

Abstract
Neuronal migration and axon growth, key events during neuronal development, require distinct changes in the cytoskeleton. Although many molecular regulators of polarity have been identified and characterized, relatively little is known about their physiological role in this process. To study the physiological function of Rac1 in neuronal development, we have generated a conditional knock-out mouse, in which Rac1 is ablated in the whole brain. Rac1-deficient cerebellar granule neurons, which do not express other Rac isoforms, showed impaired neuronal migration and axon formation both in vivo and in vitro. In addition, Rac1 ablation disrupts lamellipodia formation in growth cones. The analysis of Rac1 effectors revealed the absence of the Wiskott–Aldrich syndrome protein (WASP) family verprolin-homologous protein (WAVE) complex from the plasma membrane of knock-out growth cones. Loss of WAVE function inhibited axon growth, whereas overexpression of a membrane-tethered WAVE mutant partially rescued axon growth in Rac1-knock-out neurons. In addition, pharmacological inhibition of the WAVE complex effector Arp2/3 also reduced axon growth. We propose that Rac1 recruits the WAVE complex to the plasma membrane to enable actin remodeling necessary for axon growth.
Introduction
Neuronal differentiation requires spatial and dynamic reorganization of the cytoskeleton. In particular, finely tuned actin filament organization and microtubule dynamics are essential for neuronal migration, axon growth, and guidance (Luo, 2000; da Silva and Dotti, 2002; Dent and Gertler, 2003; Kawauchi and Hoshino, 2008; Witte and Bradke, 2008). The Rho-GTPases Cdc42, Rac, and Rho, key regulators of the cytoskeleton, are implicated in these processes (Govek et al., 2005). However, the physiological role of these molecules during neuronal development remains fragmentary.
Loss-of-function mutants of the Drosophila Rac genes show impaired axon growth, branching, and guidance (Hakeda-Suzuki et al., 2002; Ng et al., 2002). The role of mammalian Rac proteins in axon development is less clear. Rac1 and Rac3 are present in the nervous system (Didsbury et al., 1989; Bolis et al., 2003), and the expression of mutant Racs causes aberrant axon formation in mouse and chick neurons (Luo et al., 1996; Albertinazzi et al., 1998). By contrast, deletion of Rac3 or specific ablation of Rac1 in the mouse cortex does not affect axon growth (Corbetta et al., 2005; Chen et al., 2007; Gualdoni et al., 2007; Kassai et al., 2008). This suggests that axon growth may be independent of Rac activity. However, Rac3, which is also expressed in the cortex, could have a redundant function with Rac1 (Bolis et al., 2003; Corbetta et al., 2009). Thus, the physiological role of mammalian Racs in axon growth remains to be elucidated.
Apart from its debated role in axon growth, Rac1 may also be involved in regulating cell migration (Ridley, 2001a). However, this idea has been challenged in recent in vivo studies, showing that Rac1 is dispensable for the migration of many neuronal and non-neuronal cells, including fibroblasts, macrophages, leukocytes, and cortical neurons (Bokoch, 2005; Vidali et al., 2006; Wheeler et al., 2006; Kassai et al., 2008).
Among the Rho GTPases, Rac has a unique role in remodeling the actin cytoskeleton during lamellipodia formation (Nobes and Hall, 1995; Hall, 1998; Ridley, 2001b). Rac regulates actin dynamics through two main downstream effector pathways, p21-activated kinase (PAK) and the Wiskott–Aldrich syndrome protein (WASP) family verprolin-homologous protein (WAVE) (Miki et al., 1998; Sells et al., 1999; Ridley, 2001a; Burridge and Wennerberg, 2004; Stradal et al., 2004). PAK signals to the actin cytoskeleton via cofilin (Ng and Luo, 2004), whereas WAVE induces cytoskeletal changes by direct interaction with the actin-related protein 2/3 (Arp2/3) complex and profilin II (Takenawa and Miki, 2001; Pilo Boyl et al., 2007). Yet it remains unclear whether these pathways are activated during Rac1-mediated actin reorganization and axon growth in the mammalian nervous system.
To directly characterize the role of Rac1 in neuronal development, we generated a brain-specific Rac1-knock-out mouse and focused on cerebellar granule neurons (CGNs), which are devoid of other Rac isoforms (Bolis et al., 2003). We report that Rac1 positively regulates neuronal migration and polarization by modulating actin dynamics. Our data suggest that Rac1 regulates the WAVE complex and Arp2/3 to enable actin remodeling necessary for axon growth.
Materials and Methods
Mice.
All animal experiments were performed in accordance with animal handling laws. The generation of the mouse line carrying a floxed Rac1 allele has been described previously (Chrostek et al., 2006). To obtain mice lacking Rac1 in the nervous system (Rac1−/−), mice homozygous for a floxed Rac1 allele (Rac1flox/flox) were crossed with mice heterozygous for a floxed Rac1 allele (Rac1flox/wild type) and expressing Cre recombinase under the control of a Nestin promoter (Tronche et al., 1999; Graus-Porta et al., 2001). For our experiments, we used both male and female animals.
Expression constructs.
Wild-type Rac1 or Rac3 cloned into vector pcDNA3.1 were obtained from Missouri Science and Technology cDNA Resource Center (Rolla, MO). Wild-type WAVE1 cloned into pCMV6-XL5 vector was obtained from OriGene. pcDNA3.1 and pcDNA3.1/myc-His control plasmids and pEYFP-C1 transfection marker were from Invitrogen. Constitutively active Pak1 (PakT423E) was kindly provided by Dr. J. Chernoff (Fox Chase Cancer Center, Philadelphia, PA) (Sells et al., 1997). Wild-type and catalytically inactive Pak1 constructs in pCMV6Myc-CAAX expression plasmid (Pak-CAAX and PakR299-CAAX, respectively) were kindly provided by Dr. G. Bokoch (The Scripps Research Institute, La Jolla, CA) (Daniels et al., 1998). To obtain a membrane-tethered WAVE1 construct (WAVE-CAAX), cDNA encoding the wild-type WAVE1 was amplified by PCR (forward primer, 5′-AAGGATCCATGCCGCTAGTGAAAAGAAAC-3′, and reverse primer, 5′-AGCTAGCCTCCAACCAATCTACTTCATCAA-3′) from a WAVE1-containing plasmid and subcloned as BamHI–NheI fragment into the pCMV6Myc-CAAX expression plasmid. EGFP-WAVE2 plasmid has been described previously (Benesch et al., 2002).
Protein extraction and Western blotting.
Dissected cortices and cerebella from postnatal day 9 (P9) brains were resuspended in cold lysis buffer (Cytoskeleton) containing 50 mm sodium fluoride (Sigma-Aldrich), 0.5 mm AEBSF [4-(2-aminoetyl)benzensulfonyl fluoride hydrochloride] (Sigma-Aldrich), and 1 mm sodium orthovanadate (Sigma-Aldrich). The tissue was homogenized at 4°C using a micropestle (Schubert & Weiss) and centrifuged for 3 min at 10,000 rpm, and the supernatant was transferred to a fresh tube. For Western blotting, the following mouse monoclonal primary antibodies and dilutions were used: anti-Rac, clone 23A8 (1:2000; Millipore), anti-α-tubulin, clone B-5-1-2 (1:150,000; Sigma-Aldrich), anti-GAPDH (1:2000; Abcam). The rabbit polyclonal primary antibodies used were as follows: anti-PAK1 C-19 (1:5000; Santa Cruz Biotechnology), anti-phospho-PAK1 (1:1000) kindly provided by Drs. G. Schratt and M. Greenberg (Harvard Medical School, Boston, MA) (Shamah et al., 2001), anti-cofilin 1439 (1:1600) and anti-phospho-cofilin 4317 (1:600) kindly provided by Dr. J. Bamburg (Colorado State University, Fort Collins, CO) (Meberg et al., 1998), anti-pan-WAVE (1:2000), anti-Sra-1 (1:5000), and anti-Nap1 (1:2000). Goat secondary anti-mouse and anti-rabbit IgG-HRP antibodies (Santa Cruz Biotechnology) were used. The blots were developed using chemiluminescence (GE Healthcare).
Histology and immunohistochemistry.
Animals were anesthetized with a mixture of Rompun (Bayer Health Care) and ketamine (WDT), killed, and perfused with 4% paraformaldehyde (PFA) in 0.1 m PBS, pH 7.4. Brains were dissected and postfixed in 4% PFA for 1 h. Histological examination was performed on 10 μm Nissl-stained paraffin sections. For immunohistochemistry, 25 μm frozen tissue sections were permeabilized with 0.5% Triton X-100 in PBS for 30 min, blocked with 5% goat serum (Invitrogen) and 0.5% Triton X-100 in PBS for 1 h, and subsequently incubated overnight with primary antibody diluted in blocking solution. The primary antibodies used were mouse monoclonal anti-neuron-specific nuclear protein (NeuN) (1:100; Millipore Bioscience Research Reagents) and anti-L1 (1:100; Abcam) and rabbit polyclonal anti-calbindin D-28K (1:100; Millipore Bioscience Research Reagents), anti-glial fibrillary acidic protein (GFAP) (1:400; Dako), anti-active caspase-3 (1:100; Promega), anti-neurofilament (150 kDa) (1:200; Millipore Bioscience Research Reagents), anti-phosphohistone H3 (1:100; Millipore), and anti-Ki-67 (1:50; Novocastra Laboratories). The sections were incubated with appropriate secondary antibodies conjugated to Alexa Fluor 488, 568, and 350 (Invitrogen). Confocal images were acquired with an SP2 confocal microscope (Leica) in sequential scanning mode.
5-Bromo-2′-deoxyuridine labeling.
P7–P8 mice were injected intraperitoneally with 50 mg/kg 5-bromo-2′-deoxyuridine (BrdU) (250 mg/ml DMSO stock; Sigma-Aldrich). Mice were perfused 24 or 96 h after injection. After denaturing the DNA (2N HCl; 40°C; 30 min), the brain sections were immunostained using a mouse anti-BrdU monoclonal antibody, clone MoBu-1 (1:400; Acris Antibodies).
Cerebellar microexplant culture.
Microexplant cultures were prepared from the P5 external granule layer (EGL) as described previously (Nagata and Nakatsuji, 1990). Cerebellar microexplants were fixed and stained with Tuj1 and 4′,6-diamidino-2-phenylindole (DAPI) (2 μg/ml; Invitrogen).
Migration analysis.
To assess migration, the number of DAPI-labeled migratory cells surrounding the explant was counted using the Scion Image 4.0.2 software. Explants were normalized according to their size, and results were expressed as number of migratory cells per square millimeter of explant area. To evaluate the distribution of migratory cells around the explant, we measured the occupancy area defined as the area that contained >90% of migratory cells.
Quantification of cell proliferation, apoptosis, and migration.
Quantification of cell proliferation was performed by counting the number of phosphohistone H3- and Ki-67-positive cells in the EGL and molecular layer (ML) of three comparable nonadjacent midsagittal sections. Results were expressed as the number of proliferating cells per millimeter of EGL length. For quantification of cell death, the number of active caspase-3-positive cells in three comparable nonadjacent midsagittal sections was counted. Results were expressed as the number of proliferating cells per square millimeter of cerebellar surface. Quantification of cell migration was performed by counting the number of NeuN-positive cells in the ML of three comparable nonadjacent midsagittal sections. Results were expressed as the number of proliferating cells per micrometer of ML length.
DiI labeling of parallel fibers.
P15 cerebella were fixed with 4% PFA, embedded in 3% agarose in PBS, and cut into 80 μm coronal sections using a vibratome. Sections were mounted on a glass slide, and tissue was punctured with the lipophilic dye DiI (Invitrogen)-filled glass pipette (no pressure applied). The DiI-filled pipette was inserted beneath the pial surface, into the molecular layer of a cerebellum at the midline of folium pyramis vermis. To test for possible variations in the injection area, we performed 13 DiI injections in the cerebellar tissue and measured the labeled area. The mean injection diameter was 65 μm, and the SD was ±4 μm (6%). Sections were incubated in 4% PFA at room temperature for 2–4 d and subsequently analyzed for parallel fiber extension using fluorescence microscopy. Fluorescent images were processed using Adobe Photoshop and Canvas software.
Cell culture and transfections.
Primary culture of CGNs was prepared from the cerebellar cortex of P9 mice as described previously (Hatten, 1985; Ertürk et al., 2007). Briefly, CGNs were dissociated using a trypsin (10 mg/ml)/DNase (1 mg/ml) mix and triturated to remove cellular aggregates. The granule cells were plated at densities of 300–500 cells/mm2 in 6 cm tissue culture dishes containing poly-l-lysine (1 mg/ml; Sigma-Aldrich)-coated glass coverslips and Neurobasal medium (Invitrogen) supplemented with 2% B27 (Invitrogen), 2 mm glutamine (Invitrogen), and 1% penicillin–streptomycin (Invitrogen). Transfections were performed using Amaxa Nucleofector (2 × 106 cells in 100 μl cell suspension with 5 μg of DNA; Amaxa Biosystems). Various expression constructs or control plasmids were mixed with pEYFP-C1 as a transfection marker at a ratio of 4:1. Cells were analyzed 3 d after transfection.
For the RNA interference (RNAi) experiments, Nap1 short hairpin RNA (shRNA) and control vector were kindly provided by Dr. Eva Anton (University of North Carolina School of Medicine, Chapel Hill, NC) (Yokota et al., 2007). To perform RNAi experiments, CGNs were transfected with 2 μg of the Nap1 shRNA or control vector and plated into uncoated dishes to avoid cell attachment and differentiation. Twenty-four hours after plating, the cells were transferred into dishes containing coated glass coverslips and cultured as described above. Cells were analyzed after 2 d in vitro (DIV).
B16-F1 mouse melanoma cells were cultured as described previously (Steffen et al., 2004). Transfections were performed using Superfect (QIAGEN). Transfected cells were plated onto laminin (Sigma-Aldrich)-coated coverslips and fixed after 5 h.
Immunocytochemistry.
CGNs in dissociated cultures and microexplants were fixed and immunostained as described previously (Ertürk et al., 2007). B16-F1 cells were fixed and processed as described previously (Steffen et al., 2004). The following mouse monoclonal primary antibodies were used: anti-βIII-tubulin, clone Tuj1 (1:20,000; Covance), anti-Tau-1, clone PC1C6 (1:5000; Millipore), anti-cortactin, clone 4F11 (1:500; Millipore), and anti-myc 9E10 (Abcam). The rabbit polyclonal primary antibodies used were as follows: anti-phospho Ser199/204 Pak1/Pak2 (1:500; Millipore), anti-Sra-1 (1:100), anti-Nap1 (1:7) (Steffen et al., 2004), and anti-green fluorescent protein (GFP) (1:1000; Research Diagnostics). The mouse anti-Abi1 (1:50) and rabbit anti-pan-WAVE (1:100) antibodies were kindly provided by Dr. G. Scita (Department of Experimental Oncology, Istituto Europeo di Oncologia, Milan, Italy) (Innocenti et al., 2005). Secondary antibodies conjugated to Alexa Fluor 488 and rhodamine phalloidin were used (Invitrogen).
Live cell imaging and image acquisition.
For time-lapse analysis of growth cone dynamics, coverslips with neurons were kept at 36°C in custom-made cell chambers filled with HEPES-buffered HBSS. Images were acquired on an Axiovert 135TV inverted microscope (Zeiss) equipped with standard filters for GFP, Texas Red, and DAPI, using high-performance CCD camera 4912 (Cohu) and Scion Image Beta 4.0.2 software for Microsoft Windows. Adobe Photoshop 8 (Adobe Systems) and Canvas (ACD Systems) were used for processing and annotation of the images. Process lengths and growth cone areas were measured using the Scion Image software. Statistical significance (p value) was calculated using Student's t test.
For long-term time-lapse analysis of neurite formation and short-term treatments with Arp2/3 inhibitor, CGNs were imaged in 3.5 cm glass-bottom dishes to which 20 mm coverslips were adhered to the bottom with aquarium sealant. For long-term experiments, imaging was performed in culture medium. Images were acquired using a DeltaVision RT (Applied Precision) live-cell imaging setup based on an Olympus IX71 inverted microscope with a CO2-regulated incubation chamber maintained at 36°C (Solent Scientific). Imaging was performed with a 60×, numerical aperture 1.6 objective (Olympus). Images were acquired with a Photometrics CoolSnap HQ camera (Roper Scientific) using SoftWoRx 3.5.0 imaging software (Applied Precision).
F-actin assay.
For quantifications of F-actin amounts, CGNs were cultured as described above, fixed, and permeabilized at 1.5 DIV in PHEM buffer (60 mm PIPES, 25 mm HEPES, 5 mm EGTA, 1 mm MgCl) containing 0.25% glutaraldehyde, 3.7% PFA, 3.7% sucrose, and 0.1% Triton X-100 and costained for Tuj1 and phalloidin. Images were captured with 100× (1.6×) objective at below saturation (one to four frames). The average intensity of F-actin was measured in growth cones. Background intensity was measured in nearby region and subtracted. F-actin levels were normalized to controls, thereby correcting for differences in stainings from different experiments.
Drug treatment.
Cytochalasin D (Sigma-Aldrich) was dissolved in DMSO at 10 mm and applied to neuronal cultures 4 h after plating at a final concentration of 1 μm. Cells were analyzed at 1 and 2 DIV.
The Arp2/3 inhibitor CK-548 (ChemDiv) was dissolved in DMSO at 100 mm and applied to neuronal cultures 4 h after plating at a final concentration of 50 μm. After 1 DIV, another 50 μm of the inhibitor was added to the cells. Cells were fixed at 2.5 DIV and immunostained for Tuj1. The length of the longest Tuj1-stained neurite was quantified. For short-term time-lapse observations of growth cone motility, cells were cultured for 1 DIV and then imaged at 5 s intervals for 15 min before adding the Arp2/3 inhibitor (50 μm) to observe the growth cone dynamics and continuously imaged for 30 min after addition of the drug.
RhoA and Cdc42 activation assay.
To measure the activities of RhoA and Cdc42, G-LISA RhoA and Cdc42 Activation Assay Biochem kits (Cytoskeleton) were used. Results were expressed as the ratio between absorbance at 490 nm (A490) in wild-type and Rac1−/− extracts.
Results
Deletion of Rac1 in the nervous system
Rac1 depletion from the germ line results in lethality at early embryonic day 9.5 (Sugihara et al., 1998). To study the function of Rac1 in the developing nervous system, we crossed mice carrying a floxed Rac1 allele (Chrostek et al., 2006) with a mouse line expressing Cre recombinase under the control of a nestin promoter (Tronche et al., 1999; Graus-Porta et al., 2001). Both Rac1 and its highly homologous isoform Rac3 are expressed in the cortex, whereas Rac1 represents by far the predominant form in the cerebellum (Bolis et al., 2003).
To confirm that the expression of Cre leads to the excision of Rac1 and the absence of Rac isoforms in the cerebellum, cerebellar and cortical protein extracts were analyzed using a Rac1 antibody that also cross-reacts with Rac3. Cortical extracts from Rac1-deficient animals contained Rac protein, which can be attributed to the presence of Rac3 (Fig. 1A). By contrast, Rac protein was virtually absent in cerebellar extracts of P0–P9 Rac1-deficient animals (Fig. 1A) (data not shown). Lack of Rac3 expression in the cerebellum precludes a compensatory mechanism by this isoform, making the developing cerebellum uniquely suited to study the cellular role of Rac1 in neuronal development.
Figure 1.
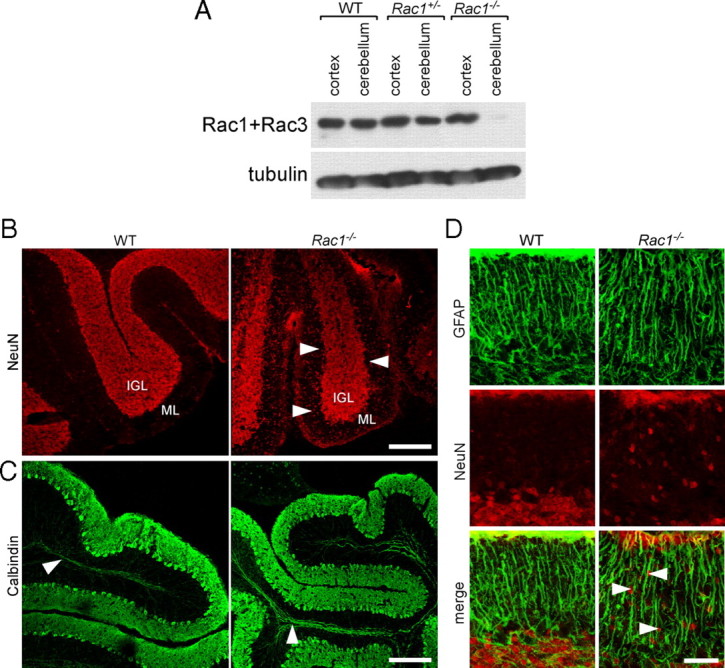
Loss of Rac1 from the nervous system results in defects in the migration of CGNs. A, Depletion of Rac1 from Rac1−/− cerebellum. P9 cerebellar and cortical protein extracts were prepared from wild-type, Rac1+/−, and Rac1−/− brains. Equal amounts of protein were subjected to Western blot analysis using an antibody recognizing Rac1 and cross-reacting with Rac3. The signal detected in cortical extracts of Rac1−/− brains represents Rac3, which is absent from cerebellar extracts. B, Ablation of Rac1 causes aberrant distribution of NeuN-positive neurons. Sagittal sections from P18 cerebella were immunostained with NeuN. Many CGNs stacked in the ML of the Rac1-null cerebellum can be observed (arrowheads). In contrast, in the wild type, the majority of CGNs have reached the IGL. Scale bar, 150 μm. C, Sagittal sections from P18 cerebella immunostained with calbindin (axonal tracts of Purkinje neurons, arrowheads). The Rac1-null cerebellum shows the typical monolayer of Purkinje neurons. Scale bar, 200 μm. D, Sagittal sections from P18 cerebella coimmunostained with NeuN and GFAP. The Rac1−/− cerebellum exhibits a normal organization of GFAP-positive Bergmann glia cells that are in proximity to NeuN-positive CGNs (arrowheads). Scale bar, 40 μm.
Newborn Rac1−/− pups were obtained at a normal mendelian ratio. Around P10, Rac1−/− littermates became distinguishable from the wild-type and heterozygous pups based on their smaller size. Rac1-deficient animals showed a resting tremor, clenching of the hindlimbs, and poor balance (data not shown). Rac1−/− animals died at P18–P21. Heterozygous Rac1+/− pups showed no obvious phenotypic changes compared with wild type.
The Rac1−/− brains were already at P9 smaller than wild-type brains (supplemental Fig. S1A, available at www.jneurosci.org as supplemental material). Morphological analysis of Nissl-stained brain sections showed that all brain regions in Rac1-deficient animals were reduced in size (supplemental Fig. S1B,C, available at www.jneurosci.org as supplemental material). Moreover, the ventricles were enlarged (supplemental Fig. S1B, arrowhead, available at www.jneurosci.org as supplemental material) (data not shown), and the neuroepithelium was thicker than in wild-type brains (supplemental Fig. S1C, available at www.jneurosci.org as supplemental material). Nissl staining did not reveal major abnormalities within the cerebral cortex and the hippocampus (supplemental Fig. S1C, available at www.jneurosci.org as supplemental material).
In contrast, we found profound changes within the cerebellum of the Rac1-deficient animals (supplemental Fig. S1B, available at www.jneurosci.org as supplemental material). First, the cerebellum was significantly reduced in size. Staining cerebellar sections with an antibody against active caspase-3 revealed a threefold increase in the number of apoptotic cells in the P0–P1 Rac1-knock-out cerebellum (supplemental Fig. S2A,B, available at www.jneurosci.org as supplemental material), which could contribute to the observed brain size reduction. Similarly, the number of apoptotic cells increased in the Rac1-null cortex at P0–P1, indicating that Rac1 and Rac3 may have non-overlapping functions in cell survival (data not shown). Later during cerebellar development, the number of apoptotic cells in wild-type and Rac1-knock-out animals was similar (supplemental Fig. S2A,B, available at www.jneurosci.org as supplemental material). We found no evidence for decreased cell proliferation that could contribute to cerebellar size reduction. Quantification of phosphohistone H3- and Ki-67-positive cells showed no differences between Rac1-knock-out and wild-type cerebella at the analyzed stages P0–P1, P8–P10, and P18 (supplemental Fig. S2C,D, available at www.jneurosci.org as supplemental material) (data not shown).
Apart from the reduction in size, the Rac1−/− cerebellum showed an enlarged fourth ventricle (supplemental Fig. S1B, arrowhead, available at www.jneurosci.org as supplemental material), a complete loss of the second, third, fourth, and fifth cerebellar lobe (supplemental Fig. S1B, lobes indicated in the wild type, available at www.jneurosci.org as supplemental material), as well as foliation defects (supplemental Fig. S1B, available at www.jneurosci.org as supplemental material). In addition, cerebellar structures were located between the brainstem and the enlarged inferior colliculus in the Rac1-null cerebellum in contrast to the wild-type brain (supplemental Fig. S1B, asterisk, available at www.jneurosci.org as supplemental material). Thus, Rac1 ablation leads to strong defects in cerebellar development and organization.
Rac1-knock-out CGNs show defects in migration
Neuronal migration is one early polarized event during neuronal development. To test whether neuronal migration is impaired in the absence of Racs, we focused on CGNs that express only Rac1. We stained cerebellar sections with antibodies against the CGN markers NeuN and Pax-6 (Yamasaki et al., 2001; Weyer and Schilling, 2003). In the P18 wild-type cerebellum, NeuN-positive CGNs were almost exclusively present in the internal granule layer (IGL) (Fig. 1B). By contrast, in the Rac1-null cerebellum, we found a fivefold increase in the number of NeuN-positive cells in the ML compared with control (1.1 ± 0.2 in Rac1-null vs 0.2 ± 0.1 cells in wild type; average ± SEM/micrometers of ML from three animals; n = 3 sections/cerebellum; p < 0.05) (Fig. 1B, arrowheads). Similarly, Pax-6-positive cells were still present in the EGL and ML of the P18 Rac1−/− cerebellum (data not shown).
In contrast to the aberrant localization of CGNs, Purkinje neurons, which express Rac3 (Bolis et al., 2003), formed their typical monolayer in the P18 Rac1-null cerebellum (Fig. 1C). We also examined the in vivo organization of Bergmann glia by immunocytochemistry using the GFAP, but found no obvious differences between wild-type and Rac1-knock-out cerebella (Fig. 1D). The majority of Rac1−/− CGNs were in close proximity to GFAP-positive glia fibers (Fig. 1D, arrowheads).
To directly analyze inward migration of CGNs, we labeled dividing granule cell precursors by injecting BrdU in P7–P8 pups (Borghesani et al., 2002). At 24 h after injection, BrdU-labeled cells resided in the EGL in both the wild-type and Rac1−/− cerebellum (Fig. 2A, arrowheads). At 96 h after injection, wild-type BrdU-labeled cells had reached the IGL, whereas many Rac1−/− BrdU-labeled cells resided within the EGL and ML (Fig. 2A, arrowheads). These results indicate a defect in migration of CGNs in the absence of Rac1.
Figure 2.
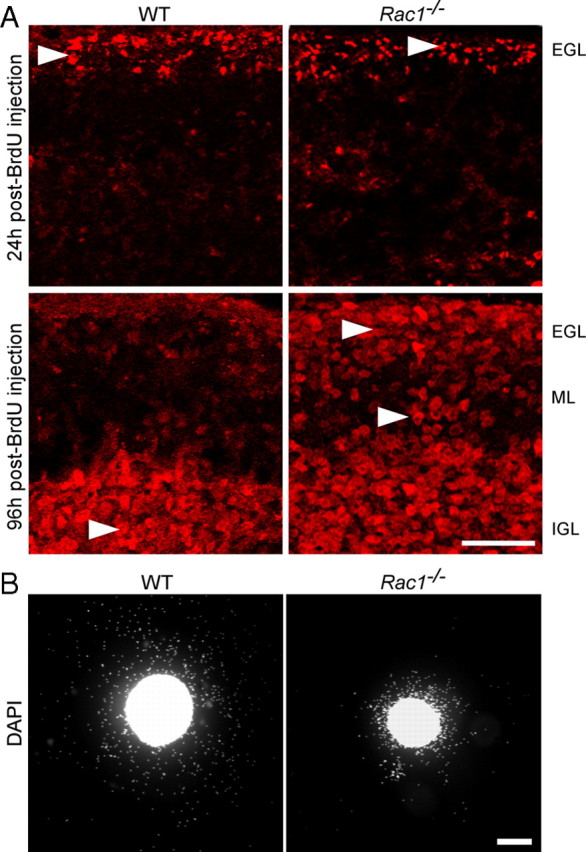
Abnormal in vivo and in vitro migration of Rac1−/− CGNs. A, Dividing cerebellar granule cell precursors were labeled by systemic injection of BrdU into P7 wild-type and Rac1-deficient mice. At 24 h after BrdU injection, the majority of BrdU-labeled granule cells are in the EGL in both the wild-type and Rac1−/− cerebellum (arrowheads). At 96 h after injection, the majority of BrdU-labeled granule cells in the wild-type cerebellum have reached the IGL (arrowhead). By contrast, in the Rac1−/− cerebellum, more BrdU-labeled granule cells (arrowheads) stayed in the EGL and ML. Scale bar, 50 μm. B, EGL explants from P5 wild-type and Rac1−/− mice were stained with DAPI after 2 DIV. Rac1−/− neurons migrate from the explant less efficiently than wild-type neurons. Scale bar, 200 μm.
Although we found Rac1-knock-out CGNs in close proximity with Bergmann glia fibers, we could not exclude the possibility that the loss of Rac1 interferes with the ability of neurons to associate and/or dissociate with the glia fibers, resulting in an impaired neuronal migration. To test whether the migration defect in the Rac1-null CGNs is cell autonomous, we studied their migration in cultured EGL microexplants (Fig. 2B). As reported previously (Nagata and Nakatsuji, 1990, 1994), we confirmed that cells migrating out of the explants are CGNs (data not shown). Their migration in microexplants occurs independently of Bergmann glia, making this system well suited to study cell-autonomous migratory cues (Nagata and Nakatsuji, 1990, 1994). After 2 DIV, we found more than a twofold decrease in the number of Rac1-knock-out granule cells migrating out of the explant compared with wild-type (913.8 ± 140 in Rac1-knock-out vs 2231 ± 322.1 cells in wild type; average ± SEM/square micrometers of explant surface from five independent cultures; n = 100 explants; p < 0.01) (Fig. 2B). Furthermore, Rac1-knock-out granule neurons migrated far shorter distances and distributed over smaller occupancy area compared with migrating wild-type neurons (0.2 ± 0.1 in Rac1-knock-out vs 0.5 ± 0.1 mm2 in wild type; average ± SEM from five independent cultures; n = 100 explants; p < 0.001). Together, our data demonstrate that Rac1 is necessary for the migration of CGNs in a cell-autonomous fashion.
Rac1-knock-out CGNs show impaired axon growth
Axon growth and migration occur simultaneously during the development of CGNs (Hatten and Heintz, 1995; Yamasaki et al., 2001). In addition, defects in the axon formation often correlate with aberrant migration of CGNs (Tomoda et al., 1999; Yamasaki et al., 2001). During radial migration, CGNs extend two axons, the parallel fibers, that reside in the ML (Hatten et al., 1997) and can be visualized with an antibody against the cell adhesion molecule L1 (Fig. 3A) (Sakurai et al., 2001). In the Rac1−/− cerebellum, only few L1-positive axons were observed (Fig. 3A, arrows). Moreover, axonal fibers in the ML visualized by neurofilament immunostaining were short and misoriented (Fig. 3B, arrows). To further analyze axonal tracts in vivo, we injected DiI in the wild-type and Rac1-knock-out cerebella (Fig. 3C). At 2 d after DiI injection, labeled axons of CGNs were visible in the wild-type (Fig. 3C, asterisks), but not in the Rac1-knock-out cerebellum. At 4 d after DiI injection, typical bipolar and parallel axonal tracts extended over distances >400 μm from the injection site in the wild-type cerebellum (Fig. 3C, asterisks). In contrast, no axonal labeling was observed in the Rac1-knock-out cerebellum. This suggests that Rac1 signaling is critically required during parallel fiber formation. Of note, Rac1-knock-out Purkinje cells, which express Rac3, do not show defects in extending their axon (Fig. 1C, arrowheads).
Figure 3.
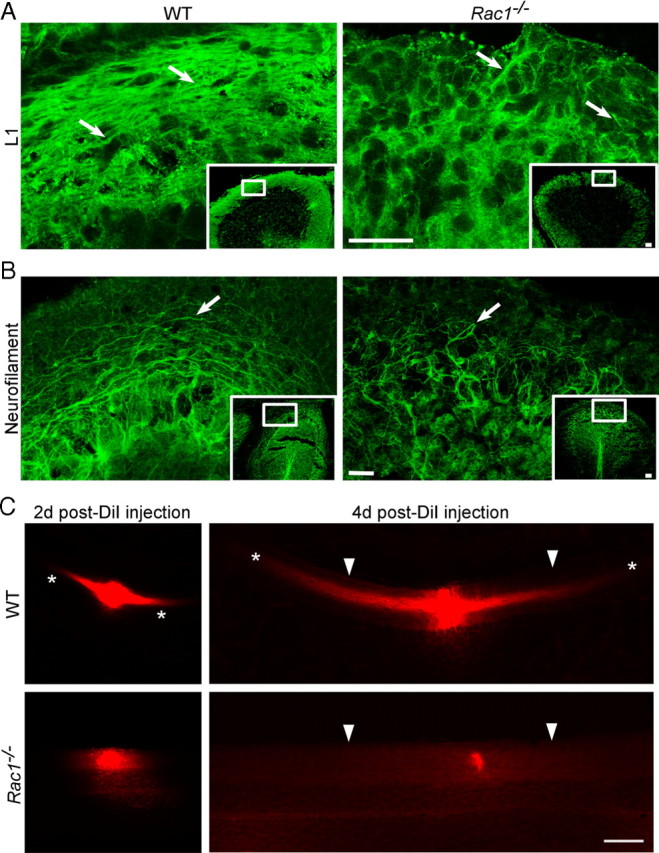
Rac1−/− neurons show deficient axon extension in vivo. A, P15 coronal sections from the middle part of the cerebellum immunostained with an antibody against L1 that visualizes parallel fibers in the ML. Images of boxed regions of the ML are depicted at higher magnification. L1 immunostaining reveals less densely packed parallel fibers in the ML of the Rac1-null cerebellum compared with wild type (arrows). Scale bars, 30 μm. B, P18 midsagittal sections immunostained with an antibody against neurofilament to visualize axons. Images of boxed regions of the ML are depicted at higher magnification. Long axonal fibers can be observed in the wild-type cerebellum and short and misrouted axons in the Rac1−/− cerebellum (arrows). Scale bars, 30 μm. C, DiI-labeled parallel fibers in P15 coronal sections of wild-type and Rac1−/− cerebella. At 2 d after DiI injection, parallel fiber labeling within the ML is visible in the wild-type cerebellum (asterisks), but not in the Rac1−/− cerebellum. At 4 d after injection, long parallel fibers project from both sides of the DiI injection place in the wild-type cerebellum, but this labeling is absent in the Rac1−/− cerebellum. The pial surface of the cerebellum is indicated by arrowheads. Note that the intensity of DiI labeling at the injection site in wild-type and Rac1−/− cerebellum is comparable (2 d after DiI injection). Scale bar, 100 μm.
To assess whether Rac1 affects axon growth in a cell-autonomous manner, when separated from spatial cues, we examined axon formation in dissociated CGNs, an established model of neuronal polarity (Hatten and Heintz, 1995; Powell et al., 1997; Zmuda and Rivas, 1998). Cultured CGNs undergo a set of well described morphological changes that resemble those occurring in vivo (Powell et al., 1997). Long neurites that develop after 2 DIV correspond to axons and can be morphologically distinguished from dendrites, whose development initiates at later stages (5–6 DIV). We found a threefold decrease in the number of Rac1−/− neurons that extended axons >100 μm after 2 DIV (Fig. 4A,B). Additionally, we assessed the ability of Rac1-knock-out CGNs to form axons. To this end, we used the Tau-1 antibody that recognizes a dephosphorylated Tau epitope that is axon specific (Mandell and Banker, 1996). We found a 50% reduced ability of Rac1-knock-out CGNs to form axons compared with wild type (supplemental Fig. S3, available at www.jneurosci.org as supplemental material). These data suggest a role of Rac1 in axon growth. Interestingly, Rac1−/− neurons showed an increase in the average number of neurites per cell. Whereas >80% of wild-type neurons had one to two neurites per cell, the majority of Rac1−/− neurons contained three or more neurites (Fig. 4A,C). Nevertheless, the average length of all neurites at 2 DIV decreased from 147.8 ± 5.1 μm in wild-type neurons to 113.4 ± 1.4 μm in Rac1-null CGNs (average ± SEM from three independent cultures; n = 100 cells/culture; p < 0,05). Time-lapse videomicroscopy analysis revealed that Rac1-null neurons initiated neurite formation at the same time as wild-type neurons (supplemental Fig. S4A,B, Videos 1, 2, available at www.jneurosci.org as supplemental material). We observed a constant growth rate of both wild-type and Rac1-null neurons during the first 24 h. Wild-type neurons exhibited a growth rate of 1.5 μm/h. Instead, Rac1−/− neurons elongated their longest neurite slowly, with a growth rate of 0.8 μm/h. After 24 h, wild-type neurons continued extending their axons further (supplemental Fig. S4B, available at www.jneurosci.org as supplemental material), whereas Rac1−/− neurons formed additional neurites (supplemental Fig. S4C, available at www.jneurosci.org as supplemental material).
Figure 4.

Reduced length and an increased number of neurites in cultured Rac1−/− CGNs can be reversed by overexpressing Rac1. A, Dissociated CGNs were immunostained after 2 DIV with an antibody against Tuj1. Rac1−/− CGNs display several shorter Tuj1-positive neurites, whereas wild-type CGNs typically exhibit one or two longer Tuj1-positive neurites. Scale bar, 25 μm. B, Quantification of the length of the longest Tuj1-stained neurite. The values represent averages ± SEM from three independent cultures (n = 100 cells/culture; ***p < 0.001; **p < 0.01). C, Quantification of the neurite number. The values represent averages ± SEM from three independent cultures (n = 100 cells/culture; *p < 0.05). D, Quantification of the length of the longest neurite in CGNs cotransfected with pEYFP-C1 and pcDNA3.1 (ctrl) or with pEYFP-C1 and pRac1 (pRac1) at 3 DIV. The values represent averages ± SEM from four independent cultures (n = 100 cells/culture; **p < 0.01). E, Quantification of the neurite number in CGNs cotransfected with pEYFP-C1 and pcDNA3.1 (ctrl) or with pEYFP-C1 and pRac1 (pRac1) at 3 DIV. The values represent averages ± SEM from three independent cultures (n = 100 cells/culture; *p < 0.05).
To examine whether the defect in axon outgrowth was specifically caused by Rac1 ablation, we coexpressed wild-type Rac1 and YFP in Rac1−/− CGNs. The length of the longest process increased by >50% on overexpression of Rac1 in Rac1−/− neurons compared with Rac1−/− neurons transfected with the control plasmid (Fig. 4D). On Rac1 overexpression, we could no longer observe a significant reduction of neurite length between wild-type and Rac1−/− CGNs (Fig. 4D). Additionally, overexpression of Rac1 in Rac1-deficient neurons decreased the average number of neurites per cell (Fig. 4E). The majority of Rac1−/− neurons transfected with Rac1 contained one to two neurites. Overexpression of Rac1 in wild-type neurons affected neither their neurite length nor their number of neurites (Fig. 4D,E). Similar to Rac1, we found that overexpressing Rac3 in Rac1-null neurons significantly increased the length of the longest process (supplemental Fig. S5C, available at www.jneurosci.org as supplemental material) and reduced neurite number (data not shown), whereas it had no effect in wild-type neurons. This suggests that Rac3 and Rac1 have overlapping functions in axon growth.
Rac1−/− neurons lack lamellipodia
To investigate whether the defect in axon formation is caused by aberrant regulation of the actin cytoskeleton, we stained cultured CGNs from wild-type and Rac1-knock-out animals with fluorescently labeled phalloidin. Lamellipodia were visualized using an antibody against cortactin (Wu and Parsons, 1993). Wild-type growth cones contained both long and thin actin protrusions, filopodia (Fig. 5A, arrowhead), and fanlike lamellipodia (Fig. 5A, arrow). By contrast, Rac1-null neurons were typically devoid of lamellipodia (Fig. 5A). Quantification of the proportion of cells bearing lamellipodia showed a 70% reduction in the Rac1-null CGNs compared with wild type (21.4 ± 4.1 in Rac1-null vs 71 ± 1.7% in wild type; average ± SEM from three independent cultures; n = 100 cells/culture; p < 0.01). Consistent with the absence of lamellipodia, we found that the growth cone surface of the Rac1-deficient CGNs reduced to 44.3 ± 1.6% of the wild-type growth cone surface (average ± SEM from three independent cultures; n = 20 cells/culture; p < 0.001). Additionally, Rac1-null neurites typically showed actin-rich filopodia at their ends (Fig. 5A, arrowheads). Examination of cultured CGNs by videomicroscopy revealed dynamic filopodia in wild-type and Rac1-knock-out neurons (Fig. 5B; supplemental Videos 3, 4, available at www.jneurosci.org as supplemental material), but the specific absence of lamellipodia dynamics in Rac1-null CGNs (supplemental Video 4, available at www.jneurosci.org as supplemental material). We also observed that the relative amount of F-actin in the Rac1-knock-out growth cones reduced to 49 ± 1% of the wild type (average ± SEM from four independent cultures; n = 100 cells/culture; p < 0.01). Thus, our data demonstrate that Rac1 is essential for lamellipodia formation and growth cone dynamics of CGNs.
Figure 5.
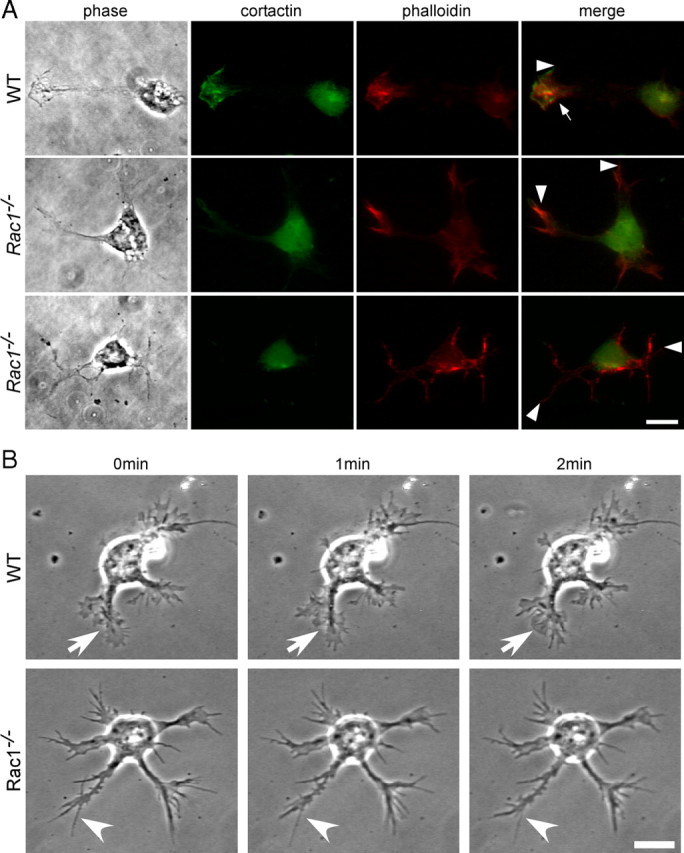
Rac1−/− CGNs lack lamellipodia and show reduced actin dynamics. A, Dissociated CGNs at 1.5 DIV were costained with rhodamine–phalloidin to visualize actin filaments and with an antibody against cortactin to visualize lamellipodia. Images from wild-type and two different Rac1−/− neurons are shown. Cortactin and phalloidin staining of a wild-type neuron reveal a lamellipodium (arrow) and a filopodium (arrowhead), whereas Rac1−/− neurons only have filopodia at their neurite tips (arrowheads) but lack lamellipodia. Scale bar, 10 μm. B, Time-lapse videomicroscopy of representative wild-type and Rac1−/− CGNs at 1.5 DIV. Three frames at 1 min intervals are shown. A dynamic lamellipodium of a wild-type growth cone is indicated by an arrow. A filopodium of a Rac1−/− growth cone is indicated by an arrowhead. Scale bar, 10 μm.
Modulating the actin cytoskeleton increases the ability of Rac1-knock-out neurons to form axons
Actin dynamics orchestrate neuronal polarization and axon growth. In fact, depolymerization of the actin cytoskeleton induces formation of multiple axons in dissociated hippocampal neurons (Bradke and Dotti, 1999, 2000; Kunda et al., 2001; Schwamborn and Püschel, 2004). We wondered whether the observed changes in the actin cytoskeleton of Rac1-null neurons are underlying defects in neuronal polarization. We therefore pharmacologically depolymerized the actin cytoskeleton and assessed axon growth in Rac1−/− CGNs (Fig. 6A–C). Cytochalasin D treatment of Rac1−/− CGNs increased by 2.4-fold the length of the longest neurite (Fig. 6A,C) and by 2-fold the ability to form Tau-1-positive axons (Fig. 6A,B). Cytochalasin D treatment of Rac1−/− CGNs also decreased their neurite number (Fig. 6D). In contrast to hippocampal neurons, we did not observe the formation of multiple axons on treatment of CGNs with cytochalasin D as previously reported (Zmuda and Rivas, 2000). These differences may stem from different intrinsic developmental properties of these two distinct neuronal populations. Together, our work reveals that depolymerization of the actin cytoskeleton by cytochalasin D abrogated the difference in axon growth and neurite number between Rac1-knock-out and wild-type CGNs.
Figure 6.

Actin depolymerization reverses Rac1-mediated defects in axon growth. A, Dissociated CGNs were treated with cytochalasin D (CytoD) or control treated with DMSO 4 h after plating. Cells were fixed at 2 DIV and immunostained for the axonal marker Tau-1. CytoD-treated Rac1−/− neurons extend longer, Tau-1-positive axons bearing a typical proximodistal staining gradient. DMSO-treated Rac1−/− neurons extend several shorter, Tau-1-negative neurites. Cytochalasin D treatment abrogates the difference in axon formation between Rac1−/− and wild-type CGNs. Scale bar, 30 μm. B, Quantification of axon formation. Cells, bearing at least one Tau-1-positive axon (proximodistal staining gradient), were scored as positive. The values represent averages ± SEM from three independent cultures (n = 100 cells/culture; **p < 0.01). C, D, Dissociated CGNs were treated with CytoD or DMSO 4 h after plating. Cells were fixed at 1 DIV and visualized by immunostaining for Tuj1. Quantification of the length of the longest Tuj1-stained neurite (C) or neurite number (D). The values represent averages ± SEM from three independent cultures (n = 100 cells/culture; *p < 0.05; ***p < 0.001).
Modest stabilization of microtubules also induces the growth of multiple axons in dissociated hippocampal neurons (Gomis-Rüth et al., 2008; Witte et al., 2008) and enhances neurite outgrowth of CGNs when plated on the inhibitory substrate myelin (Ertürk et al., 2007). However, treating Rac1−/− CGNs with the microtubule-stabilizing drug Taxol did not affect their neurite length (data not shown). Therefore, our experiments suggest that Rac1 predominantly regulates neurite outgrowth via modulating the organization of the actin cytoskeleton.
Rac1-knock-out cerebella show a decrease in PAK1 phosphorylation
Since expression of constitutively active Rac in fibroblasts causes a decrease in Rho activity (Sander et al., 1999), we examined whether there was a cross talk between Rac1 and RhoA in the cerebellum. However, neither total RhoA expression levels (data not shown) nor the levels of active RhoA in Rac1−/− cerebellar extracts were altered (Fig. 7A). We also did not find any change in Cdc42 activation in Rac1-null compared with wild-type cerebellar extracts (Fig. 7A). This indicates that defects in the Rac1−/− cerebellum are not mediated through RhoA or Cdc42 signaling.
Figure 7.

Rac1 deletion results in a reduced activity of PAK but does not change the activity of cofilin. A, ELISA-based assay for RhoA and Cdc42 activation. The results are expressed as ratios between A490 in wild-type and Rac1−/− extracts. The values represent averages ± SEM from three animals. There is no significant difference between the activity of RhoA or Cdc42 in Rac1−/− and wild-type cerebella. Cortical extracts were used as control. B, Analysis of phospho-PAK1 (p-PAK1) and PAK1 protein levels from P9 cortices and cerebella. Rac1−/− cerebellar extracts show reduced levels of p-PAK1 compared with wild-type extracts. C, Dissociated CGNs at 2 DIV were immunostained with an antibody against p-PAK1/2. A decreased level of p-PAK1/2 can be observed in neurites of Rac1−/− CGNs. By contrast, higher levels of p-PAK1/2 accumulate in a neurite of a wild-type CGN. Scale bar, 15 μm. D, Quantification of the length of the longest neurite in CGNs cotransfected with pEYFP-C1 and pcDNA3.1/myc-His A (ctrl) or pEYFP-C1 and pPAKT423E (pPAKT423E) at 3 DIV. The values represent averages ± SEM from three independent cultures (n = 100 cells/culture; *p < 0.05; **p < 0.01). Overexpression of constitutively active PAK1 (PAKT423E) in Rac1−/− CGNs does not increase neurite length. E, Analysis of phosphorylated-cofilin (p-cofilin) and cofilin protein levels from P9 cortices and cerebella. Wild-type and Rac1−/− extracts show comparable levels of p-cofilin.
We assessed potential effectors of Rac1 that regulate actin dynamics. Previous work showed that PAK1 regulates axon formation in hippocampal neurons (Jacobs et al., 2007). We observed a decrease in the phosphorylation of PAK1 at Ser198/203 in Rac1−/− cerebellar extracts (Fig. 7B), indicating a reduced catalytic activity of PAK1 (Shamah et al., 2001). The total PAK1 protein levels remained constant (Fig. 7B). Decrease in the phosphorylation of PAK1 was mainly attributable to decreased levels of phosphorylated PAK in neurites of Rac1−/− CGNs (Fig. 7C).
We attempted to rescue the growth defect of Rac1-knock-out neurons by manipulating PAK1. Transfection of a phosphomimetic PAK1 mutant (PAKT423E) caused no increase in neurite length (Fig. 7D) or number (data not shown) in Rac1-knock-out neurons, suggesting that activating PAK1 alone is not sufficient to rescue axon formation. It has been shown that membrane-targeting and kinase activity of PAK1 are required for axon formation in hippocampal neurons (Jacobs et al., 2007). However, overexpression of the membrane-targeted PAK1 construct (Pak1-CAAX) also did not rescue neurite length or axon formation in Rac1-knock-out neurons (supplemental Fig. S5D, available at www.jneurosci.org as supplemental material) (data not shown). In another study, it has been shown that overexpression of the kinase-dead, membrane-targeted form of PAK1 (PakR299-CAAX) induces neurite outgrowth in PC12 cells, suggesting that the effect of PAK1 on neurite outgrowth is independent of its kinase activity but requires membrane localization (Daniels et al., 1998). We therefore also overexpressed a PakR299-CAAX in Rac1−/− CGNs, but failed to detect an increase in neurite length or axon formation (supplemental Fig. S5E, available at www.jneurosci.org as supplemental material) (data not shown).
We also analyzed the activity of the PAK effector cofilin, an F-actin depolymerizing and severing factor that affects axon formation in hippocampal neurons (Garvalov et al., 2007; Jacobs et al., 2007) but found no detectable differences in phosphorylation of cofilin between wild-type and Rac1-knock-out cerebellar extracts (Fig. 7E). Together, our data suggest that Rac1 regulates axon growth through pathways other than PAK-cofilin.
Rac1-knock-out neurons mislocalize the WAVE complex from the growth cone plasma membrane
Overexpression studies in fibroblasts and melanoma cells have shown that the WAVE complex regulates lamellipodia formation and that Rac1 is necessary for its recruitment to the plasma membrane (Steffen et al., 2004). The WAVE complex, comprising WAVE, Sra-1, Nck-associated protein 1 (Nap1), Abelson-interacting protein 1 (Abi1), and HSPC300, has been implicated in axon development in vertebrate and invertebrate neurons (Hummel et al., 2000; Bogdan et al., 2004; Kawano et al., 2005; Yokota et al., 2007). We therefore assessed whether WAVE signaling is disturbed in Rac1-knock-out neurons. First, we investigated protein levels of the WAVE complex components Sra-1, Nap1, and WAVE, but could not detect any differences between wild-type and Rac1-knock-out brains (Fig. 8D). As WAVE recruitment to the plasma membrane is crucial for its signaling (Steffen et al., 2004; Stradal et al., 2004), we analyzed the intracellular localization of WAVE protein using a pan-WAVE antibody. In wild-type neurons, WAVE localized to the plasma membrane of lamellipodial protrusions (Fig. 8A,B). By contrast, in Rac1-knock-out neurons, WAVE was absent from the plasma membrane (Fig. 8A,B). As the vast majority of Rac1−/− neurons contained no lamellipodia (Fig. 5B), we selected for the rare Rac1−/− neurons that still contained some lamellipodia-like protrusions. Even in those neurons, WAVE, as well as other components of the WAVE complex, including Abi1, Sra-1 (Fig. 8A–C), and Nap1 (data not shown), failed to localize to the plasma membrane of the growth cone. Only 20–30% of the Rac1-knock-out neurons containing some lamellipodia remnants showed Abi1, Sra-1, and WAVE localization at the plasma membrane of the growth cone (Fig. 8C). These results suggest that Rac1 regulates the WAVE complex activity by localizing it to the plasma membrane of neuronal growth cones.
Figure 8.

Rac1−/− neurons mislocalize WAVE from the growth cone plasma membrane. A, Dissociated CGNs at 1 DIV were costained with rhodamine–phalloidin and an antibody against WAVE (pan-WAVE). Rac1−/− neurons that still contained some lamellipodia-like protrusions were examined for WAVE localization. Scale bar, 10 μm. B, Merged images of boxed regions in A are depicted at higher magnification. WAVE is absent from the edges of Rac1−/− growth cones, whereas in wild-type neurons WAVE predominantly localizes to membranes of lamellipodial protrusions (arrowhead). Scale bar, 5 μm. C, Quantification of plasma membrane localization of Abi1, Sra-1, and WAVE. CGNs, showing plasma membrane localization of the respective protein in at least one growth cone, were scored as positive. The values represent averages ± SEM from three independent cultures (n = 100 cells/culture; *p < 0.05; **p < 0.01; ***p < 0.001). D, Mislocalization of the WAVE complex in the Rac1−/− neurons is not caused by protein degradation. Western blot analysis of P9 cortical and cerebellar extracts reveals no difference in the protein amounts of the respective WAVE complex components.
The WAVE complex and its downstream effector Arp2/3 regulate axon growth
We hypothesized that Rac1-deficient neurons show defects in axon growth because the WAVE complex cannot be efficiently recruited to the plasma membrane. To test this, we generated a WAVE1 mutant fused to a Ras prenylation sequence (WAVE-CAAX). Plasma membrane localization of the WAVE-CAAX was confirmed in B16F1 mouse melanoma cells (supplemental Fig. S5A,B, available at www.jneurosci.org as supplemental material). WAVE-CAAX overexpression in Rac1-knock-out CGNs caused a 50% increase in the number of cells forming Tau-1-positive axons (Fig. 9A,B) and a 35% increase in the axon length (Fig. 9A,C). Additionally, overexpression of WAVE-CAAX in Rac1-knock-out CGNs resulted in a decreased neurite number (data not shown). We did not observe any changes in axon growth (Fig. 9A–C) or neurite number (data not shown) in wild-type CGNs on overexpression of WAVE-CAAX construct. Furthermore, we did not observe an additional increase in axon growth in Rac1-knock-out neurons when WAVE-CAAX was cotransfected with Pak299R-CAAX (supplemental Fig. S5F, available at www.jneurosci.org as supplemental material) or PAK-CAAX (data not shown).
Figure 9.

WAVE complex and its downstream effector Arp2/3 regulate axon growth downstream of Rac1. A, Dissociated CGNs were cotransfected with pEYFP-C1 and pcDNA3.1/myc-His A (ctrl) or with pEYFP-C1 and pWAVE-CAAX (pWAVE-CAAX). Cells were fixed at 3 DIV and immunostained for Tau-1 to visualize axons. On overexpression of WAVE-CAAX, Rac1−/− CGNs extend significantly more Tau-1-positive axons bearing a typical proximodistal staining gradient. Instead, Rac1−/− neurons transfected with the ctrl typically extend several shorter, Tau-1-negative neurites. Overexpression of WAVE-CAAX partially rescues the axon formation defect of Rac1−/− CGNs. Scale bar, 25 μm. B, Quantification of axon formation on transfection of pWAVE-CAAX or ctrl. Cells, bearing at least one Tau-1-positive axon (proximodistal staining gradient), were scored as positive. The values represent averages ± SEM from four independent cultures (n = 100 cells/culture; **p < 0.01; ***p < 0.001). C, Quantification of the length of the longest neurite in CGNs transfected with pWAVE-CAAX or ctrl. The values represent averages ± SEM from five independent cultures (n = 100 cells/culture; **p < 0.01). D, Time-lapse videomicroscopy of representative growth cones from wild-type CGNs cultured for 1 d and imaged before and after the addition of the Arp2/3 inhibitor (CK-548). The top panel represents four frames at 30 s intervals before the addition of CK-548, and the bottom panel, after the addition of CK-548. The additional two frames depict the growth cone dynamics 6 min before and after the inhibitor was added. Scale bar, 10 μm.
We next tested whether the WAVE complex is necessary for axon growth in CGNs. It has been shown that disrupting WAVE signaling by knocking down the WAVE complex component Nap1 leads to axon growth defects in cortical neurons (Yokota et al., 2007). Consistently, we observed a 2.1-fold decrease in length of the longest process in Nap1 shRNA-expressing neurons at 2 DIV compared with control (11.9 ± 0.7 on Nap1 shRNA expression vs 25.6 ± 1.9 μm in control; average ± SEM from three independent cultures; n = 100 cells/culture; p < 0.05).
As WAVE regulates the actin cytoskeleton through the Arp2/3 complex, we tested whether Arp2/3 is necessary for axon growth in CGNs. To this end, we treated wild-type neurons with the pharmacological Arp2/3 inhibitor CK-548 (Nolen et al., 2009). CK-548 treatment reduced the length of the longest neurite by 50% (119.7 ± 9.5 in DMSO-treated control vs 59.4 ± 10 μm in CK-548-treated cells; average ± SEM from three independent cultures; n = 100 cells/culture; p < 0.05). Time-lapse videomicroscopy analysis revealed that CK-548 treatment of wild-type CGNs drastically reduced lamellipodia formation, whereas filopodia dynamics remained intact (Fig. 9D). Together, these data suggest that Rac1 regulates axon growth through the WAVE complex, which mediates its effects onto the actin cytoskeleton through the Arp2/3 complex.
Discussion
The cytoskeleton regulates the development of the nervous system, including proliferation, migration, and axon growth. The Rho family of GTPases, orchestrators of dynamic actin and microtubular network, appears to play a key role in neuronal development. However, our knowledge about their specific physiological function in the mammalian nervous system is only fragmentary. Using a brain-specific knock-out, we studied the role of Rac1 in neuronal migration and axon growth. We focused our analysis on the cerebellum as other Rac members are widely absent from this tissue (Bolis et al., 2003) and can therefore not substitute for the functions of Rac1. Our study shows that Rac1, by regulating actin cytoskeleton dynamics in neuronal growth cones, is a physiological regulator of neuronal migration and axon growth.
Neuronal migration requires Rac1 function
Rac1 was proposed to regulate cell migration (Ridley, 2001a). However, this role has been challenged in recent in vivo studies (Bokoch, 2005; Vidali et al., 2006; Wheeler et al., 2006; Kassai et al., 2008). For example, migration of cortical neurons in cortex-restricted Rac1-knock-out mice is not impaired (Kassai et al., 2008). Here, we showed that Rac1 signaling is essential for efficient migration of one of the largest population of neurons in the brain, CGNs. Although one interpretation could be that Rac1 regulates migration in a neuron-specific manner, we favor the hypothesis that Rac3 functions in a redundant fashion with Rac1. Hence, the role of Rac1 in the cortex may be masked because of the presence of Rac3. In line with this idea, we found that Rac1-deficient Purkinje cells, which express Rac3 (Bolis et al., 2003), were able to form their typical monolayer.
Apart from regulating neuronal migration, we found that Rac1 was important for neuronal survival. Thus, our in vivo data support previous cell culture overexpression studies (Linseman et al., 2001; Le et al., 2005) and reveal antiapoptotic effects of Rac1 signaling in the cerebellum. This particular role of Rac1 may be more widespread in the nervous system because Rac1 deficiency in the forebrain also results in an increased rate of apoptosis (Chen et al., 2009). However, this study revealed a positive role of Rac1 in proliferation, whereas our study shows that Rac1 is dispensable for proliferation during cerebellar development. Together, this suggests that regulation of cell proliferation by Rac1 may be more cell type specific.
Rac1 ablation from the nervous system impairs axon growth
Previous in vitro studies suggested Rac1 as both a positive and a negative regulator of axonogenesis (Govek et al., 2005). These discrepancies may stem from differences in the cellular systems analyzed or may be attributable to the fact that most of the previous studies used dominant-negative and constitutively active mutants, which can exhibit nonspecific or nonphysiological effects resulting from perturbation of multiple Rho GTPases (Wang and Zheng, 2007).
Analysis in mice have shown that Rac1-deficient cortical neurons exhibit axon guidance defects and fail to cross the midline. However, axon growth defects could not be observed (Chen et al., 2007; Kassai et al., 2008). One potential reason for the normal polarization of the Rac1-ablated cortical neurons is that Rac3 may compensate for some Rac1-dependent functions (Bolis et al., 2003; Corbetta et al., 2009). Consistent with this view, knocking out Rac3 alone does not lead to defects in axon development (Corbetta et al., 2005; Gualdoni et al., 2007). However, downregulation of Rac1, in the Rac3-knock-out neurons, revealed changes in dendrite development and spine formation (Gualdoni et al., 2007; Corbetta et al., 2009). It is therefore possible that, in contrast to CGNs, axon formation in hippocampal neurons occurs independently of Rac1. Alternatively, some residual Rac1 activity could have still been present during axonal formation that occurs early during development (Bradke and Dotti, 2000).
Our study clearly identifies Rac1 as a positive physiological regulator of axon growth in the mammalian nervous system. In addition to the in vivo defect in axon formation in the Rac1-knock-out cerebellum, cultured Rac1−/− CGNs are disrupted in their polarization and show markedly reduced axon growth, indicating that this phenotype is cell intrinsic.
Rac1 regulates axon formation through the actin cytoskeleton
Axon outgrowth relies on coordinated regulation of the actin cytoskeleton and microtubule dynamics (da Silva and Dotti, 2002; Dent and Gertler, 2003; Witte and Bradke, 2008). Previous studies using mutant Rho-GTPases indicated that Rac1 may regulate both actin and microtubule dynamics (Kunda et al., 2001; Ng and Luo, 2004; Watabe-Uchida et al., 2006a,b). However, although we did not study microtubule dynamics in detail, staining of the microtubules did not reveal any obvious defects in the microtubule organization, and microtubule stabilization did not rescue axon formation in the Rac1-knock-out CGNs (data not shown). Instead, we observed a strong defect in the organization and dynamics of the actin cytoskeleton in the growth cone. These Rac1-induced changes of the actin cytoskeleton did not prevent neurite formation, but selectively axon growth. Furthermore, actin depolymerization reversed the defect in axon formation. This indicates that Rac1 regulates axon formation by acting predominantly through the actin cytoskeleton.
Cellular signaling pathways downstream of Rac1
Potential effectors through which Rac1 regulates actin dynamics include the PAK–cofilin pathway and the WAVE–Arp2/3 pathway (Burridge and Wennerberg, 2004). We found that PAK1, one of the upstream regulators of cofilin, was mostly inactive in Rac1-depleted cerebella. However, there was no concomitant change in cofilin activity between wild-type and Rac1-knock-out cerebellar extracts, suggesting that cofilin activity is not crucial for Rac1 signaling to the actin cytoskeleton. In line with our findings, a recent study showed that, in neutrophils, Rac2, rather than Rac1, is responsible for signaling to cofilin (Sun et al., 2007). Although we observed a reduced PAK1 activity in the Rac1-knock-out cerebellum, increasing the levels of PAK1 activity by mimicking phosphorylation or recruiting PAK1 to the plasma membrane did not rescue the axon growth defect of Rac1-knock-out CGNs. This indicates that modulating PAK1 activity cannot compensate for the absence of Rac1. Another possibility is that PAK1 acts upstream rather than downstream of Rac1 (Jacobs et al., 2007). In this context, it may be possible that the decreased level of PAK1 phosphorylation is not a direct effect, but rather mediated through other molecules that are deregulated in the absence of Rac1.
We identified the WAVE complex as a physiological target downstream of Rac1. Rac1-knock-out CGNs showed drastic changes in the localization of the WAVE complex, including WAVE, Sra-1, Nap1, and Abi1. These molecules were virtually absent from the plasma membrane of Rac1-knock-out growth cones. In addition, we found that inactivation of the WAVE complex by knocking down Nap1 or inhibition of the Arp2/3 complex mimicked the axon growth defect observed in Rac1−/− CGNs. Conversely, defects in axon growth of Rac1−/− CGNs could be partially rescued by expression of a membrane-tethered WAVE mutant. Moreover, overexpression of WAVE-CAAX construct in Rac1−/− CGNs partially rescued the increased number of neurites. We envision that Rac1 provides the spatial information where actin polymerization occurs, and lamellipodia will form by recruiting the WAVE complex to those sites of the growth cone plasma membrane. When this spatial information is missing, actin polymerization occurs at lower efficiencies, but at various sites, resulting in the protrusion of several shorter neurites instead of two long ones. Consistent with this model, downregulation of Nap1 or inhibition of Arp2/3, which affect actin polymerization, impairs axon formation but does not increase neurite number. Thus, our study supports the view that Rac1 regulates the recruitment of the WAVE complex to the plasma membrane to activate the Arp2/3 complex and that this regulation is required for axonal growth.
Rac1 regulation of WAVE signaling is not unidirectional; interestingly, binding partners of WAVE can also regulate Rac signaling, creating regulatory feedback loops. For example, the WAVE1-associated Rac GAP protein (WRP) functions as a Rac GTPase-activating protein (GAP) and inactivates Rac. Overexpression of WRP in cultured CGNs drastically reduces neurite outgrowth (Soderling et al., 2002), a phenotype that we observe in Rac1−/− CGNs. Together, these results suggest that a strict spatiotemporal regulation of Rac signaling is important for axon formation. We propose a model in which active Rac1 induces the WAVE complex to translocate to specific sites at the plasma membrane of the neuronal growth cone where it triggers changes in actin dynamics important for lamellipodia formation. The WRP–WAVE complex could then be required to downregulate Rac1 signaling. Multiple cycles of Rac1 activation and inactivation are necessary for the tuning of the actin cytoskeleton and lamellipodial dynamics, and this is instrumental for axon growth.
Here, we identified Rac1 as positive physiological regulator of axonal growth and neuronal polarization and found the WAVE complex as key effector regulating these processes in CGNs. Our previous work depicted Cdc42 and cofilin as positive regulators of polarity in hippocampal neurons (Garvalov et al., 2007). Clearly, there may be neuron type-specific differences in the molecular regulation of neuronal polarization. However, it may be possible that neuronal polarization is triggered by a multitude of regulatory pathways. Along this view, neuronal polarization could be rather a consequence of specific cytoskeletal properties that can be adjusted by different actin regulators. The challenge ahead is to precisely depict the cytoskeletal properties that enable axonal growth and neuronal polarization under physiological conditions.
Footnotes
This work was supported by the Max Planck Society and additional grants from the Deutsche Forschungsgemeinschaft. S.T. was supported by Deutsche Forschungsgemeinschaft Fellowship TA 551/1-1. F.B. is a recipient of a Career Development Award from the Human Frontier Science Program. We thank Rüdiger Klein, Peter Mayinger, Ferdinando Rossi, and Walter Witke for helpful comments and suggestions on this manuscript. We are indebted to James Bamburg, Michael Greenberg, Gerhard Schratt, and Giorgio Scita for antibodies, Jonathan Chernoff for the pCMV6M-PakT423E construct, Gary Bokoch for the Pak-CAAX constructs, and Eva Anton for the Nap1 shRNA construct. We also thank the animal facility of the Max Planck Institute of Neurobiology, Liane Meyn for technical assistance, and Christian Feller for the help with statistical analysis.
References
- Albertinazzi C, Gilardelli D, Paris S, Longhi R, de Curtis I. Overexpression of a neural-specific rho family GTPase, cRac1B, selectively induces enhanced neuritogenesis and neurite branching in primary neurons. J Cell Biol. 1998;142:815–825. doi: 10.1083/jcb.142.3.815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benesch S, Lommel S, Steffen A, Stradal TE, Scaplehorn N, Way M, Wehland J, Rottner K. Phosphatidylinositol 4,5-biphosphate (PIP2)-induced vesicle movement depends on N-WASP and involves Nck, WIP, and Grb2. J Biol Chem. 2002;277:37771–37776. doi: 10.1074/jbc.M204145200. [DOI] [PubMed] [Google Scholar]
- Bogdan S, Grewe O, Strunk M, Mertens A, Klämbt C. Sra-1 interacts with Kette and Wasp and is required for neuronal and bristle development in Drosophila. Development. 2004;131:3981–3989. doi: 10.1242/dev.01274. [DOI] [PubMed] [Google Scholar]
- Bokoch GM. Regulation of innate immunity by Rho GTPases. Trends Cell Biol. 2005;15:163–171. doi: 10.1016/j.tcb.2005.01.002. [DOI] [PubMed] [Google Scholar]
- Bolis A, Corbetta S, Cioce A, de Curtis I. Differential distribution of Rac1 and Rac3 GTPases in the developing mouse brain: implications for a role of Rac3 in Purkinje cell differentiation. Eur J Neurosci. 2003;18:2417–2424. doi: 10.1046/j.1460-9568.2003.02938.x. [DOI] [PubMed] [Google Scholar]
- Borghesani PR, Peyrin JM, Klein R, Rubin J, Carter AR, Schwartz PM, Luster A, Corfas G, Segal RA. BDNF stimulates migration of cerebellar granule cells. Development. 2002;129:1435–1442. doi: 10.1242/dev.129.6.1435. [DOI] [PubMed] [Google Scholar]
- Bradke F, Dotti CG. The role of local actin instability in axon formation. Science. 1999;283:1931–1934. doi: 10.1126/science.283.5409.1931. [DOI] [PubMed] [Google Scholar]
- Bradke F, Dotti CG. Differentiated neurons retain the capacity to generate axons from dendrites. Curr Biol. 2000;10:1467–1470. doi: 10.1016/s0960-9822(00)00807-1. [DOI] [PubMed] [Google Scholar]
- Burridge K, Wennerberg K. Rho and Rac take center stage. Cell. 2004;116:167–179. doi: 10.1016/s0092-8674(04)00003-0. [DOI] [PubMed] [Google Scholar]
- Chen L, Liao G, Waclaw RR, Burns KA, Linquist D, Campbell K, Zheng Y, Kuan CY. Rac1 controls the formation of midline commissures and the competency of tangential migration in ventral telencephalic neurons. J Neurosci. 2007;27:3884–3893. doi: 10.1523/JNEUROSCI.3509-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Melendez J, Campbell K, Kuan CY, Zheng Y. Rac1 deficiency in the forebrain results in neural progenitor reduction and microcephaly. Dev Biol. 2009;325:162–170. doi: 10.1016/j.ydbio.2008.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chrostek A, Wu X, Quondamatteo F, Hu R, Sanecka A, Niemann C, Langbein L, Haase I, Brakebusch C. Rac1 is crucial for hair follicle integrity but is not essential for maintenance of the epidermis. Mol Cell Biol. 2006;26:6957–6970. doi: 10.1128/MCB.00075-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corbetta S, Gualdoni S, Albertinazzi C, Paris S, Croci L, Consalez GG, de Curtis I. Generation and characterization of Rac3 knockout mice. Mol Cell Biol. 2005;25:5763–5776. doi: 10.1128/MCB.25.13.5763-5776.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corbetta S, Gualdoni S, Ciceri G, Monari M, Zuccaro E, Tybulewicz VL, de Curtis I. Essential role of Rac1 and Rac3 GTPases in neuronal development. FASEB J. 2009;23:1347–1357. doi: 10.1096/fj.08-121574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniels RH, Hall PS, Bokoch GM. Membrane targeting of p21-activated kinase 1 (PAK1) induces neurite outgrowth from PC12 cells. EMBO J. 1998;17:754–764. doi: 10.1093/emboj/17.3.754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- da Silva JS, Dotti CG. Breaking the neuronal sphere: regulation of the actin cytoskeleton in neuritogenesis. Nat Rev Neurosci. 2002;3:694–704. doi: 10.1038/nrn918. [DOI] [PubMed] [Google Scholar]
- Dent EW, Gertler FB. Cytoskeletal dynamics and transport in growth cone motility and axon guidance. Neuron. 2003;40:209–227. doi: 10.1016/s0896-6273(03)00633-0. [DOI] [PubMed] [Google Scholar]
- Didsbury J, Weber RF, Bokoch GM, Evans T, Snyderman R. rac, a novel ras-related family of proteins that are botulinum toxin substrates. J Biol Chem. 1989;264:16378–16382. [PubMed] [Google Scholar]
- Ertürk A, Hellal F, Enes J, Bradke F. Disorganized microtubules underlie the formation of retraction bulbs and the failure of axonal regeneration. J Neurosci. 2007;27:9169–9180. doi: 10.1523/JNEUROSCI.0612-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garvalov BK, Flynn KC, Neukirchen D, Meyn L, Teusch N, Wu X, Brakebusch C, Bamburg JR, Bradke F. Cdc42 regulates cofilin during the establishment of neuronal polarity. J Neurosci. 2007;27:13117–13129. doi: 10.1523/JNEUROSCI.3322-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomis-Rüth S, Wierenga CJ, Bradke F. Plasticity of polarization: changing dendrites into axons in neurons integrated in neuronal circuits. Curr Biol. 2008;18:992–1000. doi: 10.1016/j.cub.2008.06.026. [DOI] [PubMed] [Google Scholar]
- Govek EE, Newey SE, Van Aelst L. The role of the Rho GTPases in neuronal development. Genes Dev. 2005;19:1–49. doi: 10.1101/gad.1256405. [DOI] [PubMed] [Google Scholar]
- Graus-Porta D, Blaess S, Senften M, Littlewood-Evans A, Damsky C, Huang Z, Orban P, Klein R, Schittny JC, Müller U. Beta1-class integrins regulate the development of laminae and folia in the cerebral and cerebellar cortex. Neuron. 2001;31:367–379. doi: 10.1016/s0896-6273(01)00374-9. [DOI] [PubMed] [Google Scholar]
- Gualdoni S, Albertinazzi C, Corbetta S, Valtorta F, de Curtis I. Normal levels of Rac1 are important for dendritic but not axonal development in hippocampal neurons. Biol Cell. 2007;99:455–464. doi: 10.1042/BC20060119. [DOI] [PubMed] [Google Scholar]
- Hakeda-Suzuki S, Ng J, Tzu J, Dietzl G, Sun Y, Harms M, Nardine T, Luo L, Dickson BJ. Rac function and regulation during Drosophila development. Nature. 2002;416:438–442. doi: 10.1038/416438a. [DOI] [PubMed] [Google Scholar]
- Hall A. Rho GTPases and the actin cytoskeleton. Science. 1998;279:509–514. doi: 10.1126/science.279.5350.509. [DOI] [PubMed] [Google Scholar]
- Hatten ME. Neuronal regulation of astroglial morphology and proliferation in vitro. J Cell Biol. 1985;100:384–396. doi: 10.1083/jcb.100.2.384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatten ME, Heintz N. Mechanisms of neural patterning and specification in the developing cerebellum. Annu Rev Neurosci. 1995;18:385–408. doi: 10.1146/annurev.ne.18.030195.002125. [DOI] [PubMed] [Google Scholar]
- Hatten ME, Alder J, Zimmerman K, Heintz N. Genes involved in cerebellar cell specification and differentiation. Curr Opin Neurobiol. 1997;7:40–47. doi: 10.1016/s0959-4388(97)80118-3. [DOI] [PubMed] [Google Scholar]
- Hummel T, Leifker K, Klämbt C. The Drosophila HEM-2/NAP1 homolog KETTE controls axonal pathfinding and cytoskeletal organization. Genes Dev. 2000;14:863–873. [PMC free article] [PubMed] [Google Scholar]
- Innocenti M, Gerboth S, Rottner K, Lai FP, Hertzog M, Stradal TE, Frittoli E, Didry D, Polo S, Disanza A, Benesch S, Di Fiore PP, Carlier MF, Scita G. Abi1 regulates the activity of N-WASP and WAVE in distinct actin-based processes. Nat Cell Biol. 2005;7:969–976. doi: 10.1038/ncb1304. [DOI] [PubMed] [Google Scholar]
- Jacobs T, Causeret F, Nishimura YV, Terao M, Norman A, Hoshino M, Nikolić M. Localized activation of p21-activated kinase controls neuronal polarity and morphology. J Neurosci. 2007;27:8604–8615. doi: 10.1523/JNEUROSCI.0765-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kassai H, Terashima T, Fukaya M, Nakao K, Sakahara M, Watanabe M, Aiba A. Rac1 in cortical projection neurons is selectively required for midline crossing of commissural axonal formation. Eur J Neurosci. 2008;28:257–267. doi: 10.1111/j.1460-9568.2008.06343.x. [DOI] [PubMed] [Google Scholar]
- Kawano Y, Yoshimura T, Tsuboi D, Kawabata S, Kaneko-Kawano T, Shirataki H, Takenawa T, Kaibuchi K. CRMP-2 is involved in kinesin-1-dependent transport of the Sra-1/WAVE1 complex and axon formation. Mol Cell Biol. 2005;25:9920–9935. doi: 10.1128/MCB.25.22.9920-9935.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawauchi T, Hoshino M. Molecular pathways regulating cytoskeletal organization and morphological changes in migrating neurons. Dev Neurosci. 2008;30:36–46. doi: 10.1159/000109850. [DOI] [PubMed] [Google Scholar]
- Kunda P, Paglini G, Quiroga S, Kosik K, Caceres A. Evidence for the involvement of Tiam1 in axon formation. J Neurosci. 2001;21:2361–2372. doi: 10.1523/JNEUROSCI.21-07-02361.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le SS, Loucks FA, Udo H, Richardson-Burns S, Phelps RA, Bouchard RJ, Barth H, Aktories K, Tyler KL, Kandel ER, Heidenreich KA, Linseman DA. Inhibition of Rac GTPase triggers a c-Jun- and Bim-dependent mitochondrial apoptotic cascade in cerebellar granule neurons. J Neurochem. 2005;94:1025–1039. doi: 10.1111/j.1471-4159.2005.03252.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linseman DA, Laessig T, Meintzer MK, McClure M, Barth H, Aktories K, Heidenreich KA. An essential role for Rac/Cdc42 GTPases in cerebellar granule neuron survival. J Biol Chem. 2001;276:39123–39131. doi: 10.1074/jbc.M103959200. [DOI] [PubMed] [Google Scholar]
- Luo L. Rho GTPases in neuronal morphogenesis. Nat Rev Neurosci. 2000;1:173–180. doi: 10.1038/35044547. [DOI] [PubMed] [Google Scholar]
- Luo L, Hensch TK, Ackerman L, Barbel S, Jan LY, Jan YN. Differential effects of the Rac GTPase on Purkinje cell axons and dendritic trunks and spines. Nature. 1996;379:837–840. doi: 10.1038/379837a0. [DOI] [PubMed] [Google Scholar]
- Mandell JW, Banker GA. A spatial gradient of tau protein phosphorylation in nascent axons. J Neurosci. 1996;16:5727–5740. doi: 10.1523/JNEUROSCI.16-18-05727.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meberg PJ, Ono S, Minamide LS, Takahashi M, Bamburg JR. Actin depolymerizing factor and cofilin phosphorylation dynamics: response to signals that regulate neurite extension. Cell Motil Cytoskeleton. 1998;39:172–190. doi: 10.1002/(SICI)1097-0169(1998)39:2<172::AID-CM8>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- Miki H, Suetsugu S, Takenawa T. WAVE, a novel WASP-family protein involved in actin reorganization induced by Rac. EMBO J. 1998;17:6932–6941. doi: 10.1093/emboj/17.23.6932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagata I, Nakatsuji N. Granule cell behavior on laminin in cerebellar microexplant cultures. Brain Res Dev Brain Res. 1990;52:63–73. doi: 10.1016/0165-3806(90)90222-k. [DOI] [PubMed] [Google Scholar]
- Nagata I, Nakatsuji N. Migration behavior of granule cell neurons in cerebellar cultures I. A PKH26 labeling study in microexplant and organotypic cultures. Dev Growth Differ. 1994;36:19–27. doi: 10.1111/j.1440-169X.1994.00019.x. [DOI] [PubMed] [Google Scholar]
- Ng J, Luo L. Rho GTPases regulate axon growth through convergent and divergent signaling pathways. Neuron. 2004;44:779–793. doi: 10.1016/j.neuron.2004.11.014. [DOI] [PubMed] [Google Scholar]
- Ng J, Nardine T, Harms M, Tzu J, Goldstein A, Sun Y, Dietzl G, Dickson BJ, Luo L. Rac GTPases control axon growth, guidance and branching. Nature. 2002;416:442–447. doi: 10.1038/416442a. [DOI] [PubMed] [Google Scholar]
- Nobes CD, Hall A. Rho, rac and cdc42 GTPases: regulators of actin structures, cell adhesion and motility. Biochem Soc Trans. 1995;23:456–459. doi: 10.1042/bst0230456. [DOI] [PubMed] [Google Scholar]
- Nolen BJ, Tomasevic N, Russell A, Pierce DW, Jia Z, McCormick CD, Hartman J, Sakowicz R, Pollard TD. Characterization of two classes of small molecule inhibitors of Arp2/3 complex. Nature. 2009;460:1031–1034. doi: 10.1038/nature08231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pilo Boyl P, Di Nardo A, Mulle C, Sassoè-Pognetto M, Panzanelli P, Mele A, Kneussel M, Costantini V, Perlas E, Massimi M, Vara H, Giustetto M, Witke W. Profilin2 contributes to synaptic vesicle exocytosis, neuronal excitability, and novelty-seeking behavior. EMBO J. 2007;26:2991–3002. doi: 10.1038/sj.emboj.7601737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powell SK, Rivas RJ, Rodriguez-Boulan E, Hatten ME. Development of polarity in cerebellar granule neurons. J Neurobiol. 1997;32:223–236. doi: 10.1002/(sici)1097-4695(199702)32:2<223::aid-neu7>3.0.co;2-a. [DOI] [PubMed] [Google Scholar]
- Ridley AJ. Rho GTPases and cell migration. J Cell Sci. 2001a;114:2713–2722. doi: 10.1242/jcs.114.15.2713. [DOI] [PubMed] [Google Scholar]
- Ridley AJ. Rho family proteins: coordinating cell responses. Trends Cell Biol. 2001b;11:471–477. doi: 10.1016/s0962-8924(01)02153-5. [DOI] [PubMed] [Google Scholar]
- Sakurai T, Lustig M, Babiarz J, Furley AJ, Tait S, Brophy PJ, Brown SA, Brown LY, Mason CA, Grumet M. Overlapping functions of the cell adhesion molecules Nr-CAM and L1 in cerebellar granule cell development. J Cell Biol. 2001;154:1259–1273. doi: 10.1083/jcb.200104122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sander EE, ten Klooster JP, van Delft S, van der Kammen RA, Collard JG. Rac downregulates Rho activity: reciprocal balance between both GTPases determines cellular morphology and migratory behavior. J Cell Biol. 1999;147:1009–1022. doi: 10.1083/jcb.147.5.1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwamborn JC, Püschel AW. The sequential activity of the GTPases Rap1B and Cdc42 determines neuronal polarity. Nat Neurosci. 2004;7:923–929. doi: 10.1038/nn1295. [DOI] [PubMed] [Google Scholar]
- Sells MA, Knaus UG, Bagrodia S, Ambrose DM, Bokoch GM, Chernoff J. Human p21-activated kinase (Pak1) regulates actin organization in mammalian cells. Curr Biol. 1997;7:202–210. doi: 10.1016/s0960-9822(97)70091-5. [DOI] [PubMed] [Google Scholar]
- Sells MA, Boyd JT, Chernoff J. p21-activated kinase 1 (Pak1) regulates cell motility in mammalian fibroblasts. J Cell Biol. 1999;145:837–849. doi: 10.1083/jcb.145.4.837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shamah SM, Lin MZ, Goldberg JL, Estrach S, Sahin M, Hu L, Bazalakova M, Neve RL, Corfas G, Debant A, Greenberg ME. EphA receptors regulate growth cone dynamics through the novel guanine nucleotide exchange factor ephexin. Cell. 2001;105:233–244. doi: 10.1016/s0092-8674(01)00314-2. [DOI] [PubMed] [Google Scholar]
- Soderling SH, Binns KL, Wayman GA, Davee SM, Ong SH, Pawson T, Scott JD. The WRP component of the WAVE-1 complex attenuates Rac-mediated signalling. Nat Cell Biol. 2002;4:970–975. doi: 10.1038/ncb886. [DOI] [PubMed] [Google Scholar]
- Steffen A, Rottner K, Ehinger J, Innocenti M, Scita G, Wehland J, Stradal TE. Sra-1 and Nap1 link Rac to actin assembly driving lamellipodia formation. EMBO J. 2004;23:749–759. doi: 10.1038/sj.emboj.7600084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stradal TE, Rottner K, Disanza A, Confalonieri S, Innocenti M, Scita G. Regulation of actin dynamics by WASP and WAVE family proteins. Trends Cell Biol. 2004;14:303–311. doi: 10.1016/j.tcb.2004.04.007. [DOI] [PubMed] [Google Scholar]
- Sugihara K, Nakatsuji N, Nakamura K, Nakao K, Hashimoto R, Otani H, Sakagami H, Kondo H, Nozawa S, Aiba A, Katsuki M. Rac1 is required for the formation of three germ layers during gastrulation. Oncogene. 1998;17:3427–3433. doi: 10.1038/sj.onc.1202595. [DOI] [PubMed] [Google Scholar]
- Sun CX, Magalhães MA, Glogauer M. Rac1 and Rac2 differentially regulate actin free barbed end formation downstream of the fMLP receptor. J Cell Biol. 2007;179:239–245. doi: 10.1083/jcb.200705122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takenawa T, Miki H. WASP and WAVE family proteins: key molecules for rapid rearrangement of cortical actin filaments and cell movement. J Cell Sci. 2001;114:1801–1809. doi: 10.1242/jcs.114.10.1801. [DOI] [PubMed] [Google Scholar]
- Tomoda T, Bhatt RS, Kuroyanagi H, Shirasawa T, Hatten ME. A mouse serine/threonine kinase homologous to C. elegans UNC51 functions in parallel fiber formation of cerebellar granule neurons. Neuron. 1999;24:833–846. doi: 10.1016/s0896-6273(00)81031-4. [DOI] [PubMed] [Google Scholar]
- Tronche F, Kellendonk C, Kretz O, Gass P, Anlag K, Orban PC, Bock R, Klein R, Schütz G. Disruption of the glucocorticoid receptor gene in the nervous system results in reduced anxiety. Nat Genet. 1999;23:99–103. doi: 10.1038/12703. [DOI] [PubMed] [Google Scholar]
- Vidali L, Chen F, Cicchetti G, Ohta Y, Kwiatkowski DJ. Rac1-null mouse embryonic fibroblasts are motile and respond to platelet-derived growth factor. Mol Biol Cell. 2006;17:2377–2390. doi: 10.1091/mbc.E05-10-0955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Zheng Y. Cell type-specific functions of Rho GTPases revealed by gene targeting in mice. Trends Cell Biol. 2007;17:58–64. doi: 10.1016/j.tcb.2006.11.009. [DOI] [PubMed] [Google Scholar]
- Watabe-Uchida M, John KA, Janas JA, Newey SE, Van Aelst L. The Rac activator DOCK7 regulates neuronal polarity through local phosphorylation of stathmin/Op18. Neuron. 2006a;51:727–739. doi: 10.1016/j.neuron.2006.07.020. [DOI] [PubMed] [Google Scholar]
- Watabe-Uchida M, Govek EE, Van Aelst L. Regulators of Rho GTPases in neuronal development. J Neurosci. 2006b;26:10633–10635. doi: 10.1523/JNEUROSCI.4084-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weyer A, Schilling K. Developmental and cell type-specific expression of the neuronal marker NeuN in the murine cerebellum. J Neurosci Res. 2003;73:400–409. doi: 10.1002/jnr.10655. [DOI] [PubMed] [Google Scholar]
- Wheeler AP, Wells CM, Smith SD, Vega FM, Henderson RB, Tybulewicz VL, Ridley AJ. Rac1 and Rac2 regulate macrophage morphology but are not essential for migration. J Cell Sci. 2006;119:2749–2757. doi: 10.1242/jcs.03024. [DOI] [PubMed] [Google Scholar]
- Witte H, Bradke F. The role of the cytoskeleton during neuronal polarization. Curr Opin Neurobiol. 2008;18:479–487. doi: 10.1016/j.conb.2008.09.019. [DOI] [PubMed] [Google Scholar]
- Witte H, Neukirchen D, Bradke F. Microtubule stabilization specifies initial neuronal polarization. J Cell Biol. 2008;180:619–632. doi: 10.1083/jcb.200707042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu H, Parsons JT. Cortactin, an 80/85-kilodalton pp60src substrate, is a filamentous actin-binding protein enriched in the cell cortex. J Cell Biol. 1993;120:1417–1426. doi: 10.1083/jcb.120.6.1417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamasaki T, Kawaji K, Ono K, Bito H, Hirano T, Osumi N, Kengaku M. Pax6 regulates granule cell polarization during parallel fiber formation in the developing cerebellum. Development. 2001;128:3133–3144. doi: 10.1242/dev.128.16.3133. [DOI] [PubMed] [Google Scholar]
- Yokota Y, Ring C, Cheung R, Pevny L, Anton ES. Nap1-regulated neuronal cytoskeletal dynamics is essential for the final differentiation of neurons in cerebral cortex. Neuron. 2007;54:429–445. doi: 10.1016/j.neuron.2007.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zmuda JF, Rivas RJ. The Golgi apparatus and the centrosome are localized to the sites of newly emerging axons in cerebellar granule neurons in vitro. Cell Motil Cytoskeleton. 1998;41:18–38. doi: 10.1002/(SICI)1097-0169(1998)41:1<18::AID-CM2>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- Zmuda JF, Rivas RJ. Actin filament disruption blocks cerebellar granule neurons at the unipolar stage of differentiation in vitro. J Neurobiol. 2000;43:313–328. doi: 10.1002/1097-4695(20000615)43:4<313::aid-neu1>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]