAD-GH (BD Biosciences Clontech) or to GST using pGEX-KG and the restriction sites EcoRI and HindIII or EcoRI and XhoI. For the generation of N-terminal deletion mutants of Erbin, each one of the following 5′ primers was used for PCR: (1) hErbin-NΔ-1007: 5′-CCC CCG GGC TGC AGG AAT TCT GGC ACG AG-3′, (2) hErbin-NΔ-1080: 5′-CGT GTC CTC CAC AGC CTC T-3′, (3) hErbin-NΔ-1200: 5′-CGG AAT TCC GTA CTT CGA CAT ATT GAA-3′, or (4) hErbin-PDZ (NΔ-1261): 5′-CCC TCC ATA TAC ACA GCC C-3′ together with the 3′ primer: 5′-TTA TGA GGA AAC TTC TCG-3′. For the construction of C-terminal Erbin deletion mutants, fragments were amplified using the following as 5′ primers: 5′-CGG AAT TCG GCA CGA GCT CGT GCC GC-3′ for pGAD-GH Erbin clones and 5′-CCC CCG GGC TGC AGG AAT TCT GGC ACG AG-3′ for pGEX-KG Erbin clones, together with one of the following 3′ primers: (1) hErbin-1007-1085: 5′-TTA AGA GGC TGT GGA GGA CAC-3′, (2) hErbin-1007-1145: 5′-TTA CGA GCA CTC TGA GGT CTT G-3′, (3) hErbin-1007-1204: 5′-TTA TTC AAT ATG TCG AAG TAC-3′, (4) hErbin-ΔPDZ (1007–1266): 5′-TTA GGG CTG TGT ATA TGG AGG-3′. A series of fragments encoding overlapping parts of Erbin, spanning from the amino acid residue 1007 to the C terminus, were amplified by PCR using the following primer pairs: (1) hErbin-697-1211: 5′-CAA GAA GAT GAA AAT TTT AAC AGC-3′ and 5′-CTT TTC TAA CTT TTT GGC TTC AAT-3′, (2) hErbin-1007-1076: 5′-CCC CCG GGC TGC AGG AAT TCT GGC ACG AG-3′ and 5′-TTA TGT CGC TGG ATT GTA CTT CG-3′, (3) hErbin-1050–1151: 5′-CAG CAA GAC ATG GGG AAA TGT-3′ and 5′-TTA ATT TCA TTA ATA GAG GGT CGA GCA-3′, (4) hErbin-1120–1175: 5′-TGA TGC CAG GAT CAC AGA GA-3′ and 5′-TTA TGC ATT TGG TCT TTT TGA AGG-3′, (5) hErbin-1145-1229: 5′-GTG CTC GAC CCT CTA TTA ATG AAA T-3′ and 5′-TTA ATA ATT TGC CTG AGG CCT GA-3′, (6) hErbin-1175-1211: 5′-GCA AGG GTT GGT TCT GAG C-3′ and 5′-CTT TTC TAA CTT TTT GGC TTC AAT-3′, (7) hErbin-1201-1279: 5′-TTC GAC ATA TTG AAG CCA AAA A-3′ and 5′-TTA CTT GTT TTG CCA GTT CAT GG-3′, (8) hErbin-1212-1280: 5′-ATG CCT TTG AGT AAT GGA CAG-3′ and 5′-CTC TTG TTT TGC CAG TTC ATG-3′, (9) hErbin-1255-1371: 5′-TGA AAG TGG CCC ACC AG-3′ and 5′-TTA TGA GGA AAC TTC TCG-3′.
To create an expression plasmid for Erbin-MuSK-bd, the binding domain (amino acid residues 1175–1211) was cut out of the respective pGEX-KG construct with BamHI and HindIII and cloned into pCMV5–3xT7, which contains three times the T7 tag. The Erbin construct lacking the binding domain for MuSK (hErbin-ΔMuSK-bd) was generated by PCR using three primers: 5′-ATG ACT ACA AAA CGA AGT TTG TTT-3′ (start codon), 5′-TTA TGA GGA AAC TTC TCG TAC AA-3′ (stop codon), and 5′-CT AGT CCT TCA AAA AGA CCA AAT ATG CCT TTG AGT AAT GGA CAG-3′ (internal primer fusing amino acid residues 1174–1212). The PCR product was cloned into pCMV5-T7 using the restriction sites SalI and NotI and was verified by sequencing. The coding sequence of murine Erbin (full length and Δex22/23) was amplified using mouse hindlimb-derived first cDNA as template and cloned into pCMV5-T7 using the restriction sites SalI and NotI. Silencing of the murine Erbin (GenBank Acc. No. NM_021563), the Lano (GenBank Acc. No. NM_001146048.1), and the Scribble gene (GenBank Acc. No. NM_134089.1) was performed by plasmid-derived shRNAs. Complementary oligos (Erbin 5′-GAT CCC CTG ATG CTT TCC TTG AAT TCT TCA AGA GAG AAT TCA AGG AAA GCA TCA TTT TTG GAA A-3′ and 5′-AGC TTT TCC AAA AAT GAT GCT TTC CTT GAA TTC TCT CTT GAA GAA TTC AAG GAA AGC ATC AGG G-3′, Lano 5′-GAT CCC CGA GAG AAT CTT CTT ACA TAT TCA AGA GAT ATG TAA GAA GAT TCT CTC TTT TTA-3′ and 5′-AGC TTA AAA AGA GAG AAT CTT CTT ACA TAT CTC TTG AAT ATG TAA GAA GAT TCT CTC GGG-3′, and Scribble 5′-GAT CCC CAG CAC TTC AAG ATC TCC AAT TCA AGA GAT TGG AGA TCT TGA AGT GCT TTT TTA-3′ and 5′-AGC TTA AAA AAG CAC TTC AAG ATC TCC AAT CTC TTG AAT TGG AGA TCT TGA AGT GCT GGG-3′) were designed and used as before (Cheusova et al., 2006b). To quantify the strength of silencing the Erbin gene by luciferase assays, Erbin encoding cDNA was amplified (5′-GCC ATG TCG CTG TCT ACG AGG A-3′ and 5′-TTA AAA GTC CTG GAG GTT TTC ACA-3′) using C2C12 cell-derived first cDNA as template and cloned into pCMX-PL1-luciferase.
To create a murine Smad3 expression plasmid, total RNA was extracted from hindlimb muscles, reverse transcribed, and amplified by PCR with the following primers: 5′-TAT GGTACC GGT GAC CCT TCG GTG CCA GCC-3′ and 5′-TAT TCTAGA CTA AGA CAC ACT GGA ACA GCG-3′, and cloned into pCMV5-myc using KpnI and XbaI.
The expression plasmid for ErbB2 (pNEU rat ErbB2) was a generous gift from Hans-Rudolph Brenner (Institution of Physiology, Basel, Switzerland).
For the generation of a MuSK-specific antibody, different exposed epitopes of mouse MuSK were amplified by PCR using the following primer pairs: (1) 5′-TGC TGC CGA AGG AGG AAA GA-3′ and 5′-ATA CTC CAG GCT GAG CAA CT-3′ (juxtamembrane region, GenBank Acc. No. NM_010944.1, amino acid residues 519–572), (2) 5′-TAC TAT GGA ATG GCC CAC GA-3′ and 5′-GGC GAG GAT GTT GCC ATC TCT CA-3′ (epitope within the kinase domain, amino acid residues 805–824). Both PCR products were cloned as His-tagged fusion into pET28a (Merck), and the fusion protein is referred to as MuSK-20kd to indicate its molecular weight.
For whole-mount DIG in situ hybridization of mouse diaphragm, a DIG-labeled RNA probe, comprising part of exon 21 of mouse Erbin, was generated by amplifying (5′-TAT GGA TTC CAG CCA GGC ATG GAG AAA TGT-3′ and 5′-TAT CTC GAG CTC AAT GTG TCG TAG TAC TTG-3′) and cloning mouse Erbin (GenBank Acc. No. NM_001005868) from 3469 to 3917 bp into pGEM Teasy (Promega).
Tissue culture, transfection, extract preparation, Western blot, luciferase assays, immunoprecipitation, GST pull downs.
HEK293 cells were maintained as described previously (Cheusova et al., 2006b). C2C12 cells were maintained for proliferation in DMEM containing 20% (v/v) fetal calf serum. For differentiation, the medium was replaced by DMEM with 5% (v/v) heat-inactivated horse serum (Invitrogen). Myotubes formed after 4–6 d.
Primary skeletal muscle cells were prepared from leg muscles of neonatal mice. In brief, the tissue was minced with a razor blade, dissociated with collagenase type IV (Sigma-Aldrich) and Dispase II (Roche) in Mg2+- and Ca2+-free HBSS buffer. Digestion was stopped by adding FCS. After filtering through a cell strainer (Invitrogen) and centrifugation, cells were resuspended in 80% Ham's F10 (Ca2+-free), 20% FCS, 1% penicillin/streptomycin, and recombinant human fibroblast growth factor (5 ng/ml) (Promega). Subsequently, cells were seeded on Matrigel-coated plates (Invitrogen). After 24 h, the culture medium was replaced by 40% DMEM, 40% Ham's F10, 20% FCS, 1% penicillin/streptomycin, and recombinant human fibroblast growth factor (5 ng/ml). For electroporation, 5 × 105 primary skeletal myoblasts were used according to the manufacturer's instructions (Nucleofector, Lonza Cologne). Transfection efficiency was determined after 24 h. At the same time, primary skeletal muscle cells reached confluence and the medium was replaced by 98% DMEM, 2% horse serum, and 1% penicillin/streptomycin. After 3–4 d, the cells differentiated into myotubes.
HEK293 cells were transiently transfected using polyethylenimine (Sigma-Aldrich). Cells grown in 10 cm Petri dishes were incubated with 10 μg of plasmid DNA and 31 μl of polyethylenimine (0.05%, w/v) for 12 h. After 48 h, cells were harvested and lysed by incubating them with lysis buffer (50 mm HEPES, pH 7.5, 10% glycerol, 150 mm NaCl, 1% Triton X-100, 1.5 mm MgCl2, 1 mm EGTA, pH 8) and proteinase inhibitors (1 mm PMSF, 10 μg/μl aprotinin, 10 μg/μl leupeptin). C2C12 myoblasts were transiently transfected using polyethylenimine (Sigma-Aldrich). Cells grown in 3.5 cm Petri dishes and reaching a density of ∼90% were incubated with 3 μg of plasmid DNA and 20 μl of polyethylenimine 0.05% (w/v). After centrifugation for 10 min at 1000 rpm, C2C12 cells were incubated for 4 h with the transfection reagent. The growth medium was replaced by differentiation medium with low serum content to permit the formation of myotubes.
Monoclonal antibodies directed against the T7- (Merck), the myc- (New England Biolabs), the HA-epitope (Santa Cruz Biotechnology) or polyclonal antibodies directed against Erbin or MuSK served as primary antibodies. Horseradish-peroxidase-coupled-protein A or anti-mouse-Ig-coupled-horseradish-peroxidase was used as secondary detection reagent in Western blots using the ECL detection system (GE Healthcare). C2C12 cells were transfected for immunocytochemistry using Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions.
The production of agrin-conditioned medium was described before (Cheusova et al., 2006b). Agrin-conditioned medium was added to primary skeletal or C2C12 myotubes at 1:8 dilutions. AChR microclusters were detected without agrin treatment by staining them with rhodamine-α-bungarotoxin (Invitrogen). To induce AChR aggregates, cells were treated with soluble nerve-derived agrin48 for 16 h, stained the same way with Rh-BTX, fixed, embedded, and quantified.
For GST pull downs, bacteria were transformed with the respective constructs, grown until OD600 0.4, and induced with 1 mm IPTG for 4 h to express the GST-fusion proteins. Bacteria were collected by centrifugation, incubated in sonication buffer (50 mm NaH2PO4, 300 mm NaCl, 25 U/ml Benzonase, 10 μg/ml leupeptin, 10 μg/ml aprotinin, 1 μl/ml Triton X-100, 10 μg/ml DNaseI, 15 U/μl lysozyme) at 4°C for 30 min, lysed by sonication, and centrifuged. The supernatants containing the GST-fusion proteins were supplemented with 30 μl of equilibrated glutathione beads and incubated under constant rotation at 4°C for 2–12 h. After washing three times with 4.3 mm Na2HPO4, 1.47 mm KH2PO4, and 137 mm NaCl or alternatively with 20 mm NaCl and 2.7 mm KCl, an aliquot of the beads, now carrying the GST-fusion protein, was incubated with 25 μl up to 300 μl of extract from HEK293 cells expressing the desired protein after transient transfection. Proteins bound to the beads were analyzed by SDS-PAGE and Western blot.
For preparation of cell extracts, transiently transfected HEK293 or C2C12 cells were grown on 100 mm plates. The formation of AChR aggregates on C2C12 cells was induced by incubating them with nerve-derived agrin for 16 h. For preparation of muscle tissue extract, rat soleus and gastrocnemius muscles were frozen in liquid nitrogen and mashed. In all cases, tissue or cells were washed twice with cold PBS and lysed in lysis buffer (50 mm HEPES pH 7.5, 10% glycerol, 150 mm NaCl, 1% Triton X-100, 1.5 mm MgCl2, 1 mm EGTA) supplemented with 1 mm phenylmethylsulfonylfluoride (PMSF), 10 μg/ml aprotinin, and 10 μg/ml leupeptin. The expression level of protein was analyzed by Western blot for each extract. Next, monoclonal antibodies against the T7- (Merck) or hemagglutinin-epitope (New England Biolabs) or polyclonal sera directed to either MuSK or Erbin were added together with PBS-equilibrated protein A Sepharose CL-4B beads (GE Healthcare) and precipitated overnight. After washing the beads in 50 mm HEPES, pH 7.5, 150 mm NaCl, 1 mm EDTA, 10% glycerol, 0.1% Triton X-100, 1 mm PMSF, 10 μg/ml leupeptin, and 10 μg/ml aprotinin, the precipitated proteins were analyzed by SDS-PAGE and Western blot.
AChRε-luciferase reporter assays were performed using transiently transfected primary skeletal muscle cells or C2C12 myoblasts to investigate the role of different Erbin or MuSK mutants. The AChRε-luciferase reporter construct contains 416 nt of the 5′-untranslated region of the AChRε gene in front of the luciferase gene. Four hours after transfection of C2C12 cells with polyethylenimine (0.05% w/v), myoblasts were differentiated to myotubes, stimulated with 5 nm NRGβ1 for 24 h, and harvested, and cell extracts were prepared for luciferase assays. Primary skeletal muscle cells were transfected by Nucleofector (Lonza Cologne). To neutralize autocrine saturation of ErbB activation by endogenous neuregulin, anti-NRGβ1 antibody (30 μg/ml; Ab2, Thermo Fischer Scientific) was added to control cultures at the time when exogenous NRGβ1 was added to test cultures.
SBE4-luciferase reporter assays were performed using transiently transfected C2C12 myoblasts with a SBE4-luciferase reporter (Zawel et al., 1998) and other expression plasmids (HA-tagged hErbin-v2, HA-tagged MuSK, or myc-tagged Smad3). After transfection, cells were differentiated to myotubes, stimulated with 5 nm TGF-β (R&D Systems) for 24 h, and harvested, and cell extracts were prepared for luciferase assays.
To investigate the silencing capacity of Erbin-specific shRNA, pSuper plasmid (Oligoengine) or a pSuper plasmid bearing a short stretch of an Erbin-coding DNA was transfected into HEK293 cells. Cell extracts were prepared for luciferase assays 48 h after transfection.
RNA preparation, reverse transcription, and PCR.
Total RNA was extracted from mouse tissues or C2C12 cells with TRIzol reagent (Invitrogen) as described previously (Cheusova et al., 2006b). After reverse transcription, cDNA was used with the following mouse-specific primer pairs for quantitative PCRs using the Lightcycler–FastStart DNA Master SYBR Green Kit and the Lightcycler Thermal Cycle System (Roche Diagnostics) according to the manufacturer's instructions: (1) MuSK: 5′-GCC TTG GTT GAA GAA GTA GC-3′ and 5′-CTT GAT CCA GGA CAC AGA TG-3′; (2) AChRα: 5′-ACG CTG AGC ATC TCT GTC TT-3′ and 5′-TTG GAC TCC TGG TCT GAC TT-3′; (3) Erbin: 5′-CAG GAG AGT GTT GCC AAG AT-3′ and 5′ CAT GGC CTT GTC TAG GAG AA 3′; (4) TGF-βRI: 5′-TCT GCC ATA ACC GCA CTG TC-3′ and 5′-TCT CTG CCT CTC GGA ACC AT-3′; (5) TGF-βRII: 5′-TCA CCT ACC ACG GCT TCA CT-3′ and 5′-TCA GCT TGG CCT TGT AGA CC-3′; (6) Smad3: 5′-GAG AGT AGA GAC GCC AGT TC-3′ and 5′-AGT AGG TGA CTG GCT GTA GG-3′; (7) β-actin: 5′-TGG AAT CCT GTG GCA TCC ATG AAA-3′ and 5′-TAA AAC GCA GCT CAG TAA CAG TCC G-3′. For detection of mouse Erbin splice variants, the following primer pair was used: 5′-GCG TGT CTT CAA CGG CTT CT-3′ and 5′-ACT GTA GCC ATT AGC CTG AA-3′. The three amplified DNA fragments have been purified, subcloned into pGEM Teasy, and verified by sequencing.
Immunohistochemistry and microscopy.
C2C12 myoblasts were transiently transfected with 0.75 μg of a pSuper.GFP vector expressing a scrambled shRNA or an Erbin-, Lano-, or Scribble-specific shRNA. After transfection, the myoblasts were immediately transferred into differentiation medium. Myotubes were formed after several days, then incubated with nerve-derived agrin-conditioned medium for 16 h and stained afterward by adding Rh-BTX (1:2000, rhodamine-α-bungarotoxin; Invitrogen) to the medium for 1 h. Cells were washed with PBS, fixed in 4% PFA for 10 min, and embedded in Mowiol.
AChR cluster stability was determined as described before (Cheusova et al., 2006b).
Confocal images of primary skeletal muscle cells or C2C12 cells were taken, as described before, using the Leica confocal microscope TCS SL (Cheusova et al., 2006). The confocal stacks were shown as average projections comprising several stacks taken with an interval of 0.5 μm. The analysis of the fluorescence intensity of the cluster area in each stack was determined with the Leica confocal software TCS SL version 2.5.1347a. In total, >65 (C2C12 cells) or >50 (primary skeletal muscle cells) randomly chosen clusters for each transfection with scrambled shRNA or Erbin-, Lano-, or Scribble-specific shRNA have been measured.
A nerve-derived agrin-dependent AChR cluster was defined as an aggregation of AChRs with >5 μm in length (Weston et al., 2000). AChR microclusters were 1–5 μm in length with a surface area of 1–4.5 μm2 (Henriquez et al., 2008).
The influence on myotube formation in the presence of Smad3, MuSK, a constitutively active MuSK mutant (MuSKneuTMuSK) (Jones et al., 1999), a constitutively active erbB2 mutant (neuT) (Bargmann et al., 1986), or hErbin-v2 was analyzed by identifying transiently transfected cells expressing a cotransfected pCMX-PL2-GFPnls (Hashemolhosseini et al., 2000). GFP-positive cells were counted and the ratio between myoblasts and myotubes was determined.
For immunohistochemical analysis, all muscles were quickly frozen in prechilled isopentane. Muscles from adult mice were cryotome sectioned to 10 μm slices. Unfixed sections were rinsed with PBS and permeabilized for 5–10 min in PBS supplemented with 0.1% Triton X-100, blocked in 10% fetal calf serum and 1% bovine serum albumin for 1–2 h. Cryotome sections were stained for AChR with Alexa-α-bungarotoxin or rhodamine-α-bungarotoxin at 1:2500 dilution (Rh-BTX, Invitrogen) or incubated with anti-Erbin (1:100). Either a self-made antibody (anti-MuSK20kd; 1:500) or anti-MuSK (1:1000), which was kindly provided by Markus Ruegg (Biozentrum, Basel, Switzerland), was used as MuSK-specific antibody. Secondary antibodies conjugated to Cy2 or Cy3 immunofluorescent dye (Dianova) were used for detection of Erbin and MuSK. Sections were analyzed and documented using a Leica inverted microscope (DMIRB) equipped with a cooled MicroMax CCD camera (Princeton Instruments) or a Leica confocal microscope TCS SL equipped with Leica confocal software TCS SL version 2.5.1347a.
Results
Identification of an interaction between Erbin and MuSK
Our aim was to identify, by Y2H screens, new proteins interacting with the intracellular domain of MuSK. To this end, we linked two identical intracellular domains of MuSK together via a peptide linker (MuSK2xwt) (Fig. 1a). In the same way, a second dimer was generated for a kinase-defective MuSK variant (MuSK2xkd) (Fig. 1a). Expression of these constructs and their applicability for yeast two-hybrid screens have been described (Cheusova et al., 2006b). Although MuSK2xwt (but not MuSK2xkd) was previously shown to be autophosphorylated, now when expressed in mouse primary myotubes, neither was it able to stimulate a cotransfected AChRε-promoter Luciferase reporter construct (AChRε-Luc), nor was the formation of AChR aggregates by agrin affected (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). This is not surprising given that MuSK2xwt lacks a transmembrane region and is not localized in the membrane.
Figure 1.

Erbin interacts with MuSK. a, Scheme of wild-type MuSK and two MuSK mutants (MuSK2xwt, MuSK2xkd). The mutants are generated by linking two intracellular parts of MuSK together via a peptide linker, thereby imitating the dimeric active intracellular part of wild-type MuSK. Each of the mutants was used for yeast two-hybrid. The term kd reflects the use of a kinase-defective MuSK mutant. Ig, Ig-like domain; C6, cysteine-rich region; TM, transmembrane segment; JM, juxtamembrane region. b, Illustration of the epitopes of the human Erbin-variant2 (hErbin-v2) (Favre et al., 2001). Below the scheme of human Erbin-v2, the area of Erbin interacting with MuSK is drawn (hErbin-NΔ-1007). SID, Smad-interacting domain; WW, Trp-Trp; SH3, Src homology domain 3. c, The growth of different yeast clones on agar lacking histidine and adenine is shown. Note that yeast cells are able to grow if they contain either MuSK2xwt or MuSK2xkd as bait plasmids and the C-terminal part of human Erbin (hErbin-NΔ-1007) as prey plasmid, or if they contain p53 and SV40 large T antigen (positive control). d, e, Extracts of transiently transfected HEK293 cells were used to precipitate MuSK2xwt, MuSK2xkd, or full-length hemagglutinin (HA)-tagged MuSK. As a consequence human Erbin-v2 was coprecipitated. The amount corresponding to 1/10 of the input was ascertained by Western blot. f, g, Precipitation of endogenous Erbin or MuSK proteins from C2C12 cells or hindlimb muscle cell extracts coprecipitated MuSK or Erbin as indicated.
Among the yeast clones identified, two contained a fragment of the human LAP protein family member Erbin (hErbin-NΔ-1007) (Fig. 1b). This fragment coded for the entire C-terminal part beginning with the codon for amino acid residue 1007 and ending with the stop codon. In human tissues, several splice variants of Erbin transcripts have been described (Favre et al., 2001). Here we found the C-terminal part of human Erbin variant 2 (hErbin-v2) (Fig. 1b). Additional yeast two-hybrid interaction assays confirmed that MuSK2xwt interacts with hErbin-NΔ-1007 (Fig. 1c). The same interaction was also detected between the kinase-defective intracellular part of MuSK and hErbin-NΔ-1007 (MuSK2xkd) (Fig. 1c).
To examine whether Erbin and MuSK interact outside of yeast, we transiently transfected HEK293 cells with Erbin alone or together with T7-tagged MuSK2xwt or MuSK2xkd. Immunoprecipitating these cell extracts by using a T7-tag-specific antibody recognizing MuSK2xwt or MuSK2xkd led to coprecipitation of full-length human Erbin-v2 (Fig. 1d). When we used cell extracts of HEK293 cells transfected with full-length MuSK, we detected again coprecipitation of human Erbin-v2 (Fig. 1e). To analyze whether the interaction between Erbin and MuSK also occurs when both are expressed at physiological amounts, we performed coimmunoprecipitations using either C2C12 or hindlimb muscle extract, where Erbin and MuSK are endogenously coexpressed (Fig. 1f,g). Indeed, Erbin and MuSK were coprecipitated by their respective binding partner (Fig. 1f,g).
Erbin transcript and protein is enriched at synaptic sites of muscle fibers
Given that Erbin and MuSK interact physically, we sought to determine the spatial expression profile of Erbin in synaptic and extrasynaptic regions of skeletal muscle. To this end, we compared the transcript levels of Erbin with those of other transcripts known to accumulate at the NMJ, such as MuSK and AChRα by RT-PCR (Merlie and Sanes, 1985; Valenzuela et al., 1995). For first-strand cDNA preparation, we used total RNA extracted from synaptic and extrasynaptic regions of diaphragms of mice. As control, we used first-strand cDNA from the CNS (brain), which was reported to contain only marginal amount of MuSK transcript (Cheusova et al., 2006a; Garcia-Osta et al., 2006). As expected, the mRNAs of MuSK and AChRα were enriched in the synaptic region of the diaphragm (Fig. 2a,b). Erbin transcript quantity was significantly higher in muscle than in brain, and transcript levels were significantly higher in the synaptic than in the extrasynaptic part of diaphragms (Fig. 2c). Finally, we confirmed the local synaptic transcription of Erbin by performing whole-mount in situ hybridization on diaphragms of mice (Fig. 2d).
Figure 2.
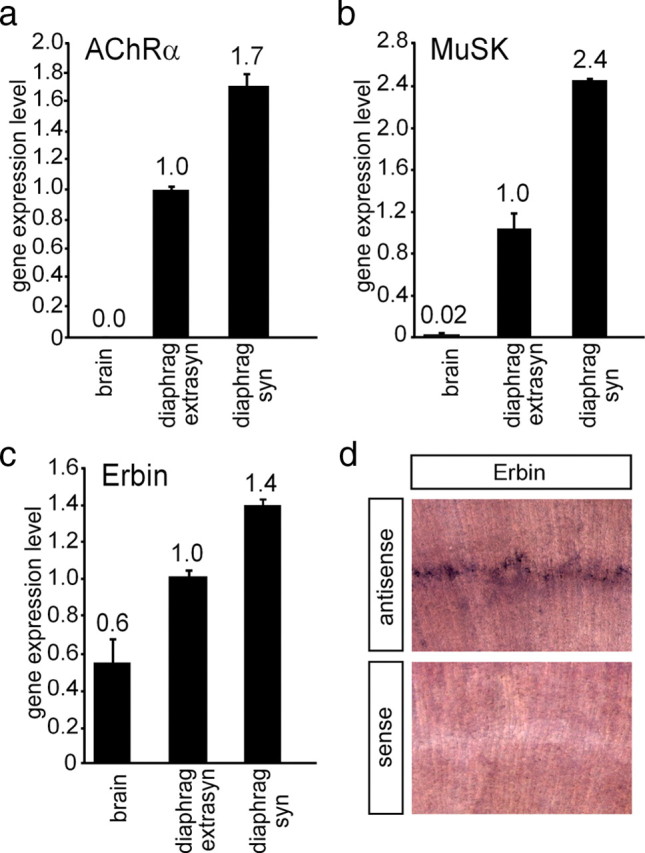
Distribution of AChRα, MuSK, and Erbin in muscle cells. The distribution of different transcripts was followed by quantitative RT-PCR in brain (used as control) and extrasynaptic and synaptic regions of diaphragm. Results are presented as graphs. a, b, AChRα and MuSK were used as controls, because their transcript levels are known to be enriched at synaptic sites of diaphragms. c, Erbin is also transcribed at a higher rate at synaptic sites of the diaphragm. Quantitative RT-PCR data were normalized to β-actin. Transcript levels in extrasynaptic parts of the diaphragm were set to 1. d, Images present diaphragm tissue of a wild-type mouse 6 d after birth stained by in situ hybridization using an antisense or a sense Erbin riboprobe, as indicated.
Next, we asked whether Erbin protein is concentrated at postsynaptic sites in vivo. Previously, Erbin was detected at postsynaptic specializations of the hindlimb muscle of mice (Huang et al., 2001). By immunofluorescent staining of cross sections of soleus muscle, we detected a significant accumulation of Erbin at the NMJs, where it colocalized with synaptic AChRs (Fig. 3a). Moreover, we showed for the first time a direct colocalization between Erbin and MuSK (Fig. 3c). Postsynaptic localization of Erbin was confirmed in two ways: (1) in chronically denervated muscle where presynaptic terminals had degenerated (Fig. 3b) and (2) by demonstrating colocalization of Erbin and AChRs at ectopic postsynaptic membranes induced by recombinant nerve-derived agrin (Fig. 3d,e).
Figure 3.
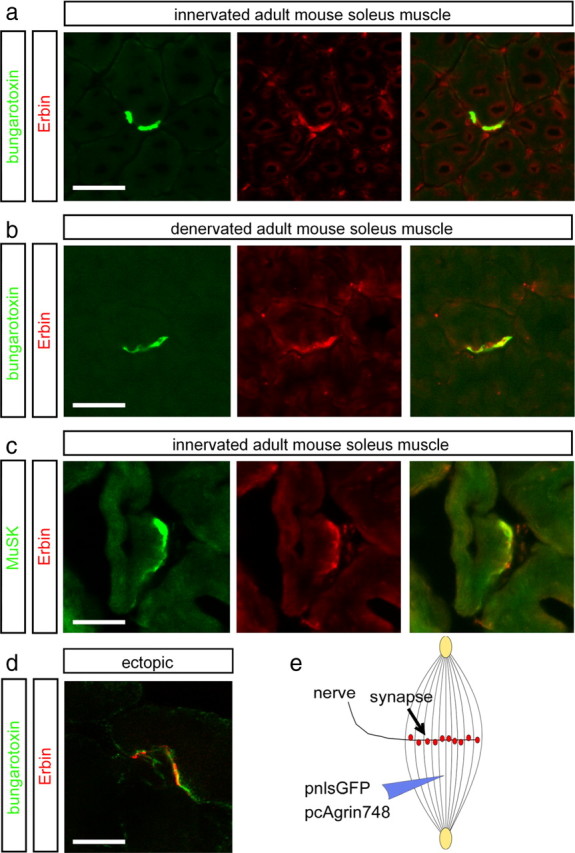
Erbin colocalizes with postsynaptic specializations in vivo. a, b, Frozen 12-μm-thick cryotome cross sections of surgically denervated (5 d after operation) or innervated (contralateral) mouse hindlimb muscles were immunoassayed with rhodamine-α-bungarotoxin (shown in red) and an Erbin-reactive antibody (shown in green). Note that the colocalization between Erbin and postsynaptic specializations is also present in the absence of the nerve. c, Direct colocalization between MuSK and Erbin was verified by staining muscle cross sections with MuSK- and Erbin-recognizing antibodies. d, To exclude a staining of terminal Schwann cells with Erbin-reactive antibodies, ectopic postsynaptic membranes were generated. A typical image showing subcellular colocalization of AChR clusters and Erbin at ectopic postsynaptic membranes is presented. e, As illustrated, nerve-derived agrin and a nuclear localized GFP (pnls-GFP) (Hashemolhosseini et al., 2000) were injected into single muscle fibers to generate ectopic postsynaptic membranes. d, Note that in d, a single projected image of a set of Z-images obtained by confocal microscopy is shown. Scale bar, 20 μm.
The Smad-interacting domain of Erbin mediates the interaction with the kinase domain of MuSK
To map the epitopes of MuSK and Erbin interacting with each other, we used a series of C-terminal truncations of MuSK (Fig. 4a). First, in yeast two-hybrid assays, we observed growth of the yeast when hErbin-NΔ-1007 was used as prey only together with the bait constructs MuSK-515-868 and MuSK-515-857. MuSK-515-868 comprises the complete intracellular part of MuSK. MuSK-515-857 contains the MuSK kinase domain without the PDZ-binding domain located at the very C terminus. Further C-terminal deletion mutants of MuSK were unable to ensure growth of the yeast (Fig. 4b). Second, we transfected expression plasmids for the same truncations of MuSK into HEK293 cells. GST pull-down experiments with a recombinant GST-tagged hErbin-NΔ-1007 protein and MuSK-containing HEK293 cell extracts confirmed only an interaction between the C-terminally truncated MuSK variants, MuSK-515-868 or MuSK-515-857, and hErbin-NΔ-1007 (Fig. 4c).
Figure 4.

The interaction between MuSK and Erbin requires the kinase domain of MuSK. a, Schematic presentation of MuSK mutants containing C-terminal truncations of their intracellular domain for the use in yeast two-hybrid experiments and GST pull-down assays. PDZ-bd, PDZ binding domain. b, The image shows the growth of yeast clones transformed with different combinations of bait and prey plasmids on agar lacking histidine and adenine. Apart from yeast containing p53 and SV40 large T antigen (positive control), only yeast clones that contained bait plasmids encoding the whole kinase domain of MuSK together with human Erbin-NΔ-1007 as prey plasmid grew. c, Western blots demonstrated that by GST pull-down studies, MuSK mutants lacking any part of their kinase domain are unable to interact with human Erbin-NΔ-1007.
Next we sought to identify the sequence of hErbin-NΔ-1007 interacting with MuSK. To this end, we generated several hErbin-NΔ-1007 deletion mutants, truncated at either their N- or C-terminal ends (Fig. 5a). First, we transfected preys consisting of each of the hErbin-NΔ-1007 mutants together with a bait plasmid encoding MuSK2xwt into yeast. Only yeast cells that contained, in addition to MuSK2xwt, either hErbin-NΔ-1007 or the Erbin fragment deleted up to amino acid residue 1200 (hErbin-NΔ-1200) grew on selective medium (Fig. 5b). When the hErbin-NΔ-1007 fragment was trimmed at its C terminus, only yeast containing Erbin starting from amino acid residue 1007 and ending at amino acid residue 1266 (hErbin-ΔPDZ) or 1204 (hErbin-1007-1204) grew on selective medium (Fig. 5c). Furthermore, we confirmed the epitopes of Erbin that interact with MuSK by GST pull downs (Fig. 5d,e). Thus, amino acids 1145–1261 were required for interaction. To further narrow down the sequence of Erbin important for this interaction, we expressed, extracted, and purified a series of overlapping GST-tagged fragments representing parts of the hErbin-NΔ-1007 protein in bacteria (Fig. 6a). We incubated them with HEK293 cell extracts containing MuSK2xwt (Fig. 6b,c). These experiments revealed that a sequence of hErbin-NΔ-1007 comprising amino acids 1175 and 1229 is required for the interaction between Erbin and MuSK (Fig. 6b,c). This sequence is encoded by the 3′-end of exon 21 and 5′-end of exon 23, because hErbin-NΔ-1007 is part of splice variant hErbin-v2, which lacks exon 22. Narrowing down hErbin-NΔ-1007 even further showed that amino acid residues 1145–1211, which are encoded only by exon 21, are necessary and sufficient for MuSK–Erbin interaction. Conspicuously, the identified MuSK binding domain nearly overlaps with a recently identified Smad-interacting domain of Erbin (Dai et al., 2007).
Figure 5.

Identification of the epitope of Erbin that interacts with MuSK. a, Illustration of full-length and different N- and C-terminal truncations of human Erbin-v2 used for yeast two-hybrid studies and GST pull downs. b, c, Growth of yeast clones containing different combinations of bait and prey plasmids on agar lacking histidine and adenine. Only yeast containing MuSK2xwt as bait plasmid and a prey plasmid that encodes at least part of the region of Erbin between amino acids 1145 and 1261 is able to grow in the absence of the auxotrophic markers. d, e, GST pull-down assays confirmed that the area of human Erbin-v2 spanning amino acids 1145–1261 is necessary for binding of MuSK.
Figure 6.

The MuSK binding domain of Erbin is encoded by exon 21. a, Drawing of full-length human Erbin-v2 and small fragments representing parts of Erbin. Each of these recombinant fragments was expressed and purified as GST fusion for pull-down assays. GST fusion proteins were incubated together with HEK293 cell extracts containing myc-tagged MuSK2xwt. b, Western blot of GST pull-down experiments immunoassayed with an anti-myc antibody to detect myc-tagged MuSK2xwt. Note that the shortest Erbin part able to pull down MuSK2xwt spans from amino acid residue 1145 to 1229. c, Western blot images of GST pull-down experiments, like in b, are presented. Here, Erbin mutants were designed such that they helped to define exactly the MuSK binding domain of Erbin. Note that amino acid residues encoded by exon 23 were completely unable to interact with MuSK2xwt. d, RT-PCRs were performed to find out whether either C2C12 cell nuclei or extrasynaptic or synaptic nuclei of diaphragm muscle fibers transcribe differently spliced Erbin variants. MB, Myoblast; MT, myotube.
We asked whether muscle cells express different isoforms of Erbin at the NMJ, some with and others without exon 21. PCR studies with specific primers and first cDNA, reverse transcribed from total RNA of C2C12 cells or murine diaphragms, revealed mainly three different muscle-specific Erbin transcripts (Fig. 6d). In addition to the hErbin-v2 transcript, C2C12 cells and diaphragm express a full-length Erbin transcript and a variant that lacks exons 22 and 23 (Fig. 6d). Note that the bands for each of the three isoforms of Erbin appeared in different ratio within the same tissue (Fig. 6d).
Next, we examined whether the area of Erbin that interacted with MuSK2xwt will also interact with full-length MuSK. First, we generated expression plasmids of three additional differently deleted Erbin variants, (1) a variant that lacks exons 22 and 23, (2) a variant where the MuSK binding domain is missing, and (3) a variant comprising only the MuSK binding domain (Fig. 7a). The expression of each variant was verified by immunodetection using cell extracts of transiently transfected HEK293 cells (Fig. 7b). Each Erbin variant was then coexpressed with full-length MuSK, and the cell extracts were used for coimmunoprecipitation studies (Fig. 7c–e). Of these fragments, all except hErbinΔMuSK-bd caused coprecipitation of full-length MuSK (Fig. 7c,d). This indicates that the MuSK binding domain of Erbin is sufficient to interact with MuSK, and that other epitopes of Erbin do not contribute to this interaction (Fig. 7d).
Figure 7.
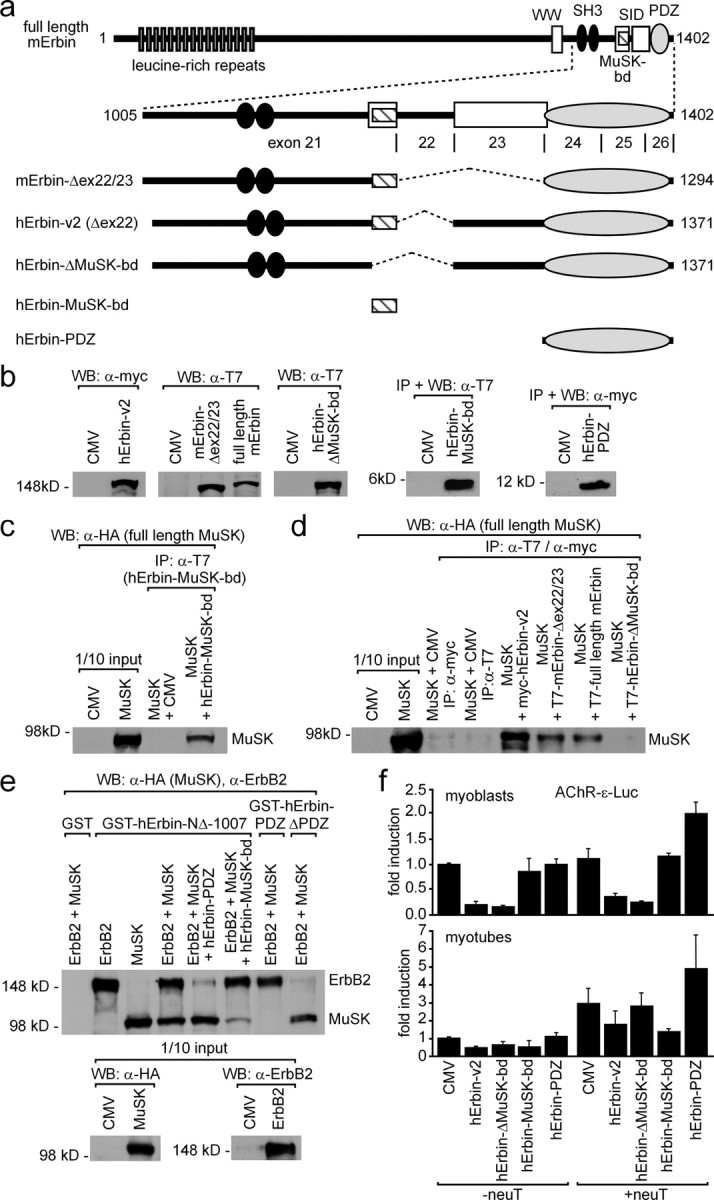
Erbin interacts concomitantly with MuSK and ErbB2. a, Scheme of full-length mouse Erbin and deletion mutants thereof. The C-terminal part of Erbin is encoded by exons 21–26. Below this scheme, mouse or human Erbin mutants lacking different single exons are depicted. Other human Erbin mutants consist only of the MuSK binding domain or the PDZ domain. b, Expression of these different mouse and human Erbin variants, as presented in a, was ascertained by Western blot. c, Precipitation of the human Erbin MuSK binding domain (hErbin-MuSK-bd) using transiently transfected HEK293 cells coprecipitated full-length MuSK as shown by Western blot. d, Extracts from transiently transfected HEK293 cells expressing different Erbin variants were used for coimmunoprecipitation studies. After precipitation of Erbin variants, precipitates were analyzed for the presence of full-length MuSK by Western blot. Note that precipitation of any human Erbin mutant that does not contain the MuSK binding domain is unable to coprecipitate MuSK. e, GST fusions of human Erbin-NΔ-1007, Erbin-PDZ, or Erbin-ΔPDZ, immobilized on glutathione beads, were incubated with HEK293 cell extracts containing full-length ErbB2 and MuSK and bound proteins were analyzed by Western blot. Note that GST-hErbin-PDZ pulled down ErbB2 but not MuSK, while GST-hErbin-ΔPDZ pulled down MuSK but not ErbB2. Further, the specificity of this binding was demonstrated by competition with either human Erbin-PDZ or Erbin-MuSK-bd, which inhibited the pull down of either ErbB2 or MuSK, respectively. f, To investigate whether overexpression of Erbin variants affects AChR gene expression, C2C12 cells were transfected three independent times by a hErbin-v2, hErbin-ΔMuSK-bd, hErbin-MuSK-bd, or hErbin-PDZ together with an AChRε-luciferase reporter. Moreover, the same transfections were done together with an additional plasmid encoding constitutively active ErbB2 (neuT). Luciferase activities were measured from myoblasts (MB) or from myotubes (MT). Note that the hErbin-MuSK-bd inhibited the reporter in myotubes that were cotransfected with neuT, but not in myoblasts.
To examine whether all three proteins ErbB2, Erbin, and MuSK concomitantly interact and form a trimeric complex, we purified GST-tagged recombinant Erbin variants, composed of hErbin-NΔ-1007, hErbin-PDZ, or hErbin-MuSK-bd, and incubated them with full-length MuSK- or ErbB2-containing HEK293 cell extracts (Fig. 7d,e). Indeed, pulling down hErbin-NΔ-1007 allowed coprecipitation of both full-length ErbB2 and MuSK (Fig. 7e). If HEK293 cell extracts contained additionally hErbin-PDZ or hErbin-MuSK-bd, the coprecipitation of ErbB2 or MuSK was abolished, respectively (Fig. 7e). As controls, human Erbin-PDZ or Erbin-ΔPDZ were able to precipitate only ErbB2 or MuSK, respectively, from an ErbB2- and MuSK-containing HEK293 cell extract (Fig. 7e).
It has been reported that neuregulin stimulates transcription of the ε-subunit of the AChR (Si et al., 1997). Erbin was previously shown to inhibit this neuregulin-dependent AChRε-subunit reporter gene stimulation (Huang et al., 2003). This inhibition was assigned to the LRR domain of Erbin (Dai et al., 2006). We wondered whether Erbin mutants either lacking or solely composed of the MuSK binding domain might influence the enhanced transcription of an AChR ε-subunit reporter gene mediated by a constitutively active ErbB2 (neuT) (Bargmann et al., 1986). C2C12 cells were transiently cotransfected with the reporter gene together with hErbin-v2 or with different Erbin mutants (Fig. 7f). Extracts were prepared 48 h after myotube formation, and luciferase activities were assayed. In myoblasts and in the absence of a neuT, enhanced AChR ε-subunit reporter expression was inhibited by Erbin and an Erbin mutant that lacks the MuSK binding domain (Fig. 7f, lower panel). Surprisingly, the Erbin MuSK binding or PDZ domain alone were unable to inhibit the AChR ε-subunit reporter. In the presence of neuT, the Erbin PDZ domain even stimulated the AChR ε-subunit reporter by more than twofold. As shown before in the absence of neuT, Erbin inhibited AChR ε-subunit reporter gene transcription in C2C12 myotubes (Fig. 7f, lower panel) (Huang et al., 2003). Moreover, the Erbin MuSK binding domain also inhibited AChR ε-subunit reporter expression. Erbin mutants that lacked the MuSK binding domain were unable to inhibit strongly the AChR ε-subunit reporter. In the presence of neuT, only the Erbin MuSK binding domain inhibited AChR ε-subunit reporter expression. In contrast, the expression level of the AChR ε-subunit reporter was highest if the PDZ domain of Erbin was cotransfected into the myotubes (Fig. 7f).
Erbin enhances aggregation of AChRs in primary skeletal myotubes and C2C12 cells
Previously, Erbin was shown to interact with ErbB2 and to be enriched at the NMJ (Borg et al., 2000; Huang et al., 2001). Having identified Erbin interacting with MuSK, thereby linking MuSK with ErbB2, we sought to investigate the biological significance of this interaction. To this end, we investigated whether ablation or overexpression of Erbin will affect AChR clustering. First, we designed several shRNA-producing vectors and demonstrated their efficiency to knock down Erbin transcript. As examined by luciferase reporter assays, one of the shRNA species reduced the amount of Erbin transcript by >80% (supplemental Fig. 2a, available at www.jneurosci.org as supplemental material). Cotransfection of Erbin expression and Erbin shRNA plasmids in HEK293 cells abolished Erbin protein expression almost completely (supplemental Fig. 2b, available at www.jneurosci.org as supplemental material). To examine the effects of different Erbin protein levels on AChR aggregation in muscle cells, we transiently transfected an Erbin expression plasmid together with pnls-GFP (Hashemolhosseini et al., 2000), or the Erbin shRNA plasmid, into murine primary skeletal myotubes or C2C12 cells (data with C2C12 cells are shown as supplemental Fig. 3, available at www.jneurosci.org as supplemental material). By addition of nerve-derived agrin to the myotubes and staining with rhodamine-labeled bungarotoxin, the influence of modulated Erbin protein amounts on AChR cluster formation was investigated by confocal microscopy. Surplus of Erbin did not influence AChR clustering (data not shown). A decreased amount of Erbin protein significantly reduced the density of aggregated AChRs (Fig. 8a). When scrambled shRNA was transfected, AChR aggregation was not affected (Fig. 8a). To see whether the size of AChR aggregates differs between muscle cells transfected with Erbin or scrambled shRNA, the length and surface area of the AChR aggregates were analyzed (Fig. 8b,c). On the one hand, no difference could be detected, arguing that the physical dimensions of AChR aggregates did not change by knockdown of Erbin. On the other hand, comparing the mean fluorescence intensity of AChR aggregates with the length or area of AChR aggregates showed that knockdown of Erbin decreased the density of AChR aggregates (Fig. 8d–f). Next, we asked whether knockdown of Erbin will affect also the density of microclusters, which were formed in the absence of agrin (Fig. 8f). In fact, we observed a lower fluorescence intensity of AChR microclusters (Fig. 8f).
Figure 8.
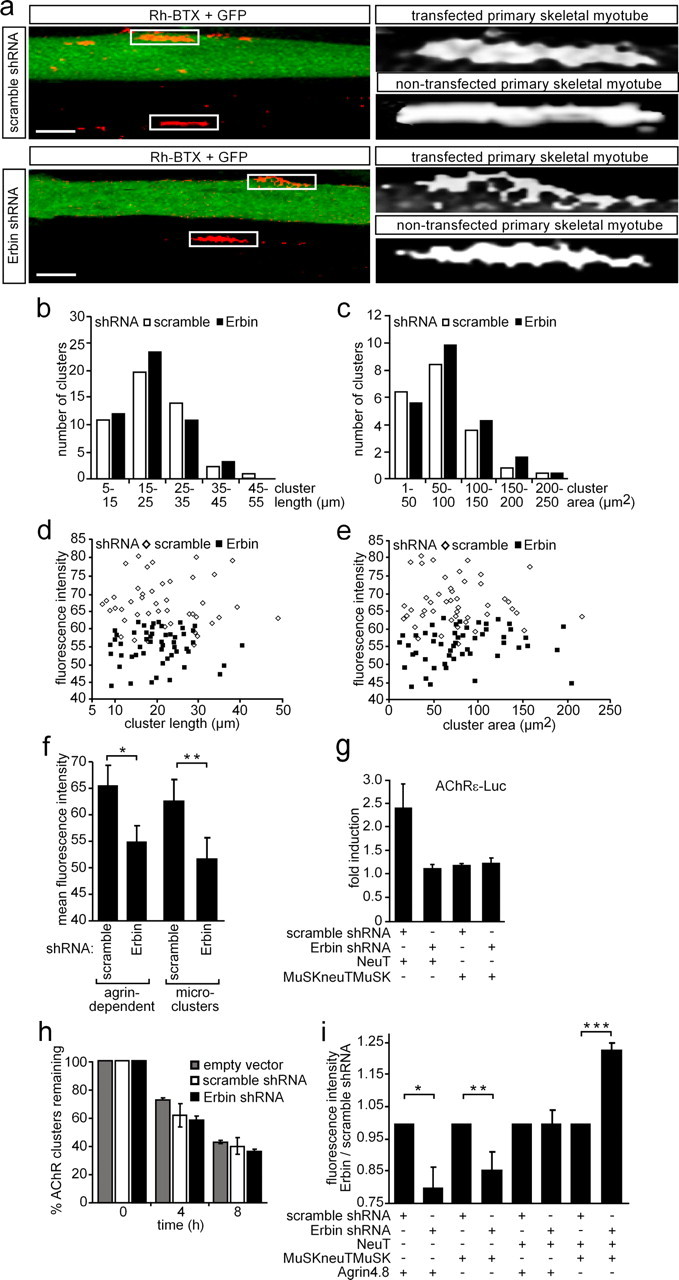
Knockdown of Erbin reduces the density of AChR aggregates. a, Primary skeletal myoblasts were transfected with shRNA (Erbin-specific or scrambled) cloned in pSuperGFPneo, differentiated to myotubes, and incubated with nerve-derived agrin. On the left, typical confocal images (compressed Z-stack) are shown. On the right, high-resolution grayscale images of AChR aggregates localized on transfected or nontransfected myotubes are shown. Note that the AChR aggregates formed in GFP-positive myotubes are less dense. Scale bar, 25 μm. b, c, Graphs represent comparisons of surface areas and lengths of AChR clusters as counted on primary skeletal myotubes after transient transfection with either scrambled or Erbin-specific shRNA (n > 65). d, e, Fluorescence intensities of AChR aggregates on primary skeletal myotubes are plotted against cluster length or surface area. Note that most of the AChR aggregates are of significantly lower density if muscle cells were transfected with Erbin-specific shRNA compared to scrambled shRNA. f, Graph summarizes data presented by d and e showing the mean fluorescence density of all analyzed AChR aggregates. Further, primary muscle cells were transfected as in a but not incubated with nerve-derived agrin for the analysis of the mean fluorescence intensities of AChR microclusters (n > 32). Note that even AChR microclusters are significantly less dense if Erbin is knocked down. Student's t test, *p < 2 × 10−11, **p value <2.4 × 10−4. g, Primary skeletal myoblasts were transiently transfected, differentiated to myotubes, and harvested for preparation of cell extracts. Luciferase activities were measured a minimum of three independent times with an AChRε-luciferase reporter and different expression plasmids. NeuT-mediated enhancement of AChRε transcription was inhibited if concomitantly Erbin was knocked down. h, To find out whether AChR cluster stability decreased in the absence of Erbin, nerve-derived agrin was depleted from cell media at the indicated times, and remaining AChR aggregates were counted at 0 (pSuper, scramble, Erbin-specific, n = 76/67/70) or after 4 (n = 80/67/82) and 8 (n = 71/72/79) hours of agrin withdrawal and presented as percentage of remaining clusters. i, Graph summarizes the ratio of mean fluorescence densities of AChR aggregates on C2C12 cells, which were transfected as indicated. After transfection of the cells with MuSKneuTMuSK, incubation of the cells with nerve-derived agrin became no longer necessary because MuSKneuTMuSK induces formation of AChR aggregates even in the absence of agrin. Note that AChR aggregates formed after knockdown of Erbin in the presence of MuSKneuTMuSK and neuT together are even more dense than aggregates formed in the presence of Erbin. Student's t test, *p < 1.2 × 10−7, **p < 8.1 × 10−9, ***p < 1.2 × 10−8.
We aimed to know whether the knockdown of Erbin will affect AChR gene expression. To this end, we transiently transfected muscle cells with AChRε-reporter, Erbin shRNA, and ErbB2 or a constitutively active MuSK (MuSKneuTMuSK) (Jones et al., 1999). It turned out that knockdown of Erbin diminished ErbB2-dependent reporter gene activity (Fig. 8g, supplemental Fig. 2, available at www.jneurosci.org as supplemental material). It was reported that MuSKneuTMuSK does not induce AChRε-luciferase reporter (Jones et al., 1999). We did not observe any change in luciferase activity when we knocked down Erbin in muscle cells transfected with MuSKneuTMuSK and AChRε-luciferase reporter (Fig. 8g, supplemental Fig. 3, available at www.jneurosci.org as supplemental material).
Altogether, we confirmed that AChR aggregates formed in the absence of Erbin have significantly less fluorescence intensity regardless of whether they are formed in the presence or absence of agrin. To find out whether AChR aggregates will disappear faster in the absence of nerve-derived agrin and Erbin, we transiently transfected C2C12 cells with either an empty plasmid, scrambled shRNA, or Erbin shRNA and measured the number of AChR clusters remaining after removal of nerve-derived agrin. The number of remaining AChR clusters decreased with time independently of the presence or absence of agrin (Fig. 8h).
Finally, we investigated whether in C2C12 myotubes the knockdown of Erbin affects the density of AChR aggregates (n > 36) formed in the presence of constitutively active mutants of ErbB2 (neuT) or MuSK (MuSKneuTMuSK). Surprisingly, expression of neuT or MuSKneuTMuSK compensated for the decrease in intensity of the AChR aggregates that were observed after knockdown of Erbin (Fig. 8i). Note that neuT expression even fully restored the density of AChR aggregates. Excitingly, AChR aggregates formed after cotransfection of neuT and MuSKneuTMuSK are of even higher density (Fig. 8i).
Erbin associates ErbB2 with MuSK and TGF-β signaling at the NMJ
Smad proteins are intracellular signaling mediators of TGF-β (ten Dijke and Heldin, 2006). Recently, it was reported that Erbin inhibits TGF-β signaling through a novel Smad-interacting domain (Dai et al., 2007). We asked whether C2C12 muscle cells express TGF-β receptors and Smad and whether this happens in a nerve-derived agrin-dependent manner. We extracted total RNA from C2C12 myoblasts or myotubes. Using quantitative RT-PCR, we detected TGF-βRI and TGF-βRII receptors and Smad3 in C2C12 cells (Fig. 9a). Both TGF-β receptors and Smad3 were transcribed at higher amount in myotubes compared with myoblasts (Fig. 9a). To further assess the role of Erbin and MuSK in TGF-β signaling, we transiently transfected C2C12 cells with an expression plasmid encoding Smad3. As reported, Smad3 inhibited myotube formation from C2C12 myoblasts (Fig. 9b) (Liu et al., 2001). Cotransfection of the C2C12 cells with hErbin-v2, MuSK, MuSKneuTMuSK, neuT, or different combinations of these plasmids released this inhibition and cells differentiated and formed myotubes (Fig. 9b). On the one hand, this finding is in agreement with previous data showing that increased Erbin expression in HEK293 cells physically sequesters Smad2/Smad3 by its Smad-interacting domain from their association with Smad4 and, thus, negatively modulates TGF-β-dependent transcriptional responses and inhibition of cell growth (Dai et al., 2007). On the other hand, even MuSK, MuSKneuTMuSK, or neuT are able to release the Smad3-mediated inhibition of myotube formation (Fig. 9b). To verify the physiological consequence of a MuSK–Erbin–Smad interaction, the influence of Erbin alone or together with MuSK on Smad2/Smad3-mediated transcriptional activation of TGF-β target genes was examined using the SBE4-Luc reporter, which contains four copies of a Smad binding site in front of the luciferase gene (Zawel et al., 1998). Transient transfection of C2C12 cells with Smad3 but not hErbin-v2 or MuSK led to a significant increase of luciferase activity (Fig. 9c). If either hErbin-v2, MuSK, or both were cotransfected with Smad3, less luciferase activity was detectable (Fig. 9c). These data imply that MuSK, Erbin, or both partially inhibit Smad3-mediated transactivation of respective target genes.
Figure 9.

MuSK impinges on the modulation of TGF-β signaling by Erbin. a, Expression profiles of TGF-β receptors I and II and Smad3 in C2C12 cells were examined by quantitative RT-PCRs and the results depicted as graphs. b, C2C12 cells were transiently transfected with pnls-GFP (Hashemolhosseini et al., 2000) and Smad3 alone, or together with human Erbin-v2, MuSK, MuSKneuTMuSK, or neuT. GFP-positive myoblasts and myotubes were counted, and their ratio was presented as a graph. Note that Smad3 inhibits C2C12 cell differentiation. Erbin, MuSK, MuSKneuTMuSK, or neuT release the Smad3-mediated inhibition of C2C12 differentiation. c, Competition between Erbin binding either Smad3 or MuSK was studied by transfecting Smad3, human Erbin-v2, and MuSK, together with a Smad3-responsible reporter (SBE4-luc) into C2C12 cells. Note that Smad3 strongly transactivates SBE4-Luc, while transfection of Erbin or MuSK together with Smad3 resulted in less SBE4-Luc-derived luciferase activity.
LAP family members Lano and Scribble but not Densin-180 are expressed at the NMJ
To find out whether other LAP family members are expressed in muscle cells, we performed RT-PCR experiments with C2C12 myoblasts and myotubes, which were incubated with either muscle- or nerve-derived agrin. We detected RT-PCR-amplified fragments from total RNA for Scribble, Lano, but not Densin-180 proving that Lano and Scribble were expressed in muscle cells (Fig. 10a). Next, by quantifying transcript levels in C2C12 cells it turned out that the transcript amount of Erbin and Lano were quite similar. Compared with Erbin or Lano, Scribble is expressed sixfold higher (Figs. 2c, 10b). We investigated whether Lano and Scribble also interact with MuSK by performing coimmunoprecipitation experiments (Fig. 10c). We detected a low amount of coprecipitated MuSK when we precipitated fusion proteins of GFP with Lano or Scribble in lysates from transiently transfected HEK293 cells (Fig. 10c). Next, we designed and examined shRNA to knockdown either Lano or Scribble. Both shRNAs turned out to knockdown GFP-Lano or GFP-Scribble very efficiently as ascertained by Western blot or fluorescence microscopy (supplemental Fig. 2c,d, available at www.jneurosci.org as supplemental material). To find out whether in the absence of Lano or Scribble aggregation of AChRs is affected, we transiently transfected Lano- or Scribble-specific shRNAs into primary skeletal myotubes or C2C12 cells (data with C2C12 cells are presented as supplemental Fig. 4, available at www.jneurosci.org as supplemental material). After myotube formation, nerve-derived agrin was added to the myotubes and then they were stained with rhodamine-labeled bungarotoxin to visualize agrin-dependent AChR aggregates. Moreover, we also examined whether the formation of AChR microclusters in the absence of agrin was disturbed. We characterized many AChR aggregates (n > 50 for primary skeletal muscle cells, n > 60 for C2C12 cells) or microclusters (n > 30) obtained from several independent transfections. Contrary to the data acquired with Erbin shRNA, AChR aggregations were affected in surface area if Lano or Scribble were knocked down (Fig. 10d–k). The aggregates were slightly smaller regarding surface area when Lano was knocked down (Fig. 10e), and bigger in surface area and length when Scribble was knocked down (Fig. 10i). On the other hand, the measured fluorescence intensities were unchanged for the agrin-dependent AChR aggregates (Fig. 10l). Furthermore, fluorescence intensities of AChR microclusters were significantly lower if Scribble, but not Lano, was knocked down (Fig. 10m).
Figure 10.
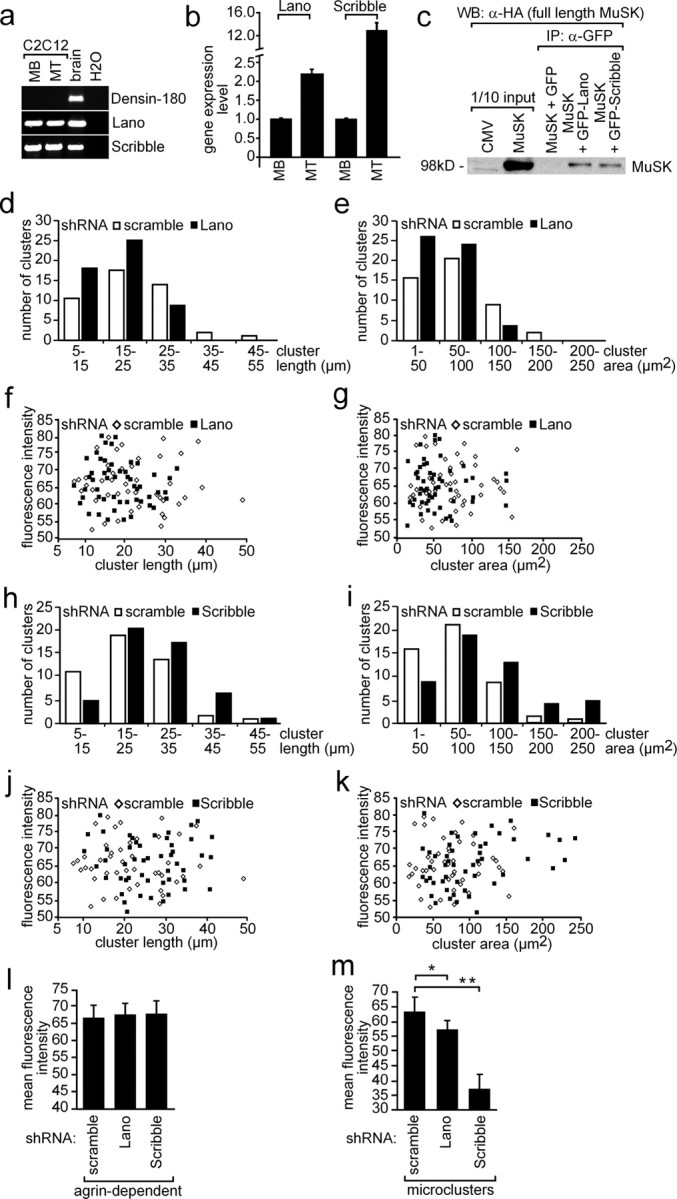
Role of LAP protein family members Lano and Scribble on AChR aggregation. a, RT-PCR data from C2C12 myoblasts and myotubes incubated with agrin or brain. Note that Scribble and Lano, but not Densin-180, are expressed in C2C12 cells. b, Expression profiling by quantitative RT-PCR demonstrated more Scribble and Lano expression in myotubes than in myoblasts. MB, Myoblast; MT, myotubes. c, Western blot of coimmunoprecipitation studies. Note that precipitation of Lano or Scribble resulted in coprecipitation of MuSK. Band intensities reflect a weak interaction between MuSK and Lano or Scribble. d, e, Scribble-specific shRNA was transiently transfected into primary skeletal muscle cells. After differentiation to myotubes, the cells were treated with nerve-derived agrin. Length and surface area of AChR aggregates were plotted against their numbers. Note that a higher number of AChR aggregates with decreased surface areas were detected if Lano was knocked down. f, g, The fluorescence intensities of AChR aggregates analyzed (d, e) were presented as graphs. h–k, Same data shown for nerve-derived AChR aggregates on primary skeletal myotubes transfected with Scribble-specific shRNA (d–g) is presented for primary skeletal muscle cells transfected with Lano-specific shRNA. l, Graph summarizes the mean fluorescence intensity of AChR aggregates shown in f, g, j, and k. m, Primary skeletal muscle cells were transiently transfected with Lano- or Scribble-specific shRNAs, differentiated to myotubes, and identified by their GFP expression. Mean fluorescence intensities of nerve-independent AChR microclusters were determined and depicted as graph. Student's t test, *p < 1.6 × 10−5, **p < 1.6 × 10−12.
Discussion
Our data demonstrate for the first time an interaction between Erbin and MuSK at the NMJ. Since MuSK bears a PDZ-binding domain at its C terminus, it was attractive to assume its interaction with the PDZ domain of Erbin, thereby linking together ErbB2 and MuSK (Jean-Paul Borg, personal communication). Previous approaches failed to detect such an interaction because it is not the PDZ domain of Erbin that interacts with MuSK (Figs. 5–7). Erbin binds MuSK by a yet unidentified domain (Fig. 7) that is positioned between SH3 epitopes and the PDZ domain (Borg et al., 2000). Erbin appears to be one of the long-awaited links between MuSK and AChR expression and clustering at the NMJ, thereby cumulating evidence for a cross talk between agrin and neuregulin signaling. Previously, only extracellular, not intracellular, synergistic effects of neuregulin and agrin on muscle acetylcholine receptor expression were observed (Li et al., 2004).
Further, the MuSK binding domain of Erbin is part of a domain involved in Smad3 binding (SID) (Dai et al., 2007). This is surprising, as the overlapping binding domains of TGF-β and MuSK implicate that their signaling also cross talks at the NMJ.
Different TGF-β members and their receptors have been found at the vertebrate NMJ, but not much is known about their role (McLennan and Koishi, 1994; Jiang et al., 2000). Previously, TGF-β members were postulated to control the onset of secondary myotube formation by preventing the fusion of late myoblasts (Sánchez and Robbins, 1994). All three known members of TGF-β were independently knocked out in mice, but turned out not to be involved in the onset of myotube formation (TGF-β1), or the skeletal muscles of the knock-out mice were not studied in detail (Kaartinen et al., 1995; Proetzel et al., 1995; Sanford et al., 1997; McLennan et al., 2000). All of this is in contrast to what is known from the development of invertebrate NMJs, where TGF-β signaling plays an essential role (Packard et al., 2003). It has been postulated that alternative TGF-β-like factors might also be involved in vertebrate NMJ formation (Packard et al., 2003). Here, expression of TGF-β receptors and Smad3 was identified in myoblasts and upregulated in myotubes (Fig. 9a). Further, the MuSK binding domain of Erbin is encoded by part of exon 21, while the SID of Erbin is located on exons 21 and 23. It seemed unlikely that the Smad binding domain is encoded by the exons 21 and 23 but not 22, because during evolution functional epitopes of proteins are in many cases known to be encoded and transmitted on single exons (Doolittle, 1995). Consequently, it might be speculated whether the SID of Erbin (1) spans from exon 21 to 23, (2) overlaps more precisely with the MuSK binding domain, or (3) is encoded only by exon 23. Here, different isoforms of Erbin were detected at the NMJ all containing the MuSK-binding domain, but not all containing the SID (Fig. 6d). Presumably, these variants would interact with MuSK, but not with Smad3.
Here, we show that Erbin has three downstream effects in muscle cells: it (1) enhances density of AChR aggregates, (2) inhibits neuregulin-mediated enhancement of AChRε transcription, and (3) interferes with Smad-mediated TGF-β effects. But which of these three effects depends on the ErbB2-Erbin-MuSK link? By using knockdown approaches, we showed that the correct density of AChR aggregates depends on the MuSK–Erbin–ErbB2 link (Fig. 8). By measuring the degree of recovery of the density of AChR aggregates using constitutively active mutants of MuSK and ErbB2 on muscle cells ablated for endogenous Erbin expression, a higher density of AChR aggregates was observed than in cells with Erbin expression (Fig. 8i). This finding also argues for a role of Erbin in regulating the correct density of AChR aggregates at the NMJ. In agreement with such a hypothesis, an Erbin mutants composed of the MuSK binding domain is inhibiting the neuT-mediated stimulation of the reporter (Fig. 7f). The ability of Smad to mediate TGF-β effects seems to depend on the MuSK–Erbin–ErbB2 crosslink in a different way, as coexpression of Erbin together with constitutively active mutants of MuSK or ErbB2 result in a block of Smad effects (Fig. 9b).
Our data mark the beginning in understanding the biological role of LAP proteins at the NMJ. By knocking down Erbin, the density of AChR aggregates and aneural microclusters on skeletal myotubes significantly decreased, but their physical dimensions remained unchanged (Fig. 8a–g, supplemental Fig. 3, available at www.jneurosci.org as supplemental material). These findings raise two questions: (1) how might Erbin regulate AChR density, and (2) what do these findings imply on the cross talk between the agrin and neuregulin signaling at the NMJ? It was recently reported that mice with inactivation of either ErbB2/ErbB4 in muscles or neuregulin in skeletal muscle, motor neurons, or both, still form functional synapses in skeletal muscles (Escher et al., 2005; Jaworski and Burden, 2006). Synapses in these mice appeared morphologically normal, but some minor changes were observed. In the absence of motor neuron- and muscle-derived neuregulin, the total number of AChRs at the synapse was slightly reduced (Jaworski and Burden, 2006). Further, in the absence of muscle-specific ErbB2/ErbB4, the endplate bands of diaphragm showed a weak reduction of AChR mRNA expression by 20–30% (Escher et al., 2005). Although these changes did not affect muscle strength in mice, it is conceivable that Erbin has a role in ErbB2-mediated AChR expression at the NMJ.
Erbin might also be involved in capturing or anchoring AChRs at the NMJ, or ensuring their absence. Although the latter appears unlikely, because Erbin is primarily located at the NMJ (Fig. 3), it was reported that neuregulin reduced the number of spontaneous or agrin-induced clusters and increased the disassembly of AChR clusters (Trinidad and Cohen, 2004). Erbin might be able to modulate neuregulin-mediated ErbB activity and thereby participate in the disassembly of AChR clusters. In contradiction to an Erbin-mediated AChR disassembly, a faster disappearance of AChR aggregates in the absence of agrin was not detected after Erbin knockdown in C2C12 cells (Fig. 8h). This does not rule out that Erbin influences AChR disassembly in microclusters.
It is attractive to speculate that Erbin is participating in anchoring all the required proteins at the NMJ in lipid rafts. Possibly, lipid rafts regulate AChR clustering by facilitating agrin–MuSK signaling and thereby promote the interaction between AChRs and rapsyn (Zhu et al., 2006). Erbin might be involved in this regulation by colocalizing MuSK, ErbB2, and AChRs at lipid rafts for signaling at the NMJ. Erbin and ErbB receptors might also be part of the proposed oligomeric complex consisting of at least MuSK and Lrp4 (Kim et al., 2008). Moreover, Erbin ensures accumulation of AChR in microclusters (Fig. 8f, supplemental Fig. 3, available at www.jneurosci.org as supplemental material). This implies that Erbin might start participating in aggregation of AChRs, even before arrival of factors from the nerve.
Other LAP family members, next to Erbin, are Densin-180 (Apperson et al., 1996), Lano (Saito et al., 2001), and Scribble (Murdoch et al., 2003). Densin-180 is associated with postsynaptic densities in rat brain and might be involved in localization of synaptic proteins (Huang et al., 2001). Scribble is a junctional protein involved in establishment and maintenance of epithelial cell polarity (Navarro et al., 2005). Here, Lano and Scribble but not Densin-180 were detected at the NMJ (Fig. 10a). All three LAP proteins are expressed at significantly higher level in myotubes compared to myoblasts (Figs. 2c, 10b). In situ hybridization demonstrates that Erbin transcription only occurs at the synaptic area. Knockdown experiments with Lano and Scribble did not affect the density of agrin-induced AChR aggregates, but their physical dimensions are changed in muscle cells (Fig. 10d,e,h,i, supplemental Fig. 4, available at www.jneurosci.org as supplemental material). Knockdown of Lano decreased, while knockdown of Scribble increased, the size of AChR aggregates but not their density (Fig. 10e,i). In the absence of agrin, AChR microclusters were of significantly lower density when Lano or Scribble was knocked down (Fig. 10m; supplemental Fig. 4, available at www.jneurosci.org as supplemental material). Additional experiments are necessary to understand why the absence of Erbin affects the density of agrin-induced AChR aggregates, while the absence of Lano or Scribble changed their physical dimensions. While NMJs are formed, AChR clusters are initially assembled into microclusters, followed by condensation and formation of large clusters. The initial aggregation of AChRs is mediated by agrin–MuSK signaling (DeChiara et al., 1996). Then, after binding of agrin to the MuSK receptor complex, rac and PAK1 are activated (Weston et al., 2000; Luo et al., 2002). LAP proteins, like Erbin might already influence initial steps of AChR aggregation. Accordingly, we observed a lowered density of AChR microclusters independent of whether Erbin, Lano, or Scribble was knocked down (Figs. 8f, 10m, supplemental Figs. 3, 4, available at www.jneurosci.org as supplemental material). Recently, it was shown that Erbin together with Merlin cooperatively regulates cell-type-specific activation of PAK2 by TGF-β (Wilkes et al., 2009). It would be interesting to investigate whether Erbin is also involved in regulation of PAK1. Our data support the view that balanced physiological amounts of LAP family members might play a crucial role in fine-tuning physical dimension and density of AChR aggregates at the NMJ.
Footnotes
This work was supported by the Deutsche Forschungsgemeinschaft (HA 3309/1-3) and the Interdisciplinary Centre for Clinical Research at the University Hospital of the University of Erlangen-Nuremberg (E2) to S.H. We thank Markus Ruegg for providing the MuSK antibody; Jean-Paul Borg for providing Erbin antibody and pEGFP-Lano and-Scribble expression plasmids; Stephan Kröger for providing agrin-releasing HEK293 cells; Michael Sendtner for help in performing ischiadicus lesions; Hans-Rudolf Brenner for providing total RNA of synaptic and extrasynaptic diaphragm, muscle sections containing ectopic postsynaptic aggregates, and MuSKneuTMuSK and neuT (ErbB2) expression plasmids. We thank Kun Wang for helpful comments on this manuscript.
References
- Altiok N, Bessereau JL, Changeux JP. ErbB3 and ErbB2/neu mediate the effect of heregulin on acetylcholine receptor gene expression in muscle: differential expression at the endplate. EMBO J. 1995;14:4258–4266. doi: 10.1002/j.1460-2075.1995.tb00100.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apperson ML, Moon IS, Kennedy MB. Characterization of densin-180, a new brain-specific synaptic protein of the O-sialoglycoprotein family. J Neurosci. 1996;16:6839–6852. doi: 10.1523/JNEUROSCI.16-21-06839.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bargmann CI, Hung MC, Weinberg RA. Multiple independent activations of the neu oncogene by a point mutation altering the transmembrane domain of p185. Cell. 1986;45:649–657. doi: 10.1016/0092-8674(86)90779-8. [DOI] [PubMed] [Google Scholar]
- Borg JP, Marchetto S, Le Bivic A, Ollendorff V, Jaulin-Bastard F, Saito H, Fournier E, Adélaïde J, Margolis B, Birnbaum D. ERBIN: a basolateral PDZ protein that interacts with the mammalian ERBB2/HER2 receptor. Nat Cell Biol. 2000;2:407–414. doi: 10.1038/35017038. [DOI] [PubMed] [Google Scholar]
- Cheusova T, Khan MA, Enz R, Hashemolhosseini S. Identification of developmentally regulated expression of MuSK in astrocytes of the rodent retina. J Neurochem. 2006a;99:450–457. doi: 10.1111/j.1471-4159.2006.04086.x. [DOI] [PubMed] [Google Scholar]
- Cheusova T, Khan MA, Schubert SW, Gavin AC, Buchou T, Jacob G, Sticht H, Allende J, Boldyreff B, Brenner HR, Hashemolhosseini S. Casein kinase 2-dependent serine phosphorylation of MuSK regulates acetylcholine receptor aggregation at the neuromuscular junction. Genes Dev. 2006b;20:1800–1816. doi: 10.1101/gad.375206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai F, Chang C, Lin X, Dai P, Mei L, Feng XH. Erbin inhibits transforming growth factor beta signaling through a novel Smad-interacting domain. Mol Cell Biol. 2007;27:6183–6194. doi: 10.1128/MCB.00132-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai P, Xiong WC, Mei L. Erbin inhibits RAF activation by disrupting the Sur-8-Ras-Raf complex. J Biol Chem. 2006;281:927–933. doi: 10.1074/jbc.M507360200. [DOI] [PubMed] [Google Scholar]
- DeChiara TM, Bowen DC, Valenzuela DM, Simmons MV, Poueymirou WT, Thomas S, Kinetz E, Compton DL, Rojas E, Park JS, Smith C, DiStefano PS, Glass DJ, Burden SJ, Yancopoulos GD. The receptor tyrosine kinase MuSK is required for neuromuscular junction formation in vivo. Cell. 1996;85:501–512. doi: 10.1016/s0092-8674(00)81251-9. [DOI] [PubMed] [Google Scholar]
- Doolittle RF. The multiplicity of domains in proteins. Annu Rev Biochem. 1995;64:287–314. doi: 10.1146/annurev.bi.64.070195.001443. [DOI] [PubMed] [Google Scholar]
- Escher P, Lacazette E, Courtet M, Blindenbacher A, Landmann L, Bezakova G, Lloyd KC, Mueller U, Brenner HR. Synapses form in skeletal muscles lacking neuregulin receptors. Science. 2005;308:1920–1923. doi: 10.1126/science.1108258. [DOI] [PubMed] [Google Scholar]
- Falls DL, Rosen KM, Corfas G, Lane WS, Fischbach GD. ARIA, a protein that stimulates acetylcholine receptor synthesis, is a member of the neu ligand family. Cell. 1993;72:801–815. doi: 10.1016/0092-8674(93)90407-h. [DOI] [PubMed] [Google Scholar]
- Favre B, Fontao L, Koster J, Shafaatian R, Jaunin F, Saurat JH, Sonnenberg A, Borradori L. The hemidesmosomal protein bullous pemphigoid antigen 1 and the integrin beta 4 subunit bind to ERBIN. Molecular cloning of multiple alternative splice variants of ERBIN and analysis of their tissue expression. J Biol Chem. 2001;276:32427–32436. doi: 10.1074/jbc.M011005200. [DOI] [PubMed] [Google Scholar]
- Garcia RA, Vasudevan K, Buonanno A. The neuregulin receptor ErbB-4 interacts with PDZ-containing proteins at neuronal synapses. Proc Natl Acad Sci U S A. 2000;97:3596–3601. doi: 10.1073/pnas.070042497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Osta A, Tsokas P, Pollonini G, Landau EM, Blitzer R, Alberini CM. MuSK expressed in the brain mediates cholinergic responses, synaptic plasticity, and memory formation. J Neurosci. 2006;26:7919–7932. doi: 10.1523/JNEUROSCI.1674-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashemolhosseini S, Moore C, Landmann L, Sander A, Schwarz H, Witzemann V, Sakmann B, Brenner HR. Electrical activity and postsynapse formation in adult muscle: gamma-AChRs are not required. Mol Cell Neurosci. 2000;16:697–707. doi: 10.1006/mcne.2000.0911. [DOI] [PubMed] [Google Scholar]
- Henriquez JP, Webb A, Bence M, Bildsoe H, Sahores M, Hughes SM, Salinas PC. Wnt signaling promotes AChR aggregation at the neuromuscular synapse in collaboration with agrin. Proc Natl Acad Sci U S A. 2008;105:18812–18817. doi: 10.1073/pnas.0806300105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang YZ, Won S, Ali DW, Wang Q, Tanowitz M, Du QS, Pelkey KA, Yang DJ, Xiong WC, Salter MW, Mei L. Regulation of neuregulin signaling by PSD-95 interacting with ErbB4 at CNS synapses. Neuron. 2000;26:443–455. doi: 10.1016/s0896-6273(00)81176-9. [DOI] [PubMed] [Google Scholar]
- Huang YZ, Wang Q, Xiong WC, Mei L. Erbin is a protein concentrated at postsynaptic membranes that interacts with PSD-95. J Biol Chem. 2001;276:19318–19326. doi: 10.1074/jbc.M100494200. [DOI] [PubMed] [Google Scholar]
- Huang YZ, Zang M, Xiong WC, Luo Z, Mei L. Erbin suppresses the MAP kinase pathway. J Biol Chem. 2003;278:1108–1114. doi: 10.1074/jbc.M205413200. [DOI] [PubMed] [Google Scholar]
- Jaworski A, Burden SJ. Neuromuscular synapse formation in mice lacking motor neuron- and skeletal muscle-derived Neuregulin-1. J Neurosci. 2006;26:655–661. doi: 10.1523/JNEUROSCI.4506-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Y, McLennan IS, Koishi K, Hendry IA. Transforming growth factor-beta 2 is anterogradely and retrogradely transported in motoneurons and up-regulated after nerve injury. Neuroscience. 2000;97:735–742. doi: 10.1016/s0306-4522(00)00084-1. [DOI] [PubMed] [Google Scholar]
- Jones G, Moore C, Hashemolhosseini S, Brenner HR. Constitutively active MuSK is clustered in the absence of agrin and induces ectopic postsynaptic-like membranes in skeletal muscle fibers. J Neurosci. 1999;19:3376–3383. doi: 10.1523/JNEUROSCI.19-09-03376.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaartinen V, Voncken JW, Shuler C, Warburton D, Bu D, Heisterkamp N, Groffen J. Abnormal lung development and cleft palate in mice lacking TGF-beta 3 indicates defects of epithelial-mesenchymal interaction. Nat Genet. 1995;11:415–421. doi: 10.1038/ng1295-415. [DOI] [PubMed] [Google Scholar]
- Kim N, Stiegler AL, Cameron TO, Hallock PT, Gomez AM, Huang JH, Hubbard SR, Dustin ML, Burden SJ. Lrp4 is a receptor for Agrin and forms a complex with MuSK. Cell. 2008;135:334–342. doi: 10.1016/j.cell.2008.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q, Esper RM, Loeb JA. Synergistic effects of neuregulin and agrin on muscle acetylcholine receptor expression. Mol Cell Neurosci. 2004;26:558–569. doi: 10.1016/j.mcn.2004.04.009. [DOI] [PubMed] [Google Scholar]
- Liu D, Black BL, Derynck R. TGF-beta inhibits muscle differentiation through functional repression of myogenic transcription factors by Smad3. Genes Dev. 2001;15:2950–2966. doi: 10.1101/gad.925901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo ZG, Wang Q, Zhou JZ, Wang J, Luo Z, Liu M, He X, Wynshaw-Boris A, Xiong WC, Lu B, Mei L. Regulation of AChR clustering by Dishevelled interacting with MuSK and PAK1. Neuron. 2002;35:489–505. doi: 10.1016/s0896-6273(02)00783-3. [DOI] [PubMed] [Google Scholar]
- McLennan IS, Koishi K. Transforming growth factor-beta-2 (TGF-beta 2) is associated with mature rat neuromuscular junctions. Neurosci Lett. 1994;177:151–154. doi: 10.1016/0304-3940(94)90889-3. [DOI] [PubMed] [Google Scholar]
- McLennan IS, Poussart Y, Koishi K. Development of skeletal muscles in transforming growth factor-beta 1 (TGF-beta1) null-mutant mice. Dev Dyn. 2000;217:250–256. doi: 10.1002/(SICI)1097-0177(200003)217:3<250::AID-DVDY3>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- Merlie JP, Sanes JR. Concentration of acetylcholine receptor mRNA in synaptic regions of adult muscle fibres. Nature. 1985;317:66–68. doi: 10.1038/317066a0. [DOI] [PubMed] [Google Scholar]
- Moscoso LM, Chu GC, Gautam M, Noakes PG, Merlie JP, Sanes JR. Synapse-associated expression of an acetylcholine receptor-inducing protein, ARIA/heregulin, and its putative receptors, ErbB2 and ErbB3, in developing mammalian muscle. Dev Biol. 1995;172:158–169. doi: 10.1006/dbio.1995.0012. [DOI] [PubMed] [Google Scholar]
- Murdoch JN, Henderson DJ, Doudney K, Gaston-Massuet C, Phillips HM, Paternotte C, Arkell R, Stanier P, Copp AJ. Disruption of scribble (Scrb1) causes severe neural tube defects in the circletail mouse. Hum Mol Genet. 2003;12:87–98. doi: 10.1093/hmg/ddg014. [DOI] [PubMed] [Google Scholar]
- Navarro C, Nola S, Audebert S, Santoni MJ, Arsanto JP, Ginestier C, Marchetto S, Jacquemier J, Isnardon D, Le Bivic A, Birnbaum D, Borg JP. Junctional recruitment of mammalian Scribble relies on E-cadherin engagement. Oncogene. 2005;24:4330–4339. doi: 10.1038/sj.onc.1208632. [DOI] [PubMed] [Google Scholar]
- Packard M, Mathew D, Budnik V. Wnts and TGF beta in synaptogenesis: old friends signalling at new places. Nat Rev Neurosci. 2003;4:113–120. doi: 10.1038/nrn1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ponomareva ON, Ma H, Vock VM, Ellerton EL, Moody SE, Dakour R, Chodosh LA, Rimer M. Defective neuromuscular synaptogenesis in mice expressing constitutively active ErbB2 in skeletal muscle fibers. Mol Cell Neurosci. 2006;31:334–345. doi: 10.1016/j.mcn.2005.10.004. [DOI] [PubMed] [Google Scholar]
- Proetzel G, Pawlowski SA, Wiles MV, Yin M, Boivin GP, Howles PN, Ding J, Ferguson MW, Doetschman T. Transforming growth factor-beta 3 is required for secondary palate fusion. Nat Genet. 1995;11:409–414. doi: 10.1038/ng1295-409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rangwala R, Banine F, Borg JP, Sherman LS. Erbin regulates mitogen-activated protein (MAP) kinase activation and MAP kinase-dependent interactions between Merlin and adherens junction protein complexes in Schwann cells. J Biol Chem. 2005;280:11790–11797. doi: 10.1074/jbc.M414154200. [DOI] [PubMed] [Google Scholar]
- Saito H, Santoni MJ, Arsanto JP, Jaulin-Bastard F, Le Bivic A, Marchetto S, Audebert S, Isnardon D, Adélaïde J, Birnbaum D, Borg JP. Lano, a novel LAP protein directly connected to MAGUK proteins in epithelial cells. J Biol Chem. 2001;276:32051–32055. doi: 10.1074/jbc.C100330200. [DOI] [PubMed] [Google Scholar]
- Sánchez A, Robbins J. Unprocessed myogenin transcripts accumulate during mouse embryogenesis. J Biol Chem. 1994;269:1587–1590. [PubMed] [Google Scholar]
- Sanes JR, Lichtman JW. Development of the vertebrate neuromuscular junction. Annu Rev Neurosci. 1999;22:389–442. doi: 10.1146/annurev.neuro.22.1.389. [DOI] [PubMed] [Google Scholar]
- Sanford LP, Ormsby I, Gittenberger-de Groot AC, Sariola H, Friedman R, Boivin GP, Cardell EL, Doetschman T. TGFbeta2 knockout mice have multiple developmental defects that are non-overlapping with other TGFbeta knockout phenotypes. Development. 1997;124:2659–2670. doi: 10.1242/dev.124.13.2659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Si J, Miller DS, Mei L. Identification of an element required for acetylcholine receptor-inducing activity (ARIA)-induced expression of the acetylcholine receptor epsilon subunit gene. J Biol Chem. 1997;272:10367–10371. doi: 10.1074/jbc.272.16.10367. [DOI] [PubMed] [Google Scholar]
- ten Dijke P, Heldin C-H. Berlin: Springer; 2006. Smad signal transduction: Smads in proliferation, differentiation and disease. [Google Scholar]
- Trinidad JC, Cohen JB. Neuregulin inhibits acetylcholine receptor aggregation in myotubes. J Biol Chem. 2004;279:31622–31628. doi: 10.1074/jbc.M400044200. [DOI] [PubMed] [Google Scholar]
- Trinidad JC, Fischbach GD, Cohen JB. The Agrin/MuSK signaling pathway is spatially segregated from the neuregulin/ErbB receptor signaling pathway at the neuromuscular junction. J Neurosci. 2000;20:8762–8770. doi: 10.1523/JNEUROSCI.20-23-08762.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valenzuela DM, Stitt TN, DiStefano PS, Rojas E, Mattsson K, Compton DL, Nuñez L, Park JS, Stark JL, Gies DR, Thomas S, Le Beau MM, Fernald AA, Copeland NG, Jenkins NA, Burden SJ, Glass DJ, Yancopoulos GD. Receptor tyrosine kinase specific for the skeletal muscle lineage: expression in embryonic muscle, at the neuromuscular junction, and after injury. Neuron. 1995;15:573–584. doi: 10.1016/0896-6273(95)90146-9. [DOI] [PubMed] [Google Scholar]
- Weston C, Yee B, Hod E, Prives J. Agrin-induced acetylcholine receptor clustering is mediated by the small guanosine triphosphatases Rac and Cdc42. J Cell Biol. 2000;150:205–212. doi: 10.1083/jcb.150.1.205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkes MC, Repellin CE, Hong M, Bracamonte M, Penheiter SG, Borg JP, Leof EB. Erbin and the NF2 tumor suppressor Merlin cooperatively regulate cell-type-specific activation of PAK2 by TGF-beta. Dev Cell. 2009;16:433–444. doi: 10.1016/j.devcel.2009.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zawel L, Dai JL, Buckhaults P, Zhou S, Kinzler KW, Vogelstein B, Kern SE. Human Smad3 and Smad4 are sequence-specific transcription activators. Mol Cell. 1998;1:611–617. doi: 10.1016/s1097-2765(00)80061-1. [DOI] [PubMed] [Google Scholar]
- Zhu D, Xiong WC, Mei L. Lipid rafts serve as a signaling platform for nicotinic acetylcholine receptor clustering. J Neurosci. 2006;26:4841–4851. doi: 10.1523/JNEUROSCI.2807-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu X, Lai C, Thomas S, Burden SJ. Neuregulin receptors, erbB3 and erbB4, are localized at neuromuscular synapses. EMBO J. 1995;14:5842–5848. doi: 10.1002/j.1460-2075.1995.tb00272.x. [DOI] [PMC free article] [PubMed] [Google Scholar]