

Abstract
c-Jun N-terminal kinases (JNKs) (comprising JNK1–3 isoforms) are members of the MAPK (mitogen-activated protein kinase) family, activated in response to various stimuli including growth factors and inflammatory cytokines. Their activation is facilitated by scaffold proteins, notably JNK-interacting protein-1 (JIP1). Originally considered to be mediators of neuronal degeneration in response to stress and injury, recent studies support a role of JNKs in early stages of neurite outgrowth, including adult axonal regeneration. However, the function of individual JNK isoforms, and their potential effector molecules, remained unknown. Here, we analyzed the role of JNK signaling during axonal regeneration from adult mouse dorsal root ganglion (DRG) neurons, combining pharmacological JNK inhibition and mice deficient for each JNK isoform and for JIP1. We demonstrate that neuritogenesis is delayed by lack of JNK2 and JNK3, but not JNK1. JNK signaling is further required for sustained neurite elongation, as pharmacological JNK inhibition resulted in massive neurite retraction. This function relies on JNK1 and JNK2. Neurite regeneration of jip1−/− DRG neurons is affected at both initiation and extension stages. Interestingly, activated JNKs (phospho-JNKs), as well as JIP1, are also present in the cytoplasm of sprouting or regenerating axons, suggesting a local action on cytoskeleton proteins. Indeed, we have shown that JNK1 and JNK2 regulate the phosphorylation state of microtubule-associated protein MAP1B, whose role in axonal regeneration was previously characterized. Moreover, lack of MAP1B prevents neurite retraction induced by JNK inhibition. Thus, signaling by individual JNKs is differentially implicated in the reorganization of the cytoskeleton, and neurite regeneration.
Introduction
Regeneration of adult neurons requires their intrinsic capacity to survive a traumatic or degenerative lesion and activation of a cell-autonomous program that will end in plastic changes in neuronal circuitry. Integration of environmental signals leads to morphological and functional changes, depending both on regulation of regeneration-related gene expression and reorganization of the cytoskeleton. The specific signaling pathways underlying neuronal regeneration are not yet fully elucidated.
c-Jun N-terminal kinases (JNKs) are members of a large group of serine/threonine-directed kinases known as mitogen-activated protein kinases (MAPKs) (Widmann et al., 1999; Davis, 2000). Mammalian JNKs are encoded by three distinct genes (jnk1, jnk2, and jnk3) giving rise to at least 10 different splice variants (Gupta et al., 1996). All variants share an epitope that needs to be dually phosphorylated for JNK activation (phospho-JNK). This is facilitated by scaffold proteins, notably the JNK-interacting proteins (JIP), that promote the sequential phosphorylation of cascade of MAP kinases (Morrison and Davis, 2003).
JNKs are best known as mediators of neuronal degeneration after stress and injury, but may be important also for neural development and survival (for review, see Waetzig et al., 2006). Recent studies support a role of JNK and JIP1 in the regulation of neurite outgrowth during development (Oliva et al., 2006; Dajas-Bailador et al., 2008). Moreover, JNKs are also implicated in axonal regeneration of adult DRG neurons, as their specific inhibition in vitro dramatically reduces neuritogenesis, without affecting neuronal survival (Kenney and Kocsis, 1998; Lindwall et al., 2004). In response to peripheral nerve injury, JNKs are rapidly activated and retrogradely transported to the nuclei where they phosphorylate and, thereby, activate transcription factors such as c-Jun and activating transcription factor-3 (ATF3) (Lindwall et al., 2004; Cavalli et al., 2005). There is also a growing evidence for a cytosolic function of JNKs during neuronal development. In fact, JNKs may influence cytoskeletal reorganization via the phosphorylation of proteins regulating microtubule stability, including doublecortin (DCX) (Gdalyahu et al., 2004), stathmin family protein (SCG10) (Tararuk et al., 2006), and microtubule-associated proteins, MAP2 and MAP1B (Chang et al., 2003).
Whereas JNK1 and JNK2 are expressed in a large variety of tissues, JNK3 expression is restricted to the brain, heart, and testis (Gupta et al., 1996; Kuan et al., 1999), suggesting that the different JNK isoforms may be involved in distinct cellular processes. This prompted us to investigate the regulation and specific function of each JNK isoform during axonal regeneration and plasticity in the adult. Thus, using pharmacological and genetic approaches (use of JNK- and JIP1-deficient mice) on the paradigm of DRG neuron regeneration, we were able to attribute specific roles to individual JNK isoforms in either initiation or extension of regenerating neurites. We have further shown that JNK isoforms are important regulators of the phosphorylation state of MAP1B, a cytoskeletal protein for which we previously demonstrated a role in guidance, branching, and retraction of regenerating axons (Bouquet et al., 2004, 2007; Stroissnigg et al., 2007).
Materials and Methods
Animals.
Genetic inactivation of JNK1-, JNK2-, JNK3-, JIP1-, and MAP1B-deficient mice (male and female) has been described in detail previously (Yang et al., 1997, 1998; Dong et al., 1998; Meixner et al., 2000; Whitmarsh et al., 2001). Adult (>2 months) homozygous deficient mice and their control wild-type littermates were used after systematic genotyping. Genotyping of individual mouse was performed by isolating DNA from the tail, and PCR amplification was performed using the following primers: jnk1 forward, 5′-CGCCAGTCCAAAATCAAGAATC-3′; 5′-GCCATTCTGGTAGAGGAAGTTTCTC-3′; jnk1 reverse, 5′-CCAGCTCATTCCTCCACTCATG-3′; jnk2 forward, 5′-GGAGCCCGATAGTATCGAGTTACC-3′; 5′-GTTAGACAATCCCAGAGGTTGTGTG-3′; jnk2 reverse, 5′-CCAGCTCATTCCTCCACTCATG-3′; jnk3 forward, 5′-CCTGCTTCTCAGAAACACCCTTC-3′; 5′-CGTAATCTTGTCACAGAAATCCCATAC-3′; jnk3 reverse, 5′-CTCCAGACTGCCTTGGGAAAA-3′; jip1 forward, 5′-CGCGGTCTCAGGTGAGCAA-3′; 5′-CTGACTAGGCCTGTAAGAC-3′; jip1 reverse, 5′-CTCCAGACTGCCTTGGGAAAA-3′; map1b forward, 5′-CTGGATTCATCGACTGTGGCCG-3′; 5′-ATAGGTGCTGTGC CTCGCCTGA-3′; map1B reverse, 5′-TTAGGAAGGCAGAGGAGCAGCC-3′. PCR products were analyzed by agarose gel electrophoresis. All experimental procedures on animals were in accordance with the European Community directive (86/609/EEC; authorization number 91-78 to F. Nothias).
Surgery.
Animals were anesthetized by intraperitoneal injection of a solution containing 7.4 mg/kg xylazine (Rompun, 2%; Bayer) and 146 mg/kg ketamine (Imalgène 500; Mérieux). For spinal cord analysis, right sciatic nerves were cut at midthigh levels and reapposed by a single 9-0 suture through the epineurium to allow nerve regeneration. The left (unoperated) spinal cords were used as normal controls, in addition to intact animals. After a survival time of 1 week after axotomy, animals were deeply anesthetized with pentobarbitol and perfused transcardially with saline (0.9% NaCl at 37°C) supplemented with heparin (50 U/ml; sanofi-aventis), followed by 4% paraformaldehyde (200 ml) in 0.1 m phosphate buffer (PB), pH 7.4. Spinal cords were dissected and cryoprotected in 30% sucrose in 0.1 m PB containing NaCl (0.9%; PB), and coronal sections (30 μm) were performed on a cryostat.
To analyze peripheral nerve regeneration in vivo, right sciatic nerves were crushed at midthigh levels with forceps for 10 s. After 3 d, animals were reanesthetized, and 3 μl of anthra[1,9-cd]pyrazol-6(2H)-one (SP600125) (50 μm in 5% DMSO/PBS; n = 2) or 5% DMSO/PBS (for control animals; n = 2) was applied onto the lesion site in a cuff. The injury site was closed, and animals were perfused as described above, 48 h after SP600125 or DMSO application. Nerves were dissected and cryoprotected in 15% sucrose in 0.1 m PB containing NaCl (0.9%; PB), and longitudinal sections (16 μm) were performed on a cryostat.
Culture of dissociated adult DRG neurons.
DRGs were dissected in F-12 medium and cut into small pieces before enzymatic dissociation by collagenase (90 min at 37°C; 4000 U/ml; Sigma-Aldrich), followed by treatments with trypsin/EDTA (Invitrogen) with DNase-1 (50 μg/ml; Sigma-Aldrich) for 10 min at 37°C, and by a final trituration using a narrowed Pasteur pipette. After multiple washes with F-12 medium, cells were resuspended in F-12 medium supplemented with N3 (Bottenstein and Sato, 1979) and penicillin/streptomycin (all reagents from Invitrogen), plated at a density of 100 cells/cm2 on precoated glass coverslips, and placed in a 37°C, 5% CO2 incubator. Coverslips were precoated with poly-l-lysine (10 μg/ml; Sigma-Aldrich) overnight at 37°C, followed by laminin (10 μg/ml; at least 3 h at 37°C; Sigma-Aldrich). To analyze the effect of JNK inhibition on neurite initiation, SP600125 (20 μm; BIOMOL International) was added to culture medium 4 h after plating (i.e., before any neurites emerged), and cell cultures were maintained for 24 and 48 h before fixation. The effect of SP600125 on neurite elongation was determined after adding the inhibitor to the culture medium 36 h after plating and further cultured for another 12 h.
Primary antibodies.
Mouse anti-β3 tubulin (Tuj-1) was obtained from Covance; mouse anti-β-actin was obtained from Sigma-Aldrich; rat anti-L1 was obtained from Millipore Bioscience Research Reagents; rabbit anti-CGRP (calcitonin gene-related peptide) was from Peninsula; rabbit anti-detyrosinated tubulin (superglu) (Gundersen et al., 1984) was a gift from Dr. G. G. Gundersen (Columbia University, New York, NY); rabbit anti-total MAP1B (Fischer and Romano-Clarke, 1990) was provided by Dr. I. Fischer (Drexel University College of Medicine, Philadelphia, PA), and mouse anti-phosphorylated MAP1B (MAP1B-P) (Nothias et al., 1996) was homemade; rabbit polyclonal anti-total stathmin (anti-peptide C) (Koppel et al., 1990) and anti-phospho-Ser25 stathmin (Gavet et al., 1998) were generously given by Dr. A. Sobel (Institut du Fer à Moulin, Paris, France); anti-phosphorylated JNK (P-JNK) was obtained from R&D Systems; mouse anti-JIP1 and mouse anti-glyceraldehyde-3-phosphate dehydrogenase (GAPDH) were obtained from Santa Cruz Biotechnology; mouse anti-JNK1 was obtained from BD Pharmingen; and anti-JNK2 and anti-JNK3 were obtained from Cell Signaling Technology.
Immunostaining.
Cultured neurons were fixed with 4% paraformaldehyde/15% sucrose in PB, pH 7.4, for 20 min. For immunocytochemistry and histochemistry, specimens were permeabilized with 0.3% Triton X-100 in PB, and nonspecific binding sites were blocked with 10% normal goat serum (NGS) in PB, before incubation with primary antibodies diluted in 5% NGS in PB. After several rinses in 5% NGS in PB, coverslips were incubated with appropriate fluorochrome-labeled secondary antibodies (Jackson ImmunoResearch), rinsed again, and mounted in Mowiol.
Morphological analysis.
Spinal cord coronal sections were immunostained for P-JNK and MAP1B-P and analyzed under confocal microscopy (SP5; Leica). Cells were plated on coverslips and visualized by tubulin staining. Images were taken on an Axiovert 200 inverted microscope equipped with an AxioCamera (Zeiss) and analyzed using NIH ImageJ (PC version: http://rsbweb.nih.gov/ij/) or Image ProPlus software (Media Cybernetics) for the following parameters: percentage of neurons with neurites, total neuritic length per neuron, and longest neurite per neuron. For analysis of the percentage of neurite-bearing neurons, cells were cultured for 24 and 48 h before fixation; all neurons on a coverslip were analyzed, and only neurites longer than one soma diameter were taken into account. For each condition, three independent experiments were performed, and per individual experiment at least 250 neurons were analyzed. For neuritic length analysis, only neurites longer than one soma diameter were manually traced and measured on size-calibrated images using Image ProPlus (three independent experiments with >50 neurons analyzed per condition). For neurite length distribution analysis, neurons were classified in different categories depending on their total neuritic length (ranging from 500 to >3500 μm) or length of their longest neurite (ranging from 50 to >400 μm), and the results were expressed as percentage of neurons in each category. Statistical significance was determined using independent t test and one-way ANOVA, and the levels of significance are indicated as follows: ***p < 0.001, **p < 0.01, *p < 0.05. Compiled data are expressed as mean ± SEM.
Time-lapse video microscopy.
For time-lapse analysis, DRG neurons were plated on poly-l-lysine/laminin-coated coverslips. After 36 h, 10 mm HEPES, pH 7.4, was added to the medium, and cultures were placed in a chamber maintained at 37°C. Individual cells were imaged with a high-speed confocal microscope (resonant scanner SP5; 8000 Hz; Leica). Movies were composed of sequential 500 ms stills, taken at 2 or 4 min intervals over periods of 100 or 180 min. ImageJ software was used for quantitative evaluation of outgrowth and retraction velocities, by manually recording the distance traveled by the growth cones during 180 min, corresponding to the periods before and after application of SP600125. For each condition, three independent experiments were performed, and 10 neurites were analyzed per individual experiment.
Preparation of cell extracts and Western blotting.
Lysis of cultures of DRG neurons plated for 48 h was preceded by washing with ice-cold PB. For MAP1B analysis, cells were lysed in denaturing extraction buffer containing 100 mm Tris-HCl, pH 6.8, 4% SDS, 20% (v/v) glycerol, 12 mm EDTA, 0.2% bromophenol blue, 0.3% DTT, and protease inhibitors (tablets from Roche Diagnostics). Samples were sonicated (3 times 20 s) and heated at 95°C for 5 min before loading on a 4.5% SDS-polyacrylamide gel. For alkaline phosphatase (AP) treatment, N2A neuroblastoma cells were cultured on poly-l-lysine for 48 h in DMEM–10% FCS (Invitrogen), at 37°C and 5% CO2. To some cultures, SP600125 (20 μm) was added for 24 h. Cells were then rinsed twice with PBS and lysed in extraction buffer (see above), and samples were sonicated, centrifuged, and divided into two aliquots. One aliquot was treated for 30 min at 37°C with 10 U of calf alkaline phosphatase (New England Biolabs) before loading, and the other was mock-treated. For JNK immunoblot analysis, cultured dissociated DRG neurons were solubilized in denaturing buffer containing 1% SDS, 1% protease inhibitor mixture, and 1 mm NaVO4 (Sigma-Aldrich) and sonicated, and proteins were separated on 10% SDS-polyacrylamide gels. After transfer, nitrocellulose membranes were blocked with 5% nonfat dry milk and incubated with primary antibodies. After thorough washing, membranes were incubated with appropriate alkaline phosphatase-conjugated secondary antibodies followed by revelation of antigens by NBT (nitroblue tetrazolium)/BCIP (5-bromo-4-chloro-3-indolyl phosphate) staining, or with fluorochrome-labeled secondary antibodies followed by detection on an infrared imaging system (Odyssey; LI-COR).
Results
Phospho-JNK is upregulated in axons undergoing plastic changes in vivo
We performed a lesion of sciatic nerve, known to induce a structural reorganization of axons within the central projection area of DRG neurons (i.e., sprouting in dorsal horn lamina II, most pronounced ∼1 week after lesion in mice) (for details, see Soares et al., 2002). We previously showed that MAP1B-P increases within these fibers, and is well suited as marker for axonal sprouting and regeneration in the adult nervous system (Soares et al., 2002, 2007). In Figure 1A, fibers in lamina II ipsilateral to the sciatic nerve lesion display a strong increase in P-JNK immunostaining, whereas the contralateral dorsal horn is almost devoid of staining. Double immunostaining for MAP1B-P and P-JNK clearly shows that high levels of P-JNK are found in fibers that sprout in response to the peripheral lesion (Fig. 1B–D). These results demonstrate that, in vivo, DRG neurons, when entering the regenerative state in response to peripheral axotomy, exhibit a cytoplasmic increase of activated JNK.
Figure 1.
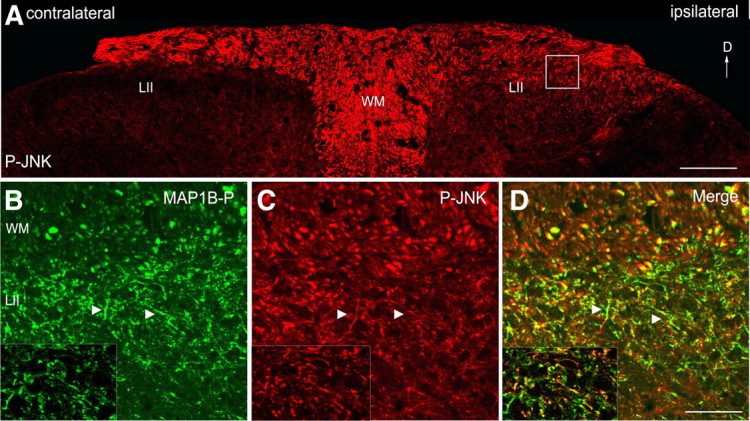
JNK activation in central primary sensory fibers undergoing sprouting in response to a peripheral lesion. Shown are confocal microscope images. A, P-JNK immunostaining (red) in dorsal horn of adult mouse spinal cord, 1 week after unilateral sciatic nerve transection. P-JNK is detected in fibers in lamina II ipsilateral to the lesion (right), compared with the contralateral side (left), devoid of P-JNK staining. B–D, Higher magnification of the area marked by a white square in A reveals colocalization of P-JNK (red) with phosphorylated MAP1B (green) within the same fibers (arrowheads). The insets show single optical plane images. D, Dorsal; LII, lamina II; WM, white matter. Scale bars: A, 100 μm; B–D, insets, 40 μm.
JNK activity is involved in both initiation and stabilization of regenerating neurons
Immunocytochemical analysis of primary cultures of adult DRG neurons, plated for 48 h, shows a distribution of P-JNK both in the nucleus and in the cytoplasm of soma and neurites of all DRG neuronal populations (Fig. 2A–D; supplemental Fig. S4A–D, available at www.jneurosci.org as supplemental material). Interestingly, P-JNK was present along distal neurite shafts and at growth cones (Fig. 2B), suggesting a local role in axonal regeneration. To test this hypothesis, we used an ATP-competitive inhibitor of all JNK isoforms, SP600125 (Bennett et al., 2001). Treatment of neurons with SP600125 4 h after plating, before neurite initiation, severely affected neuritogenesis since only 3.0 ± 0.7 and 24.9 ± 9.4% of SP600125-treated neurons had formed neurites after a culture period of 24 and 48 h, respectively, compared with untreated neurons set to 100% (Fig. 2E).
Figure 2.
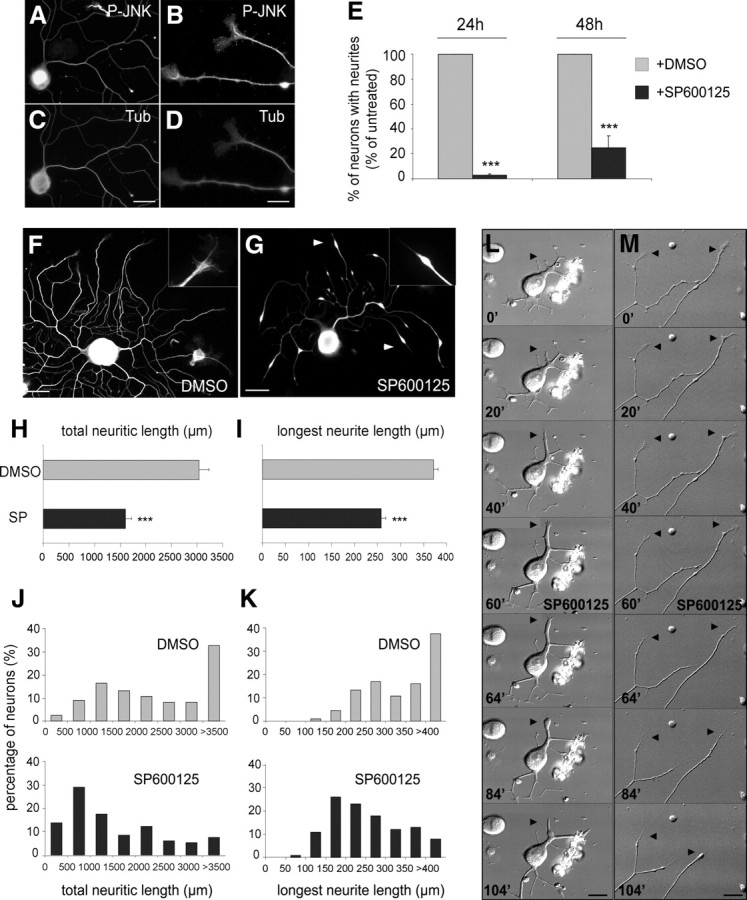
JNK activation is required for both neuritogenesis and sustained extension of regenerating neurites. A–D, P-JNK and tubulin immunolabeling of DRG neurons cultured for 48 h. P-JNK is found in soma (including the nucleus), axon shafts, and growth cones. E, Quantification of neurite-bearing neurons cultured for 24 and 48 h, without (gray bars) or with chronic SP600125 treatment (black bars), reveals that neurite formation is blocked by JNK inhibition (n ≥ 446). F, G, Neurons treated with or without SP600125 and visualized by tubulin immunostaining. JNK inhibition induces loss of growth cones and leads to retraction bulb formation and trailing remnants along neurites (arrowheads); insets show higher magnifications of a typical growth cone under control conditions and a drug treatment-induced retraction bulb. H, I, Quantitative analysis reveals that JNK inhibition decreases total neuritic and longest neurite lengths of regenerating neurons (n ≥ 55). J, K, Distribution diagrams showing the percentage of neurons exhibiting a given total neuritic length and length of the longest neurite. Whereas the majority of neurons cultured under control conditions displays a total neurite length >3500 μm and a longest neurite >400 μm, JNK inhibition induces a shift of both values toward neurons with shorter neurites. L, M, Frames of time-lapse recordings illustrating the typical response of neurites to JNK inhibition: Before SP600125 addition (t = 60 min), growth cones (arrowheads) are highly dynamic and neurites elongate steadily. Shortly after drug addition, growth cones collapse, and neurites stop extending and start to retract. Scale bars: A, C, F, G, L, M, 50 μm; B, D, 15 μm. ***p < 0.001. Error bars indicate SEM.
Given the presence of P-JNK in neurites during the whole culture period, we further investigated whether JNK activation is also required for sustained neurite extension. This was addressed by addition of JNK inhibitor for 12 h, at a time point when the majority of neurons had extended neurites (36 h after plating). JNK inhibition induced the formation of retraction bulbs and trailing remnants along neurites (Fig. 2F,G). A similar effect of SP600125 treatment was also observed in vivo on regenerating sciatic nerves after a lesion (supplemental Fig. S1, available at www.jneurosci.org as supplemental material). Whereas peripheral fibers grow linearly in condition control (supplemental Fig. S1A–D,I, available at www.jneurosci.org as supplemental material), the SP600125 addition severely disrupted and disorganized the regeneration profile (supplemental Fig. S1E–H, available at www.jneurosci.org as supplemental material) and caused the occurrence of retraction bulbs (supplemental Fig. S1J, available at www.jneurosci.org as supplemental material). This reproducible effect of SP600125 on the morphology of regenerating neurites, in vitro, was correlated with a reduction in total neurite length per neuron by 47.3% (1604.1 ± 118.6 μm for SP600125-treated vs 3041.2 ± 190.4 μm for untreated neurons) (Fig. 2H) and with a 30.6% shortening of the length of the longest neurite (258.2 ± 9.4 vs 372.1 ± 12.6 μm) (Fig. 2I). To determine whether JNK inhibition affected all growing neurites independently of their initial length, we established the distribution of neurons based on each parameter (Fig. 2J,K). Whereas a majority of untreated neurons (60%) exhibited a total neuritic length >2000 μm, this distribution was strongly shifted toward shorter neurites after SP600125 treatment: only 30% of neurons had a total neurite length >2000 μm (Fig. 2J). A similar shift was noted in the distribution of the length of the longest neurite: In control cultures, 64% of neurons show a longest neurite >300 μm versus 29% after drug treatment (Fig. 2K).
To assess the dynamics of these events, DRG neuron cultures were analyzed using time-lapse videomicroscopy (Fig. 2L,M). Image sequences, representing a neuron during the initial stages of neurite elongation (Fig. 2L) and a neuron having already extended longer neurites (Fig. 2M), are shown (see also supplemental Video 1, available at www.jneurosci.org as supplemental material). In both cases, neurites elongated steadily and possessed typical fan-shaped growth cones with filopodia and lamellipodia exploring the environment. Addition of SP600125 (at t = 60 min) caused a rapid collapse of the growth cone (within 4–8 min), followed by neurite retraction (within 12–16 min) that could be reversed by removal of the inhibitor from the medium (data not shown). JNK inhibition-induced neurite retraction was observed on both short and long neurites, corroborating the analysis of neurite length distribution (Fig. 2J,K). Moreover, the rearward movement of neurites that followed the growth cone collapse was consistent with the neurite morphology observed on fixed preparations (Fig. 2G). Together, our analysis demonstrates that JNK activity is required for both neuritogenesis and sustained extension of regenerating neurites.
Differential involvement of individual JNK isoforms in neurite initiation and extension
Since the pharmacological approach does not discriminate between individual JNK isoforms for which distinct, but also partly overlapping, functions had been described (Bogoyevitch, 2006), we examined the expression and potential contribution of each isoform to DRG neurite regeneration. Using antibodies specific for each isoform, we found that JNK1, JNK2, and JNK3 were expressed in regenerating DRG neurons (Fig. 3A, Western blot). The specificity of each antibody was demonstrated by the absence of reactive protein bands when using DRG neuron extracts from mice deficient in the corresponding JNK isoform.
Figure 3.
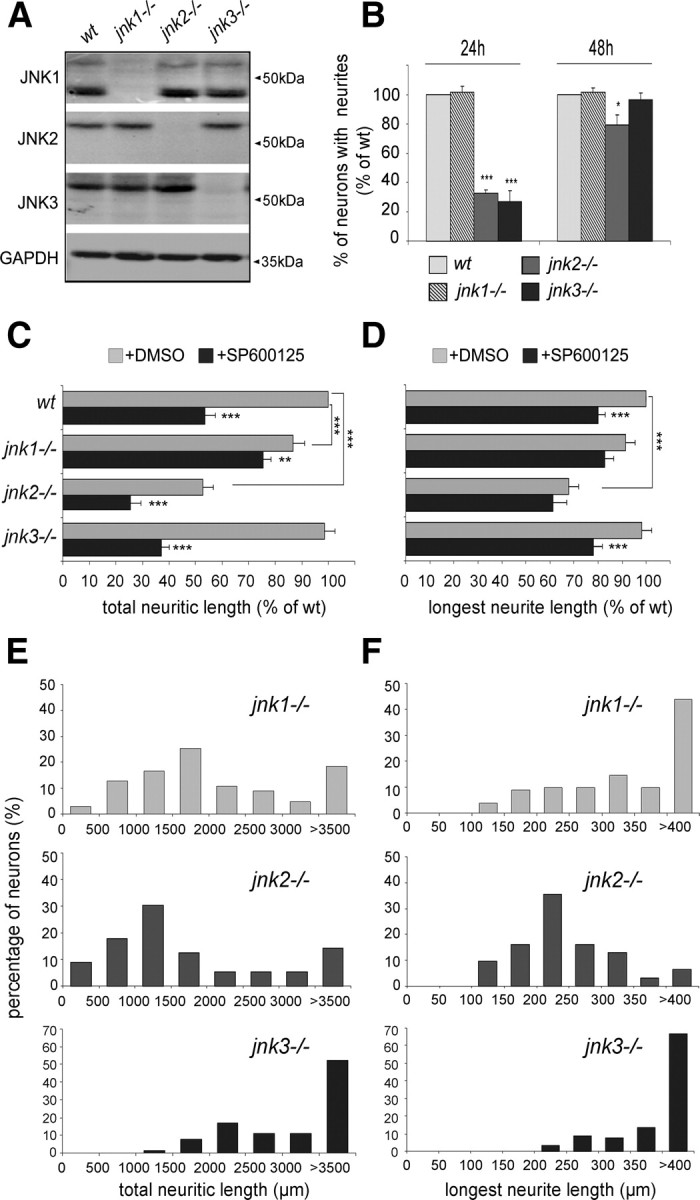
Differential involvement of JNK isoforms in neurite initiation and elongation. A, Western blots of protein extracts from wild-type and JNK-deficient mice DRG neurons, cultured 48 h, reacted with antibodies specific for individual JNK proteins. In regenerating wild-type neurons, all isoforms are expressed. B, Quantification of neurons bearing neurites after 24 and 48 h of culture: At 24 h, jnk2−/− and jnk3−/− neurons exhibit a strong decrease in the percentage of neurons with neurites compared with wild-type neurons. At 48 h, only jnk2−/− neurons still display a slight but significant difference. Neurite initiation is not affected in regenerating jnk1−/− neurons (n ≥ 297). C, D, Quantification of total neurite length and length of the longest neurite for wild-type versus JNK-deficient neurons without (gray bars) or with SP600125 treatment (black bars). Total neurite lengths for jnk1−/− and jnk2−/−, but not jnk3−/− neurons, are decreased compared with wild-type neurons; the average length of the longest neurite is significantly affected only by lack of JNK2. SP600125 treatment induces an additional reduction in total neurite length for neurons derived from all three jnk-ko mice, but induces a specific decrease in length of the longest neurite only in jnk3−/− neurons (n ≥ 50). E, F, The effect on neurite length of JNK1 and JNK2, but not JNK3 deficiency, is reflected in a leftward shift in the distribution diagrams for total neurite length; values for the length of the longest neurite per neuron display a leftward shift only for jnk2−/− neurons; those for jnk1−/− and jnk3−/− neurons are similar to wild type. ***p < 0.001; **p < 0.01; *p < 0.05. Error bars indicate SEM.
We then examined the implication of individual JNK isoforms in neuritogenesis by culturing neurons derived from jnk1−/−, jnk2−/−, and jnk3−/− mice for 24 and 48 h (Fig. 3B). At 24 h, neurite initiation of jnk1−/− neurons was similar to the one observed in wild-type neurons, whereas only 32.8 ± 2.1% of jnk2−/− and 27.1 ± 7.4% of jnk3−/− neurons had formed neurites compared with wild type. However, after 48 h in culture, no difference in the percentage of neurons bearing neurites were observed between jnk3−/− and wild-type neurons, whereas neuritogenesis of jnk2−/− neurons was partially restored but still significantly affected (79.4% of neurons with neurites). These results indicate that JNK2 and JNK3, but not JNK1, are involved in neurite initiation. Furthermore, the low percentage of jnk2−/− and jnk3−/− neurons bearing neurites at early stages was mainly attributable to a delay in neurite initiation.
To investigate the role of JNK isoforms in sustaining the extension of regenerating neurites over time, total neuritic length and length of the longest neurite per neuron were determined after culturing wild-type and JNK-deficient neurons for 48 h (Fig. 3C,D). Neurites of jnk2−/−, and to a lesser degree of jnk1−/−, neurons were significantly shorter than those of wild-type neurons (62.8 ± 6.6 and 86.6 ± 4.8% of wild-type total neuritic length, respectively), whereas neurite outgrowth from jnk3−/− neurons was not affected (Fig. 3C). A difference in the average length of the longest neurite was, however, observed only for jnk2−/− neurons (Fig. 3D). Accordingly, the distribution of total neuritic length, and length of the longest neurite, exhibited a shift toward smaller values for both jnk1−/− and jnk2−/−, but not jnk3−/− neurons (Fig. 3E,F). To understand the basis for this decrease in neurite length, we monitored the neurite outgrowth speed of DRG neurons derived from the different JNK-deficient mice. Compared with wild-type neurites that advance at 0.76 ± 0.09 μm/min, the growth speed of jnk1−/−, and to an even higher degree of jnk2−/−, neurites was significantly lower (0.57 ± 0.02 and 0.38 ± 0.02 μm/min, respectively) (Fig. 4A,B). Slower growth rates are thus responsible for jnk1−/− and jnk2−/− neurites being shorter than their wild-type or jnk3−/− counterparts noted at the end of the culture period.
Figure 4.
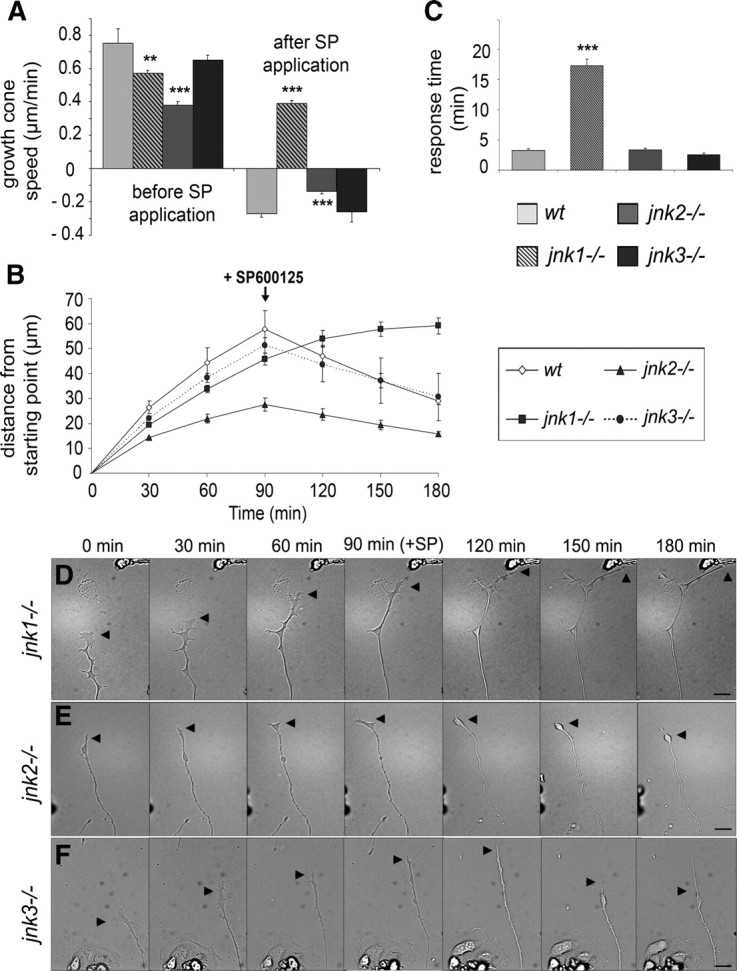
Lack of JNK1 and JNK2 affects axonal elongation rate. A, Mean speed of growth cone advancement and retraction of wild-type and JNK-deficient DRG neurons. Note that jnk1−/− and jnk2−/−, but not jnk3−/−, neurite outgrowth rates are significantly decreased compared with wild type. After SP600125 application, the retraction rate of jnk2−/− neurites is decreased compared with jnk3−/− and wild-type neurites, and no retraction of jnk1−/−neurites occurs. B, Advancement or retraction of growth cones measured 90 min before and 90 min after SP600125 application. Within a few minutes after JNK inhibition, neurites from jnk2−/− and jnk3−/− neurons retract, whereas jnk1−/− neurites continue growing during the remaining recording period. Response time for growth cones to collapse after SP600125 application: jnk2−/− and jnk3−/− growth cones collapse rapidly, in contrast to jnk1−/− growth cones. D–F, Frames of time-lapse recordings illustrating the outgrowth and typical response of neurites to JNK inhibition from jnk1−/−, jnk2−/−, and jnk3−/− DRG neurons, 90 min before and after SP600125 addition. Arrowheads, Growth cones. Scale bars, 50 μm. ***p < 0.001; **p < 0.01. Error bars indicate SEM.
Finally, we assessed the additional effect of SP600125 treatment on the elongation of neurites from the individual JNK isoform-deficient neurons. Whereas complete JNK inhibition induces a decrease in total neurite length of jnk2−/− and jnk3−/− neurons by 27.4 ± 7.1 and 61.3 ± 6.6%, respectively, it has only a lower (although significant) effect on jnk1−/− neurites (Fig. 3C,D; supplemental Fig. S2, available at www.jneurosci.org as supplemental material). Dynamic analysis, by time-lapse videomicroscopy, again revealed a clear difference in the behavior of the different JNK-deficient neurons. Thus, similarly to wild-type neurons, jnk2−/− and jnk3−/− growth cones reacted to JNK inhibition by collapsing 3.38 ± 0.23 and 2.53 ± 0.29 min after drug addition, followed by neurite retraction after 13.68 ± 0.72 and 12.73 ± 0.77 min, respectively (Fig. 4C). Whereas jnk3−/− neurites retract with a rate comparable with wild type (0.26 ± 0.06 and 0.27 ± 0.02 μm/min, respectively), the rate of jnk2−/− neurite retraction is significantly reduced (0.14 ± 0.01 μm/min) (Fig. 4A,B; supplemental Videos 3, 4, available at www.jneurosci.org as supplemental material). The response of jnk1−/− neurites was delayed since, after SP600125 treatment, the growth cone collapse occurs within 17.27 ± 1.1 min and no neurite retraction was yet observed during the 90 min that followed the drug addition (Fig. 4A–D; supplemental Video 2, available at www.jneurosci.org as supplemental material).
To summarize, our results indicate that the combination of JNK1 and JNK2 activities is crucial for neurite extension.
JIP1 is required for axonal regeneration through signaling that involves JNK
The JIP1 scaffolding protein interacts with all three JNK isoforms. It contributes to their activation in response to stress stimuli (Whitmarsh and Davis, 1998) by facilitating the phosphorylation cascade based on MAP kinases that ultimately leads to JNK activation. JIP1 appears to be involved in axonal outgrowth during development (Dajas-Bailador et al., 2008). Here, we found JIP1 to be enriched in growth cones of adult regenerating DRG neurons (Fig. 5A–C). Thus, we examined the neuritogenesis of DRG neurons from jip1−/− mice. Twenty-four hours after plating, only 32.9 ± 13.9% jip1−/− neurons were bearing neurites, compared with wild-type neurons set to 100% (supplemental Fig. S3A, available at www.jneurosci.org as supplemental material). However, after 48 h, the percentage of jip1−/− neurons bearing neurites was similar to wild type, suggesting that JIP1 contributes to neurite initiation. Although neurite initiation was restored at 48 h, jip1−/− neurons exhibit shorter neurites (by 70.4%) compared with wild-type neurons (900.4 ± 56 vs 3041.2 ± 190.4 μm) (Fig. 5E,F,H) [see also the drastic leftward shift in the length distribution graph (Fig. 5I)]. Similar reductions were observed for the length of the longest neurite (supplemental Fig. S3C, available at www.jneurosci.org as supplemental material), associated to a drastic decrease in neurite growth rate (0.27 ± 0.02 vs 0.76 ± 0.09 μm/min for wild-type neurites) (Fig. 5J–L). Moreover, the morphology of jip1−/− neurons was abnormal, their neurites being curled, and exhibiting a higher degree of terminal branching (Fig. 5F; quantitative supplemental Fig. S3B, available at www.jneurosci.org as supplemental material). We further examined DRG neurons from jip1−/− mice with respect to alterations in JNK phosphorylation. Western blot of protein extract from cultured jip1−/− DRG neurons shows an obvious decrease of the 46 kDa band corresponding mainly to phospho-JNK1 (Fig. 5D), probably related to the fact that JNK1 is the most active isoform during later phases of neurite extension (here, at 48 h of culture). Since JNK phosphorylation is not completely abolished in jip1−/− neurons, we examined a potential additional effect of SP600125 treatment on sustained neurite extension in jip1−/− neurons. Whereas the morphology of jip1−/− neurons cultured for 48 h was apparently only little affected by JNK inhibition (Fig. 5G), only a small decrease (19.5%) was noted for their total neurite length and length of the longest neurite (Fig. 5H; supplemental Fig. S3C, available at www.jneurosci.org as supplemental material) (compare with wild-type data in Fig. 2H,I). The effect of SP treatment on jip1−/− neurons was also reflected by a slight leftward shift in the distribution diagram of total neurite lengths (Fig. 5I; length of longest neurites, supplemental Fig. S3D, available at www.jneurosci.org as supplemental material). Dynamic analysis by time-lapse videomicroscopy again revealed that no neurite retraction was observed during the 90 min that follow the drug addition (Fig. 5K,L; supplemental Video 5, available at www.jneurosci.org as supplemental material).
Figure 5.
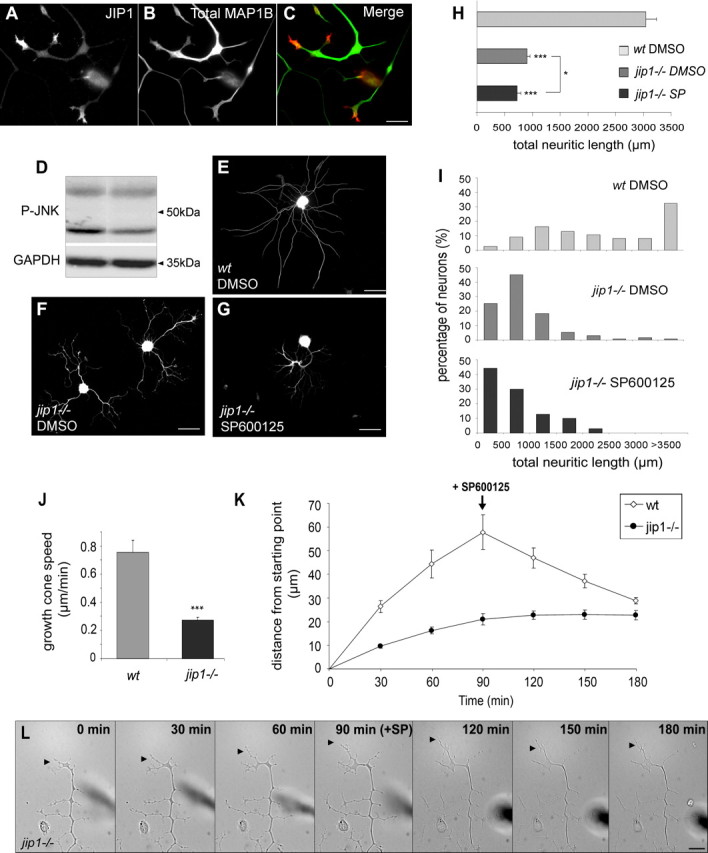
Scaffold protein JIP1 is required for neurite regeneration. A, B, and merge in C, JIP1 immunostaining is enriched in growth cones of regenerating DRG neurites, shafts of which are visualized by total MAP1B immunostaining. D, Western blot analysis of P-JNK in protein extracts from wild-type or jip1−/− DRG neurons cultured 48 h before lysis; the expression of GAPDH was monitored in the lower part of the same membrane as loading control. Lack of JIP1 results in a reduction of the 46 kDa form of P-JNK. E–G, Tubulin immunostaining of DRG neurons cultured for 48 h derived from wild-type (E), jip1−/− (F), and jip1−/− neurons with additional SP600125 treatment (G). Note the abnormal “curly” morphology of neurites from jip1−/− DRG exhibiting more terminal branching; additional SP600125 treatment has no effect on jip1−/− neurites. H, Quantification of total neuritic length of wild-type versus jip1−/− neurons reveals a difference of 70.4%; in contrast, additional SP600125 treatment has only a slight effect on neurite length of jip1−/− DRG neurons (n ≥ 55). I, Diagram showing the distribution of total neuritic lengths of wild-type versus jip1−/− DRG neurons, the latter cultured with or without additional SP600125 treatment. J, Mean speed of growth cone advancement from wild-type and JIP1-deficient DRG neurons showing that the outgrowth rate of jip1−/− neurite is drastically decreased. K, Growth cone advancement or retraction measured 90 min before and 90 min after SP600125 application, showing that no neurite retraction occurs from jip1−/− neurons during the remaining recording time, as illustrated in L, showing corresponding frames of time-lapse recordings. Arrowheads, Growth cones. Scale bars: A, C, 15 μm; E–G, 100 μm; L, 50 μm. ***p < 0.001; *p < 0.05. Error bars indicate SEM.
Our results demonstrate that JIP1 deficiency in adult DRG neurons results in reduced JNK phosphorylation and a drastically reduced neurite regrowth capacity.
JNK signaling in regenerating axons acts on the cytoskeletal protein MAP1B
It is now widely accepted that the regulation of neurite outgrowth in response to extracellular factors passes through signaling cascades that converge onto a reorganization of the neuronal cytoskeleton. Thus, extracellular signaling will ultimately trigger posttranslational modifications (such as phosphorylation) of microtubule-interacting proteins, including stabilizing and destabilizing factors, and thereby regulate microtubule dynamics. During development, MAP1B and the stathmin family members are examples for microtubule-associated proteins that might be influenced by JNK signaling. Stathmin family proteins, when activated, increase the rate of microtubule catastrophes, either through tubulin sequestration or by acting on microtubule plus ends. As shown by Tararuk et al. (2006), JNKs may negatively regulate the activity of these destabilizing proteins (e.g., SCG10) during development, resulting in maintaining microtubule stability and efficient neurite elongation.
Here, we analyzed by Western blotting the phosphorylation state of both stathmin and MAP1B in extracts from cultured adult regenerating DRG neurons deficient for each JNK isoform. Phosphorylation of stathmin was monitored using an antibody specifically recognizing stathmin protein phosphorylated on serine 25, a site known to be predominantly phosphorylated by MAPK (Leighton et al., 1993; Hayashi et al., 2006). Hypophosphorylation of stathmin was found in jnk1−/− DRG neurons, whereas no changes were noted for regenerating jnk2−/− or jnk3−/− neurons (Fig. 6A). Using the same protein extracts, we then showed a hypophosphorylation of MAP1B in jnk1−/− and jnk2−/− neurons, as indicated by a lower apparent molecular weight of the MAP1B protein band, compared with highly phosphorylated MAP1B from wild-type and jnk3−/− neurons (Fig. 6B). The downward shift in the protein band representing MAP1B is indeed characteristic of hypophosphorylation, described previously (Nothias et al., 1996; Alfei et al., 2004, Soares et al., 2007) and demonstrated here in Figure 6C: SP600125 treatment (lane 2) and dephosphorylation by AP of protein extract from cells that had not been treated with SP (lane 3) result in the same shift of the band representing MAP1B to a lower apparent molecular weight.
Figure 6.

Regulation of stathmin and MAP1B phosphorylation by JNKs: Western blot analysis with GADPH used as internal loading control. A, Relative amounts of total stathmin and stathmin phosphorylated on serine 25 (P-Ser25) in lysates of wild-type and JNK-deficient DRG neurons cultured for 48 h; note the decrease in stathmin phosphorylation in regenerating jnk1−/− neurons. B, MAP1B staining of protein extracts from wild-type and JNK-deficient DRG neurons cultured for 48 h, showing a shift in the protein band representing MAP1B from higher to lower apparent molecular weight for jnk1−/− and jnk2−/−, but not jnk3−/− neurons. This shift reflects a reduced level of MAP1B phosphorylation as demonstrated in C, MAP1B in lysates of N2A neuroblastoma cells cultured for 48 h is phosphorylated under control conditions (lane 1); experimental dephosphorylation by AP treatment of lysates (lanes 3, 4) results in the same shift to a lower molecular weight as JNK inhibition by SP600125 (SP) addition to the N2A cultures (lane 2).
These results suggest that JNK1 regulates the phosphorylation of stathmin, whereas both JNK1 and JNK2 regulate MAP1B phosphorylation.
Since the deletion of both JNK1 and JNK2 appeared to affect axonal regeneration in DRG neurons, we focused on the relationship between JNK signaling and the activity of MAP1B for which we had already shown a role in axonal regeneration. In addition, the codistribution of MAP1B-P and P-JNK in sprouting or regenerating fibers, reported here (Fig. 1A–D), suggested that MAP1B might be phosphorylated through JNK signaling during remodeling of the neuronal cytoskeleton. As shown in Figure 7A–C, MAP1B-P staining exhibits a proximo-distal gradient in regenerating neurites, from all DRG neuronal populations (small to large size) (supplemental Fig. S4E–L, available at www.jneurosci.org as supplemental material). The highest MAP1B-P levels are observed in the tips of growing neurites where stable microtubules are rare, as indicated here by the low proportion of detyrosinated tubulin. MAP1B-P, but no detyrosinated tubulin, is found at sites where dynamic “pioneer” microtubules (tyrosinated microtubules) (Bouquet et al., 2004, 2007) invade the peripheral growth cone domain (Fig. 7D–F). In contrast to total MAP1B, displaying a homogeneous distribution in all neuronal compartments (Fig. 7G) that is unchanged after JNK inhibition (Fig. 7J), the neurite localization of MAP1B-P was dramatically altered in response to JNK inhibition by SP600125 (Fig. 7K). The absence of MAP1B-P in neurites after JNK inhibition can be correlated with the strong reduction in levels of phosphorylated MAP1B after SP600125 treatment revealed by Western blotting (Fig. 6C). A role for JNK signaling in MAP1B phosphorylation was further confirmed by analysis of neurons derived from jip1−/− mice, in which we noted strong reduction in MAP1B-P levels, as shown by Western blotting (Fig. 6B) and immunocytochemistry (Fig. 7L,M).
Figure 7.
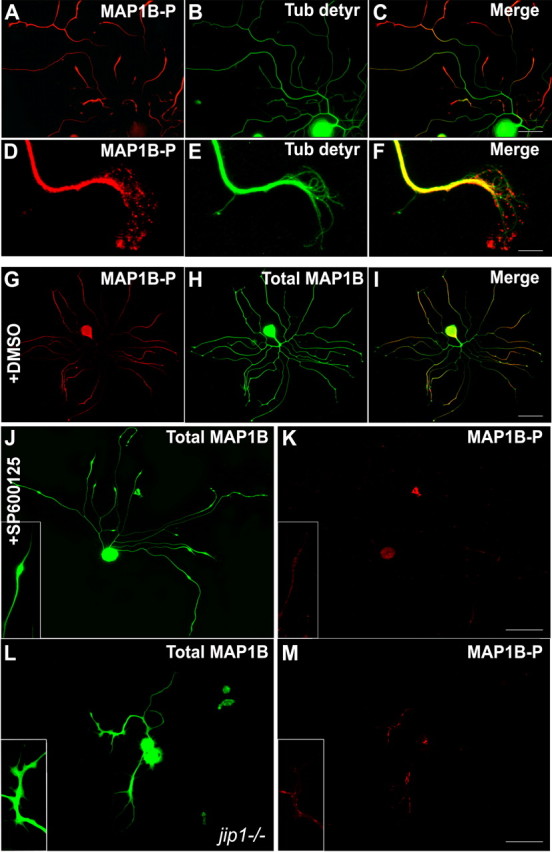
JNK inhibition affects MAP1B phosphorylation in regenerating neurons. A–C, In control DRG neurons, distal parts of neurites are enriched in MAP1B-P and characterized by low levels of detyrosinated tubulin (representing stable microtubules). D–F, Higher magnification of a growth cone showing the presence of MAP1B-P at the leading edge, where no colocalization with detyrosinated tubulin can be found. G–I, Double immunostaining reveals that total MAP1B is distributed in all parts of the neuron (green), whereas MAP1B-P (red) is mainly localized in neurites and exhibits a proximo-distal gradient. J, K, Application of SP600125, although not altering total MAP1B levels, dramatically decreases the neurite content in MAP1B-P, concomitantly with growth cone retraction (see also insets). L, M, Immunostaining for MAP1B-P, compared with total MAP1B, on regenerating jip1−/− neurons reveals that lack of JIP strongly affects the level of MAP1B phosphorylation. Scale bars: A–C, 50 μm; D–F, 10 μm; G–M, 100 μm.
Next, we examined whether the overall levels of MAP1B-P, or its proximo-distal distribution along the neurite, depended on a specific JNK isoform. Compared with wild-type neurons (Fig. 8A–C), immunocytochemical staining for MAP1B-P was markedly reduced in regenerating jnk1−/− and jnk2−/− neurons (Fig. 8D–I) but unchanged in jnk3−/− neurites (Fig. 8J–L). These results demonstrate that MAP1B phosphorylation is regulated through signaling by JNK1 and JNK2.
Figure 8.

JNK1 and JNK2 regulate MAP1B phosphorylation. Immunostaining for total MAP1B (left row) and MAP1B-P (middle row; the right row represents merge images) of regenerating neurons from wild-type (A–C) and JNK-deficient mice. MAP1B-P levels are strongly reduced in neurites elongating from jnk1−/− (D–F) and jnk2−/− (G–I) neurons, whereas both the level and the proximo-distal distribution of MAP1B-P in jnk3−/− neurites (J–L) are similar to those of wild type. Scale bars, 50 μm.
Finally, we used DRG neuron cultures derived from map1b−/− mice to investigate the functional relationship between MAP1B and JNK signaling during axonal regeneration. Whereas JNK inhibition by SP600125 treatment in wild-type neurons led to a rapid retraction of neurites (compare Fig. 2), no retraction was observed under the same conditions for map1b−/− neurons, as illustrated in Figure 9A–D. Quantitative analysis shows that JNK inhibition in map1b−/− neurons reduced their total neuritic length by only 24% (Fig. 9E) and their longest neurite by 3.6% (Fig. 9F), in contrast to wild-type neurons showing a reduction by 66.7 and 38.2%, respectively. The rather small effect of SP600125 treatment on map1b−/− neurons is also reflected in the distribution diagrams of total neurite length and longest neurite per neuron (supplemental Fig. S5, available at www.jneurosci.org as supplemental material). The response of map1b−/− neurites to SP600125 treatment was further examined by time-lapse videomicroscopy (Fig. 9G; supplemental Video 6, available at www.jneurosci.org as supplemental material). Before the addition of JNK inhibitor, map1b−/− neurites exhibit dynamic growth cones and elongate continuously. Extension and retraction of filopodia is very similar to wild-type neurons, although the main neurites of map1b−/− neurons are highly branched (Bouquet et al., 2004). On SP600125 addition, growth cone filopodia collapse (that could explain the small reduction in total neuritic length), but in contrast to wild-type neurons, no neurite retraction occurs. Thus, the neurite retraction resulting from JNK inhibition depends on MAP1B.
Figure 9.
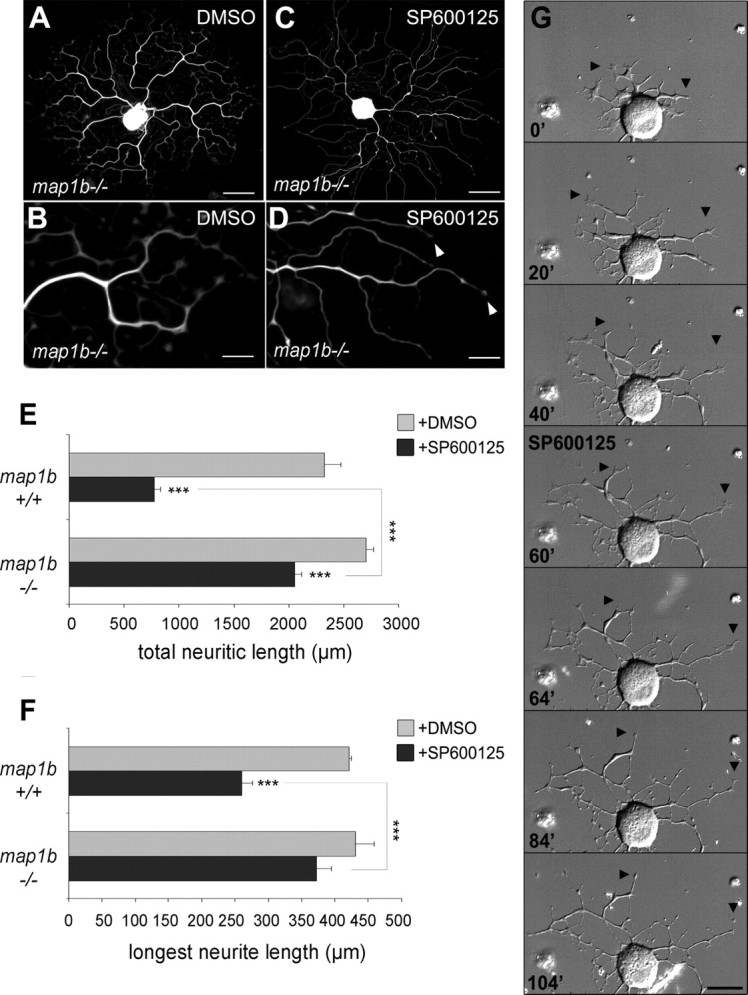
MAP1B is required for neurite retraction induced by JNK inhibition. A–D, map1b−/− neurons after 48 h in culture, with or without SP600125 treatment, visualized by tubulin immunostaining. Note the increased number of branching points on the neurite tree of map1b−/− neurons. JNK inhibition by SP600125 addition to the culture medium induces growth arrest and loss of growth cones (arrowheads), but no retraction bulbs are observed along the neurites, in contrast to wild-type neurons (compare Fig. 2G). E, F, Quantitative analysis reveals no significant differences in total neurite length and length of longest neurites between map1b+/+ and map1b−/− neurons under control conditions (gray bars); however, whereas SP600125 application (black bars) dramatically reduces total neurite length and length of longest neurites on map1b+/+ neurons, JNK inhibition has only very limited effect on map1b−/− neurites (n ≥ 52). G, Time-lapse recording illustrating the typical response of a map1b−/− neurite: After SP600125 addition (at t = 60 min), growth cones collapse, neurites stop extending, but no retraction is observed (arrowheads) (see movie in supplemental data S6, available at www.jneurosci.org as supplemental material). Scale bars: A, C, 50 μm; B, D, 15 μm; G, 25 μm. ***p < 0.001. Error bars indicate SEM.
Discussion
Using both pharmacological JNK inhibition, and mice deficient for each JNK isoform and JIP1, we provide evidence that JNK signaling is involved in two processes crucial for axonal regeneration: initiation and elongation.
All three JNK isoforms are expressed in adult DRG neurons, but their specific roles in the regeneration process obviously differ. Lack of JNK2 or JNK3, but not JNK1, delays neurite initiation. Whereas JNK1 is present mainly in the cytoplasm of neuronal processes (Coffey et al., 2002), JNK2 and JNK3 can be translocated to the nucleus (Fig. 10a) (for review, see Raivich and Makwana, 2007) and affect transcription of regeneration-associated genes such as c-jun, whose role in promoting neuronal differentiation and neurite outgrowth is well characterized. JNK2 and JNK3 may act synergistically in phosphorylating numerous transcription factors including c-Jun, junD, ATF3, and STAT3 (Kenney and Kocsis, 1998; Sheu et al., 2000; Lindwall et al., 2004). Their different functions in neurite initiation are likely attributable to different affinities for individual substrates (Kallunki et al., 1994; Gupta et al., 1996). During later phases of neurite extension, JNK2 and JNK1 (but not JNK3) are important. JNK1 deficiency only affects extension of preformed neurites, which indicates that JNK1 may play a major role in elongation by phosphorylating proteins that regulate microtubule dynamics, such as MAP1B, MAP2, and stathmin family proteins (Chang et al., 2003; Björkblom et al., 2005; Tararuk et al., 2006; present data) (see below). Moreover, during later phases of neurite regeneration, JNK1 activity appears to be particularly affected by JIP1 deletion and jnk1−/− neurites are less sensitive to additional SP600125 treatment. Only JNK2 appears to act on both neurite initiation and elongation, the latter activity being at least in part related to phosphorylation of MAP1B. Not surprisingly, neurite regeneration is most severely affected in jnk2−/− mice.
Figure 10.

Model of JNK functions in axonal regeneration. This model is based on previous studies (see Discussion) and incorporates our present findings and hypotheses. a, Axotomy provokes an activation of JNK signaling leading to phosphorylation of transcription factors (including c-jun, ATF3, and STAT3) that, in turn, will regulate the expression of regeneration-associated genes. During early stages of DRG neuron regeneration, specific activation of JNK2 and JNK3 may mediate transcriptional events required for neurite growth initiation in the “correct” time window. b, JNK activation is maintained during neurite extension. Signaling by P-JNK1 and -2, present in axon shaft and growth cone, regulates cytoplasmic effectors that finally influence microtubule (MT) dynamics. Among cytoplasmic effectors of JNKs are MT-destabilizing (stathmin) and -stabilizing proteins (MAP1B). Phosphorylation of stathmin (by JNK1) family members overcomes their destabilizing function on MT plus ends, whereas phosphorylation of MAP1B (by JNK1 and JNK2) affects its binding to MTs or bind more labile MTs. c, JNK inhibition induces retraction of previously formed axons. Nonphosphorylated stathmin sequesters tubulin dimers and thereby prevents MT polymerization and axon outgrowth. In parallel, a decrease in MAP1B phosphorylation will increase the binding of MAP1B to MTs, enhancing their stabilization and protecting them from depolymerization. Process retraction, crucial for correct axonal pathfinding, is based on two independent molecular mechanisms: Extension forces generated by the action of dynein on MTs are counterbalanced by actin–myosin contractility resulting in axon retraction. The dynamic interaction between the two opposed forces likely involves competition between MAP1B and dynein.
Although one may argue about the specificity of SP600125, our data clearly show that it does inhibit JNKs efficiently and has only very little effect on jip1−/− neurons. A previous study reported that this drug exhibited a higher affinity for JNK1 and JNK2 than for JNK3 (Bennett et al., 2001), corroborating our observations. The relatively strong effect of SP600125 seen in jnk2−/− and jnk3−/− neurites may be related, even partially, to changes in expression and/or activity of the remaining JNK isoforms. This phenomenon has been previously described for jnk2−/− mouse embryo fibroblasts in which increased JNK1 activity was noted (Jaeschke et al., 2006).
Interestingly, we did not observe any differences in neurite extension when comparing wild-type and JNK-deficient DRG neurons from embryonic day 13.5 embryos, despite a rather pronounced effect of pharmacological JNK inhibition on these neurons (data not shown). During development, lack of individual JNK proteins may be completely compensated by other isoforms, or even other signaling pathways/kinases. Accordingly, no specific function in neurite outgrowth from developing hippocampal or cortical neurons could be attributed to individual JNK isoforms (Eminel et al., 2008). The differential functions for individual JNKs, reported here, may thus be specific to adult regenerating neurites and particularly axons. Indeed, the DRG neurites examined in our study are most likely axons: they are all immunopositive for axon-specific cytoskeleton proteins such as tau and phosphorylated MAP1B (Soares et al., 2002; present data), but not stained for the dendrite marker protein MAP2. Furthermore, all neurites are immunostained by antibodies to JIP1, shown to be specifically enriched in nascent and mature axons (Dajas-Bailador et al., 2008). Lack of JIP1, in adult neurons, results in reduction in the phosphorylation and hence activation of JNK, inducing dramatic effects on neuritogenesis. However, abnormal neuritogenesis of jip1−/− neurons may not simply be a consequence of altered JNK signaling. JIP1 was shown to interact with kinesin-1 and doublecortin, thereby regulating polarization, transport, and axon outgrowth of cortical neurons (Gdalyahu et al., 2004; Horiuchi et al., 2005; Dajas-Bailador et al., 2008).
Phosphorylated JNK, and even more JIP1, are enriched in the distal part of growing neurites both during development (Whitmarsh et al., 2001; Gdalyahu et al., 2004; Tararuk et al., 2006) and axonal regeneration (present data). This is suggestive of a direct action of phospho-JNK on local molecular effectors, for which the following microtubule stability-regulating proteins are good candidates: DCX (Gdalyahu et al., 2004), tau (Yoshida et al., 2004), MAP2 and MAP1B (Chang et al., 2003), and microtubule-destabilizing protein SCG10 (Tararuk et al., 2006). Evidence for a direct action of JNKs on cytoskeleton dynamics is provided by our observation that regenerating growth cones and axon shafts respond to local application of JNK inhibitor within a few minutes, excluding transcriptional regulation. This response is associated with a rapid retraction, rather than a depolymerization of the microtubule network (Ahmad et al., 2000; Baas and Ahmad, 2001; Bouquet et al., 2007; Ertürk et al., 2007). A regulation of microtubule dynamics by JNKs in regenerating neurons is further supported by our in vivo data showing activation of JNKs in fibers undergoing lesion-induced sprouting. Increased levels of cytoplasmic phospho-JNK are maintained during the whole period of axonal remodeling, suggesting involvement of other JNK effectors, in addition to nuclear c-jun activation. Moreover, the clear association between JNK activation and MAP1B phosphorylation within sprouting fibers and, conversely, the decrease in MAP1B-P levels after JNK inhibition (and in JNK1-, JNK2-, and JIP1-deficient mice) argue for MAP1B being an effector of JNK signaling. The function of MAP1B in stabilizing axonal microtubules is regulated by phosphorylation through two mechanisms: mode II phosphorylation via casein kinase II is observed until adulthood. Mode I is developmentally regulated and implies PDPKs (proline-directed protein kinases), such as GSK-3β, Cdk-5, and MAPK (JNKs) (Ulloa et al., 1993; Kawauchi et al., 2003, 2005; Trivedi et al., 2005) (for references and review, see Riederer, 2007). Nonphosphorylated and mode II phosphorylated MAP1B is found in all compartments of neuronal cytoplasm. In contrast, mode I phosphorylated MAP1B (termed MAP1B-P) is absent from dendrites, preferentially localized to distal regions of the axon, and its distribution in growth cones resembles that of newly formed (mainly tyrosinated), highly dynamic microtubules (Baas and Black, 1990; Ahmad et al., 1993; Bouquet et al., 2004, 2007). These observations, as well as the data provided by Mack et al. (2000) showing that phosphorylated MAP1B is required for microtubule stabilization in the growth cone, would suggest that MAP1B-P is implicated in regulating microtubule dynamics in highly motile regions of neurites (Scales et al., 2009). In regenerating DRG neurites, this MAP1B phosphorylation should be mainly regulated by JNK1 and JNK2 since its level is not affected by lack of JNK3 (Fig. 10b).
Moreover, the requirement of MAP1B for neurite retraction after JNK inhibition is consistent with MAP1B being a crucial downstream effector of JNK signaling during reorganization of neuronal cytoskeleton in the adult nervous system. We previously demonstrated that MAP1B regulates growth cone turning and branching of regenerating neurites (Bouquet et al., 2004) and provided physiological evidence for its requirement in coupling microtubule and actin filament dynamics during the retraction of cell processes in response to extracellular stimuli involving the RhoA/ROCK GTPases (Bouquet et al., 2007; Stroissnigg et al., 2007). This MAP1B-dependent neurite retraction does not result in microtubule depolymerization, which indicates that axon extension and retraction are based on two interconnected molecular mechanisms: The forces driving axon extension are generated by the action of dynein on microtubules, counterbalanced by contractile forces based on actin–myosin activity leading to axon retraction (Ahmad et al., 2000). This dynamic interaction between the two opposed forces might involve a competition between MAP1B and dynein, since the microtubule-binding sequence of these two proteins is similar (Koonce and Tikhonenko, 2000) [see also discussion in the study by Stroissnigg et al. (2007)]. The function of MAP1B in axon retraction appears to be regulated by posttranslational modifications, notably nitrosylation induced by nNOS (neuronal nitric oxide synthase) activation (Stroissnigg et al., 2007). Here, we extend this notion by demonstrating that decreasing the phosphorylation level of MAP1B by JNK inhibition further confirms its involvement in neurite retraction, whereas neurite extension is obviously facilitated by JNK signaling (probably activated by Rac1) (Weston and Davis, 2002) that converges on MAP1B. Moreover, the two opposed processes (i.e., MAP1B nitrosylation leading to neurite retraction and JNK-induced phosphorylation leading to extension) are likely to be interdependent since exogenous nitric oxide negatively regulates the activity of JNKs (Pei et al., 2008). This fine-tuning of the function of MAP1B seems a good example for the convergence of a wide range of signaling pathways, activated in response to extracellular guidance cues, on the regulation of cytoskeletal dynamics. At the same time, a single signaling pathway may act on cytoskeleton-associated proteins exerting opposite functions: As we show here (Fig. 10), JNK signaling affects both the microtubule-stabilizing protein MAP1B and the destabilizing stathmin family proteins.
Together, our data clearly demonstrate the differential involvement of JNK isoforms in controlling cytoskeleton reorganization, notably by regulating the function of MAP1B. Our study therefore provides new insight into how extracellular cues will eventually regulate guidance, branching, and retraction of neurites through orchestrated rearrangement of the cytoskeleton, triggered by specific intracellular signaling cascades.
Footnotes
This work was supported by Centre National de la Recherche Scientifique, Institut National de la Santé et de la Recherche Médicale, and Université Pierre et Marie Curie (Paris 06), and by Institut de Recherche pour la Moelle Epinière et l'Encéphale, Association Française contre les Myopathies, Egide-Amadeus Programme d'Action Intégrée (F.N.), the Austrian Exchange Service, Program Amadée Project FR 12/2007 (F.P.), and Association pour la Recherche sur le Cancer Grant 3746 (H.E.). We thank Dr. Ysander von Boxberg and Dr. Jamilé Hazan for careful reading and helpful comments on this manuscript. We are very grateful to Dr. Itzhak Fischer, Dr. Greeg Gundersen, and Valérie Manceau and Dr. André Sobel for providing us with anti-MAP1B, “superglu,” and anti-stathmin antibodies, respectively. We also thank Michèle Ravaille-Veron and Marie-Noëlle Benassy (Unité 7224), and Virginie Georget and Richard Schwartzmann (imaging platform; Institut Fédératif de Recherche 83) for technical help.
References
- Ahmad FJ, Pienkowski TP, Baas PW. Regional differences in microtubule dynamics in the axon. J Neurosci. 1993;13:856–866. doi: 10.1523/JNEUROSCI.13-02-00856.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmad FJ, Hughey J, Wittmann T, Hyman A, Greaser M, Baas PW. Motor proteins regulate force interactions between microtubules and microfilaments in the axon. Nat Cell Biol. 2000;2:276–280. doi: 10.1038/35010544. [DOI] [PubMed] [Google Scholar]
- Alfei L, Soares S, Alunni A, Ravaille-Veron M, Von Boxberg Y, Nothias F. Expression of MAP1B protein and its phosphorylated form MAP1B-P in the CNS of a continuously growing fish, the rainbow trout. Brain Res. 2004;1009:54–66. doi: 10.1016/j.brainres.2004.02.043. [DOI] [PubMed] [Google Scholar]
- Baas PW, Ahmad FJ. Force generation by cytoskeletal motor proteins as a regulator of axonal elongation and retraction. Trends Cell Biol. 2001;11:244–249. doi: 10.1016/s0962-8924(01)02005-0. [DOI] [PubMed] [Google Scholar]
- Baas PW, Black MM. Individual microtubules in the axon consist of domains that differ in both composition and stability. J Cell Biol. 1990;111:495–509. doi: 10.1083/jcb.111.2.495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett BL, Sasaki DT, Murray BW, O'Leary EC, Sakata ST, Xu W, Leisten JC, Motiwala A, Pierce S, Satoh Y, Bhagwat SS, Manning AM, Anderson DW. SP600125, an anthrapyrazolone inhibitor of Jun N-terminal kinase. Proc Natl Acad Sci U S A. 2001;98:13681–13686. doi: 10.1073/pnas.251194298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Björkblom B, Ostman N, Hongisto V, Komarovski V, Filén JJ, Nyman TA, Kallunki T, Courtney MJ, Coffey ET. Constitutively active cytoplasmic c-Jun N-terminal kinase 1 is a dominant regulator of dendritic architecture: role of microtubule-associated protein 2 as an effector. J Neurosci. 2005;25:6350–6361. doi: 10.1523/JNEUROSCI.1517-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogoyevitch MA. The isoform-specific functions of the c-Jun N-terminal kinases (JNKs): differences revealed by gene targeting. Bioessays. 2006;28:923–934. doi: 10.1002/bies.20458. [DOI] [PubMed] [Google Scholar]
- Bottenstein JE, Sato GH. Growth of a rat neuroblastoma cell line in serum-free supplemented medium. Proc Natl Acad Sci U S A. 1979;76:514–517. doi: 10.1073/pnas.76.1.514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouquet C, Soares S, von Boxberg Y, Ravaille-Veron M, Propst F, Nothias F. Microtubule-associated protein 1B controls directionality of growth cone migration and axonal branching in regeneration of adult dorsal root ganglia neurons. J Neurosci. 2004;24:7204–7213. doi: 10.1523/JNEUROSCI.2254-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouquet C, Ravaille-Veron M, Propst F, Nothias F. MAP1B coordinates microtubule and actin filament remodeling in adult mouse schwann cell tips and drg neuron growth cones. Mol Cell Neurosci. 2007;36:235–247. doi: 10.1016/j.mcn.2007.07.002. [DOI] [PubMed] [Google Scholar]
- Cavalli V, Kujala P, Klumperman J, Goldstein LS. Sunday Driver links axonal transport to damage signaling. J Cell Biol. 2005;168:775–787. doi: 10.1083/jcb.200410136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang L, Jones Y, Ellisman MH, Goldstein LS, Karin M. JNK1 is required for maintenance of neuronal microtubules and controls phosphorylation of microtubule-associated proteins. Dev Cell. 2003;4:521–533. doi: 10.1016/s1534-5807(03)00094-7. [DOI] [PubMed] [Google Scholar]
- Coffey ET, Smiciene G, Hongisto V, Cao J, Brecht S, Herdegen T, Courtney MJ. c-Jun N-terminal protein kinase (JNK) 2/3 is specifically activated by stress, mediating c-Jun activation, in the presence of constitutive JNK1 activity in cerebellar neurons. J Neurosci. 2002;22:4335–4345. doi: 10.1523/JNEUROSCI.22-11-04335.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dajas-Bailador F, Jones EV, Whitmarsh AJ. The JIP1 scaffold protein regulates axonal development in cortical neurons. Curr Biol. 2008;18:221–226. doi: 10.1016/j.cub.2008.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis RJ. Signal transduction by the jnk group of MAP kinases. Cell. 2000;103:239–252. doi: 10.1016/s0092-8674(00)00116-1. [DOI] [PubMed] [Google Scholar]
- Dong C, Yang DD, Wysk M, Whitmarsh AJ, Davis RJ, Flavell RA. Defective T cell differentiation in the absence of JNK1. Science. 1998;282:2092–2095. doi: 10.1126/science.282.5396.2092. [DOI] [PubMed] [Google Scholar]
- Eminel S, Roemer L, Waetzig V, Herdegen T. c-Jun N-terminal kinases trigger both degeneration and neurite outgrowth in primary hippocampal and cortical neurons. J Neurochem. 2008;104:957–969. doi: 10.1111/j.1471-4159.2007.05101.x. [DOI] [PubMed] [Google Scholar]
- Ertürk A, Hellal F, Enes J, Bradke F. Disorganized microtubules underlie the formation of retraction bulbs and the failure of axonal regeneration. J Neurosci. 2007;27:9169–9180. doi: 10.1523/JNEUROSCI.0612-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer I, Romano-Clarke G. Changes in microtubule-associated protein MAP1B phosphorylation during rat brain development. J Neurochem. 1990;55:328–333. doi: 10.1111/j.1471-4159.1990.tb08855.x. [DOI] [PubMed] [Google Scholar]
- Gavet O, Ozon S, Manceau V, Lawler S, Curmi P, Sobel A. The stathmin phosphoprotein family: intracellular localization and effects on the microtubule network. J Cell Sci. 1998;111:3333–3346. doi: 10.1242/jcs.111.22.3333. [DOI] [PubMed] [Google Scholar]
- Gdalyahu A, Ghosh I, Levy T, Sapir T, Sapoznik S, Fishler Y, Azoulai D, Reiner O. DCX, a new mediator of the JNK pathway. EMBO J. 2004;23:823–832. doi: 10.1038/sj.emboj.7600079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gundersen GG, Kalnoski MH, Bulinski JC. Distinct populations of microtubules: tyrosinated and nontyrosinated alpha tubulin are distributed differently in vivo. Cell. 1984;38:779–789. doi: 10.1016/0092-8674(84)90273-3. [DOI] [PubMed] [Google Scholar]
- Gupta S, Barrett T, Whitmarsh AJ, Cavanagh J, Sluss HK, Dérijard B, Davis RJ. Selective interaction of JNK protein kinase isoforms with transcription factors. EMBO J. 1996;15:2760–2770. [PMC free article] [PubMed] [Google Scholar]
- Hayashi K, Pan Y, Shu H, Ohshima T, Kansy JW, White CL, 3rd, Tamminga CA, Sobel A, Curmi PA, Mikoshiba K, Bibb JA. Phosphorylation of the tubulin-binding protein, stathmin, by Cdk5 and MAP kinases in the brain. J Neurochem. 2006;99:237–250. doi: 10.1111/j.1471-4159.2006.04113.x. [DOI] [PubMed] [Google Scholar]
- Horiuchi D, Barkus RV, Pilling AD, Gassman A, Saxton WM. APLIP1, a kinesin binding JIP-1/JNK scaffold protein, influences the axonal transport of both vesicles and mitochondria in Drosophila. Curr Biol. 2005;15:2137–2141. doi: 10.1016/j.cub.2005.10.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaeschke A, Karasarides M, Ventura JJ, Ehrhardt A, Zhang C, Flavell RA, Shokat KM, Davis RJ. JNK2 is a positive regulator of the c-Jun transcription factor. Mol Cell. 2006;23:899–911. doi: 10.1016/j.molcel.2006.07.028. [DOI] [PubMed] [Google Scholar]
- Kallunki T, Su B, Tsigelny I, Sluss HK, Dérijard B, Moore G, Davis R, Karin M. JNK2 contains a specificity-determining region responsible for efficient c-jun binding and phosphorylation. Genes Dev. 1994;8:2996–3007. doi: 10.1101/gad.8.24.2996. [DOI] [PubMed] [Google Scholar]
- Kawauchi T, Chihama K, Nabeshima Y, Hoshino M. The in vivo roles of STEF/Tiam1, Rac1 and JNK in cortical neuronal migration. EMBO J. 2003;22:4190–4201. doi: 10.1093/emboj/cdg413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawauchi T, Chihama K, Nishimura YV, Nabeshima Y, Hoshino M. MAP1B phosphorylation is differentially regulated by Cdk5/p35, Cdk5/p25, and JNK. Biochem Biophys Res Commun. 2005;331:50–55. doi: 10.1016/j.bbrc.2005.03.132. [DOI] [PubMed] [Google Scholar]
- Kenney AM, Kocsis JD. Peripheral axotomy induces long-term c-Jun amino-terminal kinase-1 activation and activator protein-1 binding activity by c-Jun and junD in adult rat dorsal root ganglia in vivo. J Neurosci. 1998;18:1318–1328. doi: 10.1523/JNEUROSCI.18-04-01318.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koonce MP, Tikhonenko I. Functional elements within the dynein microtubule-binding domain. Mol Biol Cell. 2000;11:523–529. doi: 10.1091/mbc.11.2.523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koppel J, Boutterin MC, Doye V, Peyro-Saint-Paul H, Sobel A. Developmental tissue expression and phylogenetic conservation of stathmin, a phosphoprotein associated with cell regulations. J Biol Chem. 1990;265:3703–3707. [PubMed] [Google Scholar]
- Kuan CY, Yang DD, Samanta Roy DR, Davis RJ, Rakic P, Flavell RA. The JNK1 and JNK2 protein kinases are required for regional specific apoptosis during early brain development. Neuron. 1999;22:667–676. doi: 10.1016/s0896-6273(00)80727-8. [DOI] [PubMed] [Google Scholar]
- Leighton IA, Curmi P, Campbell DG, Cohen P, Sobel A. The phosphorylation of stathmin by MAP kinase. Mol Cell Biochem. 1993;127–128:151–156. doi: 10.1007/BF01076766. [DOI] [PubMed] [Google Scholar]
- Lindwall C, Dahlin L, Lundborg G, Kanje M. Inhibition of c-Jun phosphorylation reduces axonal outgrowth of adult rat nodose ganglia and dorsal root ganglia sensory neurons. Mol Cell Neurosci. 2004;27:267–279. doi: 10.1016/j.mcn.2004.07.001. [DOI] [PubMed] [Google Scholar]
- Mack TG, Koester MP, Pollerberg GE. The microtubule-associated protein MAP1B is involved in local stabilization of turning growth cones. Mol Cell Neurosci. 2000;15:51–65. doi: 10.1006/mcne.1999.0802. [DOI] [PubMed] [Google Scholar]
- Meixner A, Haverkamp S, Wässle H, Führer S, Thalhammer J, Kropf N, Bittner RE, Lassmann H, Wiche G, Propst F. MAP1B is required for axon guidance and is involved in the development of the central and peripheral nervous system. J Cell Biol. 2000;151:1169–1178. doi: 10.1083/jcb.151.6.1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison DK, Davis RJ. Regulation of MAP kinase signaling modules by scaffold proteins in mammals. Annu Rev Cell Dev Biol. 2003;19:91–118. doi: 10.1146/annurev.cellbio.19.111401.091942. [DOI] [PubMed] [Google Scholar]
- Nothias F, Fischer I, Murray M, Mirman S, Vincent JD. Expression of a phosphorylated isoform of map1b is maintained in adult central nervous system areas that retain capacity for structural plasticity. J Comp Neurol. 1996;368:317–334. doi: 10.1002/(SICI)1096-9861(19960506)368:3<317::AID-CNE1>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- Oliva AA, Jr, Atkins CM, Copenagle L, Banker GA. Activated c-Jun N-terminal kinase is required for axon formation. J Neurosci. 2006;26:9462–9470. doi: 10.1523/JNEUROSCI.2625-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pei DS, Song YJ, Yu HM, Hu WW, Du Y, Zhang GY. Exogenous nitric oxide negatively regulates c-Jun N-terminal kinase activation via inhibiting endogenous NO-induced S-nitrosylation during cerebral ischemia and reperfusion in rat hippocampus. J Neurochem. 2008;106:1952–1963. doi: 10.1111/j.1471-4159.2008.05531.x. [DOI] [PubMed] [Google Scholar]
- Raivich G, Makwana M. The making of successful axonal regeneration: genes, molecules and signal transduction pathways. Brain Res Rev. 2007;53:287–311. doi: 10.1016/j.brainresrev.2006.09.005. [DOI] [PubMed] [Google Scholar]
- Riederer BM. Microtubule-associated protein 1B, a growth-associated and phosphorylated scaffold protein. Brain Res Bull. 2007;71:541–558. doi: 10.1016/j.brainresbull.2006.11.012. [DOI] [PubMed] [Google Scholar]
- Scales TM, Lin S, Kraus M, Goold RG, Gordon-Weeks PR. Nonprimed and DYRK1A-primed GSK3β-phosphorylation sites on MAP1B regulate microtubule dynamics in growing axons. J Cell Sci. 2009;122:2424–2435. doi: 10.1242/jcs.040162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheu JY, Kulhanek DJ, Eckenstein FP. Differential patterns of ERK and STAT3 phosphorylation after sciatic nerve transection in the rat. Exp Neurol. 2000;166:392–402. doi: 10.1006/exnr.2000.7508. [DOI] [PubMed] [Google Scholar]
- Soares S, von Boxberg Y, Lombard MC, Ravaille-Veron M, Fischer I, Eyer J, Nothias F. Phosphorylated MAP1B is induced in central sprouting of primary afferents in response to peripheral injury but not in response to rhizotomy. Eur J Neurosci. 2002;16:593–606. doi: 10.1046/j.1460-9568.2002.02126.x. [DOI] [PubMed] [Google Scholar]
- Soares S, Barnat M, Salim C, von Boxberg Y, Ravaille-Veron M, Nothias F. Extensive structural remodeling of the injured spinal cord revealed by phosphorylated MAP1B in sprouting axons and degenerating neurons. Eur J Neurosci. 2007;26:1446–1461. doi: 10.1111/j.1460-9568.2007.05794.x. [DOI] [PubMed] [Google Scholar]
- Stroissnigg H, Trancíková A, Descovich L, Fuhrmann J, Kutschera W, Kostan J, Meixner A, Nothias F, Propst F. S-Nitrosylation of microtubule-associated protein 1B mediates nitric-oxide-induced axon retraction. Nat Cell Biol. 2007;9:1035–1045. doi: 10.1038/ncb1625. [DOI] [PubMed] [Google Scholar]
- Tararuk T, Ostman N, Li W, Björkblom B, Padzik A, Zdrojewska J, Hongisto V, Herdegen T, Konopka W, Courtney MJ, Coffey ET. JNK1 phosphorylation of SCG10 determines microtubule dynamics and axodendritic length. J Cell Biol. 2006;173:265–277. doi: 10.1083/jcb.200511055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trivedi N, Marsh P, Goold RG, Wood-Kaczmar A, Gordon-Weeks PR. Glycogen synthase kinase-3beta phosphorylation of MAP1B at Ser1260 and Thr1265 is spatially restricted to growing axons. J Cell Sci. 2005;118:993–1005. doi: 10.1242/jcs.01697. [DOI] [PubMed] [Google Scholar]
- Ulloa L, Avila J, Díaz-Nido J. Heterogeneity in the phosphorylation of microtubule-associated protein MAP1B during rat brain development. J Neurochem. 1993;61:961–972. doi: 10.1111/j.1471-4159.1993.tb03609.x. [DOI] [PubMed] [Google Scholar]
- Waetzig V, Zhao Y, Herdegen T. The bright side of JNKs—multitalented mediators in neuronal sprouting, brain development and nerve fiber regeneration. Prog Neurobiol. 2006;80:84–97. doi: 10.1016/j.pneurobio.2006.08.002. [DOI] [PubMed] [Google Scholar]
- Weston CR, Davis RJ. The JNK signal transduction pathway. Curr Opin Genet Dev. 2002;12:14–21. doi: 10.1016/s0959-437x(01)00258-1. [DOI] [PubMed] [Google Scholar]
- Whitmarsh AJ, Davis RJ. Structural organization of MAP-kinase signaling modules by scaffold proteins in yeast and mammals. Trends Biochem Sci. 1998;23:481–485. doi: 10.1016/s0968-0004(98)01309-7. [DOI] [PubMed] [Google Scholar]
- Whitmarsh AJ, Kuan CY, Kennedy NJ, Kelkar N, Haydar TF, Mordes JP, Appel M, Rossini AA, Jones SN, Flavell RA, Rakic P, Davis RJ. Requirement of the JIP1 scaffold protein for stress-induced JNK activation. Genes Dev. 2001;15:2421–2432. doi: 10.1101/gad.922801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Widmann C, Gibson S, Jarpe MB, Johnson GL. Mitogen-activated protein kinase: conservation of a three-kinase module from yeast to human. Physiol Rev. 1999;79:143–180. doi: 10.1152/physrev.1999.79.1.143. [DOI] [PubMed] [Google Scholar]
- Yang DD, Kuan CY, Whitmarsh AJ, Rincón M, Zheng TS, Davis RJ, Rakic P, Flavell RA. Absence of excitotoxicity-induced apoptosis in the hippocampus of mice lacking the Jnk3 gene. Nature. 1997;389:865–870. doi: 10.1038/39899. [DOI] [PubMed] [Google Scholar]
- Yang DD, Conze D, Whitmarsh AJ, Barrett T, Davis RJ, Rincón M, Flavell RA. Differentiation of CD4+ T cells to Th1 cells requires MAP kinase JNK2. Immunity. 1998;9:575–585. doi: 10.1016/s1074-7613(00)80640-8. [DOI] [PubMed] [Google Scholar]
- Yoshida H, Hastie CJ, McLauchlan H, Cohen P, Goedert M. Phosphorylation of microtubule-associated protein tau by isoforms of c-Jun N-terminal kinase (JNK) J Neurochem. 2004;90:352–358. doi: 10.1111/j.1471-4159.2004.02479.x. [DOI] [PubMed] [Google Scholar]