

Abstract
Although amyloid β (Aβ) oligomers are presumed to cause synaptic and cognitive dysfunction in Alzheimer's disease (AD), their contribution to other pathological features of AD remains unclear. To address the latter, we generated APP transgenic mice expressing the E693Δ mutation, which causes AD by enhanced Aβ oligomerization without fibrillization. The mice displayed age-dependent accumulation of intraneuronal Aβ oligomers from 8 months but no extracellular amyloid deposits even at 24 months. Hippocampal synaptic plasticity and memory were impaired at 8 months, at which time the presynaptic marker synaptophysin began to decrease. Furthermore, we detected abnormal tau phosphorylation from 8 months, microglial activation from 12 months, astrocyte activation from 18 months, and neuronal loss at 24 months. These findings suggest that Aβ oligomers cause not only synaptic alteration but also other features of AD pathology and that these mice are a useful model of Aβ oligomer-induced pathology in the absence of amyloid plaques.
Introduction
Soluble oligomers of amyloid β (Aβ) are believed to be a cause of synaptic and cognitive dysfunction in the early stages of Alzheimer's disease (AD) (Klein et al., 2001; Selkoe, 2002). This conclusion is based primarily on experimental evidence that natural and synthetic Aβ oligomers impair synaptic plasticity (Lambert et al., 1998; Walsh et al., 2002; Shankar et al., 2008) and memory (Cleary et al., 2005; Lesné et al., 2006; Shankar et al., 2008) and cause loss of synapses (Lacor et al., 2007; Shankar et al., 2007) when applied exogenously into rat cerebral ventricle, cultured brain slices, or dissociated neurons. In addition, many studies have supported this conclusion by indicating a correlation between soluble Aβ levels and synaptic and cognitive impairment in humans (Lue et al., 1999; Gong et al., 2003) as well as animal models of AD (Mucke et al., 2000; Dodart et al., 2002; Cheng et al. 2007; Matsuyama et al., 2007).
On the other hand, whether Aβ oligomers contribute to other pathological features of AD, such as abnormal tau phosphorylation, glial activation, and neuronal loss, remains unclear. Several studies have demonstrated that exogenously applied Aβ oligomers induce tau hyperphosphorylation (De Felice et al., 2008), activate astrocytes (Hu et al., 1998) and microglia (Jimenez et al., 2008), and cause neuronal death (Lambert et al., 1998; Kayed et al., 2003) in vitro. However, in animal models, these findings have never been observed before amyloid plaque deposition (for review, see Duyckaerts et al., 2008). Once Aβ deposits develop, it is difficult to distinguish which pathological features were induced by soluble Aβ oligomers or by insoluble Aβ fibrils.
We recently identified the E693Δ mutation in amyloid precursor protein (APP) in patients with AD (Tomiyama et al., 2008). This mutation produces variant Aβ lacking glutamate-22 (E22Δ). The mutant Aβ peptide does not form amyloid fibrils in vitro and patients with the mutation lack deposits of amyloid plaques (Tomiyama et al., 2008). The mutant peptide, however, readily forms abundant oligomers in vitro (Tomiyama et al., 2008) and accumulates in oligomeric forms within transfected cells (Nishitsuji et al., 2009). When injected into rat cerebral ventricle, synthetic mutant Aβ E22Δ peptide inhibits hippocampal long-term potentiation (LTP) more potently than wild-type (WT) peptide in vivo (Tomiyama et al., 2008). Exogenously applied mutant Aβ E22Δ peptide induces dose-dependent loss of synapses in mouse hippocampal slices (Takuma et al., 2008). These synaptotoxic effects of the mutant Aβ appear to reflect its propensity to undergo oligomerization. These findings suggest that the E693Δ mutation is suitable for production of an animal model of Aβ oligomers in the absence of amyloid plaques. Such a model could provide findings of critical importance in determining whether Aβ oligomers contribute to features of the pathology of AD other than synaptic alteration.
In the present study, we therefore generated transgenic (Tg) mice expressing the mutant APPE693Δ and compared their pathological features with those of WT APP (APPWT)-Tg mice. The findings presented here indicate that Aβ oligomers play pivotal roles in the pathogenesis of AD.
Materials and Methods
Antibodies.
Rabbit polyclonal antibodies to Aβ (β001) (Lippa et al., 1999) and APP (C40) (Suga et al., 2004) were prepared in our laboratory. For detection of Aβ oligomers, mouse monoclonal antibody NU-1 (Lambert et al., 2007) was used. Mouse monoclonal antibodies to tau, PHF-1 (Greenberg et al., 1992), and MC1 (Jicha et al., 1997), were kindly gifted by Dr. Peter Davies (Department of Pathology, Albert Einstein College of Medicine, Bronx, NY). Mouse monoclonal antibodies to Aβ (6E10; Signet Laboratories), synaptophysin (SVP-38; Sigma), NeuN, a marker of mature neurons (Millipore Bioscience Research Reagents), GFAP, a marker of astrocytes (Cappel, ICN Pharmaceuticals), and a rabbit polyclonal antibody to Iba-1, a marker of microglia (Wako Pure Chemical Industries), were purchased. Both β001 and 6E10 antibodies recognize full-length APP and its C-terminal fragment generated by β-cleavage (CTFβ) as well as Aβ.
Generation of Tg mice.
Tg mice expressing human APP695 with the E693Δ mutation were generated using the MoPrP.Xho vector (Borchelt et al., 1996) by the same method as described previously (Matsuyama et al., 2007). MoPrP-APP constructs were injected into B6C3F1 (C57BL/6N × C3H/HeN) embryos. The mice were backcrossed with C57BL/6 mice at least 10 generations. To elucidate the pathological effects of the E693Δ mutation, phenotypes of the APPE693Δ-Tg mice were compared with those of APPWT-Tg mice with the same mouse prion promoter (Matsuyama et al., 2007). As a positive control for immunohistochemistry, Tg2576 mice, a well known model of AD exhibiting massive amyloid deposition (Hsiao et al., 1996), were purchased from Taconic. All mice used were heterozygous for the transgene of interest. Levels of expression of human APP were determined with 6E10 antibody as described previously (Matsuyama et al., 2007). All animal experiments were approved by the committee of Osaka City University and were performed in accordance with the Guide for Animal Experimentation, Osaka City University. Every effort was made to minimize the number of animals used and their suffering.
Immunohistochemistry.
Mouse brains were fixed in 4% paraformaldehyde, embedded in paraffin, sectioned at 5 μm, and deparaffinized with xylene and ethanol. Only for Aβ staining, the sections were pretreated by boiling in 0.01N HCl (pH 2) for 10 min to expose epitopes. We found that GFAP and Iba-1 can be stained well when sections are pretreated with acidic solution, but such a treatment makes the difference between the APPE693Δ-Tg mice and control mice unclear. Therefore, we used untreated sections in staining for these markers. After being washed with 100 mm Tris-HCl, pH 7.6, 150 mm NaCl [Tris-buffered saline (TBS)], the sections to be stained with horseradish peroxidase (HRP) were treated with 0.3% H2O2 for 30 min to inactivate endogenous peroxidases. The sections were then blocked with 20% calf serum in TBS for 1 h. Aβ, tau, and neuronal and glial markers were stained with corresponding antibodies followed by biotin-labeled second antibodies (Vector Laboratories), HRP-labeled avidin-biotin complex (Vector Laboratories), and the substrate DAB (Dojindo). Synaptophysin was stained with SVP-38 antibody followed by FITC-labeled second antibody (Jackson ImmunoResearch Laboratories). Thioflavine S staining to visualize amyloid fibrils was performed as described previously (Oakley et al., 2006). The specimens were observed under a BZ-8000 fluorescence microscope (Keyence). Synaptic density in the hippocampal CA3 region was estimated by quantifying synaptophysin fluorescence intensity in 30 μm × 60 μm area using the NIH ImageJ software obtained from a public website (National Institutes of Health; http://rsb.info.nih.gov/nih-image/). Neuronal loss was evaluated by counting NeuN-positive cells remaining in the pyramidal cell layer of the hippocampal CA3 region within 900 μm from its end toward the dentate gyrus.
Immunoprecipitation/Western blotting of Aβ.
Mouse brains, not including the hindbrain, were homogenized by sonication in 4 volumes of TBS containing protease inhibitor mixture (P8340; Sigma), and fractionated by four-step ultracentrifugation including TBS, Triton X-100, SDS, and formic acid (FA) extraction (Kawarabayashi et al., 2001). In brief, the homogenates were centrifuged at 100,000 × g at 4°C for 1 h, and the supernatants were harvested. The precipitates were dissolved by sonication in the same volume (4 times tissue weight) of 1% Triton X-100/TBS containing P8340 and centrifuged again. The supernatants were harvested, and the precipitates were then dissolved in 2% SDS/TBS containing P8340 and centrifuged at 100,000 × g at room temperature for 1 h. The supernatants were harvested, and the precipitates were finally dissolved in 70% FA. After being centrifuged again, the supernatants were harvested. Then, 100 μl portions of the TBS-, Triton X-100-, and SDS-extracted fractions were diluted 10-, 10-, and 20-fold, respectively, in TBS containing P8340, while 100 μl portions of FA-extracted fractions were diluted tenfold in 1 m Tris solution (pH 11). Aβ in the samples was immunoprecipitated with 1 μg of 6E10 antibody and 10 μl of 50% protein A Sepharose (Pharmacia) at 4°C overnight. After centrifugation, the precipitates were washed three times with 1% Triton X-100/TBS and once with TBS and then boiled for 5 min in SDS sample buffer. The eluates were subjected to SDS-PAGE with 12% NuPage Bis-Tris gels (Invitrogen) and transferred to polyvinylidene difluoride membranes (Millipore). The membranes were boiled in PBS for 10 min to enhance signals, and Aβ was probed with β001 antibody followed by HRP-labeled second antibody and the chemiluminescent substrate Immobilon Western (Millipore). Signals were visualized using a LAS-3000 luminescent image analyzer (Fujifilm).
In vivo electrophysiology.
Experiments were performed with mice at 8 months of age as described previously (Matsuyama et al., 2007). Synaptic functions, including basal synaptic transmission and short-term and long-term synaptic plasticity, were examined in vivo by recording of population spikes from the granular cell body layer of the dentate gyrus in response to stimulation of the perforant path.
Behavioral tests.
Spatial reference memory in mice was assessed at 8 months of age using the Morris water maze, essentially as described previously (Iso et al., 2007). Male mice were trained to swim to the platform in a pool with a diameter of 96 cm for 6 consecutive days. Training consisted of five trials per day with intertrial intervals of 30 s. At day 7, retention of spatial memory was assessed by a probe trial consisting of a 30 s free swim in the pool without the platform. Locomotor activities of the mice were examined by an open-field test, as described previously (Iso et al., 2007).
Statistical analysis.
All data in animal experiments were expressed as the mean ± SEM. Comparisons of means among the three groups were performed with ANOVA followed by Fisher's protected least significant difference test. Differences with a p value of <0.05 were considered significant.
Results
Generation of APPE693Δ-Tg mice
Tg mice expressing the mutant APPE693Δ under the mouse prion promoter were generated using the MoPrP.Xho vector (Borchelt et al., 1996). Three lines of APPE693Δ-Tg mice were established. Western blotting of brain homogenates with the monoclonal antibody 6E10, which is specific to human APP/Aβ, revealed that lines 1, 2, and 3 possessed the highest, the lowest, and intermediate expression of the transgene, respectively. In this study, line 1, with the highest APP expression was examined. For comparison, we used two lines (lines 1 and 3) of APPWT-Tg mice which had been previously established in our laboratory using the same promoter (Matsuyama et al., 2007). Line 1 was used as a control for immunohistochemistry and behavioral tests, whereas line 3 was used for electrophysiology. We also used the well known Tg2576 mice as another control for immunohistochemistry; these mice overexpress APP harboring the Swedish mutation (K670N/M671L) (APPSW) under the hamster prion promoter (Hsiao et al., 1996).
The levels of expression of human APP in these mice were compared at 8 or 12 months of age by Western blotting with 6E10 antibody. The APPE693Δ-Tg mice expressed human APP to only half the extent of APPWT-Tg line 1 but at levels similar to those in line 3, and only 1/10 that in Tg2576 mice (Fig. 1 A). We previously reported that both lines 1 and 3 of the APPWT-Tg mice exhibit impaired synaptic plasticity at 8 months and that synaptic plasticity in Tg mice is closely related to their level of expression of APP (Matsuyama et al., 2007). In the present study, we therefore used APPWT-Tg line 3 as a control for electrophysiology. In Western blotting with an antibody to the C-terminal region of APP (C40) (Suga et al., 2004), which recognizes both human and mouse APP, the APPE693Δ-Tg mice exhibited twice the amount of APP as their non-Tg littermates (Fig. 1 B), indicating that the levels of human APP expressed in APPE693Δ-Tg mice were similar to those of endogenous mouse APP.
Figure 1.
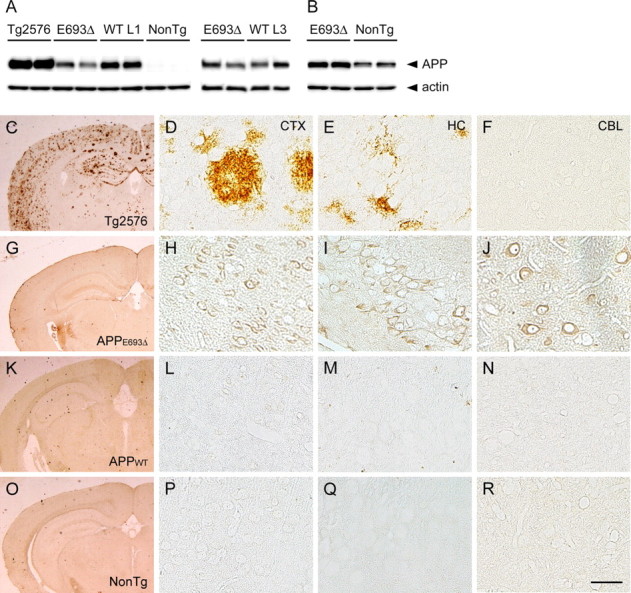
APPE693Δ-Tg mice do not exhibit extracellular amyloid deposits but do display intraneuronal Aβ accumulation. A, B, Levels of expression of human APP in APPE693Δ-Tg mice. Brain homogenates of Tg mice were subjected to Western blotting to examine levels of APP expression. A, Human APP was probed with 6E10 antibody specific to human APP/Aβ. Comparison among the Tg2576 mice, APPE693Δ-Tg mice, APPWT-Tg line 1, and non-Tg littermates was performed at 12 months, while that between the APPE693Δ-Tg mice and APPWT-Tg line 3 was performed at 8 months. B, Human and mouse APP in APPE693Δ-Tg mice and non-Tg littermates were stained at 12 months with C40 antibody recognizing the C-terminal region of human and mouse APP. C–R, Brain Aβ burden in APPE693Δ-Tg mice. Brain sections of 24-month-old Tg mice were stained with β001 antibody to the N-terminal region of Aβ. Tg2576 mice (C–F) displayed abundant amyloid plaques in cerebral cortex (D) and hippocampus (E, CA3 region) but not in cerebellum (F), whereas APPE693Δ-Tg mice (G–J), APPWT-Tg mice (K–N), and non-Tg littermates (O–R) exhibited no extracellular amyloid deposits in any regions examined; cerebral cortex (H, L, P), hippocampal CA3 region (I, M, Q), and cerebellum (J, N, R). The APPE693Δ-Tg mice did, however, exhibit intraneuronal staining of Aβ in these regions. CTX, Cerebral cortex; HC, hippocampus; CBL, cerebellum. Scale bar, 30 μm.
APPE693Δ-Tg mice exhibit accumulation of Aβ oligomers within neurons but no extracellular amyloid deposits
We initially examined brain amyloid pathology in our Tg mice by immunohistochemistry with a polyclonal antibody to the N-terminal region of Aβ (β001) (Lippa et al., 1999). At 24 months, the Tg2576 mice displayed abundant extracellular Aβ deposits in the cerebral cortex and hippocampus (Fig. 1 C–F), as previously reported (Hsiao et al., 1996). On the other hand, neither the APPE693Δ-Tg (Fig. 1 G–J) nor APP WT-Tg mice (Fig. 1 K–N) exhibited amyloid plaque in any regions we examined at the same age, similar to the non-Tg littermates (Fig. 1 O–R). We noted, however, that the APPE693Δ-Tg mice exhibited abundant intraneuronal staining in the cerebral cortex, hippocampus (particularly the CA3 region), and cerebellum. The APPWT-Tg mice did not exhibit such intracellular staining despite having higher expression of APP than the APPE693Δ-Tg mice. This finding is consistent with our previous observation that, in transfected cells, mutant Aβ E22Δ accumulated within cells more abundantly than WT Aβ (Nishitsuji et al., 2009).
Since the mutant Aβ E22Δ that accumulated in transfected cells tended to form oligomers (Nishitsuji et al., 2009), it is likely that intraneuronal Aβ in the APPE693Δ-Tg mice also forms oligomers. To test whether this is the case, brain sections from mice at various ages were stained with a monoclonal antibody NU-1 selective to Aβ oligomers (Lambert et al., 2007). As we expected, intraneuronal Aβ in the cerebral cortex and hippocampus of the APPE693Δ-Tg mice was stained by NU-1 (Fig. 2 A–H). Aβ oligomers first appeared at 8 months in both brain regions and accumulated in age-dependent fashion. The APPWT-Tg mice (Fig. 2 I,J) and non-Tg littermates (Fig. 2 K,L) exhibited no NU-1 staining even at 24 months.
Figure 2.
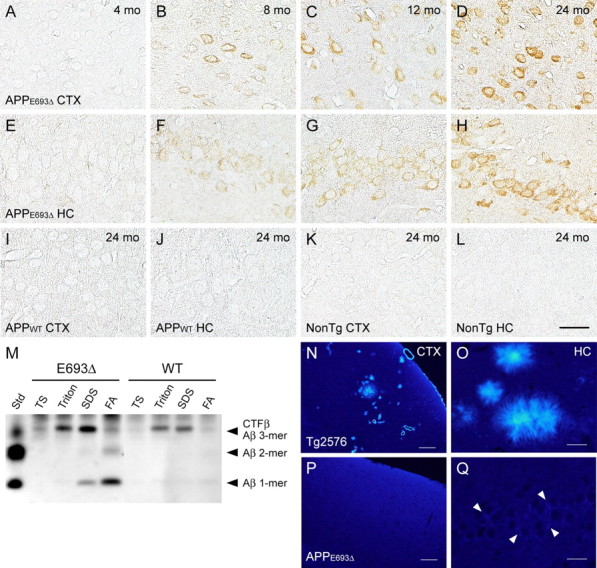
Age-dependent accumulation of intraneuronal Aβ oligomers in APPE693Δ-Tg mice. A–L, Brain sections of 4 (A, E)-, 8 (B, F)-, 12 (C, G)-, and 24 (D, H)-month-old APPE693Δ-Tg mice were stained with Aβ oligomer-selective antibody NU-1. Intraneuronal Aβ oligomers first appeared at 8 months in the cerebral cortex (A–D) and hippocampus (E–H, the CA3 region) and accumulated in age-dependent fashion. No NU-1 staining was observed in APPWT-Tg mice (I, J) or non-Tg littermates (K, L) even at 24 months; cerebral cortex (I, K) and hippocampal CA3 region (J, L). Scale bars, 30 μm. M, Oligomer formation of Aβ in APPE693Δ-Tg mice was confirmed by immunoprecipitation/Western blotting analysis. Brain homogenates of 24-month-old Tg mice were fractionated by 4-step ultracentrifugation into TBS-, Triton X-100-, SDS-, and FA-soluble fractions. Aβ in each fraction was immunoprecipitated with 6E10 and stained with β001. Aβ dimers, and possibly trimers, were detected in the FA-extracted fraction from APPE693Δ-Tg mice but only slightly in APPWT-Tg mice. Std, Standard; TS, TBS. N–Q, Brain sections of 24-month-old Tg mice were stained with the amyloid-binding dye thioflavin S. Tg2576 mice exhibited abundant extracellular staining due to parenchymal and vascular amyloid deposits in cerebral cortex (N) and hippocampus (O), whereas APPE693Δ-Tg mice exhibited no thioflavin S staining in these regions; cerebral cortex (P) and hippocampal CA3 region (Q). However, APPE693Δ-Tg mice exhibited very weak and somewhat diffuse staining within neurons (Q, arrowhead). CTX, Cerebral cortex; HC, hippocampus. Scale bars: N, P, 100 μm; O, Q, 20 μm.
Oligomer formation of Aβ in the APPE693Δ-Tg mice was confirmed by immunoprecipitation/Western blotting analysis. Brain tissues at 24 months were homogenized and fractionated by 4-step ultracentrifugation (Kawarabayashi et al., 2001). Initially, extracellular and intracellular soluble Aβ was extracted with TBS, and TBS-insoluble Aβ was sequentially extracted with two types of detergent, Triton X-100 and SDS, and finally with FA, which is commonly used to extract Aβ from amyloid plaque cores. The Aβ in TBS-, Triton X-100-, SDS-, and FA-extracted fractions was immunoprecipitated with 6E10 antibody and stained with β001 antibody. Aβ dimers and possibly trimers were detected in the APPE693Δ-Tg mice, but only slightly in the APPWT-Tg mice (Fig. 2 M). Notably, Aβ oligomers were fractionated predominantly into insoluble fractions, particularly the FA-extracted fraction. This result appears inconsistent with the prevalent view that Aβ oligomers are soluble, although the presence in AD brains of additional oligomers that could be detected only by detergent extraction has been reported (Gong et al., 2003).
The finding that Aβ oligomers were largely recovered in insoluble fractions suggests the possibility that intraneuronal Aβ in the APPE693Δ-Tg mice may form fibrils, as reported in other APP-Tg mice (Casas et al., 2004; Oakley et al., 2006). To test whether this is the case, brain sections at 24 months were stained with an amyloid-binding dye, thioflavin S. Tg2576 mice displayed abundant, strong staining due to amyloid plaques in the cerebral cortex and hippocampus (Fig. 2 N,O), whereas APPE693Δ-Tg mice exhibited no staining (Fig. 2 P,Q), except for very weak, somewhat diffuse staining within neurons in the cerebral cortex and hippocampus. Although the origin of these faint signals is unclear, we suspect that it reflects the binding of thioflavin S to Aβ oligomers rather than to Aβ fibrils, since several amyloid-binding dyes, such as Congo red, thioflavin T, and PIB, have been shown to react with Aβ oligomers as well as Aβ fibrils (Maezawa et al., 2008).
Synaptic and cognitive dysfunction in APPE693Δ-Tg mice
Aβ oligomers have been demonstrated to cause synaptic and cognitive dysfunction (Lambert et al., 1998; Walsh et al., 2002; Cleary et al., 2005; Lesné et al., 2006; Shankar et al., 2008; Tomiyama et al., 2008) and loss of synapses (Lacor et al., 2007; Shankar et al., 2007; Takuma et al., 2008) when applied exogenously. To examine whether synaptic alteration occurs in the APPE693Δ-Tg mice, we first examined their synaptic density. Brain sections from mice at various ages were stained with an antibody to the presynaptic marker synaptophysin. Compared with non-Tg littermates (Fig. 3 A–D) and APPWT-Tg mice (Fig. 3 E–H), APPE693Δ-Tg mice (Fig. 3 I–L) exhibited significant decrease in synaptophysin in the hippocampus, particularly in the CA3 region, in an age-dependent fashion from 8 months (Fig. 3 M). This timing coincides with that of intraneuronal accumulation of Aβ oligomers.
Figure 3.

Age-dependent decrease in synaptophysin in APPE693Δ-Tg mice. A–L, Brain sections of 4 (A, E, I)-, 8 (B, F, J)-, 12 (C, G, K)-, and 24 (D, H, L)-month-old Tg mice were stained with antibody to the presynaptic marker synaptophysin. All images were taken from the hippocampal CA3 region. Unlike non-Tg littermates (A–D) and APPWT-Tg mice (E–H), the APPE693Δ-Tg mice (I–L) exhibited age-dependent decrease in synaptophysin in the hippocampus from 8 months. Scale bars, 30 μm. M, Synaptophysin fluorescence intensity in 30 μm × 60 μm area of the hippocampal CA3 region was quantified using the NIH ImageJ software and shown in arbitrary units (AU). Each bar represents the mean ± SEM (n = 3). *p = 0.0258 versus NonTg; p = 0.0244 versus APPWT-Tg, **p = 0.0052 versus NonTg; p = 0.0092 versus APPWT-Tg, ***p = 0.0140 versus NonTg; p = 0.0387 versus APPWT-Tg.
We next examined synaptic function by in vivo electrophysiology at 8 months, at which age Aβ oligomers have begun to accumulate in APPE693Δ-Tg mice. In this experiment, we used line 3 of the APPWT-Tg mice as a control, as described above. Population spikes were recorded in the granular cell body layer of the dentate gyrus in response to stimulation of the perforant path. Basal synaptic transmission was tested by preparing input/output (I/O) curves with increasing stimulus intensities. No significant difference was observed among the non-Tg littermates, APPWT-Tg, and APPE693Δ-Tg mice (Fig. 4 A). Short-term synaptic plasticity was examined by measuring paired-pulse facilitation (PPF). Compared with the non-Tg littermates, both the APPWT-Tg and APPE693Δ-Tg mice exhibited significantly reduced PPF (Fig. 4 B). The reduction was larger in the APPE693Δ-Tg mice than in the APPWT-Tg mice, although the difference between them was not significant. Last, long-term synaptic plasticity was examined by measuring LTP, which was elicited by delivering high-frequency stimulation (HFS) to the perforant path. Again, significant impairment of LTP was observed in both the APPWT-Tg and APPE693Δ-Tg mice compared with the non-Tg littermates (Fig. 4 C). The initial potentiation of population spikes was suppressed more profoundly in the APPE693Δ-Tg mice than in the APPWT-Tg mice, and this stronger suppression lasted for at least 120 min, though the difference between them was not significant. Thus, synaptic plasticity was more strongly impaired in the APPE693Δ-Tg mice than in the APPWT-Tg mice.
Figure 4.

Impairment of synaptic plasticity in APPE693Δ-Tg mice. Synaptic functions of Tg mice were examined by in vivo electrophysiology at 8 months. Population spikes were recorded in the granular cell body layer of the dentate gyrus in response to stimulation of the perforant path. A, Basal synaptic transmission was examined by preparing I/O curves with increasing stimulus intensities. No significant difference was observed among the non-Tg littermates, APPWT-Tg, and APPE693Δ-Tg mice (n = 4 for each group). B, Short-term synaptic plasticity was studied by testing PPF. Compared with non-Tg littermates, both APPE693Δ-Tg and APPWT-Tg mice exhibited significantly reduced PPF; the reduction was larger in APPE693Δ-Tg mice than in APPWT-Tg mice. *p = 0.0275 versus NonTg, **p = 0.0002 versus NonTg; but not significant versus APPWT-Tg (n = 7 for APPE693Δ-Tg and NonTg; n = 5 for APPWT-Tg). C, Long-term synaptic plasticity was investigated by measuring LTP, which was elicited by delivering HFS to the perforant path. Typical population spikes at 0 and 120 min after HFS are shown. Compared with non-Tg littermates, both APPE693Δ-Tg and APPWT-Tg mice exhibited significant impairment of LTP, which was more severe in the APPE693Δ-Tg mice than the APPWT-Tg mice. *p = 0.0093 versus NonTg, **p = 0.0003 versus NonTg; but not significant versus APPWT-Tg, when compared 5–120 min after HFS (n = 5 for APPE693Δ-Tg and NonTg; n = 4 for APPWT-Tg). All values are the mean ± SEM.
We also examined cognitive function at the same age (8 months) using the Morris water maze. In this experiment, line 1 of the APPWT-Tg mice was used as a control. Before the trials, spontaneous locomotor activity of mice was measured by the open-field test and confirmed not to differ among the non-Tg littermates, APPWT-Tg, and APPE693Δ-Tg mice (data not shown). The mice were trained for 6 d to remember the location of a hidden platform in a swimming pool, and the time required to reach the platform was measured in every trial. The APPE693Δ-Tg mice exhibited significant deficits in performance, with longer escape latencies than the APPWT-Tg mice and non-Tg littermates (Fig. 5 A). The APPWT-Tg mice exhibited slightly, though not significantly, longer escape latencies than the non-Tg littermates. In a probe trial at day 7, the APPWT-Tg mice and non-Tg littermates spent ∼40% of their time in the target quadrant, whereas the APPE693Δ-Tg mice swam in that area only ∼20% of the time (Fig. 5 B). Thus, spatial reference memory of APPE693Δ-Tg mice was found to be markedly disrupted, whereas that of APPWT-Tg mice was not affected.
Figure 5.

Impairment of memory in APPE693Δ-Tg mice. Spatial reference memory of Tg mice was assessed by the Morris water maze at 8 months. A, Mice were trained to swim to the hidden platform for 6 consecutive days. Each point represents the mean latency of five trials per day ± SEM (n = 9 for APPE693Δ-Tg; n = 8 for NonTg and APPWT-Tg). The APPE693Δ-Tg mice exhibited significantly longer escape latencies than the APPWT-Tg mice and non-Tg littermates. The APPWT-Tg mice exhibited slightly, but not significantly, longer escape latencies than the non-Tg littermates. *p < 0.0001 versus NonTg; p = 0.0010 versus APPWT-Tg. B, At day 7, retention of memory was assessed by a probe trial for 30 s with the platform removed. Each bar represents the mean time occupancy ± SEM in the target quadrant (TQ), adjacent quadrants (AQ1, AQ2), or opposite quadrant (OQ). The APPE693Δ-Tg mice spent significantly shorter time in the target quadrant than the APPWT-Tg mice and non-Tg littermates did. *p = 0.0265 versus NonTg; p = 0.0149 versus APPWT-Tg.
In summary, the APPE693Δ-Tg mice exhibited synaptic alteration in parallel with intraneuronal accumulation of Aβ oligomers without formation of amyloid plaques. These features indicate that the Tg mouse is a suitable model for investigation of Aβ oligomer-induced pathology free of the effects of amyloid plaques even at old ages.
Abnormal tau phosphorylation in APPE693Δ-Tg mice
Based on the above findings, we moved to the next study on the contribution of Aβ oligomers to other features of AD pathology, such as abnormal tau phosphorylation, glial activation, and neuronal loss. Aβ oligomers have been demonstrated to cause these pathological changes in vitro when applied exogenously to cultured cells and brain slices (Hu et al., 1998; Lambert et al., 1998; Kayed et al., 2003; De Felice et al., 2008; Jimenez et al., 2008). However, it is still unclear whether Aβ oligomers have similar pathological effects in vivo. In studies of APP-Tg mice reported thus far, none of these pathological features were detected before amyloid plaque formation (Duyckaerts et al., 2008).
We first examined abnormal tau phosphorylation in APPE693Δ-Tg mice. Brain sections from mice at various ages were stained with two antibodies reactive to pathological tau: PHF-1, which is specific to the phosphorylation at Ser396/Ser404 (Greenberg et al., 1992), and MC1, which recognizes the conformational epitopes of pathological tau (Jicha et al., 1997). In the Tg2576 mice, these antibodies stained dystrophic neurites around amyloid plaques at 24 months (Fig. 6 A,I). The non-Tg littermates (data not shown) and APPWT-Tg mice (Fig. 6 B–D,J–L) exhibited no staining with these antibodies in any regions we examined even at 24 months. In contrast, the APPE693Δ-Tg mice began to exhibit PHF-1-positive and MC1-positive staining in hippocampal Mossy fibers from 8 months (Fig. 6 E,M). At 12 months, immunoreactivity was more pronounced, and the cingulum in cerebral cortex was also stained with PHF-1 but not MC1 (Fig. 6 F–H,N–P). These findings demonstrated that Aβ oligomers caused abnormal tau phosphorylation in the absence of amyloid plaques.
Figure 6.
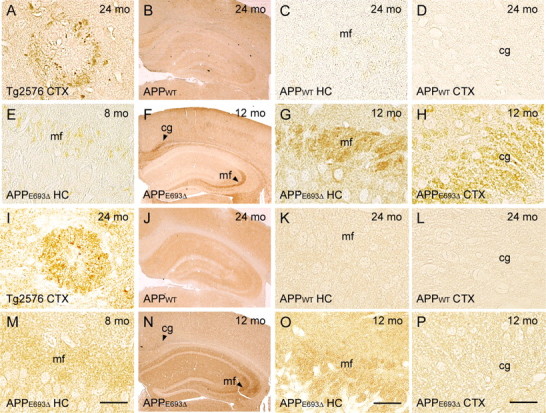
Abnormal tau phosphorylation in APPE693Δ-Tg mice. A–P, Brain sections of 8 (E, M)-, 12 (F–H, N–P)-, and 24 (A–D, I–L)-month-old Tg mice were stained with antibodies reactive to pathological tau, PHF-1 (A–H), and MC1 (I–P). These antibodies stained dystrophic neurites around amyloid plaques in the Tg2576 mice at 24 months (A, I). The APPWT-Tg mice (B–D, J–L; C, K, hippocampal CA3 region; D, L, cerebral cortex) exhibited no staining with these antibodies even at 24 months. In contrast, APPE693Δ-Tg mice (E–H, M–P) began to display PHF-1-positive and MC1-positive hippocampal mossy fibers (E, M, CA3 region) from 8 months. At 12 months, immunoreactivity was more evident (G, O, the CA3 region), and the cingulum was also stained with PHF-1 but not MC1 (H, P, the cerebral cortex). CTX, Cerebral cortex; HC, hippocampus; mf, mossy fibers; cg, cingulum. Scale bars, 30 μm.
Activation of astrocytes and microglia in APPE693Δ-Tg mice
We next examined glial activation. Brain sections from mice at various ages were stained with antibodies to GFAP and Iba-1, which are markers of astrocytes and microglia (Imai et al., 1996), respectively. At 24 months, Tg2576 mice displayed massive staining around amyloid plaques with these antibodies (Fig. 7 A,C). The non-Tg littermates exhibited no staining with these antibodies even at 24 months under our staining conditions (Fig. 7 B,D). The APPWT-Tg mice possessed no Iba-1-positive cells (Fig. 7 E–H) and only a few GFAP-positive cells (Fig. 7 M–P) in cerebral cortex and hippocampus at 24 months. In contrast, the APPE693Δ-Tg mice began to display Iba-1-positive cells at 12 months (Fig. 7 I–L) and GFAP-positive cells at 18 months (Fig. 7 Q–T) in these regions. The observed staining in the APPE693Δ-Tg mice indicates increased expression of microglial and astrocyte marker proteins, which is believed to reflect activation of these cells.
Figure 7.
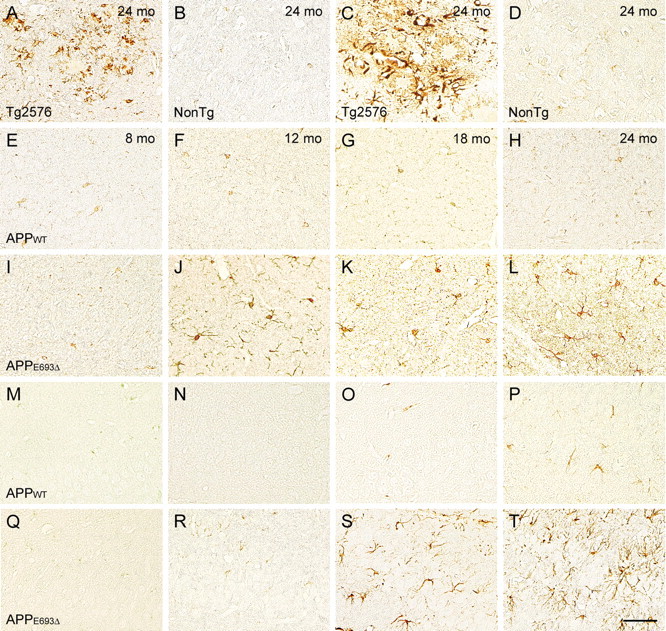
Glial activation in APPE693Δ-Tg mice. A–T, Brain sections of 8 (E, I, M, Q)-, 12 (F, J, N, R)-, 18 (G, K, O, S)-, and 24 (A–D, H, L, P, T)-month-old Tg mice were stained with antibodies to Iba-1 (A, B, E–L) and GFAP (C, D, M–T), which are markers of microglia and astrocytes, respectively. All images were taken from the hippocampal CA3 region, except those of the Tg2576 mice, which were obtained from cerebral cortex. The Tg2576 mice (A, C) at 24 months exhibited massive staining with these antibodies around amyloid plaques, while the non-Tg littermates (B, D) exhibited no staining at 24 months. The APPWT-Tg mice (E–H, M–P) possessed no Iba-1-positive cells and only a few GFAP-positive cells at 24 months. In contrast, the APPE693Δ-Tg mice (I–L, Q–T) displayed Iba-1-positive cells from 12 months and GFAP-positive cells from 18 months in both the hippocampus and cerebral cortex. Scale bar, 30 μm.
Neuronal loss in APPE693Δ-Tg mice
Finally, we examined whether neuronal loss occurs in the APPE693Δ-Tg mice. Brain sections from mice at various ages were stained with an antibody to NeuN, a marker of mature neurons (Mullen et al., 1992). Compared with non-Tg littermates (Fig. 8 A,E) and APPWT-Tg mice (Fig. 8 B,F), APPE693Δ-Tg mice (Fig. 8 C,G) exhibited a significant decrease in NeuN-positive cells in the hippocampal CA3 region at 24 months (Fig. 8 D). No apparent decrease in NeuN-positive cells was observed in the cerebral cortex at this age. The APPWT-Tg mice did not exhibit significant neuronal loss compared with the non-Tg littermates. These findings suggest that Aβ oligomers triggered the pathological cascades leading to neuronal death, in which neuronal loss occurred a long period of time after the start of intraneuronal accumulation of Aβ oligomers at 8 months. The hippocampal CA3 region appears to be particularly vulnerable to the toxic effects of Aβ oligomers.
Figure 8.

Neuronal loss in APPE693Δ-Tg mice. A–C, E–G, Brain sections of Tg mice were stained with an antibody to NeuN, a marker of mature neurons. Compared with non-Tg littermates (A, E) and APPWT-Tg mice (B, F), APPE693Δ-Tg mice (C, G) exhibited significant decrease in NeuN-positive cells in the hippocampal CA3 region, but no decrease in cerebral cortex at 24 months; hippocampal CA3 region (A–C) and cerebral cortex (E–G). No significant difference was observed between non-Tg littermates and APPWT-Tg mice at 24 months. D, NeuN-positive cells in the pyramidal cell layer of the hippocampal CA3 region were counted within 900 μm from its end toward the dentate gyrus in the photographs. *p = 0.0044 versus NonTg; p = 0.0121 versus APPWT-Tg (n = 4). CTX, Cerebral cortex; HC, hippocampus. Scale bars, 100 μm.
These findings together suggest that Aβ oligomers, which are localized within neurons in APPE693Δ-Tg mice, significantly contribute not only to synaptic alteration but also to other features of AD pathology in vivo.
Discussion
In the present study, we generated novel APP-Tg mice expressing the E693Δ mutation to test in vivo the ability of Aβ oligomers to produce the synaptic, cognitive, and neuropathological features of AD. On immunohistochemical examination, APPE693Δ-Tg mice exhibited age-dependent accumulation of Aβ oligomers within neurons in the cerebral cortex and hippocampus from 8 months, but no amyloid plaques even at 24 months. Biochemical analysis confirmed the accumulation of Aβ dimers and possibly trimers in their brains. This is consistent with our previous findings that mutant Aβ E22Δ peptides neither formed amyloid fibrils in vitro nor were deposited in amyloid plaques in AD patient brains, and instead formed abundant oligomers in vitro (Tomiyama et al., 2008) and accumulated in oligomeric forms within transfected cells (Nishitsuji et al., 2009). The enhanced formation of Aβ oligomers and the lack of amyloid plaques in our APPE693Δ-Tg mice indicate that this mouse model is suitable for study of the contribution of Aβ oligomers to the pathogenesis of AD.
We initially tested in our APPE693Δ-Tg mice whether the consensus that Aβ oligomers cause early synaptic pathology in AD is correct. Synaptic and cognitive functions were examined by in vivo electrophysiology and behavioral tests using the Morris water maze at 8 months. By this age, Aβ oligomers had begun to accumulate within neurons in the hippocampus and cerebral cortex of the APPE693Δ-Tg mice. Although their basal synaptic transmission was not affected at this age, short-term and long-term synaptic plasticity (PPF and LTP, respectively) and spatial reference memory were significantly impaired. Our control APPWT-Tg mice also exhibited weaker but significant impairment of synaptic plasticity at 8 months, despite little accumulation of Aβ oligomers. One possible explanation for these findings is that in vivo electrophysiology is a very sensitive assay, and that therefore even trace amounts of Aβ oligomers in the APPWT-Tg mice could be found to cause synaptic dysfunction. Such small amounts of Aβ oligomers may not be detected by immunohistochemistry or Western blotting, and may be insufficient to cause cognitive impairment in water maze tasks which may be less sensitive than other behavioral tasks. Another possibility is that elevated secretion of Aβ from neurons may lead to synaptic depression (Kamenetz et al., 2003; Ting et al., 2007), in which Aβ monomer, as well as oligomers, may act as a negative regulator of synaptic transmission. Alternatively, overexpression of APP in neurons may itself cause synaptic dysfunction, independent of Aβ production. We also examined whether loss of synapses occurred in our APPE693Δ-Tg mice. Immunohistochemistry for the presynaptic marker synaptophysin demonstrated that synaptic density in the hippocampus of these mice decreased in age-dependent fashion from 8 months, in parallel with accumulation of Aβ oligomers. Thus, Aβ oligomers were confirmed to cause early synaptic pathology in the absence of amyloid plaques in our APPE693Δ-Tg mice.
The next and main objective of the present study was to clarify whether Aβ oligomers contribute in vivo to features of AD pathology other than synaptic alteration. Although numerous lines of APP-Tg mice have been shown to exhibit abnormal tau phosphorylation in the brain, this was detected only after amyloid plaque formation and within the dystrophic neurites surrounding plaques (Duyckaerts et al., 2008). We examined our APPE693Δ-Tg mice for tau pathology using immunohistochemistry. In the absence of amyloid plaques, the mice displayed abnormal tau phosphorylation in the hippocampus and cerebral cortex from 8 months, in parallel with intraneuronal accumulation of Aβ oligomers. Tau hyperphosphorylation was detected in hippocampal mossy fibers and the cingulum, which connect granule cells of the dentate gyrus and the hippocampal CA3 region and the cingulate gyrus and entorhinal cortex, respectively. These neural connections have been thought to play important roles in learning, memory, and consciousness, and to be altered early in AD (Arendt, 2004; Villain et al., 2008).
Clustering of activated astrocytes and microglia around amyloid plaques is another feature of AD pathology thought to be involved in neurodegeneration via cytokine and chemokine release from these cells. Activation of astrocytes and/or microglia has been observed in many APP-Tg mice, though again this was only after amyloid plaque formation and was in the vicinity of plaques (Duyckaerts et al., 2008). On immunohistochemical examination, the APPE693Δ-Tg mice exhibited activation of microglia from 12 months and activation of astrocytes from 18 months in the cerebral cortex and hippocampus. Although we did not confirm the presence of extracellular Aβ oligomers in APPE693Δ-Tg mouse brain, the observed glial activation and recruitment may be caused by overflow of extracellular diffusible Aβ oligomers from neurons (Oddo et al., 2006). Alternatively, glial cells might have been activated and recruited by cytokines or chemokines released from glial cells which neighbored and were in contact with aberrant neurons containing Aβ oligomers.
The most striking feature of AD pathology is neuronal loss. In APP-Tg mice, the occurrence of neuronal loss has in several lines been reported only after intense development of amyloid plaques (Duyckaerts et al., 2008). On immunohistochemical examination, we found that our APPE693Δ-Tg mice, despite their lack of amyloid plaques, exhibited significant neuronal loss in the hippocampal CA3 region at 24 months. To our knowledge, this is the first report that neuronal loss was induced by Aβ oligomers alone in vivo in the absence of amyloid plaques. No neuronal loss was observed at younger ages or in other brain regions, suggesting that Aβ oligomer-induced neuronal death requires long exposure of cells to Aβ oligomers in vivo and occurs in cells vulnerable to the toxic effects of Aβ oligomers. This is consistent with our observation that, in patients with the E693Δ mutation, mild atrophy began to occur initially in the hippocampus several years after the onset of AD (H. Shimada, S. Ataka, T. Tomiyama, J. Takeuchi, H. Takechi, H. Mori, and T. Miki, unpublished observations).
Our findings imply that intracellular Aβ plays important roles in synaptic alteration and subsequent neuropathology. A similar relationship between intracellular Aβ and synaptic pathology has been reported in other Tg mice. In the triple transgenic 3xTg-AD mice, synaptic and cognitive dysfunction were found to be correlated with the accumulation of intraneuronal Aβ in the hippocampus (4 months) before amyloid plaque formation (12 months) (Oddo et al., 2003; Billings et al., 2005). In arcAβ mice, cognitive dysfunction was observed at 6 months, after the intraneuronal accumulation of Aβ (3 months) but before amyloid plaque formation (7 months) (Knobloch et al., 2007). Furthermore, in AD and Tg2576 mouse brains, morphological alterations of synapses occurred in association with intraneuronal accumulation of Aβ (Takahashi et al., 2002), and some of the intraneuronal Aβ formed oligomers (Takahashi et al., 2004). We also previously observed that in AD brains, synaptophysin was decreased around neurons containing Aβ oligomers (Ishibashi et al., 2006). Many other studies of patients with AD (Gouras et al., 2000; Fernández-Vizarra et al., 2004) and Down syndrome (Mori et al., 2002) and of APP-Tg mice (Casas et al., 2004; Lord et al., 2006; Oakley et al., 2006) have demonstrated that intraneuronal accumulation of Aβ is an early pathological change observed before amyloid plaque formation. However, we cannot exclude the possibility that the pathological changes we observed were induced by extracellular soluble Aβ oligomers. It appears possible that both extracellular and intracellular Aβ oligomers contribute to the pathology of AD.
Notably, Aβ oligomers predominantly accumulated in insoluble fractions, particularly the FA-extracted fraction, in APPE693Δ-Tg mice. This finding appears inconsistent with the prevalent view that Aβ oligomers are soluble. However, detergent-insoluble features of intracellular Aβ have also been demonstrated in 3xTg-AD mice (Billings et al., 2005). It may be that the soluble fraction of intracellular Aβ oligomers is in an equilibrium with the insoluble fraction, and that the E22Δ mutation in Aβ tends to shift the equilibrium toward the insoluble fraction. It is unclear whether these insoluble Aβ oligomers form fibrils within cells. While very weak thioflavin S staining was observed in neurons in the APPE693Δ-Tg mice, this staining may have reflected its binding to Aβ oligomers rather than Aβ fibrils. Alternatively, intraneuronal thioflavin S staining may reflect the presence of fibrillar aggregates of hyperphosphorylated tau. In our previous study, Aβ was found to localize largely at endosomes, and to a lesser extent in the ER, Golgi, lysosomes, and autophagosomes in cultured cells (Nishitsuji et al., 2009). In AD and Tg2576 mouse brains, Aβ was found to accumulate in multivesicular bodies, which are a type of endosome (Takahashi et al., 2002). Thus, endosomes may be the main site of accumulation of insoluble Aβ. A study to determine the subcellular localization of Aβ oligomers in our APPE693Δ-Tg mice is ongoing.
In summary, we found that Aβ oligomers caused not only synaptic alteration but also abnormal tau phosphorylation, microglial activation, astrocyte activation, and neuronal loss in vivo in the absence of amyloid plaques. Our findings provide a new insight into the pathogenesis of AD, that amyloid plaque formation is not an absolute requirement for the onset and development of AD. Instead, Aβ oligomers play pivotal roles throughout the progression of AD. It is true that we have not yet succeeded in forming neurofibrillary tangles in these mice and thus need to refine this model. Nevertheless, our Tg mice are, at present, the only animal model of Aβ oligomer-induced pathology avoiding the effects of amyloid plaques even at old ages, and are thus a valuable means of investigation of the pathological and physiological roles of Aβ oligomers and for evaluation of strategies for treatment of AD that specifically target Aβ oligomers.
Footnotes
This study was supported by Grants-in-Aid for Scientific Research on Priority Areas - Research on Pathomechanisms of Brain Disorders from the Ministry of Education, Culture, Sports, Science and Technology of Japan, nos. 17300114, 18023033, 20023026, and 20023026; by Grants-in-Aid for Comprehensive Research on Dementia from the Ministry of Health, Labour and Welfare, Japan; and in part by the Alzheimer's Association (IIRG-09-132098). We thank Dr. David R. Borchelt (Department of Neuroscience, McKnight Brain Institute, University of Florida, Gainesville, FL) for providing the MoPrP.Xho vector. We also thank Yuki Yamashita for technical assistance and Drs. Haruhiko Akiyama, Tetsuaki Arai, and Kenji Ikeda for helpful discussion.
The authors declare no competing financial interests.
References
- Arendt T. Neurodegeneration and plasticity. Int J Dev Neurosci. 2004;22:507–514. doi: 10.1016/j.ijdevneu.2004.07.007. [DOI] [PubMed] [Google Scholar]
- Billings LM, Oddo S, Green KN, McGaugh JL, LaFerla FM. Intraneuronal Aβ causes the onset of early Alzheimer's disease-related cognitive deficits in transgenic mice. Neuron. 2005;45:675–688. doi: 10.1016/j.neuron.2005.01.040. [DOI] [PubMed] [Google Scholar]
- Borchelt DR, Davis J, Fischer M, Lee MK, Slunt HH, Ratovitsky T, Regard J, Copeland NG, Jenkins NA, Sisodia SS, Price DL. A vector for expressing foreign genes in the brains and hearts of transgenic mice. Genet Anal. 1996;13:159–163. doi: 10.1016/s1050-3862(96)00167-2. [DOI] [PubMed] [Google Scholar]
- Casas C, Sergeant N, Itier JM, Blanchard V, Wirths O, van der Kolk N, Vingtdeux V, van de Steeg E, Ret G, Canton T, Drobecq H, Clark A, Bonici B, Delacourte A, Benavides J, Schmitz C, Tremp G, Bayer TA, Benoit P, Pradier L. Massive CA1/2 neuronal loss with intraneuronal and N-terminal truncated Aβ42 accumulation in a novel Alzheimer transgenic model. Am J Pathol. 2004;165:1289–1300. doi: 10.1016/s0002-9440(10)63388-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng IH, Scearce-Levie K, Legleiter J, Palop JJ, Gerstein H, Bien-Ly N, Puoliväli J, Lesné S, Ashe KH, Muchowski PJ, Mucke L. Accelerating amyloid-β fibrillization reduces oligomer levels and functional deficits in Alzheimer disease mouse models. J Biol Chem. 2007;282:23818–23828. doi: 10.1074/jbc.M701078200. [DOI] [PubMed] [Google Scholar]
- Cleary JP, Walsh DM, Hofmeister JJ, Shankar GM, Kuskowski MA, Selkoe DJ, Ashe KH. Natural oligomers of the amyloid-β protein specifically disrupt cognitive function. Nat Neurosci. 2005;8:79–84. doi: 10.1038/nn1372. [DOI] [PubMed] [Google Scholar]
- De Felice FG, Wu D, Lambert MP, Fernandez SJ, Velasco PT, Lacor PN, Bigio EH, Jerecic J, Acton PJ, Shughrue PJ, Chen-Dodson E, Kinney GG, Klein WL. Alzheimer's disease-type neuronal tau hyperphosphorylation induced by Aβ oligomers. Neurobiol Aging. 2008;29:1334–1347. doi: 10.1016/j.neurobiolaging.2007.02.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodart JC, Bales KR, Gannon KS, Greene SJ, DeMattos RB, Mathis C, DeLong CA, Wu S, Wu X, Holtzman DM, Paul SM. Immunization reverses memory deficits without reducing brain Aβ burden in Alzheimer's disease model. Nat Neurosci. 2002;5:452–457. doi: 10.1038/nn842. [DOI] [PubMed] [Google Scholar]
- Duyckaerts C, Potier MC, Delatour B. Alzheimer disease models and human neuropathology: similarities and differences. Acta Neuropathol. 2008;115:5–38. doi: 10.1007/s00401-007-0312-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernández-Vizarra P, Fernández AP, Castro-Blanco S, Serrano J, Bentura ML, Martínez-Murillo R, Martínez A, Rodrigo J. Intra- and extracellular Aβ and PHF in clinically evaluated cases of Alzheimer's disease. Histol Histopathol. 2004;19:823–844. doi: 10.14670/HH-19.823. [DOI] [PubMed] [Google Scholar]
- Gong Y, Chang L, Viola KL, Lacor PN, Lambert MP, Finch CE, Krafft GA, Klein WL. Alzheimer's disease-affected brain: presence of oligomeric Aβ ligands (ADDLs) suggests a molecular basis for reversible memory loss. Proc Natl Acad Sci U S A. 2003;100:10417–10422. doi: 10.1073/pnas.1834302100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gouras GK, Tsai J, Naslund J, Vincent B, Edgar M, Checler F, Greenfield JP, Haroutunian V, Buxbaum JD, Xu H, Greengard P, Relkin NR. Intraneuronal Aβ42 accumulation in human brain. Am J Pathol. 2000;156:15–20. doi: 10.1016/s0002-9440(10)64700-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenberg SG, Davies P, Schein JD, Binder LI. Hydrofluoric acid-treated τPHF proteins display the same biochemical properties as normal τ. J Biol Chem. 1992;267:564–569. [PubMed] [Google Scholar]
- Hsiao K, Chapman P, Nilsen S, Eckman C, Harigaya Y, Younkin S, Yang F, Cole G. Correlative memory deficits, Aβ elevation, and amyloid plaques in transgenic mice. Science. 1996;274:99–102. doi: 10.1126/science.274.5284.99. [DOI] [PubMed] [Google Scholar]
- Hu J, Akama KT, Krafft GA, Chromy BA, Van Eldik LJ. Amyloid-beta peptide activates cultured astorocytes: morphological alterations, cytokine induction and nitric oxide release. Brain Res. 1998;785:195–206. doi: 10.1016/s0006-8993(97)01318-8. [DOI] [PubMed] [Google Scholar]
- Imai Y, Ibata I, Ito D, Ohsawa K, Kohsaka S. A novel gene iba1 in the major histocompatibility complex class III region encoding an EF hand protein expressed in a monocytic lineage. Biochem Biophys Res Commun. 1996;224:855–862. doi: 10.1006/bbrc.1996.1112. [DOI] [PubMed] [Google Scholar]
- Ishibashi K, Tomiyama T, Nishitsuji K, Hara M, Mori H. Absence of synaptophysin near cortical neurons containing oligomer Aβ in Alzheimer's disease brain. J Neurosci Res. 2006;84:632–636. doi: 10.1002/jnr.20952. [DOI] [PubMed] [Google Scholar]
- Iso H, Simoda S, Matsuyama T. Environmental change during postnatal development alters behaviour, cognitions and neurogenesis of mice. Behav Brain Res. 2007;179:90–98. doi: 10.1016/j.bbr.2007.01.025. [DOI] [PubMed] [Google Scholar]
- Jicha GA, Bowser R, Kazam IG, Davies P. Alz-50 and MC-1, a new monoclonal antibody raised to paired helical filaments, recognize conformational epitopes on recombinant tau. J Neurosci Res. 1997;48:128–132. doi: 10.1002/(sici)1097-4547(19970415)48:2<128::aid-jnr5>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
- Jimenez S, Baglietto-Vargas D, Caballero C, Moreno-Gonzalez I, Torres M, Sanchez-Varo R, Ruano D, Vizuete M, Gutierrez A, Vitorica J. Inflammatory response in the hippocampus of PS1M146L/APP751SL mouse model of Alzheimer's disease: age-dependent switch in the microglial phenotype from alternative to classic. J Neurosci. 2008;28:11650–11661. doi: 10.1523/JNEUROSCI.3024-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamenetz F, Tomita T, Hsieh H, Seabrook G, Borchelt D, Iwatsubo T, Sisodia S, Malinow R. APP processing and synaptic function. Neuron. 2003;37:925–937. doi: 10.1016/s0896-6273(03)00124-7. [DOI] [PubMed] [Google Scholar]
- Kawarabayashi T, Younkin LH, Saido TC, Shoji M, Ashe KH, Younkin SG. Age-dependent changes in brain, CSF, and plasma amyloid β protein in the Tg2576 transgenic mouse model of Alzheimer's disease. J Neurosci. 2001;21:372–381. doi: 10.1523/JNEUROSCI.21-02-00372.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kayed R, Head E, Thompson JL, McIntire TM, Milton SC, Cotman CW, Glabe CG. Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science. 2003;300:486–489. doi: 10.1126/science.1079469. [DOI] [PubMed] [Google Scholar]
- Klein WL, Krafft GA, Finch CE. Targeting small Aβ oligomers: the solution to an Alzheimer's disease conundrum? Trends Neurosci. 2001;24:219–224. doi: 10.1016/s0166-2236(00)01749-5. [DOI] [PubMed] [Google Scholar]
- Knobloch M, Konietzko U, Krebs DC, Nitsch RM. Intracellular Aβ and cognitive deficits precede β-amyloid deposition in transgenic arcAβ mice. Neurobiol Aging. 2007;28:1297–1306. doi: 10.1016/j.neurobiolaging.2006.06.019. [DOI] [PubMed] [Google Scholar]
- Lacor PN, Buniel MC, Furlow PW, Clemente AS, Velasco PT, Wood M, Viola KL, Klein WL. Aβ oligomer-induced aberrations in synapse composition, shape, and density provide a molecular basis for loss of connectivity in Alzheimer's disease. J Neurosci. 2007;27:796–807. doi: 10.1523/JNEUROSCI.3501-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert MP, Barlow AK, Chromy BA, Edwards C, Freed R, Liosatos M, Morgan TE, Rozovsky I, Trommer B, Viola KL, Wals P, Zhang C, Finch CE, Krafft GA, Klein WL. Diffusible, nonfibrillar ligands derived from Aβ1-42 are potent central nervous system neurotoxins. Proc Natl Acad Sci U S A. 1998;95:6448–6453. doi: 10.1073/pnas.95.11.6448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert MP, Velasco PT, Chang L, Viola KL, Fernandez S, Lacor PN, Khuon D, Gong Y, Bigio EH, Shaw P, De Felice FG, Krafft GA, Klein WL. Monoclonal antibodies that target pathological assemblies of Aβ. J Neurochem. 2007;100:23–35. doi: 10.1111/j.1471-4159.2006.04157.x. [DOI] [PubMed] [Google Scholar]
- Lesné S, Koh MT, Kotilinek L, Kayed R, Glabe CG, Yang A, Gallagher M, Ashe KH. A specific amyloid-β protein assembly in the brain impairs memory. Nature. 2006;440:352–357. doi: 10.1038/nature04533. [DOI] [PubMed] [Google Scholar]
- Lippa CF, Ozawa K, Mann DM, Ishii K, Smith TW, Arawaka S, Mori H. Deposition of β-amyloid subtypes 40 and 42 differentiates dementia with Lewy bodies from Alzheimer diseae. Arch Neurol. 1999;56:1111–1118. doi: 10.1001/archneur.56.9.1111. [DOI] [PubMed] [Google Scholar]
- Lord A, Kalimo H, Eckman C, Zhang XQ, Lannfelt L, Nilsson LN. The Arctic Alzheimer mutation facilitates early intraneuronal Aβ aggregation and senile plaque formation in transgenic mice. Neurobiol Aging. 2006;27:67–77. doi: 10.1016/j.neurobiolaging.2004.12.007. [DOI] [PubMed] [Google Scholar]
- Lue LF, Kuo YM, Roher AE, Brachova L, Shen Y, Sue L, Beach T, Kurth JH, Rydel RE, Rogers J. Soluble amyloid β peptide concentration as a predictor of synaptic change in Alzheimer's disease. Am J Pathol. 1999;155:853–862. doi: 10.1016/s0002-9440(10)65184-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maezawa I, Hong HS, Liu R, Wu CY, Cheng RH, Kung MP, Kung HF, Lam KS, Oddo S, Laferla FM, Jin LW. Congo red and thioflavin-T analogs detect Aβ oligomers. J Neurochem. 2008;104:457–468. doi: 10.1111/j.1471-4159.2007.04972.x. [DOI] [PubMed] [Google Scholar]
- Matsuyama S, Teraoka R, Mori H, Tomiyama T. Inverse correlation between amyloid precursor protein and synaptic plasticity in transgenic mice. Neuroreport. 2007;18:1083–1087. doi: 10.1097/WNR.0b013e3281e72b18. [DOI] [PubMed] [Google Scholar]
- Mori C, Spooner ET, Wisniewsk KE, Wisniewski TM, Yamaguch H, Saido TC, Tolan DR, Selkoe DJ, Lemere CA. Intraneuronal Aβ42 accumulation in Down syndrome brain. Amyloid. 2002;9:88–102. [PubMed] [Google Scholar]
- Mucke L, Masliah E, Yu GQ, Mallory M, Rockenstein EM, Tatsuno G, Hu K, Kholodenko D, Johnson-Wood K, McConlogue L. High-level neuronal expression of Aβ1-42 in wild-type human amyloid protein precursor transgenic mice: synaptotoxicity without plaque formation. J Neurosci. 2000;20:4050–4058. doi: 10.1523/JNEUROSCI.20-11-04050.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullen RJ, Buck CR, Smith AM. NeuN, a neuronal specific nuclear protein in vertebrates. Development. 1992;116:201–211. doi: 10.1242/dev.116.1.201. [DOI] [PubMed] [Google Scholar]
- Nishitsuji K, Tomiyama T, Ishibashi K, Ito K, Teraoka R, Lambert MP, Klein WL, Mori H. The E693Δ mutation in amyloid precursor protein increases intracellular accumulation of amyloid β oligomers and causes endoplasmic reticulum stress-induced apoptosis in cultured cells. Am J Pathol. 2009;174:957–969. doi: 10.2353/ajpath.2009.080480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oakley H, Cole SL, Logan S, Maus E, Shao P, Craft J, Guillozet-Bongaarts A, Ohno M, Disterhoft J, Van Eldik L, Berry R, Vassar R. Intraneuronal β-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer's disease mutations: potential factors in amyloid plaque formation. J Neurosci. 2006;26:10129–10140. doi: 10.1523/JNEUROSCI.1202-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oddo S, Caccamo A, Shepherd JD, Murphy MP, Golde TE, Kayed R, Metherate R, Mattson MP, Akbari Y, LaFerla FM. Triple-transgenic model of Alzheimer's disease with plaques and tangles: intracellular Aβ and synaptic dysfunction. Neuron. 2003;39:409–421. doi: 10.1016/s0896-6273(03)00434-3. [DOI] [PubMed] [Google Scholar]
- Oddo S, Caccamo A, Smith IF, Green KN, LaFerla FM. A dynamic relationship between intracellular and extracellular pools of Aβ. Am J Pathol. 2006;168:184–194. doi: 10.2353/ajpath.2006.050593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selkoe DJ. Alzheimer's disease is a synaptic failure. Science. 2002;298:789–791. doi: 10.1126/science.1074069. [DOI] [PubMed] [Google Scholar]
- Shankar GM, Bloodgood BL, Townsend M, Walsh DM, Selkoe DJ, Sabatini BL. Natural oligomers of the Alzheimer amyloid-β protein induce reversible synapse loss by modulating an NMDA-type glutamate receptor-dependent signaling pathway. J Neurosci. 2007;27:2866–2875. doi: 10.1523/JNEUROSCI.4970-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shankar GM, Li S, Mehta TH, Garcia-Munoz A, Shepardson NE, Smith I, Brett FM, Farrell MA, Rowan MJ, Lemere CA, Regan CM, Walsh DM, Sabatini BL, Selkoe DJ. Amyloid-β protein dimers isolated directly from Alzheimer's brains impair synaptic plasticity and memory. Nat Med. 2008;14:837–842. doi: 10.1038/nm1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suga K, Tomiyama T, Mori H, Akagawa K. Syntaxin 5 interacts with presenilin holoproteins, but not with their N- or C-terminal fragments, and affects β-amyloid peptide production. Biochem J. 2004;381:619–628. doi: 10.1042/BJ20040618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi RH, Milner TA, Li F, Nam EE, Edgar MA, Yamaguchi H, Beal MF, Xu H, Greengard P, Gouras GK. Intraneuronal Alzheimer Aβ42 accumulates in multivesicular bodies and is associated with synaptic pathology. Am J Pathol. 2002;161:1869–1879. doi: 10.1016/s0002-9440(10)64463-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi RH, Almeida CG, Kearney PF, Yu F, Lin MT, Milner TA, Gouras GK. Oligomerization of Alzheimer's β-amyloid within processes and synapses of cultured neurons and brain. J Neurosci. 2004;24:3592–3599. doi: 10.1523/JNEUROSCI.5167-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takuma H, Teraoka R, Mori H, Tomiyama T. Amyloid β E22Δ variant induces synapse alteration in mouse hippocampal slices. Neuroreport. 2008;19:615–619. doi: 10.1097/WNR.0b013e3282fb78c4. [DOI] [PubMed] [Google Scholar]
- Ting JT, Kelley BG, Lambert TJ, Cook DG, Sullivan JM. Amyloid precursor protein overexpression depresses excitatory transmission through both presynaptic and postsynaptic mechanisms. Proc Natl Acad Sci U S A. 2007;104:353–358. doi: 10.1073/pnas.0608807104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomiyama T, Nagata T, Shimada H, Teraoka R, Fukushima A, Kanemitsu H, Takuma H, Kuwano R, Imagawa M, Ataka S, Wada Y, Yoshioka E, Nishizaki T, Watanabe Y, Mori H. A new amyloid β variant favoring oligomerization in Alzheimer's-type dementia. Ann Neurol. 2008;63:377–387. doi: 10.1002/ana.21321. [DOI] [PubMed] [Google Scholar]
- Villain N, Desgranges B, Viader F, de la Sayette V, Mézenge F, Landeau B, Baron JC, Eustache F, Chételat G. Relationships between hippocampal atrophy, white matter disruption, and gray matter hypometabolism in Alzheimer's disease. J Neurosci. 2008;28:6174–6181. doi: 10.1523/JNEUROSCI.1392-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh DM, Klyubin I, Fadeeva JV, Cullen WK, Anwyl R, Wolfe MS, Rowan MJ, Selkoe DJ. Naturally secreted oligomers of amyloid β protein potently inhibit hippocampal long-term potentiation in vivo . Nature. 2002;416:535–539. doi: 10.1038/416535a. [DOI] [PubMed] [Google Scholar]