

Abstract
Weakly electric fish can enhance the detection and localization of important signals such as those of prey in part by cancellation of redundant spatially diffuse electric signals due to, e.g., their tail bending. The cancellation mechanism is based on descending input, conveyed by parallel fibers emanating from cerebellar granule cells, that produces a negative image of the global low-frequency signals in pyramidal cells within the first-order electrosensory region, the electrosensory lateral line lobe (ELL). Here we demonstrate that the parallel fiber synaptic input to ELL pyramidal cell undergoes long-term depression (LTD) whenever both parallel fiber afferents and their target cells are stimulated to produce paired burst discharges. Paired large bursts (4-4) induce robust LTD over pre–post delays of up to ±50 ms, whereas smaller bursts (2-2) induce weaker LTD. Single spikes (either presynaptic or postsynaptic) paired with bursts did not induce LTD. Tetanic presynaptic stimulation was also ineffective in inducing LTD. Thus, we have demonstrated a form of anti-Hebbian LTD that depends on the temporal correlation of burst discharge. We then demonstrated that the burst-induced LTD is postsynaptic and requires the NR2B subunit of the NMDA receptor, elevation of postsynaptic Ca2+, and activation of CaMKIIβ. A model incorporating local inhibitory circuitry and previously identified short-term presynaptic potentiation of the parallel fiber synapses further suggests that the combination of burst-induced LTD, presynaptic potentiation, and local inhibition may be sufficient to explain the generation of the negative image and cancellation of redundant sensory input by ELL pyramidal cells.
Introduction
It is generally believed that long-lasting changes in synaptic efficacy are an essential cellular component underlying activity-dependent adaptive modification of neuronal function. A causal link between synaptic plasticity and altered systems level neural processing has, however, been difficult to establish. We describe here a form of anti-Hebbian long-term depression (LTD) in a feedback circuit of the medullary electrosensory nucleus [electrosensory lateral line lobe (ELL)] of the weakly electric fish Apteronotus leptorhynchus. This form of LTD requires specific temporal correlations of presynaptic and postsynaptic spike bursts. Based on the coding properties of bursts in this sensory system, we hypothesize that LTD in the ELL feedback pathway, together with local circuit short-term synaptic dynamics, implements a mechanism that adaptively cancels spatially redundant low-frequency electrosensory signals.
A. leptorhynchus emits a high-frequency (species range: ∼600–1000 Hz) electric organ discharge (EOD) that is sensed by electroreceptors distributed over its body surface (Carr et al., 1982). P-units respond to amplitude modulations (AMs) of the EOD caused by objects such as prey or by interfering EODs of conspecifics (Maler, 2007). As the fish moves past prey, spatially localized low-frequency AMs are induced and these guide the fish precisely toward the prey target (Nelson and Maciver, 1999). Tail movements and interaction of fish with similar EOD frequencies also induce AMs whose frequency range overlaps that induced by prey (Bastian, 1996a).
In vivo studies (Bastian, 1996a, 1998b) have shown that activation of ELL pyramidal cells by correlated sensory stimulation of their receptive fields (RFs) and electrical or natural stimulation of their indirect feedback input [dorsal molecular layer (DML) PFs] leads to short-term anti-Hebbian depression of the feedback input. This depression is believed to be part of the cellular basis of a mechanism that cancels spatially redundant low-frequency electrosensory signals (Bastian et al., 2004). These studies did not address either the required precision of the correlated input and feedback signals or the possible importance of their temporal ordering. In the ELL of mormyrid fish (an unrelated family of electric fish) a precisely ordered relation between presynaptic input and postsynaptic spiking is essential for such cancellation (Bell et al., 1997; Han et al., 2000).
Anti-Hebbian depression depends on Ca2+ influx (Bastian, 1998b). One possible source of Ca2+ is NMDARs expressed by ELL pyramidal cells (Bottai et al., 1997; Berman et al., 2001), especially NR2B-containing NMDARs (Harvey-Girard et al., 2007).
We therefore investigated both the temporal dynamics of the anti-Hebbian cancellation mechanism and its cellular basis in an in vitro ELL preparation. We tested the effects of correlated discharge of pyramidal cells and PFs and found that specific burst patterns were able to induce LTD of pyramidal cell EPSPs evoked by PF stimulation. We also found that this form of LTD requires NMDARs (NR1/NR2B subunits) located on dendritic spines of ELL pyramidal cells, elevation of postsynaptic Ca2+ levels, and activation of CaMKIIβ. We propose that this LTD is the cellular basis of long-lasting negative image formation in the apteronotid ELL.
Materials and Methods
ELL slice preparation
ELL slices were prepared as previously described (Berman et al., 1997; Berman and Maler, 1998b,c). Briefly, fish were anesthetized with 0.2% 3-aminobenzoic ethyl ester (MS-222; Sigma) in oxygenated water. The ELL was removed, and slices of 350 μm in its transverse plane were prepared on a vibratome while the ELL was immersed in ice-cold artificial CSF (ACSF) containing the following (in mm): 124 NaCl, 3 KCl, 0.75 KH2PO4, 2 CaCl2, 1.5 MgSO4, 24 NaHCO3, and 10 d-glucose containing 1 mm kynurenic acid. Slices were maintained in an interface chamber in ACSF at room temperature for 30 min to 1 h before recording. All protocols were approved by the University of Ottawa Animal Care Committee.
Stimulation and recording
Intracellular recordings were made using sharp microelectrodes (80–150 MΩ) filled with 3 m KAc with tips placed in the pyramidal cell layer of the ELL centrolateral segment (CLS) (Maler, 2007); we chose the CLS since it has been the focus of numerous in vivo studies related to plasticity of feedback connections (Bastian, 1995, 1996a,b, 1998a; Chacron et al., 2003; Bastian et al., 2004). Intracellular recordings were amplified and filtered (DC-1 kHz, Axoclamp-2B; Molecular Devices), digitized at 5 kHz (ITC-16; Instrutech), and acquired using Pulse Control (Instrutech) and Igor Pro (Wavemetrics) software running on a Macintosh G3 computer (Apple).
Parallel fibers (PFs) in the DML of the ELL (Maler, 2007) were stimulated through a stimulus isolation unit (100 μs pulses, 100 mV; Digitimer) using a gold-coated tungsten microelectrode (0.8–1.2 MW; FHC). In all protocols described below, parallel fibers were stimulated every 4 s to avoid the induction of presynaptic potentiation (Oswald et al., 2002; Lewis and Maler, 2004). One hundred presynaptic stimulus pulses were delivered as controls while postsynaptic responses were recorded. Injection of intracellular current (pulses of 1.5 nA for 1 ms) was used to evoke postsynaptic action potentials. Then, 110 pre–post paired stimulations were delivered at 0.25 Hz with both the number of stimuli and their relative timing varied as described below. This was followed by stimulation of the presynaptic PFs (360 times) to test for changes of the synaptic response. We quantified the synaptic responses by measuring the peak amplitude from baseline. The baseline was taken as the mean membrane potential over a 50 ms window recorded just previous to stimulus onset.
We tried different types of pre–post pairing stimulation protocols (Morrison et al., 2008). First, we tested standard spike timing-dependent plasticity protocols (named here 1-1 protocols) with delays from −100 to +100 ms (presynaptic stimulation time − postsynaptic spike time). We then tested presynaptic bursts paired with postsynaptic bursts. We delivered presynaptic and postsynaptic bursts containing four 100 μs pulses at 100 Hz (referred to as 4-4 protocols). We varied the delay between the “preburst” and the “postburst” from −100 ms (i.e., post 100 ms before pre) to +100 ms (i.e., post 100 ms after pre) from the starting spikes within each burst. We also tested other pairing protocols at varying delays, as one presynaptic and four postsynaptic stimulations (1-4), four presynaptic coupled to one presynaptic stimulation (4-1), and two presynaptic paired with two postsynaptic stimulations (2-2).
Throughout control and test stimulations, a paired-pulse facilitation (PPF) protocol with a delay of 20 ms between pulses was performed every tenth stimulation to assess possible changes of presynaptic efficacy. At the end of the plasticity protocol band-limited Gaussian white noise current (0–60 Hz) was injected (Oswald et al., 2004). Coherence of the evoked spike train and noise stimulus was used to determine whether recording was from a basilar pyramidal (E cell) or a nonbasilar pyramidal cell (I cell); in the CLS, E cells are high pass, whereas I cells are low pass (Ellis et al., 2007; Mehaffey et al., 2008). Twenty-three cells were identified by their filtering properties as E cells (Bastian et al., 2004; Ellis et al., 2007), 11 were identified as I cells, and 5 remained unidentified (data not shown). No differences were seen among the cell classes for any of the protocols, so the data were pooled across cell types.
Previous in vivo experiments showed that redundant electrosensory signals are cancelled by ELL pyramidal cells with a process involving synaptic depression of their indirect feedback inputs (DML PFs) (Bastian, 1996a, 1998a). We reproduced in vitro these in vivo experiments in the following way: (1) a single PF test pulse was delivered at 1 Hz while EPSPs were recorded (repeated 30 times); (2) while we continued to deliver the test pulse as before, a tetanic train of six PF stimulations (15 ms ISI) was delivered 250 ms after the test pulse (repeated 120 times); (3) stimulation continued as before [test pulse as in (2) plus tetanic train] except that during the tetanic train a negative current was injected for 100 ms to hyperpolarize the cell (repeated 60 times); and (4) finally, the same protocol as in (2) was repeated (60 times). During these experiments, pyramidal cell firing rate was monitored for comparison with in vivo firing rate as previously reported for this protocol (Bastian, 1998a).
Drug applications
NMDA receptors containing the NR2B subunit are expressed in ELL pyramidal cells and are the major contributor to the PF-evoked EPSPs (Berman et al., 1997, 2001; Harvey-Girard and Dunn, 2003; Harvey-Girard et al., 2007). We therefore bath applied ifenprodil (30 μm), an antagonist of NR2B-containing NMDA receptors, to assess the involvement of NMDA-Rs containing this subunit in the induction of synaptic plasticity. Our previous work has demonstrated that the apteronotid NR2B subunit contains the ifenprodil binding site (Harvey-Girard and Dunn, 2003).
ELL pyramidal cells express high levels of CaMKIIb (Maler and Hincke, 1999); we therefore bath applied the CaMKII antagonists KN-62 (3.3 μm) (Wang and Maler, 1998) and KN-93 (3.3 μm), as well as KN-92 (3.3 μm) as a negative control (Valverde et al., 2005; Hansel et al., 2006), to assess the possible contribution of this kinase to the induction of the synaptic plasticity we observed. During our experiments, KN-92 and KN-93, but not KN-62, increased cell excitability and spontaneous spiking. We therefore hyperpolarized the cells during recording to reduce the number of spontaneous spikes and eliminated any remaining spikes before data analysis. The increased excitability is presumably due to the extracellular block of wide range of voltage-gated K+ channels by KN-92 and KN-93 (Rezazadeh et al., 2006). KN-62 did not increase cell excitability and therefore gave cleaner results.
Bastian (1998a) has demonstrated that postsynaptic Ca2+ is required for the in vivo induction of plasticity in ELL pyramidal cells. We therefore also assessed the role of postsynaptic Ca2+ in the induction of synaptic plasticity in vitro by including BAPTA (50 mm) in the recording pipette.
Effects of the drug applications on the evoked EPSPs were monitored for 20 min before starting the pairing protocol.
Modeling the interactions of long-term depression with short-term plasticity and inhibition
To assess the functional implications of decreasing postsynaptic strength on the long time scales associated with LTD, we consider how the net parallel fiber inputs change in the context of negative image generation during sinusoidal spatially global stimulus presentation (Bastian, 1995; Nelson and Paulin, 1995; Bastian et al., 2004). The parallel fiber inputs consist of a direct excitatory component and a disynaptic inhibitory component. Previous studies have developed an experimentally based model of the dynamics underlying these inputs (Lewis and Maler, 2002a, 2004; Lewis et al., 2007). This original model described excitation in terms of the presynaptic dynamics as well as inhibition. Here, we consider the effects of changing the postsynaptic excitatory conductance. A basic description of the model is provided in the following sections; the details, along with the motivation and associated data, can be found in previous studies (Lewis and Maler, 2002a, 2004; Lewis et al., 2007).
We consider a conductance-based single-neuron (leaky integrate-and-fire) model (Dayan and Abbott, 2001). In addition to a leak conductance, the neuron receives both excitatory and inhibitory inputs as follows:
and
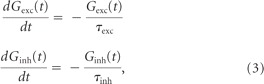 |
with threshold V thresh = −65 mV; reset V reset = −70 mV; leak conductance go = 100 μS; leak reversal potential Vo = −70 mV; constant current input, Idc = 0.4 nA; excitatory and inhibitory reversal potentials V exc = 0 mV and V inh = −80 mV; and excitatory and inhibitory time constants τexc = 5 ms and τinh = 10 ms. The total synaptic conductance G exc and G inh (and hence total synaptic current, I syn) are due to the combined effects of 120 independent excitatory inputs and 120 independent inhibitory inputs, respectively.
The presynaptic excitatory inputs also exhibit short-term facilitation (F) and depression (D) and are modeled using the following standard approach (Dayan and Abbott, 2001; Lewis et al., 2007):
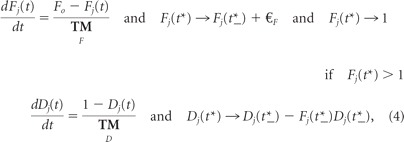 |
where t* is the time of a spike in parallel fiber j and t*_ is the time just before, Fo equals 0.05, ΔF equals 0.23, and the facilitation and depression time constants are τF = 79 ms and τD = 83 ms. Note that while all 120 inputs are described by the same parameters, they are independent in their firing times (Poisson spike trains); Fj and Dj describe the facilitation and depression levels for input fiber j.
The dynamics of G exc and G inh are described by Equation 3, except when a presynaptic input spike arrives, at which time they are updated using the following instantaneous update rules:
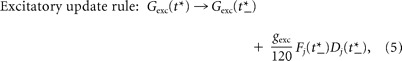 |
Excitatory update rule.
When an excitatory input j fires at a time t*, G exc is increased by a discrete amount (Eq. 5) and then decays back to zero with time constant τexc (Eq. 3). This update is performed every time any of the 120 excitatory inputs fire, using the values of Fj and Dj just before their own updates (denoted by time t*_); thus, the product FjDj determines the presynaptic strength. The mean rate of firing in each of the excitatory inputs is denoted by re, and G exc is the excitatory postsynaptic conductance (G exc = 400 μS, unless otherwise stated).
Inhibitory update rule.
The method for updating G inh mimics spontaneous synaptic activity and asynchronous inputs due to disynaptic inhibition. In this case, when an inhibitory input j fires at a time t** (independent of excitatory input times t*), G inh is increased by a constant amount (Eq. 6). Following the update, G inh decays back to zero with time constant τinh (Eq. 3). The inhibitory postsynaptic conductance is given by G inh = 520 μS. The dynamics of disynaptic inhibition are accounted for by setting the mean firing rate ri of each of the 120 inhibitory inputs (independent Poisson spike trains) to be proportional to re. This relationship is calculated explicitly in previous studies and is well described as follows as long as changes in re are slower than ∼2 Hz:
Both excitatory and inhibitory inputs exhibit random Poisson firing activity with mean rates re and ri, respectively.
We simulate the experimental conditions involved in the generation of a negative image by a sinusoidal rate modulation of the parallel fibers (Lewis et al., 2007). In doing so, we assume that a sinusoidal spatially global sensory input is represented accurately in the parallel fiber activity (Bastian et al., 2004). A negative image results when the pyramidal cell activity (variations in membrane potential or spike output) is out of phase with the input modulation. We quantify the neural responses in terms of the phase histogram and the vector strength (Bastian et al., 2004; Lewis et al., 2007). All simulations used an Euler integration method (time step = 0.2 ms) and were implemented in Igor Pro (Wavemetrics).
Results
Electroreceptors respond to the low-frequency AMs of the EOD caused by objects such as prey and this sensory input guides the fish precisely toward the prey target (Maler, 2007). Tail bending induces AMs that overlap in frequency content with those induced by prey (Bastian, 1996a). The difference between these classes of signals is that the tail-bending-induced AMs affect electroreceptors over the entire body surface (a global signal), whereas those induced by prey are spatially localized.
While P-units respond well to tail-movement-associated AMs, their central targets, ELL pyramidal cells, do not (Bastian, 1996a). This lack of response is due to an adaptive mechanism that generates a negative image of a global sensory input to cancel it (Bastian, 1996b, 1998a). Adaptive cancellation is due to PF feedback as well as proprioceptive descending input to the apical dendrites of pyramidal cells (Bastian, 1995; Chacron et al., 2003; Bastian et al., 2004); these PFs emanate from a caudal mass of cerebellar granule cells [eminentia granularis posterior (EGp)] (Sas and Maler, 1987).
This work raised an apparent paradox. Tetanic stimulation of PFs in vivo (Bastian, 1996b, 1998a) induces depression of the evoked EPSPs in pyramidal cells, but they recover rapidly and do not show LTD. However, pyramidal cells fail to respond to a low-frequency global amplitude modulation (Bastian et al., 2004) or to tail bending (Bastian, 1996a). Blockade of the EGp feedback within ELL reinstates the cells' response (Bastian et al., 2004), suggesting that the cancellation signal is very long lasting and is therefore presumably generated by long-term plasticity. We therefore investigated both the temporal dynamics of the anti-Hebbian cancellation mechanism and its cellular basis in an in vitro ELL preparation in order to determine whether long-lasting LTD can be elicited, and whether such depression might be capable of explaining the long-lasting cancellation observed in vivo.
Temporal spike patterning and synaptic depression of PF–pyramidal cell synapses
In this section, we explore the temporal stimulation patterns required to induce LTD at PF synapses onto ELL pyramidal cells. Given earlier work on mormyrid electric fish (Bell et al., 1997; Han et al., 2000), we first examined the effect of a classic spike timing-dependent plasticity protocol. As described below, this protocol failed to induce any form of plasticity. Given our previous work demonstrating the role of spike bursts in encoding low-frequency electrosensory signals (Oswald et al., 2004, 2007; Ellis et al., 2007; Mehaffey et al., 2008; Marsat et al., 2009), we then used a variety of presynaptic and postsynaptic burst-pairing protocols in order to determine which, if any, might be capable of inducing long-term synaptic plasticity. In addition, we also investigated whether the induced plasticity was specific to the stimulated synapses.
Absence of spike timing-dependent plasticity at feedback synapses to pyramidal cells
EPSPs from ELL pyramidal cells were recorded by sharp electrode while DML PFs were electrically stimulated (once/4 s) (Fig. 1 A). Such a low stimulus frequency was required to avoid short-term presynaptic potentiation at these synapses (Lewis and Maler, 2004). Stimulus artifact (decayed within 1.5–2 ms) and evoked EPSP (peak ∼5–7 ms) were well separated and easy to distinguish (Fig. 1 B). Furthermore, the stimulus electrode was gold coated and the stimulus artifact was monitored (Fig. 1 C) to ensure long-term stimulation stability. Experimental cases showing >5% change in the stimulus artifact were discarded.
Figure 1.
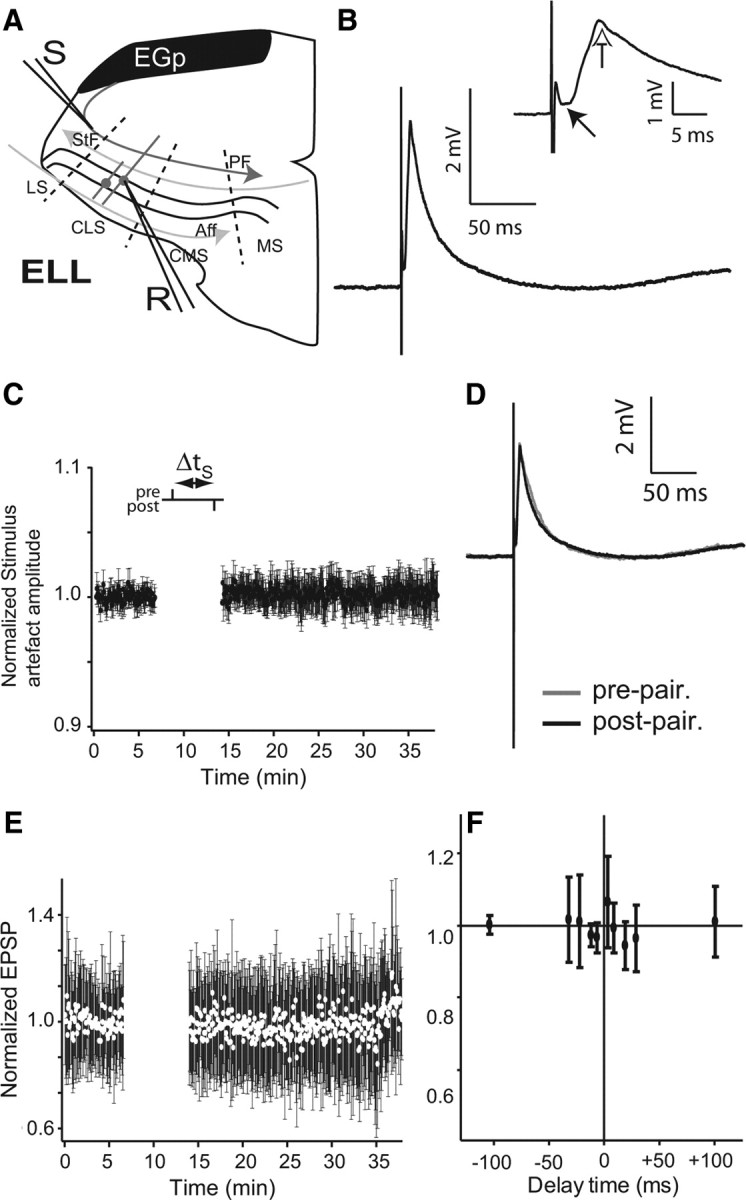
Absence of spike timing-dependent-plasticity in ELL pyramidal cells with single-pulse paired stimulation. A, Schema showing the ELL brain slice with recording and stimulation arrangement. Stimulation with a bipolar electrode (S) of PFs (the indirect feedback pathway) was done every 4 s. A sharp electrode (R) recorded EPSPs in somata of superficial or intermediate pyramidal cells in the CLS. Aff, Afferent inputs; LS, lateral segment; MS: medial segment; StF: stratum fibrosum. B, Trace average (100 traces) illustrating a single-pulse-evoked EPSP and the lack of a strong IPSP. Inset, Black arrow points to the clear separation between the stimulus artifact and EPSP onset (1.5–2 ms) and the white arrow points to the EPSP peak at a latency of 5–7 ms with respect to stimulus onset. C, Normalized stimulus artifact amplitude during STDP protocol to demonstrate its stability throughout the entire experimental protocol. D, EPSP averages before (gray) and after (black) single-pulse pairing. E, Normalized EPSP peak amplitude averages during STDP protocol at a delay (Δt s) of +10 ms (n = 5). F, Summary of the effects of varying the delay between presynaptic and postsynaptic stimulation. All time points are normalized (to the first EPSP) and are the mean EPSP amplitude of five cells, except ±100 ms, which has only n = 2. Delay times are as follows: ±5, ±10, ±20, ±30, and ±100 ms. Error bars show the SD in C, E, and F.
EPSPs evoked by low-frequency PF stimulation (1/4 s) were stable for the 100 prepairing control stimuli (Fig. 1 D,E) and the prepairing stimulation shown in subsequent figures; this stability was observed to persist for up to 90 min of unpaired stimulation (n = 2, data not shown). We paired single presynaptic stimuli and postsynaptic spikes (110 pairings; 1.5 nA pulse for 1 ms) at delays ranging from 0 to ±100 ms. Cells were slightly hyperpolarized so as to limit spiking to that evoked by intracellular current injection and not PF stimulation. EPSP failures were not seen. We never observed any consistent change in either the early (AMPA and NMDA receptor-mediated) or late (NMDA-R-mediated) phases of the evoked EPSP (Fig. 1 E) (Berman et al., 1997, 2001; Harvey-Girard et al., 2007). Peak values of the postpairing EPSPs were, for all pairing intervals, nearly identical to those observed during prepairing stimulation (Fig. 1 F) (postpairing:prepairing ratio = 0.994 ± 0.043). This conclusion held for neurons exhibiting both low (I cells)- and high (E cells)-frequency tuning.
Parallel fibers exhibit robust PPF (Bastian, 1998a; Lewis and Maler, 2004). No statistically significant changes in PPF were observed (prepairing: 1.959 ± 0.715; postpairing: 2.405 ± 1.442; n = 13; t test, p < 0.01) suggesting that this stimulation protocol did not induce presynaptic potentiation (Lewis and Maler, 2004) that might mask a postsynaptic depression. We conclude that, unlike the case for the mormyrid ELL, the indirect feedback input to ELL pyramidal cells of A. leptorhynchus do not exhibit classic STDP involving singles spikes in the presynaptic and postsynaptic neurons.
Burst-induced anti-Hebbian long-term depression
ELL pyramidal cells burst in response to depolarizing current injection; the bursts are induced by depolarizing spike afterpotentials (DAPs) that, in turn, are due to somatic spikes backpropagating into their apical dendrites (Turner et al., 1994, 1996). In the centrolateral ELL and centromedial segments (CMS) bursts code for low-frequency prey-like sensory signals, whereas isolated spikes can encode high-frequency global communication-like signals (Oswald et al., 2004, 2007; Ellis et al., 2007; Mehaffey et al., 2008; Marsat et al., 2009). Further, low-frequency input to a pyramidal cell's RF induces bursting (Chacron and Bastian, 2008; Marsat et al., 2009). Cancellation of this signal requires low-frequency spatially diffuse (global) activation of the pyramidal cell's nonclassic RF (Chacron et al., 2003), and this is conveyed by the indirect cerebellar (EGp) PF feedback pathway to their apical dendrites (Bastian et al., 2004). The spiking pattern of the EGp granule cells to such stimulation is unknown; patch recordings from mammalian cerebellar granule cells demonstrate that they can burst in response to sensory stimulation (D'Angelo et al., 2001; Chadderton et al., 2004; Rancz et al., 2007). We therefore investigated whether burst discharge was required to induce synaptic plasticity of the indirect PF feedback pathway. Spike intervals within a burst are typically ≤10 ms (Turner et al., 1994; Oswald et al., 2007); we chose interspike intervals of 10 ms to fit Oswald's ISI upper limit for burst definition ms (Oswald et al., 2007).
High-frequency stimulation of PFs evoke long-lasting compound EPSPs whose late phase is NMDAR dependent (Berman et al., 1997, 2001; Harvey-Girard et al., 2007). In many systems such activation of NMDARs is sufficient to induce long-term plasticity (Andersen et al., 2006). We therefore first verified that presynaptic bursts (four pulse bursts at 100 Hz with 4 s between bursts) alone were unable to induce synaptic plasticity depression or potentiation (n = 2; ratio of poststimulation to prestimulation EPSP amplitude was 0.981 ± 0.139) (Fig. 2 A). We next investigated whether STDP protocols, but using bursts instead of isolated spikes, might induce plasticity. Four pulse bursts were delivered both presynaptically and postsynaptically (100 Hz: ISI = 10 ms) at various pairing delays.
Figure 2.
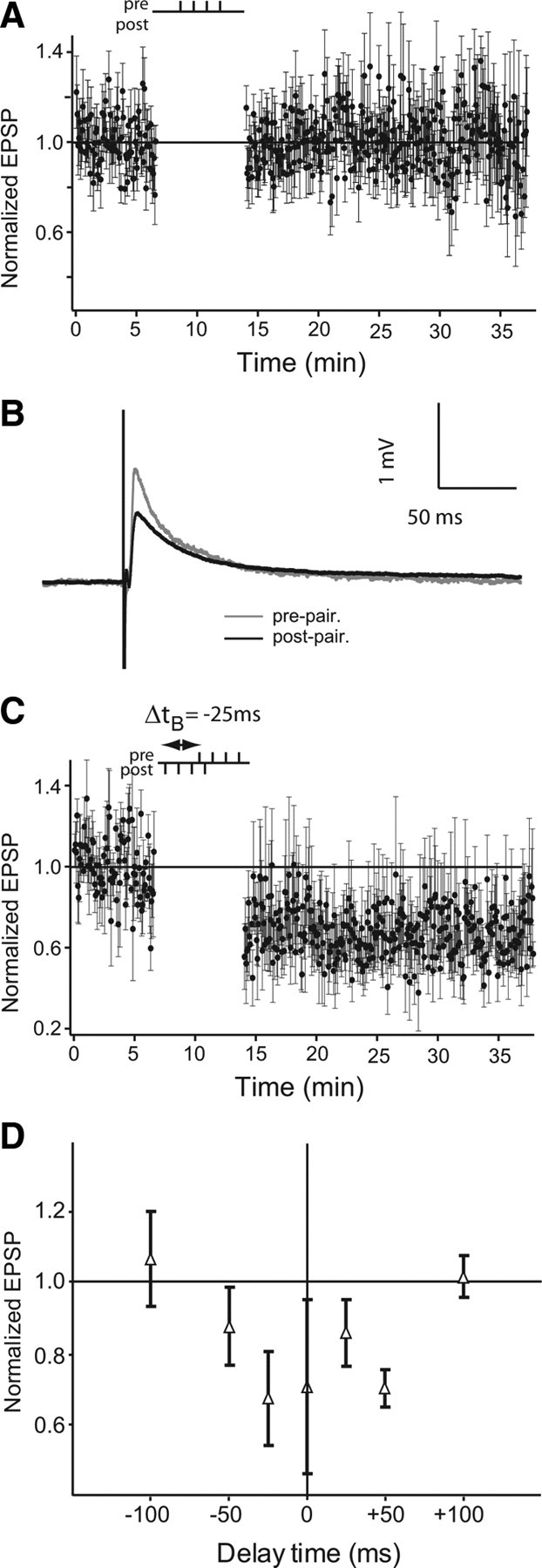
Burst-induced long-term depression in ELL pyramidal cells. A, Normalized EPSP peak amplitude averages during control of only presynaptic burst stimulation (4 pulses at 100 Hz) (4-0; n = 2). B, EPSP averages before burst pairing (4 presynaptic and 4 postsynaptic stimulations; gray) and after pairing (black). C, Normalized EPSP peak amplitude averages during pairing protocol at a delay (Δt B) of −25 ms (n = 5). Δt B is defined as the time delay between first pulse of both presynaptic and postsynaptic stimulations. D, The effect of varying Δt B. All time points are normalized EPSP amplitude averages of five to seven cells, except ±100 ms, which has n = 4. Error bars show the SD in A, C, and D.
Figure 2, B and C, displays a recording of one cell stimulated with a burst-pairing protocol at Δt B = −25 ms. As shown in Figure 2 C, Δt B (delay time between bursts) was defined as the time between the first spike of the presynaptic and postsynaptic bursts. EPSP amplitude is strongly reduced after burst-pairing (Fig. 2 B). Burst-induced depression was long-lasting and remained stable for up to 90 min after the burst-pairing protocol (data not shown). As shown by the time window for burst-induced depression in Figure 2 D, we performed burst-pairing at Δt B ranging from −100 to +100 ms. It should be noted that for Δt B = ±25 ms there is an overlap of 5 ms between presynaptic and postsynaptic stimulus trains, whereas for Δt B = ±50 ms they are separated by 20 ms. Circles show mean normalized EPSP amplitude after burst-pairing and the error bars display their SDs (Fig. 2 D) (32 cells total from −50 to +50 ms, and n = 4 for ±100 ms). Overall, strong burst-induced depression was expressed in DML synapses of pyramidal cells at delay times ranging from −50 to +50 ms (overall average for values ranging from −50 to +50 ms was 0.763 ± 0.163). The lack of plasticity at pairing intervals of ±100 ms clearly demonstrates that neither low-frequency presynaptic burst stimulation nor postsynaptic burst spiking are, by themselves, capable of inducing LTD in the PF feedback pathway. It should be noted that tetanic stimulation (unpaired with postsynaptic spiking) of DML parallel fibers does induce postsynaptic depression in vivo (Bastian, 1998a), whereas low-frequency stimulation of these fibers in vitro induces a presynaptic enhancement (Lewis and Maler, 2004). However, both these forms of synaptic plasticity were relatively short lasting (∼2 and 20 min, respectively).
Burst-induced depression was displayed by both E- and I-type pyramidal cells; its time window was always symmetrical and it was always depressing. Therefore, whereas burst-induced depression does require a precise temporal correlation of patterned spikes, it is a symmetric plasticity rule and therefore different from the classic temporally asymmetric STDP of the mormyrid ELL (Bell et al., 1997).
During pairing protocols, we also tested PPF every tenth stimulation during the prepairing controls and postpairing tests to determine whether burst-induced depression might be associated with changes in presynaptic efficacy (Manabe et al., 1993; Han et al., 2000; Lewis and Maler, 2004) in the PF feedback pathway. During STDP protocols, PPF ratios are 1.959 ± 0.715 and 2.405 ± 1.442 for controls and postpairing tests, respectively, whereas during burst-induced depression (±50 ms), PPF ratios are 1.951 ± 0.823 and 2.213 ± 0.695, respectively (STDP n = 15; burst-induced depression n = 17). These values are not significantly different, suggesting that presynaptic strength is not altered after depression induced by coupled bursts.
Presynaptic and postsynaptic bursts are required for anti-Hebbian long-term depression
To determine what is the minimal requirement for burst-induced LTD, we performed other pairing protocols. Fig. 3 A displays the EPSP traces obtained in one cell stimulated with a presynaptic burst of four spikes coupled to a single postsynaptic action potential (4-1; Δt B = 0 ms). A presynaptic burst paired with a single postsynaptic spike (4-1) and postsynaptic burst paired with a single presynaptic stimulation (1-4) gave similar results in a preliminary analysis. We therefore decided to pool the results obtained for further analysis. Overall, no synaptic changes (normalized EPSP average is 0.999 ± 0.065, n = 15) (Fig. 3 B) were seen with any coupling delay (Δt B of −20, 0, +50 ms) with 4-1 or 1-4 pairing protocols. These results demonstrate that burst–induced LTD requires paired bursts of both presynaptic and postsynaptic neurons. PPF did not change during 4-1 and 1-4 protocols (before pairing: 1.613 ± 0.381; after pairing: 1.5751 ± 0.3039, n = 10) suggesting a lack of presynaptic potentiation (Lewis and Maler, 2004) that might mask a postsynaptic depression. These results demonstrate that single postsynaptic action potential or single presynaptic stimulation are insufficient to induce LTD even if they are coupled to a burst of four spikes. Paired presynaptic and postsynaptic bursts are required to induce synaptic depression.
Figure 3.

Absence of LTD when pairing burst and single stimulations. A, EPSP averages before (gray) and after (dashed black) burst pairing for 4-1 (4 presynaptic stimulations and 1 postsynaptic stimulation) at a delay (Δt B) of 0 ms. B, Normalized EPSP peak amplitude averages during pairing protocol at a delay (Δt B) of 0 ms (n = 15) for 4-1 (4 presynaptic stimulations and 1 postsynaptic stimulation) and 1-4 (1 presynaptic stimulation and 4 postsynaptic stimulations). Δt B is defined as the time between first pulse of both presynaptic and postsynaptic stimulations. Error bars show the SD.
We also tested whether pairing doublets (2-2), the smallest possible bursts, could induce depression (Fig. 4 A). Coupled presynaptic and postsynaptic doublets were not effective at inducing synaptic depression at delays larger than ±10 ms (Fig. 4 C). At these large time delays and Δt B of 0 ms, EPSP amplitude remained unchanged (overall normalized EPSP average 0.992 ± 0.168). However, paired presynaptic and postsynaptic doublets appeared to slightly decrease the EPSP amplitude at very short time delay Δt B (0.905 ± 0.129 at −5 ms and 0.817 ± 0.053 at +5 ms) (Fig. 4 B,C) (n = 5 at any time delay). PPF during the prepairing controls and the postpairing tests remained stable during all the different conditions (larger time delays and 0 ms: prepairing PPF 2.07 ± 0.327 and postpairing 1.922 ± 0.278, n = 15; for time delays of ±5 ms: 2.413 ± 0.183 and postpairing PPF 2.287 ± 0.180, n = 6). The stability of PPF suggests that the presynaptic strength is not changed after burst-induced depression. These results show that the synaptic depression caused by paired doublets is very weak at best and will operate only over a very narrow (±5 ms) delay window.
Figure 4.
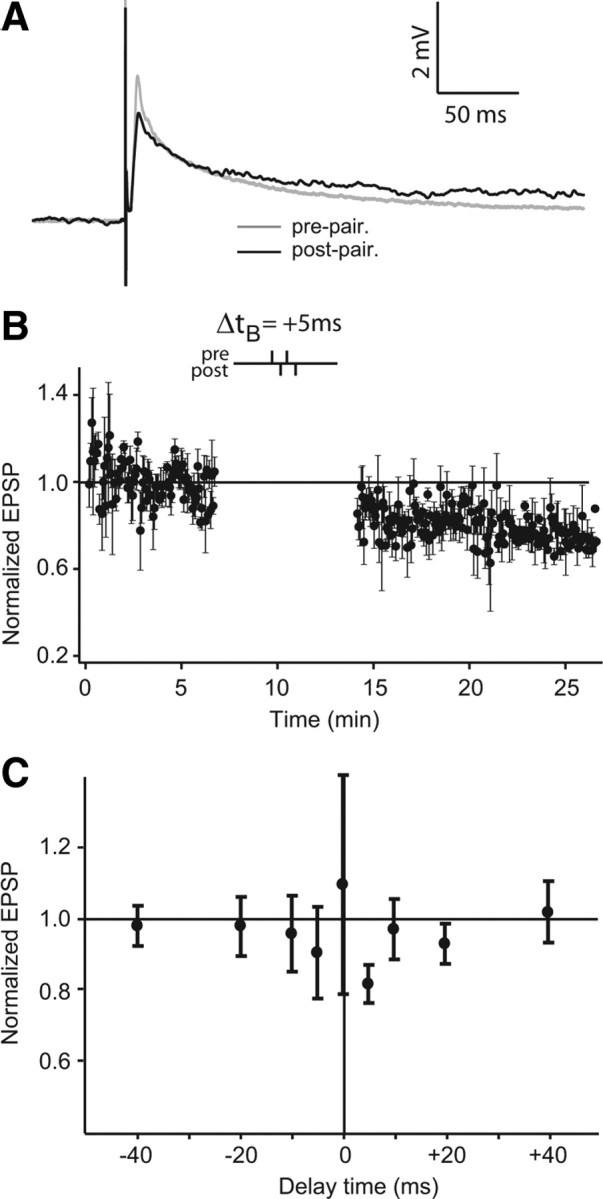
Weak depression induced by short bursts. A, EPSP averages before (gray) and after (black) burst pairing for 2-2 (2 presynaptic and 2 postsynaptic stimulations) at a delay of +5 ms. B, Normalized EPSP peak amplitude averages during pairing protocol at a delay (Δt B) of +5 ms (n = 5). Δt B is defined as the time between first pulse of both presynaptic and postsynaptic stimulations. C, The effect of varying Δt B. All time points are normalized EPSP amplitude averages of five cells. Delay times are as follows: 0, ±5, ±10, ±20, and ±40 ms. Error bars show the SD in B and C.
Burst-induced depression is site specific
In many systems, including the mormyrid ELL, synaptic plasticity has been shown to be specific to the stimulation site (Han et al., 2000; Bi and Poo, 2001). To determine whether burst-induced depression is also site specific we used two stimulating electrodes in the DML separated by at least 150 μm (Fig. 5 A) and alternately stimulated each of them (alternating stimulation every two seconds; every 4 s at each individual site). We then delivered paired stimulation with a 4-4 protocol (Δt B = +25 ms) at the S1 site while the S2 site remained unpaired, getting only presynaptic stimulation. The paired S1 site EPSPs after pairing were 0.783 ± 0.140 of the prepaired value (Fig. 5 B1,C), whereas the S2 site EPSPs (normalized to the mean of the control stimulation) were 1.015 ± 0.114 (n = 3) (Fig. 5 B2,C). In two cases we were able to then apply the paired stimulation protocol at the S2 site. The S2 normalized EPSPs underwent a similar level of depression after pairing (0.820 ± 0.114), whereas the S1 EPSPs were unchanged, i.e., they remained depressed (data not shown). These data clearly demonstrate that burst-induced depression is specific to the site of paired stimulation.
Figure 5.
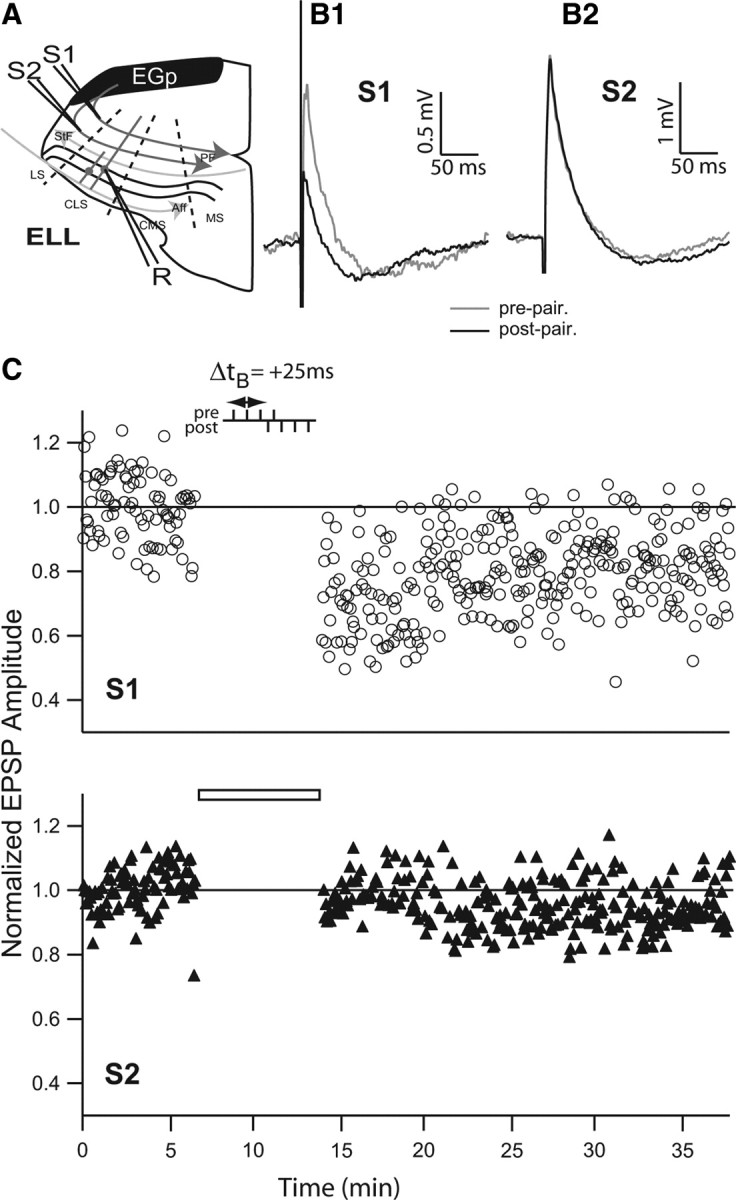
Site specificity of burst-induced LTD. A, Schema showing ELL and recording/stimulation conformation. The stimulation electrodes (S1 and S2) stimulated the PFs at two sites separated by 150 μm. Stimuli were alternately delivered at S1 and S2 sites every 2 s (or 4 s at each site). A sharp electrode (R) recorded somatic EPSPs in the CLS. Aff, Afferent inputs; LS, lateral segment; MS, medial segment; StF, stratum fibrosum. B1, EPSP averages before (gray) and after (black) burst pairing for stimulations at site S1. B2, EPSP averages before (gray) and after (black) burst pairing for stimulations at site S2. C, Normalized EPSP peak amplitude averaged during burst-pairing protocol at stimulation site S1 (burst-pairing stimulation time delay, Δt B = +25 ms) and site S2 (blank bar shows absence of pairing).
Burst stimulation of hyperpolarized cells does not induce potentiation or reverse burst-induced depression in vitro
In several systems (Jörntell and Ekerot, 2002; Fujino and Oertel, 2003; Fino et al., 2005), including ELL of mormyrid fish (Han et al., 2000), STDP and/or LTP/LTD is easily reversible depending on the stimulus pattern. In apteronotid ELL pyramidal cells, in vivo electrical stimulation of parallel fibers paired with stimulation of the cell's RF with naturalistic sensory input induced reversible short-term plasticity (Bastian, 1996, 1998). Specifically, parallel fiber stimulation trains associated with RF input that inhibited pyramidal cells induced an increase in ELL pyramidal cell firing rate response to PF stimulation that lasted ∼30 s. Conversely, PF stimulation trains paired with RF excitation decreased their firing rate response to PF stimulation alone (Bastian, 1998). We reasoned that, during in vitro recordings of ELL pyramidal cells, RF-evoked excitation and inhibition were equivalent to depolarization and hyperpolarization, respectively (Bastian et al., 2004; Oswald et al., 2004, 2007). We therefore hyperpolarized ELL pyramidal cells while stimulating with presynaptic bursts to see whether we could induce potentiation. Hyperpolarization paired with presynaptic bursts did not affect synaptic strength (0.988 ± 0.082, n = 5) (Fig. 6 A).
Figure 6.
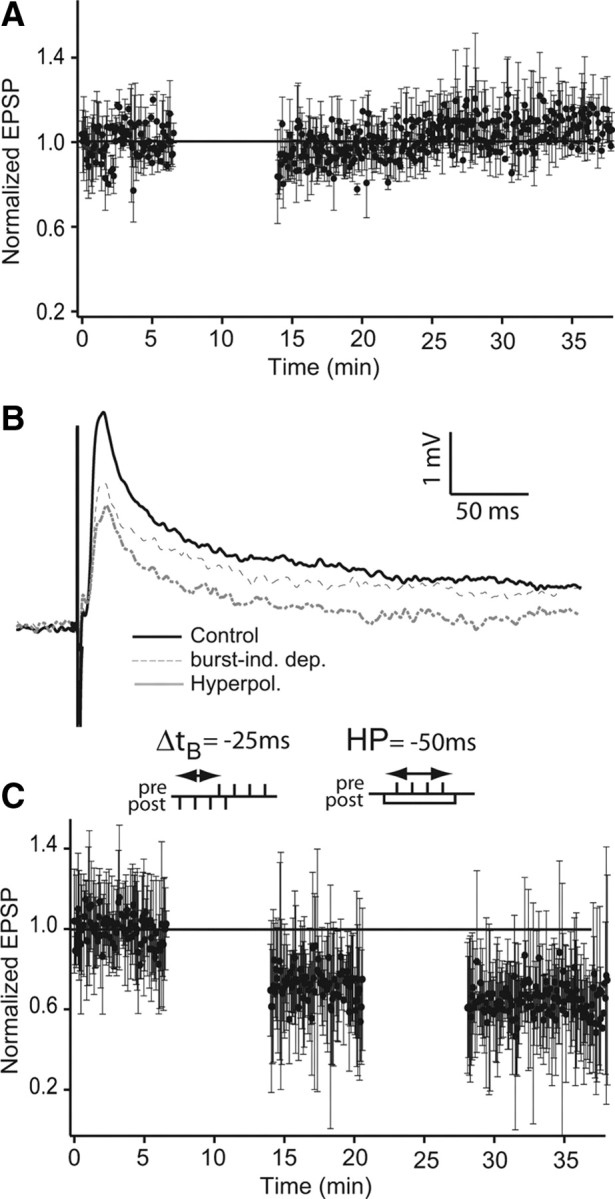
Hyperpolarization does not reverse burst-induced depression. A, Presynaptic burst stimulation paired with postsynaptic hyperpolarization does not induce potentiation. Normalized EPSP peak amplitude following a protocol consisting of presynaptic stimulation (4 spikes, 100 Hz) paired with postsynaptic hyperpolarization (current injection of −0.5 nA for 50 ms; n = 4). B, C, Presynaptic burst stimulation paired with postsynaptic hyperpolarization does not reverse burst-pairing-induced LTD. B, EPSP averages (n = 5) before burst pairing (solid black line), after burst pairing (dashed line), and after subsequent pairing of postsynaptic hyperpolarization and presynaptic bursts (gray line). C, Normalized EPSP peak amplitude during pairing protocol. One hundred presynaptic single-pulse stimulations were delivered every 4 s and control EPSP amplitudes were measured. Then, 110 burst-pairing stimulations (not shown: 4 postsynaptic spikes paired with 4 presynaptic spikes; 100 Hz and at a delay, Δt B, of −25 ms; see left inset) were given followed by 100 presynaptic stimulations (every 4 s). Finally, a current of −0.5 nA was injected in the postsynaptic cell to prevent action potential firing (50 ms) while stimulating with a presynaptic burst (not shown: 4 spikes at 100 Hz, 10 ms after hyperpolarization starts; repeated 110 times; see right inset) followed by 100 single-pulse presynaptic stimulations (every 4 s). In C, error bars show the SD.
Because burst-induced LTD is symmetrical, we could not use a reversed stimulus pairing (Han et al., 2000) to reverse the synaptic depression. We therefore attempted reversing LTD by pairing hyperpolarization and presynaptic burst stimulation (Fig. 6 B,C). Initially, burst-induced LTD was obtained as previously shown (time delay Δt B = −25 ms), and showed a strong reduction in EPSP amplitude (Fig. 6 B, dashed trace, C) (0.687 ± 0.048). Afterward, pairing hyperpolarization (50 ms pulse current injection of −0.5 nA: this was to prevent spiking) and presynaptic bursts (four stimuli at 100 Hz) did not induce any recovery of the EPSP amplitude (Fig. 6 B, gray trace, C) (0.641 ± 0.043). These results demonstrate that potentiation or de-depression of DML synaptic input to ELL pyramidal cells cannot be induced by pairing hyperpolarization and presynaptic bursts. However, we cannot exclude the possibility that, given the lack of knowledge of the natural firing patterns of the cerebellar granule cells providing ELL feedback, we have not yet found the proper protocol to induce potentiation or de-depression at these synapses.
Molecular basis for burst-induced depression
In the subsequent three sections, we explore the cellular requirements for the burst-induced depression described above. Specifically we focus on (1) the previously identified requirement for increases in postsynaptic Ca2+ levels for depression of PF synaptic input to pyramidal cells (Bastian, 1998b), (2) the role of the strongly expressed NR1/NR2B NMDA receptor (Harvey-Girard et al., 2007), and (3) the role of the CaMKIIβ previously shown to be present in ELL pyramidal cells (Maler and Hincke, 1999).
Postsynaptic calcium influx is required for burst-induced depression
Postsynaptic calcium influx is required for induction of long-term potentiation and long-term depression induced by stimulation trains or spike timing-dependent plasticity (Bliss and Collingridge, 1993; Yang et al., 1999; Han et al., 2000; Lisman et al., 2002; Collingridge et al., 2004; Dan and Poo, 2006). In apteronotid fish, postsynaptic calcium influx at PF synapses of ELL pyramidal cells is necessary for in vivo induction short-term synaptic depression (Bastian, 1998b). We therefore used intracellular application of BAPTA, a Ca2+ chelating agent, to evaluate the possible role of postsynaptic Ca2+ in burst-induced depression.
After 20 min of BAPTA application in the recording electrode, the peak amplitude of the EPSPs remained unchanged (0.993 ± 0.131 mV; n = 4) (Fig. 7 A). During a 4-4 protocol (delay of −25 ms or +25 ms; n = 3 for each condition), BAPTA application completely blocked the induction of depression (Fig. 7 B2,C), whereas controls show strong depression (Fig. 7 B1). The normalized EPSP amplitude average with BAPTA was 1.003 ± 0.040 after pre–post burst pairing, whereas the control normalized EPSP amplitude was 0.746 ± 0.169 (ANOVA, Tukey HSD test, p < 0.01). These results demonstrate that elevation of postsynaptic Ca2+ in pyramidal cells during paired bursts is required for burst-induced LTD. These results also show that postsynaptic Ca2+ elevation is required whatever the sign of the pairing delay: both presynaptic bursts preceding or following postsynaptic bursts require postsynaptic Ca2+ for the induction of LTD by paired bursts.
Figure 7.

Application of postsynaptic BAPTA prevents burst-pairing-induced LTD. A, Effect of BAPTA application (50 mm) in the recording pipette. EPSP averages before (gray) and after 20 min (black). BAPTA does not affect the baseline EPSP. B1, EPSP averages before (gray) and after (black) burst pairing for control (no BAPTA, n = 6). B2, EPSP averages before (gray) and after (black) burst pairing with BAPTA application (n = 6). Note that BAPTA blocks induction of LTD by burst pairing. C, Normalized EPSP peak amplitude averaged during burst-pairing protocol with BAPTA at pairing stimulation time delay, Δt B = −25 ms. Calibration bars in A are used also in B1 and B2.
NR2B-containing NMDARs are required for induction of depression by paired bursts
Postsynaptic calcium influx through NMDARs mediates LTP and LTD induction during unpaired presynaptic stimulation (Pláteník et al., 2000; Shouval et al., 2002; Castellani et al., 2005) and during STDP in mammals (Caporale and Dan, 2008) and mormyrid fish (Han et al., 2000). NMDARs are tetramers made of two mandatory NR1 subunits and two NR2 subunits (A–D) that determine many electrophysiological properties and signaling protein interactions (Dingledine et al., 1999; Yamakura and Shimoji, 1999). Recent data using pharmacological or genetic approaches suggest that the bidirectionality of synaptic plasticity might be due to subpopulations of NMDARs: NR2A-containing NMDARs have been hypothesized to be required for LTP induction, and NR2B-containing NMDARs for the induction of LTD (Liu et al., 2004; Massey et al., 2004; Kim et al., 2005; Weitlauf et al., 2005; Li et al., 2007). However, because of the low specificity of the NR2A antagonist, these hypotheses remain controversial (Berberich et al., 2005; Weitlauf et al., 2005).
In apteronotid ELL, we have shown that NMDA receptors and the NR1 subunit are strongly expressed in DML and associated with the spines of pyramidal cell apical dendrites (Maler and Monaghan, 1991; Berman et al., 2001). As might therefore be expected, a large component of the feedback-evoked EPSPs of ELL pyramidal cells is blocked by the general NMDAR antagonists CPP and APV (Berman et al., 1997; Berman and Maler, 1998a). Furthermore, NR2B subunit mRNA transcripts are highly expressed in superficial and intermediate pyramidal cells and NR2B-containing NMDARs are predominant in PF feedback input EPSPs (Harvey-Girard et al., 2007). Finally, apteronotid NR2B contains the ifenprodil binding site (Harvey-Girard and Dunn, 2003) and ifenprodil, a specific antagonist against NR2B-containing NMDARs, blocks apteronotid NR2B-containing NMDARs with similar affinity as in mammals (Harvey-Girard and Dunn, 2003).
We therefore used ifenprodil to determine whether NR2B containing NMDARs were required for burst-induced depression. Twenty-minute bath application of ifenprodil strongly reduced both early and late phases of the EPSPs evoked by parallel fiber stimulation, as expected from our previous experiments (Berman and Maler, 1998a; Harvey-Girard et al., 2007) (Fig. 8 A). Ifenprodil completely blocked the induction of the long-term depression (n = 11, 4-4 pairing protocol, ±25 ms) (Fig. 8 B2), whereas control experiments showed strong depression (Fig. 8 B1). Figure 8 C displays a stimulation protocol at pre–post delay of −25 ms during ifenprodil application showing that ifenprodil completely blocked burst-induced LTD. Depression induced by a 4-4 protocol with pre–post delay of +25 ms was also blocked by ifenprodil (data not shown); we therefore pooled both sets of data. Normalized change of EPSP amplitude average during ifenprodil application was 1.024 ± 0.068, whereas normalized EPSP amplitude average of controls was 0.774 ± 0.031 (Fig. 8 C) (ANOVA, Tukey HSD test, p < 0.01). These data showed that NR2B-containing NMDARs are necessary for induction of long-term depression in the PF feedback pathway of ELL pyramidal cells. In light of the BAPTA results, we propose that NR2B-containing NMDARs act by increasing Ca2+ influx, which then acts as the trigger for burst-induced LTD.
Figure 8.

Application of ifenprodil blocks burst-induced depression. A, EPSP averages before (gray) and after (black) 20 min of ifenprodil bath application. Note reduction of both early and late phases of the baseline EPSP consistent with previous reports on the preponderance of NR1/NR2B receptors associated with the PF feedback pathway. B1, EPSP averages before (gray) and after (black) burst pairing for control. B2, EPSP averages before (gray) and after (black) burst pairing during ifenprodil application. Note that ifenprodil blocks induction of LTD by burst pairing. C, Normalized EPSP peak amplitude averaged during burst-pairing protocol with ifenprodil at pairing time delay, Δt B = −25 ms. Error bars show the SD in C. Calibration bars in A are used also in B1 and B2.
Postsynaptic CaMKIIβ is required for burst-induced depression
In mammals, depending on its dynamics, calcium influx through NMDARs can induce activation of Ca2+/calmodulin-dependent protein kinase II (CaMKII). CaMKII is considered as a molecular switch that can lead to LTP (Graupner and Brunel, 2007) by controlling signaling processes such as AMPA receptor surface expression (Lisman and Zhabotinsky, 2001; Lisman et al., 2002). CaMKII binds directly to NMDAR NR2B subunit at synapses enhancing its ability to remain activated during LTP (Strack and Colbran, 1998; Leonard et al., 1999; Bayer et al., 2001).
In the apteronotid ELL, two isoforms of CaMKII are expressed: CaMKIIα and CaMKIIβ. CaMKIIα is restricted to presynaptic terminals of the direct feedback pathway to the proximal apical dendrites of pyramidal cells (ventral molecular layer, VML), whereas CaMKIIβ is enriched in soma and apical dendrites (both proximal and distal, VML and DML) of pyramidal cells (Maler and Hincke, 1999). Posttetanic potentiation (PTP) in the direct feedback pathway is blocked by extracellular application of KN62, a CaMKII antagonist, whereas intracellular application of KN62 or a CaMKII inhibitory peptide had no effect, showing the effect of presynaptic CaMKIIα of the direct feedback pathway (Wang and Maler, 1998). However, PTP (presynaptic) of the PF feedback pathway in the DML was not affected by extracellular application of KN62 (Wang and Maler, 1998) consistent with the lack of CaMKIIα in these fibers.
We therefore investigated whether NMDAR-induced Ca2+ elevation might induce long-term depression by activating postsynaptic CaMKIIβ in pyramidal cells. Twenty-minute bath application of KN-62 (3.3 μm) did not change EPSP amplitude and shape (Fig. 9 A). In presence of KN-62, a 4-4 burst pairing stimulation (n = 5, Δt B = −25 ms) was not able to induce depression for 24 min after pairing (ratio of prestimulation to poststimulation EPSP amplitude = 1.022 ± 0.253) (Fig. 9 B,E). Similar results were obtained at +25 ms time delay (n = 3; 1.014 ± 0.182; data not shown). Similar results were obtained at both positive and negative pairing delays with another CaMKII antagonist, KN-93 (3.3 μm, n = 5; ratio = 0.989 ± 0.192). Burst-induced LTD was, however, still present after application of the negative control isomer, KN-92 (3.3 μm) (Fig. 9 D,F) (n = 3). On average, control (depressed) normalized EPSP amplitudes were 0.774 ± 0.031, whereas KN-62 and KN-93 (data combined) normalized EPSPs were 1.070 ± 0.088 (Fig. 9 G) (n = 13; data were significantly different, ANOVA, Tukey HSD test, p < 0.01) and KN-92 normalized amplitudes were 0.833 ± 0.010 (Fig. 9 G) (KN-92 and CaMKII antagonist results were significantly different, ANOVA, Tukey HSD test, p < 0.05). These results demonstrate that CaMKIIβ is an essential intermediate between activation of NMDAR (NR2B subunit)-dependent Ca2+ influx and induction of depression by paired bursts.
Figure 9.
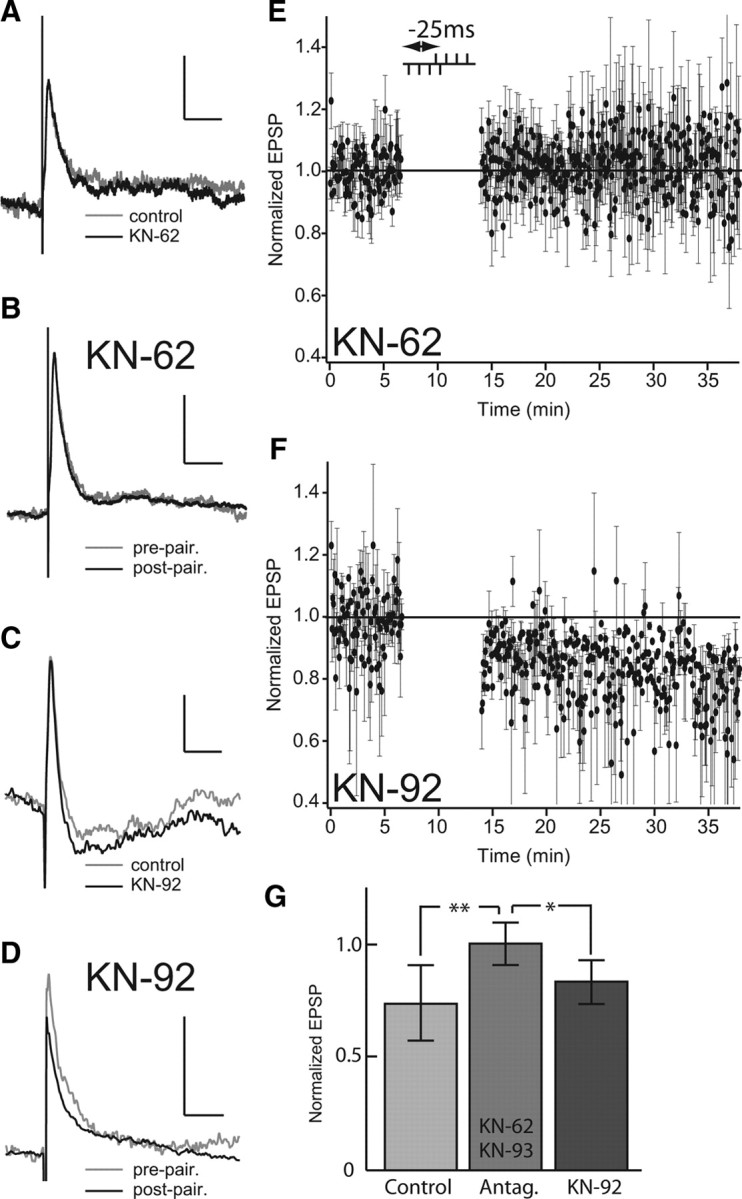
Application of CaMKII antagonists block burst-induced depression. A, Effect of KN-62 on EPSP showing EPSP averages before (gray) and after (black) 20 min bath application. CaMKII antagonists do not affect the baseline EPSP. B, Effect of KN-62 on the mean EPSP during burst-pairing protocol before (gray) and after (black) burst pairing. Note that KN-62 blocks induction of LTD by burst pairing. C, Effect of KN-92 on EPSP showing EPSP averages before (gray) and after (black) 20 min bath application. Note that KN-92, an inactive enantiomer of KN-93, does not affect the PF-evoked EPSP. D, Effect of KN-92 on EPSP averages during burst-pairing protocol before (gray) and after (black) burst pairing. Note that KN-92 permits induction of LTD by burst pairing. E, Normalized EPSP peak amplitude averaged during burst-pairing protocol (Δt B = −25 ms) with KN-62 in the bath. F, Normalized EPSP peak amplitude averaged during burst-pairing protocol (Δt B = −25 ms) with KN-92 in the bath. G, Normalized EPSP peak amplitude averaged after burst-induced depression for control (n = 14; light gray), for the two CaMKII antagonists used (KN-62 and KN-93; n = 13; medium gray), and for KN-92 application (n = 3; dark gray). Error bars show the SD in E–G. *p < 0.05; **p < 0.01.
Paired bursts induce LTD of both the early (primarily AMPA receptor-mediated) and late (primarily NMDA receptor-mediated) phases of the PF-evoked EPSP (Fig. 2 A) (Berman et al., 1997; Harvey-Girard et al., 2007). However, our data do not allow us to determine whether the depression of the NMDA receptor component is direct or due to a primary reduction of the AMPA receptor-mediated EPSP and a secondary effect due to the voltage sensitivity of the NMDA receptor component. In either case the reduction of the NMDA receptor component of the EPSP suggests that burst-induced depression is a self-limiting process since it will lead to decreasing Ca2+ influx and therefore reduced depression (Bastian et al., 2004).
An in vivo synaptic depression stimulus protocol does not induce long-term depression in vitro
Bastian (1995) initially demonstrated that the electrosensory input induced by tail bending (A. leptorhynchus) evoked a strong response in electroreceptors but failed to modulate the discharge of ELL pyramidal cells. Bastian therefore concluded that there existed a mechanism (negative image) within the ELL that canceled this spatially redundant (global) low-frequency input. The cancellation was always present upon first presentation of a tail-bending stimulus, and the negative image was therefore a long-lasting phenomenon [see also Bastian et al. (2004) for a similar result]. Bastian and coworkers subsequently performed a lengthy series of experiments in vivo to investigate how such long-lasting cancellation might be induced in ELL pyramidal cells. Bastian was able to demonstrate synaptic depression of the pyramidal cell indirect feedback inputs (DML PFs) that was induced by postsynaptic Ca2+ influx via NMDA receptors and required PF firing (not bursting) correlated with pyramidal cell activity (not bursting). The induced depression was hypothesized to part of the cancellation mechanism. However, the induced in vivo synaptic depression decayed in ∼30 s (Bastian, 1996a, 1998a). As mentioned above, subsequent experiments revealed the specific role of pyramidal cell spike bursting in encoding low-frequency signals (Oswald et al., 2004); burst stimulation had not been used by Bastian. One legitimate question is therefore to ask whether the brief induced depression seen in vivo by Bastian (no burst stimulation) can be elicited in an in vitro preparation and whether it is long-lasting as is the burst-induced LTD we have demonstrated in vitro. This is a control experiment in the sense that (1) if we were to induce LTD with the Bastian protocol, it would imply that bursting was not essential for anti-Hebbian depression; and (2) some (unknown) difference between in vivo and in vitro conditions was responsible for the short- versus long-lasting depression, respectively. If this control experiment produced a negative result—the Bastian stimulation protocol was not able to induce LTD—it would strengthen our contention that burst stimulation pairing was part of the cellular substrate of the cancellation (negative image) observed in vivo (see below).
We therefore reproduced Bastian's in vivo experiment in our slice preparation (Fig. 10) (n = 5). First, a single PF test pulse was delivered at 1 Hz while EPSPs were recorded (repeated 30 times) (Fig. 10 A1). Second, while continuing to deliver the test pulse, a tetanic train of seven PF stimuli (15 ms ISI) was delivered 250 ms after each test pulse (repeated 120 times) (Fig. 10 A2,A3). Third, stimuli were similar as before (test pulse plus tetanic train) except that during the tetanic train negative current was injected for 100 ms to hyperpolarize the cell (repeated 60 times) (Fig. 10 A4) (black bar displays current injection). Finally, the stimulus protocol without hyperpolarization was repeated (60 times) (Fig. 10 A5) (similar to Fig. 10 A2,A3). Figure 10 C displays average EPSPs of the test stimulations taken at different stages of the protocol (Fig. 10 D), whereas Fig. 10 E shows the normalized instantaneous firing rate during the tetanic train: the colors in panel C match the trace colors in panels A1–A5 and the bar colors in panels D and E.
Figure 10.
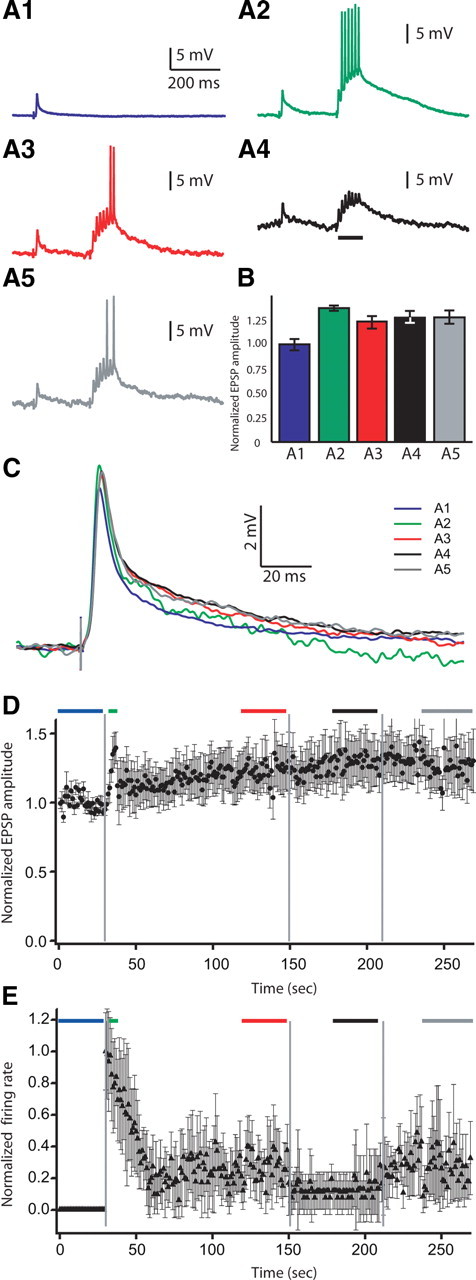
An in vivo stimulus protocol with tetanic stimulation but no burst pairing (Bastian, 1998a) does not induce LTD in vitro. A, Examples of recorded traces during stimulus protocol. EPSP elicited by a single test stimulation (A1). EPSP elicited by test stimulation followed by presynaptic tetanic train (7 spikes; 15 ms ISI) at the beginning (A2) and at the end (A3) of the tetanic train application. A4, EPSP elicited by a single test stimulation followed by presynaptic tetanic train coupled to negative current injection to hyperpolarize the cell. A5, EPSP elicited by test stimulation followed by presynaptic tetanic train. B, Normalized EPSP peak amplitude averaged during pairing protocol at the different protocol stages (n = 5). C, EPSP averages during pairing protocol at the different protocol stages. D, Normalized EPSP peak amplitude averaged during pairing protocol at the different protocol stages (n = 5). Note that this protocol induces modest enhancement of the evoked EPSP and not depression. E, Normalized firing rate of recorded cells averaged during pairing protocol at the different stages of the protocol (n = 5). Colors of traces and bars match everywhere. Blue, EPSP elicited by control test stimulation (A1, B–E); green, at the beginning of tetanic train stimulation application (A2, B–E); red, at the end of tetanic train stimulation application (A3, B–E); black: during tetanic train coupled to negative current injection (A4, B–E); gray, tetanic stimulation application done at the end of the protocol (A5, B–E). Error bars show the SD in B, D, and E.
Initially, when we started applying the tetanic train a fast increase in EPSP amplitude was seen (Fig. 10 D, green) (1.373 ± 0.023 After this initial peak, EPSP amplitude quickly decreased almost to baseline and slowly enhanced until it reached a slightly enhanced plateau (Fig. 10 D, red) (1.233 ± 0.065). This EPSP amplitude enhancement is most likely the presynaptic potentiation of PF inputs that happens at this stimulus frequency range (1 Hz and more) (Bastian et al., 2004; Lewis and Maler, 2004). Hyperpolarizing the cell had no effect on the EPSP amplitude (Fig. 10 D, black) (1.272 ± 0.058). Stopping current injection had no effect as well (Fig. 10 D, gray) (1.285 ± 0.070). Bastian's protocol (in vivo) induces only short-term depression (decays in 30 s)(Bastian, 1996a, 1998a). Applying the in vivo protocol used by Bastian to an in vitro slice preparation does not induce LTD. We hypothesize that the failure of the Bastian protocol to induce LTD either in vivo or in vitro is due to the lack of reliable pairing of presynaptic and postsynaptic bursts. We cannot explain the lack of even short-term synaptic depression during in vitro stimulation with the Bastian protocol, although it may in part be related to a greater in vitro induction of PF presynaptic potentiation compared with short-term postsynaptic depression.
We also recorded the evoked firing rate during tetanic stimulations. At the beginning, all neurons were complete silent (Fig. 10 E, blue bar). During the initial tetanic train applications, the neuron firing rate increased to a maximum. To better visualize the time dependence of tetanic stimulation-evoked firing rate, we first normalized the firing rate responses, for each cell, by dividing the firing rate response to each tetanus by the firing rate response to the initial tetanus; this results in a unit-free ratio that is then averaged over all the cells. Inspection of the normalized firing rates (Fig. 10 D) revealed that there was an initial peak response for the first four presentations (green bar; this was observed for each cell; data not shown) followed by a decline to a steady state. We therefore took our firing rate estimates over the initial four presentations and during the steady states after the various perturbations.
The normalized average firing rate of the first four presentations was 0.973 ± 0.270 (Fig. 10 E, green) (the normalized firing rate of response to the initial train was set to 1 for each cell, which represented ∼6 spikes during the initial train). The initial peak firing rate decayed and reached a plateau within 30 s (0.247 ± 0.192 from 121 to 150 s) (Fig. 10 E, red). Negative current injection further reduced firing rate (0.121 ± 0.127; 181–210 s) (Fig. 10 E, black). When the injected current was removed, the firing rate bounced back to the plateau value it had reached previously (0.270 ± 0.210; 241–270 s) (Fig. 10 E, gray) and therefore was far lower than the spiking response to the initial tetani. These results appear to be contradictory: tetanic stimulation of PFs induces an increase of their evoked EPSP in pyramidal cells (likely presynaptic) but also reduces the pyramidal cell firing rate in response to the same PF stimulation. We hypothesize that the firing rate reduction is due to stimulation of a local ELL interneuron—the vml cell (Maler, 1979). The vml interneuron is GABAergic, synapses mainly on the proximal apical dendrite of pyramidal cells (Fig. 11 A) (Maler and Mugnaini, 1994) and uses GABA-A receptors (Berman and Maler, 1998b,c). Mehaffey et al. (2005) have shown that mimicking vml cell activation (via activation of GABA-A receptors on pyramidal cell proximal apical dendrites) reduces the pyramidal cell firing rate response (gain) to positive current injection. This effect was in turn demonstrated to be due to vml cell-induced shunting of backpropagating action potentials. Further experimental work will be required to determine the interaction between paired burst-induced LTD and these complicated network interactions.
Figure 11.
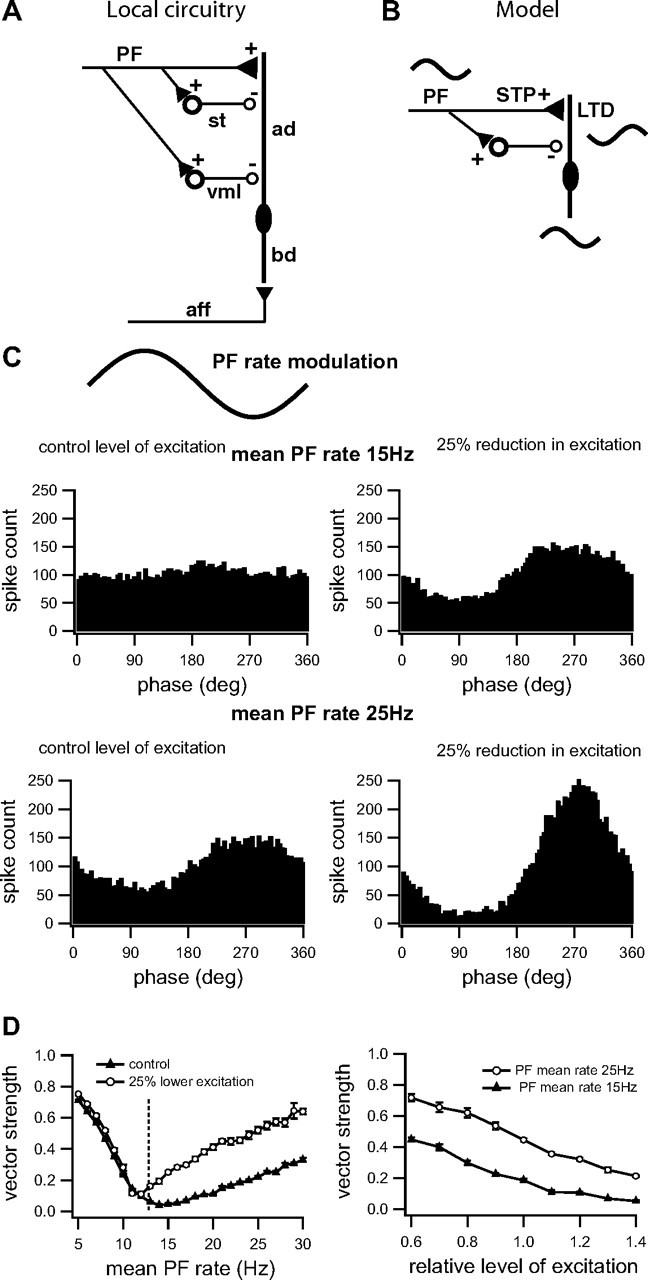
Interactions between postsynaptic depression, short-term plasticity, and disynaptic inhibition determine the strength of the negative image. A, Schematic illustration of ELL intrinsic feedback circuitry showing a superficial pyramidal cell with its basilar dendrite (bd) receiving electrosensory afferent inputs (aff). The indirect feedback pathway coming from EGp via PFs projects to apical dendrites of pyramidal cells (ad) and two types of GABAergic inhibitory interneurons: stellate cells (st) and vml cells (vml). Stellate cells project to distal apical dendrites, whereas vml cells project mainly to proximal apical dendrites of pyramidal cells. B, Schematic illustration of the simplified model of feedback to an ELL pyramidal cell. The pyramidal cell receives low-frequency sinusoidal input via its basilar dendrite. This input is also fed back to the cell's apical dendrites via PFs. The model simplifies the feedback inhibition by incorporating only one inhibitory interneuron and ignoring the distinction between the stellate and vml cells. The PF to pyramidal cell synapses show short-term plasticity (STP); when the strength of the PF to pyramidal cell synapses is reduced by LTD, the STP causes the inhibitory component to dominate and the response of the pyramidal cell to feedback input becomes inverted and therefore forms a negative image that can cancel the sensory input. C, Effect of depressing postsynaptic excitation on model neuron responses to PF frequency modulation (1 Hz; inset; simulated 1 Hz PF firing rate modulation) at two different mean rates, 15 Hz (top) and 25 Hz (bottom). It is assumed here that these neurons are receiving a direct electrosensory input (via P-units) in phase with the PF rate modulation; see supplemental Figures 1 and 2, available at www.jneurosci.org as supplemental material, for an analysis of the effects of conduction and synaptic delays in the feedback pathway. Responses are quantified with phase histograms (spike times during the modulation cycle). Left panels show responses for a control level of excitation (g exc = 400 μS) (Lewis et al., 2007) and right panels show changes resulting from the decreased excitation (g exc = 300 μS) that might be expected following burst-induced depression. Note that local inhibitory circuitry associated with the PF feedback inverts the phase of the PF firing rate modulation so that it can now act as a negative image for the electrosensory input. D, Effect of mean PF rate and relative level of excitation on the vector strength of the model neuron responses. We illustrate averages over five simulated trials (with SEM indicated). Left panel shows vector strength for two different levels of excitatory conductance (as in B; g exc = 400 μS, square symbols and g exc = 300 μS, circle symbols); dotted line shows the PF rate at which there is a shift to a negative image (pyramidal cell firing rate response 180° out of phase with the electrosensory input). Note that vector strength (amplitude of negative image) increases with mean PF firing rate and that it is always greater for lower levels of postsynaptic conductance (putatively due to burst-induced depression). Right panel shows vector strength as a function of relative level of excitatory conductance (a value of 1 corresponds to g exc = 400 μS) for two different mean PF rates, 15 Hz (square symbols) and 25 Hz (round symbols). Note again that vector strength (amplitude of negative image) increases with decreasing strength of PF-evoked excitation (putatively due to burst-induced depression) and that it is always greater for higher mean PF firing rates. Thus, the burst-induced depression of PF input to pyramidal cells can produce a strong negative image; furthermore, the strength of this cancellation signal can be sensitively regulated by the mean PF firing rate.
The work of Berman and Maler (1998c) has demonstrated that the PF stimulation evoked disynaptic IPSPs that entirely overlapped the evoked monosynaptic EPSPs and so could not be directly seen in isolation (Fig. 1 B). The paired burst-induced LTD that we have described above might therefore be interpreted as due to one of the following: (1) a direct decrease of the PF-evoked EPSP amplitude, (2) an increase of the disynaptic GABA-A IPSP leading to a reduction of the overlapping EPSP, or (3) a combination of these effects. We are not able to experimentally investigate possible long-term potentiation of the PF-evoked disynaptic inhibition (because it overlaps the EPSP). However, we note that the NMDA receptors required for LTD induction are located on the spines of the pyramidal cell's apical dendrites (Berman et al., 2001); in contrast the GABAergic synapses are located on the dendritic shafts (Maler et al., 1981; Maler and Mugnaini, 1994). The most parsimonious hypothesis is therefore that burst-pairing-induced LTD is due to the NMDA receptor (NR2B subunit)-induced Ca2+ influx acting locally within the dendritic spine to cause a decrease in the evoked EPSP. We cannot and do not wish to exclude the possibility that LTD might also caused in part by an enhancement of disynaptic IPSPs as suggested by Bastian's in vivo experiments (this was a short-term enhancement and not fully characterized) (Bastian, 1998a). Since we could not directly investigate the latter possibilities [(2) and (3)] we decided to use modeling to determine whether the simplest hypothesis—burst-pairing-induced LTD is due to the reduction of PF-evoked EPSP amplitude—is capable of accounting for the in vivo cancellation of spatially redundant low-frequency input. As discussed below our modeling results reveal that the simplest hypothesis (LTD of EPSPs) is sufficient to account for the in vivo results although they cannot, of course, exclude a role for potentiation of inhibition. We note that this conclusion is identical to that reached for the cancellation mechanism described in the mormyrid ELL (Han et al., 2000). Anti-Hebbian depression of PF synaptic input (demonstrated in vitro) is sufficient to account for the cancellation seen in vivo (Roberts and Bell, 2000). However, a theoretical analysis also demonstrated that the potentiation of associated inhibitory input (not experimentally shown) made the cancellation more robust—cancellation was induced more rapidly and was able to work over a wider range of signal intensities (Roberts, 2000).
Interaction of postsynaptic depression, short-term plasticity, and disynaptic inhibition
Cancellation of a low-frequency global input to an E cell requires a reduction of PF excitatory input (and therefore a shift to net disynaptic inhibition) during the positive phase of the input signal (the opposite is true for an I cell but, for simplicity, we consider only the E cell here). We hypothesize that paired burst-induced LTD is responsible for this effect. Cancellation of the negative phase of the input signal would appear to require potentiation of PF feedback excitation to counteract the signal-evoked decrease in the pyramidal cell discharge rate (Bastian, 1996a,b; Bastian et al., 2004); this suggests that pairing PF stimulation with a hyperpolarization of a (nonspiking) pyramidal cell should induce potentiation of the PF-evoked EPSP. In vivo experiments have shown that, whereas such pairing can relieve the depression caused by tetanic stimulation of PFs, it does not induce potentiation above the baseline EPSP (Bastian, 1998a). Application of intracellular BAPTA blocks the initial depression as well as the partial recovery due to hyperpolarization (Bastian, 1998b) and does not therefore imply an independent potentiation mechanism. Low-frequency stimulation of PFs induces a presynaptically mediated enhancement (Lewis and Maler, 2004); however, this potentiation is relatively short lasting (∼20 min). Further this enhancement does not require PF bursting and so cannot be a direct inverse of the LTD we have described. We have shown (see above, Burst stimulation of hyperpolarized cells) that pairing postsynaptic hyperpolarization with burst stimulation does not induce potentiation. It would therefore seem that the indirect feedback pathway cannot completely cancel the entire low-frequency global AM.
The depression of PF input to pyramidal cells does, however, not occur in isolation: PF input to pyramidal cells exhibit short-term plasticity (primarily facilitation) (Lewis and Maler, 2002a, 2004) and also evoked disynaptic inhibition of pyramidal cells via local interneurons (Bastian, 1993; Maler and Mugnaini, 1994; Berman and Maler, 1998a; Lewis and Maler, 2002b). The effect of burst-induced LTD of PF-evoked EPSPs is thus filtered by the local short-term dynamics of PF afferents and inhibition; we therefore explored whether LTD alone might account for the cancellation of both phases of global low-frequency signals (Lewis et al., 2007).
In this section, we adopt a previous modeling approach (Lewis et al., 2007) to explore the role of burst-induced LTD in negative image generation. This previous study has shown how negative image generation can occur through a shift in balance between excitation and inhibition. In this case, the dynamics were attributed to presynaptic mechanisms (Lewis et al., 2007). Here, we show that similar shifts can occur through changes in excitatory postsynaptic conductance of a magnitude similar to that observed during burst-induced LTD. We use a simplified version of the disynaptic inhibitory circuitry (Fig. 11 A,B) and do not take into account more complex effects related to pyramidal cell dynamics (see above) (Mehaffey et al., 2005).
The model previously developed was based on experimental data from the CMS. We therefore first confirmed that the existing model of short-term plasticity of PF input was also appropriate for CLS. Random stimulation of PF input to pyramidal cells was performed as previously described (Lewis and Maler, 2004); the previous model was able to successfully predict the resulting sequence of EPSPs (n = 2 cells; fitting errors 10 and 8%, with Fo = 0.17 and 0.18, respectively; data not shown). The model of Lewis and Maler (2004) therefore carries over to CLS. This is an expected result given that the model involves only PFs and local interneurons; the same PF population innervates all ELL maps (Sas and Maler, 1987) and the same interneurons are present in the molecular layer of these maps. We also confirmed that short-term plasticity did not change after induction of LTD (n = 2 cells; fitting errors 6 and 11%, with Fo = 0.19 and 0.18, respectively; data not shown); this is consistent with the presynaptic expression of short-term plasticity and the postsynaptic expression of LTD (see above). With these preliminary results in hand, we could now use the model of Lewis et al. (2007) to examine possible interactions of PF short-term plasticity and local inhibition induction with postsynaptic LTD.
The PF inputs to the model neuron are driven by a sinusoidal rate modulation (±5 Hz) with mean rates of 15 Hz and 25 Hz (schematically shown, Fig. 8 B, top panel); this is meant to mimic the response of EGp granule cells to the firing rate modulation of deep pyramidal cells evoked by natural sinusoidal stimuli (Bastian et al., 2004). In the control condition (postsynaptic excitatory conductance, G exc = 400 μS), two different postsynaptic firing responses are shown in the phase histograms depending on the mean PF rate (Fig. 8 B, left panels). For PF mean rate of 15 Hz, very little modulation in the neural response occurs, whereas for a PF mean rate of 25 Hz, a negative image (i.e., out-of-phase response) is observed. This dependence on mean PF rate is a result of the presynaptic effects described previously (Lewis et al., 2007). In addition to these effects however, even relatively small decreases in G exc (mimicking the LTD described in previous sections) can also exert an influence on the neural response, either creating a negative image or strengthening it, depending on the initial state (Fig. 8 B, right panels). This is not due to a specific choice of model parameters. Figure 8 C, left, shows the vector strength at various PF mean firing rates (the dotted vertical line indicates the approximate PF rate at which the shift to a negative image occurs) for the control and depressed condition. Although a postsynaptic depression has little effect for low PF rates when inhibition is minimal, for higher PF rates, it always enhances the negative image (increased vector strength). Similarly, increases in vector strength (negative image only) are found for various levels of depression (i.e., relative levels of excitation where 1.0 is the control) (Fig. 8 C, right panel). Therefore, when the depression induced by feedback is embedded in the ELL local circuitry together with the experimentally observed short-term synaptic dynamics, it appears sufficient to account for the cancellation of both negative and positive phases of a global low-frequency signal. The key feature responsible for this result appears to be the frequency dependence of excitatory–inhibitory balance, which is determined by the short-term presynaptic enhancement of PF synaptic transmission as well as by the postsynaptic conductance (Lewis and Maler, 2002a, 2004; Lewis et al., 2007).
We hypothesize that the combination of paired burst-induced LTD together with local circuit dynamics are sufficient to account for the physiologically observed long-term cancellation of spatially redundant (predictable) signals. However, our model was run in open-loop mode and with sinusoidal modulations of PF firing rate. During in vivo stimulation closed loop conditions apply with attendant feedback delays due to conduction and synaptic delays. Furthermore, pyramidal cells of the CLS can also cancel the low-frequency component (<20 Hz) of global random amplitude modulations (Chacron et al., 2003; Bastian et al., 2004; Krahe et al., 2008); in this case the phases of the different frequency components are randomized. Additional in vivo experiments and more detailed modeling will be required to validate our proposed link between cellular and local network dynamics and the ability of the large scale ELL feedback network to adaptively cancel low-frequency input.
Discussion
A. leptorhynchus uses its electric sense for prey capture and navigation (Nelson and Maciver, 1999; Cowan and Fortune, 2007; Snyder et al., 2007) by detecting the spatially localized low-frequency AMs of their electric field (electric images) caused by the presence of objects (Nelson and Maciver, 1999; MacIver et al., 2001; Snyder et al., 2007). AMs in the same frequency range can also be caused by spatially diffuse (global) signals due to tail bending (Bastian, 1996a) or conspecifics with similar EOD frequencies (Maler, 2007). These global signals interfere with prey detection and this species has evolved mechanisms to eliminate such interference. In particular, a subset of CLS pyramidal cells [superficial and intermediate pyramidal cells (SPCs)] are tuned to low-frequency local AMs (prey-like signals) but filter out such signals when they are presented globally (Chacron et al., 2003; Bastian et al., 2004)—these cells cancel spatially redundant low-frequency input yet respond to local input with the same frequency content. This cancellation is achieved by an adaptive filter that learns the correlation between simultaneous input to an SPC's RF and its nonclassic surround (Chacron et al., 2003); this learning process generates a negative image of the signal that summates and cancels the sensory input (Bastian et al., 2004). The adaptive filter is implemented by an indirect descending pathway (feedback and proprioceptive afferents) (Sas and Maler, 1987; Bastian, 1996a) to the ELL whose final stage consists of parallel fibers emanating from the EGp (Bastian et al., 2004).
We have demonstrated that, in the most plastic map of the ELL (CLS) (Krahe et al., 2008), pairing large bursts of presynaptic and postsynaptic spikes (4-4) within a ±50 ms time window induces anti-Hebbian LTD. Pairing presynaptic and postsynaptic doublets (2-2) caused a small synaptic depression with a narrow time window (5 ms). Presynaptic or postsynaptic bursts alone, as well as paired spikes (1-1) or paired single spikes and bursts (1-4, 4-1), do not induce synaptic depression. We hypothesize that burst-induced depression underlies the adaptive cancellation of redundant low-frequency electrosensory input. The requirement for postsynaptic bursts provides a direct link with natural spike patterns that occur during adaptive cancellation. Spike bursts in the CLS and CMS code specifically for low-frequency signals (Oswald et al., 2004) (Marsat et al., 2009). Burst discharge is initiated by the depolarizing afterpotentials (DAPs) that follow back-propagation of spikes up the SPC's apical dendritic tree Section (Turner et al., 2002); bursts are therefore also associated with depolarization of the dendritic targets of PF feedback. Global (but not local) signals activate the PFs (Chacron et al., 2003; Bastian et al., 2004) and can therefore evoke EPSPs on the apical dendrites in temporal proximity to their depolarization by back-propagating spikes. We propose that the temporal correlation of dendritic depolarization (bursts-low-frequency signals) and PF discharge (global signals) causes LTD. Single spikes can encode both low and high-frequency AMs (Oswald et al., 2004) and high-frequency global AMs can occur during electrocommunication behavior (Benda et al., 2006). The lack of anti-Hebbian LTD after single-spike pairings may therefore be a cellular mechanism that permits CLS SPCs to maintain responsiveness to communication signals. This suggests a threshold-type mechanism underlies burst-induced LTD and differentiates it from recent extensions of classic STDP models (Pfister and Gerstner, 2006; Morrison et al., 2008). In these models, multiple spikes essentially add-up according to a STDP-rule that is based on single paired spikes. In burst-induced LTD, it appears that there is a nonlinear effect. The involvement of Ca2+ and CaMKII suggests that this nonlinear threshold is mediated by a molecular switch [see following discussion and Oswald et al. (2002) and Tanaka and Augustine (2008)].
Paired bursts cause only LTD of the PF-evoked EPSPs. In contrast, cancellation of low-frequency input requires a negative image. At the cellular level, we can equate the negative image as a hyperpolarization (inhibition) during the positive (excitatory) phase of the stimulus and depolarization (possibly potentiated PF-evoked EPSPs) during its negative phase. However, a reversal of sign, from depolarization to hyperpolarization (EPSPs to IPSPs) does not occur after our burst-pairing protocols, and we were not able to induce potentiation of PF-evoked EPSPs. The in vitro burst-pairing-induced anti-Hebbian LTD demonstrated in this paper therefore cannot, by itself, account for the negative image cancellation mechanism observed in vivo.
We propose that short-term PF plasticity and local inhibitory circuitry provide the link between PF burst-induced LTD and adaptive cancellation (negative image formation). Parallel fibers exhibit both facilitation and PKA-dependent short-term enhancement (Lewis and Maler, 2002a, 2004); however, high-frequency (>16 Hz) stimulation of PFs strongly activates local GABAergic interneurons (Maler and Mugnaini, 1994) that convert the enhanced presynaptic transmission into effective postsynaptic inhibition (Lewis and Maler, 2002a). Lewis et al. (2007), using a realistic model of the network and synaptic dynamics, already demonstrated that it could invert the phase of PF rate-modulated discharge. Here we demonstrate (Fig. 11) that weak LTD (25% EPSP reduction) can indeed generate a strong negative image when incorporated into the model network. These results suggest that the combination of burst-induced LTD, presynaptic potentiation, and local inhibition may be sufficient to explain the generation of the negative image and cancellation of redundant sensory input by ELL pyramidal cells. A more detailed computational analysis will be required to incorporate realistic feedback delays into this model.
Burst-induced depression requires activation of the NR2B subunit of the NMDAR, which is the most abundant NMDAR of SPCs and responsible for most of the PF-evoked EPSP (Harvey-Girard et al., 2007). An elevation of postsynaptic Ca2+ and activation of CaMKIIβ are also required for burst-induced LTD. These cellular events are clearly consistent with the temporal sequence of the presynaptic bursts preceding the postsynaptic ones. The requirement for presynaptic bursts is likely due to (1) PF presynaptic facilitation that increases EPSP amplitude over successive pulses (Lewis and Maler, 2001) and (2) the strong long-lasting (>50 ms) NMDAR component evoked by tetanic stimulation of PFs (Berman et al., 2001). We have no direct evidence that EGp granule cells respond to low-frequency input with spike bursts but such burst responses were reported for mammalian granule cells (D'Angelo et al., 2001; Chadderton et al., 2004; Rancz et al., 2007). Subsequent postsynaptic bursts might be required because of the voltage dependence of the PF-evoked EPSPs (Berman et al., 2001). We hypothesize that a four spike postsynaptic burst will produce a large postsynaptic depolarization via summating backpropagating spikes. Together these events should result in a strong activation of the NMDAR (Harvey-Girard et al., 2007) leading to a large Ca2+ influx and subsequent activation of CaMKIIβ. The downstream cascades that translate CaMKIIβ activation (and any additional NR2B/Ca2+-activated processes) into LTD are unknown though, since LTD is rapidly induced, these cascades presumably act locally within the stimulated dendritic branch and/or spine.
It is difficult, at present, to understand how postsynaptic bursts preceding presynaptic ones can mediate LTD since there is no obvious mechanism by which coincident postsynaptic depolarization can occur. However, one possibility could be that CLS SPCs possess dendritic voltage-gated Ca2+ channels that are activated by backpropagating spike bursts (R. Turner, personal communication). A second possibility could come from the complex dynamics of Mg2+ unblock of NMDARs (Vargas-Caballero and Robinson, 2003, 2004; Clarke and Johnson, 2008). The slow Mg2+ unblock could keep NMDARs in an “openable” state during a long period after a postsynaptic burst, until the presynaptic burst releases glutamate to allow Ca2+ influx through NMDARs. Furthermore, both possibilities of Ca2+ influx could also cause release of Ca2+ from intracellular stores [ryanodine receptors (Zupanc et al., 1992)]; this extended Ca2+ transient might allow for an overlap with subsequent presynaptic inputs.
Mormyrid fish have independently evolved an active electric sense and must also cancel predictable sensory input to better detect salient signals (Bell et al., 1997, 2008; Sawtell et al., 2007). One type of neuron in the mormyrid ELL shows synaptic depression via an anti-Hebbian STDP rule (Han et al., 2000), which involves NMDARs, dendritic spiking, and Ca2+ influx (Bell et al., 1999, 2008; Han et al., 2000; Sawtell et al., 2007). In mormyrids, the depression could be reversed by unpaired stimulation (Bell et al., 1997; Han et al., 2000), which was not possible in apteronotids. We assume that the different learning rules for mormyrid (STDP) and apteronotid (correlative burst depression) are the consequence of both the nature of the sensory input—regular sampling with a sinusoidal EOD (apteronotid) versus discrete sampling at irregular EOD intervals (mormyrid), as well as the availability of temporally exact predictive signal (corollary discharge) in mormyrids but not apteronotid.
The dorsal cochlear nucleus (DCN) and ELL (apteronotid and mormyrid) are cerebellum-like structures and share many anatomical and physiological similarities (Bell et al., 2008; Tzounopoulos and Kraus, 2009). DCN and ELL may face similar processing problems: it has been proposed that DCN might filter out irrelevant proprioceptive induced changes in acoustic signals and/or redundant signals related to respiration or self-vocalization (Tzounopoulos and Kraus, 2009). DCN fusiform cells receive cochlear auditory afferent inputs (basal dendrites) and excitatory and inhibitory feedback inputs from PFs (Tzounopoulos and Kraus, 2009). At the PF synapses, fusiform cells show a Hebbian STDP rule, whereas local inhibitory interneurons use an anti-Hebbian rule (Tzounopoulos et al., 2004). However, it is not yet possible to determine the signals conveyed by the PF inputs to DCN neurons, and consequently difficult to compare the computational effects of the various plasticity rules on processing in DCN versus ELL (gymnotid or mormyrid).
More-detailed theoretical analyses will be required to connect electrosensory and auditory system-level constraints to the DCN/ELL differences in cellular plasticity rules. Such analyses might also shed light on the more general relation between diverse underlying cellular mechanisms and the functional effects of different LTD rules on information transfer in neural networks (Egger et al., 1999; Phares et al., 2003; Birtoli and Ulrich, 2004; Tzounopoulos et al., 2004, 2007; Letzkus et al., 2006; Caporale and Dan, 2008).
Footnotes
This work was supported by a grant from the Canadian Institutes of Health Research (L.M.).
References
- Andersen P, Morris R, Amaral D, Bliss T, O'Keefe J. The hippocampus book. New York: Oxford UP; 2006. [Google Scholar]
- Bastian J. The role of amino acid neurotransmitters in the descending control of electroreception. J Comp Physiol A. 1993;172:409–423. doi: 10.1007/BF00213523. [DOI] [PubMed] [Google Scholar]
- Bastian J. Pyramidal-cell plasticity in weakly electric fish: a mechanism for attenuating responses to reafferent electrosensory inputs. J Comp Physiol A. 1995;176:63–73. doi: 10.1007/BF00197753. [DOI] [PubMed] [Google Scholar]
- Bastian J. Plasticity in an electrosensory system. I. General features of a dynamic sensory filter. J Neurophysiol. 1996a;76:2483–2496. doi: 10.1152/jn.1996.76.4.2483. [DOI] [PubMed] [Google Scholar]
- Bastian J. Plasticity in an electrosensory system. II. Postsynaptic events associated with a dynamic sensory filter. J Neurophysiol. 1996b;76:2497–2507. doi: 10.1152/jn.1996.76.4.2497. [DOI] [PubMed] [Google Scholar]
- Bastian J. Plasticity in an electrosensory system. III. Contrasting properties of spatially segregated dendritic inputs. J Neurophysiol. 1998a;79:1839–1857. doi: 10.1152/jn.1998.79.4.1839. [DOI] [PubMed] [Google Scholar]
- Bastian J. Modulation of calcium-dependent postsynaptic depression contributes to an adaptive sensory filter. J Neurophysiol. 1998b;80:3352–3355. doi: 10.1152/jn.1998.80.6.3352. [DOI] [PubMed] [Google Scholar]
- Bastian J, Chacron MJ, Maler L. Plastic and nonplastic pyramidal cells perform unique roles in a network capable of adaptive redundancy reduction. Neuron. 2004;41:767–779. doi: 10.1016/s0896-6273(04)00071-6. [DOI] [PubMed] [Google Scholar]
- Bayer KU, De Koninck P, Leonard AS, Hell JW, Schulman H. Interaction with the NMDA receptor locks CaMKII in an active conformation. Nature. 2001;411:801–805. doi: 10.1038/35081080. [DOI] [PubMed] [Google Scholar]
- Bell CC, Han VZ, Sugawara Y, Grant K. Synaptic plasticity in a cerebellum-like structure depends on temporal order. Nature. 1997;387:278–281. doi: 10.1038/387278a0. [DOI] [PubMed] [Google Scholar]
- Bell CC, Han VZ, Sugawara Y, Grant K. Synaptic plasticity in the mormyrid electrosensory lobe. J Exp Biol. 1999;202:1339–1347. doi: 10.1242/jeb.202.10.1339. [DOI] [PubMed] [Google Scholar]
- Bell CC, Han V, Sawtell NB. Cerebellum-like structures and their implications for cerebellar function. Annu Rev Neurosci. 2008;31:1–24. doi: 10.1146/annurev.neuro.30.051606.094225. [DOI] [PubMed] [Google Scholar]
- Benda J, Longtin A, Maler L. A synchronization-desynchronization code for natural communication signals. Neuron. 2006;52:347–358. doi: 10.1016/j.neuron.2006.08.008. [DOI] [PubMed] [Google Scholar]
- Berberich S, Punnakkal P, Jensen V, Pawlak V, Seeburg PH, Hvalby Ø, Köhr G. Lack of NMDA receptor subtype selectivity for hippocampal long-term potentiation. J Neurosci. 2005;25:6907–6910. doi: 10.1523/JNEUROSCI.1905-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berman N, Dunn RJ, Maler L. Function of NMDA receptors and persistent sodium channels in a feedback pathway of the electrosensory system. J Neurophysiol. 2001;86:1612–1621. doi: 10.1152/jn.2001.86.4.1612. [DOI] [PubMed] [Google Scholar]
- Berman NJ, Maler L. Inhibition evoked from primary afferents in the electrosensory lateral line lobe of the weakly electric fish (Apteronotus leptorhynchus) J Neurophysiol. 1998a;80:3173–3196. doi: 10.1152/jn.1998.80.6.3173. [DOI] [PubMed] [Google Scholar]
- Berman NJ, Maler L. Interaction of GABAB-mediated inhibition with voltage-gated currents of pyramidal cells: computational mechanism of a sensory searchlight. J Neurophysiol. 1998b;80:3197–3213. doi: 10.1152/jn.1998.80.6.3197. [DOI] [PubMed] [Google Scholar]
- Berman NJ, Maler L. Distal versus proximal inhibitory shaping of feedback excitation in the electrosensory lateral line lobe: implications for sensory filtering. J Neurophysiol. 1998c;80:3214–3232. doi: 10.1152/jn.1998.80.6.3214. [DOI] [PubMed] [Google Scholar]
- Berman NJ, Plant J, Turner RW, Maler L. Excitatory amino acid receptors at a feedback pathway in the electrosensory system: implications for the searchlight hypothesis. J Neurophysiol. 1997;78:1869–1881. doi: 10.1152/jn.1997.78.4.1869. [DOI] [PubMed] [Google Scholar]
- Bi G, Poo M. Synaptic modification by correlated activity: Hebb's postulate revisited. Annu Rev Neurosci. 2001;24:139–166. doi: 10.1146/annurev.neuro.24.1.139. [DOI] [PubMed] [Google Scholar]
- Birtoli B, Ulrich D. Firing mode-dependent synaptic plasticity in rat neocortical pyramidal neurons. J Neurosci. 2004;24:4935–4940. doi: 10.1523/JNEUROSCI.0795-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bliss TV, Collingridge GL. A synaptic model of memory: long-term potentiation in the hippocampus. Nature. 1993;361:31–39. doi: 10.1038/361031a0. [DOI] [PubMed] [Google Scholar]
- Bottai D, Dunn RJ, Ellis W, Maler L. N-methyl-d-aspartate receptor 1 mRNA distribution in the central nervous system of the weakly electric fish Apteronotus leptorhynchus . J Comp Neurol. 1997;389:65–80. doi: 10.1002/(sici)1096-9861(19971208)389:1<65::aid-cne5>3.0.co;2-c. [DOI] [PubMed] [Google Scholar]
- Caporale N, Dan Y. Spike timing-dependent plasticity: a Hebbian learning rule. Annu Rev Neurosci. 2008;31:25–46. doi: 10.1146/annurev.neuro.31.060407.125639. [DOI] [PubMed] [Google Scholar]
- Carr CE, Maler L, Sas E. Peripheral organization and central projections of the electrosensory nerves in gymnotiform fish. J Comp Neurol. 1982;211:139–153. doi: 10.1002/cne.902110204. [DOI] [PubMed] [Google Scholar]
- Castellani GC, Quinlan EM, Bersani F, Cooper LN, Shouval HZ. A model of bidirectional synaptic plasticity: from signaling network to channel conductance. Learn Mem. 2005;12:423–432. doi: 10.1101/lm.80705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chacron MJ, Bastian J. Population coding by electrosensory neurons. J Neurophysiol. 2008;99:1825–1835. doi: 10.1152/jn.01266.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chacron MJ, Doiron B, Maler L, Longtin A, Bastian J. Non-classical receptive field mediates switch in a sensory neuron's frequency tuning. Nature. 2003;423:77–81. doi: 10.1038/nature01590. [DOI] [PubMed] [Google Scholar]
- Chadderton P, Margrie TW, Häusser M. Integration of quanta in cerebellar granule cells during sensory processing. Nature. 2004;428:856–860. doi: 10.1038/nature02442. [DOI] [PubMed] [Google Scholar]
- Clarke RJ, Johnson JW. Voltage-dependent gating of NR1/2B NMDA receptors. J Physiol. 2008;586:5727–5741. doi: 10.1113/jphysiol.2008.160622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collingridge GL, Isaac JT, Wang YT. Receptor trafficking and synaptic plasticity. Nat Rev Neurosci. 2004;5:952–962. doi: 10.1038/nrn1556. [DOI] [PubMed] [Google Scholar]
- Cowan NJ, Fortune ES. The critical role of locomotion mechanics in decoding sensory systems. J Neurosci. 2007;27:1123–1128. doi: 10.1523/JNEUROSCI.4198-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dan Y, Poo MM. Spike timing-dependent plasticity: from synapse to perception. Physiol Rev. 2006;86:1033–1048. doi: 10.1152/physrev.00030.2005. [DOI] [PubMed] [Google Scholar]
- D'Angelo E, Nieus T, Maffei A, Armano S, Rossi P, Taglietti V, Fontana A, Naldi G. Theta-frequency bursting and resonance in cerebellar granule cells: experimental evidence and modeling of a slow K+-dependent mechanism. J Neurosci. 2001;21:759–770. doi: 10.1523/JNEUROSCI.21-03-00759.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dayan P, Abbott LF. Theoretical neuroscience: computational and mathematical modeling of neural systems. Cambridge, MA: MIT Press; 2001. [Google Scholar]
- Dingledine R, Borges K, Bowie D, Traynelis SF. The glutamate receptor ion channels. Pharmacol Rev. 1999;51:7–61. [PubMed] [Google Scholar]
- Egger V, Feldmeyer D, Sakmann B. Coincidence detection and changes of synaptic efficacy in spiny stellate neurons in rat barrel cortex. Nat Neurosci. 1999;2:1098–1105. doi: 10.1038/16026. [DOI] [PubMed] [Google Scholar]
- Ellis LD, Mehaffey WH, Harvey-Girard E, Turner RW, Maler L, Dunn RJ. SK channels provide a novel mechanism for the control of frequency tuning in electrosensory neurons. J Neurosci. 2007;27:9491–9502. doi: 10.1523/JNEUROSCI.1106-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fino E, Glowinski J, Venance L. Bidirectional activity-dependent plasticity at corticostriatal synapses. J Neurosci. 2005;25:11279–11287. doi: 10.1523/JNEUROSCI.4476-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujino K, Oertel D. Bidirectional synaptic plasticity in the cerebellum-like mammalian dorsal cochlear nucleus. Proc Natl Acad Sci U S A. 2003;100:265–270. doi: 10.1073/pnas.0135345100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graupner M, Brunel N. STDP in a bistable synapse model based on CaMKII and associated signaling pathways. PLoS Comput Biol. 2007;3:e221. doi: 10.1371/journal.pcbi.0030221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han VZ, Grant K, Bell CC. Reversible associative depression and nonassociative potentiation at a parallel fiber synapse. Neuron. 2000;27:611–622. doi: 10.1016/s0896-6273(00)00070-2. [DOI] [PubMed] [Google Scholar]
- Hansel C, de Jeu M, Belmeguenai A, Houtman SH, Buitendijk GH, Andreev D, De Zeeuw CI, Elgersma Y. alphaCaMKII Is essential for cerebellar LTD and motor learning. Neuron. 2006;51:835–843. doi: 10.1016/j.neuron.2006.08.013. [DOI] [PubMed] [Google Scholar]
- Harvey-Girard E, Dunn RJ. Excitatory amino acid receptors of the electrosensory system: the NR1/NR2B N-methyl-d-aspartate receptor. J Neurophysiol. 2003;89:822–832. doi: 10.1152/jn.00629.2002. [DOI] [PubMed] [Google Scholar]
- Harvey-Girard E, Dunn RJ, Maler L. Regulated expression of N-methyl-d-aspartate receptors and associated proteins in teleost electrosensory system and telencephalon. J Comp Neurol. 2007;505:644–668. doi: 10.1002/cne.21521. [DOI] [PubMed] [Google Scholar]
- Jörntell H, Ekerot CF. Reciprocal bidirectional plasticity of parallel fiber receptive fields in cerebellar Purkinje cells and their afferent interneurons. Neuron. 2002;34:797–806. doi: 10.1016/s0896-6273(02)00713-4. [DOI] [PubMed] [Google Scholar]
- Kim MJ, Dunah AW, Wang YT, Sheng M. Differential roles of NR2A- and NR2B-containing NMDA receptors in Ras-ERK signaling and AMPA receptor trafficking. Neuron. 2005;46:745–760. doi: 10.1016/j.neuron.2005.04.031. [DOI] [PubMed] [Google Scholar]
- Krahe R, Bastian J, Chacron MJ. Temporal processing across multiple topographic maps in the electrosensory system. J Neurophysiol. 2008;100:852–867. doi: 10.1152/jn.90300.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonard AS, Lim IA, Hemsworth DE, Horne MC, Hell JW. Calcium/calmodulin-dependent protein kinase II is associated with the N-methyl-d-aspartate receptor. Proc Natl Acad Sci U S A. 1999;96:3239–3244. doi: 10.1073/pnas.96.6.3239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letzkus JJ, Kampa BM, Stuart GJ. Learning rules for spike timing-dependent plasticity depend on dendritic synapse location. J Neurosci. 2006;26:10420–10429. doi: 10.1523/JNEUROSCI.2650-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis JE, Maler L. Neuronal population codes and the perception of object distance in weakly electric fish. J Neurosci. 2001;21:2842–2850. doi: 10.1523/JNEUROSCI.21-08-02842.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis JE, Maler L. Dynamics of electrosensory feedback: short-term plasticity and inhibition in a parallel fiber pathway. J Neurophysiol. 2002a;88:1695–1706. doi: 10.1152/jn.2002.88.4.1695. [DOI] [PubMed] [Google Scholar]
- Lewis JE, Maler L. Blurring of the senses: common cues for distance perception in diverse sensory systems. Neuroscience. 2002b;114:19–22. doi: 10.1016/s0306-4522(02)00220-8. [DOI] [PubMed] [Google Scholar]
- Lewis JE, Maler L. Synaptic dynamics on different time scales in a parallel fiber feedback pathway of the weakly electric fish. J Neurophysiol. 2004;91:1064–1070. doi: 10.1152/jn.00856.2003. [DOI] [PubMed] [Google Scholar]
- Lewis JE, Lindner B, Laliberté B, Groothuis S. Control of neuronal firing by dynamic parallel fiber feedback: implications for electrosensory reafference suppression. J Exp Biol. 2007;210:4437–4447. doi: 10.1242/jeb.010322. [DOI] [PubMed] [Google Scholar]
- Li R, Huang FS, Abbas AK, Wigström H. Role of NMDA receptor subtypes in different forms of NMDA-dependent synaptic plasticity. BMC Neurosci. 2007;8:55. doi: 10.1186/1471-2202-8-55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisman J, Schulman H, Cline H. The molecular basis of CaMKII function in synaptic and behavioural memory. Nat Rev Neurosci. 2002;3:175–190. doi: 10.1038/nrn753. [DOI] [PubMed] [Google Scholar]
- Lisman JE, Zhabotinsky AM. A model of synaptic memory: a CaMKII/PP1 switch that potentiates transmission by organizing an AMPA receptor anchoring assembly. Neuron. 2001;31:191–201. doi: 10.1016/s0896-6273(01)00364-6. [DOI] [PubMed] [Google Scholar]
- Liu L, Wong TP, Pozza MF, Lingenhoehl K, Wang Y, Sheng M, Auberson YP, Wang YT. Role of NMDA receptor subtypes in governing the direction of hippocampal synaptic plasticity. Science. 2004;304:1021–1024. doi: 10.1126/science.1096615. [DOI] [PubMed] [Google Scholar]
- MacIver MA, Sharabash NM, Nelson ME. Prey-capture behavior in gymnotid electric fish: motion analysis and effects of water conductivity. J Exp Biol. 2001;204:543–557. doi: 10.1242/jeb.204.3.543. [DOI] [PubMed] [Google Scholar]
- Maler L. The posterior lateral line lobe of certain gymnotoid fish: quantitative light microscopy. J Comp Neurol. 1979;183:323–363. doi: 10.1002/cne.901830208. [DOI] [PubMed] [Google Scholar]
- Maler L. Neural strategies for optimal processing of sensory signals. Prog Brain Res. 2007;165:135–154. doi: 10.1016/S0079-6123(06)65009-7. [DOI] [PubMed] [Google Scholar]
- Maler L, Hincke MT. Distribution of calcium/calmodulin-dependent kinase 2 in the brain of Apteronotus leptorhynchus . J Comp Neurol. 1999;408:177–203. doi: 10.1002/(sici)1096-9861(19990531)408:2<177::aid-cne4>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]
- Maler L, Monaghan D. The distribution of excitatory amino acid binding sites in the brain of an electric fish, Apteronotus leptorhynchus . J Chem Neuroanat. 1991;4:39–61. doi: 10.1016/0891-0618(91)90031-7. [DOI] [PubMed] [Google Scholar]
- Maler L, Mugnaini E. Correlating gamma-aminobutyric acidergic circuits and sensory function in the electrosensory lateral line lobe of a gymnotiform fish. J Comp Neurol. 1994;345:224–252. doi: 10.1002/cne.903450206. [DOI] [PubMed] [Google Scholar]
- Maler L, Sas EK, Rogers J. The cytology of the posterior lateral line lobe of high-frequency weakly electric fish (Gymnotidae): dendritic differentiation and synaptic specificity in a simple cortex. J Comp Neurol. 1981;195:87–139. doi: 10.1002/cne.901950107. [DOI] [PubMed] [Google Scholar]
- Manabe T, Wyllie DJ, Perkel DJ, Nicoll RA. Modulation of synaptic transmission and long-term potentiation: effects on paired pulse facilitation and EPSC variance in the CA1 region of the hippocampus. J Neurophysiol. 1993;70:1451–1459. doi: 10.1152/jn.1993.70.4.1451. [DOI] [PubMed] [Google Scholar]
- Marsat G, Proville RD, Maler L. Transient signals trigger synchronous bursts in an identified population of neurons. J Neurophysiol. 2009;102:714–723. doi: 10.1152/jn.91366.2008. [DOI] [PubMed] [Google Scholar]
- Massey PV, Johnson BE, Moult PR, Auberson YP, Brown MW, Molnar E, Collingridge GL, Bashir ZI. Differential roles of NR2A and NR2B-containing NMDA receptors in cortical long-term potentiation and long-term depression. J Neurosci. 2004;24:7821–7828. doi: 10.1523/JNEUROSCI.1697-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehaffey WH, Doiron B, Maler L, Turner RW. Deterministic multiplicative gain control with active dendrites. J Neurosci. 2005;25:9968–9977. doi: 10.1523/JNEUROSCI.2682-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehaffey WH, Maler L, Turner RW. Intrinsic frequency tuning in ELL pyramidal cells varies across electrosensory maps. J Neurophysiol. 2008;99:2641–2655. doi: 10.1152/jn.00028.2008. [DOI] [PubMed] [Google Scholar]
- Morrison A, Diesmann M, Gerstner W. Phenomenological models of synaptic plasticity based on spike timing. Biol Cybern. 2008;98:459–478. doi: 10.1007/s00422-008-0233-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson ME, Maciver MA. Prey capture in the weakly electric fish Apteronotus albifrons: sensory acquisition strategies and electrosensory consequences. J Exp Biol. 1999;202:1195–1203. doi: 10.1242/jeb.202.10.1195. [DOI] [PubMed] [Google Scholar]
- Nelson ME, Paulin MG. Neural simulations of adaptive reafference suppression in the elasmobranch electrosensory system. J Comp Physiol A. 1995;177:723–736. doi: 10.1007/BF00187631. [DOI] [PubMed] [Google Scholar]
- Oswald AM, Lewis JE, Maler L. Dynamically interacting processes underlie synaptic plasticity in a feedback pathway. J Neurophysiol. 2002;87:2450–2463. doi: 10.1152/jn.00711.2001. [DOI] [PubMed] [Google Scholar]
- Oswald AM, Chacron MJ, Doiron B, Bastian J, Maler L. Parallel processing of sensory input by bursts and isolated spikes. J Neurosci. 2004;24:4351–4362. doi: 10.1523/JNEUROSCI.0459-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oswald AM, Doiron B, Maler L. Interval coding. I. Burst interspike intervals as indicators of stimulus intensity. J Neurophysiol. 2007;97:2731–2743. doi: 10.1152/jn.00987.2006. [DOI] [PubMed] [Google Scholar]
- Pfister JP, Gerstner W. Triplets of spikes in a model of spike timing-dependent plasticity. J Neurosci. 2006;26:9673–9682. doi: 10.1523/JNEUROSCI.1425-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phares GA, Antzoulatos EG, Baxter DA, Byrne JH. Burst-induced synaptic depression and its modulation contribute to information transfer at Aplysia sensorimotor synapses: empirical and computational analyses. J Neurosci. 2003;23:8392–8401. doi: 10.1523/JNEUROSCI.23-23-08392.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pláteník J, Kuramoto N, Yoneda Y. Molecular mechanisms associated with long-term consolidation of the NMDA signals. Life Sci. 2000;67:335–364. doi: 10.1016/s0024-3205(00)00632-9. [DOI] [PubMed] [Google Scholar]
- Rancz EA, Ishikawa T, Duguid I, Chadderton P, Mahon S, Häusser M. High-fidelity transmission of sensory information by single cerebellar mossy fibre boutons. Nature. 2007;450:1245–1248. doi: 10.1038/nature05995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rezazadeh S, Claydon TW, Fedida D. KN-93 (2-[N-(2-hydroxyethyl)]-N-(4-methoxybenzenesulfonyl)]amino-N-(4-chlorocinn amyl)-N-methylbenzylamine), a calcium/calmodulin-dependent protein kinase II inhibitor, is a direct extracellular blocker of voltage-gated potassium channels. J Pharmacol Exp Ther. 2006;317:292–299. doi: 10.1124/jpet.105.097618. [DOI] [PubMed] [Google Scholar]
- Roberts PD. Modeling inhibitory plasticity in the electrosensory system of mormyrid electric fish. J Neurophysiol. 2000;84:2035–2047. doi: 10.1152/jn.2000.84.4.2035. [DOI] [PubMed] [Google Scholar]
- Roberts PD, Bell CC. Computational consequences of temporally asymmetric learning rules: II. Sensory image cancellation. J Comput Neurosci. 2000;9:67–83. doi: 10.1023/a:1008938428112. [DOI] [PubMed] [Google Scholar]
- Sas E, Maler L. The organization of afferent input to the caudal lobe of the cerebellum of the gymnotid fish Apteronotus leptorhynchus . Anat Embryol (Berl) 1987;177:55–79. doi: 10.1007/BF00325290. [DOI] [PubMed] [Google Scholar]
- Sawtell NB, Williams A, Bell CC. Central control of dendritic spikes shapes the responses of Purkinje-like cells through spike timing-dependent synaptic plasticity. J Neurosci. 2007;27:1552–1565. doi: 10.1523/JNEUROSCI.5302-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shouval HZ, Bear MF, Cooper LN. A unified model of NMDA receptor-dependent bidirectional synaptic plasticity. Proc Natl Acad Sci U S A. 2002;99:10831–10836. doi: 10.1073/pnas.152343099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snyder JB, Nelson ME, Burdick JW, Maciver MA. Omnidirectional sensory and motor volumes in electric fish. PLoS Biol. 2007;5:e301. doi: 10.1371/journal.pbio.0050301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strack S, Colbran RJ. Autophosphorylation-dependent targeting of calcium/calmodulin-dependent protein kinase II by the NR2B subunit of the N-methyl-d-aspartate receptor. J Biol Chem. 1998;273:20689–20692. doi: 10.1074/jbc.273.33.20689. [DOI] [PubMed] [Google Scholar]
- Tanaka K, Augustine GJ. A positive feedback signal transduction loop determines timing of cerebellar long-term depression. Neuron. 2008;59:608–620. doi: 10.1016/j.neuron.2008.06.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner RW, Maler L, Deerinck T, Levinson SR, Ellisman MH. TTX-sensitive dendritic sodium channels underlie oscillatory discharge in a vertebrate sensory neuron. J Neurosci. 1994;14:6453–6471. doi: 10.1523/JNEUROSCI.14-11-06453.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner RW, Plant JR, Maler L. Oscillatory and burst discharge across electrosensory topographic maps. J Neurophysiol. 1996;76:2364–2382. doi: 10.1152/jn.1996.76.4.2364. [DOI] [PubMed] [Google Scholar]
- Turner RW, Lemon N, Doiron B, Rashid AJ, Morales E, Longtin A, Maler L, Dunn RJ. Oscillatory burst discharge generated through conditional backpropagation of dendritic spikes. J Physiol Paris. 2002;96:517–530. doi: 10.1016/S0928-4257(03)00007-X. [DOI] [PubMed] [Google Scholar]
- Tzounopoulos T, Kraus N. Learning to encode timing: mechanisms of plasticity in the auditory brainstem. Neuron. 2009;62:463–469. doi: 10.1016/j.neuron.2009.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tzounopoulos T, Kim Y, Oertel D, Trussell LO. Cell-specific, spike timing-dependent plasticities in the dorsal cochlear nucleus. Nat Neurosci. 2004;7:719–725. doi: 10.1038/nn1272. [DOI] [PubMed] [Google Scholar]
- Tzounopoulos T, Rubio ME, Keen JE, Trussell LO. Coactivation of pre- and postsynaptic signaling mechanisms determines cell-specific spike-timing-dependent plasticity. Neuron. 2007;54:291–301. doi: 10.1016/j.neuron.2007.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valverde RH, Tortelote GG, Lemos T, Mintz E, Vieyra A. Ca2+/calmodulin-dependent protein kinase II is an essential mediator in the coordinated regulation of electrocyte Ca2+-ATPase by calmodulin and protein kinase A. J Biol Chem. 2005;280:30611–30618. doi: 10.1074/jbc.M501880200. [DOI] [PubMed] [Google Scholar]
- Vargas-Caballero M, Robinson HP. A slow fraction of Mg2+ unblock of NMDA receptors limits their contribution to spike generation in cortical pyramidal neurons. J Neurophysiol. 2003;89:2778–2783. doi: 10.1152/jn.01038.2002. [DOI] [PubMed] [Google Scholar]
- Vargas-Caballero M, Robinson HP. Fast and slow voltage-dependent dynamics of magnesium block in the NMDA receptor: the asymmetric trapping block model. J Neurosci. 2004;24:6171–6180. doi: 10.1523/JNEUROSCI.1380-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang D, Maler L. Differential roles of Ca2+/calmodulin-dependent kinases in posttetanic potentiation at input selective glutamatergic pathways. Proc Natl Acad Sci U S A. 1998;95:7133–7138. doi: 10.1073/pnas.95.12.7133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weitlauf C, Honse Y, Auberson YP, Mishina M, Lovinger DM, Winder DG. Activation of NR2A-containing NMDA receptors is not obligatory for NMDA receptor-dependent long-term potentiation. J Neurosci. 2005;25:8386–8390. doi: 10.1523/JNEUROSCI.2388-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamakura T, Shimoji K. Subunit- and site-specific pharmacology of the NMDA receptor channel. Prog Neurobiol. 1999;59:279–298. doi: 10.1016/s0301-0082(99)00007-6. [DOI] [PubMed] [Google Scholar]
- Yang H, Wanner IB, Roper SD, Chaudhari N. An optimized method for in situ hybridization with signal amplification that allows the detection of rare mRNAs. J Histochem Cytochem. 1999;47:431–446. doi: 10.1177/002215549904700402. [DOI] [PubMed] [Google Scholar]
- Zupanc GK, Airey JA, Maler L, Sutko JL, Ellisman MH. Immunohistochemical localization of ryanodine binding proteins in the central nervous system of gymnotiform fish. J Comp Neurol. 1992;325:135–151. doi: 10.1002/cne.903250202. [DOI] [PubMed] [Google Scholar]