

Abstract
Norrin is a secreted protein that binds to frizzled 4 and controls development of capillaries in retina and inner ear. We provide evidence that Norrin has distinct neuroprotective properties that are independent from its effects on vascular development. The function of Norrin was investigated in a mouse model of excitotoxic retinal ganglion cell (RGC) damage after intravitreal injection of NMDA, and in cultured Müller glia or immortalized RGC-5 cells. Intravitreal injection of Norrin significantly increased the number of surviving RGC axons in the optic nerve and decreased apoptotic death of retinal neurons following NMDA-mediated damage. This effect could be blocked by adding dickkopf (DKK)-1, an inhibitor of the Wnt/β-catenin signaling pathway. Treatment of eyes with combined Norrin/NMDA activated Wnt/β-catenin signaling and increased the retinal expression of leukemia inhibitory factor and endothelin-2, as well as that of neurotrophic growth factors such as fibroblast growth factor-2, brain-derived neurotrophic factor, lens epithelium-derived growth factor, and ciliary neurotrophic factor. A similar activation of Wnt/β-catenin signaling and an increased expression of neurotrophic factors was observed in cultured Müller cells after treatment with Norrin, effects that again could be blocked by adding DKK-1. In addition, conditioned cell culture medium of Norrin-treated Müller cells increased survival of differentiated RGC-5 cells. We conclude that Norrin has pronounced neuroprotective properties on retinal neurons with the distinct potential to decrease the damaging effects of NMDA-induced RGC loss. The effects of Norrin involve activation of Wnt/β-catenin signaling and subsequent induction of neurotrophic growth factors in Müller cells.
Introduction
Norrin is a secreted 131 aa protein with a cysteine knot motif (Meitinger et al., 1993). Mutations in the NDP gene that encodes Norrin cause Norrie disease, an X-linked retinal dysplasia that presents with congenital or early childhood blindness (Berger et al., 1992; Meindl et al., 1992). Ndpy/− mutant mice with a targeted disruption of Ndp (Berger et al., 1996) show complete absence of intraretinal capillaries, a persistent hyaloid vasculature, and a continuous loss of vessels within the stria vascularis of the cochlea (Richter et al., 1998; Rehm et al., 2002; Luhmann et al., 2005; Ohlmann et al., 2005). βB1-Crystallin-Norrin transgenic mice with ocular overexpression of ectopic Norrin under control of the lens-specific βB1-crystallin promoter have been developed, and in mixed βB1-Crystallin-Norrin/Ndpy/− mice, the vascular phenotype is completely rescued (Ohlmann et al., 2005). Norrin binds with high-affinity to the frizzled (Fzd) 4 receptor and activates in cooperation with its coreceptor the low-density lipoprotein receptor-related protein (Lrp) 5 the classical Wnt/β-catenin signaling pathway (Xu et al., 2004; Smallwood et al., 2007). Recent studies indicate that the tetraspanin TSPAN12 is also associated with the Fzd 4/Lrp5 receptor complex enhancing specifically the activation of Norrin/β-catenin signaling, but not that of Wnt/β-catenin (Junge et al., 2009). There is considerable evidence that Norrin, Fzd 4, and Lrp5 constitute an essential signaling system that controls the formation of retinal capillaries during development (Ye et al., 2009). In support of this are recent findings that show that Norrin promotes vascular regrowth after oxygen-induced retinal vessel loss and suppresses retinopathy in mice (Ohlmann et al., 2010).
In addition to vascular changes, Ndpy/− mutant mice develop an early loss of retinal ganglion cells (RGCs) (Richter et al., 1998; Ohlmann et al., 2005). The structural changes correlate with loss of function, as Ndpy/− mice show severe alterations in their ocular electroretinogram and develop sensorineural deafness (Ruether et al., 1997; Rehm et al., 2002; Ohlmann et al., 2005). βB1-Crystallin-Norrin mice, which overexpress Norrin in the eye, show a higher proliferation of retinal progenitor cells and are born with thicker retinae than wild-type littermates (Ohlmann et al., 2005). Since Norrin has been shown to decrease apoptosis in immortalized RGCs (RGC-5) (Lin et al., 2009), and because of the substantial evidence that Wnt/β-catenin signaling has protective effects on neurodegeneration both in vitro and in vivo (Li et al., 2007; Leng et al., 2008; Toledo et al., 2008), we wondered whether Norrin might mediate neuroprotective effects on retinal neurons that are independent from its action on retinal endothelial cells and capillary formation.
Norrin is expressed in Müller cells (Ye et al., 2009) and in retinal neurons during development (Berger et al., 1996) as well as in the adult retina (Bernstein and Wong, 1998; Hartzer et al., 1999). Since loss of RGCs is an early and very characteristic finding in the eyes of Ndpy/− mutant mice, we hypothesized that Norrin could be part of an endogenous protective system that is important for RGC survival during development and after injury, and that acts via Wnt/β-catenin signaling. To test this hypothesis, we examined the effects of treatment with Norrin on RGC death following excitotoxic damage after intravitreal injection of NMDA (Siliprandi et al., 1992; Li et al., 1999). Our results provide evidence for a potent neuroprotective role of Norrin for RGC survival upon injury that involves Wnt/β-catenin signaling, activation of Müller glia, and an increased synthesis of neuroprotective factors in the damaged retina.
Materials and Methods
All procedures in this study conformed to the tenets of the Declaration of Helsinki, the National Institutes of Health Guidelines on the Care and Use of Animals in Research, and the ARVO Statement for the Use of Animals in Ophthalmic and Vision Research.
Expression and purification of recombinant human Norrin.
Recombinant human Norrin was purified as described previously (Ohlmann et al., 2010). In brief, the cDNA of human Norrin was cloned into the SecTag2 plasmid (Invitrogen) and transfected into HEK 293 EBNA cells. Protein purification of Norrin was performed by affinity chromatography using heparin agarose (Sigma). Eluted fractions were analyzed by Western blot analyses using goat anti-human Norrin antibodies (R&D Systems) and by silver staining of a SDS-polyacrylamide gel according to standard protocols. Fractions that were highly enriched with Norrin and without detectable amounts of contaminating proteins were dialyzed overnight against PBS using a dialyzing membrane with a 2 kDa cutoff (Spectra/Por). Protein content was measured on a semiquantitative SDS-polyacrylamide gel loaded with known amounts of bovine serum albumin (BSA), and visualized by silver staining according to standard protocols.
NMDA-induced retinal damage.
NMDA-mediated retinal damage was induced as reported previously (Siliprandi et al., 1992). Briefly, adult C57BL/6 mice were deeply anesthetized with ketamine (120 mg/kg of body weight, i.m.) and xylazine (8 mg/kg of body weight, i.m.), and the ocular surface was disinfected by an iodine tincture. To induce retinal damage, 3 μl of NMDA (10 mm dissolved in PBS) (Sigma) were injected into the vitreous body of one eye while the fellow was treated with NMDA (10 mm) and Norrin (5 ng/μl). In some animals, 3 μl of NMDA (10 mm), Norrin (5 ng/μl), and dickkopf-1 (DKK-1) (5 μg/μl) (R&D Systems) were injected into one eye, and NMDA (10 mm) and Norrin (5 ng/μl) in the fellow eye. Other experiments included injection of PBS in one eye and Norrin (5 ng/μl) or NMDA (10 mm) in the fellow eye. Mice were killed 24 h or 3 weeks after injection, and eyes as well as optic nerves were isolated. For light and electron microscopy, eyes and optic nerves were fixed in 2% paraformaldehyde/2.5% glutaraldehyde overnight, and for terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling (TUNEL) in 4% paraformaldehyde for 4 h. For expression analyses, retinae were dissected and stored at −80°C until further analyses.
Isolation and characterization of Müller cells.
Isolation and initiation of primary cultures from purified Müller cells were performed as previously described (Hicks and Courtois, 1990). In brief, eyes of 8- to 10-d-old Wistar rats were enucleated and placed in DMEM containing 10% fetal bovine serum (FBS), gentamicin (20 μg/ml), penicillin (100 U/ml), and streptomycin (100 μg/ml) overnight. At the next day, eyes were incubated for 30 min at 37°C in DMEM containing 0.1% trypsin (PAA) and 70 U/ml collagenase (Sigma), followed by an additional incubation in DMEM with 10% FBS. Subsequently, the retina was dissected from the rest of the ocular tissue and chopped into 1 mm2 fragments. Six to eight retinal fragments were transferred into a 35 mm cell culture dish, covered with a sterile glass coverslip, and incubated in DMEM containing 10% FBS, gentamicin (20 μg/ml), penicillin (100 U/ml), and streptomycin (100 μg/ml) at 37°C in humidified 5% CO2. Medium remained unchanged for 5–6 d, and was then changed every third day. When cell outgrowth reached local semiconfluency, retinal aggregates and debris were removed by forcibly pipetting medium onto the dish. This procedure was repeated until all aggregates were removed, as judged by examination under an inverse phase-contrast microscope (Carl Zeiss). After isolated Müller cells reached confluency, the cells were passaged two times before the experiments were performed. The absence of neuronal, fibroblastic, astrocytic, retinal pigment epithelial, ciliary epithelial, endothelial, and microglial cell contamination was confirmed by quantitative real-time RT-PCR using cDNA from cultured Müller cells and retinae of rats as positive control. Moreover, RT-PCR and immunohistochemistry for specific transcripts of Müller cells in culture such as carbonic anhydrase, glutamine synthetase, vimentin, and GFAP were performed (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). Cultured Müller cells were incubated with Norrin (40 ng/ml), DKK-1 (100 ng/ml), and/or NMDA (50 or 100 μm). To incubate RGC-5 with Norrin conditioned cell culture medium, Müller cells were starved overnight without FBS and were then treated with Norrin (40 ng/ml) for 7 h. After washing with PBS, fresh DMEM without FBS was added for additional 8 h and finally used as Norrin conditioned cell culture medium.
Cell culture, differentiation, and WST-1 viability assay of RGC-5.
Transformed RGC-5 cells (Krishnamoorthy et al., 2001) were kindly provided by Neeraj Agarwal (University of North Texas, Fort Worth, TX). RGC-5 cells were cultured in DMEM (PAA) containing 1 g/L glucose with l-glutamine, supplemented with 10% FBS, 100 U/ml penicillin, and 100 μg/ml streptomycin. Cells were incubated at 37°C in humidified 5% CO2. Before viability assays, differentiation of RGC-5 was induced by staurosporine as described previously (Frassetto et al., 2006). In brief, up to 45,000 cells/well were plated onto a 96-well plate. After attachment, cells were washed twice with PBS and incubated with 0.4 μm staurosporine in unsupplemented DMEM for 1 h. For recovery, RGC-5 cells were incubated in supplemented DMEM for additional 16 h. After differentiation, cells were incubated for 24 h in DMEM without FBS with or without Norrin or conditioned cell culture medium of Müller cells as indicated. Cell viability was analyzed using WST-1 dye according to manufacturer's instructions (Roche). After incubation of RGC-5, the medium was removed and unsupplemented cell culture medium containing WST-1 was added for 30 min. Extinction was measured at 450 nm using an ELISA plate reader (Tecan).
Light and electron microscopy.
Eyes and optic nerves were washed overnight in cacodylate buffer, postfixed with OsO4, dehydrated, and embedded in Epon according to standard protocols (Roth). One-micrometer semithin sections of eyes, displaying the entire retina from the ora serrata of one side via the optic nerve to the ora serrata of the other side, were stained with fuchsin/methylene blue, whereas sections of optic nerves were stained with paraphenylenediamine according to standard protocols and analyzed by light microscopy using an Axiovision microscope (Carl Zeiss). For quantification, the total number of axons in optic nerves and of neuronal perikarya in the ganglion cell layer per sagittal section and the area of the inner plexiform layer (IPL) per sagittal section were measured using the Axiovision software 3.0 (Carl Zeiss) and calculated as relative number of retinal axons, RGCs per 100 μm of retina, or area of IPL per retinal length (in square micrometers per micrometer). For transmission electron microscopy, samples were rinsed in 0.1 m cacodylate buffer, postfixed in a mixture of 1% OsO4 and 0.8% potassium ferrocyanide in 0.1 m cacodylate buffer for 2 h at 4°C. Specimens were then dehydrated in a graded series of ethanol and embedded in Epon (Serva). Ultrathin sections were mounted on uncoated copper grids, stained with uranyl acetate and lead citrate, and examined on a Zeiss EM 10A electron microscope.
Immunohistochemistry and TUNEL.
For immunohistochemistry, cultured Müller cells were fixed with 4% PFA for 15 min. Before overnight incubation at 4°C with rabbit anti-β-catenin antibodies (1:100; Cell Signaling Technology), anti-glutamine synthetase antibodies (1:500; Sigma), anti-GFAP antibodies (1:40; Signet), and anti-vimentin antibodies (1:20; Dako), samples were incubated in 3% BSA solution for 30 min. After three washes (10 min each) with PBS, samples were treated for 1 h with Alexa 488 Fluor-labeled goat anti-rabbit antibodies (1:1000; Invitrogen). After three additional washes, sections were mounted in fluorescent mounting medium containing 1:50 DAPI (Vector Laboratories), and analyzed on an Axiovision fluorescent microscope (Carl Zeiss). Apoptotic retinal cells were detected by TUNEL assay using the Deadend Fluorometric TUNEL system (Promega). After preparation, eyes were fixed for 4 h in 4% paraformaldehyde and embedded in paraffin according to standard procedures. TUNEL of paraffin sections was performed according to the manufacturer's instructions. Slides were analyzed on an Axiovision fluorescent microscope (Carl Zeiss). For generation of confocal images, an LSM 710 (Carl Zeiss) was used. For quantification, the total number of TUNEL-positive neurons as well as the total number of nuclei in the retinal ganglion cell layer were quantified in the entire retina in sagittal sections and calculated as the percentage of TUNEL-positive neurons per sagittal section.
RNA isolation, cDNA synthesis, and real-time RT-PCR analyses.
After starving in serum-free medium, confluent Müller cells were incubated with Norrin (20 and 40 ng/ml) and/or DKK-1 (100 ng/ml) for 7 or 24 h and harvested from 35 mm cell culture dishes in Trizol (Invitrogen). After differentiation (see above), RGC-5 cells were treated with DKK-1 (100 ng/ml) and/or Norrin (40 ng/ml) for 7 h and subjected to RNA isolation using Trizol. After injection of 3 μl of PBS, NMDA (10 mm), and/or Norrin (5 μg/μl), total RNA from mouse retinae was extracted using Trizol at the time points indicated and according to the manufacturer's recommendations. The integrity of the obtained RNA was confirmed by gel electrophoresis. First-strand cDNA synthesis was prepared from total RNA using the iScript cDNA Synthesis Kit (Bio-Rad) according to manufacturer's instructions. Quantitative real-time RT-PCR analyses were performed using the Bio-Rad iQ5 Real-Time PCR Detection System. Taq polymerase (Hot Star; Qiagen) was used for PCR according to the manufacturer's protocol. PCR was performed in a volume of 25 μl, consisting of 2.5 μl of 10× PCR buffer, 2.0–2.5 μl of MgCl2 (25 mm), 0.5 μl of dNTPs (10 mm each; Promega), 0.5 μl of Taq (5 U/μl), 0.5 μl of primer mix (20 μm each), and 2.5 μl of 1× SYBR green I solution (Sigma-Aldrich). The temperature profile was denaturation at 95°C for 10 s and annealing and extension at 60°C for 40 s for 40 cycles. All PCR primers (Table 1) were purchased from Invitrogen and span exon–intron boundaries. For quantification, the housekeeping genes GAPDH for murine retinae, RPL32 for RGC-5, and CypA for Müller cells from rats were used simultaneously. Results were quantitatively analyzed using Bio-Rad iQ5 Standard-Edition (version 2.0.148.60623) software. Semiquantitative RT-PCR analyses for Müller cell transcripts were performed in a final volume of 50 μl, consisting of 5.0 μl of 10× PCR buffer, 2.0 μl of MgCl2 (25 mm), 0.5 μl of dNTPs (10 mm each), 0.5 μl of Taq (5 U/μl; all from Bioline), and 1.0 μl of primer mix (10 pmol of each; Invitrogen). The thermal cycle profile was initial denaturation at 94°C for 2 min, followed by 30 cycles of 30 s at 94°C, 45 s of annealing at 55°C, and 90 s of extension at 72°C. After the last cycle, the extension time was 10 min.
Table 1.
Primers used for RT-PCR amplification
| Product | Species | Accession no. | Forward primer | Position | Reverse primer | Position |
|---|---|---|---|---|---|---|
| β-Catenin | Mouse | NM_007614.2 | CCTTGGATATCGCCAGGAT | 2330–2348 | TGGCCGTATCCACCAGAG | 2372–2389 |
| BDNF | Mouse | NM_007540.4 | AGTCTCCAGGACAGCAAAGC | 614–633 | TGCAACCGAAGTATGAAATAACC | 685–707 |
| CNTF | Mouse | NM_170786.2 | TTGATTCCACAGGCACAAAA | 42–61 | CCCTGCCTGACTCAGAGGT | 85–103 |
| Edn2 | Mouse | NM_007902.2 | ACCTCCTCCGAAAGCTGAG | 502–520 | TTTCTTGTCACCTCTGGCTGTA | 556–577 |
| FGF2 | Mouse | NM_008006.2 | CGGCTCTACTGCAAGAACG | 285–303 | TGCTTGGAGTTGTAGTTTGACG | 371–392 |
| GAPDH | Mouse | NM_008084.2 | TGTCCGTCGTGGATCTGAC | 763–781 | CCTGCTTCACCACCTTCTTG | 818–837 |
| GFAP | Mouse | NM_010277.2 | ACAGACTTTCTCCAACCTCCAG | 1148–1169 | CCTTCTGACACGGATTTGGT | 1191–1210 |
| LEDGF | Mouse | NM_133948.4 | GGCCAGCAGTAAGACAAAGC | 1765–1784 | TGAAGTCTGCCGACCTAGTTAT | 1836–1857 |
| LIF | Mouse | NM_008501.2 | AAACGGCCTGCATCTAAGG | 172–190 | AGCAGCAGTAAGGGCACAAT | 245–264 |
| Norrin | Mouse | NM_010883.2 | CCCACTGTACAAATGTAGCTCAA | 729–751 | AGGACACCAAGGGCTCAGA | 802–820 |
| VEGF120 | Mouse | NM_001025250.2 | GGAGAGATGAGCTTCCTACAGCA | 743–765 | CTGAACAAGGCTCACAGTCATTTT | 888–905 |
| VEGF164 | Mouse | NM_001025257.2 | GGAGAGATGAGCTTCCTACAGCA | 743–765 | CCTTGGCTTGTCACATTTTTCT | 808–829 |
| β-Catenin | Rat | NM_053357.2 | GACCACAAGCAGAGTGCTGA | 1020–1039 | ACTCGGGTCTGTCAGGTGAG | 1109–1128 |
| BDNF | Rat | NM_012513.3 | AGCGCGAATGTGTTAGTGGT | 3372–3391 | GCAATTGTTTGCCTCTTTTTCT | 3416–3437 |
| FGF2 | Rat | NM_019305.2 | CGGTACCTGGCTATGAAGGA | 770–789 | GCGTTCAAAGAAGAAACACTCTT | 825–847 |
| Brn3b | Rat | NM_134355.1 | CAGCAGTTCCAGCAGCAGT | 453–471 | CATCCAGACCGCCGAATA | 545–562 |
| CD31 | Rat | NM_031591.1 | TGGATAATTGCCACTCTCACC | 1595–1615 | TTACCCGACAGGATGGAGAT | 1675–1694 |
| CNTF | Rat | NM_013166.1 | GACCTGACTGCTCTTATGGAATCT | 165–188 | GCCTGGAGGTTCTCTTGGA | 295–313 |
| Cyclophilin A | Rat | NM_017101.1 | AGCACTGGGGAGAAAGGATT | 160–179 | CCATTATGGCGTGTGAAGTC | 238–257 |
| GDNF | Rat | NM_019139.1 | CTAAGATGAAGTTATGGGATGTCG | 45–68 | CTTCGAGAAGCCTCTTACCG | 121–140 |
| Grm6 | Rat | NM_022920.1 | CGCAACTAGTCATCACCTTCG | 2233–2253 | CCCCAGCCATGCTATCAC | 2280–2297 |
| Iba1 | Rat | NM_017196.3 | CCGAGGAGACGTTCAGTTACTC | 388–409 | TGGCTTCTGGTGTTCTTTGTT | 480–500 |
| LEDGF | Rat | NM_175765.2 | CAAGCAGGAGGAACAAATGG | 1042–1061 | TTCTGGCTTCTTTCCTTCATC | 1082–1103 |
| LIF | Rat | NM_022196.2 | TGCCAATGCCCTCTTTATTT | 174–193 | CCGATACAGCTCGACCAACT | 302–321 |
| Opsin | Rat | NM_033441.1 | CCATCTACAACCCAATCATCTACA | 979–1002 | CTCCCAGTGGATTCTTGCCG | 1052–1071 |
| PEDF | Rat | NM_177927.2 | GGACTCTGATCTCAACTGCAAG | 809–830 | AAGTTCTGGGTCACCGTCAG | 882–901 |
| RPL32 | Rat | NM_013226.2 | ATCTGTTTTGCGGCATCAT | 39–57 | TTCCGCCAGTTTCGCTTA | 148–165 |
| Syntaxin1 | Rat | NM_053788.2 | CAAAGTTCGCTCCAAGCTAAA | 292–312 | GACGAGCGGTTCAGACCTT | 342–360 |
| Tyrosinase | Rat | NM_001107535.1 | TACTCAGCCCAGCGTCCTT | 849–867 | GGCTATTATACTCTTCTGATCTGCTG | 892–917 |
| VEGF164 | Rat | NM_001110336.1 | GCCAGCACATAGGAGAGATGAGC | 407–429 | CAAGGCTCACAGTGATTTTCTGG | 481–503 |
| α-Actin | Rat | NM_031004.2 | ATCACCAACTGGGACGACA | 289–307 | GCAGAGCATAGCCCTCATAG | 558–577 |
| CA | Rat | NM_019291.1 | ACTCCCAGGACTTTGCAGTG | 223–242 | GAGTGGTCAGAGAGCCAGGA | 587–606 |
| GFAP | Rat | NM_017009.2 | GAGAGGGACAATCTCA | 413–428 | TGTGAGGTCTGCAAACTTGG | 771–790 |
| GS | Rat | NM_017073.3 | TGAACAAAGGCATCAAGCAG | 146–165 | CCTGTTCCATTCCAAACCAG | 507–526 |
| Vimentin | Rat | NM_031140.1 | AGGTGGATCAGCTCACCAAT | 559–578 | TTGGCAGCCACACTTTCATA | 906–925 |
Protein preparation, Western and dot blot analyses.
After starvation without FBS overnight, confluent cultured Müller cells were incubated with Norrin (40 ng/ml) for 3–24 h. Müller cells were harvested in RIPA buffer (150 mm NaCl, 1% NP-40, 0.5% deoxycholic acid, 0.1% SDS, and 50 mm Tris), incubated on ice for 10 min, and pelleted by centrifugation. The supernatant was used for further analyses. In addition, conditioned cell culture medium of Müller cells was collected and subjected to dot blot analyses. For analyses of retinal proteins, eyes were dissected 24 h after intravitreal injection of 3 μl of PBS, NMDA (10 mm), and/or Norrin (5 ng/μl). Isolated retinae were homogenized in RIPA buffer, and insoluble constituents were removed by centrifugation. Differentiated RGC-5 cells (see above) were incubated in cell culture medium with or without 40 ng/ml Norrin. After incubation for 3 h, nuclear proteins of RGC-5 cells were isolated as described previously (Ohlmann et al., 2010). For β-catenin Western blot analyses, up to 20 μg of protein from cultured Müller cells, retinae, or RGC-5 cells were subjected to a 10% SDS-PAGE and transferred onto a PVDF membrane (Roche) by semidry blotting. After blocking with 5% low-fat milk in PBS-T, membranes were incubated overnight with rabbit anti-β-catenin antibodies (Cell Signaling Technology), diluted 1:1000 in 5% BSA in PBS-T. After washing, membranes were hybridized with HRP-conjugated chicken anti-rabbit antibodies using a 1:2000 dilution in PBS-T with 5% BSA. For visualization, membranes were incubated in Immobilon HRP substrate (Millipore) and visualized on a BAS 3000 Imager work station (Fujifilm). As loading control, HRP-conjugated anti-GAPDH antibodies were used (Rockland). For analyses of CNTF and FGF2 protein levels, 100 μl of conditioned cell culture medium were applied onto slots of a Minifold-SRC96-Dot Blot device (Schleicher and Schüll) and transferred onto a PVDF membrane via vacuum filtration. After blocking with 5% low-fat milk in PBS-T, membranes were incubated overnight with rabbit anti-FGF2 or rabbit anti-CNTF antibodies (Santa Cruz Biotechnology), both diluted 1:10,000 in 0.5% low-fat milk in PBS-T. As secondary antibody, HRP-conjugated chicken anti-rabbit was used in a 1:20,000 dilution in 0.5% BSA in PBS-T. Visualization was performed as described above. Finally, membranes were stained with Coomassie Blue as loading control. Western and dot blot signals were evaluated by densitometry with the Aida Image Analyzer v.4.06 software (Raytest).
Statistics.
All results are expressed as mean ± SEM. Comparisons between the mean variables of two groups were made by a two-tailed Student's t test. p values <0.05 were considered to be statistically significant.
Results
Norrin protects against NMDA-mediated excitotoxic neuronal damage
To investigate whether Norrin protects against NMDA-mediated neuronal damage, we analyzed the structures of optic nerves and retinae following an intravitreal injection of NMDA with and without Norrin. Since the amount of RGCs that are damaged by NMDA-induced excitotoxicity is dose dependent in the mouse eye (Li et al., 1999), we tested several doses in preliminary experiments and finally decided to inject 10 mm NMDA, which caused a substantial but not complete loss of RGC axons in the optic nerve of treated eyes. Three weeks after treatment with NMDA alone, a pronounced loss of axons and the formation of an extensive glial scarring were observed in semithin sections through optic nerves of treated eyes (Fig. 1A,B). In contrast, in optic nerves of mice that were injected with NMDA in combination with Norrin (Fig. 1C,D), the loss of axons and the area of glial scarring appeared to be considerably smaller than in eyes treated with NMDA alone (Fig. 1A,B). In PBS-injected control eyes, the structure of optic nerves was essentially normal (supplemental Fig. 2, available at www.jneurosci.org as supplemental material). By transmission electron microscopy, the remaining axons of both NMDA- and NMDA plus Norrin-treated animals were structurally intact, containing mitochondria, microtubules, and neurofilaments (Fig. 1E). Several axons that stained as intense dark spots by light microscopy (Fig. 1B,D) consisted of myelin whorls as seen by transmission electron microscopy (Fig. 1F), indicating axonal damage and ongoing remodeling processes. The quantitative analysis of the total number of axons in eyes injected with NMDA alone showed that 11,150 ± 858 axons (mean ± SEM) remained in the optic nerves 3 weeks after treatment, clearly indicating a substantial and highly significant loss of retinal ganglion cells when compared with the 41,754 ± 746 axons (mean ± SEM) in optic nerves from eyes that received PBS only. In contrast, in eyes that had been injected with the combination of NMDA and Norrin, almost 80% more optic nerve axons were present 3 weeks after treatment (Fig. 1G), a difference that was statistically significant (p < 0.05). To analyze whether the loss of optic nerve axons after NMDA treatment correlated with a loss of RGC perikarya, sagittal sections through eyes treated with NMDA or NMDA in combination with Norrin were analyzed. In marked correlation with the amount of axonal degeneration in the optic nerves, ∼55% more neuronal perikarya (3.64 ± 0.59 per 100 μm of retina) were observed in the ganglion cell layer of eyes treated with NMDA and Norrin than in eyes that received NMDA only (Fig. 2A,B) (2.35 ± 0.18 per 100 μm of retina, p < 0.025, n = 8). Although we were not able to differentiate RGCs from displaced amacrine cells in the ganglion cell layer, the results strongly indicated that the amount of axonal loss in the optic nerve correlated with a loss of RGC perikarya. To analyze whether the IPL, where synapses between RGCs and bipolar cells are located, is also affected by NMDA treatment, the total IPL area as seen in sagittal sections from eyes injected with NMDA or with NMDA in combination with Norrin was analyzed (Fig. 2C,D). In eyes treated with the combination of NMDA and Norrin, the area of the IPL from central to peripheral retina was ∼35% larger (38.2 ± 3.2 μm2 per μm of retina length) than in eyes injected with NMDA only (27.6 ± 1.2 μm2 per μm of retina length), an effect that was highly significant (p < 0.01, n = 8). Overall, the results strongly indicated the distinct potential of Norrin to suppress NMDA-induced excitotoxic damage of retinal neurons, including that of RGCs and their axons.
Figure 1.
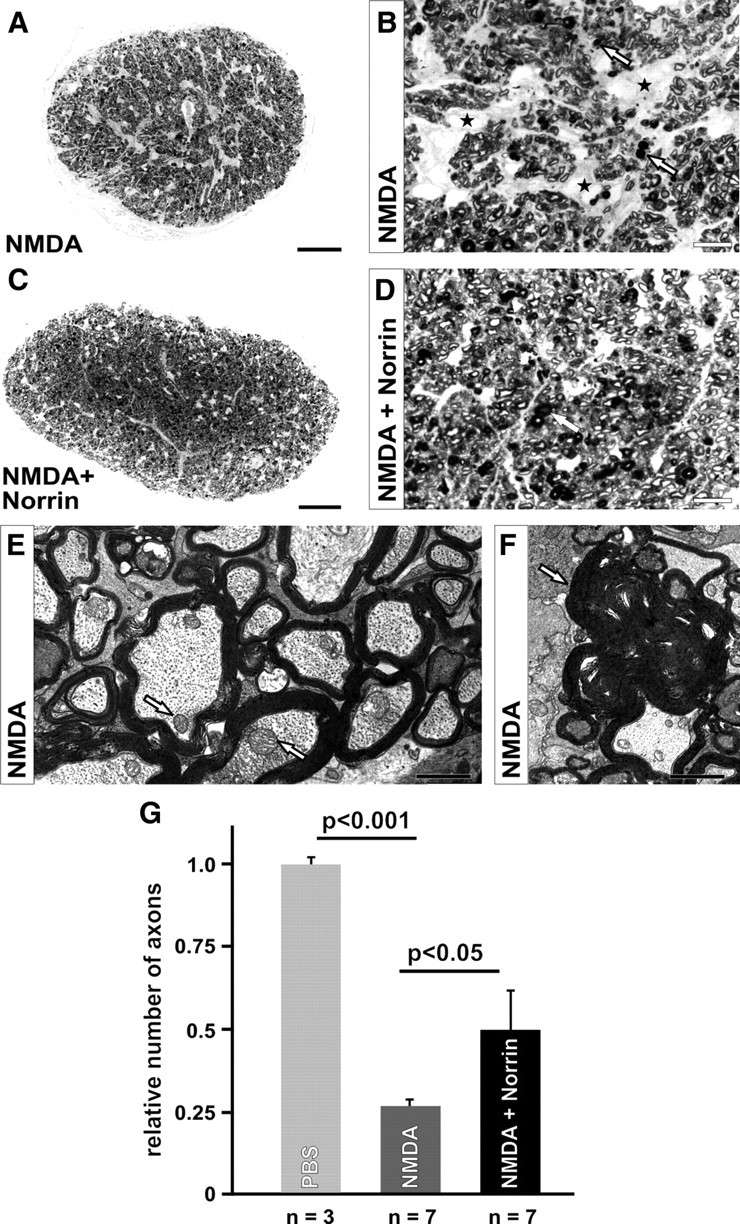
Norrin protects against NMDA-mediated loss of retinal ganglion cells. A–D, Representative sagittal semithin sections through optic nerves (A–D) of eyes 3 weeks after intravitreal injection of 3 μl of NMDA (10 mm) (A, B) or NMDA (10 mm) plus Norrin (5 ng/μl) (C, D) in the fellow eye. Three weeks after treatment with NMDA alone, a substantial loss of axons and the formation of an extensive glial scarring (asterisks) is observed in the optic nerve of the NMDA-treated eye (A, B). In contrast, in the optic nerve of the eye injected with NMDA in combination with Norrin (C, D), the loss of axons and the area of glial scarring are considerably smaller. Several axons exhibit a more intense staining of their myelin sheath, indicating axonal degeneration (arrows). E, F, Transmission electron microscopy of remaining axons of both NMDA- and NMDA plus Norrin-treated animals show structurally intact morphology with mitochondria (arrows), microtubules, and neurofilaments (E). Several axons that stain as intense dark spots by light microscopy (arrows, B, D) are degenerated and consist of myelin whorls (arrow, F). G, The total number of optic nerve axons was quantified and plotted as the relative number of optic nerve axons (mean ± SEM). Scale bars: A, C, 50 μm; B, D, 10 μm; E, 1 μm; F, 1.5 μm.
Figure 2.
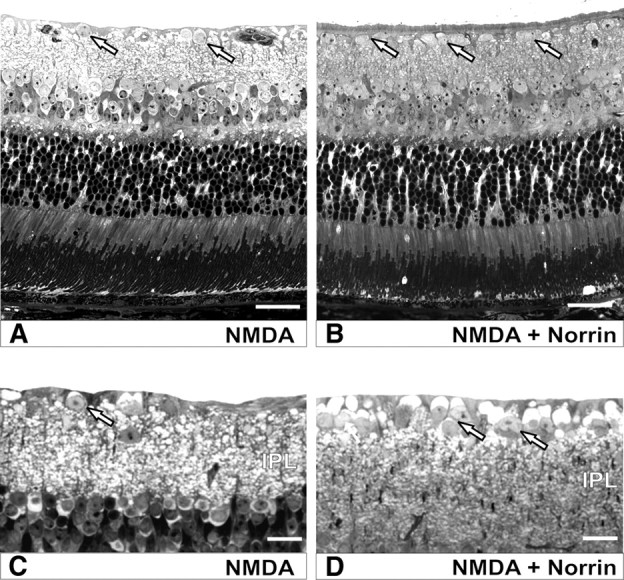
Norrin protects against NMDA-mediated loss of neurons in the retinal ganglion cell layer. Representative sagittal semithin sections through retinae of eyes 3 weeks after intravitreal injection of 3 μl of NMDA (10 mm) (A, C) or NMDA (10 mm) plus Norrin (5 ng/μl) (B, D) in the fellow eye. Eyes injected with NMDA alone (A, C) show less perikarya in the ganglion cell layer (arrows) and a thinner IPL than eyes injected with NMDA plus Norrin (B, D).
Norrin inhibits NMDA-induced apoptosis of RGCs
Neuronal cell death following NMDA treatment results from apoptosis (Li et al., 1999). To investigate whether the protective effects of Norrin on NMDA-mediated neuronal damage involve a decrease in NMDA-mediated apoptosis, nuclear fragmented DNA was investigated by TUNEL in sagittal sections of retinae 24 h following injection of NMDA plus Norrin or NMDA alone. In eyes that were injected with NMDA only, numerous TUNEL-positive cells were detected in both ganglion cell and inner nuclear layer (Fig. 3B). In contrast, in retinae of eyes injected with the combination of Norrin and NMDA, the number of TUNEL-positive cells was markedly smaller in both retinal layers (Fig. 3C). Negative controls that received PBS only showed a few apoptotic cells in the outer nuclear layer (Fig. 3A). Quantitative analysis confirmed that in eyes treated with NMDA plus Norrin, the number of TUNEL-positive neurons in the ganglion cell layer (44.4 ± 7.1%) was >30% smaller than in eyes injected with NMDA only (66.5 ± 8.6%), an effect that was highly significant (p < 0.01, n = 5). Based on the results obtained with TUNEL experiments, we concluded that the protective effects of Norrin on NMDA-mediated neuronal cell death result from a decrease in neuronal apoptosis.
Figure 3.
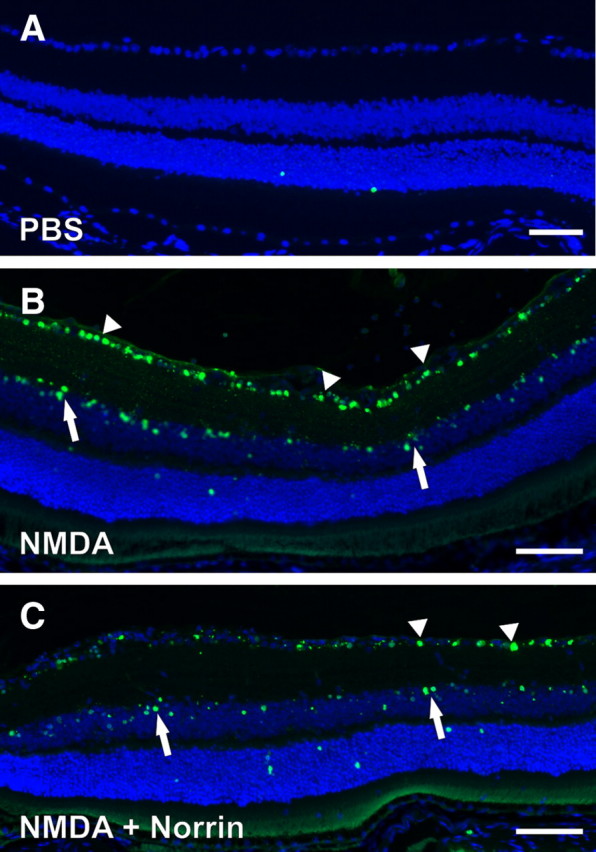
Norrin inhibits NMDA-mediated apoptosis of retinal neurons. Representative TUNEL staining (green) of sagittal sections through retinae 24 h after intravitreal injection of 3 μl of PBS (A) as well as NMDA (10 mm) in one eye (B) or NMDA (10 mm) plus Norrin (5 ng/μl) (C) in the fellow eye. After injection of PBS, only a few labeled cells in the outer nuclear layer were detected (A). In the retina of the eye injected with the combination of Norrin and NMDA (C), the number of TUNEL-positive cells is markedly smaller in ganglion cell layer (arrowheads) and inner nuclear layer (arrows) than in the retina treated with NMDA only (B). Blue, DAPI staining; scale bars in A–C, 50 μm.
Neuroprotective effects of Norrin are mediated via the Wnt/β-catenin signaling pathway
Since Norrin binds to Fzd 4 and is activating the canonical Wnt/β-catenin pathway (Xu et al., 2004; Smallwood et al., 2007), we next wanted to know whether the effects of Norrin on neuronal survival following NMDA-induced damage are also mediated via this pathway. To this end, DKK-1, an inhibitor of the Wnt/β-catenin pathway (Zorn, 2001), was injected together with Norrin and NMDA into the vitreous body of experimental animals, while the fellow eye received NMDA plus Norrin. Similar to our previous experiments, we detected a moderate reduction of axons and minor areas of glial scarring in optic nerves of eyes that had been treated with NMDA plus Norrin (Fig. 4A). In contrast, in eyes in which DKK-1 had been added to NMDA plus Norrin, broad glial scars and a marked loss of optic nerve axons were present (Fig. 4B), an effect that was quite similar to that seen in previous experiments in which eyes had been treated with NMDA alone. The quantitative analysis confirmed our qualitative assessment and showed that in optic nerves of eyes injected with NMDA plus Norrin, 69 ± 5.8% more axons were present than in eyes that were additionally treated with DKK-1, a result that was essentially comparable to that seen in the previous analysis of optic nerve axon number in NMDA- or NMDA plus Norrin-injected eyes (Fig. 1G) and that was highly significant (p < 0.01; n = 4). Since DKK-1 would not only inhibit signaling of recombinant Norrin, but also that of endogenous Wnt proteins in the retina, we next wanted to exclude that the effects of DKK-1 are due an inhibition of a putative neuroprotective action of retinal Wnt signaling. To this end, NMDA with DKK-1 was injected into the vitreous body of one eye, while the fellow eye was treated with NMDA only. Optic nerves from both eyes showed a dramatic reduction in the number of optic nerve axons, and no significant difference in the number of optic nerve axons was found between eyes treated with NMDA alone and those treated with NMDA in combination with DKK-1. In summary, the experiments involving DKK-1 strongly suggested that Norrin protects RGCs via activation of the Wnt/β-catenin signaling pathway.
Figure 4.
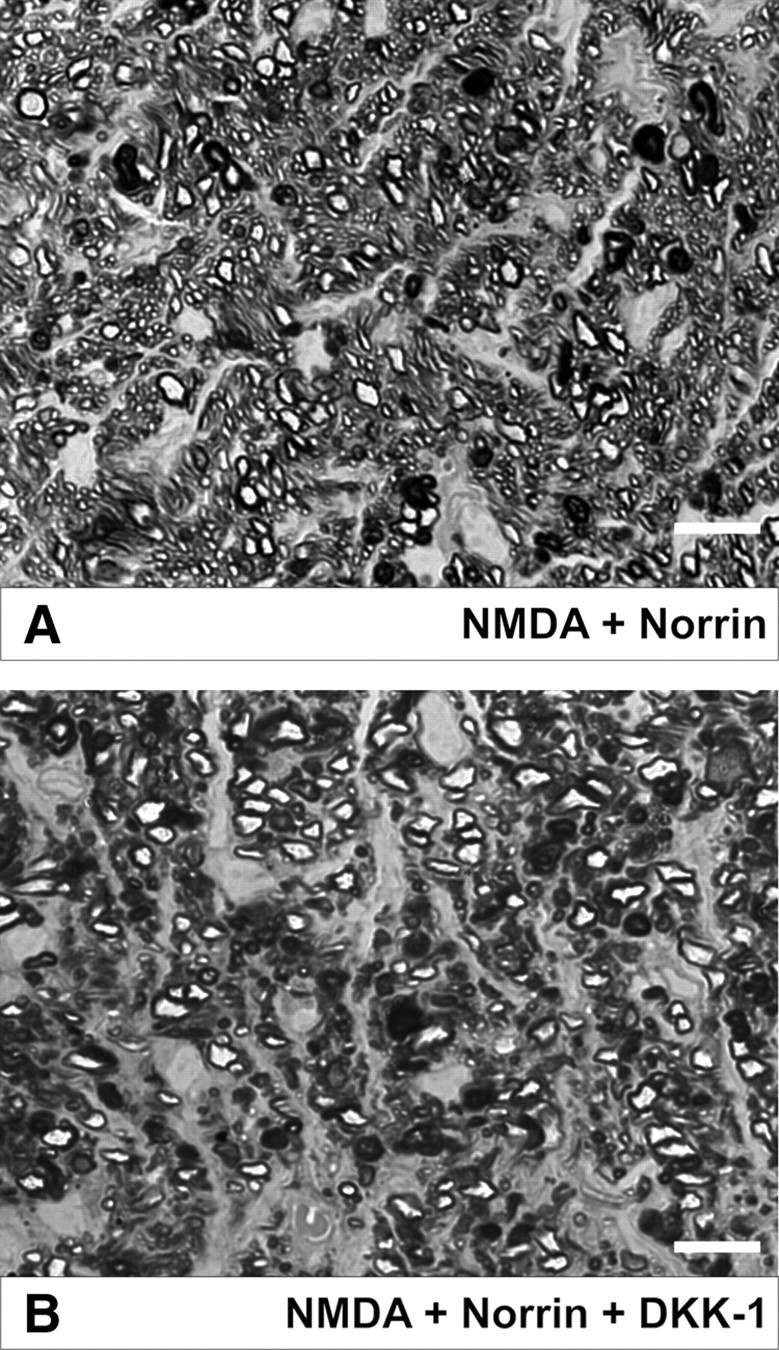
Norrin mediates its protective effect via the classical Wnt/β-catenin signaling pathway. Representative sagittal semithin sections of optic nerves from eyes 3 weeks after intravitreal injections of NMDA (10 mm) and Norrin (5 ng/μl) (A) or NMDA (10 mm), Norrin (5 ng/μl), and DKK-1 (5 μg/μl) (B) in the fellow eye. A, A moderate reduction of axons and minor areas of glial scarring are present in the optic nerve of an eye that has been treated with NMDA plus Norrin. B, In the optic nerve of an eye in which DKK-1 has been added to NMDA plus Norrin, broad glial scars and a marked loss of optic nerve axons are present. Scale bars: A, B, 10 μm.
Norrin activates the Wnt/β-catenin pathway in retinae and is expressed after NMDA injection
Next we wanted to confirm in parallel experiments that recombinant Norrin activates the Wnt/β-catenin pathway in normal retinae and in those treated with NMDA. Again eyes received an intravitreal injection of Norrin or NMDA, while the fellow eye was treated with an injection of either PBS or NMDA with Norrin. By Western blot analyses of retinal proteins from eyes that had received PBS, only a weak signal for β-catenin was detectable (Fig. 5A). In fellow eyes that had received an injection of NMDA, the signal for retinal β-catenin was readily detectable and more intense than that of PBS-injected eyes (Fig. 5A). Densitometry of several experiments revealed a moderate 1.5-fold increase of β-catenin levels after injection of NMDA when compared with PBS-injected eyes (Fig. 5B), indicating that NMDA itself can activate a moderate Wnt/β-catenin signaling in retinae. However, after injection of Norrin a pronounced major signal for β-catenin at ∼95 kDa was observed in comparison to PBS controls (Fig. 5A). In addition, three faster migrating minor bands were observed in proteins from eyes treated with Norrin (Fig. 5A), very likely indicating some minor proteolytic cleavage (Van de Craen et al., 1999). Relative densitometry analysis of multiple experiments confirmed that retinal β-catenin levels of eyes injected with Norrin were ∼3.0-fold higher than those in eyes injected with PBS alone, a difference that was statistically significant (Fig. 5B) (p < 0.05). Since NMDA itself activates Wnt/β-catenin signaling, we next analyzed whether the additional injection of Norrin further increases retinal β-catenin levels. Again a substantial signal for β-catenin was detected after intravitreal injection of NMDA that was markedly increased after the additional treatment with Norrin (Fig. 5A). Relative densitometry analysis revealed a 1.8-fold increase of retinal β-catenin level after NMDA plus Norrin injection when compared with NMDA-treated eyes and a 2.8-fold increase when compared with PBS controls (Fig. 5B). Since the difference in the amounts of retinal β-catenin was higher between PBS-treated and Norrin-treated eyes than between NMDA- and NMDA plus Norrin-treated eyes, we wanted to know whether the injection of NMDA alone induces the expression of endogenous retinal Norrin that could activate the Wnt/β-catenin pathway by itself. To this end, we performed quantitative real time RT-PCR experiments with RNA from eyes treated with PBS and compared it with RNA from NMDA-injected eyes. By quantitative real-time RT-PCR, the mRNA expression of Norrin showed a highly significant increase (p < 0.001, n = 3), which was up to fivefold after treatment with NMDA as compared to eyes that received PBS only. Overall, the experiments confirmed that recombinant Norrin activates Wnt/β-catenin signaling in the normal and NMDA-injured retina. In addition, the results indicate that Wnt/β-catenin signaling is activated following NMDA-injury alone, albeit to a lesser extent than after treatment with recombinant Norrin, an effect that might be mediated by an increased expression of endogenous Norrin.
Figure 5.

Norrin activates the Wnt/β-catenin pathway in normal and injured retinae. Western blot analyses (A) and relative densitometry (B) of β-catenin in retinal proteins after intravitreal injection of 3 μl of PBS, Norrin (5 ng/μl), NMDA (10 mm), or NMDA (10 mm) plus Norrin (5 ng/μl) (mean ± SEM, n = 4, *p < 0.01).
Norrin increases activation of Müller cells and enhances Edn2/Lif signaling in injured retinae
NMDA-induced RGC damage causes an activation of Müller cells, which release tumor necrosis factor-α, which contributes to neuronal cell death (Lebrun-Julien et al., 2009). On the other hand, in experimental models of photoreceptor damage, there is considerable evidence that activated Müller cells produce and release neurotrophic growth factors that critically support survival of photoreceptors (Rattner and Nathans, 2005; Joly et al., 2008). We therefore wondered whether the mechanism by which Norrin protects RGCs from apoptotic NMDA-induced damage depends on a modification of Müller cell signaling. To this end, we first examined the expression of mRNA for GFAP, a marker for Müller cell activation in RNA from treated retinae. In virtually all models studied, retinal Müller cells increase the expression of GFAP in response to neuronal degenerative processes in the retina (Bringmann et al., 2006). As expected, retinal mRNA for GFAP increased significantly (p < 0.01) in RNA from NMDA-injected eyes as compared to eyes that had been injected with PBS (Fig. 6A). Moreover, the combined injection of NMDA and Norrin induced an additional significant (p < 0.01) increase in the expression of GFAP mRNA as compared to eyes injected with NMDA alone (Fig. 6A), indicating an enhancement of Müller cell activation by Norrin in the NMDA-damaged retina. Injection of Norrin alone did not change GFAP mRNA expression in treated retinae, suggesting that Norrin does not increase GFAP expression by itself, but rather needs additional signals that derive from the injured retina to induce Müller cell activation (Fig. 6A). Next we investigated the retinal expression of leukemia inhibitory factor (Lif), which is a Müller cell-derived neuronal survival factor that is required for Müller cell activation in mouse models of hereditary photoreceptor damage (Joly et al., 2008). After intravitreal injection of Norrin, a moderate albeit not significant increase of Lif mRNA expression was observed (Fig. 6B). However, the injection of NMDA significantly (p < 0.001) increased the expression of Lif mRNA in retinal RNA up to 5.8-fold as compared to PBS-injected controls (Fig. 6B). The substantial induction of Lif mRNA that had been observed after NMDA treatment further increased up to 9.3-fold in eyes that were treated with combined NMDA and Norrin, an effect that was again highly significant (Fig. 6B) (p < 0.01). There is evidence that Lif induces the expression of endothelin-2 (Edn2) in photoreceptors and that Edn2 signals onto Müller cells to promote their activation (Joly et al., 2008). Again, the injection of Norrin alone did not alter Edn2 mRNA expression in treated retinae (Fig. 6C). In contrast, intravitreal injection of NMDA significantly (p < 0.001) increased the expression of Edn2 mRNA in retinal RNA up to 5.6-fold as compared to PBS-injected controls (Fig. 6C). The induction of Edn2 mRNA that had been observed after NMDA treatment further increased up to 13.6-fold in eyes that were treated with combined NMDA and Norrin, an effect that was again highly significant (Fig. 6C) (p < 0.001). In summary, our data strongly suggested that Norrin not only enhances activation of Müller cells upon retinal injury, but also induces the expression of Lif and Edn2 mRNA in the retina, indicating that the Lif/Edn2 signaling pathway is involved in Norrin-mediated protection of RGCs following excitotoxic damage.
Figure 6.
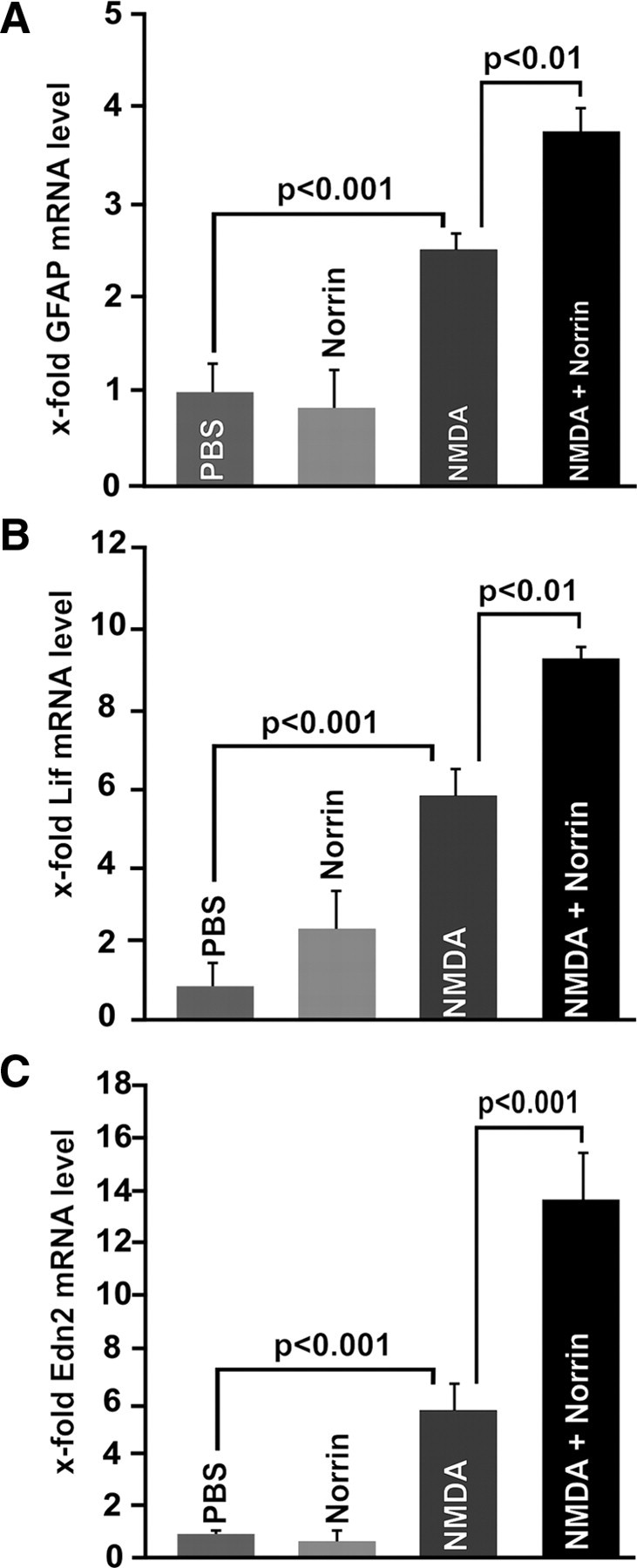
Norrin activates Müller cells and enhances Lif/Edn2 signaling in injured retinae. Quantitative real-time RT-PCR for GFAP (A), Lif (B), and Edn2 (C) mRNA in RNA from retinae 7 h after injection of 3 μl of PBS, Norrin (5 ng/μl), NMDA (10 mm), or NMDA plus Norrin (5 ng/μl) (mean ± SEM, n ≥ 6).
Norrin induces the expression of neuroprotective factors in the retina
Since activated Müller glia is known to secrete several neurotrophic factors, we next investigated by quantitative real-time RT-PCR the expression of mRNA for fibroblast growth factor-2 (FGF2), brain-derived neurotrophic growth factor (BDNF), ciliary neurotrophic growth factor (CNTF), vascular endothelial growth factor (VEGF), and lens epithelium-derived growth factor (LEDGF), in RNA isolated from retinae of treated animals. In eyes that were treated with combined NMDA/Norrin, no changes in mRNA expression for the splice variants VEGF 164 and 120 were detected when compared to eyes that were injected with NMDA alone (Fig. 7A). In contrast, a significant (p < 0.01) increase in mRNA expression was observed for LEDGF, BDNF, FGF2, and CNTF after injection of NMDA plus Norrin when compared to injection of NMDA alone (Fig. 7A). Because of the strong neuroprotective potential of FGF2 in the retina, we further analyzed whether the increase in mRNA for FGF2 leads to an increase in FGF2 translation. By dot blot analysis of multiple experiments, a significant (p < 0.01, n = 7) up to 3.8-fold increase in the amounts of FGF2 was observed in proteins from retinae of mice treated with NMDA plus Norrin when compared to eyes injected with NMDA alone (Fig. 7B). Next we examined whether Norrin alone induces the expression of neuroprotective factors in the retina, or whether an additional stress signal such as NMDA injury is needed. To this end the expression of mRNA for neurotrophic growth factors was analyzed in retinal RNA from animals that were injected with PBS in one eye and Norrin in the fellow eye. Following treatment with Norrin, no change in retinal expression for VEGF 164, LEDGF, BDNF, and CNTF mRNA was detected when compared to eyes that received PBS only (Fig. 7C). The expression of the splice variant VEGF 120 was significantly reduced in Norrin-treated eyes. In contrast, Norrin significantly (p < 0.01) induced up to 1.9-fold the expression of FGF2 mRNA as compared to PBS controls (Fig. 7C), albeit the mRNA levels were ∼50% lower than those of NMDA- or NMDA plus Norrin-treated eyes (Fig. 7A). By dot blot analyses of multiple experiments, a significant (p < 0.01, n = 7) 2.0-fold increase in the amounts of retinal FGF2 was observed in retinal proteins from Norrin-treated animals when compared with eyes of animals that were injected with PBS alone (Fig. 7D). We concluded that Norrin mediates its neuroprotective properties, at least partially, via the induction of neurotrophic growth factors. Moreover, the expression of growth factors following treatment with Norrin is substantially increased in retinae that have been injured by NMDA-treatment.
Figure 7.

Norrin induces expression of neurotrophic factors in the retina. A, C, Quantitative real-time RT-PCR for neurotrophic factors 7 h after injection of 3 μl of NMDA (10 mm), of NMDA (10 mm) plus Norrin (5 ng/μl) (A; mean ± SEM, n ≥ 4, *p < 0.01, **p < 0.001), or of PBS or Norrin (5 ng/μl) (C; mean ± SEM, n ≥ 4, *p < 0.01). B, D, Dot blot analyses for FGF2 levels in proteins from NMDA- (10 mm), NMDA (10 mm) plus Norrin- (5 ng/μl) (B), PBS-, or Norrin- (5 ng/μl) (D) treated retinae 24 h after injection.
Norrin activates the Wnt/β-catenin pathway and induces β-catenin mRNA expression in Müller cells
Next we wanted to test whether Müller cells are a direct target of Norrin. We isolated Müller cells from the retinae of rat eyes and first examined the purity of established primary cultures by immunohistochemical analyses for specific markers of Müller cells or retinal neurons. By semiquantitative RT-PCR, mRNA for typical Müller cell transcripts such as carboanhydrase, glutamine synthetase, and vimentin was detected in RNA from cultured Müller cells (supplemental Fig. 1A, available at www.jneurosci.org as supplemental material) and in RNA from fresh rat retinae that were used as positive control (supplemental Fig. 1A, available at www.jneurosci.org as supplemental material). Expression of GFAP mRNA was only detected in RNA from cultured Müller cells, but not in that from fresh retinae (supplemental Fig. 1A, available at www.jneurosci.org as supplemental material). Subsequently we continued with quantitative real-time RT-PCR for specific transcripts of RGCs, microglia, endothelial cells, bipolar neurons, photoreceptors, amacrine neurons, retinal pigmented epithelium, and pigmented ciliary epithelial cells in RNA of Müller cell cultures and compared the results with that obtained in RNA from fresh retinae (supplemental Fig. 1B, available at www.jneurosci.org as supplemental material). Neither mRNA for Brn3b, a marker for RGCs, nor for Grm6, a marker for bipolar neurons, could be detected in RNA isolated from Müller cell cultures. The same was true for Iba1, a marker for microglia, and for tyrosinase, a marker for RPE and pigmented ciliary epithelial cells (supplemental Fig. 1B, available at www.jneurosci.org as supplemental material). In contrast, mRNA for all of these proteins could be readily detected in RNA from fresh retinae. mRNA for syntaxin 1, a marker for amacrine neurons, was detectable in RNA from Müller cells, as was mRNA for CD31, a marker for vascular endothelial cells. Still, the amounts of mRNA for syntaxin 1 and CD31 in RNA of Müller cell cultures was minimal, if compared with that detected in RNA from fresh retinae. Finally, immunohistochemical stainings were performed for vimentin, glutamine synthetase, and GFAP, which showed immunoreactivity for all of these proteins in >80% of cells (supplemental Fig. 1C–E, available at www.jneurosci.org as supplemental material). In summary, we were able to isolate primary cell cultures that were highly enriched in Müller cells.
Next we analyzed whether Norrin can activate the Wnt/β-catenin pathway in primary Müller cell cultures. To this end, we investigated the intracellular accumulation of β-catenin by immunohistochemistry. An incubation of cultured Müller cells with Norrin for 3 h induced a substantial accumulation of β-catenin in the nucleus and perinuclear cytoplasm (Fig. 8B, arrows) as well as in region of focal contacts (Fig. 8B, arrowheads) when compared with untreated control cells (Fig. 8A). The results obtained with immunohistochemistry correlated with that obtained by Western blot analyses. After treatment with Norrin, the amounts of β-catenin in proteins from cultured Müller cells increased up to 1.6-fold as compared to untreated control cells (Fig. 8C). To analyze whether Norrin not only causes an accumulation of β-catenin, but also increases the expression of β-catenin mRNA, we investigated by quantitative real-time RT-PCR the expression of β-catenin mRNA in RNA from cultured Müller cells. After incubation of the cells with Norrin for 7 h, a highly significant increase (p < 0.01) of β-catenin mRNA was observed in RNA of treated cells, which was nearly twofold when compared to RNA of untreated control cells (Fig. 8D). The increase in β-catenin mRNA could be blocked completely by an additional incubation with DKK-1 (Fig. 8D). In summary, the results indicated that Norrin can activate the Wnt/β-catenin pathway in Müller cells. The activation of Wnt/β-catenin signaling not only induces the intracellular accumulation of β-catenin, but also increases the transcription of β-catenin mRNA.
Figure 8.

Norrin activates the Wnt/β-catenin pathway and induces β-catenin mRNA expression in Müller cells. A, B, Immunohistochemistry for β-catenin (green) in Müller cells after incubation with Norrin (40 ng/ml) for 3 h. By confocal microscopy, an intense accumulation of β-catenin is seen in nucleus and perinuclear cytoplasm of Norrin-treated Müller cells (arrows, B) when compared with untreated controls (A). In addition, β-catenin accumulates in regions of focal contacts after incubation with Norrin (arrowheads, B), whereas in control cells, only weak staining is observed (A). Blue, DAPI staining. Scale bars: A, B, 20 μm. C, Western blot analysis of β-catenin in total protein from Müller cells after treatment with Norrin (40 ng/ml) for 3 h. D, Quantitative real-time RT-PCR for β-catenin mRNA in RNA from Müller cells after incubation with DKK-1 and/or Norrin for 7 h, and in untreated controls (mean ± SEM; n > 3; *p < 0.05; **p < 0.01).
Norrin induces the expression of neuroprotective growth factors in cultured Müller cells via the Wnt/β-catenin pathway
To investigate whether Müller cells are the likely source for the enhanced expression of LEDGF, BDNF, FGF2, and CNTF in the retina after combined injection of NMDA and Norrin, the expression of neurotrophic factors was analyzed in cultured Müller cells after treatment with Norrin. In Müller cell cultures that were treated with Norrin, no significant changes in mRNA expression for the splice variant VEGF 164, for GDNF (glial cell line-derived neurotrophic factor), or for LEDGF was detected when compared to RNA of untreated control cultures (Fig. 9A). In contrast, significant changes in mRNA levels were observed for Lif, pigment epithelium-derived growth factor (PEDF), BDNF, FGF2, and CNTF after incubation for 7 h with Norrin (Fig. 9A). To exclude that NMDA has any effects on growth factor expression by itself, Müller cells were incubated with NMDA and compared with untreated controls. No changes in mRNA expression for BDNF, FGF-2, and CNTF were observed (data not shown). To analyze whether Norrin mediates the induction of neurotrophic growth factors via the Wnt/β-catenin pathway, cells were additionally incubated with DKK-1. After coincubation with DKK-1, the mRNA levels for BDNF, FGF2, and CNTF were reduced significantly (Fig. 9A). Next we investigated by dot blot analysis whether the increase in mRNA levels for FGF2 and CNTF following treatment with Norrin also results in an increase of translation and secretion of both growth factors in the culture medium. After incubation for 24 h with Norrin, we detected a distinct and significant increase in the amounts of FGF2 up to 2.2-fold (p < 0.05, n = 7) and CNTF up to 3.7-fold (p < 0.001, n = 7) in culture medium of Müller cells when compared to medium from untreated control cells (Fig. 9B,C). Our data obtained in cultured Müller cells strongly suggested that Müller glia is the source for the increased expression of neurotrophic growth factors following treatment with Norrin and subsequent activation of the Wnt/β-catenin signaling pathway.
Figure 9.

Norrin induces the expression of neurotrophic growth factors in cultured Müller cells via the Wnt/β-catenin pathway. A, Quantitative real-time RT-PCR for neurotrophic growth factors in RNA of Müller cells after incubation for 7 h with DKK-1(100 ng/ml) and/or Norrin (40 ng/ml), and in untreated controls (mean ± SEM; n ≥ 4; *p < 0.05; **p < 0.01). B, C, Dot blot analyses of proteins from cell culture medium of Müller cells for FGF2 (B) and CNTF (C) after incubation for 24 h with Norrin (40 ng/ml) and in untreated controls.
Norrin mediates neuroprotective properties on immortalized RGCs in vitro
To analyze whether Norrin has also direct neuroprotective properties in vitro, differentiated immortalized RGC-5 cells derived from rat retinal ganglion cells (Krishnamoorthy et al., 2001) were incubated with Norrin (Fig. 10A) or Norrin with DKK-1 (Fig. 10B). First RGC-5 cells were kept in culture medium without FBS, which caused a distinct decrease in cell viability when compared to control cultures kept in 10% FBS (Fig. 10A). When Norrin was added to RGC-5 cultures kept in growth factor-deprived serum-free culture medium, the number of surviving cells increased by ∼20% (Fig. 10A), an effect that was highly significant (p < 0.001). To investigate whether the protective effect of Norrin is also mediated via an activation of the Wnt/β-catenin pathway, cells were treated with combined Norrin/DKK-1. Treatment of differentiated RGC-5 cells with combined Norrin/DKK-1 did not decrease cell survival when compared with cells that were incubated with Norrin only (Fig. 10B), strongly indicating that the direct neuroprotective effect of Norrin on RGC-5 cells in vitro is not mediated via the Wnt/β-catenin signaling pathway. The observation correlated with Western blot analyses from nuclear proteins of Norrin-treated differentiated RGC-5 cells that did not show any difference in the amounts of β-catenin levels when compared to untreated controls (Fig. 10C). To analyze whether Norrin mediates its neuroprotective properties on differentiated RGC-5 cells via an induction of neurotrophic growth factors, mRNA for FGF2, BDNF, CNTF, LEDGF, and PEDF was analyzed after incubation with Norrin. In control RGC-5 cultures, mRNA for several growth factors except that of BDNF was detected in substantial amounts by quantitative real-time RT-PCR. After treatment of RGC-5 cells with Norrin, neither growth factor showed significant changes regarding the amounts of its mRNA when compared to control cultures (Fig. 10D). Since Norrin induces the expression of neurotrophic growth factors in cultured Müller cells, we next wondered whether treatment of RGC-5 cells with conditioned cell culture medium obtained from Müller cells stimulated with Norrin has similar effects on survival of RGC-5 cells. After incubation in conditioned cell culture medium derived from control Müller cells that had not been treated with Norrin, the viability of differentiated RGC-5 increased up to 20% when compared to control cultures that only received normal medium without FBS (Fig. 10E). The positive effect on cell survival further increased significantly by nearly 20% when conditioned cell culture medium of Norrin-treated Müller cells was used (Fig. 10E). In summary, our observations indicate direct and indirect neuroprotective properties of Norrin on immortalized RGCs in vitro. Direct effects are not mediated via an activation of the classical Wnt/β-catenin signaling. Indirect effects involve Norrin-induced neurotrophic factors in Müller cells that protect immortalized RGC-5 from apoptotic cell death.
Figure 10.

Norrin induces survival in cultured RGC-5 cells. A, B, Quantification of cell number in differentiated RGC-5 cells after incubation for 24 h in serum-free medium with and without Norrin (A; mean ± SEM of 3 independent experiments; n ≥ 48; ***p < 0.001) or with 40 ng/ml Norrin with or without 100 ng/ml DKK-1 (B; mean ± SEM of 3 independent experiments; n ≥ 32; **p < 0.001). C, Western blot analysis for β-catenin in nuclear proteins from differentiated RGC-5 cells after incubation with Norrin (40 ng/ml) for 3 h. D, Quantitative real-time RT-PCR for neurotrophic growth factors in RNA of differentiated RGC-5 cells after incubation for 7 h with 20 or 40 ng/ml Norrin, and in untreated controls (mean ± SEM; n ≥ 4). E, Quantification of cell number in differentiated RGC-5 cells after incubation for 24 h in serum-free medium or after treatment with serum-free conditioned (cond.) cell culture medium of Müller cells that were preincubated with and without 40 ng/ml Norrin (mean ± SEM of 3 independent experiments; n ≥ 28; **p < 0.01; ***p < 0.001).
Discussion
We conclude that Norrin has distinct neuroprotective properties that save retinal neurons from excitotoxic damage, an effect that is mediated via activation of the Wnt/β-catenin signaling pathway and that involves an increased expression of Müller cell-derived neuroprotective growth factors. This conclusion rests upon (1) the capability of Norrin to substantially increase survival of RGCs and their axons following excitotoxic damage, (2) the observations that the Wnt/β-catenin signaling pathway is activated in fresh retinae and cultured Müller cells following treatment with Norrin, (3) the potential of DKK-1 to block the neuroprotective effects of Norrin upon excitotoxic damage, and finally (4) the finding that treatment with Norrin leads to an increase in the expression of neurotrophic growth factors in damaged retinae and in cultured Müller cells.
Treatment with NMDA causes excitotoxicity of neurons by hyperactivation of NMDA-type glutamate receptors, which results in a massive Ca2+ influx that subsequently propagates proapoptotic signaling cascades (Shen et al., 2006). In the retina, RGCs and amacrine cells are particularly sensitive to excitotoxicity, and excess glutamate has been proposed to underlie common neurodegenerative disorders of the eye, including glaucoma and retinal artery occlusion (Seki and Lipton, 2008). In our study, we used two complementary techniques to quantify neuronal cell death following NMDA-mediated injury: counting of the total number of optic nerve axons as a measure of the number of surviving RGCs, and TUNEL of neurons on retinal cross sections. In the mouse eye, neuronal cell loss following topical injection of NMDA is dose dependent (Li et al., 1999). Lebrun-Julien et al. (2009) injected 20 mm NMDA into the vitreous body of C57BL/6 mice, and observed a loss of >90% of RGCs 24 h after treatment. We injected a lower dose (10 mm) and observed a remaining number of ∼11,000 RGCs axons 3 weeks after treatment. Compared with PBS-injected eyes that had ∼42,000 RGC axons, excitotoxicity induced by NMDA-treatment caused a loss of >70% of optic nerve axons. The number of surviving axons increased by ∼80% when Norrin was added to NMDA, an effect that was blocked completely upon adding DKK-1 to combined NMDA/Norrin. DKK-1 antagonizes Wnt signaling by acting as an inhibitory ligand of the Lrp5/6 coreceptor, which prevents the formation of Wnt-Fzd-Lrp5/6 complexes, and blocks the canonical Wnt/β-catenin pathway (Zorn, 2001). Norrin binds with high affinity to Fzd 4 and activates the canonical Wnt/β-catenin pathway (Xu et al., 2004; Smallwood et al., 2007). It seems reasonable to assume that the neuroprotective effects observed in the present study depend on a Norrin-mediated activation of Wnt/β-catenin signaling that is blocked by adding DKK-1. Norrin treatment leads to a significant reduction in the number of TUNEL-positive neurons in the NMDA-injured retina, strongly indicating that the number of neurons undergoing apoptotic cell death decreases following Norrin-mediated activation of Wnt/β-catenin signaling. It is of interest to note that the number of optic nerve axons in PBS-treated animals (41,754 ± 746) was somewhat lower than that observed by others in untreated C57BL/6 mice, which counted 45,400 ± 4000 (Bonfanti et al., 1996) or 55,500 ± 1300 (Strom and Williams, 1998) optic nerve axons, respectively. Injury caused by an intravitreal injection of PBS might already cause some damage to RGCs and their axons.
Our data indicate that several protective processes are initiated in the NMDA-injured retina upon treatment with Norrin that each contribute to neuronal survival and protection from apoptotic cell death. The processes include activation of Müller glia, which appears to be a very important component of Norrin-mediated neuroprotection. In virtually all models of neuronal degenerative processes in the retina that have been studied, Müller cells become activated and very characteristically increase the expression of GFAP as hallmark of their activation (Bringmann et al., 2006). Accordingly, we observed an increase in Müller cell GFAP expression after NMDA-mediated excitotoxic damage corroborating previous findings (Lebrun-Julien et al., 2009). There is evidence from recent studies that activation of Müller cells depends on an extensive intraretinal signaling system. Components of this signaling system have been characterized in animal models of acute and chronic experimental photoreceptor degeneration and involve Lif, Edn2, and FGF2 (Rattner and Nathans, 2005; Joly et al., 2008). According to the model proposed by Joly et al. (2008), Müller cells appear to be able to sense neuronal damage in the retina by an as yet unidentified mechanism and respond with the expression of Lif, which in turn induces the upregulation of Edn2 in photoreceptors. Edn2 functions as general stress signal that signals back to Müller cells to induce their activation (Rattner and Nathans, 2005), and activated Müller cells in turn express substantial amounts of FGF2, which contributes to prevent neuronal cell death. In support of this model, neither GFAP nor Edn2 are upregulated upon photoreceptor damage in Lif−/− mice in which FGF2 is not upregulated following photoreceptor damage and neuronal cell death is strongly accelerated (Joly et al., 2008). Our results indicate that the Lif/Edn2 system is also upregulated upon excitotoxic damage of the inner retina. Moreover, treatment with Norrin further augments the induction of Lif, Edn2, and GFAP in the NMDA-injured retina, an effect that very likely contributes to an increased activation of Müller glia, and to the substantial increase in the retinal expression of neurotrophic factors that we observed in eyes treated with combined NMDA/Norrin.
The factors that were found to be upregulated in their retinal expression included LEDGF, BDNF, FGF2, and CNTF, and have all been implicated in paracrine pathways of neuroprotection of retinal neurons in various models of degeneration, including excitotoxic damage of the inner retina (Mey and Thanos, 1993; Unoki and LaVail, 1994; Wen et al., 1995; Gao and Hollyfield, 1996; Cayouette et al., 1998; Ogata et al., 2001; Inomata et al., 2003; Schuettauf et al., 2004; Joly et al., 2007). Since all of these factors were also upregulated in their expression in cultured Müller glia following treatment with Norrin, we assume that Müller cells are their major source in retinae of NMDA-injured eyes treated with Norrin. In support of this assumption is our observation that treatment with DKK-1 almost completely prevented the effects of Norrin on the increased mRNA expression of neurotrophic factors in cultured Müller cells, similar to its prevention of the protective effects of Norrin upon RGC damage in vivo. Norrin induced an accumulation of β-catenin in cultured Müller cells and its translocation into the nucleus, and increased the expression of β-catenin mRNA, an effect that again was blocked by adding DKK-1. The presence of the required cellular pathways to activate canonical Wnt/β-catenin signaling in Müller cells has been shown previously by treating cultured cells with Wnt3a, or with LiCl or SB216763, which are both chemical activators of Wnt signaling (Yi et al., 2007). It is of interest to note that Norrin did also induce the expression of Lif in cultured Müller cells, an observation that indicates that Norrin and the Wnt/β-catenin signaling pathway act upstream of the Lif/Edn2 intraretinal signaling system. The neuroprotective effects of Norrin appear to be considerably more important during events of retinal injury than in the normal eye, where Norrin upregulated neither the expression of Lif to levels that were significant, nor the expression of other neuroprotective factors, with the notable exception of FGF-2. The expression of Norrin, which very likely occurs in Müller cells (Ye et al., 2009), was considerably upregulated in the NMDA-damaged retina. The molecular mechanisms that are causative for this upregulation remain unclear at the moment.
While our results clearly argue for an important role of Müller cell-derived neurotrophic factors in mediating the neuroprotective effects of Norrin, we also investigated whether Norrin has direct protective effects on RGCs since Lin et al. (2009) reported that Norrin significantly reduced staurosporine-induced cell death of RGC-5 cells, an effect that was only incompletely blocked by DKK-1. In our experiments, we observed a comparable positive role of Norrin on RGC-5 survival. The effect, which was not dose dependent, was not inhibited by DKK-1, though, and did not involve an accumulation of β-catenin, clearly arguing against a role of Wnt/β-catenin signaling. An involvement of neurotrophic factors in the direct effects of Norrin on RGC-5 survival appears to equally unlikely, as the expression of such factors was not increased upon treatment of RGC-5 with Norrin, and neurotrophic factor-enriched conditioned Müller cell culture medium was additive to the direct effects of Norrin. While the molecular mechanisms of the direct effects of Norrin on RGC-5 survival in vitro are unclear at the moment, it appears to be highly unlikely that they play a major role in the protective effects of Norrin on NMDA-mediated excitotoxic damage of RGCs in the retina, which could be completely blocked upon adding DKK-1.
Our results show Norrin is part of an endogenous protective system in the retina that is important for RGC survival during and after injury, and that appears to act largely independently from the effects of Norrin on vascular development and repair (Ye et al., 2009; Ohlmann et al., 2010). Norrin and the molecules involved in its signaling pathway appear to be promising targets to develop strategies that increase RGC survival not only in experimental animal models, but also in patients with RGC damage following ischemia or glaucoma.
Footnotes
This work was supported by Research Unit (Forschergruppe) FOR 1075 (TP 7) of the Deutsche Forschungsgemeinschaft and by Freifrau von Nauendorf Stiftung, Wiesbaden, Germany. The excellent technical help of Angelika Pach, Margit Schimmel, and Elke Stauber is greatly appreciated.
References
- Berger W, van de Pol D, Warburg M, Gal A, Bleeker-Wagemakers L, de Silva H, Meindl A, Meitinger T, Cremers F, Ropers HH. Mutations in the candidate gene for Norrie disease. Hum Mol Genet. 1992;1:461–465. doi: 10.1093/hmg/1.7.461. [DOI] [PubMed] [Google Scholar]
- Berger W, van de Pol D, Bächner D, Oerlemans F, Winkens H, Hameister H, Wieringa B, Hendriks W, Ropers HH. An animal model for Norrie disease (ND): gene targeting of the mouse ND gene. Hum Mol Genet. 1996;5:51–59. doi: 10.1093/hmg/5.1.51. [DOI] [PubMed] [Google Scholar]
- Bernstein SL, Wong P. Regional expression of disease-related genes in human and monkey retina. Mol Vis. 1998;4:24. [PubMed] [Google Scholar]
- Bonfanti L, Strettoi E, Chierzi S, Cenni MC, Liu X-H, Martinou J-C, Maffei L, Rabacchi SA. Protection of retinal ganglion cells from natural and axotomy-induced cell death in neonatal transgenic mice overexpressing bcl-2. J Neurosci. 1996;16:4186–4194. doi: 10.1523/JNEUROSCI.16-13-04186.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bringmann A, Pannicke T, Grosche J, Francke M, Wiedemann P, Skatchkov SN, Osborne NN, Reichenbach A. Muller cells in the healthy and diseased retina. Prog Retin Eye Res. 2006;25:397–424. doi: 10.1016/j.preteyeres.2006.05.003. [DOI] [PubMed] [Google Scholar]
- Cayouette M, Behn D, Sendtner M, Lachapelle P, Gravel C. Intraocular gene transfer of ciliary neurotrophic factor prevents death and increases responsiveness of rod photoreceptors in the retinal degeneration slow mouse. J Neurosci. 1998;18:9282–9293. doi: 10.1523/JNEUROSCI.18-22-09282.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frassetto LJ, Schlieve CR, Lieven CJ, Utter AA, Jones MV, Agarwal N, Levin LA. Kinase-dependent differentiation of a retinal ganglion cell precursor. Invest Ophthalmol Vis Sci. 2006;47:427–438. doi: 10.1167/iovs.05-0340. [DOI] [PubMed] [Google Scholar]
- Gao H, Hollyfield JG. Basic fibroblast growth factor: increased gene expression in inherited and light-induced photoreceptor degeneration. Exp Eye Res. 1996;62:181–189. doi: 10.1006/exer.1996.0022. [DOI] [PubMed] [Google Scholar]
- Hartzer MK, Cheng M, Liu X, Shastry BS. Localization of the Norrie disease gene mRNA by in situ hybridization. Brain Res Bull. 1999;49:355–358. doi: 10.1016/s0361-9230(99)00071-4. [DOI] [PubMed] [Google Scholar]
- Hicks D, Courtois Y. The growth and behaviour of rat retinal Muller cells in vitro. 1. An improved method for isolation and culture. Exp Eye Res. 1990;51:119–129. doi: 10.1016/0014-4835(90)90063-z. [DOI] [PubMed] [Google Scholar]
- Inomata Y, Hirata A, Koga T, Kimura A, Singh DP, Shinohara T, Tanihara H. Lens epithelium-derived growth factor: neuroprotection on rat retinal damage induced by N-methyl-D-aspartate. Brain Res. 2003;991:163–170. doi: 10.1016/j.brainres.2003.08.011. [DOI] [PubMed] [Google Scholar]
- Joly S, Pernet V, Chemtob S, Di Polo A, Lachapelle P. Neuroprotection in the juvenile rat model of light-induced retinopathy: evidence suggesting a role for FGF-2 and CNTF. Invest Ophthalmol Vis Sci. 2007;48:2311–2320. doi: 10.1167/iovs.06-1205. [DOI] [PubMed] [Google Scholar]
- Joly S, Lange C, Thiersch M, Samardzija M, Grimm C. Leukemia inhibitory factor extends the lifespan of injured photoreceptors in vivo. J Neurosci. 2008;28:13765–13774. doi: 10.1523/JNEUROSCI.5114-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Junge HJ, Yang S, Burton JB, Paes K, Shu X, French DM, Costa M, Rice DS, Ye W. TSPAN12 regulates retinal vascular development by promoting Norrin- but not Wnt-induced FZD4/beta-catenin signaling. Cell. 2009;139:299–311. doi: 10.1016/j.cell.2009.07.048. [DOI] [PubMed] [Google Scholar]
- Krishnamoorthy RR, Agarwal P, Prasanna G, Vopat K, Lambert W, Sheedlo HJ, Pang IH, Shade D, Wordinger RJ, Yorio T, Clark AF, Agarwal N. Characterization of a transformed rat retinal ganglion cell line. Brain Res Mol Brain Res. 2001;86:1–12. doi: 10.1016/s0169-328x(00)00224-2. [DOI] [PubMed] [Google Scholar]
- Lebrun-Julien F, Duplan L, Pernet V, Osswald I, Sapieha P, Bourgeois P, Dickson K, Bowie D, Barker PA, Di Polo A. Excitotoxic death of retinal neurons in vivo occurs via a non-cell-autonomous mechanism. J Neurosci. 2009;29:5536–5545. doi: 10.1523/JNEUROSCI.0831-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leng Y, Liang MH, Ren M, Marinova Z, Leeds P, Chuang DM. Synergistic neuroprotective effects of lithium and valproic acid or other histone deacetylase inhibitors in neurons: roles of glycogen synthase kinase-3 inhibition. J Neurosci. 2008;28:2576–2588. doi: 10.1523/JNEUROSCI.5467-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li HL, Wang HH, Liu SJ, Deng YQ, Zhang YJ, Tian Q, Wang XC, Chen XQ, Yang Y, Zhang JY, Wang Q, Xu H, Liao FF, Wang JZ. Phosphorylation of tau antagonizes apoptosis by stabilizing beta-catenin, a mechanism involved in Alzheimer's neurodegeneration. Proc Natl Acad Sci U S A. 2007;104:3591–3596. doi: 10.1073/pnas.0609303104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Schlamp CL, Nickells RW. Experimental induction of retinal ganglion cell death in adult mice. Invest Ophthalmol Vis Sci. 1999;40:1004–1008. [PubMed] [Google Scholar]
- Lin S, Cheng M, Dailey W, Drenser K, Chintala S. Norrin attenuates protease-mediated death of transformed retinal ganglion cells. Mol Vis. 2009;15:26–37. [PMC free article] [PubMed] [Google Scholar]
- Luhmann UF, Lin J, Acar N, Lammel S, Feil S, Grimm C, Seeliger MW, Hammes HP, Berger W. Role of the Norrie disease pseudoglioma gene in sprouting angiogenesis during development of the retinal vasculature. Invest Ophthalmol Vis Sci. 2005;46:3372–3382. doi: 10.1167/iovs.05-0174. [DOI] [PubMed] [Google Scholar]
- Meindl A, Berger W, Meitinger T, van de Pol D, Achatz H, Dörner C, Haasemann M, Hellebrand H, Gal A, Cremers F. Norrie disease is caused by mutations in an extracellular protein resembling C-terminal globular domain of mucins. Nat Genet. 1992;2:139–143. doi: 10.1038/ng1092-139. [DOI] [PubMed] [Google Scholar]
- Meitinger T, Meindl A, Bork P, Rost B, Sander C, Haasemann M, Murken J. Molecular modelling of the Norrie disease protein predicts a cystine knot growth factor tertiary structure. Nat Genet. 1993;5:376–380. doi: 10.1038/ng1293-376. [DOI] [PubMed] [Google Scholar]
- Mey J, Thanos S. Intravitreal injections of neurotrophic factors support the survival of axotomized retinal ganglion cells in adult rats in vivo. Brain Res. 1993;602:304–317. doi: 10.1016/0006-8993(93)90695-j. [DOI] [PubMed] [Google Scholar]
- Ogata N, Wang L, Jo N, Tombran-Tink J, Takahashi K, Mrazek D, Matsumura M. Pigment epithelium derived factor as a neuroprotective agent against ischemic retinal injury. Curr Eye Res. 2001;22:245–252. doi: 10.1076/ceyr.22.4.245.5506. [DOI] [PubMed] [Google Scholar]
- Ohlmann A, Scholz M, Goldwich A, Chauhan BK, Hudl K, Ohlmann AV, Zrenner E, Berger W, Cvekl A, Seeliger MW, Tamm ER. Ectopic norrin induces growth of ocular capillaries and restores normal retinal angiogenesis in Norrie disease mutant mice. J Neurosci. 2005;25:1701–1710. doi: 10.1523/JNEUROSCI.4756-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohlmann A, Seitz R, Braunger B, Seitz D, Bösl MR, Tamm ER. Norrin promotes vascular regrowth after oxygen-induced retinal vessel loss and suppresses retinopathy in mice. J Neurosci. 2010;30:183–193. doi: 10.1523/JNEUROSCI.3210-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rattner A, Nathans J. The genomic response to retinal disease and injury: evidence for endothelin signaling from photoreceptors to glia. J Neurosci. 2005;25:4540–4549. doi: 10.1523/JNEUROSCI.0492-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rehm HL, Zhang DS, Brown MC, Burgess B, Halpin C, Berger W, Morton CC, Corey DP, Chen ZY. Vascular defects and sensorineural deafness in a mouse model of Norrie disease. J Neurosci. 2002;22:4286–4292. doi: 10.1523/JNEUROSCI.22-11-04286.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richter M, Gottanka J, May CA, Welge-Lüssen U, Berger W, Lütjen-Drecoll E. Retinal vasculature changes in Norrie disease mice. Invest Ophthalmol Vis Sci. 1998;39:2450–2457. [PubMed] [Google Scholar]
- Ruether K, van de Pol D, Jaissle G, Berger W, Tornow RP, Zrenner E. Retinoschisislike alterations in the mouse eye caused by gene targeting of the Norrie disease gene. Invest Ophthalmol Vis Sci. 1997;38:710–718. [PubMed] [Google Scholar]
- Schuettauf F, Vorwerk C, Naskar R, Orlin A, Quinto K, Zurakowski D, Dejneka NS, Klein RL, Meyer EM, Bennett J. Adeno-associated viruses containing bFGF or BDNF are neuroprotective against excitotoxicity. Curr Eye Res. 2004;29:379–386. doi: 10.1080/02713680490517872. [DOI] [PubMed] [Google Scholar]
- Seki M, Lipton SA. Targeting excitotoxic/free radical signaling pathways for therapeutic intervention in glaucoma. Prog Brain Res. 2008;173:495–510. doi: 10.1016/S0079-6123(08)01134-5. [DOI] [PubMed] [Google Scholar]
- Shen Y, Liu XL, Yang XL. N-methyl-D-aspartate receptors in the retina. Mol Neurobiol. 2006;34:163–179. doi: 10.1385/MN:34:3:163. [DOI] [PubMed] [Google Scholar]
- Siliprandi R, Canella R, Carmignoto G, Schiavo N, Zanellato A, Zanoni R, Vantini G. N-methyl-D-aspartate-induced neurotoxicity in the adult rat retina. Vis Neurosci. 1992;8:567–573. doi: 10.1017/s0952523800005666. [DOI] [PubMed] [Google Scholar]
- Smallwood PM, Williams J, Xu Q, Leahy DJ, Nathans J. Mutational analysis of Norrin-Frizzled4 recognition. J Biol Chem. 2007;282:4057–4068. doi: 10.1074/jbc.M609618200. [DOI] [PubMed] [Google Scholar]
- Strom RC, Williams RW. Cell production and cell death in the generation of variation in neuron number. J Neurosci. 1998;18:9948–9953. doi: 10.1523/JNEUROSCI.18-23-09948.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toledo EM, Colombres M, Inestrosa NC. Wnt signaling in neuroprotection and stem cell differentiation. Prog Neurobiol. 2008;86:281–296. doi: 10.1016/j.pneurobio.2008.08.001. [DOI] [PubMed] [Google Scholar]
- Unoki K, LaVail MM. Protection of the rat retina from ischemic injury by brain-derived neurotrophic factor, ciliary neurotrophic factor, and basic fibroblast growth factor. Invest Ophthalmol Vis Sci. 1994;35:907–915. [PubMed] [Google Scholar]
- Van de Craen M, Berx G, Van den Brande I, Fiers W, Declercq W, Vandenabeele P. Proteolytic cleavage of beta-catenin by caspases: an in vitro analysis. FEBS Lett. 1999;458:167–170. doi: 10.1016/s0014-5793(99)01153-9. [DOI] [PubMed] [Google Scholar]
- Wen R, Song Y, Cheng T, Matthes MT, Yasumura D, LaVail MM, Steinberg RH. Injury-induced upregulation of bFGF and CNTF mRNAS in the rat retina. J Neurosci. 1995;15:7377–7385. doi: 10.1523/JNEUROSCI.15-11-07377.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Q, Wang Y, Dabdoub A, Smallwood PM, Williams J, Woods C, Kelley MW, Jiang L, Tasman W, Zhang K, Nathans J. Vascular development in the retina and inner ear: control by Norrin and Frizzled-4, a high-affinity ligand-receptor pair. Cell. 2004;116:883–895. doi: 10.1016/s0092-8674(04)00216-8. [DOI] [PubMed] [Google Scholar]
- Ye X, Wang Y, Cahill H, Yu M, Badea TC, Smallwood PM, Peachey NS, Nathans J. Norrin, frizzled-4, and Lrp5 signaling in endothelial cells controls a genetic program for retinal vascularization. Cell. 2009;139:285–298. doi: 10.1016/j.cell.2009.07.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi H, Nakamura RE, Mohamed O, Dufort D, Hackam AS. Characterization of Wnt signaling during photoreceptor degeneration. Invest Ophthalmol Vis Sci. 2007;48:5733–5741. doi: 10.1167/iovs.07-0097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zorn AM. Wnt signalling: antagonistic Dickkopfs. Curr Biol. 2001;11:R592–R595. doi: 10.1016/s0960-9822(01)00360-8. [DOI] [PubMed] [Google Scholar]