

Abstract
Amyotrophic lateral sclerosis–Parkinson dementia complex (ALS-PDC) is a neurodegenerative disease with ALS, parkinsonism, and Alzheimer's symptoms that is prevalent in the Guam population. β-N-Methylamino alanine (BMAA) has been proposed as the toxic agent damaging several neuronal types in ALS-PDC, including substantia nigra pars compacta dopaminergic (SNpc DAergic) neurons. BMAA is a mixed glutamate receptor agonist, but the specific pathways activated in DAergic neurons are not yet known. We combined electrophysiology, microfluorometry, and confocal microscopy analysis to monitor membrane potential/current, cytosolic calcium concentration ([Ca2+]i) changes, cytochrome-c (cyt-c) immunoreactivity, and reactive oxygen species (ROS) production induced by BMAA. Rapid toxin applications caused reversible membrane depolarization/inward current and increase of firing rate and [Ca2+]i in DAergic neurons. The inward current (IBMAA) was mainly mediated by activation of metabotropic glutamate receptor 1 (mGluR1), coupled to transient receptor potential (TRP) channels, and to a lesser extent, AMPA receptors. Indeed, mGluR1 (CPCCOEt) and TRP channels (SKF 96365; Ruthenium Red) antagonists reduced IBMAA, and a small component of IBMAA was reduced by the AMPA receptor antagonist CNQX. Calcium accumulation was mediated by mGluR1 but not by AMPA receptors. Application of a low concentration of NMDA potentiated the BMAA-mediated calcium increase. Prolonged exposure to BMAA caused significant modifications of membrane properties, calcium overload, cell shrinkage, massive cyt-c release into the cytosol and ROS production. In SNpc GABAergic neurons, BMAA activated only AMPA receptors. Our study identifies the mGluR1-activated mechanism induced by BMAA that may cause the neuronal degeneration and parkinsonian symptoms seen in ALS-PDC. Moreover, environmental exposure to BMAA might possibly also contribute to idiopathic PD.
Introduction
The amyotrophic lateral sclerosis–Parkinson dementia complex (ALS-PDC) is a rare form of neuronal degeneration, occurring among the Chamorro people of Guam (Reed and Brody, 1975; Garruto and Yase, 1986; Spencer et al., 1986). It has been proposed to be caused by exposure to the non-protein amino acid β-N-methylamino-l-alanine (BMAA) (Spencer et al., 1986, 1987). The recent discovery that BMAA can be produced by cyanobacteria throughout the world, including symbionts and free-living cyanobacteria (Cox et al., 2003; Ince and Codd, 2005), has increased the interest for this toxin involvement in other neurodegenerative diseases. Indeed, BMAA has been found in British water bodies (Metcalf et al., 2008), in South Africa freshwater impoundments (Esterhuizen and Downing, 2008) and in Peruvian highland lakes (Johnson et al., 2008), suggesting that it may be of concern not only for specific populations of Western Pacific Islands but also for the world-wide population. Moreover, BMAA can accumulate in bacteria, plant, animal, and human tissues as free amino acid as well as bound within proteins (Murch et al., 2004). This creates an endogenous reservoir of neurotoxin within the human body, capable of slow release into cerebral and other tissues during protein catabolism for years or decades after its consumption. In vivo (Spencer et al., 1987; Smith and Meldrum, 1990; Rakonczay et al., 1991; Chang et al., 1993; Santucci et al., 2009) and in vitro studies have reported that BMAA acts as a glutamate agonist onto spinal motoneurons (Rao et al., 2006) and cortical (Lobner et al., 2007) and cerebellar (Staton and Bristow, 1997) neurons. However, a detailed description of its actions on substantia nigra pars compacta (SNpc) dopaminergic cells (DAergic) is still lacking. This would be of particular interest, as it would help to understand mechanisms of neuronal degeneration involved not only in ALS-PDC, but also in sporadic Parkinson's disease (PD) (Hanna and Bhatia, 1997; Beal, 1998; Ince and Codd, 2005). To this regard, the neuronal loss within the SNpc that characterizes PD may be due either to genetic (Klein and Schlossmacher, 2007; Bogaerts et al., 2008) and/or environmental factors (Le Couteur et al., 2002; Di Monte, 2003; Greenamyre et al., 2003). There is evidence that in PD, glutamate may cooperate to induce nigral excitotoxicity (Rodriguez et al., 1998) and, in addition to NMDA receptors, metabotropic receptors (mGluRs) possibly mediate neuronal death (Vernon et al., 2005, 2007). Indeed, mGluR1 activation promotes numerous processes, such as activation of inward cationic currents (Guatteo et al., 1999; Tozzi et al., 2003), inhibition of potassium channels, and direct influx of calcium ions, which leads to inappropriate activation of kinases, proteases, and phospholipases and finally to energy deficits (Salińska et al., 2005).
Therefore, the aim of the present study was to identify the BMAA-induced electrophysiological changes, intracellular calcium modifications, and toxic effects on SNpc neurons. We found that BMAA preferentially activates an mGluR1-mediated pathway on SNpc DAergic neurons and that its prolonged application causes significant functional alterations inducing also massive cyt-c release from mitochondria and reactive oxygen species (ROS) production. Indeed, these excitotoxic effects were mitigated by mGluR1 antagonists, the mitochondrial transition pore inhibitor cyclosporine A, and the antioxidant 1,4-dithiothreitol.
Materials and Methods
Tissue preparation.
All procedures were approved by the Animal Ethics Committee of the University of Rome Tor Vergata. Preparation of rat midbrain slices was performed as described previously (Lacey et al., 1989; Mercuri et al., 1995). Briefly, Wistar rats (P18–P22) (Charles River Laboratories) were deeply anesthetized with halothane and decapitated. The brain was rapidly removed and horizontal slices (thickness 250 μm), containing the substantia nigra pars compacta (SNpc) and the ventral tegmental area (VTA), were cut with a vibratome (VT 1000s, Leica) in ice-cold modified artificial CSF (ACSF). This solution contained the following (in mm): 126 NaCl, 2.5 KCl, 1.2 MgCl2, 1.2 NaH2PO4, 2.4 CaCl2, 10 glucose, and 24 NaHCO3, 290 mOsm−1, and was gassed with 95% O2–5% CO2, pH 7.4. After 30 min recovery in 95% O2–5% CO2-gassed ACSF at 34°C, a single slice was transferred to a recording chamber (volume 0.6 ml) and completely submerged with continuously flowing (2.5 ml · min−1) ACSF at 34–35°C.
Patch-clamp recordings.
The recording chamber was mounted on the stage of an upright microscope (Olympus BX51WI) equipped with a CCD camera (Till Photonics) and monochromator (Polychrome IV, Till Photonics) for microfluorometry. Individual neurons were visualized through a 40× water-immersion objective (Olympus) using an infrared differential interference contrast (IR-DIC) system. Whole-cell patch-clamp recordings, combined with microfluorometry, were performed on SNpc neurons localized in a region of tightly packed neurons adjacent to the medial terminal nucleus of the accessory optic tract. DAergic neurons were electrophysiologically identified based on a slow and regular spontaneous firing (1–5 Hz), hyperpolarizing/outward response to dopamine (30 μm), prominent Ih in response to hyperpolarizing voltage steps (Mercuri et al., 1995) and, in current-clamp mode, a depolarizing sag when negative currents were applied (Grillner and Mercuri, 2002). The GABAergic interneurons in the SNpc showed a high regular spontaneous firing at rest (25 Hz) (Lacey et al., 1989; Berretta et al., 2000; Gernert et al., 2004), no response to dopamine application, modest Ih and sag potential in response to hyperpolarizing voltage and current pulses, respectively.
Patch pipettes (2–5 MΩ), pulled with a PP 83 Narishige puller, were filled with a solution containing the following (in mm): K-gluconate (115), CaCl2 (4), MgCl2 (2), KCl (10), EGTA (10), HEPES (10), Mg2-ATP (2), and Na3-GTP (0.3), pH 7.3, osmolarity 280 mOsm. For microfluorometry, we used a pipette solution containing the following (in mm): K-gluconate (135), CaCl2 (0.1), MgCl2 (2), KCl (10), EGTA (0.75), and 0.25 fura-2 pentapotassium salt (Invitrogen), pH 7.3, osmolarity 280 mOsm.
Whole-cell voltage-clamp or current-clamp experiments were performed with an Axopatch 200 B amplifier (Molecular Devices), filtered at 1 kHz, and digitized at 10 kHz. In voltage clamp, holding potential (Vhold) was −60 mV. No hyperpolarizing holding currents were applied in current-clamp recordings except, in the prolonged treatment experiments, for membrane resistance (Rm) and number of evoked spikes measurements, in which the cells were held at −50 mV. Rm changes were calculated at the end of the hyperpolarizing potentials produced by injecting a negative current pulse (−30 pA, 700 ms duration), whereas the number of spikes were calculated in response to a depolarizing current step (+100 pA, 700 ms).
BMAA (3 mm, or 1 mm in Fig. 9) and glutamate (300 μm), dissolved in ACSF, were pressure applied (10 psi, 0.5–1.0 s), through a patch electrode connected to a Pneumatic Pico-pump PV 800 (WPI). The puffing electrode was positioned above the slice in close proximity to the recorded neuron and the agonist-mediated inward currents (IBMAA; IGLU) were elicited every 2 min. In experiments illustrated in Figures 1A–F and 7, BMAA was bath-applied (0.1–10 mm). The current/voltage (I–V) relationship of BMAA-induced current (IBMAA) was measured using a ramp (1.5 s, from −65 mV to +70 mV) executed before (control ramp) and at the peak of IBMAA (BMAA ramp, 3 mm, 2 s). The net IBMAA was calculated as digital subtraction between the control and the BMAA ramps. For these experiments, K-gluconate in the pipette solution was replaced with Cs-methanesulfonate (pH 7.3 adjusted with CsOH) and QX-314 (5 mm) was added. The extracellular solution was initially as follows (in mm): NaCl, 141; KCl, 2.5; MgCl2, 1.2; CaCl2, 2.4; NaOH, 9; glucose, 10; and HEPES, 20. Before applying ramps, we blocked Na+, K+, Ih, Ca2+, AMPA, and GABAA conductances by replacing this solution with one containing the following (in mm): NaCl, 126; 4-aminopyridine, 5; KCl, 2.5; MgCl2, 1.2; CsCl, 2; CaCl2, 1.4; CoCl2, 1; HEPES, 20; NaOH, 5; glucose, 10; tetrodotoxin, 0.001; picrotoxin, 0.1; nifedipine, 0.01; CNQX, 0.01; and CdCl2, 0.1. Both the HEPES-buffered solutions were equilibrated with 100% O2.
Figure 9.

Activation of NMDA receptors increases BMAA-induced calcium modifications. A, Simultaneous recordings of IBMAA (upper traces) and [Ca2+]i (lower traces) in control and during bath application of NMDA (20 μm). BMAA concentration in the puff pipette was 1 mm. B, Histograms showing cumulative data of IBMAA (black bar, n = 5) and [Ca2+]i (hatched bar, n = 5) in the presence of 20 μm NMDA, normalized to control response before NMDA application, in the same cells (dotted line). Note that NMDA did not affect IBMAA (p = 0.36), but it increased the BMAA-induced [Ca2+]i accumulations. *p < 0.05.
Figure 1.

BMAA-induced firing frequency increase, membrane depolarization, inward current, and [Ca2+]i modification in DAergic neurons. A, C, Example traces showing transient firing rate increase in response to 0.1, 0.3, and 1 mm BMAA bath applications (1.5–2 min). B, D, Plots showing mean changes of firing frequency (FF) and membrane potential, respectively, in response to short (1.5–2 min, 0.1–10 mm, black bars) and long (30 min, 0.1 mm, white bars) BMAA bath applications. These effects were due to activation of an inward current (IBMAA) as revealed by voltage-clamp recordings (Vhold = −60 mV) (E, F). Means ± SEM of the plots B, D, and F are shown in supplemental Table 1 (available at www.jneurosci.org as supplemental material). G, Fast BMAA application(1–1.5 s, arrow) induced an inward current (top) that was associated with calcium transients (bottom). H, Top, Averaged calcium trace (n = 13) and mean calcium values (bottom) measured at the times indicated by the corresponding letters in the trace. #p < 0.0001.
Figure 7.
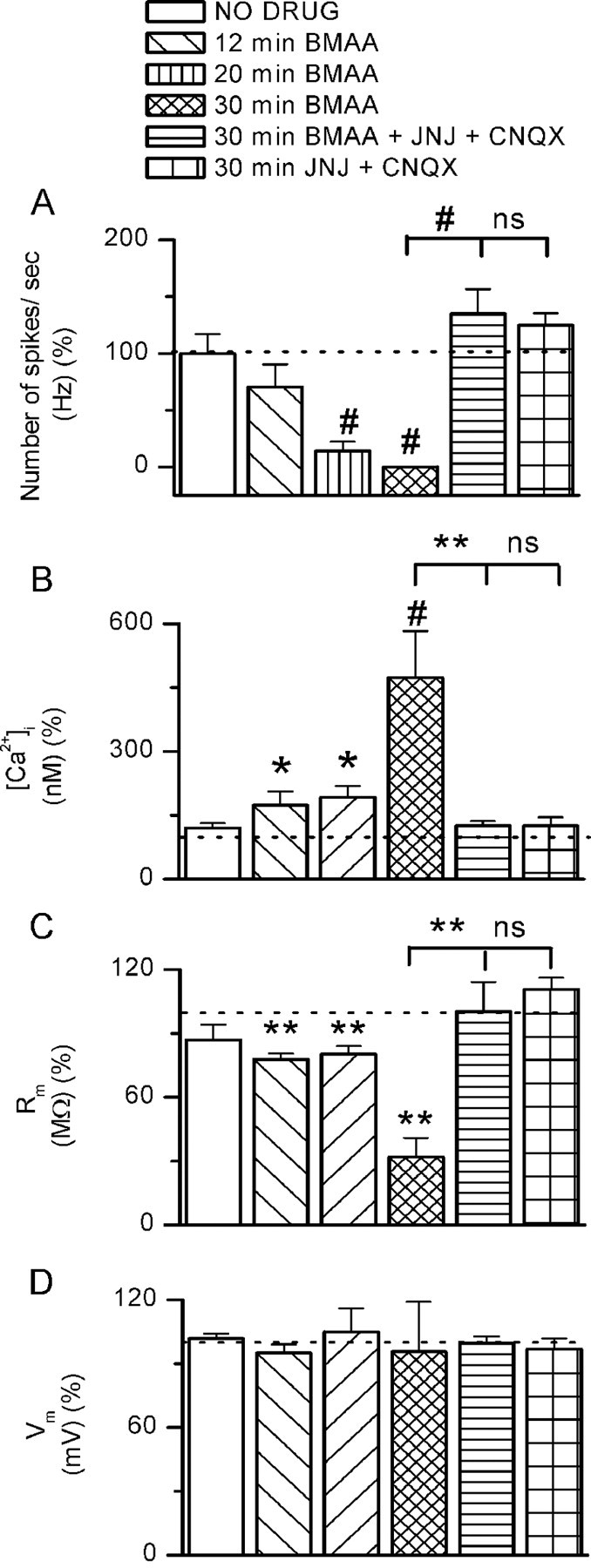
Effects of BMAA long treatments on DAergic cell viability. Histograms showing the number of action potentials in response to depolarizing step (100 pA, 700 ms) (A), [Ca2+]i (B), membrane resistance Rm (C), and membrane potentials Vm (D) in five experimental conditions: neurons exposed to BMAA for 12, 20, and 30 min, to BMAA + JNJ + CNQX for 30 min, and to JNJ + CNQX for 30 min. The levels of significance have been obtained by comparing each experimental condition with no drug. Other comparisons are indicated in the figure. Each parameter was measured after 20 min of BMAA washout. *p < 0.05, **p < 0.01, #p < 0.0001.
Data were acquired using Clampex/AxoScope softwares (version 9, Molecular Devices). Measurements (every 2 min) of Rm were performed in voltage clamp using −5 mV command pulses (20–30 ms). Off-line data analysis was performed using Origin (OriginLab) and Clampfit (Molecular Devices) softwares.
Microfluorometry.
Neurons were filled with a solution containing the Ca2+-sensitive dye fura-2 pentapotassium salt (Invitrogen) by diffusion from the patch pipette. UV excitation light (340 and 380 nm) was provided by the monochromator (Till Photonics). Emitted light passed a barrier filter (440 nm), was detected by the CCD camera (Till Photonics) and images were acquired at 2 or 5 s intervals using TillVision software. Fluorescence changes expressed as “ratio” (r) were calculated from the formula r = (F340soma − F340bg)/(F380soma − F380bg), where F340 and F380 were the fluorescence emitted at the excitation wavelengths 340 and 380 nm, respectively, for the soma and background (bg), measured from a region >100 μm away from the soma (Guatteo et al., 1998). Ratio values were converted into ion concentrations using the method of Grynkiewicz et al. (1985). The rmin and rmax values (Grynkiewicz et al., 1985) were obtained in situ by exposing perforated cells (1 μm ionomycin) to Ca2+-free (0 mm calcium, 1 mm EGTA) and 1 mm Ca2+-containing solutions.
Confocal microscopy.
Slice preparation was identical to that described for electrophysiological recordings. After cutting, slices were transferred to a holding chamber and left to recover for 30 min (33°C) in standard ACSF medium saturated with an O2/CO2 gas mixture. Slices were then placed in seven chambers containing standard saturated ACSF. In the first chamber (CTRL), no extra compounds were added; in the second (BMAA), we added BMAA (3 mm), and in the others, we added BMAA plus CPCCOEt (100 μm), CNQX (10 μm), JNJ (1 μm), a mixture of JNJ plus CNQX, or cyclosporine A (CsA, 10 μm). After 1 h treatment, slices from all the groups were left to recover (5 h) in separate chambers filled with standard saturated ACSF gassed with 95%O2, 5% CO2. For ROS detection experiments (supplemental Fig. S3, available at www.jneurosci.org as supplemental material), we divided slices into 3 groups: CTRL, BMAA, and BMAA plus 1,4-dithiothreitol (DTT; 500 μm). After 1 h BMAA treatment, hydroethidine (Invitrogen; 315 nm) was added for 30 min. For hypoxia experiments, after 30 min of recovery, slices were divided into two groups: the first (CTRL) consisted of slices kept for 30 min in the standard ACSF gassed with 95%O2, 5% CO2; the second (hypoxia) was placed in a chamber containing the ACSF saturated with a 95% N2 and 5% CO2 for 30 min.
To perform post hoc labeling of the recorded cells, biocytin (5 mm, free base; Sigma) was added in the pipette solution.
Slices from the different experimental settings were fixed overnight in 4% paraformaldehyde and, after 3 washing in phosphate buffer (PB), transferred to 30% sucrose/PB at 4°C for 48 h. They were then cut into 35-μm-thick horizontal sections with a freezing microtome and collected in PB. The sections with biocytin-loaded cells were incubated for 1 h at room temperature (RT) in PB-containing Cy2-conjugated Streptavidin (1:200; Jackson ImmunoResearch Laboratories) and 0.3% Triton X-100. The following primary antibodies were then used: goat anti-tyrosine hydroxylase (TH; 1:500; Santa Cruz Biotechnology), mouse anti-cyt-c (1:100; Promega), rabbit anti-STIM1, or rabbit anti-Orai1 (1:400; ProSci). Following overnight incubation with primary antibodies, sections were incubated for 2 h at RT in a mixture of secondary antibodies including the following: Cy5-conjugated donkey anti-goat IgG, Cy3-conjugated donkey anti-rabbit IgG, or Cy2-conjugated donkey anti-mouse IgG (1:100; all from Jackson ImmunoResearch Laboratories). Sections were air dried and coverslipped with Gel/Mount (Biomeda). Images were acquired through a confocal laser scanning microscope (Leica SP5, Leica Microsystems) providing four excitations: 405, 488, 543, and 633 nm. Plates were generated adjusting the contrast and brightness of digital images (Corel Draw, 9). Qualitative and/or quantitative observations were limited to SNpc neurons, identified by shape, cellular density and proximity to the medial terminal nucleus of the accessory optic tract. Analysis was conducted in five subsections obtained from each 250-μm-thick slice (5 slices/group).
Electrophysiological and microfluorometry data analysis.
Data were analyzed with Origin (version 6, OriginLab), expressed as mean ± SEM, and compared using one- or two-population Student's t test. Changes in firing frequency and cell membrane depolarization were calculated as the difference between the peak and baseline levels. Baseline levels were measured 10–20 s before BMAA application, and peak levels during 10–20 s just before washout of the drug. Current areas (see Fig. 5D, inset) were quantified using an integral function (OriginLab) of the area between the current trace and the baseline. In all measurements (including densitometry and cell size measurements), significance levels were indicated as *p < 0.05; **p < 0.01; #p < 0.0001.
Figure 5.

BMAA is not a substrate of EAATs. Traces showing IBMAA (A) and IGLU (B), evoked every 2 min, during dl-TBOA bath application (100 μm). C, D, Histograms of mean IBMAA (white bars, n = 6) and IGLU (black bars, n = 3) amplitude and area, respectively, calculated as indicated in the corresponding insets. Data are expressed as percentage of controls. *p < 0.05, **p < 0.01.
Densitometric analysis of fluorescence images.
Quantification of fluorescence (cyt-c immunoreactivity and ROS generation) was performed in TH-immunoreactive cells by densitometric analysis. After background subtraction, cell-associated signals were quantified by manually outlining individual neurons and measuring the mean pixel density (mean fluorescence) with the ImageJ software (NIH; http://rsb.info.nih.gov/ij/). Mean fluorescence of individual cells (F) was normalized to the total cellular surface (A). Quantification was done on 200 cells (5 slices/group). Statistical analysis was performed by means of one-way ANOVA, followed by Bonferroni post hoc test with p < 0.05 as minimal level for significance.
Neuron size measurement.
Digital images of the TH-immunoreactive SNpc neurons from 5 sections were captured using a 63× objective and 0.7 zoom factor. Neurons with clearly distinguishable nucleus, nucleolus, and cytoplasmic borders were sampled. Cell surface analysis was done by outlining the soma borders, excluding dendrites and axon. Approximately 200 cells were measured in each experimental group (5 slices/group). The areas were calculated using ImageJ. Statistical analysis was performed as described above.
Drugs.
6-Cyano-7-nitroquinoxaline-2,3-dione (CNQX), d-2-amino-5-phosphono-pentanoic acid (APV), 7(hydroxyimino)cyclopropal[b] chromen-1a-carboxylate (CPCCOEt), dl-threo-β-benzyloxyaspartic acid (dl-TBOA), (5S,10R)-(+)-5-methyl-10,11-dihydro-5H-dibenzo[a,d] cyclohepten-5,10-imine maleate (MK801), 2-methyl-6-(phenylethynyl) pyridine hydrochloride (MPEP), 1-[2-(4-methoxyphenyl)propoxy] ethyl-1H-imidazole hydrochloride (SKF 96365), ammoniated ruthenium oxychloride [Ruthenium Red (RR)], cyclopiazonic acid (CPA), 1-[(5-chloro-1H-benzimidazol-2-yl)carbonyl]-4-methylpiperazine maleate (JNJ 16259685), and picrotoxin were obtained from Tocris Cookson. Tetrodotoxin was from Alomone Laboratories. Dopamine hydrochloride (DA), β-N-methylamino-l-alanine (BMAA), glutamic acid, cyclosporine A (CsA), nifedipine, 4-aminopyridine, cesium chloride, cobalt chloride, and cadmium chloride were from Sigma-Aldrich. 1,4-Dithiothreitol (DTT) was from Boehringer Mannheim and hydroethidine from Invitrogen.
Results
Actions of BMAA on the membrane properties and [Ca2+]i in SNpc neurons
In the view that BMAA could be the toxic agent causing damage and eventually cell death of DAergic and other neuronal types in ALS/PDC as well as in sporadic PD, we studied its effects in single SNpc DAergic neurons. We first investigated the effects of BMAA on neuronal firing activity (Fig. 1A–D) in current-clamp mode, to detect their typical spontaneous tonic action potential discharge (Grillner and Mercuri, 2002). Short-lasting bath application (1.5–2 min) of BMAA (0.1–10 mm) increased the spontaneous firing frequency in a dose-dependent manner (Fig. 1A–C) and caused a rapid and reversible membrane depolarization (Fig. 1A,C,D).
To determine whether continuous presence of BMAA in the extracellular fluid, in low concentration, induces progressive alteration of neuronal activity, we applied 100 μm BMAA for 30 min. In this experimental condition, the drug produced significantly larger depolarization of the membrane potential and increase in firing frequency than during short-lasting application (Fig. 1B,D, white bars) (p < 0.05).
Voltage-clamp recordings revealed that BMAA-induced membrane depolarization was due to an inward current (IBMAA), whose mean amplitude ranged from 20.8 ± 3.9 pA to 1004 ± 160.3 pA (Fig. 1F), in response to increasing BMAA concentrations (0.1–10 mm) (Fig. 1E,F). The inward current caused by high dosages (3 and 10 mm) of BMAA was often followed by an outward current (Fig. 1E).
Measures of IBMAA and the associated [Ca2+]i dynamics were performed by applying BMAA to the recorded neuron by pressure ejection, at 2 min interval (3 mm) (see Figs. 1G,H, 2–6, 9). Using this experimental approach, we obtained fast transient IBMAAs of mean amplitude 454.5 ± 34.6 pA (n = 73), which were accompanied by reversible and transient [Ca2+]i increases of 139.3 ± 23.7 nm (n = 13). Calcium concentration changes originated from a resting level of 87.1 ± 5.2 nm, peaked at 226.4 ± 23.3 nm (p < 0.0001, paired t test) (Fig. 1H) (n = 13) and rapidly returned to the baseline (p = 0.3).
Figure 2.

A, C, IBMAA is mediated by the activation of mGluR1. Example traces of IBMAA (A) and IGLU (C) on expanded time scale, in control and in the presence of glutamate receptor antagonists, were aligned on their rising phase. B, D, Histograms of IBMAA and IGLU amplitudes, expressed as percentage of controls, in the presence of glutamate receptor antagonists. CPCCOEt (A1) and, to a lesser extent, CNQX (A2) significantly reduced IBMAA (B, n = 24 for both). APV, MK801 (A3), and MPEP (A4) did not modify IBMAA (B, n = 6, 4, and 7, respectively). IGLU was significantly affected by all ionotropic (CNQX, MK801, APV, and CNQX plus MK801) receptor antagonists (C1, C2, D1, n = 10, 5, 6, 5, respectively). The IGLU sensitivity to mGluR antagonists was assessed in the presence of CNQX plus MK801 (D2). CPCCOEt (C3) significantly reduced IGLU (D2, n = 7), whereas MPEP (C4) left IGLU unaffected (D2, n = 3). **p < 0.01, #p < 0.0001.
Figure 3.
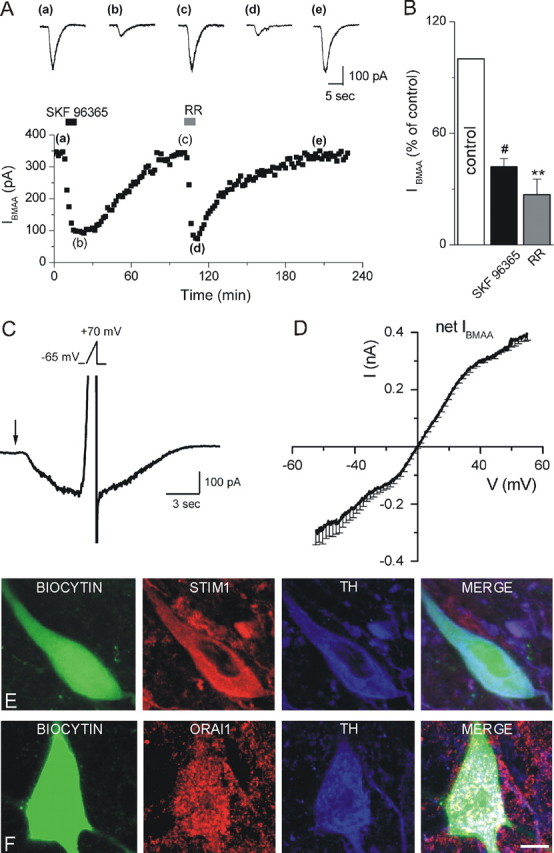
TRPCs mediate IBMAA. A, Time course of IBMAA amplitude in response to perfusion with SKF 96365 (100 μm) and RR (20 μm). Top, Raw traces of IBMAA acquired at the times indicated by the corresponding letters in the plot. B, Histogram of IBMAA amplitude, expressed as percentage of control (white bar), following exposure to SKF 96365 and RR (n = 8 and 6, respectively). C, IBMAA recorded from a DAergic neuron (Vhold = −65 mV) in response to a BMAA puff (3 mm, 2 s, arrow). At the peak of the inward current a voltage ramp (Vramp, −65 to +70 mV, 1.5 s, see inset) is executed. D, Mean I–V of the net IBMAA obtained from four cells. E, F, Confocal laser scanning microscope images of SNpc DAergic neurons loaded with biocytin (green) and stained with TH (blue) antibody. E, Triple labeling with STIM1 (red) antibody. F, Triple labeling with Orai1 (red) antibody. As shown in the merge images, biocytin-loaded TH-positive neurons are immunopositive for both STIM1 (E) and Orai1 (F). Scale bar: 6 μm. **p < 0.01, #p < 0.0001.
Figure 4.

BMAA-induced [Ca2+]i increase is sensitive to mGluR and TRPC channel blockers. A–C, Simultaneous recordings of currents (upper traces) and [Ca2+]i (lower traces) show pairs of BMAA responses in control solutions, following bath application of the drugs CPCCOEt (100 μm) (A), CNQX (10 μm) (B), SKF 96365 (100 μm) (C), and washout. Histograms show cumulative data of IBMAA and [Ca2+]i in the presence of the drugs CPCCOEt (A), CNQX (B), SKF 96365 (C), and washout normalized to the control response (white bar), measured in the same cell. *p < 0.05, **p < 0.01, #p < 0.0001.
Figure 6.

Responses of GABAergic neurons to BMAA. A, Example traces of IBMAA and calcium changes during BMAA puff applications (2 min interval). Note that current amplitude was significantly smaller than in DAergic cells (p < 0.05) (Fig. 1G). B, Histogram showing mean [Ca2+]i before (basal) (102.9 ± 10.6 nm), during (BMAA) (110.3 ± 13.9 nm), and after (post puff) (104.8 ± 11 nm) BMAA application (n = 4). C, Bottom, Time course of IBMAA amplitude in response to application of CPCCOEt (100 μm) and CNQX (10 μm). Top, Raw traces of IBMAA acquired at the times indicated by the corresponding letters in the plot. D, Histogram of IBMAA amplitude, expressed as percentage of control (white bar), following exposure to CPCCOEt and CNQX (n = 7 and 9, respectively). #p < 0.0001.
Glutamate receptors activated by BMAA
Based on the evidence that the β-carbamate form of BMAA, produced in bicarbonate-containing medium, has a chemical structure similar to glutamate (Myers and Nelson, 1990; Brownson et al., 2002), we aimed to determine which glutamate receptors activated IBMAA by using selective glutamate receptors antagonists (Fig. 2A,B). We compared the relative contribution of different receptors to the total current activated by BMAA (3 mm) with the one induced by glutamate (300 μm) (Fig. 2C,D). We assessed glutamate concentration, puff duration and electrode position to obtain similar IBMAA and IGLU amplitudes. In the presence of the selective mGluR1 antagonist CPCCOEt (100 μm), IBMAA was reduced to 41.6 ± 3.6% of control (Fig. 2A1,B) (p < 0.0001, n = 24). We also tested the effect of another selective and potent mGluR1 antagonist, JNJ 16259685 (1 μm) (Lavreysen et al., 2004), which reduced IBMAA to 3 ± 3.2% of control (n = 3, data not shown). However, the antagonistic effect of this drug did not washout within 1 h. In the presence of the selective AMPA receptor antagonist CNQX (10 μm), IBMAA was slightly but significantly reduced to 93.1 ± 2% of control (Fig. 2A2,B) (p < 0.01, n = 24). The NMDA receptor antagonists MK801 (10 μm) and APV (50 μm) and the mGluR5 antagonist MPEP (10 μm) did not significantly affect IBMAA amplitude (Fig. 2A3,A4,B). Indeed, in the presence of APV or MK801, the current was 98.7 ± 2.2% of control (p = 0.6, n = 6) and 104 ± 8.8% of control (p = 0.7, n = 4), respectively, and in the presence of MPEP, IBMAA was 95.4 ± 4.9% of control (p = 0.4, n = 7). These data demonstrate that IBMAA is activated mainly by mGluR1 and, to a lesser extent, by AMPA receptors. We refer to this mGluR1-dependent response of BMAA as “classical.”
We then compared the effects of glutamate and BMAA on SNpc DAergic cells. Differently from IBMAA, IGLU was reduced by either ionotropic or metabotropic type 1 receptor antagonists (Fig. 2C,D1,D2). In particular, CNQX (10 μm) reduced IGLU to 42.5 ± 4.2% of control (p < 0.0001, n = 10) (Fig. 2C1,D1), and APV and MK801 reduced IGLU to 75.5 ± 2.8% (p < 0.01, n = 6) and 70.4 ± 1.1% of control (p < 0.0001, n = 5), respectively (Fig. 2C2,D1). To isolate a pure mGluR-mediated current, we blocked all ionotropic components by applying CNQX (10 μm) with MK801 (10 μm) (Fig. 2C3,C4,D2). In this condition IGLU was reduced to 29.6 ± 7.3% of control (p < 0.0001, n = 7) (Fig. 2D1). The mGluR1 and mGluR5 antagonists CPCCOEt (100 μm) and MPEP (10 μm) reduced the remaining current to 77 ± 2.7% (p < 0.01, n = 7) and 88.7 ± 2.8% (p = 0.06, n = 3) of control, respectively.
In a small population of neurons (15/88), BMAA caused an inward current that was mostly insensitive to the mGluR1 antagonist CPCCOEt. In these cells, IBMAA was mediated by AMPA receptors, as it was strongly reduced by CNQX (10 μm, data not shown) but was not by the kainate receptor antagonist GYKI (300 μm). Interestingly, the electrophysiological properties of these neurons (firing frequency, sensitivity to DA, and presence of a time- and voltage-dependent inward current, Ih) fulfilled the criteria typical of DAergic neurons (data not shown).
The following focuses on the classical BMAA-induced responses.
TRPC-like channels mediate the BMAA-induced inward current
It has been previously shown in DAergic neurons that mGluR1s couple, in a G-protein-dependent manner, to transient receptor potential (TRP) channels, mostly type C1, C3, and C5 (Riccio et al., 2002; Tozzi et al., 2003). Thus, we sought to determine whether IBMAA is also mediated by TRPC-like channels by perfusing the nonselective TRP channel blockers SKF 96365 (100 μm) and RR (20 μm) (Fig. 3A,B). Figure 3A shows the time course of IBMAA amplitude, elicited every 2 min, in control (a, c) and during SKF 96365 (b) and RR (d) bath application. Both TRP antagonists caused a marked reduction of IBMAA amplitude to 42.1 ± 4.3% (p < 0.0001, n = 8) and 27 ± 8.3% of control (p < 0.01, n = 6), respectively.
To further test the hypothesis that IBMAA is due to the activation of a mixed cationic current, likely TRPC channels, we measured the I–V relationship of IBMAAs by using voltage ramps (Fig. 3C, see Materials and Methods). The current had a reversal potential of ∼1.0 mV (n = 4) (Fig. 3D), consistent with the opening of a mixed cationic channel (Okada et al., 1998; Strubing et al., 2001; Tozzi et al., 2003; Faber et al., 2006).
It has been recently demonstrated that TRPC channels require STIM1 and/or Orai1 activation, to act as store-operated calcium channels (SOCs) (Kim et al., 2009; Liao et al., 2009) (for review, see Cahalan, 2009). Therefore, we made whole-cell recordings from SNpc DAergic neurons with biocytin-containing pipettes and characterized IBMAA by using mGluR1, AMPA, and TRPC antagonists. We then conducted immunohistochemistry for TH, STIM1, and Orai1 (n = 20). All the biocytin-labeled cells were TH, STIM1, and Orai1 immunoreactive (Fig. 3E,F). Thus, the expression of STIM1 and Orai1 proteins in biocytin-labeled TH-immunoreactive neurons might suggest their putative involvement, together with TRPC channels, in the mGluR1-mediated responses. However, the functional interaction between STIM1, TRPC, and Orai1 proteins is still questioned (DeHaven et al., 2009) and needs further investigation.
Intracellular calcium changes in response to BMAA application
As reported in Figure 1G, IBMAA is also associated with transient [Ca2+]i elevation. We then explored whether the [Ca2+]i rise was dependent on activation of membrane receptors as mGluR1 and AMPA, and whether the opening of TRPC-like channels is required (Fig. 4). Therefore, we recorded IBMAA (upper traces) and calcium changes (bottom traces) in fura-2-loaded DAergic neurons before, during, and after the application of CPCCOEt (100 μm) (Fig. 4A), CNQX (10 μm) (Fig. 4B), and SKF 96365 (100 μm) (Fig. 4C). IBMAA was reduced by all drugs to 21.1 ± 4.2% (p < 0.0001, n = 7), 75 ± 7% (p < 0.05, n = 5), and 54.1 ± 4.1% of control (p < 0.0001, n = 7), respectively. In contrast, [Ca2+]i was not affected by CNQX (93.3 ± 13.2% of control, p = 0.63, n = 6) (Fig. 4B), suggesting that while the contribution of AMPA receptors to IBMAA is small but significant, the AMPA-mediated changes in [Ca2+]i are negligible. Interestingly, the BMAA-induced [Ca2+]i increase was largely dependent on both mGluR1 and TRPC-like channel activation, since their antagonists strongly reduced calcium signals (CPCCOEt: 26.6 ± 6.6% of control, p < 0.0001, n = 7; SKF 96365: 38 ± 10.3% of control, p < 0.01, n = 7, respectively) (Fig. 4A,C). All drugs washed out upon removal from bathing solution.
Coapplication of CPCCOEt and SKF 96365 did not cause additive effect on IBMAA and [Ca2+]i decrease [current: 48. 7 ± 5.8% of control (supplemental Fig. S1A, available at www.jneurosci.org as supplemental material); calcium: 27 ± 3.8% of control (supplemental Fig. S1B, available at www.jneurosci.org as supplemental material)], and the values were not significantly different from those obtained in the presence of the two compounds separately. Thus, mGluR1 activation and TRPC-like channel opening are two subsequent steps in the same pathway. Moreover, in the presence of CPA (30 μm), a reversible inhibitor of sarcoplasmic reticulum Ca2+-ATPase, BMAA-induced calcium was reduced to 12 ± 5.7% of control (p = 0.001, n = 5, data not shown).
To verify that BMAA does not produce any additional effects other than those due to mGluR1-TRPC and AMPA receptor activation, we measured IBMAA and calcium levels in the presence of mGluR1, TRPC, and AMPA receptor antagonists (CPCCOEt, SKF 96365, and CNQX, respectively). In the presence of this mixture (supplemental Fig. S1A,B, available at www.jneurosci.org as supplemental material), BMAA responses were abolished, excluding the possibility that other mechanisms might be involved (current: 10 ± 1% of control; calcium: 7.5 ± 2.5% of control). These data suggest that BMAA activates mGluR1-TRPC pathway as previously described for the selective mGluR1 agonist (S)-3,5-dihydroxyphenylglycine (DHPG) (Tozzi et al., 2003). Indeed, direct activation of mGluR1 with DHPG (100 μm) completely occluded BMAA effects (supplemental Fig. S2, available at www.jneurosci.org as supplemental material) (current: 31 ± 9.9% of control; calcium: 2 ± 0.5% of control).
Involvement of EAA transporters in BMAA-induced currents
We evaluated the possibility that excitatory amino acid transporters (EAATs) could contribute to the clearance of BMAA from the extracellular medium affecting amplitude and/or duration of IBMAA in dopamine neurons. Therefore, we studied the effect of the potent inhibitor of EAATs, dl-TBOA (100 μm) (Shimamoto et al., 1998) on IBMAA amplitude and area (see Materials and Methods) (Fig. 5A). First, we assessed the effect of dl-TBOA on glutamate-mediated inward currents (IGLU, 300 μm) (Fig. 5B), which are known to be highly modulated by EAATs (Tong and Jahr, 1994). As expected, dl-TBOA significantly increased IGLU amplitude and area to 448 ± 18.6% (p < 0.01, n = 3) and to 753 ± 127% of control (p < 0.05, n = 3), respectively (Fig. 5C,D, black bars). In contrast, IBMAA was largely unaffected by the EAAT inhibitor (Fig. 5C,D, white bars). Indeed, in the presence of dl-TBOA, IBMAA amplitude was 90.6 ± 8.7% (p = 0.3, n = 6) and IBMAA area was 101.8 ± 7.8% of control (p = 0.8, n = 6).
BMAA activates AMPA receptors in GABAergic neurons of the SNpc
It has been demonstrated that BMAA activates different types of glutamate receptors in diverse brain areas (Spencer et al., 1987; Copani et al., 1990, 1991; Allen et al., 1993; Pai et al., 1993; Staton and Bristow, 1998; Rao et al., 2006; Lobner et al., 2007). To this regard, we aimed to explore its effect onto the “secondary” cells of the SNpc, presumably GABAergic neurons (Fig. 6) (Lacey et al., 1989). We found that, in these cells, BMAA evoked an inward current (Fig. 6A, upper panel) (mean amplitude: 166 ± 20 pA; n = 9) significantly smaller than in DAergic cells. This difference was not due to cell size since IBMAA, normalized to cell capacitance, was also significantly different (IBMAA in DAergic cells 5.18 ± 0.7 pA/pF, n = 16; in GABAergic cells 2.56 ± 0.5 pA/pF, n = 6, p < 0.05; data not shown). Moreover, in GABAergic cells, IBMAA was reduced by CNQX (10 μm; 33.4 ± 2.8% of control, p < 0.0001, n = 9) but not by CPCCOEt (100 μm; 97.4 ± 5.2% of control, p = 0.64, n = 7) (Fig. 6C,D) and was not associated with [Ca2+]i increase [Fig. 6A (lower panel),B] (p = 0.14, paired t test, n = 4).
Prolonged exposure to BMAA cause significant changes in the properties of DAergic neurons
Based on the evidence that BMAA can accumulate within the human body (Murch et al., 2004), we sought to determine whether prolonged exposure to the toxin could alter DAergic neuron functional properties. To test this hypothesis, we monitored different cellular parameters before and after BMAA (3 mm) bath applications of 12, 20, and 30 min. The evaluations were done 20 min after BMAA washout, and were expressed as percentage of the control value before BMAA application. The examined parameters included the following: number of spikes evoked by a depolarizing current step (Fig. 7A), [Ca2+]i levels (Fig. 7B), membrane resistance Rm (Fig. 7C), and membrane potential changes Vm (Fig. 7D). The same parameters were also measured in a control population of cells recorded for 50 min, but not exposed to the toxin (no-drug group) (Fig. 7A–D, white bar). All the experiments were performed in current-clamp mode.
In the no-drug group, all the parameters remained unchanged up to 50 min of recording: spike frequency was 99.8 ± 17.8% of control (p = 0.99, n = 11); [Ca2+]i was 119.9 ± 11% of control (p = 0.11, n = 8); Rm was 86.9 ± 7.2% of control (p = 0.11, n = 8); and Vm was 101.8 ± 2.2% of control (p = 0.44, n = 13). Except for Vm (Fig. 7D), all the parameters were significantly modified by prolonged BMAA exposures (12, 20, or 30 min). Particularly, spike frequency (Fig. 7A) was reduced to 70.4 ± 20.2% of control (p = 0.2, n = 6) after 12 min, to 14.5 ± 8% of control (p < 0.0001, n = 10) after 20 min, and to 0 ± 0.3% of control (p < 0.0001, n = 4) after 30 min. Rm (Fig. 7C) was reduced to 77.8 ± 2.7% of control (p < 0.01, n = 5) after 12 min, to 80.2 ± 3.9% of control (p < 0.01, n = 5) after 20 min, and to 32 ± 9% of control (p < 0.01, n = 4) after 30 min. [Ca2+]i (Fig. 7B) increased to 173.9 ± 32.6% of control (p < 0.05, n = 10) after 12 min, to 192.5 ± 26.6% of control (p < 0.01, n = 12) after 20 min, and to 473.5 ± 109.3% of control (p < 0.0001, n = 4) after 30 min. Since all the parameters were measured 20 min after BMAA washout, we considered these changes as functional evidence of neuronal damage.
To additionally confirm that BMAA exclusively activates mGluR1-TRPC and AMPA receptors, slices were treated for 30 min with BMAA in the presence of JNJ 16259685 and CNQX (Fig. 7A–D). In this condition, the BMAA-induced effects were prevented. Indeed, spike frequency was 134.8 ± 21.7% (Fig. 7A) (p = 0.3, n = 4), [Ca2+]i was 124.6 ± 10.8% (Fig. 7B) (p = 0.8, n = 4), Rm was 100.3 ± 13.8% (Fig. 7C) (p = 0.37, n = 4), and Vm was 99.7 ± 3.2% (Fig. 7D) (p = 0.66, n = 4) of control. The antagonists alone (without BMAA) left the parameters unchanged (Fig. 7A–D).
BMAA induces cytochrome-c release, which is prevented by mGluR1 and mPTP antagonists, and activates ROS production
It has been proposed that mitochondrial Ca2+ overload leads to cyt-c release due to the opening of the mitochondrial permeability transition pore (mPTP) (Hajnóczky et al., 2003; Orrenius et al., 2003) and that glutamate receptor-mediated Ca2+ entry triggers cyt-c release from mitochondria inducing neuronal degeneration (Tang et al., 2005). In view of these data, we evaluated the toxic effects of BMAA by morphological analysis and immunohistochemical quantification of cyt-c release in TH-positive cells.
BMAA exposure (3 mm, 1 h) resulted in clear cell shrinkage in DAergic neurons. Indeed, morphological analyses showed that TH-positive neurons were significantly smaller in the BMAA-exposed than in the CTRL groups (Fig. 8H). One-way ANOVA analysis, performed on DAergic neuron cell size, among CTRL, BMAA, BMAA + CPCCOEt, BMAA + CNQX, BMAA + JNJ, BMAA + JNJ + CNQX, and BMAA + CsA (an mPTP blocker) groups showed a significant overall effect of treatment (F = 15.48; p < 0.0001). Post hoc comparison demonstrated that BMAA group data were significantly different from CTRL (p < 0.0001), BMAA + JNJ (p < 0.01), BMAA + JNJ + CNQX (p < 0.0001), and BMAA + CsA groups (p < 0.0001). No significant differences were observed between the BMAA and BMAA + CPCCOEt, BMAA and BMAA + CNQX, or BMAA + CPCCOEt and BMAA + CNQX groups (all p > 0.05). Furthermore, significant differences were observed between BMAA + CPCCOEt or BMAA + CNQX and BMAA + CNQX + JNJ or BMAA + CsA data (p < 0.01) (Fig. 8H). On the other hand, the differences between CTRL and BMAA + CNQX + JNJ or CTRL and BMAA + CsA or BMAA + CNQX + JNJ and BMAA + CsA groups were not significant (p > 0.05). These latter comparisons indicate that blocking either mGluR1 and AMPA receptors or mPTP (through CsA) prevents BMAA-induced neuronal damage (Fig. 8H).
Figure 8.
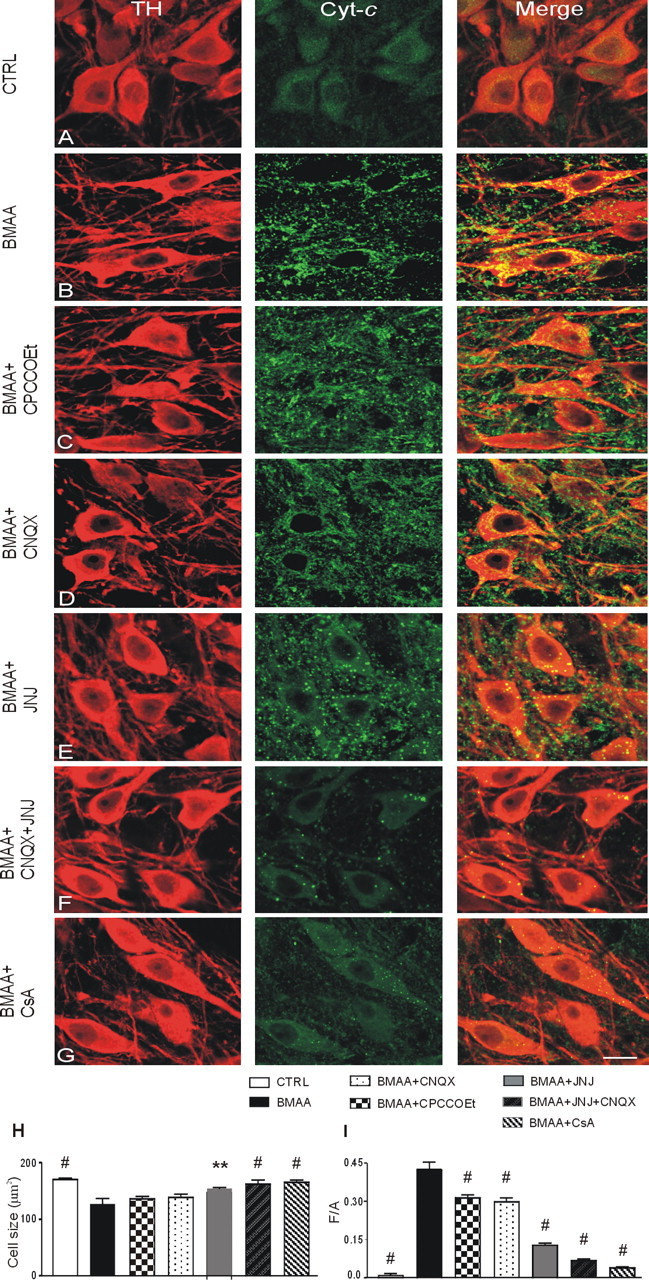
Prolonged exposure to BMAA causes neuronal shrinkage and massive cyt-c release into the cytosol of SNpc DAergic neurons. A–G, cyt-c and TH double-labeling confocal images of control (CTRL; A) and BMAA- (B), BMAA + CPCCOEt- (C), BMAA + CNQX- (D), BMAA + JNJ- (E), BMAA + CNQX + JNJ- (F), and BMAA + CsA- (G) treated slices. H, I, Histograms of soma cell size of TH-immunoreactive neurons (in square micrometers) (H) and densitometric values of cyt-c release expressed as mean fluorescence of individual cells normalized to total cellular surface (F/A) (I) in CTRL, BMAA, BMAA + CPCCOEt, BMAA + CNQX, BMAA + JNJ, BMAA + CNQX + JNJ, and BMAA + CsA groups. Data are reported as means ± SEM (n = 200 cells per group). Statistical comparison versus BMAA group, **p < 0.01, #p < 0.0001. Scale bar: 18 μm.
We further evaluated the ability of different ionotropic and metabotropic glutamate receptor antagonists to prevent BMAA-induced changes. The cyt-c immunoreactivity (IR) was almost absent in the CTRL group (Fig. 8A), while after BMAA treatment, it dramatically increased (Fig. 8B). Compared to BMAA treatment alone, BMAA + CPCCOEt and BMAA + CNQX treatments presented a small reduction of cyt-c IR intensity (Fig. 8C,D). JNJ 16259685 treatment presented a better protection to the BMAA-induced effects (Fig. 8E). Furthermore, the greatest protection was achieved when a combination of JNJ and CNQX was used or when BMAA was coapplied with CsA. These latter conditions almost completely prevented the BMAA effects on cyt-c IR (Fig. 8F,G).
One-way ANOVA performed on the cyt-c F/A values of CTRL, BMAA, BMAA + CPCCOEt, BMAA + CNQX, BMAA + JNJ, BMAA + JNJ + CNQX, and BMAA + CsA showed a significant overall effect of treatment (F = 133.84; p < 0.0001). Post hoc comparisons demonstrated that BMAA data were significantly different from those of all the other groups considered (p < 0.0001). Notably, the entity of BMAA-induced cyt-c release (F/A ratio = 0.43) was similar to that caused by hypoxia (F/A ratio = 0.5, data not shown) in DAergic neurons of SNpc, a condition known to irreversibly impair neuronal function (Geracitano et al., 2005). The comparisons between BMAA + CPCCOEt or BMAA + CNQX and BMAA + JNJ groups (p < 0.0001) or between BMAA + JNJ and BMAA + JNJ + CNQX groups (p < 0.01) were also significant (Fig. 8I). No significant differences were observed between BMAA + CPCCOEt and BMAA + CNQX, between CTRL and BMAA + CNQX + JNJ, between CTRL and BMAA + CsA, or between BMAA + CNQX + JNJ and BMAA + CsA groups (p > 0.05) (Fig. 8I). These latter data indicate that the combined antagonism of mGluR1 and AMPA receptors, as well as the combination of BMAA and CsA treatments, completely prevents BMAA-induced neuronal damage.
Furthermore, to determine the involvement of mitochondrial stress in BMAA toxicity, slices were treated with the ROS indicator hydroethidine (Anantharam et al., 2007; Liu et al., 2009; Zhang et al., 2009) to detect the production of superoxide following BMAA exposure (supplemental Fig. S3, available at www.jneurosci.org as supplemental material). BMAA caused a marked increase of the hydroethidine fluorescence in TH-positive cells compared to CTRL, indicating an augmentation of ROS production. Indeed, this increase was significantly reduced by treatment with the antioxidant 1,4-dithiothreitol (DTT, 500 μm). One-way ANOVA performed on the ROS F/A index of CTRL, BMAA, and BMAA + DTT showed a significant overall effect of treatment (F = 129.66; p < 0.0001). Post hoc comparisons demonstrated that BMAA data were significantly different from CTRL (p < 0.001) and BMAA + DTT groups (p < 0.001). Furthermore, the comparison between BMAA + DTT and CTRL was also significant (p < 0.05) (supplemental Fig. S3, available at www.jneurosci.org as supplemental material).
BMAA and the “multiple-hits hypothesis”
Although several gene mutations have been proposed as risk factors for PD development, the low penetrance of some of them suggests that PD may be due to the interaction between multiple factors. The “multiple hits hypothesis” (Sulzer, 2007) combines toxic stress, as mitochondrial dysfunction, with decrease of neuroprotective responses and exposure to environmental neurotoxic agents.
To this regard, it has been demonstrated in cortical cell cultures that low concentrations of BMAA, in combination with other insults (β-amyloid, NMDA, and MPP+), induces neuronal death (Lobner et al., 2007). To test this hypothesis in midbrain slice preparation, we puffed 1 mm BMAA (instead of 3 mm) with and without bath application of 20 μm NMDA for 8–10 min. In this condition, BMAA caused a significantly larger elevation of [Ca2+]i than in control, without affecting membrane current (Fig. 9A,B). Indeed, [Ca2+]i increased to 338.4 ± 79.6% of control (p < 0.05), whereas IBMAA remained 92.4 ± 7.4% of control (p = 0.36, n = 5 for both). These results indicate that calcium loading induced by lower BMAA concentrations can be potentiated in stressed neurons, such as during exposure to NMDA.
Discussion
The interest toward BMAA as putative toxic agent damaging DAergic cells has grown considerably due to the discovery that this toxin is not only present in the Guamanian islands but appears to be ubiquitous throughout the world (Cox et al., 2003; Esterhuizen and Downing, 2008; Johnson et al., 2008; Metcalf et al., 2008). This natural amino acid binds to proteins in human tissues and thus might represent an endogenous reservoir constantly released during protein catabolism (Murch et al., 2004). For this reason, only recent studies properly quantified the concentration of BMAA in postmortem brain tissues of patients (Murch et al., 2004; Pablo et al., 2009), using validated methods able to detect either the free or the protein-bound BMAA portions. Taking into account the findings by Duncan et al. (1991), reporting that 10–30 μg/g BMAA in the rat brain corresponds approximately to the concentration of ≤0.25 mm, it can be concluded that BMAA concentration in the human brain of patients is in the low millimolar range. The deleterious mechanisms of the toxin could underlie neurological conditions such as ALS-PDC and, potentially, idiopathic Parkinson's disease. Few studies have investigated the effects of BMAA on nigrostriatal DAergic neurons (Lindstrom et al., 1990; Santiago et al., 2006). However, BMAA toxicity was evaluated indirectly, by measuring the ability of these cells to uptake or release dopamine. In the present study, we investigated the direct effects of BMAA onto single DAergic neurons of the SNpc in an in vitro slice preparation. We found that brief BMAA applications, at a concentration similar to that found in ALS-PDC brains (Murch et al., 2004; Pablo et al., 2009), induced reversible and reproducible membrane depolarizations/inward currents, firing increase, and Ca2+ accumulation. In contrast, prolonged exposures (>12 min) caused functional impairment, cellular shrinkage, calcium overload, cyt-c release, and ROS production that did not return to control values for up to 20 min or several hours after BMAA washout.
In the majority of cells tested, IBMAA was principally mediated by activation of mGluR1-TRPC and, to a lesser extent, AMPA receptors. In contrast, Ca2+ accumulation was strongly dependent on mGluR1-TRPC but not AMPA receptor activation. Indeed, BMAA-induced Ca2+ increase was strongly reduced by CPA, indicating that this amino acid activates a mGluR1-induced Ca2+ release from endoplasmic reticulum.
Moreover, BMAA-induced responses (IBMAA and [Ca2+]i increase) were abolished by a mixture of mGluR1, TRPC, and AMPA receptor antagonists, and occluded by the selective mGluR1 agonist DHPG. Therefore, it is unlikely that BMAA activates additional mechanisms. The contribution of mGluR5 to BMAA responses is minimal or absent since the specific antagonist MPEP was without effect. When comparing the pharmacology of similar IGLU and IBMAA amplitudes, we found that IGLU, in DAergic cells, was highly sensitive to both ionotropic and mGluR1 antagonists, further suggesting that, despite the structural similarity between BMAA and glutamate, the former is preferentially an mGluR1 agonist.
We have previously reported that activation of mGluR1 by DHPG, caused an inward current in DAergic neurons (Guatteo et al., 1999) that was mediated by TRPC-like channels (Tozzi et al., 2003). Here we found that also IBMAA is due to activation of TRPC-like channels, through mGluR1. This conclusion is supported by the following evidence: (1) IBMAA was reduced by the nonselective TRPC channels blockers SKF 96365 and RR; (2) the IBMAA reversal potential was ∼0 mV, consistent with the activation of a TRPC-mediated mixed cation current (Okada et al., 1998; Strubing et al., 2001; Tozzi et al., 2003); (3) Orai1 and STIM1, proteins that are likely to interact with TRPC channels downstream of G-protein-coupled receptor activation (Wang et al., 2008; Kim et al., 2009; Klejman et al., 2009; Liao et al., 2009) (for review, see Cahalan, 2009), were expressed in biocytin-loaded neurons; (4) CPCCOEt and SKF 96365 did not have additive effects on IBMAA, demonstrating that mGluR1 activation and TRPC-like channel opening are two subsequent steps in the BMAA-induced pathway.
Interestingly, GABAergic SNpc cells responded to BMAA with an AMPA-mediated inward current that was not associated with an intracellular calcium increase. A similar drug action was also observed in a minority of DAergic neurons. BMAA has been previously reported to potently activate mGluRs in hippocampal, striatal, and cerebellar neurons (Copani et al., 1990, 1991; Manzoni et al., 1991; Staton and Bristow, 1998; Lobner et al., 2007). Alternatively, it predominantly activates NMDA receptors in cortical neurons (Weiss et al., 1989; Lobner et al., 2007) and AMPA/kainate receptors in spinal motoneurons (Rao et al., 2006). Although a likely explanation for the action of BMAA on different glutamate receptors in different cell types is not available yet, these results provide the idea that the responses to BMAA may depend on isoforms of glutamate receptors, expressed on each cell type, that are preferentially bound by the amino acid. The BMAA-induced cellular response could also depend on the particular localization of mGluRs on the plasmalemmal membrane (Hubert et al., 2001) that renders these receptors more accessible to the drug.
Despite BMAA being a natural glutamate agonist, our experiments using dl-TBOA suggest that it is not a substrate for EAATs and thus, it does not enter DAergic neurons through these carriers. Therefore, it is unlikely that the neurotoxic effects of BMAA are either mediated by its intracellular transportation through EAATs or defects in their expression (Wilson and Shaw, 2007).
An important clinical implication of our results is the observation that, when the toxin was applied for >12 min, the DAergic neurons showed a drop in membrane resistance, reduction in the number of evoked action potentials, and [Ca2+]i increase, which were prevented by mGluR1 and AMPA receptor antagonists. Moreover, BMAA-treated TH-positive cells showed an increased ROS production, prevented by the antioxidant DTT, and a release of cyt-c from mitochondria. In this regard, it has been reported a higher frequency of somatic mutations in mitochondrial genome in Guam ALS and PD patients than in Guam controls, suggesting that mitochondria dysfunction may contribute, as genetic susceptibility, to ALS-PDC disease (Lynch et al., 2008). Interestingly, application of the potent mGluR1 antagonist JNJ 16259685 considerably reduced cyt-c release, suggesting that mGluR1 are the principal mediators of the BMAA-induced mitochondrial stress. An almost complete neuroprotection was achieved when BMAA was applied in the presence of JNJ 16259685 and CNQX, or CsA, a potent mPTP inhibitor (Ferrand-Drake et al., 2003; Precht et al., 2005). Indeed, cyt-c immunoreactivity and cellular swelling were similar to those in control cells. This supports also an additional contribution to cellular damage by AMPA-like receptors. Indeed, when AMPA and low-potency mGluR1 antagonists (CNQX and CPCCOEt, respectively) were applied separately, the BMAA-induced cyt-c release was only partially prevented. These results suggest that AMPA receptor activation may participate in the mechanism of BMAA toxicity by producing neuronal depolarization (via sodium influx), which in turn promotes activation of voltage-gated Na+ and Ca2+ channels (Berman and Murray, 1996; de Erausquin, 2004).
The pathological alterations caused by BMAA in in vitro conditions agree with an in vivo study showing a loss of TH immunoreactivity following intranigral injections of this compound (Lindström et al., 1990). Indeed, the persistent activation of mGluR1 could cause a derangement of ionic homeostasis, by releasing Ca2+ from intracellular stores, elevating [Na+]i through the opening of TRPC-like channels (Guatteo et al., 1999; Tozzi et al., 2003), and activation of voltage-dependent Na+ and Ca2+ channels. This prolonged intracellular ionic dysregulation implicates mGluRs as central players in the injury of DAergic neurons (Choi, 1994; Lu et al., 1996), leading to inappropriate activation of kinases, proteases, and phospholipases and to energy deficits (Salińska et al., 2005). Moreover, the excessive elevation of [Ca2+]i might trigger mitochondrial calcium overload (Werth and Thayer, 1994), overproduction of ROS (Lafon-Cazal et al., 1993; Dugan et al., 1995; Reynolds and Hastings, 1995; Carriedo et al., 1998, 2000), and release of cyt-c from mitochondria (Orrenius et al., 2003). In line with the idea that mGluRs activate processes leading to excitotoxic neuronal death in the majority of DAergic neurons, it has been recently reported that functional antagonism of group I mGluRs (mGluR1 and mGluR5) is neuroprotective in an animal model of PD (rats treated with 6-hydroxydopamine) (Vernon et al., 2005, 2007).
It has been reported that BMAA causes neuronal death only at high millimolar concentrations (Spencer et al., 1987; Santiago et al., 2006; Buenz and Howe, 2007). In line with this, we have reported that BMAA, in the low millimolar range, caused electrophysiological and mitochondrial dysfunction in DAergic cells. Although we did not study late-onset deleterious effects of lower BMAA concentrations (due to the limitation of an in vitro slice preparation), we found that prolonged BMAA exposure (30 min) at low concentration (100 μm) caused larger depolarization and firing frequency increases than short-lasting applications. This suggests that the continuous presence of BMAA in the extracellular fluid, even in low concentration, may induce significant alteration of neuronal activity.
This conclusion is in line with the observation that BMAA can bind to proteins in the brain and can be released back as a free amino acid during protein catabolism years after consumption (Murch et al., 2004).
Moreover, according to the previous evidence on cortical neurons, that micromolar BMAA concentration is able to potentiate insults caused by other toxic agents (β-amyloid, NMDA, and MPP+; Lobner et al., 2007), we found that coapplication of BMAA and NMDA produced larger Ca2+ accumulations than BMAA alone. This suggests a potential synergistic effect of this environmental amino acid with preexisting states of neuronal stress. Therefore, since this toxin alters cellular activity, it may cooperate in causing dysfunction and late-onset injury to metabolically compromised DAergic cells, such as those surviving in PD patients (“multiple hits” hypothesis) (Sulzer, 2007). It is worth mentioning that neuropathology of ALS-PDC patients reveals Lewy's bodies and α-synuclein-positive inclusions in SNpc DAergic neurons similar to those found in PD patients (Hirano et al., 1966).
Since we have shown that BMAA, which has a human exposure more widespread than previously thought, may exacerbate physiological events caused by excitatory inputs to DAergic neurons and initiate excitotoxic pathological mechanisms by mGluRs activation, these receptors could be one of the promising targets to correct neuronal dysfunction of the DAergic system in the more common, sporadic form of Parkinson's disease.
Footnotes
The study was supported by grants from the Ministero della Salute (to N.B.M.). We are grateful to Dr. Peter S. Freestone and Prof. Janusz Lipski for their comments on the manuscript.
References
- Allen CN, Spencer PS, Carpenter DO. Beta-N-methylamino-L-alanine in the presence of bicarbonate is an agonist at non-N-methyl-D-aspartate type receptors. Neuroscience. 1993;54:567–574. doi: 10.1016/0306-4522(93)90228-8. [DOI] [PubMed] [Google Scholar]
- Anantharam V, Kaul S, Song C, Kanthasamy A, Kanthasamy AG. Pharmacological inhibition of neuronal NADPH oxidase protects against 1-methyl-4-phenylpyridinium (MPP+)-induced oxidative stress and apoptosis in mesencephalic dopaminergic neuronal cells. Neurotoxicology. 2007;28:988–997. doi: 10.1016/j.neuro.2007.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beal MF. Excitotoxicity and nitric oxide in Parkinson's disease pathogenesis. Ann Neurol. 1998;44:S110–S114. doi: 10.1002/ana.410440716. [DOI] [PubMed] [Google Scholar]
- Berman FW, Murray TF. Characterization of glutamate toxicity in cultured rat cerebellar granule neurons at reduced temperature. J Biochem Toxicol. 1996;11:111–119. doi: 10.1002/(SICI)1522-7146(1996)11:3<111::AID-JBT2>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- Berretta N, Bernardi G, Mercuri NB. Alpha(1)-adrenoceptor-mediated excitation of substantia nigra pars reticulata neurons. Neuroscience. 2000;98:599–604. doi: 10.1016/s0306-4522(00)00135-4. [DOI] [PubMed] [Google Scholar]
- Bogaerts V, Theuns J, van Broeckhoven C. Genetic findings in Parkinson's disease and translation into treatment: a leading role for mitochondria? Genes Brain Behav. 2008;7:129–151. doi: 10.1111/j.1601-183X.2007.00342.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brownson DM, Mabry TJ, Leslie SW. The cycad neurotoxic amino acid, β-N-methylamino-L-alanine (BMAA), elevates intracellular calcium levels in dissociated rat brain cells. J Ethnopharmacol. 2002;82:159–167. doi: 10.1016/s0378-8741(02)00170-8. [DOI] [PubMed] [Google Scholar]
- Buenz EJ, Howe CL. Beta-methylamino-alanine (BMAA) injures hippocampal neurons in vivo. Neurotoxicology. 2007;28:702–704. doi: 10.1016/j.neuro.2007.02.010. [DOI] [PubMed] [Google Scholar]
- Cahalan MD. STIMulating store-operated Ca2+ entry. Nat Cell Biol. 2009;11:669–677. doi: 10.1038/ncb0609-669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carriedo SG, Yin HZ, Sensi SL, Weiss JH. Rapid Ca2+ entry through Ca2+-permeable AMPA/kainate channels triggers marked intracellular Ca2+ rises and consequent oxygen radical production. J Neurosci. 1998;18:7727–7738. doi: 10.1523/JNEUROSCI.18-19-07727.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carriedo SG, Sensi SL, Yin HZ, Weiss JH. AMPA exposures induce mitochondrial Ca2+ overload and ROS generation in spinal motor neurons in vitro. J Neurosci. 2000;20:240–250. doi: 10.1523/JNEUROSCI.20-01-00240.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang YC, Chiu SJ, Kao KP. beta-N-methylamino-L-alanine (L-BMAA) decrease brain glutamate receptor number and induces behavioural changes in rats. Chin J Physiol. 1993;36:79–84. [PubMed] [Google Scholar]
- Choi DW. Calcium and excitotoxic neuronal injury. Ann N Y Acad Sci. 1994;747:162–171. doi: 10.1111/j.1749-6632.1994.tb44407.x. [DOI] [PubMed] [Google Scholar]
- Copani A, Canonico PL, Nicoletti F. Beta-N-methylamino-L-alanine (L-BMAA) is a potent agonist of ‘metabolotropic’ glutamate receptors. Eur J Pharmacol. 1990;181:327–328. doi: 10.1016/0014-2999(90)90100-k. [DOI] [PubMed] [Google Scholar]
- Copani A, Canonico PL, Catania MV, Aronica E, Bruno V, Ratti E, van Amsterdam FT, Gaviraghi G, Nicoletti F. Interaction between beta-N-methylamino-L-alanine and excitatory amino acid receptors in brain slices and neuronal cultures. Brain Res. 1991;558:79–86. doi: 10.1016/0006-8993(91)90716-9. [DOI] [PubMed] [Google Scholar]
- Cox PA, Banack SA, Murch SJ. Biomagnification of cyanobacterial neurotoxins and neurodegenerative disease among the Chamorro people of Guam. Proc Natl Acad Sci U S A. 2003;100:13380–13383. doi: 10.1073/pnas.2235808100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Erausquin GA. Transactivation of cell death signals by glutamate transmission in dopaminergic neurons. Crit Rev Neurobiol. 2004;16:107–119. doi: 10.1615/critrevneurobiol.v16.i12.120. [DOI] [PubMed] [Google Scholar]
- DeHaven WI, Jones BF, Petranka JG, Smyth JT, Tomita T, Bird GS, Putney JW., Jr TRPC channels function independently of STIM1 and Orai1. J Physiol. 2009;587:2275–2298. doi: 10.1113/jphysiol.2009.170431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Monte DA. The environment and Parkinson's disease: is the nigrostriatal system preferentially targeted by neurotoxins? Lancet Neurol. 2003;2:531–538. doi: 10.1016/s1474-4422(03)00501-5. [DOI] [PubMed] [Google Scholar]
- Dugan LL, Sensi SL, Canzoniero LM, Handran SD, Rothman SM, Lin TS, Goldberg MP, Choi DW. Mitochondrial production of reactive oxygen species in cortical neurons following exposure to N-methyl-d-aspartate. J Neurosci. 1995;15:6377–6388. doi: 10.1523/JNEUROSCI.15-10-06377.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duncan MW, Villacreses NE, Pearson PG, Wyatt L, Rapoport SI, Kopin IJ, Markey SP, Smith QR. 2-Amino-3-(methylamino)-propanoic acid (BMAA) pharmacokinetics, blood-brain barrier permeability in the rat. J Pharmacol Exp Ther. 1991;258:27–35. [PubMed] [Google Scholar]
- Esterhuizen M, Downing TG. β-N-methylamino-L-alanine (BMAA) in novel South African cyanobacterial isolates. Ecotoxicol Environ Saf. 2008;71:309–313. doi: 10.1016/j.ecoenv.2008.04.010. [DOI] [PubMed] [Google Scholar]
- Faber ESL, Sedlak P, Vidovic M, Sah P. Synaptic activation of transient receptor potential channels by metabotropic glutamate receptors in the lateral amygdala. Neuroscience. 2006;137:781–794. doi: 10.1016/j.neuroscience.2005.09.027. [DOI] [PubMed] [Google Scholar]
- Ferrand-Drake M, Zhu C, Gidö G, Hansen AJ, Karlsson JO, Bahr BA, Zamzami N, Kroemer G, Chan PH, Wieloch T, Blomgren K. Cyclosporin A prevents calpain activation despite increased intracellular calcium concentrations, as well as translocation of apoptosis-inducing factor, cytochrome c and caspase-3 activation in neurons exposed to transient hypoglycemia. J Neurochem. 2003;85:1431–1442. doi: 10.1046/j.1471-4159.2003.01794.x. [DOI] [PubMed] [Google Scholar]
- Garruto RM, Yase Y. Neurodegenerative disorders of the western pacific: the search for mechanisms of pathogenesis. Trends Neurosci. 1986;9:368–374. [Google Scholar]
- Geracitano R, Tozzi A, Berretta N, Florenzano F, Guatteo E, Viscomi MT, Chiolo B, Molinari M, Bernardi G, Mercuri NB. Protective role of hydrogen peroxide in oxygen-deprived dopaminergic neurones of the rat substantia nigra. J Physiol. 2005;568:97–110. doi: 10.1113/jphysiol.2005.092510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gernert M, Fedrowitz M, Wlaz P, Löscher W. Subregional changes in discharge rate, pattern, and drug sensitivity of putative GABAergic nigral neurons in the kindling model of epilepsy. Eur J Neurosci. 2004;20:2377–2386. doi: 10.1111/j.1460-9568.2004.03699.x. [DOI] [PubMed] [Google Scholar]
- Greenamyre JT, Betarbet R, Sherer TB. The rotenone model of Parkinson's disease: genes, environment and mitochondria. Parkinsonism Relat Disord. 2003;9:S59–S64. doi: 10.1016/s1353-8020(03)00023-3. [DOI] [PubMed] [Google Scholar]
- Grillner P, Mercuri NB. Intrinsic membrane properties and synaptic inputs regulating the firing activity of the dopamine neurons. Behav Brain Res. 2002;130:149–169. doi: 10.1016/s0166-4328(01)00418-1. [DOI] [PubMed] [Google Scholar]
- Grynkiewicz G, Poenie M, Tsien RY. A new generation of Ca2+ indicators with greatly improved fluorescence properties. J Biol Chem. 1985;260:3440–3450. [PubMed] [Google Scholar]
- Guatteo E, Mercuri NB, Bernardi G, Knöpfel T. Intracellular sodium and calcium homeostasis during hypoxia in dopamine neurons of rat substantia nigra pars compacta. J Neurophysiol. 1998;80:2237–2243. doi: 10.1152/jn.1998.80.5.2237. [DOI] [PubMed] [Google Scholar]
- Guatteo E, Mercuri NB, Bernardi G, Knöpfel T. Group I metabotropic glutamate receptors mediate an inward current in rat substantia nigra dopamine neurons that is independent from calcium mobilization. J Neurophysiol. 1999;82:1974–1981. doi: 10.1152/jn.1999.82.4.1974. [DOI] [PubMed] [Google Scholar]
- Hajnóczky G, Davies E, Madesh M. Calcium signaling and apoptosis. Biochem Biophys Res Commun. 2003;304:445–454. doi: 10.1016/s0006-291x(03)00616-8. [DOI] [PubMed] [Google Scholar]
- Hanna MG, Bhatia KP. Movement disorders and mitochondrial dysfunction. Curr Opin Neurol. 1997;10:351–356. doi: 10.1097/00019052-199708000-00012. [DOI] [PubMed] [Google Scholar]
- Hirano A, Malamud N, Elizan TS, Kurland LT. Amyotrophic lateral sclerosis and parkinsonism dementia complex on Guam. Further pathologic studies. Arch Neurol. 1966;15:35–51. doi: 10.1001/archneur.1966.00470130039004. [DOI] [PubMed] [Google Scholar]
- Hubert GW, Paquet M, Smith Y. Differential subcellular localization of mGluR1a and mGluR5 in the rat and monkey substantia nigra. J Neurosci. 2001;21:1838–1847. doi: 10.1523/JNEUROSCI.21-06-01838.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ince PG, Codd GA. Return of the cycad hypothesis-does the amyotrophic lateral sclerosis/parkinsonism dementia complex (ALS/PDC) of Guam have new implications for global health? Neuropathol Appl Neurobiol. 2005;31:345–353. doi: 10.1111/j.1365-2990.2005.00686.x. [DOI] [PubMed] [Google Scholar]
- Johnson HE, King SR, Banack SA, Webster C, Callanaupa WJ, Cox PA. Cyanobacteria (Nostoc commune) used as a dietary item in the Peruvian highlands produce the neurotoxic amino acid BMAA. J Ethnopharmacol. 2008;118:159–165. doi: 10.1016/j.jep.2008.04.008. [DOI] [PubMed] [Google Scholar]
- Kim MS, Zeng W, Yuan JP, Shin DM, Worley PF, Muallem S. Native store-operated Ca2+ influx requires the channel function of ORAI1 and TRPC1. J Biol Chem. 2009;284:9733–9741. doi: 10.1074/jbc.M808097200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein C, Schlossmacher MG. Parkinson disease, 10 years after its genetic revolution: multiple clues to a complex disorder. Neurology. 2007;69:2093–2104. doi: 10.1212/01.wnl.0000271880.27321.a7. [DOI] [PubMed] [Google Scholar]
- Klejman ME, Gruszczynska-Biegala J, Skibinska-Kijek A, Wisniewska MB, Misztal K, Blazejczyk M, Bojarski L, Kuznicki J. Expression of STIM1 in brain and puncta-like co-localization of STIM1 and ORAI1 upon depletion of Ca2+ store in neurons. Neurochem Int. 2009;54:49–55. doi: 10.1016/j.neuint.2008.10.005. [DOI] [PubMed] [Google Scholar]
- Lacey MG, Mercuri NB, North RA. Two cell types in rat substantia nigra zona compacta distinguished by membrane properties and the actions of dopamine and opioids. J Neurosci. 1989;9:1233–1241. doi: 10.1523/JNEUROSCI.09-04-01233.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lafon-Cazal M, Pietri S, Culcasi M, Bockaert J. NMDA-dependent superoxide production and neurotoxicity. Nature. 1993;364:535–537. doi: 10.1038/364535a0. [DOI] [PubMed] [Google Scholar]
- Lavreysen H, Wouters R, Bischoff F, Nóbrega Pereira S, Langlois X, Blokland S, Somers M, Dillen L, Lesage AS. JNJ16259685, a highly potent, selective and systemically active mGlu1 receptor antagonist. Neuropharmacology. 2004;47:961–972. doi: 10.1016/j.neuropharm.2004.08.007. [DOI] [PubMed] [Google Scholar]
- Le Couteur DG, Muller M, Yang MC, Mellick GD, McLean AJ. Age-environmental and gene-environmental interactions in the pathogenesis of Parkinson's disease. Rev Environ Health. 2002;17:51–64. doi: 10.1515/reveh.2002.17.1.51. [DOI] [PubMed] [Google Scholar]
- Liao Y, Plummer NW, George MD, Abramowitz J, Zhu MX, Birnbaumer L. A role of ORAI in TRPC-mediated Ca2+ entry suggests that a TRPC:ORAI complex may mediate store and receptor operated Ca2+ entry. Proc Natl Acad Sci U S A. 2009;106:3202–3206. doi: 10.1073/pnas.0813346106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindström H, Luthman J, Mouton P, Spencer P, Olson L. Plant-derived neurotoxic amino acids (beta-N-oxalylamino-L-alanine and beta-N-methylamino-L-alanine): effects on central monoamine neurons. J Neurochem. 1990;55:941–949. doi: 10.1111/j.1471-4159.1990.tb04582.x. [DOI] [PubMed] [Google Scholar]
- Liu J, Yu Z, Guo S, Lee SR, Xing C, Zhang C, Gao Y, Nicholls DG, Lo EH, Wang X. Effects of neuroglobin overexpression on mitochondrial function and oxidative stress following hypoxia/reoxygenation in cultured neurons. J Neurosci Res. 2009;87:164–170. doi: 10.1002/jnr.21826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lobner D, Piana PM, Salous AK, Peoples RW. Beta-N-methylamino-L-alanine enhances neurotoxicity through multiple mechanisms. Neurobiol Dis. 2007;25:360–366. doi: 10.1016/j.nbd.2006.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu YM, Yin HZ, Chiang J, Weiss JH. Ca2+-permeable AMPA/kainate and NMDA channels: high rate of Ca2+ influx underlies potent induction of injury. J Neurosci. 1996;16:5457–5465. doi: 10.1523/JNEUROSCI.16-17-05457.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch D, Wanglund C, Spathis R, Chan CW, Reiff DM, Lum JK, Garruto RM. The contribution of mitochondrial dysfunction to a gene-environment model of Guamanian ALS and PD. Mitochondrion. 2008;8:109–116. doi: 10.1016/j.mito.2007.09.002. [DOI] [PubMed] [Google Scholar]
- Manzoni OJ, Prezeau L, Bockaert J. Beta-N-methylamino-L-alanine is a low-affinity agonist of metabotropic glutamate receptors. Neuroreport. 1991;2:609–611. doi: 10.1097/00001756-199110000-00014. [DOI] [PubMed] [Google Scholar]
- Mercuri NB, Bonci A, Calabresi P, Stefani A, Bernardi G. Properties of the hyperpolarization-activated cation current Ih in rat midbrain dopaminergic neurons. Eur J Neurosci. 1995;7:462–469. doi: 10.1111/j.1460-9568.1995.tb00342.x. [DOI] [PubMed] [Google Scholar]
- Metcalf JS, Banack SA, Lindsay J, Morrison LF, Cox PA, Codd GA. Co-occurrence of β-N-methylamino-L-alanine, a neurotoxic amino acid with other cyanobacterial toxins in British waterbodies, 1990–2004. Environmental Microbiology. 2008;10:702–708. doi: 10.1111/j.1462-2920.2007.01492.x. [DOI] [PubMed] [Google Scholar]
- Murch SJ, Cox PA, Banack SA. A mechanism for slow release of biomagnified cyanobacterial neurotoxins and neurodegenerative disease in Guam. Proc Natl Acad Sci U S A. 2004;101:12228–12231. doi: 10.1073/pnas.0404926101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers TG, Nelson SD. Neuroactive carbamate adducts of beta-N-methylamino-L-alanine and ethylenediamine. Detection and quantitation under physiological conditions by 13C NMR. J Biol Chem. 1990;265:10193–10195. [PubMed] [Google Scholar]
- Okada T, Shimizu S, Wakamori M, Maeda A, Kurosaki T, Takada N, Imoto K, Mori Y. Molecular cloning and functional characterization of a novel receptor-activated TRP Ca2+ channel from mouse brain. J Biol Chem. 1998;273:10279–10287. doi: 10.1074/jbc.273.17.10279. [DOI] [PubMed] [Google Scholar]
- Orrenius S, Zhivotovsky B, Nicotera P. Regulation of cell death: the calcium-apoptosis link. Nat Rev Mol Cell Biol. 2003;4:552–565. doi: 10.1038/nrm1150. [DOI] [PubMed] [Google Scholar]
- Pablo J, Banack SA, Cox PA, Johnson TE, Papapetropoulos S, Bradley WG, Buck A, Mash DC. Cyanobacterial neurotoxin BMAA in ALS and Alzheimer's disease. Acta Neurol Scand. 2009;120:216–225. doi: 10.1111/j.1600-0404.2008.01150.x. [DOI] [PubMed] [Google Scholar]
- Pai KS, Shankar SK, Ravindranath V. Billion-fold difference in the toxic potencies of two excitatory plant amino acids, L-BOAA and L-BMAA: biochemical and morphological studies using mouse brain slices. Neurosci Res. 1993;17:241–248. doi: 10.1016/0168-0102(93)90051-q. [DOI] [PubMed] [Google Scholar]
- Precht TA, Phelps RA, Linseman DA, Butts BD, Le SS, Laessig TA, Bouchard RJ, Heidenreich KA. The permeability transition pore triggers Bax translocation to mitochondria during neuronal apoptosis. Cell Death Differ. 2005;12:255–265. doi: 10.1038/sj.cdd.4401552. [DOI] [PubMed] [Google Scholar]
- Rakonczay Z, Matsuoka Y, Giacobini E. Effects of L-beta-N-methylamino-L-alanine (L-BMAA) on the cortical cholinergic and glutamatergic systems of the rat. J Neurosci Res. 1991;29:121–126. doi: 10.1002/jnr.490290114. [DOI] [PubMed] [Google Scholar]
- Rao SD, Banack SA, Cox PA, Weiss JH. BMAA selectively injures motor neurons via AMPA/kainate receptor activation. Exp Neurol. 2006;201:244–252. doi: 10.1016/j.expneurol.2006.04.017. [DOI] [PubMed] [Google Scholar]
- Reed DM, Brody JA. Amyotrophic lateral sclerosis and parkinsonism-dementia on Guam, 1945–1972: I. Descriptive epidemiology. Am J Epidemiol. 1975;101:287–301. doi: 10.1093/oxfordjournals.aje.a112097. [DOI] [PubMed] [Google Scholar]
- Reynolds IJ, Hastings TG. Glutamate induces the production of reactive oxygen species in cultured forebrain neurons following NMDA receptor activation. J Neurosci. 1995;15:3318–3327. doi: 10.1523/JNEUROSCI.15-05-03318.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riccio A, Medhurst AD, Mattei C, Kelsell RE, Calver AR, Randall AD, Benham CD, Pangalos MN. mRNA distribution analysis of human TRPC family in CNS and peripheral tissues. Brain Res Mol Brain Res. 2002;109:95–104. doi: 10.1016/s0169-328x(02)00527-2. [DOI] [PubMed] [Google Scholar]
- Rodriguez MC, Obeso JA, Olanow CW. Subthalamic nucleus-mediated excitotoxicity in Parkinson's disease: a target for neuroprotection. Ann Neurol. 1998;44:S175–S188. doi: 10.1002/ana.410440726. [DOI] [PubMed] [Google Scholar]
- Salińska E, Danysz W, Łazarewicz JW. The role of excitotoxicity in neurodegeneration. Folia Neuropathol. 2005;43:322–339. [PubMed] [Google Scholar]
- Santiago M, Matarredona ER, Machado A, Cano J. Acute perfusion of BMAA in the rat's striatum by in vivo microdialysis. Toxicol Lett. 2006;167:34–39. doi: 10.1016/j.toxlet.2006.08.005. [DOI] [PubMed] [Google Scholar]
- Santucci S, Zsürger N, Chabry J. Β-N-methylamino-L-alanine induced in vivo retinal cell death. J Neurochem. 2009;109:819–825. doi: 10.1111/j.1471-4159.2009.06022.x. [DOI] [PubMed] [Google Scholar]
- Shimamoto K, Lebrun B, Yasuda-Kamatani Y, Sakaitani M, Shigeri Y, Yumoto N, Nakajima T. DL-threo-beta-benzyloxyaspartate, a potent blocker of excitatory amino acid transporters. Mol Pharmacol. 1998;53:195–201. doi: 10.1124/mol.53.2.195. [DOI] [PubMed] [Google Scholar]
- Smith SE, Meldrum BS. Receptor site specificity for the acute effects of beta-N-methylamino-alanine in mice. Eur J Pharmacol. 1990;187:131–134. doi: 10.1016/0014-2999(90)90350-f. [DOI] [PubMed] [Google Scholar]
- Spencer PS, Nunn PB, Hugon J, Ludolph AC, Roy DN. Motorneurone disease on Guam: possible role of a food neurotoxin. Lancet. 1986;1:965. doi: 10.1016/s0140-6736(86)91059-7. [DOI] [PubMed] [Google Scholar]
- Spencer PS, Nunn PB, Hugon J, Ludolph AC, Ross SM, Roy DN, Robertson RC. Guam amyotrophic lateral sclerosis-parkinsonism-dementia linked to a plant excitant neurotoxin. Science. 1987;237:517–522. doi: 10.1126/science.3603037. [DOI] [PubMed] [Google Scholar]
- Staton PC, Bristow DR. The dietary excitotoxins beta-N-methylamino-L-alanine and beta-N-oxalylamino-L-alanine induce necrotic- and apoptotic-like death of rat cerebellar granule cells. J Neurochem. 1997;69:1508–1518. doi: 10.1046/j.1471-4159.1997.69041508.x. [DOI] [PubMed] [Google Scholar]
- Staton PC, Bristow DR. Role of group III metabotropic glutamate receptors in excitotoxin-induced cerebellar granule cell death. J Neurochem. 1998;71:1280–1288. doi: 10.1046/j.1471-4159.1998.71031280.x. [DOI] [PubMed] [Google Scholar]
- Strübing C, Krapivinsky G, Krapivinsky L, Clapham DE. TRPC1 and TRPC5 form a novel cation channel in mammalian brain. Neuron. 2001;29:645–655. doi: 10.1016/s0896-6273(01)00240-9. [DOI] [PubMed] [Google Scholar]
- Sulzer D. Multiple hit hypotheses for dopamine neuron loss in Parkinson's disease. Trends Neurosci. 2007;30:244–250. doi: 10.1016/j.tins.2007.03.009. [DOI] [PubMed] [Google Scholar]
- Tang TS, Slow E, Lupu V, Stavrovskaya IG, Sugimori M, Llinás R, Kristal BS, Hayden MR, Bezprozvanny I. Disturbed Ca2+ signaling and apoptosis of medium spiny neurons in Huntington's disease. Proc Natl Acad Sci U S A. 2005;102:2602–2607. doi: 10.1073/pnas.0409402102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong G, Jahr CE. Block of glutamate transporters potentiates postsynaptic excitation. Neuron. 1994;13:1195–l203. doi: 10.1016/0896-6273(94)90057-4. [DOI] [PubMed] [Google Scholar]
- Tozzi A, Bengtson CP, Longone P, Carignani C, Fusco FR, Bernardi G, Mercuri NB. Involvement of transient receptor potential-like channels in responses to mGluR-I activation in midbrain dopamine neurons. Eur J Neurosci. 2003;18:2133–2145. doi: 10.1046/j.1460-9568.2003.02936.x. [DOI] [PubMed] [Google Scholar]
- Vernon AC, Palmer S, Datla KP, Zbarsky V, Croucher MJ, Dexter DT. Neuroprotective effects of metabotropic glutamate receptor ligands in a 6-hydroxydopamine rodent model of Parkinson's disease. Eur J Neurosci. 2005;22:1799–1806. doi: 10.1111/j.1460-9568.2005.04362.x. [DOI] [PubMed] [Google Scholar]
- Vernon AC, Zbarsky V, Datla KP, Croucher MJ, Dexter DT. Subtype selective antagonism of substantia nigra pars compacta group I metabotropic glutamate receptors protects the nigrostriatal system against 6-hydroxydopamine toxicity in vivo. J Neurochem. 2007;103:1075–1091. doi: 10.1111/j.1471-4159.2007.04860.x. [DOI] [PubMed] [Google Scholar]
- Wang Y, Deng X, Hewavitharana T, Soboloff J, Gill DL. Stim, ORAI and TRPC channels in the control of calcium entry signals in smooth muscle. Clin Exp Pharmacol Physiol. 2008;35:1127–1133. doi: 10.1111/j.1440-1681.2008.05018.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss JH, Koh JY, Choi DW. Neurotoxicity of beta-N-methylamino-L-alanine (BMAA) and beta-N-oxalylamino-L-alanine (BOAA) on cultured cortical neurons. Brain Res. 1989;497:64–71. doi: 10.1016/0006-8993(89)90970-0. [DOI] [PubMed] [Google Scholar]
- Werth JL, Thayer SA. Mitochondria buffer physiological calcium loads in cultured rat dorsal root ganglion neurons. J Neurosci. 1994;14:348–356. doi: 10.1523/JNEUROSCI.14-01-00348.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson JM, Shaw CA. Late appearance of glutamate transporter defects in a murine model of ALS-parkinsonism dementia complex. Neurochem Int. 2007;50:1067–1077. doi: 10.1016/j.neuint.2006.09.017. [DOI] [PubMed] [Google Scholar]
- Zhang L, Huang D, Kondo M, Fan E, Ji H, Kou Y, Ou B. Novel high-throughput assay for antioxidant capacity against superoxide anion. J Agric Food Chem. 2009;57:2661–2667. doi: 10.1021/jf8033368. [DOI] [PubMed] [Google Scholar]