

Abstract
Gonadotropin-releasing hormone (GnRH) neurons release GnRH in a pulsatile manner to control fertility in all mammals. The mechanisms underlying burst firing in GnRH neurons, thought to contribute to pulsatile GnRH release, are not yet understood. Using minimally invasive, dual electrical–calcium recordings in acute brain slices from GnRH-Pericam transgenic mice, we find that the soma/proximal dendrites of GnRH neurons exhibit long-duration (∼10 s) calcium transients that are perfectly synchronized with their burst firing. These transients were found to be generated by calcium entry through voltage-dependent L-type calcium channels that was amplified by inositol-1,4,5-trisphosphate receptor-dependent store mechanisms. Perforated-patch current- and voltage-clamp electrophysiology coupled with mathematical modeling approaches revealed that these broad calcium transients act to control two slow afterhyperpolarization currents (sIAHP) in GnRH neurons: a quick-activating apamin-sensitive sIAHP that regulates both intraburst and interburst dynamics, and a slow-onset UCL2077-sensitive sIAHP that regulates only interburst dynamics. These observations highlight a unique interplay between electrical activity, calcium dynamics, and multiple calcium-regulated sIAHPs critical for shaping GnRH neuron burst firing.
Introduction
Calcium entry following the action potential has multiple effects on neuronal behavior including the activation of hyperpolarizing potassium channels that help shape the action potential and subsequent firing of the neuron (Berridge, 1998). Traditionally, these afterhyperpolarizations (AHPs) have been divided into fast (fAHP), medium (mAHP), and slow (sAHP) components mediated principally by large-conductance (BK; KCa1.1), small-conductance (SK; KCa2.1–2.3) and molecularly uncharacterized Ca2+-activated potassium channels, respectively (Sah and Faber, 2002; Vogalis et al., 2003; Maylie et al., 2004; Pedarzani and Stocker, 2008). The BK channel, with a decay of <50 ms, contributes to repolarization of the action potential (Salkoff et al., 2006), whereas the predominantly SK-mediated mAHP, typically with a decay of several hundred ms, controls spike discharge frequency (Sah and Faber, 2002; Pedarzani and Stocker, 2008). The precise identity of channels underlying the sAHP have remained enigmatic but their slow decay kinetics, in the order of seconds, have made them attractive targets for regulating spike frequency adaptation (SFA) and rhythmic firing patterns (Nicoll, 1988; Sah and Faber, 2002; Goldberg and Wilson, 2005; Wilson and Goldberg, 2006). It is increasingly apparent that individual neuronal phenotypes use different combinations of Ca2+-dependent potassium channels to generate their AHP and firing characteristics (Yamada et al., 2004; Power and Sah, 2008).
The gonadotropin-releasing hormone (GnRH) neurons represent the final output cells of the neuronal network controlling fertility in mammals. The cell bodies of these neurons are dispersed within the basal forebrain but project axons to a tightly circumscribed area in the basal hypothalamus, from which they release GnRH in an episodic manner. This drives pulsatile gonadotropin secretion from the pituitary gland that is essential for gonadal function (Herbison, 2006). Although the mechanisms driving GnRH neuron burst firing remain unknown, it is likely that, taking inference from other neuroendocrine cell types, the generation of bursting patterns of electrical activity in GnRH neurons underlie episodic GnRH release (Leng and Brown, 1997; Moenter et al., 2003; Herbison, 2006).
We recently generated a transgenic mouse line in which the genetically encodable Pericam Ca2+ indicator (Nagai et al., 2001) was targeted selectively to GnRH neurons (Jasoni et al., 2007). Real-time imaging of intracellular Ca2+ concentration ([Ca2+]i) in adult GnRH neurons in acute brain slices from these mice revealed a subpopulation of neurons exhibiting spontaneous long-duration (∼10 s) Ca2+ transients, a phenomenon not previously reported in the mature brain (Jasoni et al., 2007). Using noninvasive dual electrical recording–Ca2+ imaging approaches we find here that these Ca2+ transients occur only in burst firing GnRH neurons and result from voltage-dependent membrane Ca2+ entry amplified by Ca2+-induced Ca2+ release from inositol-1,4,5-trisphosphate receptor (IP3R)-dependent stores. Electrophysiological and modeling approaches revealed the presence of large calcium-activated (∼20 s) AHPs in GnRH neurons generated by fast activating, long-duration apamin-sensitive channels and slow activating, long-duration UCL2077 (Shah et al., 2006)-sensitive channels. These two channels had distinct roles in regulating GnRH neuron burst firing with the apamin-sensitive sIAHP controlling both the structure of firing within bursts and interburst interval (IBI) while UCL-sensitive sIAHP determined solely interburst dynamics.
Materials and Methods
Animals and slice preparation.
Adult (6–12 weeks old, male or female) homozygous GnRH-Pericam (Jasoni et al., 2007) and GnRH-GFP (Spergel et al., 1999) transgenic mice were used for all studies and housed according to standard laboratory conditions. The University of Otago Animal Ethics Committee approved all experimentation. Brain slices were prepared as reported previously (Han et al., 2005; Jasoni et al., 2007). In brief, mice were killed between 09:00–12:00 h, brains quickly dissected and cut on a vibratome into 200-μm-thick coronal slices containing the rostral preoptic area, and maintained in oxygenated artificial CSF (ACSF) containing the following (in mm): 118 NaCl, 3 KCl, 2.5 CaCl2, 1.2 MgCl2, 5 HEPES, 25 NaHCO3, 11 d-glucose, pH 7.3, 300∼310 mOsm, at 30°C for at least 60 min before imaging. Slices were then placed on the stage of an Olympus BX51 upright microscope equipped with a slice chamber and perfused with oxygenated ACSF solution at a rate of 3–4 ml/min. Recordings were undertaken at room temperature (21−23°C) unless indicated otherwise.
Dual Ca2+–electrical recordings.
The electrical properties and firing behavior of individual GnRH neurons are identical in GnRH-Pericam and GnRH-EGFP transgenic mice (K. Lee and A. E. Herbison, unpublished data). [Ca2+]i was monitored in individual GnRH neurons as reported previously (Jasoni et al., 2007; Romanò et al., 2008) with slight modifications. In brief, excitation was performed by exposing slices to 415 nm wavelength for 100 ms every second using Sutter λ DG-4 high-speed filter unit. Images were filtered at 525 nm and acquired using Metafluor 7.1 software to control the DG-4 and liquid-cooled ORCA-ER CCD camera (Hamamatsu) unit. Fluorescence intensities of emitted light from both a region of interest on the soma and the same area from the background were exported and analyzed with Origin Pro 7.5 to calculate the change of the intracellular calcium concentration. Calcium transients are presented as follows: [Ca2+]i = [F(t) − Frest(t)]/Frest(t), where F(t) and Frest(t) represent the soma and the background fluorescence intensity at time t, respectively. Values were then normalized into a percentage scale against [Ca2+]i at starting time. Cell-attached recordings of action currents (representing action potentials) from GnRH-Pericam neurons were made using glass pipettes (outer diameter: 1.5 mm; inner diameter: 1.17; tip resistance 5–7 MΩ) filled with 140 mm NaCl solution (Perkins, 2006). By applying a slight suction onto the cell membrane of Pericam-positive GnRH neurons, a loose seal (20–30 MΩ) was formed and recordings undertaken with a Multiclamp 700A amplifier (Molecular Devices) and Clampex 10.2 software. Action current signals (50–400 pA amplitude) were sampled at 1 kHz speed and filtered at 10 kHz with a Bessel filter. Electrical and Ca2+ recordings were synchronized by using Metafluor to control pClamp.
Perforated-patch-clamp recordings.
Gramicidin-perforated-patch recordings of GnRH neurons were undertaken as reported previously (Han et at 2005). In brief, adult male GnRH-EGFP mice were decapitated and 200-μm-thick coronal slices containing the rostral preoptic area prepared in a same way used for slice preparation from GnRH-Pericam mice. Slices were viewed with a fixed-stage upright microscope (BX51WI, Olympus) and either fluorescence illumination, using the reflected light fluorescence illuminator (BX-RFA, Olympus) and filter (U-MWIBA2, BA510–550, Olympus), or Nomarski differential interference contrast optics. Gramicidin (Sigma) was dissolved in dimethylsulfoxide (Sigma) to a concentration of 10 mg/ml and then diluted in the pipette solution [containing the following (in mm): 135 K-gluconate, 5 NaCl, 1 CaCl2, 1 MgCl2, 10 HEPES, 0.2 EGTA, 5 MgATP, 0.1 Na2GTP, pH 7.3, 286 mOsm] to a final concentration of 30–60 μg/ml just before use and sonicated for 2–3 min. Following formation of tight gigaohm seal, access resistance was monitored at regular intervals and experiments undertaken when access resistance had stabilized (10–30 min) between 30 and 100 MΩ. The patch configuration typically went into “whole-cell” mode when access resistance fell below 20 MΩ. Perforated-patch-clamp recordings were performed using a Multiclamp 700A. The tip resistance of the electrodes was 4–7 MΩ. Signals were sampled online using a Digidata 1322A interface (Molecular Devices) connected to a PC. Signals were filtered (10 kHz, Bessel filter of Multiclamp 700A) before digitizing at a rate of 1 kHz. Acquisition and subsequent analysis of the acquired data were performed using the Clampex 10.2 suite of software (Molecular Devices). Calculated junction potential of 14.8 mV was adjusted. Pipette capacitance and membrane capacitance were compensated.
Treatments.
All drugs (Tocris Biosciences) were applied to the ACSF bathing solution at the following established concentrations: apamin 300 nm (Liu and Herbison, 2008), UCL2077 10 μm (Shah et al., 2006), 2-aminoethoxydiphenylborate (2-APB) 100 μm (Jasoni et al., 2007), cyclopiazonic acid (CPA) 30 μm (Power and Sah, 2008) and tetrodotoxin (TTX) 0.5 μm (Sim et al., 2001). Apamin and TTX were dissolved in water while the others were dissolved initially in dimethylsulfoxide resulting in a final dimethylsulfoxide concentration in the perfusion solution of 0.03 to 0.1%.
Statistical analysis.
Correlation between single dependent and multiple independent variables was assessed with multiple regression method. Differences between the means were tested by Wilcoxon signed-ranks test unless indicated otherwise. Data are presented as mean ± SEM.
Mathematical model of GnRH neuron calcium dynamics and electrical activity.
The equation for membrane potential (V) is given by the following:
 |
where Cm is the membrane capacitance, Iinj represents a stochastically modeled synaptic injection current and Iionic = Inaf + Inap + Ikdr + Ikir + Ikm + Ical + Icat + sIAHP-SK + sIAHP-UCL + Ileak.
Inaf represents the fast sodium current; Inap a persistent sodium current; Ikdr, Ikir, and Ikm denote delayed rectifier, inward rectifier, and M-type potassium currents, respectively; Ical and Icat are L- and T-type calcium currents, respectively. Equations for these currents are taken from LeBeau et al. (2000) and Roberts et al. (2009). The two pacemaker currents are an SK-type [Ca2+]i-activated K+ current (sIAHP-SK), and a slow [Ca2+]i-activated afterhyperpolarization current (sIAHP-UCL). Ileak is the passive membrane leakage current.
Conventional Hodgkin–Huxley-type models are used for all currents, as follows:
and
 |
where c is the cytoplasmic concentration of free intracellular calcium. Equations for Nkdr and Nkm, or Hnaf have the following form:
 |
while all other gating variables are set to their steady-state values. The steady-state functions are as follows:
and
The time constants τx (units in milliseconds) are as follows:
 |
 |
and
The equation for sIAHP-UCL is as follows:
The following reaction scheme governs the sIAHP-UCL subunit transitions:

where OUCL and O*UCL are two open states and SUCL is the closed state.
The equation for Ileak is as follows:
The equations governing Ca2+ handling are as follows:
 |
and
 |
where c is the cytoplasmic free calcium concentration, and ce is the concentration of calcium in the endoplasmic reticulum (ER) (Li et al., 1994; Keener and Sneyd, 2008). The individual Ca2+ flux terms are as follows:
 |
 |
 |
and
 |
To model the IPR, we used the study by Sneyd and Dufour (2002).
The voltage submodel affects the calcium submodel via two calcium currents (Ical and Icat) which act as inputs to the calcium submodel. On the other hand, cytosolic calcium affects the voltage submodel via the calcium dependence of sIAHP-SK and sIAHP-UCL. Parameter values for the voltage submodel are (mostly) taken from LeBeau et al. (2000) and Roberts et al. (2009). The exceptions are the rate constants in the Markov model of the sIAHP-UCL current. These parameters were chosen so as to predict the correct IBI. All the parameter values for the calcium submodel are taken from previous work and are shown in Table 1 (Sneyd and Dufour, 2002; Keener and Sneyd, 2008).
Table 1.
Parameter values for mathematical model
| Parameter | Value | Parameter | Value |
|---|---|---|---|
| Cm | 16 pF | gnaf | 190 nS |
| gnap | 20 nS | gkdr | 2 nS |
| gkir | 0.02 nS | gkm | 8 nS |
| gcal | 0.05 nS | gcat | 2.1 nS |
| gsk | 0.3 nS | gUCL | 950 nS |
| gleak | 0.0389 nS | Vna | 60 mV |
| Vk | −80 mV | Vca | 100 mV |
| Vleak | 100 mV | k11 | 1 × 10−7 |
| k−11 | 1.2 | k22 | 0.5 |
| k33 | 3 × 10−5 | Ksk | 1 μm |
| nsk | 2 | a1 | 1 × 10−4 |
| a2 | 35 ms | a3 | 300 ms |
| a4 | 7 ms | a5 | 35 ms |
| α | 4.8 × 10−3 μm·μm·ms−1·pA−1 | Jer | 4 × 10−7 ms−1 |
| Kf | 1.92 × 10−4 ms−1 | VNaCa | 3.5 × 10−4 μm · ms−1 |
| Vp | 4.2 × 10−3 μm · ms−1 | Kp | 0.425 s |
| γ | 27 | KNaCa | 0.05 s |
| β | 2 × 10−5 ms | Prate | 1 |
| IP3 | 0.3 μm | ρ | 0.5 |
| δ1 | 1 × 10−9 | δ2 | 1 × 10−8 |
Iinj is set as a uniform distribution from 0.02 to 0.08 pA. Wiener processes are applied on variables OUCL and O*UCL with noise SDs of δ1 and δ2, respectively. These values were chosen by trial and error so as to obtain approximately correct bursting statistics. However, no detailed fit to the statistics was done.
Results
The GnRH-Pericam transgenic mouse has the advantage of enabling real-time measurement of [Ca2+]i in the soma and proximal dendrites of GnRH neurons in situ (Fig. 1a) without requiring loading of the cells or altering their internal milieu. The ratiometric Pericam exhibits a monophasic Ca2+ dependence with a dissociation constant (Kd) of 1.7 μm and Hill constant of 1.1 (Nagai et al., 2001). Initially, we performed minimally invasive loose-cell patch to make dual recordings of [Ca2+]i and action currents from adult GnRH neurons in the acute brain slice preparation. Recordings made for a minimum of 10 min from 217 adult (6–12 weeks old) GnRH neurons (56 male, 161 diestrous female) revealed three types of [Ca2+]i-firing relationships. The first (41%) comprised of “silent” GnRH neurons that had no action currents and exhibited a flat [Ca2+]i profile (Fig. 1b). When challenged with 20 mm potassium, silent GnRH neurons became active firing a train of action potentials that was associated with the generation of a Ca2+ transient followed by a subsequent depression in [Ca2+]i (Fig. 1b). The second type of cell was tonically active firing at a relatively constant rate (mean 2.6 ± 0.3 Hz, range 1.0–3.5 Hz) but was rare (n = 7, 3%) and displayed a flat [Ca2+]i profile (Fig. 1c). The final subpopulation (56%) of GnRH neurons exhibited burst firing for which there was perfect one-to-one correlation between bursts and Ca2+ transients (Fig. 1d). No differences in the percentages of the three different populations were found between male and female mice.
Figure 1.
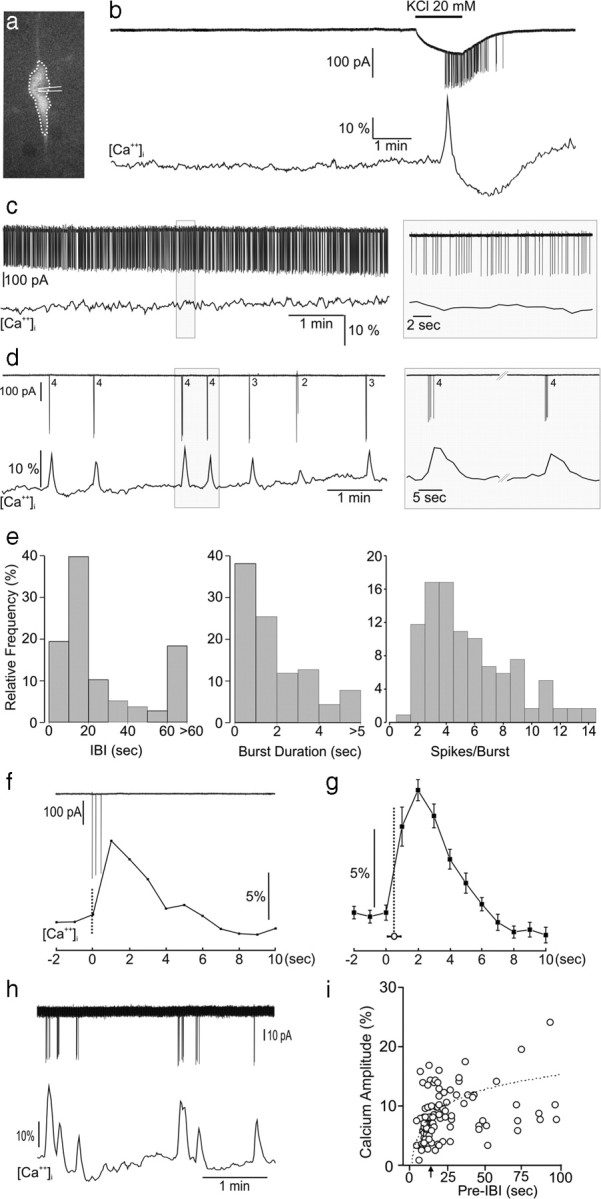
Relationship between action potential spiking and intracellular calcium concentrations [Ca2+]i in adult GnRH neurons. a, Fluorescent GnRH neuron in a brain slice of a GnRH-Pericam transgenic mouse indicating the soma/proximal dendrite area of interest (dotted line) for [Ca2+]i imaging and cell-attached recording electrode. b, Real-time dual electrical (action currents; top)–Ca2+ (bottom) recording from a silent GnRH neuron subsequently challenged with 20 mm KCl that generates a burst of firing and associated Ca2+ transient. c, Dual recording from a continuously active GnRH neuron. d, Dual recording from a burst firing GnRH neuron showing the typical perfect correlation between each burst (numbers = number spikes in burst) and occurrence of Ca2+ transients. e, Parameters of burst firing in GnRH neurons, IBI. f, Higher-temporal-resolution view of relationship between a three-spike burst and Ca2+ transient. g, Average of burst-transient timing by setting Ca2+ datum point immediately before the transient rise (defined as 3 × SD) as t = 0 and determining the time between that and the initiation of the burst (mean ± SEM; vertical dotted line). h, i, A significant correlation existed between IBI and the amplitude of the subsequent Ca2+ transient (r = 0.49, p < 0.001, n = 106; Spearman rank correlation) with transients occurring within ∼15 s (arrow) of the last burst having reduced amplitudes.
The relationship between burst firing and [Ca2+]i profile was evaluated in detail by analyzing 119 bursts from 13 randomly selected burst firing GnRH neurons. Analyses of GnRH neuron burst firing revealed a mean (±SEM) IBI of 40 ± 6 s, but with a bimodal-type distribution (Fig. 1e), mean burst duration of 2.2 ± 0.2 s with 90% of bursts being shorter than 5 s (Fig. 1e), mean spikes per burst of 5.7 ± 0.3 with 90% bursts having <10 spikes (Fig. 1e), and a mean interspike interval of 497 ± 51 ms. The associated Ca2+ transients had a mean Trise (baseline +10% to peak maximum) of 3.1 ± 0.2 s, Tdecay (peak maximum to decay baseline + 10%) of 5.1 ± 0.2 s, duration of 8.2 ± 0.4 s, and amplitude of 10.4 ± 0.7% baseline (Fig. 1f,g). Although limited by the 1 Hz [Ca2+]i imaging resolution, temporal correlations between burst onset and calcium onset indicated that Ca2+ transients followed burst initiation (Fig. 1f,g). Multiple regression analysis between the five burst parameters and three calcium parameters (Trise, Tdecay, amplitude) revealed only two significant associations: (1) Tdecay was positively associated with burst duration (β = 0.66, adjusted r2 = 0.50, p < 0.01) and (2) Ca2+ amplitude was associated with the pre-IBI (β = 0.60, adjusted r2 = 0.44, p < 0.01) such that an IBI of <15 s resulted in a transient with reduced amplitude (Fig. 1i), also observed in the raw traces (Fig. 1h). These results show that, as long as bursts arise with an interval of >15 s, Ca2+ transients of normal amplitude occur and greatly outlast the duration of the burst.
Dependence of Ca2+ transients on electrical activity and both internal and external calcium sources
To ensure that bursts did indeed initiate Ca2+ transients, GnRH neurons were either treated with TTX (0.5 μm) or depolarized through the patch electrode. TTX quickly abolished firing and the occurrence of Ca2+ transients in all 10 GnRH neurons tested and was reversible after extensive washing (Fig. 2a). In a further three GnRH neurons, direct depolarization through the patch electrode was found to initiate Ca2+ transients only when the neuron was induced to fire action currents (Fig. 2b).
Figure 2.

Burst firing drives calcium transients in GnRH neurons. a, Blocking voltage-dependent sodium channels with TTX stops both action potential firing and Ca2+ transients. b, Electrical stimulation applied through the cell-attached patch electrode initiates a Ca2+ transient only when action potentials are generated (compare right and left).
We evaluated the roles of both external and internal Ca2+ in generating Ca2+ transients in GnRH neurons. First, we reduced [Ca2+] in the extracellular space by replacing Ca2+ in the normal ACSF with equimolar Mg2+. This resulted in a gradual run down in the amplitude and Trise of Ca2+ transients with the area of calcium peaks decreasing from 94 ± 19 to 34 ± 33%.s (n = 6, p < 0.01, Dunn's multiple-comparison test) at 10 min (Fig. 3a–c) and was reversible (88 ± 38) with washout (Fig. 3a). The reduction and eventual loss of sharp Ca2+ transients correlated with a failure of the normal bursting pattern observed in GnRH neurons (Fig. 3b) with a 57% decrease in IBI (25.0 ± 3.9 s to 10.9 ± 1.5 s; p < 0.01), a threefold increase in burst duration (2.8 ± 0.3 s to 9.7 ± 2.0 s; p < 0.05) and an increase in spikes per burst (6.2 ± 0.5 vs 18.3 ± 3.6 spikes/burst; p < 0.05) (Fig. 3c). The role of L-type voltage-dependent calcium channels in calcium entry was examined by treating burst-firing GnRH neurons with 100 μm nifedipine. In all cases (n = 6), nifedipine resulted in a complete block of calcium transients and dysregulated burst firing (data not shown).
Figure 3.

Calcium transients derive from both extracellular and internal sources. a, Representative example showing the effect of switching to a zero [Ca2+] ACSF on burst firing (top) and Ca2+ transients (bottom). Insets for each experimental period are shown below. b, Expanded traces showing the relationship between burst firing structure and Ca2+ transients in control and calcium-free conditions. c, Mean (±SEM) data showing the effect of calcium-free ACSF on transient amplitude, interburst interval, and burst structure. *p < 0.05; **p < 0.01; Wilcoxon signed-ranks test; n = 6 each group. d, 2-APB, the IP3R blocker, suppresses both Ca2+ transients and burst firing. e, SERCA pump inhibitor CPA suppressed Ca2+ transient amplitude and resulted in disorganized burst firing.
The roles of internal stores were assessed by treating GnRH neurons with 100 μm 2-APB and 30 μm CPA; antagonists at IP3Rs and the sarcoplasmic reticulum Ca2+-ATPase pump, respectively. 2-APB was found to block the calcium transients and, unexpectedly, burst firing (n = 11) (Fig. 3d). Four to 5 min after treatment of GnRH neurons (n = 7) with CPA, Ca2+ transient amplitude was suppressed and burst firing became disorganized (Fig. 3e); the IBI decreased significantly from 57.6 ± 12.7 s to 29.5 ± 6.2 s in the presence of CPA (p = 0.016) but burst duration (2.9 ± 1.0 vs 3.3 ± 1.4 s), spikes/burst (5.7 ± 1.6 vs 6.3 ± 2.2) and intraburst frequency (3.7 ± 0.7 vs 5.5 ± 2.2 Hz) were not significantly different.
These observations revealed that Ca2+ transients in GnRH neurons were dependent upon action potential generation and both internal and external pools of calcium; indicating that Ca2+ transients in GnRH neurons arise from L-type voltage-dependent membrane Ca2+ flux initiating Ca2+-induced Ca2+ release from IP3R-dependent internal stores. Importantly, the loss of this calcium amplification mechanism resulted in dysregulated burst firing suggesting that one role of the Ca2+ transient was to shape burst dynamics in GnRH neurons.
Modeling of Ca2+ dynamics in GnRH neurons
In an attempt to better understand the mechanisms underlying electrical bursting and calcium transients and their interactions in GnRH neurons, we constructed a mathematical model (Fig. 4a). Electrical spiking results from the interaction of Na+ and K+ channels while a voltage-gated Ca2+ channel allows for the entry of Ca2+ during the burst. Ca2+ coming in from outside then stimulates the release of additional Ca2+ from the ER, via IP3-gated Ca2+ channels, resulting in an amplified Ca2+ transient. Prior studies in GnRH neurons have shown that an apamin-sensitive SK channel controls spike frequency adaptation in these cells (Kato et al., 2006; Liu and Herbison, 2008). This channel (sIAHP-SK) was modeled to help terminate electrical bursting, while another slow Ca2+-sensitive K+ current (sIAHP-UCL), was necessary to set the IBI in GnRH neurons. The UCL nomenclature was used due to subsequent results (vide infra).
Figure 4.
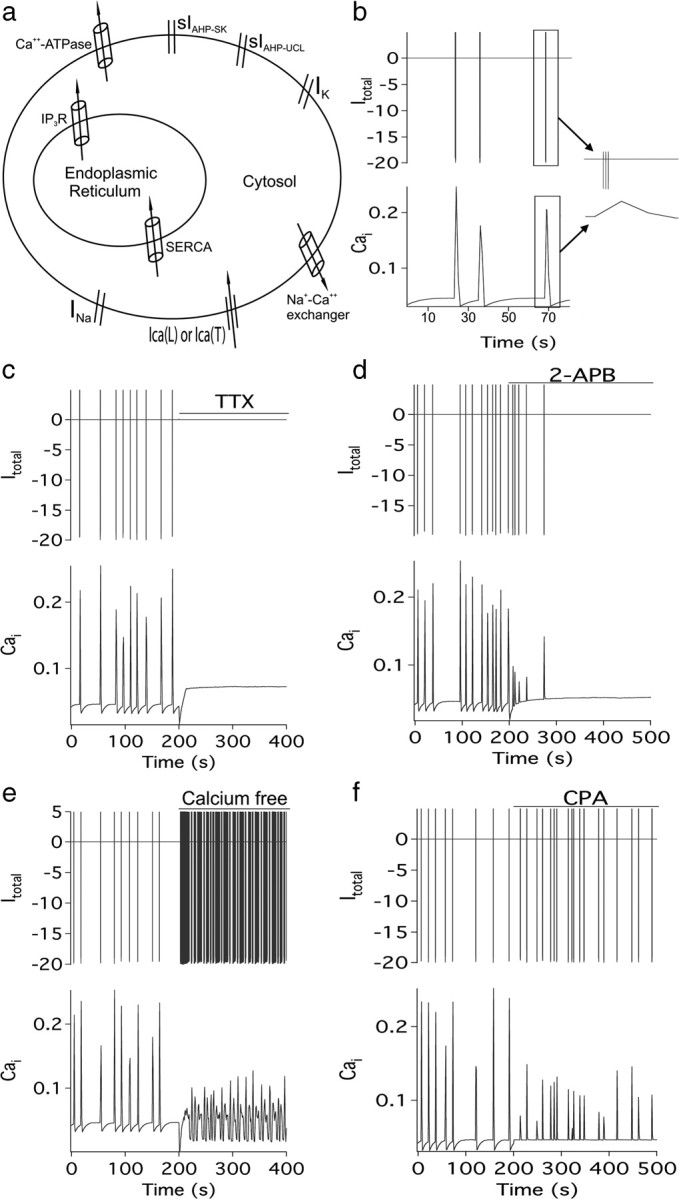
Mathematical model of GnRH neuron calcium and electrical behavior. a, Schematic showing the key channels and pumps located in the ER and plasma membrane that comprise the model (see Materials and Methods for full description). b, Baseline behavior of the model showing the relationship between burst firing (top) and [Ca2+]i (bottom). The inset shows the structure of a typical burst. Subsequent panels show the effects of TTX (Inap and Inaf set to zero) (c) and 2-APB ([IP3] set to zero, SERCA pump set to 30% and Ca2+-ATPase set to 25%) (d). The precise effects of 2-APB on calcium pumps are not known, and thus these figures of 30 and 25% were chosen so as to obtain approximately the observed behavior. However, the results are not sensitive to the values chosen. e, Calcium free (Ical set to 40%; Icat set to 30%). Experimental data indicate that calcium entry is not completely eliminated by the calcium-free solution, although the precise extent to which calcium influx is decreased is not known. Again, the figures of 40 and 30% are chosen so as to obtain approximately the correct behavior, and the model is insensitive to the exact numbers used. f, Effect of CPA (SERCA set to zero) on firing and [Ca2+]i.
Figure 4 shows simulations from the model. Electrical spiking and calcium transients are highly synchronized (Fig. 4b) and eliminated by either TTX or 2-APB (compare Fig. 4c,d to Figs. 2a, 3d). Low extracellular Ca2+ or CPA-evoked emptying of calcium stores causes a rundown of the Ca2+ transients and a loss of regular bursting activity (Fig. 4e,f). Two things in particular should be noted. First, we found it was impossible to explain the entire set of experimental results without the existence of sIAHP-UCL; the model predicted that such a channel would be found in this cell type, which turned out to be the case (Fig. 5). Furthermore, we used the model to predict the behavior that would be observed if sIAHP-UCL were to be blocked, a prediction that we confirmed experimentally. Second, the model provides an explanation for the puzzling experimental result that 2-APB eliminates electrical spiking, even though it is the spiking that is causing the Ca2+ transients. When 2-APB was modeled to inhibit only the IP3R we obtained the same result as that seen with CPA. However, when 2-APB was modeled to inhibit the plasma membrane calcium ATPase and sarcoendoplasmic reticulum Ca2+-ATPase (SERCA) pump as well as the IP3R [as is thought to occur (Missiaen et al., 2001; Peppiatt et al., 2003)], we observed the same behavior as that seen in experimental recordings (Fig. 3d). This results from an increase in intracellular calcium that eliminates electrical spiking via the activation of sIAHP-UCL (Fig. 4d).
Figure 5.

The AHP in GnRH neurons is mediated by two sIAHPs. a, Perforated-patch, voltage-clamp traces from two GnRH neurons showing the evoked IAHP and its modulation by 300 nm apamin and 10 μm UCL2077. The top cell received apamin then UCL2077 while the bottom cell received the blockers in the reverse order. Each curve is the mean of two repetitions. b, Group (mean ± SEM) data showing fold decrease in IAHP by apamin alone and then combined with UCL2077 (left) or UCL2077 alone and combined with apamin (right; **p < 0.01; Dunnett's multiple-comparison test vs control; n = 5 each group). c, Derived dynamics of apamin- and UCL2077-sensitive currents generated by averaging subtracted curves from individual cells (n = 8). The kinetics of both apamin-sensitive and UCL2077-sensitive currents were well described by exponential rising and decay functions (black lines; with r = 0.89 and r = 0.87, respectively). d, Effects of apamin or UCL2077 on evoked (400 pA for 5 ms at 20 Hz) AHP measured in current-clamp mode. Apamin evoked a large afterdepolarization potential and variable (p > 0.05) suppressive effects on the AHP. UCL blocked the AHP only. e, Effect of apamin (top) and UCL2077 (bottom) on intraburst dynamics induced by 30 pA current for 2 s. f, Group data (mean ± SEM, n = 5 each) showing effects of apamin and UCL2077 on number of spikes per burst. **p < 0.01; Wilcoxon signed-ranks test.
GnRH neurons exhibit long AHPs mediated by apamin- and UCL2077-sensitive channels
The modeling studies predicted that two slow Ca2+-activated potassium channels were required to explain the prior correlations observed between burst firing and Ca2+ transients in GnRH neurons. To examine these currents and their functions in GnRH neurons, we undertook voltage- and current-clamp recordings from GFP-tagged GnRH neurons in the acute brain slice. Despite the expression of Pericam selectively in almost all GnRH neurons (Jasoni et al., 2007), the level of recordable Pericam fluorescence is low so that typically only one or two cells can be tested in any one animal. To ensure a higher chance of obtaining a tight gigaohm seal for the clamping experiments, we recorded from GFP-tagged GnRH neurons in the acute brain slice (Spergel et al., 1999). Pilot experiments undertaken in whole-cell revealed that this mode of recording was inappropriate for examining burst firing in GnRH neurons probably due to the extensive dialysis of cell milieu by the electrode; the AHP was not evident and normal burst firing including SFA was difficult to observe. As such, we used a less invasive approach using perforated patch, by which we were able to observe normal burst firing dynamics in GnRH neurons. Using a protocol established previously to fully activate the AHP in GnRH neurons (Liu and Herbison, 2008), we confirmed the presence of a large, slowly decaying AHP (peak amplitude: 36 ± 9 pA; charge: 873 ± 322 pC; Tdecay 22 ± 3 s, n = 10) (Fig. 5a). As the kinetics of the Ca2+-dependent sIAHP is sensitive to temperature (Lee et al., 2005) we performed the same perforated-patch voltage-clamp recordings at higher temperatures (32−34°C) but again found a large, slowly decaying AHP (peak amplitude: 42 ± 5 pA; charge: 437 ± 146 pC; Tdecay 14 ± 3 s, n = 9, data not shown).
Previous studies in GnRH neurons have shown that 70–90% of the IAHP is mediated by apamin-sensitive channels, with the identity of the remaining sIAHP current unknown. The only available antagonist of the sIAHP is the UCL2077 compound that has been demonstrated to be a selective blocker of the sIAHP in hippocampal pyramidal cells, with minimal effects on SK channels and the mIAHP (Shah et al., 2006). The relatively poor solubility of UCL2077 results in 10 μm being the highest concentration that can be used. Perforated patch voltage-clamp recordings were made from 10 GnRH neurons. In five GnRH neurons in which apamin was tested first, 300 nm apamin caused a complete block of IAHP in two cells (data not shown) with the other three GnRH neurons exhibiting a partial blockade (Fig. 5a) with the remaining current eliminated by 10 μm UCL2077 (Fig. 5a). On average, apamin achieved a 72% inhibition of IAHP (range 24–100%) and the combination of apamin and UCL2077 resulted in a 90% blockade of IAHP in these five GnRH neurons (Fig. 5b). In five GnRH neurons in which UCL2077 was tested first, all cells responded and exhibited a mean 64% suppression in IAHP (range 23–93%) following UCL2077 (Fig. 5a,b) and this was enhanced to a near complete 96% suppression on addition of apamin (Fig. 5a,b). Data from the eight GnRH neurons exhibiting both currents were used to determine the kinetics of the currents underlying the IAHP (Fig. 5c). The apamin-sensitive current appeared almost instantaneously after the stimulation and reached a peak of 34 pA with a two-phase decay of 0.3 s followed by 19.0 s (total charge 352 pC) (Fig. 5c). In contrast, the UCL-sensitive current peaked at 14.2 s (amplitude 20 pA; total charge 634 pC) with τrise of 9.7 s and τdecay of 21.0 s (Fig. 5c). The control vehicle (ACSF containing 0.1–0.03% dimethylsulfoxide) did not alter the IAHP.
The effects of apamin and UCL2077 on the membrane excitability of 10 GnRH neurons were examined using a burst stimulation protocol [400 pA for 5 ms at 20 Hz (Chu and Moenter, 2006)] in current-clamp mode. This evoked a slow 3.2 ± 1.9 mV AHP (τdrop = 1.8 ± 0.5 s; τrecovery = 16.2 ± 5.4 s) (Fig. 5d). The effects of 300 nm apamin to suppress the AHP were variable between cells but, as a group, had no significant effect (60 ± 38% of the control, p = 0.37, n = 5) (Fig. 5d). This was likely due to the ability of apamin to allow the appearance of a prominent slow afterdepolarizing potential (sADP) in GnRH neurons (4.9 ± 2.3 mV, n = 5) (Fig. 5d) (Chu and Moenter, 2006; Liu and Herbison, 2008) that counteracts the ability to observe any suppressive effect of apamin on the AHP. This possibility was confirmed by showing a significant decrease in sAHP by apamin when the sADP was more strongly suppressed by injecting a higher level of current (1500 pA for 10 ms at 20 Hz). In the voltage-clamp mode, sIAHP was significantly suppressed from 146.9 ± 22.8 pA · s to 56.0 ± 19.3 pA · s (p = 0.008, n = 8). As a consequence, the AHP was significantly depressed from −73 ± 21 mV · s to −0.1 ± 15.8 mV · s (p = 0.007, n = 8) in the current-clamp mode. In contrast to apamin, 10 μm UCL2077 significantly reduced the AHP to 22 ± 10% of the control (p = 0.03, n = 5) (Fig. 5d) but had no effect on ADP magnitude.
These observations show that the long-lasting AHP in GnRH neurons is generated by two pharmacologically and kinetically distinct sIAHP currents.
The two sAHP currents in GnRH neurons regulate different aspects of burst firing
Testing the effects of apamin (sIAHP-SK set to zero) and UCL2077 [sIAHP-UCL set to 50%; 10 μm UCL2077 is reported to be the IC50 for blocking the sIAHP in hippocampal cells (Shah et al., 2006)] in the mathematical model of Ca2+ dynamics in GnRH neurons predicted that apamin would slow burst firing and reduce SFA while UCL2077 would decreased the IBI and have little effect upon intraburst firing dynamics (data not shown).
Current-clamp recordings were undertaken to evaluate the effects of apamin and UCL2077 on evoked and endogenous SFA in GnRH-GFP neurons. As shown previously in GnRH neurons (Liu and Herbison, 2008), a 30 pA current injection protocol for 2 s initiates a burst of action potentials exhibiting strong SFA (Fig. 5e). Treatment with 300 nm apamin resulted in threefold increase in spike number and clear reduction in SFA (n = 5) (Fig. 5e,f). In contrast, 10 μm UCL2077 (10 μm) had no significant effect on the number of spikes or SFA (Fig. 5e,f). To examine the effects of apamin and UCL2077 on the patterning of endogenous bursting and calcium dynamics in GnRH neurons, noninvasive dual electrical-Ca2+ recordings were undertaken in GnRH-Pericam mice. Apamin significantly slowed the rate of burst induction (Fig. 6a,b) (9.6 ± 2.3 bursts vs 5.0 ± 1.2 bursts, p = 0.04, n = 7) and exhibited the same effects on intraburst dynamics as seen in evoked responses (Fig. 5f). Apamin induced more spikes per burst with loss of SFA, and a modest increase in intraburst frequency (Fig. 6b). As indicated by our previous correlational analysis, Ca2+ transients (amplitude, Trise, or Tdecay) were not altered significantly by changes in burst firing induced by apamin. In contrast to apamin, treatment with UCL2077 resulted in a marked shortening of IBI (5.3 ± 1.2 bursts/10 min in control vs 11.3 ± 2.8 bursts/10 min with UCL2077; p = 0.035, n = 6) (Fig. 6c,d) with no significant effects on intraburst spiking dynamics (burst duration, spikes/burst, intraburst frequency) (Fig. 6d). As predicted by the relationship determined above between IBI and transient amplitude (Fig. 1h,i), the markedly decreased IBI in the presence of UCL2077 was associated with a significant decrease of the amplitude of calcium transients (13.5 ± 1.3% baseline to 9.0 ± 1.3% baseline, n = 7, p = 0.015).
Figure 6.
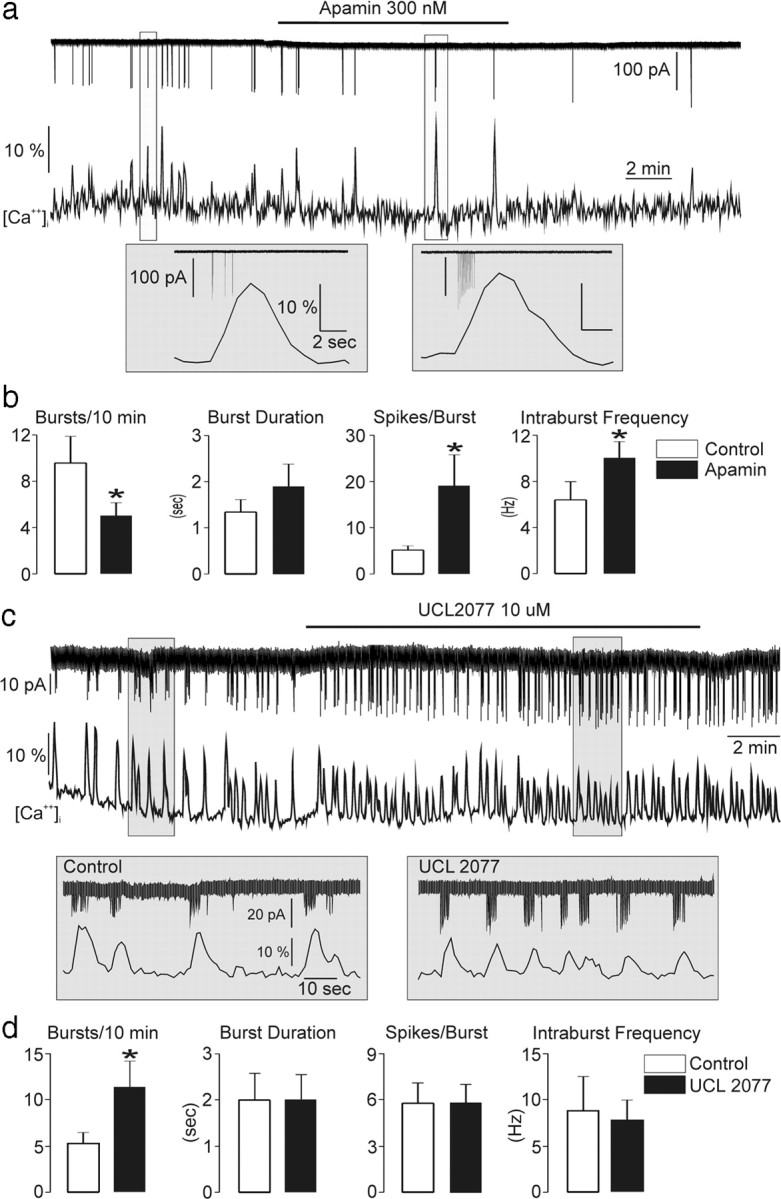
The apamin- and UCL2077-sensitive currents differentially modulate GnRH neuron burst firing dynamics. a, Representative trace showing the effect of apamin on burst firing and Ca2+ transients. Insets for each experimental period are shown below. b, Mean (±SEM) data showing effects of apamin on burst dynamics in GnRH neurons (*p < 0.05; Wilcoxon signed-ranks test; n = 7). c, Representative trace showing the effect of UCL2077 on burst firing and Ca2+ transients. Insets for each experimental period are shown below. d, Mean (±SEM) data showing effects of UCL2077 on burst firing dynamics in GnRH neurons. *p < 0.05; Wilcoxon signed-ranks test; n = 7.
Discussion
These studies demonstrate that the slow Ca2+ transients observed in adult GnRH neurons represent a faithful signature of their burst firing. Initiated by voltage-dependent Ca2+ entry across the plasma membrane and amplified by Ca2+-induced Ca2+ release from internal stores, the ∼10 s duration Ca2+ transients greatly outlast the ∼2-s-long action potential bursts of GnRH neurons. While such Ca2+ transients will likely have multiple roles within the GnRH neuron, we show here that one important task is to confine burst firing dynamics to typically five spikes per burst with an IBI of 15–20 s. This is achieved by the Ca2+ transient regulating two pharmacologically distinct channels underlying the sAHP of GnRH neurons; an apamin-sensitive channel controlling both intraburst and interburst dynamics and a UCL2077-sensitive channel controlling interburst dynamics.
The broad Ca2+ transient in GnRH neurons is initiated by voltage-dependent calcium entry across the membrane. Spontaneous transients were (1) abolished by tetrodotoxin, (2) initiated by 20 mm potassium, and (3) evoked by direct depolarization of the cell only when action potentials were generated. Calcium entry through voltage-gated calcium channels would, however, be too brief to explain the ∼3 s rise kinetics of the observed Ca2+ transient. It was not surprising, therefore, to find that both 2-APB and CPA suppressed Ca2+ transients in GnRH neurons indicating an important role for intracellular stores. Together these observations indicate that IP3R-dependent Ca2+-induced Ca2+ release is used to amplify the brief Ca2+ membrane flux into a prolonged Ca2+ transient. Specific voltage-dependent Ca2+ channels are associated with the activation of different Ca2+-activated potassium channels in striatal neurons (Goldberg and Wilson, 2005) and compatible with this, we found that L-type calcium channels are responsible for store-amplified calcium influx in GnRH neurons. Interestingly, the amplitude and kinetics of individual Ca2+ transients were relatively homogeneous and found to be independent of most burst parameters including spike number (r = 0.10); indeed, the occasional burst comprised of only one spike could generate a normal-sized Ca2+ transient equivalent to that of a five-spike burst. This suggests that stores release a certain “quanta” of Ca2+ in response to a range of plasma membrane Ca2+ influx. However, as has been noted previously (Power and Sah, 2005), store release can fail if activated with insufficient time for refilling. This is likely occurring in GnRH neurons, for which an IBI of <15 s resulted in the subsequent Ca2+ transient having a reduced amplitude. Thus, as long as individual bursts are spaced sufficiently, spontaneous burst firing in GnRH neurons results in very robust, prolonged Ca2+ transients.
Roles for SK, KCNQ, and molecularly unidentified Ca2+-actvated potassium channels have being described for generating the AHP in neurons (Sah and Faber, 2002; Gu et al., 2005; Pedarzani and Stocker, 2008; Tzingounis and Nicoll, 2008). In nearly all neurons examined, the SK channel is implicated in mediating the majority of the mAHP with little direct role in the sAHP (Bond et al., 2004; Pedarzani and Stocker, 2008). The one exception to this is the GnRH neuron, for which the apamin-sensitive current persists for several seconds in both mice and rats (Kato et al., 2006; Spergel, 2007; Liu and Herbison, 2008). As GnRH neurons express mRNA for all three SK channels (Bosch et al., 2002; Kato et al., 2006) it is very likely that SK channels underlie the apamin-sensitive currents (sIAHP-SK) in these cells. However, it is unclear why the sIAHP-SK decay is so prolonged in these cells and best modeled by a two phase dynamic comprised of a rapid (∼0.3 s) and a long decay (∼19 s). The initial decay is compatible with the mAHP current mediated by the SK channel recorded in other neurons, but the latter slow phase appears unique to GnRH neurons. In other neurons, SK channel activation and decay tends to faithfully follow the cytosolic [Ca2+]i resulting from voltage-dependent Ca2+ entry (Sah and Faber, 2002). This remains the situation even when IP3R-dependent stores are involved in generating Ca2+ transients regulating SK currents (Yamada et al., 2004; Gulledge and Stuart, 2005). This would not explain the situation fully in GnRH neurons, however, in which the long ∼8 s Ca2+ transients are only lasting for approximately half the duration of the sIAHP-SK current (∼20 s). The reasons for such prolonged currents in GnRH neurons are not known and may result, in part, from the existence of membrane-associated Ca2+ microdomains, unusual binding affinities of Ca2+ to calcium sensors and buffers (Sah and Clements, 1999; Tzingounis et al., 2007) and/or unique patterns of kinase-dependent channel phosphorylation (Maingret et al., 2008).
Prompted by the curious results we obtained with 2-APB inhibiting burst firing, we initiated a mathematical modeling exercise to help understand the likely complex Ca2+ regulation of GnRH neuron electrical excitability. In undertaking this exercise, it became readily apparent that our initial results could not be explained solely by modeling known or even slow SK channel function and that an independent, longer duration calcium-activated channel suppressing burst onset was required. This suggested the presence of the unidentified Ca2+-activated potassium channel underlying the sAHP as has been described in several cell types (Sah and Faber, 2002). The UCL2077 compound has been demonstrated to be a selective blocker of the sAHP in hippocampal pyramidal cells, with minimal effects on SK channels and the mIAHP (Shah et al., 2006). We find here that 10 μm UCL2077 is extremely effective in blocking nearly all of the remaining apamin-insensitive component of the sAHP current in GnRH neurons. In hippocampal neurons, 10 μm UCL2077 (the highest concentration that can be used in vitro) is thought to be the IC50 for the sAHP current (Shah et al., 2006) but it would appear to be much more effective here in GnRH neurons. While it remains possible that UCL2077 could have a small impact upon SK channels (Shah et al., 2006), this was not apparent in the present studies as apamin and UCL2077 exhibited very different, and even opposite, effects upon the sADP, sIAHP amplitude, SFA and GnRH neuron burst firing. Such observations indicate that UCL2077 is not modulating SK channel activity and demonstrate distinct roles for sIAHP-SK and sIAHP-UCL in regulating GnRH neuron excitability. The UCL2077-sensitive current was found to have a slow onset (τrise = 9.7 s) and very slow single exponential decay (τdecay = 21 s). As for sIAHP-SK, the duration of sIAHP-UCL in GnRH neurons is very prolonged compared with sAHP and sIAHP-UCL reported in other neuronal phenotypes and this may reflect unique Ca2+ handling in GnRH neurons or even the expression of particular subtypes of the as yet uncharacterized channels underlying the sIAHP.
Treatment with apamin revealed that sIAHP-SK is responsible for SFA adaptation in GnRH neurons and normally restricts GnRH neuron bursts to ∼5 spikes/burst. In contrast, sIAHP-UCL had little or no effect upon burst structure but exerted a powerful restraining influence upon the timing of bursts. Recent studies have highlighted likely interactions between SK and sIAHP channels in the regulation of neuronal firing in the striatum and amygdala (Power and Sah, 2008; Goldberg et al., 2009). Although we find some evidence for interaction in terms of regulating IBI, burst dynamics are clearly dependent on only sIAHP-SK. This likely to be due to the activation kinetics of sIAHP-UCL (τrise ∼10 s) being too slow to contribute to the structure of bursts that only last ∼2 s.
As with other neuroendocrine output neurons, it seems likely that the bursting behavior of GnRH neurons is key for pulsatile GnRH secretion (Leng and Brown, 1997; Moenter et al., 2003; Herbison, 2006) but the mechanisms responsible for burst firing had not been delineated until now. We show here that a critical feature for burst firing by GnRH neurons is their ability to exhibit prolonged Ca2+ transients that act, in turn, to control very slow IAHPs that regulate intraburst and interburst properties. It is interesting to note that magnocellular oxytocin and vasopressin neurons can exhibit somewhat similar bursting behaviors to GnRH neurons but do not exhibit prolonged calcium transients or use calcium-dependent potassium channels in the same manner (Li and Hatton, 1997; Ghamari-Langroudi and Bourque, 2004; Teruyama and Armstrong, 2005). The dynamics of Ca2+ transients in GnRH neurons are unique in the mature nervous system as is the existence of a very slow AHP generated by two sIAHPs in these cells. That one of these currents is apamin sensitive is surprising given prior data indicating that SK channels underlie currents with a duration of 100 ms (Pedarzani and Stocker, 2008), rather than many seconds.
While the ionic mechanism elucidated here is demonstrated to constrain GnRH neuron burst firing to ∼5 action potentials per burst every 20 s, it remains that the initiating mechanism for a burst is unknown. The GnRH neurons exhibit a slow ADP (Chu and Moenter, 2006) that may contribute, as might hyperpolarization-activated channels (Kelly and Wagner, 2002). Indeed, one plausible scenario underpinning GnRH neuron burst firing would be that calcium-activated sIAHPs act to regulate the balance between sAHPs and sADPs (Brown and Bourque, 2006) responsible for initiating the next burst of firing. The present findings help define the ionic basis for burst firing in the GnRH neurons and outline a unique calcium sIAHP mechanism for achieving this.
Footnotes
These studies were supported by the New Zealand Marsden Fund and Health Research Council.
References
- Berridge MJ. Neuronal calcium signaling. Neuron. 1998;21:13–26. doi: 10.1016/s0896-6273(00)80510-3. [DOI] [PubMed] [Google Scholar]
- Bond CT, Herson PS, Strassmaier T, Hammond R, Stackman R, Maylie J, Adelman JP. Small conductance Ca2+-activated K+ channel knock-out mice reveal the identity of calcium-dependent afterhyperpolarization currents. J Neurosci. 2004;24:5301–5306. doi: 10.1523/JNEUROSCI.0182-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosch MA, Kelly MJ, Rønnekleiv OK. Distribution, neuronal colocalization, and 17beta-E2 modulation of small conductance calcium-activated K(+) channel (SK3) mRNA in the guinea pig brain. Endocrinology. 2002;143:1097–1107. doi: 10.1210/endo.143.3.8708. [DOI] [PubMed] [Google Scholar]
- Brown CH, Bourque CW. Mechanisms of rhythmogenesis: insights from hypothalamic vasopressin neurons. Trends Neurosci. 2006;29:108–115. doi: 10.1016/j.tins.2005.11.005. [DOI] [PubMed] [Google Scholar]
- Chu Z, Moenter SM. Physiologic regulation of a tetrodotoxin-sensitive sodium influx that mediates a slow afterdepolarization potential in gonadotropin-releasing hormone neurons: possible implications for the central regulation of fertility. J Neurosci. 2006;26:11961–11973. doi: 10.1523/JNEUROSCI.3171-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghamari-Langroudi M, Bourque CW. Muscarinic receptor modulation of slow afterhyperpolarization and phasic firing in rat supraoptic nucleus neurons. J Neurosci. 2004;24:7718–7726. doi: 10.1523/JNEUROSCI.1240-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberg JA, Wilson CJ. Control of spontaneous firing patterns by the selective coupling of calcium currents to calcium-activated potassium currents in striatal cholinergic interneurons. J Neurosci. 2005;25:10230–10238. doi: 10.1523/JNEUROSCI.2734-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberg JA, Teagarden MA, Foehring RC, Wilson CJ. Nonequilibrium calcium dynamics regulate the autonomous firing pattern of rat striatal cholinergic interneurons. J Neurosci. 2009;29:8396–8407. doi: 10.1523/JNEUROSCI.5582-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu N, Vervaeke K, Hu H, Storm JF. Kv7/KCNQ/M and HCN/h, but not KCa2/SK channels, contribute to the somatic medium after-hyperpolarization and excitability control in CA1 hippocampal pyramidal cells. J Physiol. 2005;566:689–715. doi: 10.1113/jphysiol.2005.086835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gulledge AT, Stuart GJ. Cholinergic inhibition of neocortical pyramidal neurons. J Neurosci. 2005;25:10308–10320. doi: 10.1523/JNEUROSCI.2697-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han SK, Gottsch ML, Lee KJ, Popa SM, Smith JT, Jakawich SK, Clifton DK, Steiner RA, Herbison AE. Activation of gonadotropin-releasing hormone (GnRH) neurons by kisspeptin as a neuroendocrine switch for the onset of puberty. J Neurosci. 2005;25:11349–11356. doi: 10.1523/JNEUROSCI.3328-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herbison AE. Physiology of the GnRH neuronal network. In: Neill JD, editor. Knobil and Neill's physiology of reproduction. Ed 3. San Diego: Academic; 2006. pp. 1415–1482. [Google Scholar]
- Jasoni CL, Todman MG, Strumia MM, Herbison AE. Cell type-specific expression of a genetically encoded calcium indicator reveals intrinsic calcium oscillations in adult gonadotropin-releasing hormone neurons. J Neurosci. 2007;27:860–867. doi: 10.1523/JNEUROSCI.3579-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato M, Tanaka N, Usui S, Sakuma Y. The SK channel blocker apamin inhibits slow afterhyperpolarization currents in rat gonadotropin-releasing hormone neurones. J Physiol. 2006;574:431–442. doi: 10.1113/jphysiol.2006.110155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keener J, Sneyd J. Mathematical physiology. Ed 2. New York: Springer; 2008. [Google Scholar]
- Kelly MJ, Wagner EJ. GnRH neurons and episodic bursting activity. Trends Endocrinol Metab. 2002;13:409–410. doi: 10.1016/s1043-2760(02)00698-7. [DOI] [PubMed] [Google Scholar]
- LeBeau AP, Van Goor F, Stojilkovic SS, Sherman A. Modeling of membrane excitability in gonadotropin-releasing hormone-secreting hypothalamic neurons regulated by Ca2+-mobilizing and adenylyl cyclase-coupled receptors. J Neurosci. 2000;20:9290–9297. doi: 10.1523/JNEUROSCI.20-24-09290.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JC, Callaway JC, Foehring RC. Effects of temperature on calcium transients and Ca2+-dependent afterhyperpolarizations in neocortical pyramidal neurons. J Neurophysiol. 2005;93:2012–2020. doi: 10.1152/jn.01017.2004. [DOI] [PubMed] [Google Scholar]
- Leng G, Brown D. The origins and significance of pulsatility in hormone secretion from the pituitary. J Neuroendocrinol. 1997;9:493–513. doi: 10.1046/j.1365-2826.1997.00615.x. [DOI] [PubMed] [Google Scholar]
- Li YX, Rinzel J, Keizer J, Stojilković SS. Calcium oscillations in pituitary gonadotrophs: comparison of experiment and theory. Proc Natl Acad Sci U S A. 1994;91:58–62. doi: 10.1073/pnas.91.1.58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Hatton GI. Ca2+ release from internal stores: role in generating depolarizing after-potentials in rat supraoptic neurones. J Physiol. 1997;498:339–350. doi: 10.1113/jphysiol.1997.sp021862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Herbison AE. Small-conductance calcium-activated potassium channels control excitability and firing dynamics in gonadotropin-releasing hormone (GnRH) neurons. Endocrinology. 2008;149:3598–3604. doi: 10.1210/en.2007-1631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maingret F, Coste B, Hao J, Giamarchi A, Allen D, Crest M, Litchfield DW, Adelman JP, Delmas P. Neurotransmitter modulation of small-conductance Ca2+-activated K+ channels by regulation of Ca2+ gating. Neuron. 2008;59:439–449. doi: 10.1016/j.neuron.2008.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maylie J, Bond CT, Herson PS, Lee WS, Adelman JP. Small conductance Ca2+-activated K+ channels and calmodulin. J Physiol. 2004;554:255–261. doi: 10.1113/jphysiol.2003.049072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Missiaen L, Callewaert G, De Smedt H, Parys JB. 2-Aminoethoxydiphenyl borate affects the inositol 1,4,5-trisphosphate receptor, the intracellular Ca2+ pump and the non-specific Ca2+ leak from the non-mitochondrial Ca2+ stores in permeabilized A7r5 cells. Cell Calcium. 2001;29:111–116. doi: 10.1054/ceca.2000.0163. [DOI] [PubMed] [Google Scholar]
- Moenter SM, DeFazio AR, Pitts GR, Nunemaker CS. Mechanisms underlying episodic gonadotropin-releasing hormone secretion. Front Neuroendocrinol. 2003;24:79–93. doi: 10.1016/s0091-3022(03)00013-x. [DOI] [PubMed] [Google Scholar]
- Nagai T, Sawano A, Park ES, Miyawaki A. Circularly permuted green fluorescent proteins engineered to sense Ca2+ Proc Natl Acad Sci U S A. 2001;98:3197–3202. doi: 10.1073/pnas.051636098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicoll RA. The coupling of neurotransmitter receptors to ion channels in the brain. Science. 1988;241:545–551. doi: 10.1126/science.2456612. [DOI] [PubMed] [Google Scholar]
- Pedarzani P, Stocker M. Molecular and cellular basis of small- and intermediate-conductance, calcium-activated potassium channel function in the brain. Cell Mol Life Sci. 2008;65:3196–3217. doi: 10.1007/s00018-008-8216-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peppiatt CM, Collins TJ, Mackenzie L, Conway SJ, Holmes AB, Bootman MD, Berridge MJ, Seo JT, Roderick HL. 2-Aminoethoxydiphenyl borate (2-APB) antagonises inositol 1,4,5-trisphosphate-induced calcium release, inhibits calcium pumps and has a use-dependent and slowly reversible action on store-operated calcium entry channels. Cell Calcium. 2003;34:97–108. doi: 10.1016/s0143-4160(03)00026-5. [DOI] [PubMed] [Google Scholar]
- Perkins KL. Cell-attached voltage-clamp and current-clamp recording and stimulation techniques in brain slices. J Neurosci Methods. 2006;154:1–18. doi: 10.1016/j.jneumeth.2006.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Power JM, Sah P. Intracellular calcium store filling by an L-type calcium current in the basolateral amygdala at subthreshold membrane potentials. J Physiol. 2005;562:439–453. doi: 10.1113/jphysiol.2004.076711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Power JM, Sah P. Competition between calcium-activated K+ channels determines cholinergic action on firing properties of basolateral amygdala projection neurons. J Neurosci. 2008;28:3209–3220. doi: 10.1523/JNEUROSCI.4310-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts CB, O'Boyle MP, Suter KJ. Dendrites determine the contribution of after depolarization potentials (ADPs) to generation of repetitive action potentials in hypothalamic gonadotropin releasing-hormone (GnRH) neurons. J Comput Neurosci. 2009;26:39–53. doi: 10.1007/s10827-008-0095-5. [DOI] [PubMed] [Google Scholar]
- Romanò N, Lee K, Abrahám IM, Jasoni CL, Herbison AE. Nonclassical estrogen modulation of presynaptic GABA terminals modulates calcium dynamics in gonadotropin-releasing hormone neurons. Endocrinology. 2008;149:5335–5344. doi: 10.1210/en.2008-0424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sah P, Clements JD. Photolytic manipulation of [Ca2+]i reveals slow kinetics of potassium channels underlying the afterhyperpolarization in hippocampal pyramidal neurons. J Neurosci. 1999;19:3657–3664. doi: 10.1523/JNEUROSCI.19-10-03657.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sah P, Faber ES. Channels underlying neuronal calcium-activated potassium currents. Prog Neurobiol. 2002;66:345–353. doi: 10.1016/s0301-0082(02)00004-7. [DOI] [PubMed] [Google Scholar]
- Salkoff L, Butler A, Ferreira G, Santi C, Wei A. High-conductance potassium channels of the SLO family. Nat Rev Neurosci. 2006;7:921–931. doi: 10.1038/nrn1992. [DOI] [PubMed] [Google Scholar]
- Shah MM, Javadzadeh-Tabatabaie M, Benton DC, Ganellin CR, Haylett DG. Enhancement of hippocampal pyramidal cell excitability by the novel selective slow-afterhyperpolarization channel blocker 3-(triphenylmethylaminomethyl)pyridine (UCL2077) Mol Pharmacol. 2006;70:1494–1502. doi: 10.1124/mol.106.026625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sim JA, Skynner MJ, Herbison AE. Heterogeneity in the basic membrane properties of postnatal gonadotropin-releasing hormone neurons in the mouse. J Neurosci. 2001;21:1067–1075. doi: 10.1523/JNEUROSCI.21-03-01067.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sneyd J, Dufour JF. A dynamic model of the type-2 inositol trisphosphate receptor. Proc Natl Acad Sci U S A. 2002;99:2398–2403. doi: 10.1073/pnas.032281999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spergel DJ. Calcium and small-conductance calcium-activated potassium channels in gonadotropin-releasing hormone neurons before, during, and after puberty. Endocrinology. 2007;148:2383–2390. doi: 10.1210/en.2006-1693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spergel DJ, Krüth U, Hanley DF, Sprengel R, Seeburg PH. GABA- and glutamate-activated channels in green fluorescent protein-tagged gonadotropin-releasing hormone neurons in transgenic mice. J Neurosci. 1999;19:2037–2050. doi: 10.1523/JNEUROSCI.19-06-02037.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teruyama R, Armstrong WE. Enhancement of calcium-dependent afterpotentials in oxytocin neurons of the rat supraoptic nucleus during lactation. J Physiol. 2005;566:505–518. doi: 10.1113/jphysiol.2005.085985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tzingounis AV, Nicoll RA. Contribution of KCNQ2 and KCNQ3 to the medium and slow afterhyperpolarization currents. Proc Natl Acad Sci U S A. 2008;105:19974–19979. doi: 10.1073/pnas.0810535105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tzingounis AV, Kobayashi M, Takamatsu K, Nicoll RA. Hippocalcin gates the calcium activation of the slow afterhyperpolarization in hippocampal pyramidal cells. Neuron. 2007;53:487–493. doi: 10.1016/j.neuron.2007.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogalis F, Storm JF, Lancaster B. SK channels and the varieties of slow after-hyperpolarizations in neurons. Eur J Neurosci. 2003;18:3155–3166. doi: 10.1111/j.1460-9568.2003.03040.x. [DOI] [PubMed] [Google Scholar]
- Wilson CJ, Goldberg JA. Origin of the slow afterhyperpolarization and slow rhythmic bursting in striatal cholinergic interneurons. J Neurophysiol. 2006;95:196–204. doi: 10.1152/jn.00630.2005. [DOI] [PubMed] [Google Scholar]
- Yamada S, Takechi H, Kanchiku I, Kita T, Kato N. Small-conductance Ca2+-dependent K+ channels are the target of spike-induced Ca2+ release in a feedback regulation of pyramidal cell excitability. J Neurophysiol. 2004;91:2322–2329. doi: 10.1152/jn.01049.2003. [DOI] [PubMed] [Google Scholar]