

Abstract
FLICE-inhibitory protein (FLIP) is an endogenous inhibitor of the signaling pathway triggered by the activation of death receptors. Here, we reveal a novel biological function for the long form of FLIP (FLIP-L) in neuronal differentiation, which can be dissociated from its antiapoptotic role. We show that FLIP-L is expressed in different regions of the mouse embryonic nervous system. Immunohistochemistry of mouse brain sections at different stages reveals that, in neurons, FLIP is expressed early during the embryonic neuronal development (embryonic day 16) and decreases at later stages (postnatal days 5–15), when its expression is essentially detected in glial cells. FLIP-L overexpression significantly enhances neurotrophin-induced neurite outgrowth in motoneurons, superior cervical ganglion neurons, and PC12 cells. Conversely, the downregulation of FLIP-L protein levels by specific RNA interference significantly reduces neurite outgrowth, even in the presence of the appropriate neurotrophin stimulus. Moreover, NGF-dependent activation of two main intracellular pathways involved in the regulation of neurite outgrowth, extracellular signal-regulated kinases (ERKs) and nuclear factor κB (NF-κB), is impaired when endogenous FLIP-L is downregulated, although TrkA remains activated. Finally, we demonstrate that FLIP-L interacts with TrkA, and not with p75NTR, in an NGF-dependent manner, and endogenous FLIP-L interacts with TrkB in whole-brain lysates from embryonic day 15 mice embryos. Altogether, we uncover a new role for FLIP-L as an unexpected critical player in neurotrophin-induced mitogen-activated protein kinase/ERK- and NF-κB-mediated control of neurite growth in developing neurons.
Introduction
Neurotrophins, which include nerve growth factor (NGF), brain-derived neurotrophic factor (BDNF), and neurotrophins 3 and 4/5, are by far the best characterized family of neurotrophic factors (Lewin and Barde, 1996) and are essential for the development of the nervous system. Each neurotrophin can bind to two different types of receptors: the Trk receptor tyrosine kinase (TrkA, TrkB, or TrkC) and the p75 neurotrophin receptor (p75NTR), a member of the tumor necrosis factor receptor superfamily (Dechant and Barde, 2002). In addition to their effects on neuronal cell survival, neurotrophins also regulate axonal and dendritic guidance and growth as well as synaptic plasticity (McAllister, 1999; Chao, 2003). During ligation of neurotrophins, Trk and p75NTR receptors activate the mitogen-activated protein kinase (MAPK)/extracellular signal-regulated kinase (ERK) and the nuclear factor κB (NF-κB) pathways, inducing both neuronal survival and differentiation (Wood, 1995; Yoon et al., 1998; Hamanoue et al., 1999; Foehr et al., 2000a; Wooten et al., 2001; Sole et al., 2004).
Recently, we have provided evidence that the differentiation induced by neurotrophins is controlled by the short isoform of Fas apoptosis inhibitory molecule (FAIM-S) (Sole et al., 2004). The long form of the FLICE-inhibitory protein (FLIP-L), another antagonist of death receptors (DRs), is an endogenous inhibitor of the Fas-associated death domain (FADD)/caspase-8 proapoptotic signaling pathway. FLIP-L is a widely expressed molecule that confers resistance to DR-induced apoptosis in a wide range of cellular models (Irmler et al., 1997; Thome et al., 1997; Yeh et al., 2000). Although the antiapoptotic role of FLIP-L is well established in the immune system, its implication as a DR antagonist in neurons is not conclusive. FLIP-L serves as a modulator between apoptotic and proliferative responses in the immune system (Tseveleki et al., 2004). For instance, in T lymphocytes, FLIP-L interacts with different adaptor proteins to activate ERK and NF-κB signaling pathways (Kataoka et al., 1998, 2000). Thus, FLIP-L is a protein with dual roles displaying antiapoptotic or proliferation/differentiation properties, at least in the immune system. In the nervous system, FLIP-L might control apoptosis of embryonic motoneurons during development (Raoul et al., 1999). More recently, it has been reported that FLIP-L protects neurons against in vivo ischemia and in vitro glucose deprivation-induced cell death (Taoufik et al., 2007). Nonetheless, a potential role of FLIP-L in neuronal differentiation has never been addressed.
In the present work, we show that FLIP-L is expressed in different regions of the nervous system, predominantly at the early embryonic stages. The overexpression of FLIP-L protects motoneurons from apoptosis induced by BDNF deprivation but not superior cervical ganglion (SCG) neurons nor differentiated PC12 cells from NGF deprivation. Moreover, in all those neuronal models, the overexpression of FLIP-L markedly increases neurotrophin-induced neurite outgrowth. Furthermore, the decrease of the endogenous levels of FLIP-L significantly reduces neurite outgrowth, even in the presence of the appropriate neurotrophin stimulus. In PC12 cells, FLIP-L is necessary for the activation of MAPK/ERK and NF-κB signaling pathways after NGF treatment. However, the knockdown of FLIP-L does not affect the kinase activity of TrkA. We also substantiate for the first time that FLIP-L binds the neurotrophin receptor TrkA, but not p75NTR, during NGF stimulation. Furthermore, endogenous FLIP-L also binds TrkB in whole-brain lysates from embryonic day 15 (E15) mice embryos. These findings uncover a new role for FLIP-L as a crucial factor in neurotrophin-induced control of neurite growth.
Materials and Methods
Reagents.
All biochemical reagents were obtained from Sigma-Aldrich unless otherwise indicated. Purified recombinant human NGF was obtained from Genentech. Recombinant human BDNF was obtained from Alomone Labs. PD98059 (2′-amino-3′-methoxyflavone) was purchased from Calbiochem/Merck Biosciences.
Cell culture.
PC12 cells were grown on 100 mm collagen-coated tissue culture dishes (Falcon Discovery Labware; BD Biosciences) in DMEM supplemented with 6% heat-inactivated fetal calf serum, 6% heat-inactivated horse serum (Invitrogen), 10 mm HEPES, 20 U/ml penicillin, and 20 μg/ml streptomycin. SCG neurons were isolated as described previously (Davies et al., 1993). Briefly, they were dissected from postnatal day 1 (P1) CD1 mice, trypsinized (0.25% trypsin for 25 min at 37°C), and mechanically dissociated. Neurons were plated in defined, serum-free medium on a polyornithine/laminin substratum in 35 mm tissue culture dishes and were incubated at 37.5°C in a humidified 3.5% CO2 incubator for 3–4 h to allow the cells to attach before ballistic transfection. Spinal cord motoneurons (MTNs) were purified from E12.5 CD1 mice according to Arce et al. (1998) and Comella et al. (1994).
Plasmids.
Mouse FLIP-L, rat FAIM-S, human TrkA, p75NTR, and super-repressor IκBα (SR-IκBα) cDNAs were expressed under the control of a cytomegalovirus constitutive promoter in the pcDNA3 expression vector (Invitrogen). Enhanced green fluorescent protein (EGFP) plasmid was obtained from Clontech/BD Biosciences. The NF-κB-dependent reporter vector (κB HIV–LTR–Luciferase) (Rodriguez et al., 1996) was kindly provided by Dr. Ron Hay (University of St. Andrews, Fife, UK). Hemagglutinin (HA)-tagged TrkA and p75NTR constructs are from Dr. Jorge Moscat (Centro de Biología Molecular “Severo Ochoa,” Madrid, Spain). FLIP-L cDNA was purchased from Imagenes. FAIM-S and FLIP-L cDNAs were tagged with 3xFLAG and subcloned into the pcDNA3 mammalian expression vector. FLIP-L–FLAG and FAIM-S–FLAG cDNAs were also subcloned into the pEIGW vector, giving rise to the lentiviral FLIP-L–FLAG and FAIM-S–FLAG overexpressing constructs.
For RNA interference (RNAi) experiments, constructs were generated in the pSUPER.retro.puro plasmid (OligoEngine) using specific oligonucleotides for mouse and rat FLIP (sequence data available from GenBank under accession numbers NM_207653 and NM_001033864, respectively, indicated by capital letter, as follows: RNAi n°1 (R1), forward, gatccccGAAGGAGATGATGCTCTTCttcaagagaGAAGAGCATCATCTCCTTCttttt; reverse, agctaaaaaGAAGGAGATGATGCTCTTCtctcttgaaGAAGAGCATCATCTCCTTCggg; or RNAi n°2 (R2), forward, gatccccCATCCACAGAATAGACTTGttcaagagaCAAGTCTATTCTGTGGATGttttt; reverse, agctaaaaaCATCCACAGAATAGACTTGtctcttgaaCAAGTCTATTCTGTGGATGggg.
A scrambled sequence (Scr) from RNAi R1 was obtained using the following oligonucleotides: scrambled, forward, gatccccGCATATGCGTACGGATTAGttcaagagaCTAATCCGTACGCATATGCttttt; reverse, agctaaaaaGCATATGCGTACGGATTAGtctcttgaaCTAATCCGTACGCATATGCggg. Oligonucleotides were obtained from Sigma-Aldrich and cloned between BglII and HindIII sites of the pSUPER.retro.puro vector, under the control of the DNA Pol III promoter of H1. pWPTS–EGFP, pLVTHM, pSPAX2, and pMD2G plasmids were kindly provided by Dr. Trono (University of Geneva, Geneva, Switzerland). Lentiviral constructs were achieved by digestion at the EcoRI–ClaI sites to replace H1 promoter in the lentiviral vector pLVTHM with H1–short hairpin RNA cassette from pSUPER. The pLVTHM plasmid was subsequently used for lentiviral knockdown experiments. The plasmid pEIGW was constructed by replacing the EGFP nucleotide sequence between the EF1α promoter and the woodchuck post-transcriptional regulatory element in pWPTS–EGFP with the internal ribosomal entry site (IRES)–EGFP cassette from pIRES2–EGFP (Clontech), as described by Zuliani et al. (2006). The pEIGW plasmid was subsequently used for lentiviral overexpression experiments.
Lentiviral production.
Lentiviruses were propagated as described previously (Naldini et al., 1996; Zufferey et al., 1998). Briefly, human embryonic kidney 293T (HEK293T) cells were seeded at a density of 2.5 × 106 cells in 0.1% gelatin-coated 100 mm dishes. The following day, cells were transfected with 20 μg of pLVTHM- or pEIGW-derived constructs, 13 μg of pSPAX2, and 7 μg of pM2G. The transfection was routinely performed by the calcium phosphate transfection method (Cullen, 1987). Cells were allowed to produce lentiviruses for 48 h. After 48 h, the medium was centrifuged at 1200 × g for 5 min, and the supernatant was filtered using 45 μm filters. Lentiviruses were concentrated at 50,000 × g for 90 min and then resuspended in 20 μl of PBS containing 1% BSA. Lentiviruses were stored at −80°C. Biological titers of the viral preparations expressed as a number of transducing units (TU) per milliliter were determined by transducing HEK293T cells in limiting dilutions. After 48 h of incubation, the percentage of GFP-positive cells was counted, and viruses at 5 × 108 to 1 × 109 TU/ml were used in the experiments. After 4 h, medium was changed and infection efficiency was monitored in each experiment by direct counting of GFP-positive cells. The percentage of infected cells reached at least 90%.
Cell transfection and infection.
Unless infected with lentiviruses, PC12 cells were transfected with the desired plasmid using Lipofectamine2000 reagent (Invitrogen) following the instructions of the manufacturer. Selection of pools of transfected cells was performed by adding G418 to the culture medium at a final concentration of 500 μg/ml.
Ballistic transfection of SCG neuronal cultures with the experimental construct plasmids (pEGFP and pSUPER–scrambled control or pSUPER–FLIP RNAi R1) was performed as described by Sole et al. (2004).
Motoneurons were transiently transfected using the Lipofectamine2000 reagent. Cells were cotransfected with pEGFP (Clontech/BD Biosciences) and pSUPER–RNAi R1, pSUPER–RNAi R2, or pSUPER–scrambled, using a 1:3 molar ratio (pEGFP/desired plasmid) according to the experimental conditions.
Lentiviral infection was performed when required, usually to assess the effects of FLIP-L knockdown by RNAi strategy or to overexpress FLIP-L. A time course analysis was performed to check the efficiency of both overexpression or knockdown of FLIP-L. FLIP-L levels were assessed by Western blot, and lentiviruses efficiently induced FLIP-L protein overexpression or reduced protein levels after 2 and 3 d of infection, respectively, in all three models.
Neurite measurement.
Neurite length and number of branching points were performed following the protocol described by Sole et al. (2004). Transfected EGFP-labeled SCG neurons and MTNs were treated according to the experimental conditions. After the indicated time, between 70 and 100 neurons per condition were visualized and digitally acquired using an Axioplan Carl Zeiss laser-scanning confocal microscope. Sholl analysis was performed on SCG images (Sholl, 1953). By means of especially designed MATLAB script, concentric and digitally generated rings 30 μm apart were centered on the cell soma, and the number of neurites intersecting each ring was automatically counted as described previously (Gutierrez and Davies, 2007). Total neurite length and number of branch points were also measured. Pairwise comparisons were made using the Student's t test. For multiple comparisons, ANOVA was performed followed by Fisher's post hoc test.
Cell death quantification.
PC12 cells transfected with the indicated plasmids were treated with basal medium supplemented with NGF (100 ng/ml) for 3 d before removing the neurotrophin. The percentage of apoptotic cells showing nuclear condensation type II after 48 h of deprivation was quantified using Hoechst staining, as reported previously (Yuste et al., 2001).
MTNs were counted as described previously (Pérez-García et al., 2004) with some modifications. Briefly, MTNs were cotransfected after plating and washed 12 h later, and the different experimental conditions were performed for deprived or BDNF-maintained neurons (10 ng/ml; Alomone Labs). Neuronal apoptosis is expressed as the percentage of apoptotic EGFP-labeled MTNs showing nuclear condensation type II quantified using Hoechst staining.
SCG neuronal cultures were cotransfected with the indicated plasmid plus EGFP after plating and treated or not with NGF (10 ng/ml). Cell death was quantified as described above for MTNs.
Western blot.
Cells were rinsed with ice-cold PBS, pH 7.2, and resuspended in prewarmed (95°C) 2% SDS/125 mm Tris, pH 6.8, lysis buffer. Protein concentration was quantified by a modified Lowry assay (DC protein assay; Bio-Rad).
The different cell lysates obtained (5–20 μg of protein) were resolved in SDS-polyacrylamide gels and transferred onto polyvinylidene difluoride (PVDF) Immobilon-P membranes (Millipore Corporation). After blocking with TBS/0.1% Tween 20 containing 5% nonfat dry milk, the membranes were probed with the appropriate primary antibodies against the phosphorylated forms of ERK1 and ERK2, panERK, anti-IκBα (Cell Signaling Technology), anti-FLIP (Dave-2) antibody (Alexis Biochemicals), anti-pan-Trk (α-203) generated by our group (according to Martin-Zanca et al., 1989), anti-rat TrkA, anti-TrkB, anti-phospho-Tyr (4G10) (Millipore Corporation), anti-HA (clone 3F10; Roche), or anti α-tubulin (clone B-5-1-2) (Sigma).
After incubation with specific peroxidase-conjugated secondary antibodies, the membranes were developed with the EZ-ECL chemiluminescence detection kit (Biological Industries) or SuperSignal (Pierce/Thermo Fisher Scientific).
Immunohistochemistry.
E16, P5, P15, and adult OF-1 mice (Charles River) were used for the immunohistochemistry experiments. First, the animals were deeply anesthetized with a mixture of Ketolar (Parke-Davies/Pfizer)/Rompun (Bayer) and perfused with 4% paraformaldehyde. The brains were dissected, cryoprotected, and frozen. Coronal sections of 30–50 μm were obtained. Sections were incubated overnight with specific rabbit antibody against FLIP (1:500; Santa Cruz Biotechnology). Primary antibody was visualized by sequential incubation with biotinylated secondary antibodies (1:200; Vector Laboratories) and the streptavidin–peroxidase complex (1:400; GE Healthcare). Peroxidase was developed with 0.05% diaminobenzidine and H2O2. The sections were mounted onto gelatinized slides, dehydrated, and coverslipped with DPX.
Sections from E16 embryos were used for double-immunofluorescence studies. After blocking, sections were incubated overnight with rabbit antibody detecting FLIP (1:500; Santa Cruz Biotechnology) combined with mouse monoclonal antibody against neuronal-specific nuclear protein (NeuN) (1:100; Millipore Bioscience Research Reagents) or GFAP (1:500; Millipore Corporation). Primary antibodies were visualized using secondary Alexa Fluor-conjugated antibodies. Sections were mounted and viewed under confocal microscopy.
Coimmunoprecipitation experiments.
For coimmunoprecipitation experiments, a total of 5 × 106 PC12 cells or 3 × 106 HEK293T cells stably or transiently transfected, respectively, with human TrkA, were plated in 100 mm tissue culture dishes and infected with lentiviral particles containing the pEIGW–FLIP-L–FLAG, pEIGW–FAIM-S–FLAG or the empty vector. When indicated, HA-tagged TrkA or p75NTR were transfected in HEK293T cells using the calcium phosphate method. At 24 h after infection, cells were serum starved for 12 h and stimulated or not with 100 ng/ml NGF for 15 min. Cellular lysates were obtained using immunoprecipitation (IP) lysis buffer [20 mm Tris, pH 7.4, 140 mm NaCl, 10% glycerol, 1 mm sodium orthovanadate, 25 mm NaF, 40 mm β-glycerophosphate, 1% NP-40, and 1× EDTA-free Complete protease inhibitor cocktail (Roche)]. All lysates were quantified by a modified Lowry assay (DC protein assay; Bio-Rad). For FLAG immunoprecipitation experiments, 1 mg of total protein was taken and adjusted with the IP lysis buffer to achieve a final concentration of 1 μg/μl. Next, 1 mg of lysates were subjected to immunoprecipitation using 40 μl of FLAG-specific monoclonal (mAb) M2-coupled agarose beads (Sigma) overnight at 4°C in an orbital shaker. Then, beads were washed five times with IP lysis buffer and eluted for 30 min at 4°C with 50 μl of TBS containing 150 ng/ml of 3xFLAG competitor peptide (Sigma). After a short spin, supernatants were carefully taken, Laemmli's loading buffer was added, and SDS-PAGE was performed.
For the assessment of TrkA activation, 250 μg of protein were immunoprecipitated using the anti-pan-Trk (α-203). Immunocomplexes were collected with protein A-Sepharose by orbital incubation for 1 h and were washed three times with IP lysis buffer. Beads were suspended in 40 μl of Laemli's loading buffer and boiled, and samples were loaded onto an 8% SDS-polyacrylamide gel. Finally, the membrane was blotted with the anti-phospho-Tyr 4G10 antibody.
For the endogenous interaction of FLIP-L with TrkB in whole-brain lysates, total brain was obtained from E15 Swiss Webster embryos. Brain was minced in small pieces and homogenized in IP lysis buffer using a Polytron homogenizer. Cellular lysates as well as brain extracts were cleared by centrifugation at 16,000 × g and quantified by a modified Lowry assay (DC protein assay; Bio-Rad). Two milligrams of total protein were incubated with 5 μg of anti-FLIP (Dave-2) antibody overnight at 4°C. As a control, a rat monoclonal antibody anti-HA (rat IgG) was used under the same conditions. Immunocomplexes were collected with protein G-Sepharose by orbital shaking for 1 h and washed three times with IP lysis buffer. Beads were suspended in 40 μl of Laemli's loading buffer and boiled, and samples were loaded onto an 8% SDS-polyacrylamide gel. As input controls, 25–50 μg of the lysates were blotted with anti-FLIP (Dave-2), anti-TrkA, anti-TrkB, anti-HA (clone 3F10), or anti-FLAG antibodies, as indicated.
NF-κB activity.
PC12 cells were infected with lentiviral particles containing the sequence encoding for the RNAi R1 against FLIP or its scrambled sequence (Scr). After 2 d, cells were seeded in the adequate plates and transfected with an NF-κB-dependent reporter vector (LTR KB). After 24 h, cells were serum starved for 12 h and stimulated with 100 ng/ml NGF for the indicated time. Cell lysates were obtained following the instructions of the manufacturer (Luciferase Assay System kit; Promega) and as described by Gozzelino et al. (2008).
Statistical analysis.
All the experiments were repeated at least three times. Values were expressed as mean ± SEM. Student's t test was used to determine the statistical significance of differences in neurite length, branching points numbers, and luciferase activity (p ≤ 0.05 was considered significant). For multiple comparisons, ANOVA was performed followed by Fisher's post hoc test.
Results
FLIP-L is expressed in the nervous system and is upregulated during neuronal differentiation
To determine whether FLIP isoforms are present in the nervous system, we analyzed the protein expression profile in different mouse tissues at both embryonic (E16) and adult stages. An immunoreactive band of 55 kDa corresponding to FLIP-L was detected in lysates from different brain regions such as spinal cord, cerebellum, and cortex, as well as in heart, liver, and kidney (Fig. 1A). The short isoform of FLIP of 25 kDa was not detected in the nervous system tissues, whereas it was expressed in the non-neuronal tissues (data not shown). To study whether FLIP-L is regulated during the development of the nervous system, we performed immunoblots of cortical tissue lysates of mouse brain at different stages. FLIP-L levels were low at E12.5 when compared with the peak of expression that was detected at E16. Subsequently, FLIP-L levels decreased during the early postnatal stages and stabilized in the adult (Fig. 1B). To determine whether FLIP-L expression is regulated during the differentiation process induced by neurotrophic factors, we used PC12 cells cultured in the presence of NGF. As shown in Figure 1C, NGF induced an increase in FLIP-L after 1 d of stimulation compared with the levels of unstimulated cells. Then, FLIP-L protein amounts decreased gradually after 3, 5, and 7 d of neurotrophin stimulation (Fig. 1C). The NGF-induced upregulation of FLIP-L is dependent on the MAPK/ERK pathway activation, because it can be prevented when cells are cultured in the presence of the MAP kinase kinase (MEK) inhibitor PD98059 (Fig. 1D, top). The efficiency of PD98059 for inhibiting MAPK/ERK pathway is supported by the analysis of the phosphorylation status of ERK1/2 by Western blot. As shown in the bottom of Figure 1D, PD98059 inhibits ERK1/2 phosphorylation at both acute NGF stimulation time (5 min) and longer time periods such as 1 and 2 d.
Figure 1.

FLIP-L is expressed in the nervous system and is upregulated during neuronal differentiation in vivo and in vitro. A, Embryonic (E16) and adult (Ad) mouse tissue lysates (20 μg) were analyzed by SDS-PAGE/immunoblot using anti-FLIP Dave-2 antibody (Alexis Biochemicals) (top). SC, Spinal cord; Cb, cerebellum; Cx, Cortex; Ht, heart; Lv, liver; Kd, kidney. B, Cortical brain lysates of mice at different embryonic and postnatal stages were processed, and FLIP-L immunoblot was performed. C, Time course of FLIP-L protein expression in PC12 cells subjected to NGF-induced differentiation. For loading control, the membranes were stained with Naphtol Blue (NB). D, After serum deprivation during 12 h, PC12 cells pretreated or not with 50 μm PD98059 were treated with NGF for 5 min (bottom) or 1 and 2 d (top and bottom). Total protein extracts were analyzed by Western blotting using an anti-phospho-ERK1/2 antibody (bottom) or an anti-FLIP (Dave-2) antibody for detection of FLIP-L upregulation after NGF treatment (top). Note that FLIP-L was not upregulated after 1 d of NGF treatment when cells were cultured in the presence of PD98059 (top). Membranes were stripped and reprobed with an anti-pan-ERK antibody to control protein loading.
To characterize the expression pattern of FLIP during the development of the CNS, we performed immunohistochemistry using the FLIP-specific antibody. FLIP-labeled cells were detected in most brain regions throughout development (Fig. 2). In the embryonic cerebral cortex, labeled cells were located in the cortical plate and in the subplate, corresponding to postmitotic neurons in these layers (Fig. 2A). At P5, the cell bodies of layers II–III cortical neurons exhibited weak FLIP immunostaining (Fig. 2B). In addition, some cells located throughout all cortical layers, corresponding to GFAP-immunoreactive glial cells, were FLIP immunopositive (Fig. 2B, arrowheads, J–L, arrows). At postnatal and adult stages, the expression of FLIP appeared to be restricted to glial-like cells in the cerebral cortex (Fig. 2C). The cerebellum also shows an intense FLIP immunolabeling. FLIP was expressed in migratory Purkinje cells in the embryonic cerebellum (E16) (Fig. 2D). At early postnatal stages (P5), the cell bodies of Purkinje neurons occupying the incipient Purkinje cell layer expressed FLIP (Fig. 2E). In addition, at P15 and in adult mice, some aligned glial cells, GFAP positive, located just under the Purkinje cell layer, and scattered in the granule cell layer, were positive for FLIP immunostaining (Fig. 2F) (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). To confirm that FLIP was expressed in neurons at embryonic stages, sections from E16 mice that were double immunolabeled with FLIP antibodies and the neuronal marker NeuN showed an almost complete colocalization of both markers (Fig. 2G–I). Therefore, we conclude that FLIP is predominantly expressed in developing neurons during the embryonic and early postnatal stages of the CNS. Later FLIP is mainly expressed in GFAP-immunoreactive glial cells (Fig. 2J–L). The specificity of the antibody used for FLIP immunohistochemistry was validated in the SCGs that were cotransfected with the pEGFP plasmid and the overexpression FLIP-L plasmid or a plasmid containing an RNAi targeting FLIP-L (supplemental Fig. 2A,B, available at www.jneurosci.org as supplemental material). Indeed, this FLIP antibody was able to detect FLIP-L overexpression as well as its knockdown compared with nontransfected control neurons expressing endogenous FLIP. Moreover, we ensured that the variations in FLIP immunolabeling did not constitute an artifact because of the cotransfection of FLIP-L or its RNAi together with the pEGFP plasmid (supplemental Fig. 2C, available at www.jneurosci.org as supplemental material).
Figure 2.
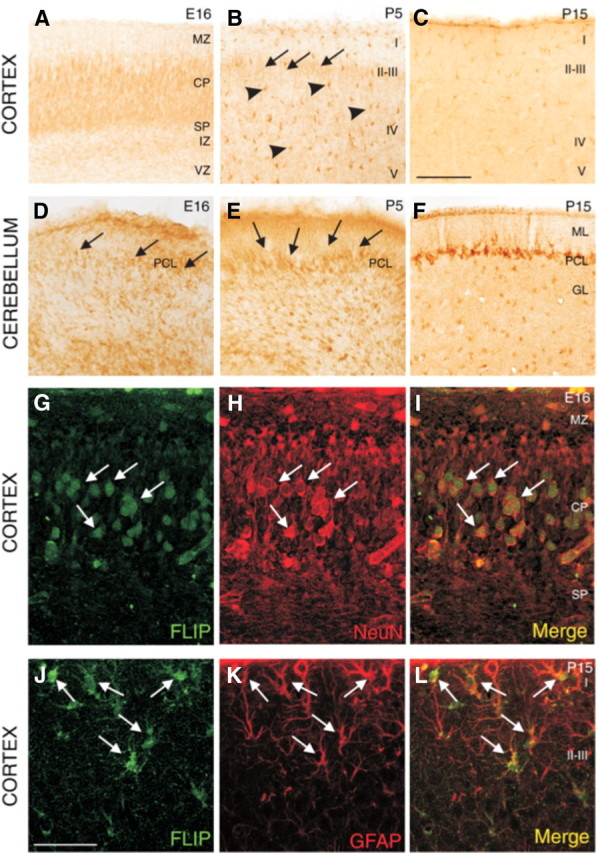
Distribution of FLIP-immunoreactive cells during the development of mice brain. A–C, In the cerebral cortex, many cells show immunohistochemical labeling. A, E16 neurons contain FLIP in the cortical plate (CP) and subplate (SP). B, At P5, some neurons in layers II–III present weak immunostaining (arrows); in contrast, at this age, some cells located throughout all cortical layers are FLIP immunopositive (arrowheads). C, At P15, FLIP-positive cells present in all cortical layers show an astrocytic morphology. D–F, In the cerebellum, the soma of some Purkinje cells shows immunohistochemical staining at embryonic (arrows in D) and early postnatal mice (arrows in E). At later postnatal stages, cells located just under the Purkinje cell layer and scattered in the granule cell layer show FLIP immunostaining (F). G–I, Immunofluorescence colocalization (arrows) of FLIP and NeuN (specific neuronal marker) in cortical plate at E16. J–L, Double immunostaining with antibodies detecting FLIP and GFAP in cerebral cortex at P15 showed the expression of FLIP in some GFAP-positive astrocytes (arrows). MZ, Marginal zone; IZ, intermediate zone; VZ, ventricular zone; ML, molecular layer; PCL, Purkinje cell layer; GL, granule cell layer. Scale bars: (in C) B–F, 100 μm; (in J), G–L, 50 μm.
FLIP-L protects motoneurons, but neither differentiated PC12 cells nor SCG neurons, from cell death induced by neurotrophin deprivation
To determine whether FLIP-L exerts a protective role against apoptosis induced by neurotrophin deprivation, we used three different neuronal models that require neurotrophin support for in vitro survival. Differentiated PC12 cells and SCG neurons need NGF, whereas motoneurons are dependent on the presence of BDNF in the culture media. NGF or BDNF withdrawal leads to cell death in SCG and PC12 or motoneurons, respectively. Once confirmed that FLIP-L is normally expressed in these cells (data not shown), we wanted to ascertain the relevance of this antiapoptotic protein in preventing neurotrophin deprivation-induced cell death. First, we performed survival experiments in motoneurons overexpressing FLIP-L. To this end, motoneurons were transiently transfected with pEGFP and either FLIP-L or the empty pcDNA3 vector and grown in the presence of BDNF or basal medium (Fig. 3A). Because of the partial transfection efficiency (∼30%), only EGFP-positive transfected cells are considered. The percentage of apoptotic motoneurons was calculated 24 h later by nuclear Hoechst staining. FLIP-L-transfected motoneurons treated with basal medium showed a significant protection from apoptosis compared with empty vector-transfected neurons (Fig. 3A). Apoptosis observed in FLIP-L-transfected neurons treated with basal medium did not significantly differ from that observed in BDNF-treated neurons (Fig. 3A).
Figure 3.

A, Cultured mouse MTNs were transfected using Lipofectamine2000 with an EGFP expression plasmid together with pcDNA3 or pcDNA3–FLIP-L (FLIP-L) vectors. Cells were treated with 10 ng/ml BNDF (+) or basal medium (−) for 24 h. Percentage of apoptotic EGFP-labeled MTNs showing nuclear condensation type II was quantified using Hoechst staining. B, Ballistic transfections of cultured mouse SCG neurons with an EGFP expression plasmid together with the pcDNA3 control vector (pcDNA3), pcDNA3–FLIP-L (FLIP-L), or pcDNA3–Bcl-xL (Bcl-xL) plasmids were performed. After 24 h of incubation with 10 ng/ml NGF (+) or basal medium (−), percentage of apoptotic EGFP-labeled SCGs was quantified. C, PC12 cells stably transfected with pcDNA3, Bcl-xL or FLIP-L were treated with basal medium without serum supplemented with 100 ng/ml NGF for 3 d before trophic factor deprivation. Percentage of apoptotic cells showing type II nuclear condensation after 48 h of NGF deprivation was quantified. Significant differences are indicated (*p < 0.001, t test). Right, Total lysates were obtained and analyzed by Western blot to assess FLIP-L levels in PC12 stably transfected using anti-FLIP (Dave-2). The membrane was reblotted with anti-tubulin as a loading control.
Subsequently, we performed similar experiments with the NGF-dependent neuronal models. SCG neurons were transiently transfected with an EGFP expression plasmid together with the pcDNA3 control vector, FLIP-L, or Bcl-xL plasmids (Fig. 3B). Bcl-xL was used as an already established antiapoptotic protein controlling mitochondrial-driven neuronal cell death during NGF withdrawal (Lindenboim et al., 1998; Putcha et al., 2002). After 24 h of incubation with NGF (10 ng/ml), cells were maintained with NGF or with basal medium, and the number of apoptotic EGFP-labeled neurons was counted 24 h later (Fig. 3B). NGF-deprived, FLIP-L-transfected SCG neurons showed a marked decrease in cell survival similar to that observed in NGF-deprived empty vector-transfected neurons, whereas Bcl-xL overexpression was able to protect SCGs from apoptosis induced by NGF deprivation (Fig. 3B). Similar results were obtained using PC12 cells, our third neuronal model (Fig. 3C). PC12 cells stably transfected with FLIP-L, Bcl-xL, or the empty vector were grown in the presence of NGF for 3 d and then NGF deprived for 48 h. Neurons displaying type II chromatin condensation were visualized by nuclear Hoechst staining and quantified. Percentage of cell death increased to 45% in both empty vector- and FLIP-L-transfected PC12 cells deprived of NGF (Fig. 3C). As found in SCGs cultures, PC12 cells transfected with Bcl-xL survived NGF deprivation compared with pcDNA3-transfected cells (Fig. 3B,C). These results demonstrate that, whereas FLIP-L has an important role in prevention of cell death after BDNF deprivation of motoneurons, it is unable to prevent apoptosis after NGF withdrawal in both differentiated PC12 cells and SCG neurons. Moreover, our results with the overexpression of Bcl-xL suggest that apoptosis induced by trophic factor deprivation in NGF-dependent neurons is driven through the mitochondrial-dependent pathway, as reported previously (Lindenboim et al., 1998; Putcha et al., 2002).
FLIP-L overexpression increases neurotrophin-induced neurite outgrowth in motoneurons, sympathetic neurons, and PC12 cells
Despite the absence of a general antiapoptotic role in neurotrophin deprivation-induced cell death, we noticed that cells transfected with FLIP-L and maintained in the presence of NGF or BNDF displayed a marked increase in neurite outgrowth compared with empty vector-transfected neurons. Therefore, we decided to further investigate whether FLIP-L overexpression might have a role in the regulation of neurite outgrowth. Primary cultures of motoneurons overexpressing FLIP-L showed a marked increase in neurite outgrowth compared with the empty vector-transfected cells (Fig. 4A). In addition, FLIP-L-transfected SCG neurons showed a significant increase in the neurite arbor when compared with empty vector-transfected neurons (Fig. 4B). Moreover, PC12 cells stably transfected with FLIP-L also displayed a significant increase in neurite outgrowth in the presence of NGF for 3 d when compared with pcDNA3-transfected cells (708.00 ± 55.24 vs 156.92 ± 14.14 μm for control cells) (Fig. 5A).
Figure 4.

A, Cultured mouse MTNs were transfected with an EGFP expression plasmid together with the control vector pcDNA3 (Ctrl) or pcDNA3–FLIP-L (FLIP-L) vectors. After 48 h of incubation with Neurobasal medium alone (NF) or supplemented with 10 ng/ml BDNF (BDNF), images were acquired and analyzed. Representative micrographs (scale bars, 100 μm) and quantification of neurite outgrowth are shown. Significant differences are indicated (*p < 0.001, t test). B, Ballistic transfections of cultured SCG neurons with an EGFP expression plasmid together with the control vector pcDNA3 (Ctrl) or pcDNA3–FLIP-L (FLIP-L) plasmids were performed 3 h after plating. After 24 h of incubation or not with 10 ng/ml NGF (NGF), EGFP-labeled SCG were visualized and digitally acquired. Representative micrographs depict the increased neurite arbor size and complexity (FLIP-L vs Ctrl), and quantification of neurite outgrowth is presented. Scale bars, 50 μm. Significant differences are indicated (*p < 0.001, ANOVA test).
Figure 5.

A, Phase-contrast micrographs of PC12 stably transfected with pcDNA3 empty vector (Control, top row) or pcDNA3–FLIP-L (FLIP, bottom row). PC12 cells were treated with complete medium (Serum, left column) or with NGF 100 ng/ml (right column) for 3 d. Scale bar, 100 μm. B, PC12 cells stably transfected with empty vector (Neo) or with SR-IκBα. Total cell lysates were analyzed by Western blot using an antibody against IκBα (top). Membranes were reprobed with anti-tubulin to normalize the loading content per lane (bottom). C, Stable Neo or SR-IκBα PC12 cells were infected with lentiviral particles either empty (Ctrl) or containing FLIP-L (FLIP-L). Two days later, cells were serum deprived, then left untreated (no NGF) or pretreated or not with 50 μm PD98059, and stimulated with 100 ng/ml NGF for 1 d, after which total neurite length was measured. Significant differences (NGF+PD98059-treated vs NGF-treated; Ctrl or FLIP-L Neo-transfected cells) are indicated (*p < 0.001, t test).
To assess the relevance of the MAPK/ERK and NF-κB pathways in FLIP-L-induced neurite outgrowth, total neurite length was measured in PC12 cells stably transfected with empty vector pcDNA3 (Neo) or pcDNA3 encoding for SR-IκBα, a form of IκBα carrying serine-to-alanine mutations at residues 32 and 36 (Fig. 5B). These mutations prevent IκBα phosphorylation and subsequent proteasome-mediated degradation, thereby impairing release and nuclear translocation of NF-κB (Rodriguez et al., 1996). Stable PC12 cells were subsequently infected with lentiviral particles either empty (Ctrl) or containing FLIP-L (FLIP-L) (Fig. 5C). Two days later, cells were serum deprived, then left untreated (no NGF), or treated with 100 ng/ml NGF for 1 d. The increase in neurite outgrowth induced by NGF stimulation of FLIP-L-overexpressing PC12 cells is significantly inhibited during blockade of the MAPK/ERK pathway by the MEK inhibitor PD98059 (Fig. 5C). In contrast, after 1 d of NGF stimulation, the total neurite length significantly decreases in SR-IκBα-overexpressing cells, even when FLIP-L is overexpressed (Fig. 5C). The expression of the dominant-negative forms of IκB kinase α (IKKα) (pcDNA3–IKKα-DN) or IKKβ (pcDNA3–IKKβ-DN) also provokes a reduction in neurite length compared with pcDNA3-transfected cells (data not shown). However, this impairment in neurite outgrowth attributable to the overexpression of the SR-IκBα alone or the MEK inhibitor alone in FLIP-L-overexpressing cells remains partial. Interestingly, neurite outgrowth is completely abrogated when either control (Ctrl) or FLIP-L PC12 cells stably expressing SR-IκBα are additionally treated with the MEK inhibitor PD98059 (Fig. 5C). Together, our results point out that FLIP-L overexpression is able to promote neurotrophin-induced neurite outgrowth in all three neuronal cultures tested (Figs. 4, 5). This novel function of FLIP-L is mediated by the activation of both NF-κB and MAPK/ERK pathways and is independent of its classical role as an inhibitor of the extrinsic apoptotic pathway.
Endogenous FLIP-L knockdown impairs neurotrophin-induced differentiation
We wanted to establish the role of endogenous FLIP-L in the regulation of the neurotrophin-mediated neurite outgrowth process. Mouse motoneurons were transfected with EGFP alone or along with FLIP-L RNAi-encoding vector R1, R2, or the scrambled sequence and were cultured in the presence of BDNF for 3 d. Efficiency of FLIP-L knockdown in these neurons was confirmed by Western blot (Fig. 6B). Neuron images were captured using epifluorescence microscopy (Fig. 6A), and the neurite length was measured by tracing neurite arbors (Fig. 6C). Motoneurons transfected with FLIP-L RNAi R1, R2, or a combination of both RNAi (R1+R2) displayed a dramatic threefold decrease in neurite length (Fig. 6A,C). However, cell survival in complete medium was not affected by FLIP-L-specific knockdown (data not shown). Alternatively, knockdown of FLIP-L in mouse SCG neurons, in the presence of NGF, showed nearly a 50% reduction in both neurite length (695.60 ± 26.41 vs 1162.26 ± 52.83 μm for control cells) and branching point measurements (2.79 ± 0.19 vs 5.13 ± 0.30 for control cells) (Fig. 6D). Likewise, Sholl analysis that provides a graphic representation of neurite branching with distance from the cell body evidenced a marked decrease in the number of neurite intersections in FLIP-L RNAi R1-transfected SCG neurons when compared with scrambled RNAi-transfected neurons (Fig. 6E). Comparable results were obtained with undifferentiated PC12 cells infected with FLIP-L RNAi or scrambled (Scr) lentiviral particles. FLIP-L RNAi is able to reduce the endogenous FLIP-L protein levels in these cultures (Fig. 7A). Figure 7, B and C, shows quantification and representative micrographs of the decrease in neurite length observed in NGF-treated PC12 cells knocked down for FLIP-L when compared with the control. The effective reduction of FLIP-L levels gives rise to almost threefold reduction in neurite outgrowth after 5 d of NGF treatment (Fig. 7B). Reduction of FLIP-L endogenous levels does not impede NGF-induced survival or cell cycle arrest (data not shown). These results point out that NGF-mediated survival and cell cycle arrest are FLIP-L-independent processes and, more interestingly, that endogenous FLIP-L expression is required for neurite outgrowth induced by neurotrophic factors in PC12 cells, SCG neurons, and motoneurons.
Figure 6.
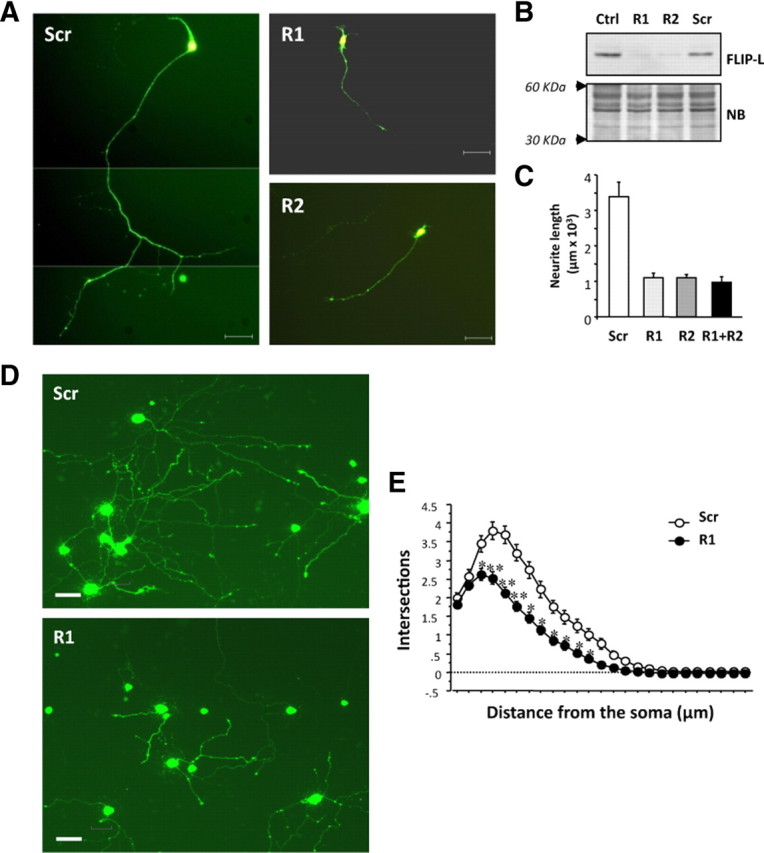
A, Mouse motoneurons were cotransfected with EGFP and pSUPER.retro.puro vector either empty (Ctrl) or with different RNAi against FLIP-L (R1 or R2) or a scrambled sequence (Scr). Cells were maintained in BDNF-supplemented Neurobasal medium for 4 d. Micrographs showing representative GFP-labeled motoneurons proved that these cells express the RNAi transcript. Scale bars, 50 μm. B, Total lysates were obtained and analyzed by Western blot to check for FLIP-L levels using anti-FLIP-L and then the membrane was stained with Naphtol Blue (NB) as a loading control. C, Quantification of the neurite outgrowth performed by tracing neurite arbors and measuring neurite length. Differences are significant between scrambled and RNAi-transfected neurons (*p < 0.001, t test). D, Mouse SCG neurons were cotransfected with EGFP and the pSUPER.retro.puro vector with either RNAi R1 against FLIP-L (R1) or the scrambled RNAi sequence (Scr). At 3 d after transfection, EGFP-labeled SCG were visualized and digitally acquired. Representative images of NGF-maintained SCGs after FLIP-L knockdown are shown. Scale bars, 50 μm. E, Measure of the number of intersections between each neurite and virtual concentric circles from soma to the growth cone. Significant differences are indicated (*p < 0.05, **p < 0.001, t test).
Figure 7.

A, PC12 cells were infected with lentiviral particles containing the sequence of an RNAi against FLIP-L (RNAi) or a scrambled sequence (Scr). After 3 d, total extracts were probed with anti-FLIP (Dave-2) antibody by Western blot, and the membrane was reprobed with anti-tubulin as a loading control. B, PC12 cells infected with FLIP-L RNAi or scrambled (Scr) lentiviral particles were incubated or not with 100 ng/ml NGF for 5 d, and total neurite length was measured. Significant differences are indicated (*p < 0.001, t test). C, Representative images of NGF-induced differentiation of PC12 cells as described in B after 5 d of treatment are shown. Scale bars, 100 μm. D, After 3 d of PC12 infection with lentiviral particles containing the sequence of the RNAi against FLIP-L or a scrambled sequence (Scr), cells were serum starved for 12 h, then stimulated with 100 ng/ml NGF for the indicated times. Protein (250 μg) was immunoprecipitated using the anti-pan-Trk (α-203) antibody, and the membrane was blotted with the anti-phospho-Tyr (4G10) antibody. As input controls, lysates were blotted with anti-FLIP (Dave-2) to ensure efficient knockdown and anti-TrkA. Lysates from the same experiment were analyzed by Western blot (WB) to assess the phosphorylation of ERK1/2. The membrane was stripped and reprobed with an anti-pan-ERK antibody to control protein loading. E, After 2 d of infection with lentiviruses containing FLIP-L RNAi or scrambled sequences, PC12 cells were plated in adequate plates and transfected with an NF-κB-dependent reporter vector (LTR KB). After 24 h, cells were serum starved for 12 h, then stimulated for 5 h with 100 ng/ml NGF. Luciferase activity was measured in the cell lysates using a Luciferase Assay System (Promega). Significant differences are indicated (*p < 0.05, t test).
FLIP-L regulates both ERK and NF-κB activation induced by NGF
Having ascertained the role of FLIP-L in neurite formation in different neuronal types, we pursued the dissection of the signaling pathways involved in this process. The MAPK/ERK cascade is considered as one of the main intracellular pathway mediating neurite outgrowth induced by the activation of neurotrophin receptors, such as TrkA, TrkB, TrkC, and p75NTR (Encinas et al., 1999; Chao, 2003). To determine whether FLIP-L affected the differentiation signaling pathway induced by NGF stimulation of its receptor, we assessed the levels of both TrkA and ERK1/2 activation in PC12 cells after FLIP-L knockdown in a time course of NGF stimulation. PC12 cells infected with Scr or FLIP-L RNAi for 3 d were subjected to serum starvation for 12 h and then stimulated for the indicated times with NGF (Fig. 7D). To explore whether FLIP-L affects NGF-induced TrkA activation, TrkA was immunoprecipitated and immunocomplexes were blotted using an anti-phospho-tyrosine antibody. The knockdown of FLIP-L did not affect the activation of TrkA by tyrosine phosphorylation, which is clearly visible after 5 min of NGF stimulation. Lysates from the same experiment were analyzed by Western blot to assess the phosphorylation of ERK1/2. In scrambled RNAi-infected PC12 cells, ERK1/2 phosphorylation peaks after 5 min of NGF stimulation then progressively decreases over time. However, FLIP-L RNAi-infected PC12 cells exhibit a marked impairment in NGF-induced ERK phosphorylation compared with scrambled RNAi-infected cells (Fig. 7D). To determine whether endogenous FLIP-L is involved in NGF-mediated NF-κB activation, the other main pathway involved in neurotrophin-dependent neuritogenic process (Sole et al., 2004), PC12 cells were infected with lentiviral particles carrying FLIP-L RNAi R1 or its scrambled control and incubated for 2 d for an effective knockdown. Then, cells were transiently transfected with an NF-κB-dependent luciferase reporter construct. At 24 h later, cells were stimulated for 5 h with 100 ng/ml NGF, and luciferase activity was measured as described in Material and Methods. As shown in Figure 7E, FLIP-L RNAi diminishes NGF-induced luciferase activity near to its basal levels, indicating that FLIP-L is a mediator of NF-κB activation. Altogether, these results show that FLIP-L does not affect the activation of TrkA and confirm the importance of endogenous FLIP-L in the activation of both the MAPK/ERK and the NF-κB pathways after NGF-induced neurite outgrowth.
FLIP-L interacts with TrkA in an NGF-dependent manner
Finally, we seek out at which level FLIP-L is acting, upstream ERK and NF-κB activation. FLIP-L is a well documented DR antagonist that prevents cell death by binding to the adaptor FADD at the death inducing signaling complex (DISC) level by the so-called death effector domains present in both FADD and FLIP-L (Yu et al., 2008). Conversely, the MAPK/ERK and NF-κB pathways that are engaged by NGF require the activation of both TrkA and p75NTR, respectively (Chao, 2003). Taking all this into consideration, it would be likely that an interaction exists between FLIP-L and TrkA, p75NTR, or even both receptors. We have demonstrated recently that FAIM-S, a death receptor antagonist that also promotes NGF-induced neurite outgrowth, interacts with both TrkA and p75NTR after NGF stimulation (Sole et al., 2004). To determine whether the same scenario applies for FLIP-L, we used HEK293T cells transfected with the NGF receptors TrkA or p75NTR, both tagged with HA, together with FLAG-tagged FLIP-L or FAIM-S. After 2 d of transfection and 15 min of NGF treatment, cells were lysed and FLAG immunoprecipitation was performed. Figure 8A demonstrates that, unlike FAIM-S, FLIP-L specifically interacts with TrkA but not with p75NTR during NGF stimulation. Furthermore, to validate these results in a neuronal model, PC12 cells stably transfected with TrkA were infected with lentiviral particles containing FLAG-tagged FLIP-L. After 24 h of infection, cells were stimulated or not with NGF for 15 min and then were lysed, and Trk immunoprecipitation was performed using a pan-Trk antibody (Fig. 8B). Although lysates from cells cultured in the absence of NGF failed to show any interaction, TrkA coimmunoprecipitates with FLIP-L only during neurotrophin stimulation, indicating an NGF-dependent interaction between FLIP-L and TrkA. This result was confirmed by the reverse coimmunoprecipitation to consolidate the interaction between FLIP-L and TrkA. FLAG-tagged FLIP-L was immunoprecipitated from lysates of PC12 cells stably transfected with TrkA, infected with lentiviral particles containing FLIP-L or empty-vector, and stimulated or not with NGF for 15 min (Fig. 8C). Once more, the interaction of TrkA with FLIP-L proved to be NGF dependent. Finally, we demonstrated that endogenous FLIP-L interacted with BDNF receptor TrkB in embryonic mice whole-brain lysates (E15) (Fig. 8D). Altogether, these results demonstrate for the first time that, in the nervous system, FLIP-L possesses a novel function distinct from its classical antiapoptotic role, mediated by its interaction with the TrkA and TrkB neurotrophin receptors that do not belong to the death receptor family. After NGF-dependent FLIP-L interaction with TrkA, the MAPK/ERK and NF-κB pathways are activated, thereby promoting neurite growth.
Figure 8.

A, HEK293T cells were transfected with FAIM-S, FLIP-L, TrkA, or p75 tagged with FLAG or HA as described in the figure. After 48 h, NGF stimulation was used in all conditions (100 ng/ml, 15 min). FLAG-tagged FLIP-L and FAIM-S were immunoprecipitated using FLAG-specific mAb M2-coupled agarose beads (Sigma). The immunoprecipitates (eluted fractions) were resolved by SDS-PAGE, and proteins were transferred onto PVDF membranes and immunoblotted with HA antibody (top). A fraction of the cell lysates was blotted with anti-FLAG or anti-HA as input controls (bottom). WB, Western blot. B, PC12 stably transfected with pcDNA3–TrkA were infected with lentiviral particles containing FLIP-L–FLAG. At 24 h after infection, cells were serum starved for 12 h, then stimulated (+) or not (−) with 100 ng/ml NGF for 15 min. Lysates were performed and subjected to immunoprecipitation using anti-pan-Trk α-203 antibody, before immunoblotting with anti-TrkA and anti-FLIP (Dave-2) (top). A fraction of the cell lysates was blotted with anti-TrkA and anti-FLIP (Dave-2) as input controls (bottom). C, PC12 cells stably transfected with pcDNA3–TrkA were infected with lentiviral particles containing the FLIP-L–Flag (+) or the empty vector (−). At 24 h after infection, cells were serum starved for 12 h, then stimulated (+) or not (−) with 100 ng/ml NGF for 15 min. Lysates were performed and subjected to immunoprecipitation as described in A and immunoblotted with anti-TrkA (top). A fraction of the cell lysates was blotted with anti-TrkA and anti-FLIP (Dave-2) (bottom). D, For the endogenous interaction of FLIP-L with TrkB in whole-brain lysates, 2 mg of total protein were immunoprecipitated with anti-FLIP (Dave-2) and immunoblotted with anti-TrkB (top) and anti-FLIP (Dave-2) (bottom). As control, a rat monoclonal antibody anti-HA (Rat IgG) was used.
Discussion
In the present work, we uncover the role of FLIP-L in neurotrophin-mediated neurite outgrowth in diverse neuronal models, which is independent of its classical antiapoptotic function. Although the relevance of the antiapoptotic protein FLIP has been described extensively in many cellular models (Thome and Tschopp, 2001), its function in the nervous system has been poorly documented. Raoul et al. (1999) have suggested that motoneurons maintained in the presence of neurotrophins for >3 d became resistant to Fas activation as a result of FasL downregulation and FLIP upregulation. However, the requirement of FLIP for gaining resistance to Fas was not further investigated in that model. Conversely, Wu et al. (2004) reported that FLIP-L can prevent Fas-mediated but not NGF withdrawal-induced cell death in PC12 cells. These observations support the differential responses we observed concerning the antiapoptotic function of FLIP-L in the different neuronal populations we studied. It might be speculated that regulation of caspase activity could be differential between cellular compartments. Ivins et al. (1998) showed that hippocampal neurons treated with β-amyloid display localized caspase activation leading to neurite degeneration, although the death signals may not affect cell viability. According to our findings, it would be of interest to study whether FLIP-L might act locally as an inhibitor of caspase-8 protecting from local neurite degeneration, or rather as a proneuritogenic factor, in that model of compartmented neuronal cultures.
To avoid uncontrolled cell and tissue damage, apoptosis is tightly controlled by inhibitors, including the DR antagonists. Notably in the nervous system, recent advances have been made concerning the role of FAIM and Lifeguard (FAIM2) in the antagonism of apoptosis induced by FasL or tumor necrosis factor α (TNFα) (Sole et al., 2004; Fernández et al., 2007; Segura et al., 2007). Particularly, we have demonstrated that the two splice variants of FAIM [the long form of FAIM (FAIM-L) and FAIM-S] display diverse functions in the nervous system. Whereas FAIM-L blocks Fas and TNFα-induced apoptosis in differentiated PC12 cells, motoneurons, cortical neurons (Segura et al., 2007), and dopaminergic neurons (Yu et al., 2008), FAIM-S overexpression enhances neurite outgrowth in differentiated PC12 cells and primary neurons, mainly through the activation and modulation of the NF-κB pathway (Sole et al., 2004). In contrast to its established role in regulating cell survival in the immune system, we revealed a novel role for FAIM-S in the control of neurite growth in the developing nervous system. FAIM-S binds neurotrophin receptors TrkA and p75NTR during NGF stimulation, thereby enhancing differentiation (Sole et al., 2004). Similarities and differences can be drawn from the findings we substantiated for FAIM and now for FLIP. First, both proteins display a dual role in the nervous system. FAIM needs two splice variants to achieve its different roles: FAIM-L is antiapoptotic, whereas FAIM-S is pro-neuritogenic. However, the long form of FLIP is able to perform a pro-differentiation role, as well as an antiapoptotic function depending on the cellular context. Second, FAIM-S and FLIP-L both promote neurite outgrowth during NGF differentiation by mechanisms depending on ERK (FLIP-L) and NF-κB activation (both FAIM-S and FLIP-L). Finally, both FAIM-S and FLIP-L interact with NGF receptors, only in the presence of the neurotrophin. However, whereas FAIM-S interacts with both NGF receptors TrkA and p75NTR, FLIP-L only interacts with TrkA or with the TrkB receptor in whole-brain lysates of E15 mice embryos. Therefore, we report in the present work that FLIP-L is a dual protein in both the nervous system and, as reported previously, the immune system.
It has been reported that both FLIP and caspase-8 are necessary for survival and proliferation of activated T lymphocytes. Elevated expression levels of FLIP have a dual effect blocking the apoptotic signaling cascade, on the one hand, and stimulating signaling pathways that lead to cellular proliferation, on the other hand (Peter, 2004; Budd et al., 2006). However, there has been no conclusive evidence that FLIP mediates these two functions through distinct molecular mechanisms (Lens et al., 2002; Tai et al., 2004). It is plausible that, after stimulation by neurotrophins, neurons would acquire a differentiated phenotype as a survival strategy. Additionally, neurons that would not attain a neuronal phenotype suitable for reaching the sources of trophic factors would undergo apoptosis. Therefore, FLIP-L might potentiate the survival of the neuron, allowing for the time necessary to reach its target. However, our results do not support this hypothesis because the overexpression of FLIP-L protects only motoneurons from neurotrophin deprivation. Moreover, in the three neuronal models tested, the overexpression of FLIP-L promotes neurite outgrowth, regardless of whether it antagonizes neurotrophin deprivation-induced apoptosis or not. Consequently, the pro-neuritogenic function of FLIP-L seems to be more general than its antiapoptotic function, at least in the nervous system. Finally, the reduction of the endogenous levels of FLIP-L was not toxic in any of the cellular models studied, nor did it affect proliferation of PC12 cells (data not shown). All these observations allow us to confirm that we found a novel function for FLIP-L in the nervous system, clearly separated from the antiapoptotic function it might exert in a specific neuronal cell type.
In CD3-activated T lymphocytes, increased levels of FLIP-L lead to the activation of the MAPK/ERK and NF-κB signaling pathways through the recruitment of TNF receptor-associated factors 1 and 2, receptor-interacting protein, and Raf-1 (Kataoka et al., 2000). Our findings go in the same direction because knockdown of FLIP-L significantly reduced the phosphorylation of ERK1/2 as well as NF-κB activation. Indeed, we demonstrated that both pathways are necessary for FLIP-L-mediated neurite outgrowth. The inhibition of the MAPK/ERK or NF-κB pathway leads to a partial although significant inhibition, whereas when MAPK/ERK and NF-κB pathways are simultaneously blocked, the neurite outgrowth is totally abrogated, regardless of FLIP-L expression levels. Then, FLIP-L requires both MAPK/ERK and NF-κB pathway to enhance NGF-induced neuritogenesis.
Although NGF is the ligand of both TrkA and p75NTR, coimmunoprecipitation experiments demonstrated that FLIP-L is able to interact only with TrkA in an NGF-dependent manner. It has also been documented that, although both NGF receptors independently activate the NF-κB pathway (Foehr et al., 2000a,b), the main stimulus is initiated by p75NTR, with TrkA exerting a slightly inhibiting effect (Mamidipudi et al., 2002). So far, FLIP has always been linked to the DISC complex, which is formed during activation of the DRs. However, our results show that, at least in the nervous system, FLIP-L could be part of another multiprotein complex that is not necessarily composed by DISC members. First, the activation stimulus of our models does not implicate any DR but rather engages neurotrophin receptors TrkB, TrkA, and p75NTR that promote neuronal survival and neurite growth. Second, the interaction of FLIP-L and TrkA is dependent on the NGF stimulus. However, these coimmunoprecipitation experiments should be cautiously interpreted because the interaction might be indirect. In any case, the activation of the TrkA receptor promotes the formation of a novel complex in which FLIP-L is included. Moreover, this protein is relevant for the correct activation of the MAPK/ERK and NF-κB, pathways triggered during TrkA ligation, because FLIP-L downregulation drastically affects the activation of both pathways. The p75NTR receptor seems not to be implicated in FLIP-L-mediated signaling, but we should not exclude that it might be modulated by FLIP-L, although in an indirect way.
We provide evidence that, although cell death, survival, proliferation, and differentiation/growth processes are finely regulated, the frontier between two opposite cellular processes (i.e., apoptosis/neurite growth) can be tenuous. In other words, they can be controlled by the same molecule(s). In that sense, it might be envisioned that the specific function of a given protein might depend on the intracellular molecular environment, as well as the spatiotemporal conditions. It has been established in the immune system that FLIP-L acts as a multifunctional protein that switches off DR-activated signaling pathways by either blocking caspase-8 activation or recruiting adaptor proteins and kinases that initiate pathways leading to proliferation or differentiation (Kataoka et al., 2000). In the nervous system, FLIP-L promotes neurite outgrowth but acts as an antiapoptotic protein in certain cellular models (i.e., motoneurons). Most interestingly, the expression pattern of FLIP-L during the embryonic development or in NGF-induced differentiation of PC12 cells, as well as its tissular expression pattern in mouse brain, further supports its novel role in the nervous system. At E16, the peak of expression of FLIP-L in the brain cortex is neuronal, whereas the distribution and morphology of FLIP-positive cells corresponds with glial-like cells at later stages. We might hypothesize that, early at E16, FLIP-L plays a pro-differentiation role in the cortex, allowing neurite outgrowth and branching processes, redistributing at later stages to the glia, in which it can participate in antagonizing apoptosis induced by death ligands (i.e., TNFα or FasL). These ligands can be produced by microglia or astrocytes in certain pathological situations such as cerebral ischemia (Kaushal and Schlichter, 2008). Nonetheless, the characterization of the role of FLIP in glial or microglial cells requires additional experimental work. In summary, we have uncovered a new unexpected role for FLIP-L, a death receptor antagonist, as a crucial factor in neurotrophin-induced neurite growth capable of interacting with TrkB or TrkA in a neurotrophin-dependent manner.
Footnotes
This work was funded by Ministerio de Sanidad y Consumo (Centro de Investigación Biomédica en Red sobre Enfermedades Neurodegenerativas (CIBERNED) Grant CB06/05/1104), Ministerio de Educación y Ciencia Grant SAF2007-60287, Generalitat de Catalunya (Suport als Grups de Recerca Consolidats) (J.X.C.), Ministerio de Ciencia e Innovación Grants BFU2008-01328 (V.J.Y.) and BFU2008-3980 (E.S.), and Fundació Caixa Catalunya Obra Social (E.S.). C.S. was supported by postgraduate fellowships from Ministerio de Educación y Ciencia and Fondo de Investigación Sanitaria. V.I.-G. holds a postgraduate fellowship from the Generalitat de Catalunya. R.S.M. and V.J.Y. are under the Juan de la Cierva and Ramon y Cajal programs, respectively, from the Ministerio de Educación y Ciencia (Spain), cofinanced by the European Social Fund. We thank D. Trono (Geneva, Switzerland) for providing the lentiviral plasmids and Dr. Joaquin Lopez-Soriano and Patricia Ortega for excellent technical support.
References
- Arce V, Pollock RA, Philippe JM, Pennica D, Henderson CE, deLapeyrière O. Synergistic effects of schwann- and muscle-derived factors on motoneuron survival involve GDNF and cardiotrophin-1 (CT-1) J Neurosci. 1998;18:1440–1448. doi: 10.1523/JNEUROSCI.18-04-01440.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budd RC, Yeh WC, Tschopp J. cFLIP regulation of lymphocyte activation and development. Nat Rev Immunol. 2006;6:196–204. doi: 10.1038/nri1787. [DOI] [PubMed] [Google Scholar]
- Chao MV. Neurotrophins and their receptors: a convergence point for many signalling pathways. Nat Rev Neurosci. 2003;4:299–309. doi: 10.1038/nrn1078. [DOI] [PubMed] [Google Scholar]
- Comella JX, Sanz-Rodriguez C, Aldea M, Esquerda JE. Skeletal muscle-derived trophic factors prevent motoneurons from entering an active cell death program in vitro. J Neurosci. 1994;14:2674–2686. doi: 10.1523/JNEUROSCI.14-05-02674.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cullen BR. Use of eukaryotic expression technology in the functional analysis of cloned genes. Methods Enzymol. 1987;152:684–704. doi: 10.1016/0076-6879(87)52074-2. [DOI] [PubMed] [Google Scholar]
- Davies AM, Lee KF, Jaenisch R. p75-deficient trigeminal sensory neurons have an altered response to NGF but not to other neurotrophins. Neuron. 1993;11:565–574. doi: 10.1016/0896-6273(93)90069-4. [DOI] [PubMed] [Google Scholar]
- Dechant G, Barde YA. The neurotrophin receptor p75(NTR): novel functions and implications for diseases of the nervous system. Nat Neurosci. 2002;5:1131–1136. doi: 10.1038/nn1102-1131. [DOI] [PubMed] [Google Scholar]
- Encinas M, Iglesias M, Llecha N, Comella JX. Extracellular-regulated kinases and phosphatidylinositol 3-kinase are involved in brain-derived neurotrophic factor-mediated survival and neuritogenesis of the neuroblastoma cell line SH-SY5Y. J Neurochem. 1999;73:1409–1421. doi: 10.1046/j.1471-4159.1999.0731409.x. [DOI] [PubMed] [Google Scholar]
- Fernández M, Segura MF, Solé C, Colino A, Comella JX, Ceña V. Lifeguard/neuronal membrane protein 35 regulates Fas ligand-mediated apoptosis in neurons via microdomain recruitment. J Neurochem. 2007;103:190–203. doi: 10.1111/j.1471-4159.2007.04767.x. [DOI] [PubMed] [Google Scholar]
- Foehr ED, Lin X, O'Mahony A, Geleziunas R, Bradshaw RA, Greene WC. NF-κB signaling promotes both cell survival and neurite process formation in nerve growth factor-stimulated PC12 cells. J Neurosci. 2000a;20:7556–7563. doi: 10.1523/JNEUROSCI.20-20-07556.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foehr ED, Bohuslav J, Chen LF, DeNoronha C, Geleziunas R, Lin X, O'Mahony A, Greene WC. The NF-kappaB-inducing kinase induces PC12 cell differentiation and prevents apoptosis. J Biol Chem. 2000b;275:34021–34024. doi: 10.1074/jbc.C000507200. [DOI] [PubMed] [Google Scholar]
- Gozzelino R, Sole C, Llecha N, Segura MF, Moubarak RS, Iglesias-Guimarais V, Perez-Garcia MJ, Reix S, Zhang J, Badiola N, Sanchis D, Rodriguez-Alvarez J, Trullas R, Yuste VJ, Comella JX. BCL-XL regulates TNF-alpha-mediated cell death independently of NF-kappaB, FLIP and IAPs. Cell Res. 2008;18:1020–1036. doi: 10.1038/cr.2008.76. [DOI] [PubMed] [Google Scholar]
- Gutierrez H, Davies AM. A fast and accurate procedure for deriving the Sholl profile in quantitative studies of neuronal morphology. J Neurosci Methods. 2007;163:24–30. doi: 10.1016/j.jneumeth.2007.02.002. [DOI] [PubMed] [Google Scholar]
- Hamanoue M, Middleton G, Wyatt S, Jaffray E, Hay RT, Davies AM. p75-mediated NF-kappaB activation enhances the survival response of developing sensory neurons to nerve growth factor. Mol Cell Neurosci. 1999;14:28–40. doi: 10.1006/mcne.1999.0770. [DOI] [PubMed] [Google Scholar]
- Irmler M, Thome M, Hahne M, Schneider P, Hofmann K, Steiner V, Bodmer JL, Schröter M, Burns K, Mattmann C, Rimoldi D, French LE, Tschopp J. Inhibition of death receptor signals by cellular FLIP. Nature. 1997;388:190–195. doi: 10.1038/40657. [DOI] [PubMed] [Google Scholar]
- Ivins KJ, Bui ET, Cotman CW. Beta-amyloid induces local neurite degeneration in cultured hippocampal neurons: evidence for neuritic apoptosis. Neurobiol Dis. 1998;5:365–378. doi: 10.1006/nbdi.1998.0228. [DOI] [PubMed] [Google Scholar]
- Kataoka T, Schröter M, Hahne M, Schneider P, Irmler M, Thome M, Froelich CJ, Tschopp J. FLIP prevents apoptosis induced by death receptors but not by perforin/granzyme, B, chemotherapeutic drugs, and gamma irradiation. J Immunol. 1998;161:3936–3942. [PubMed] [Google Scholar]
- Kataoka T, Budd RC, Holler N, Thome M, Martinon F, Irmler M, Burns K, Hahne M, Kennedy N, Kovacsovics M, Tschopp J. The caspase-8 inhibitor FLIP promotes activation of NF-kappaB and Erk signaling pathways. Curr Biol. 2000;10:640–648. doi: 10.1016/s0960-9822(00)00512-1. [DOI] [PubMed] [Google Scholar]
- Kaushal V, Schlichter LC. Mechanisms of microglia-mediated neurotoxicity in a new model of the stroke penumbra. J Neurosci. 2008;28:2221–2230. doi: 10.1523/JNEUROSCI.5643-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lens SM, Kataoka T, Fortner KA, Tinel A, Ferrero I, MacDonald RH, Hahne M, Beermann F, Attinger A, Orbea HA, Budd RC, Tschopp J. The caspase 8 inhibitor c-FLIP(L) modulates T-cell receptor-induced proliferation but not activation-induced cell death of lymphocytes. Mol Cell Biol. 2002;22:5419–5433. doi: 10.1128/MCB.22.15.5419-5433.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewin GR, Barde YA. Physiology of the neurotrophins. Annu Rev Neurosci. 1996;19:289–317. doi: 10.1146/annurev.ne.19.030196.001445. [DOI] [PubMed] [Google Scholar]
- Lindenboim L, Haviv R, Stein R. Bcl-xL inhibits different apoptotic pathways in rat PC12 cells. Neurosci Lett. 1998;253:37–40. doi: 10.1016/s0304-3940(98)00602-8. [DOI] [PubMed] [Google Scholar]
- Mamidipudi V, Li X, Wooten MW. Identification of interleukin 1 receptor-associated kinase as a conserved component in the p75-neurotrophin receptor activation of nuclear factor-kappa B. J Biol Chem. 2002;277:28010–28018. doi: 10.1074/jbc.M109730200. [DOI] [PubMed] [Google Scholar]
- Martin-Zanca D, Oskam R, Mitra G, Copeland T, Barbacid M. Molecular and biochemical characterization of the human trk proto-oncogene. Mol Cell Biol. 1989;9:24–33. doi: 10.1128/mcb.9.1.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAllister AK. Subplate neurons: a missing link among neurotrophins, activity, and ocular dominance plasticity? Proc Natl Acad Sci U S A. 1999;96:13600–13602. doi: 10.1073/pnas.96.24.13600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naldini L, Blömer U, Gage FH, Trono D, Verma IM. Efficient transfer, integration, and sustained long-term expression of the transgene in adult rat brains injected with a lentiviral vector. Proc Natl Acad Sci U S A. 1996;93:11382–11388. doi: 10.1073/pnas.93.21.11382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pérez-García MJ, Ceña V, de Pablo Y, Llovera M, Comella JX, Soler RM. Glial cell line-derived neurotrophic factor increases intracellular calcium concentration. Role of calcium/calmodulin in the activation of the phosphatidylinositol 3-kinase pathway. J Biol Chem. 2004;279:6132–6142. doi: 10.1074/jbc.M308367200. [DOI] [PubMed] [Google Scholar]
- Peter ME. The flip side of FLIP. Biochem J. 2004;382:e1–e3. doi: 10.1042/BJ20041143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Putcha GV, Harris CA, Moulder KL, Easton RM, Thompson CB, Johnson EM., Jr Intrinsic and extrinsic pathway signaling during neuronal apoptosis: lessons from the analysis of mutant mice. J Cell Biol. 2002;157:441–453. doi: 10.1083/jcb.200110108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raoul C, Henderson CE, Pettmann B. Programmed cell death of embryonic motoneurons triggered through the Fas death receptor. J Cell Biol. 1999;147:1049–1062. doi: 10.1083/jcb.147.5.1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez MS, Wright J, Thompson J, Thomas D, Baleux F, Virelizier JL, Hay RT, Arenzana-Seisdedos F. Identification of lysine residues required for signal-induced ubiquitination and degradation of I kappa B-alpha in vivo. Oncogene. 1996;12:2425–2435. [PubMed] [Google Scholar]
- Segura MF, Sole C, Pascual M, Moubarak RS, Perez-Garcia MJ, Gozzelino R, Iglesias V, Badiola N, Bayascas JR, Llecha N, Rodriguez-Alvarez J, Soriano E, Yuste VJ, Comella JX. The long form of Fas apoptotic inhibitory molecule is expressed specifically in neurons and protects them against death receptor-triggered apoptosis. J Neurosci. 2007;27:11228–11241. doi: 10.1523/JNEUROSCI.3462-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sholl DA. Dendritic organization in the neurons of the visual and motor cortices of the cat. J Anat. 1953;87:387–406. [PMC free article] [PubMed] [Google Scholar]
- Sole C, Dolcet X, Segura MF, Gutierrez H, Diaz-Meco MT, Gozzelino R, Sanchis D, Bayascas JR, Gallego C, Moscat J, Davies AM, Comella JX. The death receptor antagonist FAIM promotes neurite outgrowth by a mechanism that depends on ERK and NF-kappaB signaling. J Cell Biol. 2004;167:479–492. doi: 10.1083/jcb.200403093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tai TS, Fang LW, Lai MZ. c-FLICE inhibitory protein expression inhibits T-cell activation. Cell Death Differ. 2004;11:69–79. doi: 10.1038/sj.cdd.4401316. [DOI] [PubMed] [Google Scholar]
- Taoufik E, Valable S, Müller GJ, Roberts ML, Divoux D, Tinel A, Voulgari-Kokota A, Tseveleki V, Altruda F, Lassmann H, Petit E, Probert L. FLIP(L) protects neurons against in vivo ischemia and in vitro glucose deprivation-induced cell death. J Neurosci. 2007;27:6633–6646. doi: 10.1523/JNEUROSCI.1091-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thome M, Tschopp J. Regulation of lymphocyte proliferation and death by FLIP. Nat Rev Immunol. 2001;1:50–58. doi: 10.1038/35095508. [DOI] [PubMed] [Google Scholar]
- Thome M, Schneider P, Hofmann K, Fickenscher H, Meinl E, Neipel F, Mattmann C, Burns K, Bodmer JL, Schröter M, Scaffidi C, Krammer PH, Peter ME, Tschopp J. Viral FLICE-inhibitory proteins (FLIPs) prevent apoptosis induced by death receptors. Nature. 1997;386:517–521. doi: 10.1038/386517a0. [DOI] [PubMed] [Google Scholar]
- Tseveleki V, Bauer J, Taoufik E, Ruan C, Leondiadis L, Haralambous S, Lassmann H, Probert L. Cellular FLIP (long isoform) overexpression in T cells drives Th2 effector responses and promotes immunoregulation in experimental autoimmune encephalomyelitis. J Immunol. 2004;173:6619–6626. doi: 10.4049/jimmunol.173.11.6619. [DOI] [PubMed] [Google Scholar]
- Wood JN. Regulation of NF-kappaB activity in rat dorsal root ganglia and PC12 cells by tumour necrosis factor and nerve growth factor. Neurosci Lett. 1995;192:41–44. doi: 10.1016/0304-3940(95)11603-t. [DOI] [PubMed] [Google Scholar]
- Wooten MW, Seibenhener ML, Mamidipudi V, Diaz-Meco MT, Barker PA, Moscat J. The atypical protein kinase C-interacting protein p62 is a scaffold for NF-kappaB activation by nerve growth factor. J Biol Chem. 2001;276:7709–7712. doi: 10.1074/jbc.C000869200. [DOI] [PubMed] [Google Scholar]
- Wu S, Zhou L, Rose M, Xiao X, Graham SH. c-FLIP-L recombinant adeno-associated virus vector infection prevents Fas-mediated but not nerve growth factor withdrawal-mediated cell death in PC12 cells. Brain Res Mol Brain Res. 2004;122:79–87. doi: 10.1016/j.molbrainres.2003.12.007. [DOI] [PubMed] [Google Scholar]
- Yeh WC, Itie A, Elia AJ, Ng M, Shu HB, Wakeham A, Mirtsos C, Suzuki N, Bonnard M, Goeddel DV, Mak TW. Requirement for Casper (c-FLIP) in regulation of death receptor-induced apoptosis and embryonic development. Immunity. 2000;12:633–642. doi: 10.1016/s1074-7613(00)80214-9. [DOI] [PubMed] [Google Scholar]
- Yoon SO, Casaccia-Bonnefil P, Carter B, Chao MV. Competitive signaling between TrkA and p75 nerve growth factor receptors determines cell survival. J Neurosci. 1998;18:3273–3281. doi: 10.1523/JNEUROSCI.18-09-03273.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu LY, Saarma M, Arumäe U. Death receptors and caspases but not mitochondria are activated in the GDNF- or BDNF-deprived dopaminergic neurons. J Neurosci. 2008;28:7467–7475. doi: 10.1523/JNEUROSCI.1877-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuste VJ, Bayascas JR, Llecha N, Sánchez-López I, Boix J, Comella JX. The absence of oligonucleosomal DNA fragmentation during apoptosis of IMR-5 neuroblastoma cells: disappearance of the caspase-activated DNase. J Biol Chem. 2001;276:22323–22331. doi: 10.1074/jbc.M100072200. [DOI] [PubMed] [Google Scholar]
- Zufferey R, Dull T, Mandel RJ, Bukovsky A, Quiroz D, Naldini L, Trono D. Self-inactivating lentivirus vector for safe and efficient in vivo gene delivery. J Virol. 1998;72:9873–9880. doi: 10.1128/jvi.72.12.9873-9880.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuliani C, Kleber S, Klussmann S, Wenger T, Kenzelmann M, Schreglmann N, Martinez A, del Rio JA, Soriano E, Vodrazka P, Kuner R, Groene HJ, Herr I, Krammer PH, Martin-Villalba A. Control of neuronal branching by the death receptor CD95 (Fas/Apo-1) Cell Death Differ. 2006;13:31–40. doi: 10.1038/sj.cdd.4401720. [DOI] [PubMed] [Google Scholar]