

Abstract
One of the pathological hallmarks of Alzheimer's disease (AD) is deposition of extracellular amyloid-β (Aβ) peptide, which is generated from the cleavage of amyloid precursor protein (APP). Accumulation of Aβ is thought to associate with the progressive neuronal death observed in AD. However, the precise signaling mechanisms underlying the action of Aβ in AD pathophysiology are not completely understood. Here, we report the involvement of the transcription factor signal transducer and activator of transcription 3 (STAT3) in mediating Aβ-induced neuronal death. We find that tyrosine phosphorylation of STAT3 is elevated in the cortex and hippocampus of APP/PS1 transgenic mice. Treatment of cultured rat neurons with Aβ or intrahippocampal injection of mice with Aβ both induces tyrosine phosphorylation of STAT3 in neurons. Importantly, reduction of either the expression or activation of STAT3 markedly attenuates Aβ-induced neuronal apoptosis, suggesting that STAT3 activation contributes to neuronal death after Aβ exposure. We further identify Tyk2 as the tyrosine kinase that acts upstream of STAT3, as Aβ-induced activation of STAT3 and caspase-3-dependent neuronal death can be inhibited in tyk2−/− neurons. Finally, increased tyrosine phosphorylation of STAT3 is also observed in postmortem brains of AD patients. Our observations collectively reveal a novel role of STAT3 in Aβ-induced neuronal death and suggest the potential involvement of Tyk2/STAT3 signaling in AD pathophysiology.
Introduction
Alzheimer's disease (AD) is a devastating neurodegenerative disease that is characterized by progressive loss of cognitive functions, resulting in memory loss and dementia. Pathological hallmarks of AD include intracellular neurofibrillary tangles and extracellular amyloid plaques (Hardy et al., 1998). Although the precise cause of AD remains elusive, it has been suggested that neuronal loss in AD is attributed to the accumulation of toxic protein β-amyloid (Aβ), the constituent of extracellular plaques observed in AD patients (Hardy and Selkoe, 2002). Aβ is a cleavage product derived from amyloid precursor protein (APP). On sequential cleavage by aspartyl proteases β-secretase and γ-secretase, APP generates various peptide species, including the toxic form Aβ that is prone to oligomerization, leading to the formation of amyloid plaques (De Strooper and Annaert, 2000). Indeed, a number of mutations identified in familial cases of AD have been mapped to APP and a component of the γ-secretase presenilin 1 (PS1), which apparently favor the generation of Aβ (Duyckaerts et al., 2008). Consistent with this observation, transgenic mice harboring mutations in APP and PS1 are associated with elevated Aβ level, appearance of senile plaques, and behavioral deficits such as memory loss (Arendash et al., 2001; Eriksen and Janus, 2007). Furthermore, treatment of cortical neurons with aggregated Aβ peptides triggers neuronal apoptosis, hence supporting a pathogenic role of Aβ in AD (Estus et al., 1997).
Despite the abundance of evidence pointing to an essential role of Aβ in AD pathophysiology, our knowledge on the mechanisms underlying the action of Aβ remains limited. Emerging studies suggest that various intracellular signaling pathways are deregulated in AD brains or during Aβ-induced neuronal apoptosis (Buxbaum et al., 1990; Grant and Pant, 2002). For example, activation of stress-related kinases c-Jun N-terminal kinase (JNK) and p38 is associated with neuronal death in AD mouse model (Savage et al., 2002). Glycogen synthase kinase 3 (GSK-3) has also been implicated in Aβ-induced neurotoxicity (Ryder et al., 2004). Deregulation of distinct signaling pathways leads to aberrant phosphorylation of cellular proteins and has a profound effect on the progression of AD (Ferrer et al., 2005; Hooper et al., 2008).
In the current study, we report the identification of signal transducer and activator of transcription 3 (STAT3) as a potential key player in AD pathophysiology. STAT3 is a transcription factor that is typically associated with cytokine signaling during neuronal differentiation, inflammation, and malignancies. Interestingly, we found that tyrosine phosphorylation of STAT3, which is required for the activation of this transcription factor, is markedly elevated in neurons treated with Aβ in vitro or in vivo as well as in the brains of APP/PS1 transgenic mice. Inhibition of STAT3 activation or reduced STAT3 expression significantly attenuates Aβ-induced neuronal cell death. Moreover, activation of a tyrosine kinase Tyk2 is required for the Aβ-induced tyrosine phosphorylation of STAT3 and neuronal cell death. Notably, elevation of STAT3 tyrosine phosphorylation is evident in postmortem samples of AD brains. These observations collectively raise an intriguing possibility that STAT3 signaling is involved in neuronal apoptosis observed in AD patients.
Materials and Methods
Short interfering RNAs, antibodies, and chemical inhibitors.
For STAT3 knockdown, double-stranded 25 nt RNA duplexes for rat STAT3 were designed (Stealth RNA-mediated interference; Invitrogen). The short interfering RNA (siRNA) sequences are as follows: STAT3 siRNA, 5′-GGAAAUUUAACAUUCUGGGCACGAA; STAT3 scrambled siRNA, 5′-GGAUUUCAAUUAGUCCGGCAAAGAA. To generate pSUPER-STAT3 RNA interference (RNAi) constructs, double-stranded oligonucleotides encoding STAT3 short hairpin RNA (shSTAT3) were subcloned into BglII- and HindIII-digested pSUPER vector. The short hairpin RNA (shRNA) targets the mRNA sequences of both rat and mouse STAT3, with the target sequence 5′-GTCAGGTTGCTGGTCAAAT-3′. The RNAi-resistant STAT3 expression construct was generated by introducing five silent mutations into the cDNA sequence that is targeted by shSTAT3, without changing the amino acid sequence. Amyloid-β protein fragment 25–35 (Aβ25-35) and Aβ1-40 were from Sigma-Aldrich, whereas Aβ1-42 and Aβ42-1 were from Invitrogen. Mouse monoclonal antibody specific for APP was purchased from Abcam, whereas rabbit antibodies against STAT1, caspase-3 (8G10), and phospho-STAT1 (Tyr701) were from Cell Signaling Technology, and STAT3 antibody was from Santa Cruz. Mouse monoclonal antibody specific for Aβ was purchased from Dako, NeuN from Millipore Bioscience Research Reagents, GFAP from Cell Signaling Technology, Tyk2 from BD Biosciences, β-tubulin from Santa Cruz, and rat monoclonal antibody specific for presenilin-1 from Millipore Bioscience Research Reagents. For detection of STAT3 phosphorylation at Tyr705 residue, three antibodies including a rabbit polyclonal phosphotyrosine STAT3 antibody, a rabbit monoclonal phosphospecific antibody (D3A7), and a mouse monoclonal phosphospecific antibody (3E2) were purchased from Cell Signaling Technology. Similar results were obtained with all these specific antibodies. STAT3 inhibitor peptide, which is a STAT3 Src homology 2 domain-binding phosphopeptide that inhibits the formation of STAT3 dimer, Jak inhibitor 1, 4-amino-5-(4-chlorophenyl)-7-(t-butyl)pyrazolo[3,4-d]pyrimidine (PP2), and (E)-2-cyano-3-(3,4-dihydrophenyl)-N-(phenylmethyl)-2-propenamide (AG490) were purchased from Calbiochem. Cycloheximide and 4,6-diamidino-2-phenylindole (DAPI) were purchased from Sigma-Aldrich.
Animals, mice, and human brain samples.
APP751SL/PS1M146L double-transgenic mice were generated by crossing PS-1 (HMG PS-1 M146L) homozygous mice with hemizygous APP (Thy1 APP751 SL) transgenic mice (Wirths et al., 2001). APP695 with Swedish mutation K595N/M596L and PS1ΔE9 double-transgenic mice (APP/PS1) were purchased from The Jackson Laboratory (Borchelt et al., 1996; Lee et al., 1997). We are grateful to Prof. K. Shimoda (Kyushu University Graduate School of Medical Sciences, Fukuoka, Japan) for the tyk2−/− mice (Shimoda et al., 2000). Wild-type (WT) and transgenic mice of different stages (n > 4/type/age) were anesthetized with pentobarbital (50 mg/kg) and perfused transcardially with 0.9% NaCl at 37°C followed by ice-cold 4% paraformaldehyde. Brain tissues were removed and fixed in 4% paraformaldehyde in phosphate buffer for 24 h, and then embedded in paraffin for immunohistochemical studies.
For intrahippocampal injection, ∼4-month-old male C57BL/6J mice were anesthetized with 4% chloral hydrate. Aggregated Aβ1-42 peptide (1.5 μl; 250 μm) was injected into the hippocampal CA1 region of the right hemisphere of mice stereotactically (coordinates: anteroposterior, −2.7 mm from the bregma; lateral, −2.5 mm; dorsoventral, −1.9 mm) with a 10 μl Hamilton syringe driven by a minipump (Motorized Stereotaxic Injector; Stoelting) at an injection speed of 0.15 μl/min (Stéphan et al., 2001). The needle was kept in the injection site for another 5 min and then slowly withdrawn. Animals that were injected with the Aβ42-1 or vehicle served as controls. At 20 d after injection, the mouse brains were collected and subjected to immunohistochemical and Western blot analysis.
Hippocampal sections of seven patients (two males and five females ranging from 72 to 96 years of age) with sporadic late-onset AD and corresponding age-matched controls were fixed by immersion in 4% paraformaldehyde in phosphate buffer for 24 h and embedded in paraffin.
Cell culture and transfection.
Primary cortical neuron cultures were prepared from embryonic day 18 rat embryos. Cortical neurons were plated on poly-d-lysine-coated culture plates in Neurobasal medium containing B27 supplement, penicillin (50 U/ml), and streptomycin (100 μg/ml). Before the treatment with Aβ, cultured neurons at 7 d in vitro (DIV) were washed with Neurobasal medium twice, kept in the same medium for 2 h, and then treated with Aβ (40 μm) for various time intervals. PC12 cells were transfected with siRNAs or with DNA expression constructs using Lipofectamine 2000 (Invitrogen) according to the manufacturer's instruction.
Preparation of brain extracts, cell extracts, and Western blot analysis.
Brain parts including cortex, hippocampus, cerebellum, and striatum were dissected from APP/PS1 transgenic mice of various stages (n > 8/type/age) and kept on dry ice. Frozen brain tissues were homogenized in homogenizing buffer (25 mm Tris-HCl, pH 7.4, 150 mm NaCl, 1 mm EDTA, pH 7.4, 50 mm NaF) with various protease inhibitors. The supernatants were kept for Western blotting and the pellets were reextracted using RIPA buffer [150 mm NaCl, 1% (v/v) Nonidet P-40, 0.5% deoxycholic acid, 0.1% (w/v) SDS] containing various protease inhibitors [2 μg/ml aprotinin, 1 mm phenylmethylsulfonyl fluoride, 5 mm benzamidine, 1 mm NaOV (sodium orthovanadate), and 10 μg/ml soybean trypsin inhibitor]. Primary cultured cortical neurons or PC12 cells were harvested and lysed in RIPA buffer containing protease inhibitors. The proteins separated by SDS-PAGE were then transferred onto nitrocellulose membrane (Micron Separations) and probed with various antibodies. Proteins were visualized using the Supersignal West Pico Chemiluminescent Substrate kit (Pierce).
Immunohistochemical analysis.
Paraffin human brain slices were boiled in 1 mm EDTA buffer, pH 8.0, for 5 min for antigen retrieval. DAB or 3-amino-9-ethylcarbazole (AEC) staining was processed according to manufacturer's protocol (R&D Systems). For immunofluorescence staining, brain sections were blocked with 4% goat serum in PBS containing 0.4% Triton X-100 for 20 min at room temperature. The sections were incubated with primary antibody (anti-NeuN, 1:400; or anti-GFAP, 1:200) and anti-phospho-STAT3 (Tyr705) (1:100) at 4°C overnight, followed by Alexa Fluor 488-conjugated anti-rabbit and Alexa Fluor 568-conjugated anti-mouse IgG (Invitrogen; 1:500) at room temperature for 2 h. The slices were then washed and mounted with anti-fade reagent (Invitrogen) and analyzed under fluorescence microscope (Nikon TE2000E). Cultured cortical neurons were fixed with 4% paraformaldehyde at room temperature for 30 min, and then blocked with 4% goat serum in PBS containing 0.4% Triton X-100 for 20 min at room temperature. Immunocytochemical analysis was performed using anti-NeuN monoclonal antibody (1:500) and anti-phospho-STAT3 (Tyr705) polyclonal antibody (1:200) at 4°C overnight. The cells were labeled with Alexa Fluor 488- or Alexa Fluor 568-conjugated anti-rabbit or anti-mouse IgG (Invitrogen; 1:1000) at room temperature for 1 h, and then mounted and analyzed under confocal microscope (Olympus BX61).
Cell viability assay and proteolytic activity assay of caspase-3.
Cell viability was quantified by its ability to reduce tetrazolium salt 3-(4,5-dimethylthiazol-2-y)-2,5-diphenyl tetrasodium bromide (MTT) to a colored formazan products (Cell Proliferation Kit I; Roche) according to the manufacturer's instruction. Briefly, MTT reagent (5 mg/ml in Dulbecco's PBS), which is taken up only by viable cells, was added to the cells at 1/10 volume of the medium. Cells were incubated at 37°C for 4 h. MTT solubilization buffer (0.01 m HCl; 10% SDS) of twofold volume was added to the cells, followed by incubation in the dark at 37°C for 24 h. The absorbance was measured at 570 nm with a MRX-TC Revelation microplate reader (Dynex Technologies). Cell viability was expressed as the percentage of absorbance obtained in control cultures.
Activity of caspase-3 was quantified using CaspACE Assay System, Fluorometric kit (Promega) according to the manufacturer's protocol. Briefly, the cells were lysed, and the activity of caspase-3 in cell extracts was determined by fluorometric assay. The fluorescence emission of the 7-amino-4-methylcoumarin (AMC), results from a caspase-3-specific cleavage of the substrate peptide DEVD labeled with fluorochrome AMC, was measured using SpectraMaxGemini with an excitation wavelength of 360 nm and emission wavelength of 460 nm. Protein assay was performed as the normalization control.
Promoter luciferase assay and quantitative reverse transcription-PCR.
PC12 cells were transfected with reporter constructs, pSTAT3-TA-Luc or pGAS, as previously described (Fu et al., 2004). The cells were differentiated with NGF, and then treated with Aβ. Total RNA was isolated from Aβ-treated and untreated cortical neurons using RNeasy mini kit (QIAGEN) according to the manufacturer's instruction. First-strand cDNA was synthesized from 5 μg of total RNA using the SuperScript III first-strand synthesis kit (Invitrogen) and random hexamer system (Roche). Quantification of the target genes was performed with Power SYBR Green PCR master mix kit in Bio-Rad MX3000P real-time PCR system, according to the manufacturer's instructions. Triplicate quantitative reverse transcription-PCRs were done for each sample. The PCR amplification cycles were as follows: initial denaturation, 95°C for 15 min; followed by 40 cycles with denaturation, 95°C for 20 s, and annealing–extension, 60°C for 35 s. The specificity of the SYBR Green PCR signal was confirmed by melting curve analysis. Acquired data were analyzed by LightCycler 2000 software 3.5 (Roche). The Ct value of each gene was normalized against that of GAPDH. Primer sequences were as follows: rat inducible nitric oxide synthase (iNOS), 5′-gctacacttccaacgcaaca and 5-catggtgaacacgttcttgg; mouse iNOS, 5′-gtggtgacaagcacatttgg and 5′-aaggccaaacacagcatacc; and mouse tumor necrosis factor-related apoptosis-inducing ligand (TRAIL), 5′-cagcacttcaggatgatggt and 5′-gctgcttcatctcgttggtg.
Image analysis and statistical analysis.
For measuring the fold change of tyrosine phosphorylation of STAT3, grayscale intensity of the immunoreactive bands was measured using NIH ImageJ software. The ratio of the tyrosine-phosphorylated STAT3 over total amount of STAT3 was calculated. For statistical comparison, statistical analysis of the data was performed with Student's t test, one-way or two-way ANOVA using GraphPad Prism software as indicated. A value of p < 0.05 was considered statistically significant.
Results
Phosphorylation of STAT3 at Tyr705 is elevated in brain tissues of different transgenic mouse models of AD
To identify the aberrantly activated signaling molecules potentially involved in AD pathophysiology, we first screened for activation of signaling proteins in a transgenic mouse model of Aβ accumulation (APP751SL/PS1M146L), which displays distinct pathological characteristics. Lysates of cortices from these mice of 9 months of age were subjected to Western blot analysis (supplemental Fig. S1, available at www.jneurosci.org as supplemental material). Consistent with previous observations (Xiang et al., 2002; Otth et al., 2003; Ricardo et al., 2009), enhanced phosphorylation of JNK, Rb, and GSK-3 was observed in the brains of these transgenic mice. Intriguingly, we found that phosphorylation of the transcription factor STAT3 at Tyr705 was also consistently increased. Previous studies have revealed that tyrosine phosphorylation at this specific site is required for the transcriptional activity of STAT3 (Levy and Darnell, 2002). Interestingly, we found that increased phosphorylation of STAT3 at Tyr705 could be detected specifically in the cortex and hippocampus, but not in the cerebellum of APP751SL/PS1M146L transgenic mice (Fig. 1A). Similar results were observed using different antibodies specific for phosphotyrosine STAT3 (data not shown). This finding suggests that tyrosine phosphorylation of STAT3 is specifically elevated in brain regions that are affected in mouse models of Aβ accumulation. Consistent with this notion, increased tyrosine phosphorylation of STAT3 was similarly observed in another transgenic mouse model of Aβ accumulation, APP695 with Swedish mutation and PS1ΔE9 double-transgenic mice (APPswe/PS1ΔE9; hereon APP/PS1). Temporally, STAT3 phosphorylation at Tyr705 was first detected in the cortex of 4-month-old APP/PS1 transgenic mice (Fig. 1B). The tyrosine phosphorylation of STAT3 became more robust in older APP/PS1 transgenic mice and persisted for up to 18 months. The regulation of STAT3 phosphorylation therefore appears to occur before the deposition of amyloid plaques, which can be observed in the brain of APP/PS1 transgenic mice at ∼6 months (Garcia-Alloza et al., 2006).
Figure 1.
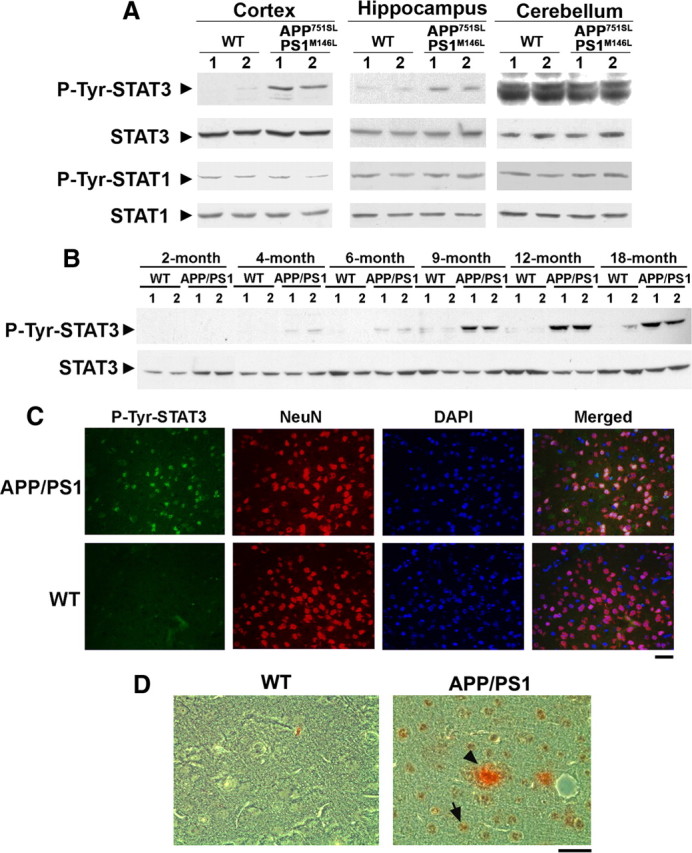
Tyrosine phosphorylation of STAT3 is elevated in cortex and hippocampus of AD mouse models. A, Cortices, hippocampi, and cerebelli from 12-month-old APP751SL/PS1M146L double-transgenic mouse brains were lysed and subjected to Western blot analysis using p-Tyr705 STAT3 (p-Tyr-STAT3), p-Tyr701 STAT1 (p-Tyr-STAT1), and their corresponding total antibodies. B, Cortices from different ages of APPswe/PS1ΔE9 (designated as APP/PS1) transgenic mice were homogenized, and lysates were subjected to Western blot analysis for p-Tyr-STAT3 and STAT3. C, Paraffin sections of 9-month-old APP/PS1 transgenic mouse brain cortices were stained using p-Tyr-STAT3 antibody, DAPI, and the neuron-specific marker NeuN as indicated. Scale bar, 50 μm. D, Paraffin sections of 12-month-old APP/PS1 transgenic mouse cortices were stained using p-Tyr-STAT3 antibody (brown; DAB) and Aβ antibody (red; AEC). Scale bar, 50 μm. The arrow indicated the p-Tyr STAT3-positive cells, whereas the arrowhead depicted the Aβ deposit.
To determine whether the elevated tyrosine phosphorylation of STAT3 observed in APP/PS1 transgenic mice is localized to neurons or astroglial cells, we performed immunostaining on paraffin sections of cortices from WT and APP/PS1 mice. In agreement with our Western blot analysis, Tyr705-phosphorylated STAT3 was barely detectable in the cortex of WT mice, whereas an increase in p-Tyr705-STAT3 staining was observed in the APP/PS1 cortex (Fig. 1C). Majority of the p-Tyr705-STAT3 staining was colocalized with the nuclei of NeuN-positive neurons in APP/PS1 mouse cortex (Fig. 1C). Prominent tyrosine phosphorylation of STAT3 and amyloid plaques (as indicated by Aβ staining) are depicted in Figure 1D.
Aβ exposure induces tyrosine phosphorylation of STAT3 in cortical neurons
Since Aβ generation is believed to cause neuronal dysfunctions in APP/PS1 mice, we examined whether the tyrosine phosphorylation of STAT3 detected in the cortex of APP/PS1 mice is induced by Aβ. Treatment of cultured cortical neurons at 7 DIV with Aβ25-35 triggered an increase in the phosphorylation of STAT3 at Tyr705 (Fig. 2A,B). Induction of STAT3 tyrosine phosphorylation was observed as early as 30 min and was sustained until 4 h of Aβ treatment (Fig. 2A). Furthermore, the tyrosine phosphorylation of STAT3 was concentrated in the nuclei of type III β-tubulin-positive neurons upon Aβ treatment (Fig. 2B), suggesting that Aβ induces STAT3-dependent transcriptional activity. Importantly, we found that intrahippocampal injection of mice with Aβ1-42 induced tyrosine phosphorylation of STAT3 in the cortex and hippocampus (Fig. 2C,D). Twenty days after injection of Aβ1-42, Aβ deposition was observed in the hippocampus and the cortex regions near the injection site, whereas Aβ staining was not detected in the brains of control mice injected with Aβ42-1 (supplemental Fig. S2A,B, available at www.jneurosci.org as supplemental material). In addition, astrogliosis and microgliosis surrounding the Aβ deposition was observed in Aβ1-42-injected brains (Fig. 2D) (data not shown). Consistent with Western blot analysis, immunohistochemistry revealed that prominent tyrosine phosphorylation of STAT3 was observed in the nuclei of neurons in cortex after injection with Aβ1-42, whereas injection of the control peptide Aβ42-1 did not stimulate the activation of STAT3 in mouse brains (Fig. 2D; supplemental Fig. S2C, available at www.jneurosci.org as supplemental material).
Figure 2.

Tyrosine phosphorylation of STAT3 is increased in cortical neurons or in mouse brains in response to Aβ insult. A, Primary cortical neurons at 7 DIV were treated with Aβ25-35 (40 μm; H2O served as control) for various time periods as indicated. Cell lysates were collected and subjected to immunoblotting using p-Tyr-STAT3 and total STAT3 antibodies. B, Primary cortical neurons treated with Aβ25-35 for 1 h were fixed and immunostained using p-Tyr-STAT3 and β-tubulin-III (β-Tub III) antibodies. Scale bar, 10 μm. C, D, Tyrosine phosphorylation of STAT3 increased in mouse brains upon injection of Aβ1-42. A single intrahippocampal injection of Aβ1-42 or Aβ42-1 in mice was performed. C, Lysates of mouse cortex and hippocampus at 20 d after injection were collected and subjected to Western blot analysis for p-Tyr-STAT3 and STAT3. D, Cortex sections were collected at 20 d after injection and stained for p-Tyr-STAT3, DAPI, NeuN, or the astrocytic marker GFAP as indicated. Scale bar, 50 μm.
Knockdown of STAT3 or inhibition of STAT3 activity protects the neurons from Aβ-induced apoptosis
Since Aβ stimulation has been observed to induce neuronal death in cortical neurons (Ueda et al., 1994), we were interested to examine whether the Aβ-dependent phosphorylation of STAT3 at Tyr705 leads to neuronal apoptosis. To study the role of STAT3 in Aβ-induced neuronal death, the expression of STAT3 was knocked down in PC12 cells. Western blot analysis revealed that expression of STAT3 protein was reduced by ∼80% in PC12 cells transfected with STAT3 siRNA (Fig. 3A). The cells were then differentiated by NGF for 4 d and subsequently treated with Aβ25-35 peptide (hereon Aβ). In agreement with previous studies (Vaisid et al., 2008), treatment of PC12 cells with Aβ induced cell death (∼50%). However, Aβ treatment did not reduce cell viability in STAT3 siRNA-transfected cells (Fig. 3B), indicating that knockdown of STAT3 protected the PC12 cells against Aβ-induced cell death. To confirm the specific role of STAT3 in Aβ-induced cell death, we depleted STAT3 expression in PC12 cells using another shRNA construct, and performed rescue experiment by cotransfecting the STAT3 shRNA construct together with the construct encoding shRNA-resistant STAT3. Expression of STAT3Res was resistant to STAT3 knockdown mediated by the STAT3 shRNA when coexpressed in PC12 cells (Fig. 3C). Whereas knockdown of STAT3 by the shRNA construct protected PC12 cells from Aβ-induced neurotoxicity, expression of STAT3Res construct restored the Aβ-induced cell death in STAT3-depleted PC12 cells (Fig. 3D). This finding further verified that the Aβ-induced reduction of neuronal death is dependent on the expression of STAT3. We then asked whether the effect of STAT3 inhibition on neurotoxicity could be observed in cortical neurons on Aβ exposure. Treatment of cortical neurons with either Aβ25-35 or Aβ1-42 resulted in reduction of neuronal viability (Fig. 3E,F; supplemental Fig. S3, available at www.jneurosci.org as supplemental material). Changes that are indicative of apoptosis could be observed in these neurons, including nuclear chromatin condensation or fragmentation pattern, after staining with the fluorescent DNA binding dye, DAPI (data not shown). To further elucidate the role of STAT3 in Aβ-induced neuronal death, cultured cortical neurons were pretreated with STAT3 inhibitor peptide, which blocks the dimerization and hence activation of STAT3, before Aβ exposure (Fu et al., 2004). We found that this peptide could effectively protect cortical neurons from Aβ25-35- or Aβ1-42-induced apoptosis, as indicated by its ability to attenuate Aβ-induced reduction in cell viability (Fig. 3E,F). Thus, blockade of STAT3 activity conferred resistance to neurotoxicity induced by Aβ peptide fragment. The involvement of STAT3 activation in Aβ-induced neuronal apoptosis was further elucidated by measuring the activity of caspase-3. Addition of STAT3 inhibitor peptide protected the neurons from Aβ-dependent cleavage and activation of caspase-3 (Fig. 3G,H). Together, these findings demonstrate that STAT3 activation is involved in Aβ-induced neuronal death.
Figure 3.

Knockdown of STAT3 ameliorates Aβ-induced neuronal apoptosis. A–D, Knockdown of STAT3 reduced neuronal cell death induced by Aβ25-35 treatment, whereas expression of RNAi-resistant STAT3 restored the neurotoxic effect of Aβ. PC12 cells were transfected with STAT3 siRNA (A, B) or with STAT3 shRNA construct alone or together with shRNA-resistant STAT3 as indicated (C, D). A, C, Western blot analysis for STAT3 was performed at 3 d after transfection of PC12 cells (α-tubulin serves as a loading control). B, D, After transfection, PC12 cells were differentiated with NGF for 4 d, and then exposed to Aβ25-35 for 24 h. The cell viability was assessed by MTT assay. B, Data are represented as mean ± SD (n = 3) (***p < 0.005, Aβ vs Con in scrambled siRNA condition; ANOVA). D, Data are represented as mean ± SEM (n ≥ 3) (*p < 0. 05, Aβ vs Con treatment; ANOVA). E–H, Blocking STAT3 activation protected the cortical neurons from Aβ-induced apoptosis. E, F, Primary cortical neurons at 7 DIV were pretreated with cell-permeable STAT3 inhibitor peptide (200 μg/ml) for 30 min before Aβ treatment. The cell viability of neurons after treatment with Aβ25-35 (E) or Aβ1-42 (F) was measured by MTT assay (*p < 0.05, Aβ vs Con in the absence of STAT3 inhibitor; ANOVA). G, H, Inhibition of STAT3 activation attenuates the increase of caspase-3 activity in Aβ25-35-treated neurons. G, The cell lysates were analyzed by immunoblotting using caspase-3 antibody. H, Quantification of the caspase-3 activity. Caspase-3 activity was determined by measuring the hydrolysis of fluorometric substrate Ac-DEVD-MCA (***p < 0.005 Aβ vs Con in the absence of STAT3 inhibitor; ##p < 0.01, STAT3 inhibitor vs control on Aβ exposure; ANOVA).
Aβ treatment induces STAT3-dependent transcriptional activity
We next asked whether Aβ treatment also affects STAT3 transcriptional activity, which is known to be dependent on phosphorylation at Tyr705. To examine this possibility, PC12 cells were transfected with a luciferase construct linked to the STAT3-response enhancer element (pSTAT3-Luc). Another luciferase construct linked to the STAT1-response enhancer element (pGAS-Luc) served as control. The transfected cells were then treated with Aβ or NGF, which is known to increase the transcriptional activity of both STAT1 and STAT3. We found that, whereas NGF treatment induced the promoter activity of STAT3 and STAT1, Aβ peptide only induced the promoter activity of STAT3 (Fig. 4A). Thus, our observations suggest that Aβ treatment not only induces tyrosine phosphorylation of STAT3 but also increases its transcriptional activity.
Figure 4.

Inhibition of STAT3 activation reduces Aβ-induced gene transcription. A, Aβ25-35 treatment increased the transcriptional activity of STAT3 in PC12 cells. PC12 cells were transiently transfected with a STAT3 (pSTAT3-TA-Luc) or STAT1 (pGAS-TA-Luc) reporter gene construct and an internal control plasmid (β-gal-pCMV). Luciferase activity was measured and normalized against the β-gal activity in the samples. Promoter activity was expressed as the ratio of luciferase activity. Data are represented as mean ± SD (n = 4) (***p < 0.005, Aβ or NGF treatment vs Con in pSTAT3-transfected condition; ###p < 0.005, NGF vs Con in pGAS-transfected condition; ANOVA). B, Cortical neurons were pretreated with STAT3 inhibitor peptide for 30 min, followed by Aβ25-35 (40 μm) treatment for 6 h. Total RNA was collected and reverse transcribed. The cDNA was subjected to real-time PCR analysis using iNOS-specific primers. Data are represented as mean ± SEM; n = 3. Results were analyzed with ANOVA (**p < 0.01, Aβ vs Con; #p < 0.05, STAT3 inhibitor vs control upon Aβ exposure). C, iNOS and TRAIL mRNA expression increased in APP/PS1 mouse forebrains. Forebrains of APP/PS1 mice at 6 months were collected and the mRNA expression of iNOS or TRAIL from wild-type and APP/PS1 mice was determined by quantitative PCR. Data are represented as mean ± SEM; n = 3. Results were analyzed with Student's t test (***p < 0.005).
In light of the elevated transcription activity of STAT3 in response to Aβ treatment, we were interested to identify the target genes downstream of STAT3 activation that may contribute to Aβ-induced neuronal apoptosis. Among the known target genes of STAT3, we found that the transcription of iNOS is elevated in Aβ-challenged neurons. However, pretreatment with STAT3 inhibitor peptide essentially abolished the Aβ-induced iNOS gene expression, indicating that STAT3 activation is necessary for Aβ-induced iNOS upregulation (Fig. 4B). Moreover, an increase in the gene expression of iNOS and another STAT target gene TRAIL was similarly observed in APP/PS1 mice, whereas mRNA level of an antiapoptotic gene myeloid cell leukemia-1 (Mcl-1) protein remained relatively unchanged (Fig. 4C) (data not shown). These findings supported the notion that transcriptional activity of STAT3 was induced by Aβ in vivo.
Tyk2 is required for Aβ-induced tyrosine phosphorylation of STAT3 and neuronal apoptosis
To identify the upstream regulators of STAT3 activation in response to Aβ stimulation, the effect of different inhibitors on Aβ-induced STAT3 phosphorylation at Tyr705 was examined. Interestingly, JAK inhibitor 1, which exhibits a potent inhibitory effect on various tyrosine kinases upstream of STAT3 including JAK1, JAK2, JAK3, and Tyk2, completely inhibited the Aβ-induced tyrosine phosphorylation of STAT3 (Fig. 5A). However, JAK2 inhibitor AG490 failed to attenuate Aβ-induced Tyr705-phosphorylation of STAT3, suggesting that Aβ-induced STAT3 phosphorylation was mediated by JAK family members other than JAK2. A Src kinase inhibitor, PP2, or a protein synthesis inhibitor, cycloheximide, did not inhibit STAT3 tyrosine phosphorylation induced by Aβ, indicating that Aβ-induced phosphorylation of STAT3 did not occur via the Src pathway, nor did it require protein synthesis.
Figure 5.

Tyk2 is required for the Aβ-induced tyrosine phosphorylation of STAT3 and neuronal cell death. A, Primary cortical neurons were pretreated for 2 h with various inhibitors including a potent inhibitor of JAK (JAK inhibitor 1), a JAK2 inhibitor (AG490), a Src tyrosine kinase inhibitor (PP2), and a protein synthesis inhibitor [cycloheximide (Cyclohex)], followed by treatment with Aβ25-35 for 4 or 24 h. The cell lysates were immunoblotted with p-Tyr-STAT3 and total STAT3 antibodies. B, Primary cortical neurons were treated with Aβ25-35. The cell lysates were coimmunoprecipitated using Tyk2 or JAK2 antibodies, and then subjected to Western blot analysis using phosphotyrosine antibody (p-Tyr). C–F, Primary cortical neurons prepared from tyk2−/− mice were treated with Aβ25-35. C, The cell lysates were subjected to immunoblotting using antibodies against p-Tyr-STAT3, total STAT3, caspase-3, or α-tubulin as indicated. Quantification analysis of fold change in p-Tyr-STAT3 (D) and cleaved caspase-3 (E) (*p < 0.05, ***p < 0.005, Aβ treatment for various time period vs 0 h in WT neurons; #p < 0.05, ###p < 0.005, tyk2−/− neurons vs WT neurons at specific time periods; ANOVA). F, Cell viability of tyk2−/− neurons after treatment with Aβ was measured by MTT assay (###p < 0.001, tyk2−/− vs WT neurons on Aβ treatment; ANOVA). Error bars indicate SEM.
To verify that JAK family member regulates Aβ-induced STAT3 activation, we treated cortical neurons with Aβ for 1 h and examined activation of the JAK family members Tyk2 and JAK2. Endogenous Tyk2 or JAK2 were coimmunoprecipitated by specific antibodies, and activation of kinases was detected by phosphotyrosine antibody. In agreement with our observation that JAK2 inhibitor had negligible effect on Aβ-induced STAT3 activation, we found that Aβ failed to trigger JAK2 activation. In contrast, tyrosine phosphorylation of Tyk2 was induced in Aβ-treated neurons (Fig. 5B), suggesting that Tyk2 contributes to Aβ-induced STAT3 activation. We then examined the role of Tyk2 in Aβ-induced neuronal apoptosis using cortical neurons prepared from tyk2−/− mice. Interestingly, whereas phosphorylation of STAT3 at Tyr705 was induced in Aβ-treated wild-type neurons, the increase was abolished in tyk2−/− neurons (Fig. 5C,D). Remarkably, the Aβ-triggered cleavage of caspase-3 and cell death was both attenuated in tyk2−/− neurons (Fig. 5C,E,F). Our observations collectively reveal that Tyk2 mediates STAT3 activation in Aβ-treated cells and that Tyk2–STAT3 signaling pathway is required for Aβ-induced neuronal apoptosis.
Tyrosine phosphorylation of STAT3 was detected in the postmortem brains of AD patients
Given our observations on the increased STAT3 Tyr705 phosphorylation in brains of APP/PS1 mice and the induction of STAT3 activation by Aβ, we were interested to determine whether Tyr705 phosphorylation of STAT3 is also observed in the brains of AD patients. To address this possibility, postmortem hippocampus brain slices from human control and AD patients were stained by antibody against p-Tyr705-STAT3. Similar to our observation in APP/PS1 mice, whereas low p-Tyr-STAT3 reactivity was detected in the postmortem control human brain sections (∼10% of the cells were positive for p-Tyr-STAT3 staining), phosphorylation of STAT3 at Tyr705 was clearly observed in the brains of AD patients (∼31% of the cells) (Fig. 6A). In addition, we found that the majority of the Tyr705-phosphorylated STAT3 colocalized with NeuN-positive neurons (∼70%) when compared with GFAP-positive glial cells (∼7%) (Fig. 6B). These findings are consistent with our observation on APP/PS1 transgenic mice, revealing that elevated Tyr705 phosphorylation and hence activation of STAT3 occurs predominantly in neurons in postmortem brains of AD patients.
Figure 6.
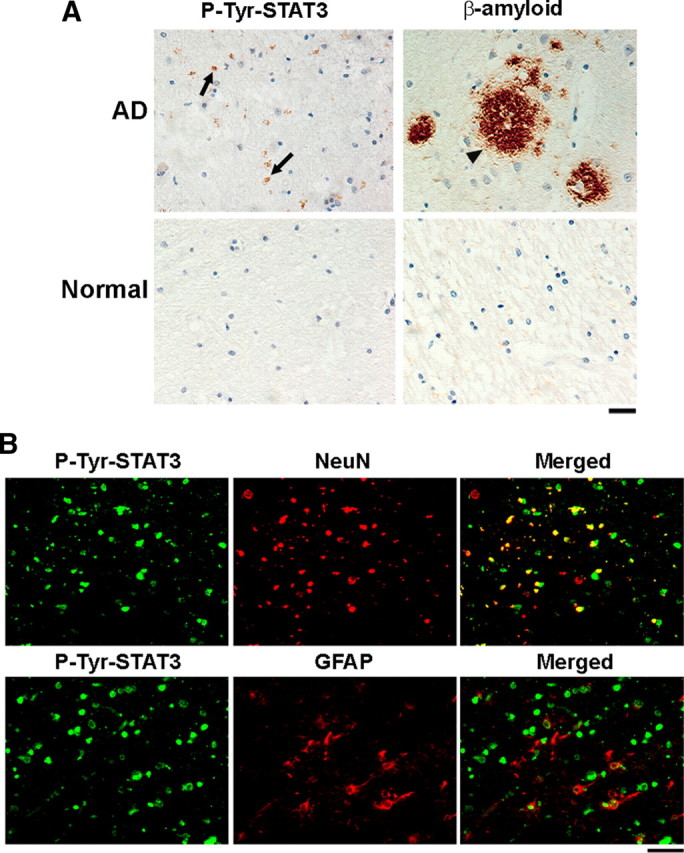
Tyrosine phosphorylation of STAT3 is increased in the hippocampus of Alzheimer's disease patients. A, Paraffin sections of postmortem brains from AD patients were immunostained with Aβ and p-Tyr-STAT3 antibodies. Cell nuclei were stained with hematoxylin. The arrows indicated the p-Tyr STAT3-positive cells, and the arrowhead indicated the β-amyloid deposit (n = 5 for the normal and n = 7 for the AD patient group). Scale bar, 50 μm. B, Paraffin sections of postmortem brains from AD patients were costained with p-Tyr-STAT3 antibody and NeuN or GFAP antibodies. Scale bar: left panels, 50 μm.
Discussion
The sequential events leading to the demise of neurons in AD have been extensively studied, yet the precise cause of neuronal death in AD is not well understood. Explicating the signaling mechanisms that underlie neuronal apoptosis during AD is therefore critical for unraveling the pathophysiology of the disease. In this study, we found that STAT3 signaling is activated in neurons of AD mutant mice or in mice injected with Aβ. The effect is specific for STAT3 because tyrosine phosphorylation and hence transcription activity of STAT1 is not elevated in these mouse brain tissues (Fig. 1). More importantly, similar activation of STAT3 is observed in postmortem brain of AD patients. Furthermore, inhibition of STAT3 activity or absence of Tyk2 expression markedly attenuates Aβ-induced neuronal apoptosis. Collectively, these findings suggest that Tyk2/STAT3-mediated signaling is involved in Aβ-induced neuronal apoptosis and raise the possibility that STAT3 is an important player in the progression of AD.
It has been well established that STAT3 regulates various cellular processes, including cell proliferation, differentiation, and survival. STAT3 has been found to be essential for embryonic development, since deletion of the STAT3 gene leads to embryonic lethality (Akira, 2000). However, STAT3 activation has been reported in various pathological events that lead to cell death, such as ischemic injury or in degenerating retina (Suzuki et al., 2005; Samardzija et al., 2006). How activation of STAT3 might lead to neuronal death in AD brain remains to be elucidated. Since Aβ results in a robust activation of STAT3 in cultured cortical neurons, stimulation of the target receptor on neurons by Aβ can directly trigger the STAT3-mediated signaling pathway. It is noteworthy that inflammation has been suggested to be one of the important pathways that underlies neuronal death during the progression of AD. In addition to enhanced microgliosis and astrogliosis, an increase in inflammation and cytokine release has been observed in AD brains. In particular, inflammatory cytokines, including tumor necrosis factor-α and interleukins, have been reported to be elevated in the brain and plasma of AD patients (Solfrizzi et al., 2006) and are important for AD progression (Schwab and McGeer, 2008; Ricardo et al., 2009). Indeed, cytokines released from glia have been suggested to cause neuronal death, although the mechanisms remain unclear (Mrak and Griffin, 2005; Farfara et al., 2008). We therefore speculate that STAT3 activation in neurons is triggered by the activation of cytokine receptors on neurons in response to the increased release of cytokine(s) from surrounding activated glial cells. In this context, it is noteworthy that Tyk2 has been linked to the biological response to type I interferons (Rani et al., 2007). Together with the observed activation of Tyk2–STAT3 signaling in Aβ-challenged cells, it is plausible that Tyk2–STAT3 signaling is triggered by the enhanced release of cytokines in AD brains, thereby contributing to neuronal death.
Upon phosphorylation at Tyr705, STAT3 dimerizes and translocates to the nucleus to enhance gene transcription through binding to the promoters of its candidate target genes. Since STAT3 activation is observed in apoptotic neurons, and inhibition of STAT3 function protects against apoptosis, it is likely that STAT3 contributes to the transcription of proapoptotic genes in response to Aβ. A number of target genes containing the STAT3 response element have been identified, including iNOS and BACE1 (Wen et al., 2008). The blockade of STAT3 dimerization inhibits the upregulation of iNOS transcript induced by Aβ, further supporting the notion that STAT3 transcriptional activity is important for the Aβ-induced gene transcription. It is noteworthy that Aβ could stimulate NO production in astrocytes and microglia through induction of iNOS expression (Akama et al., 1998; Hu et al., 1999). Activation of STAT3 may therefore mediate the increase in NO release in neurons of AD brain, which has been suggested to scavenge O2− and concurrently increases the cytotoxic species ONOO−, which mediates neuronal damage in AD brain (Chabrier et al., 1999).
In addition to the tyrosine phosphorylation at Tyr705, maximal transcriptional activity of STAT3 depends on the phosphorylation at Ser727 (Wen et al., 1995). It has recently been reported that serine phosphorylation of STAT3 and its transcriptional activation is required for the upregulation of BACE1 transcription during amyloidogenic processing of APP (Wen et al., 2008). However, downregulation of STAT3 tyrosine phosphorylation is observed in cultured hippocampal neurons after prolonged treatment with Aβ or in hippocampi of aged AD mouse models (>28 months) (Chiba et al., 2009). We speculate that STAT3 phosphorylation and its transcriptional activity are coordinately regulated during the progression of AD and that a precise temporal and spatial regulation of STAT3 signaling is critical in various pathological stages of the disease.
In conclusion, we have identified Tyk2–STAT3 activation as a new pathway for Aβ-dependent neuronal death in models of AD. Moreover, we show that iNOS is a potential downstream target of STAT3-mediated signaling in Aβ-challenged neurons. Further studies to elucidate the involvement of cytokine-mediated toxicity in AD pathophysiology and progression of the disease will provide important insights on the development of therapeutic agents against AD.
Footnotes
This study was supported in part by Research Grants Council of Hong Kong Grants HKUST6421/05M, 6444/06M, 661007, and HKUST 1/06C; Area of Excellence Scheme of the University Grants Committee Grant AoE/B-15/01; and the Hong Kong Jockey Club. N.Y.I. and K.-O.L. were recipients of the Croucher Foundation Senior Research Fellow and Research Fellow, respectively. We are grateful to Profs. Yung Hou Wong and Robert Qi for their expert advice. We thank Drs. Yu Pong Ng and Yi Xiong, Busma Butt, Winnie Chien, Venus Lee, Li Wang, Annie Li, Yuming Zhou, William Chau, Kwok-Wang Hung, and Tao Ye for their excellent technical assistance. We also thank Dr. Zelda Cheung for critical reading of this manuscript and members of the Ip Laboratory for many helpful discussions.
References
- Akama KT, Albanese C, Pestell RG, Van Eldik LJ. Amyloid β-peptide stimulates nitric oxide production in astrocytes through an NFκB-dependent mechanism. Proc Natl Acad Sci U S A. 1998;95:5795–5800. doi: 10.1073/pnas.95.10.5795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akira S. Roles of STAT3 defined by tissue-specific gene targeting. Oncogene. 2000;19:2607–2611. doi: 10.1038/sj.onc.1203478. [DOI] [PubMed] [Google Scholar]
- Arendash GW, King DL, Gordon MN, Morgan D, Hatcher JM, Hope CE, Diamond DM. Progressive, age-related behavioral impairments in transgenic mice carrying both mutant amyloid precursor protein and presenilin-1 transgenes. Brain Res. 2001;891:42–53. doi: 10.1016/s0006-8993(00)03186-3. [DOI] [PubMed] [Google Scholar]
- Borchelt DR, Davis J, Fischer M, Lee MK, Slunt HH, Ratovitsky T, Regard J, Copeland NG, Jenkins NA, Sisodia SS, Price DL. A vector for expressing foreign genes in the brains and hearts of transgenic mice. Genet Anal. 1996;13:159–163. doi: 10.1016/s1050-3862(96)00167-2. [DOI] [PubMed] [Google Scholar]
- Buxbaum JD, Gandy SE, Cicchetti P, Ehrlich ME, Czernik AJ, Fracasso RP, Ramabhadran TV, Unterbeck AJ, Greengard P. Processing of Alzheimer β/A4 amyloid precursor protein: modulation by agents that regulate protein phosphorylation. Proc Natl Acad Sci U S A. 1990;87:6003–6006. doi: 10.1073/pnas.87.15.6003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chabrier PE, Demerlé-Pallardy C, Auguet M. Nitric oxide synthases: targets for therapeutic strategies in neurological diseases. Cell Mol Life Sci. 1999;55:1029–1035. doi: 10.1007/s000180050353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiba T, Yamada M, Sasabe J, Terashita K, Shimoda M, Matsuoka M, Aiso S. Amyloid-beta causes memory impairment by disturbing the JAK2/STAT3 axis in hippocampal neurons. Mol Psychiatry. 2009;14:206–222. doi: 10.1038/mp.2008.105. [DOI] [PubMed] [Google Scholar]
- De Strooper B, Annaert W. Proteolytic processing and cell biological functions of the amyloid precursor protein. J Cell Sci. 2000;113:1857–1870. doi: 10.1242/jcs.113.11.1857. [DOI] [PubMed] [Google Scholar]
- Duyckaerts C, Potier MC, Delatour B. Alzheimer disease models and human neuropathology: similarities and differences. Acta Neuropathol. 2008;115:5–38. doi: 10.1007/s00401-007-0312-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eriksen JL, Janus CG. Plaques, tangles, and memory loss in mouse models of neurodegeneration. Behav Genet. 2007;37:79–100. doi: 10.1007/s10519-006-9118-z. [DOI] [PubMed] [Google Scholar]
- Estus S, Tucker HM, van Rooyen C, Wright S, Brigham EF, Wogulis M, Rydel RE. Aggregated amyloid-β protein induces cortical neuronal apoptosis and concomitant “apoptotic” pattern of gene induction. J Neurosci. 1997;17:7736–7745. doi: 10.1523/JNEUROSCI.17-20-07736.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farfara D, Lifshitz V, Frenkel D. Neuroprotective and neurotoxic properties of glial cells in the pathogenesis of Alzheimer's disease. J Cell Mol Med. 2008;12:762–780. doi: 10.1111/j.1582-4934.2008.00314.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrer I, Gomez-Isla T, Puig B, Freixes M, Ribé E, Dalfó E, Avila J. Current advances on different kinases involved in tau phosphorylation, and implications in Alzheimer's disease and tauopathies. Curr Alzheimer Res. 2005;2:3–18. doi: 10.2174/1567205052772713. [DOI] [PubMed] [Google Scholar]
- Fu AK, Fu WY, Ng AK, Chien WW, Ng YP, Wang JH, Ip NY. Cyclin-dependent kinase 5 phosphorylates signal transducer and activator of transcription 3 and regulates its transcriptional activity. Proc Natl Acad Sci U S A. 2004;101:6728–6733. doi: 10.1073/pnas.0307606100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Alloza M, Robbins EM, Zhang-Nunes SX, Purcell SM, Betensky RA, Raju S, Prada C, Greenberg SM, Bacskai BJ, Frosch MP. Characterization of amyloid deposition in the APPswe/PS1dE9 mouse model of Alzheimer disease. Neurobiol Dis. 2006;24:516–524. doi: 10.1016/j.nbd.2006.08.017. [DOI] [PubMed] [Google Scholar]
- Grant P, Pant HC. Topographic regulation of kinase activity in Alzheimer's disease brains. J Alzheimers Dis. 2002;4:269–281. doi: 10.3233/jad-2002-4402. [DOI] [PubMed] [Google Scholar]
- Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science. 2002;297:353–356. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- Hardy J, Duff K, Hardy KG, Perez-Tur J, Hutton M. Genetic dissection of Alzheimer's disease and related dementias: amyloid and its relationship to tau. Nat Neurosci. 1998;1:355–358. doi: 10.1038/1565. [DOI] [PubMed] [Google Scholar]
- Hooper C, Killick R, Lovestone S. The GSK3 hypothesis of Alzheimer's disease. J Neurochem. 2008;104:1433–1439. doi: 10.1111/j.1471-4159.2007.05194.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu S, Ali H, Sheng WS, Ehrlich LC, Peterson PK, Chao CC. Gp-41-mediated astrocyte inducible nitric oxide synthase mRNA expression: involvement of interleukin-1β production by microglia. J Neurosci. 1999;19:6468–6474. doi: 10.1523/JNEUROSCI.19-15-06468.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee MK, Borchelt DR, Kim G, Thinakaran G, Slunt HH, Ratovitski T, Martin LJ, Kittur A, Gandy S, Levey AI, Jenkins N, Copeland N, Price DL, Sisodia SS. Hyperaccumulation of FAD-linked presenilin 1 variants in vivo. Nat Med. 1997;3:756–760. doi: 10.1038/nm0797-756. [DOI] [PubMed] [Google Scholar]
- Levy DE, Darnell JE., Jr Stats: transcriptional control and biological impact. Nat Rev Mol Cell Biol. 2002;3:651–662. doi: 10.1038/nrm909. [DOI] [PubMed] [Google Scholar]
- Mrak RE, Griffin WS. Glia and their cytokines in progression of neurodegeneration. Neurobiol Aging. 2005;26:349–354. doi: 10.1016/j.neurobiolaging.2004.05.010. [DOI] [PubMed] [Google Scholar]
- Otth C, Mendoza-Naranjo A, Mujica L, Zambrano A, Concha II, Maccioni RB. Modulation of the JNK and p38 pathways by cdk5 protein kinase in a transgenic mouse model of Alzheimer's disease. Neuroreport. 2003;14:2403–2409. doi: 10.1097/00001756-200312190-00023. [DOI] [PubMed] [Google Scholar]
- Rani MR, Pandalai S, Shrock J, Almasan A, Ransohoff RM. Requirement of catalytically active Tyk2 and accessory signals for the induction of TRAIL mRNA by IFN-beta. J Interferon Cytokine Res. 2007;27:767–779. doi: 10.1089/jir.2007.0005. [DOI] [PubMed] [Google Scholar]
- Ricardo BM, Leonel ER, Jorge AF, Rodrigo OK. The role of neuroimmunomodulation in Alzheimer's disease. Ann N Y Acad Sci. 2009;1153:240–246. doi: 10.1111/j.1749-6632.2008.03972.x. [DOI] [PubMed] [Google Scholar]
- Ryder J, Su Y, Ni B. Akt/GSK3β serine/threonine kinases: evidence for a signalling pathway mediated by familial Alzheimer's disease mutations. Cell Signal. 2004;16:187–200. doi: 10.1016/j.cellsig.2003.07.004. [DOI] [PubMed] [Google Scholar]
- Samardzija M, Wenzel A, Aufenberg S, Thiersch M, Remé C, Grimm C. Differential role of Jak-STAT signaling in retinal degenerations. FASEB J. 2006;20:2411–2413. doi: 10.1096/fj.06-5895fje. [DOI] [PubMed] [Google Scholar]
- Savage MJ, Lin YG, Ciallella JR, Flood DG, Scott RW. Activation of c-Jun N-terminal kinase and p38 in an Alzheimer's disease model is associated with amyloid deposition. J Neurosci. 2002;22:3376–3385. doi: 10.1523/JNEUROSCI.22-09-03376.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwab C, McGeer PL. Inflammatory aspects of Alzheimer disease and other neurodegenerative disorders. J Alzheimers Dis. 2008;13:359–369. doi: 10.3233/jad-2008-13402. [DOI] [PubMed] [Google Scholar]
- Shimoda K, Kato K, Aoki K, Matsuda T, Miyamoto A, Shibamori M, Yamashita M, Numata A, Takase K, Kobayashi S, Shibata S, Asano Y, Gondo H, Sekiguchi K, Nakayama K, Nakayama T, Okamura T, Okamura S, Niho Y, Nakayama K. Tyk2 plays a restricted role in IFN alpha signaling, although it is required for IL-12-mediated T cell function. Immunity. 2000;13:561–571. doi: 10.1016/s1074-7613(00)00055-8. [DOI] [PubMed] [Google Scholar]
- Solfrizzi V, D'Introno A, Colacicco AM, Capurso C, Todarello O, Pellicani V, Capurso SA, Pietrarossa G, Santamato V, Capurso A, Panza F. Circulating biomarkers of cognitive decline and dementia. Clin Chim Acta. 2006;364:91–112. doi: 10.1016/j.cca.2005.06.015. [DOI] [PubMed] [Google Scholar]
- Stéphan A, Laroche S, Davis S. Generation of aggregated β-amyloid in the rat hippocampus impairs synaptic transmission and plasticity and causes memory deficits. J Neurosci. 2001;21:5703–5714. doi: 10.1523/JNEUROSCI.21-15-05703.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki S, Yamashita T, Tanaka K, Hattori H, Sawamoto K, Okano H, Suzuki N. Activation of cytokine signaling through leukemia inhibitory factor receptor (LIFR)//gp130 attenuates ischemic brain injury in rats. J Cereb Blood Flow Metab. 2005;25:685–693. doi: 10.1038/sj.jcbfm.9600061. [DOI] [PubMed] [Google Scholar]
- Ueda K, Fukui Y, Kageyama H. Amyloid β protein-induced neuronal cell death: neurotoxic properties of aggregated amyloid β protein. Brain Res. 1994;639:240–244. doi: 10.1016/0006-8993(94)91736-1. [DOI] [PubMed] [Google Scholar]
- Vaisid T, Kosower NS, Elkind E, Barnoy S. Amyloid beta peptide toxicity in differentiated PC12 cells: calpain-calpastatin, caspase, and membrane damage. J Neurosci Res. 2008;86:2314–2325. doi: 10.1002/jnr.21670. [DOI] [PubMed] [Google Scholar]
- Wen Y, Yu WH, Maloney B, Bailey J, Ma J, Marié I, Maurin T, Wang L, Figueroa H, Herman M, Krishnamurthy P, Liu L, Planel E, Lau LF, Lahiri DK, Duff K. Transcriptional regulation of beta-secretase by p25/cdk5 leads to enhanced amyloidogenic processing. Neuron. 2008;57:680–690. doi: 10.1016/j.neuron.2008.02.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen Z, Zhong Z, Darnell JE., Jr Maximal activation of transcription by Stat1 and Stat3 requires both tyrosine and serine phosphorylation. Cell. 1995;82:241–250. doi: 10.1016/0092-8674(95)90311-9. [DOI] [PubMed] [Google Scholar]
- Wirths O, Multhaup G, Czech C, Blanchard V, Tremp G, Pradier L, Beyreuther K, Bayer TA. Reelin in plaques of β-amyloid precursor protein and presenilin-1 double-transgenic mice. Neurosci Lett. 2001;316:145–148. doi: 10.1016/s0304-3940(01)02399-0. [DOI] [PubMed] [Google Scholar]
- Xiang Z, Ho L, Valdellon J, Borchelt D, Kelley K, Spielman L, Aisen PS, Pasinetti GM. Cyclooxygenase (COX)-2 and cell cycle activity in a transgenic mouse model of Alzheimer's disease neuropathology. Neurobiol Aging. 2002;23:327–334. doi: 10.1016/s0197-4580(01)00282-2. [DOI] [PubMed] [Google Scholar]