

Abstract
With a multitude of substrates, γ-secretase is poised to control neuronal function through a variety of signaling pathways. Presenilin 1 (PS1) is an integral component of γ-secretase and is also a protein closely linked to the etiology of Alzheimer's disease (AD). To better understand the roles of γ-secretase and PS1 in normal and pathological synaptic transmission, we examined evoked and spontaneous neurotransmitter release in cultured hippocampal neurons derived from PS1 knock-out (KO) mice. We found no changes in the size of evoked synaptic currents, short-term plasticity, or apparent calcium dependence of evoked release. The rate of spontaneous release from PS1 KO neurons was, however, approximately double that observed in wild-type (WT) neurons. This increase in spontaneous neurotransmission depended on calcium influx but did not require activation of voltage-gated calcium channels or presynaptic NMDA receptors or release of calcium from internal stores. The rate of spontaneous release from PS1 KO neurons was significantly reduced by lentivirus-mediated expression of WT PS1 or familial AD-linked M146V PS1, but not the D257A PS1 mutant that does not support γ-secretase activity. Treatment of WT neuronal cultures with γ-secretase inhibitor mimicked the loss of PS1, leading to a selective increase in spontaneous release without any change in the size of evoked synaptic currents. Together, these results identify a novel role for γ-secretase in the control of spontaneous neurotransmission through modulation of low-level tonic calcium influx into presynaptic axon terminals.
Introduction
Identifying the role of presenilin 1 (PS1) in normal neuronal function is of particular interest because mutations in the gene encoding this protein are the most common identified cause of inherited Alzheimer's disease (AD) (Tanzi and Bertram, 2001). PS1 is an integral component of γ-secretase, a protease with numerous cleavage targets—including amyloid precursor protein and Notch (De Strooper, 2003)—and γ-secretase activity in hippocampal neurons lacking PS1 is only approximately a fifth of the levels found in wild-type (WT) neurons (De Strooper et al., 1998). PS1 has also been found to regulate calcium release from internal stores (endoplasmic reticulum) through interactions with InsP3 receptors (Cheung et al., 2008), ryanodine receptors (Rybalchenko et al., 2008), and/or by forming PS1 leak channels in the membrane of the endoplasmic reticulum (Tu et al., 2006). PS1 may further modulate intracellular calcium levels by mediating calcium influx through voltage-gated calcium channels (VGCCs) (Cook et al., 2005). Exactly how γ-secretase activity and PS1-mediated regulation of calcium dynamics influence neuronal function in general, and synaptic transmission in particular, has yet to be determined.
Although conditional PS1 loss in adult cerebral cortex had no effect on evoked field EPSPs recorded in hippocampal slices (Feng et al., 2001; Yu et al., 2001; but see Dewachter et al., 2008), loss of PS1 was reported to increase the frequency of spontaneous neurotransmitter release in cultured cortical neurons derived from PS1 knock-out (KO) mice (Parent et al., 2005). These results suggest that PS1 might play a selective role in action potential-independent release, but the mechanisms responsible for this remain to be identified.
To characterize the effects on synaptic transmission when PS1 is eliminated in more detail, we performed whole-cell voltage-clamp experiments using autaptic hippocampal neurons cultured from PS1 KO mice. Evoked neurotransmitter release was normal in PS1 KO neurons, whereas the rate of spontaneous neurotransmitter release was significantly elevated compared with WT controls. These high levels of spontaneous release required influx of extracellular calcium, but did not depend on voltage-gated calcium channels, presynaptic NMDA receptors, or release from internal stores. Virally mediated expression of WT or familial AD-linked mutant PS1 significantly reduced spontaneous release rates, whereas expression of a catalytically inactive mutant PS1 failed to rescue a wild-type phenotype in PS1 KO neurons. Together, these results suggest that γ-secretase activity selectively regulates spontaneous neurotransmitter release by modulating tonic calcium influx into presynaptic terminals.
Materials and Methods
Neuronal cultures.
Neurons isolated from the hippocampi of embryonic (day 16) PS1−/− mice and their WT and heterozygous (HET) wombmates (Shen et al., 1997) were cultured on small microislands of permissive substrate, as previously described (Stevens and Sullivan, 2003). Experiments were performed according to the guidelines for the care and use of animals approved by the Institutional Animal Care and Use Committee at the University of Washington.
Viral constructs.
At 2–3 d in culture, hippocampal neurons were infected with lentivirus encoding one of the following: (1) WT human PS1 and, as a separate protein, enhanced green fluorescent protein (EGFP) to provide a real-time visual reporter of infection; (2) familial AD-linked M146V PS1 and EGFP; (3) catalytically inactive D257A PS1 and EGFP; or (4) EGFP alone as a control for viral infection and EGFP expression. Point mutations in PS1 were introduced using the QuikChange PCR strategy (Stratagene) and confirmed by sequencing. Lenti virions were generated according to the Invitrogen ViraPower Lentiviral Expression System manual using the lentivirus plasmid pRRL-cPPT-CMV-X-PRE-SIN (Barry et al., 2001). Expression of WT and mutant PS1 was confirmed by Western blot (see below). All experiments were performed in accordance with University of Washington Environmental Health and Safety approved protocols.
Western blot.
Protein lysates from cultured primary neurons derived from WT or PS1 KO embryonic mice were prepared for Western blot analyses in standard Laemmli sample buffer (Bio-Rad). Total protein concentrations for each lysate were determined by the BCA method (Pierce). Proteins were resolved by SDS-PAGE on a 4–20% Tris-glycine gradient gel. Resolved proteins were transferred to nitrocellulose and Western blotted with polyclonal rabbit antisera (provided by G. Schellenberg, University of Pennsylvania, Philadelphia, PA) that specifically recognizes the N-terminal domain of PS1 (Yang and Cook, 2004). Protein bands were detected using horseradish peroxidase-conjugated secondary antibodies (Sigma) in conjunction with chemiluminescence (Pierce).
Electrophysiology.
Whole-cell voltage-clamp recordings of EPSCs were obtained from isolated autaptic neurons after 13–16 d in microisland culture using Axopatch 200B and Multiclamp 700A amplifiers (Molecular Devices). All experiments included measurements obtained from at least two different culture preparations, and within each preparation were always performed on age-matched neurons. The standard extracellular solution contained the following: 119 mm NaCl, 5 mm KCl, 2.5 mm CaCl2, 1.5 mm MgCl2, 30 mm glucose, 20 mm HEPES, and 1 μm glycine. Recording pipettes of 2–5 MΩ were filled with 148.5 mm potassium gluconate, 9 mm NaCl, 1 mm MgCl2, 10 mm HEPES, and 0.2 mm EGTA. The chloride concentration in these solutions facilitates the distinction between excitatory (glutamatergic) and inhibitory (GABAergic) autaptic neurons. Peak amplitudes of evoked responses were measured using custom-written software. The relative amplitudes of synaptic currents carried by AMPA receptors (AMPARs) and NMDA receptors (NMDARs) were measured by recording in external recording solution lacking magnesium (to relieve NMDAR blockade at hyperpolarized membrane potentials), first in the absence and then in the presence of the AMPAR antagonist CNQX (10 μm); the peak current in the absence of CNQX is almost entirely mediated by AMPARs (our unpublished observations), whereas the peak current in the presence of CNQX is mediated by NMDARs. For each neuron, the IAMPAR/INMDAR ratio was calculated as the peak amplitude of the response in the absence of CNQX divided by the peak amplitude in the presence of CNQX. CNQX and APV were obtained from Tocris Bioscience; ryanodine, thapsigargin, 2-APB (2-aminoethyl diphenyl borinate), and SKF 96365 (1-[2-[3-(4-methoxyphenyl)propoxy]-2-(4-methoxyphenyl)ethyl]-1H-imidazole hydrochloride) from Ascent Scientific; xestospongin C from Cayman Chemical; and l-685,458 from Calbiochem. Series resistance was monitored, and only cells with stable series resistance were included in the data analysis. During recordings of evoked responses, series resistance was compensated ∼85% to reduce errors in measuring large currents.
Spontaneous miniature EPSCs (mEPSCs) were recorded continuously over 10 s periods for several minutes, starting 3–6 min after making a whole-cell configuration. Peak amplitudes of spontaneous mEPSCs were measured offline semiautomatically with custom-written software by using an adjustable amplitude threshold. All deflections from baseline greater than threshold were detected. Selected events were then visually examined; any spurious events were manually rejected and any missed events were flagged for inclusion in the mean amplitude and frequency calculations. For each neuron, ∼200 mEPSCs were analyzed. mEPSC frequencies were calculated by dividing the total number of mEPSC events by the total time sampled.
For the repetitive stimulation protocol, a 20 Hz train of 40 stimuli (1 ms steps from −60 mV to +30 mV) was delivered every 60 s and each EPSC in the train was normalized to the peak amplitude of the first EPSC. To measure recovery from depression, an additional test stimulus was delivered 1.5 s after the end of the train.
Data analysis.
All data are presented as mean ± SEM and n refers to the number of cells. Data were analyzed with Graphpad Prism software using one-way ANOVA followed by Tukey's post hoc tests to permit direct comparison between all groups, or with Student's t test, as appropriate.
Results
Lack of PS1 does not alter evoked neurotransmission or short-term plasticity
Synaptic strength was quantified in autaptic hippocampal neurons by evoking an action current and recording the resultant EPSC under whole-cell voltage clamp at −60 mV (Fig. 1A). Differences between average EPSC amplitudes of WT, HET, and PS1 KO neurons recorded in standard extracellular solution containing 2.5 mm calcium were not statistically significant (WT = 6855 ± 710 pA, n = 30; HET = 7191 ± 1091 pA, n = 23; PS1 KO = 7210 ± 1043, n = 29) (Fig. 1B), indicating that neurons lacking PS1 display normal evoked synaptic strengths. To ensure that the failure of PS1 deficiency to alter evoked transmission in PS1 KO neurons was not due to a ceiling effect on neurotransmitter release, EPSC amplitude was also measured in lower (1.5 mm) external calcium. For all neurons, decreasing the external calcium concentration from 2.5 to 1.5 mm led to a comparable decrease in evoked EPSC amplitude, and the average EPSC amplitudes of PS1 KO and HET neurons were not significantly different from WT in 1.5 mm external calcium (Fig. 1B). In addition to showing that loss of PS1 has no effect on evoked neurotransmission over a range of release probability conditions, these experiments also indicate that the calcium sensitivity of the vesicle release machinery is normal in PS1 KO neurons.
Figure 1.
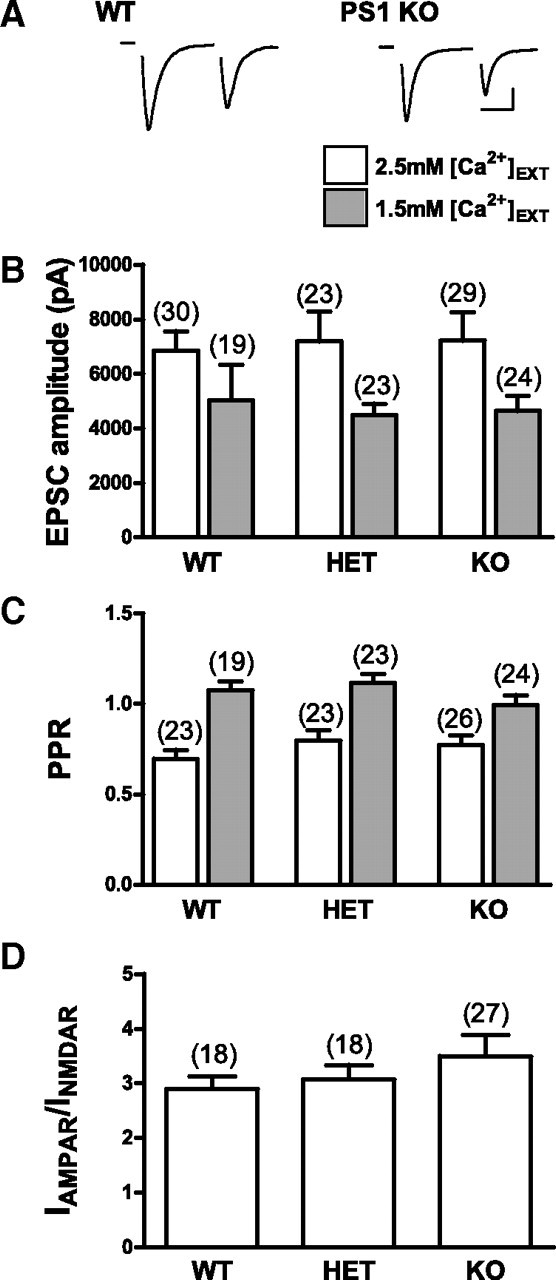
Evoked excitatory neurotransmission, short-term plasticity, and AMPAR/NMDAR ratio are not altered in PS1 KO neurons. A, Representative pairs of EPSCs recorded from WT and PS1 KO autaptic mouse neurons. Delivery of a 1 ms step depolarization to 40 mV initiates an unclamped presynaptic action current (blanked for clarity) followed by an EPSC. Scale bars, 2000 pA, 20 ms. B, Average EPSC size was not significantly different in WT, HET, or PS1 KO neurons in 2.5 or 1.5 mm external Ca2+ [Ca2+]EXT. C, There were no significant differences in the PPR, measured as the relative sizes of the first and second responses to a pair of stimuli delivered 50 ms apart, in WT, HET, or PS1 KO neurons in either 2.5 or 1.5 mm external Ca2+. D, The AMPAR/NMDAR ratio was the same for WT, HET, and PS1 KO neurons. The number of neurons in each group is indicated and plotted values represent the mean ± SEM.
To quantify short-term plasticity of evoked transmission, 40 stimuli delivered at 20 Hz were used to evoke a train of EPSCs. There were no significant differences in the paired-pulse ratio (PPR; the ratio of the second EPSC amplitude to the first) between groups (WT = 0.70 ± 0.05, n = 23; HET = 0.80 ± 0.06, n = 23; PS1 KO = 0.77 ± 0.05, n = 26) (Fig. 1C). Reducing external calcium to 1.5 mm led to comparable increases in the PPR of all groups, and there were still no statistically significant differences between WT, HET, and PS1 KO neurons (Fig. 1C). In addition, no statistically significant differences were observed in the relative amplitudes of EPSCs that occurred later in the train, nor in the extent to which the EPSC recovered 1.5 s after the train (data not shown), suggesting no changes in the overall dynamics of evoked neurotransmitter release, in the size of readily releasable pool, or in the rate at which the readily releasable pool refills after emptying.
It was previously reported that neurons lacking PS1 had specific reductions in NMDA receptor-mediated transmission, measured using field potential recordings from hippocampal slices (Dewachter et al., 2008; but see Zhang et al., 2009). To determine whether loss of PS1 reduced NMDA receptor-mediated currents in our cultured neurons, we measured the relative amplitudes of synaptic currents carried by AMPARs and NMDARs by recording in external recording solution lacking magnesium in the absence and presence of the AMPAR antagonist CNQX (10 μm). There were no statistically significant differences between WT, HET, and PS1 KO groups in their AMPAR/NMDAR ratio of synaptic currents (WT = 2.9 ± 0.2, n = 18; HET = 3.1 ± 0.3, n = 18; PS1 KO = 3.5 ± 0.4, n = 27) (Fig. 1D). It may be that the decrease in NMDAR-mediated transmission seen in adult hippocampal slices derived from PS1 KO mice (Dewachter et al., 2008) reflects a compensatory mechanism that develops over several months, and is not an immediate and direct effect of eliminating PS1.
Together, these data suggest that basic aspects of evoked synaptic transmission are not dependent on PS1.
Neurons lacking PS1 display an increased frequency of spontaneous mEPSCs
In addition to action potential-dependent neurotransmitter release, another component of synaptic transmission is the ongoing, spontaneous, action potential-independent release of individual presynaptic vesicles. To characterize this component of synaptic transmission, we recorded spontaneous mEPSCs from WT, HET, and PS1 KO neurons at a holding potential of −70 mV. We found that the majority of PS1 KO neurons exhibited a significantly higher frequency of mEPSCs compared with HET and WT neurons (WT = 4.6 ± 0.6 Hz, n = 27; HET = 3.8 ± 0.6 Hz, n = 23; PS1 KO = 8.8 ± 1.3 Hz, n = 32; ANOVA, p < 0.0001) (Fig. 2A,B). A similarly enhanced frequency of mEPSCs was also seen in conventional (nonisland) cultures of PS1 neurons (WT = 2.0 ± 0.4 Hz, n = 10; PS1 KO = 7.6 ± 1.4 Hz, n = 14; t test, p < 0.004), consistent with results from mass-cultured cortical neurons (Parent et al., 2005), verifying that this phenotype is not unique to hippocampal autapses. Interestingly, a small population of PS1 KO neurons displayed mEPSC frequencies that were quite similar to WT frequencies (Fig. 2A, cumulative probability plot; B, fourth trace). This could reflect the existence of two distinct types of excitatory neurons in these hippocampal cultures, only one of which displays enhanced vesicle release in the absence of PS1. Another possibility is that, in any individual neuron, the observed enhanced release of vesicles is not happening 100% of the time. Importantly, the lack of phenotype expression in a PS1 KO neuron did not appear to be linked to any other facet of transmission that we studied (e.g., there was no relationship between amplitude of evoked response and mEPSC frequency in PS1 KO neurons). The average amplitude of mEPSCs was not significantly different across groups (WT = 23.0 ± 1.7 pA, n = 27; HET = 23.4 ± 1.6 pA, n = 23; PS1 KO = 21.3 ± 1.2 pA, n = 32) (Fig. 2C), indicating that PS1 is not necessary for proper regulation of the number of AMPARs at individual postsynaptic sites.
Figure 2.

PS1 KO neurons exhibit a significantly higher frequency of spontaneous mEPSCs compared with WT or HET neurons. A, Cumulative probability distribution plot of mEPSC frequency for PS1 KO (●), HET (*), and WT (○) neurons. Each point on the graph represents the average frequency of an individual neuron. Notice that although a portion of neurons from all three groups had equally low frequencies, a large fraction of PS1 KO neurons had a higher and broader range of frequencies that are distinct from WT and HET. Inset, Bar graph of the same data showing mean ± SEM. ANOVA, p < 0.0001; **Tukey's multiple comparison test, p < 0.01 between PS1 KO and WT, and between PS1 KO and HET. B, Representative traces of spontaneous mEPSCs depicting the range of frequencies recorded from three WT (top) and three PS1 KO (bottom) neurons. Notice that the lowest frequency example trace for KO (KO 1) is similar to the lowest frequency WT trace (WT 1), but that the highest frequency KO trace (KO 3) is distinct from highest frequency WT trace (WT 3). C, Cumulative probability distribution plot of mEPSC amplitude for PS1 KO (●), HET (*), and WT (○) neurons. Each point on the graph represents the average mEPSC amplitude of an individual neuron. Notice that the distribution of averaged mEPSC amplitudes is similar across groups. Inset, Bar graph of the same data showing mean ± SEM. For insets, the number of neurons in each group is indicated above each bar.
An increase in mEPSC frequency could be explained by an increase in the number of synapses received by the postsynaptic neuron and/or an increase in the probability of spontaneous vesicle release from a given number of synapses. It is unlikely that the increase in mEPSC frequency was due to an increase in number of synapses, because an increase in number of synapses would be expected to result in larger evoked EPSC amplitudes (given that mEPSC amplitudes are unchanged), which we did not observe (Fig. 1B). The high mEPSC frequency phenotype, therefore, is most likely due to an increase in the probability of spontaneous vesicle release. Next, we focused on the mechanisms by which PS1 deficiency enhances spontaneous release of vesicles.
The increase in mEPSC frequency observed in PS1 KO neurons is calcium-dependent
PS1 is known to play a role in regulating calcium release from the endoplasmic reticulum (LaFerla, 2002; Stutzmann, 2005; Bezprozvanny and Mattson, 2008). This led us to ask whether the increase in mEPSCs observed in PS1 KO neurons could be the result of aberrant regulation of intracellular calcium levels. To test this, mEPSCs were recorded from WT and PS1 KO neurons with or without the calcium chelator EGTA added to the internal recording solution. We found that 10 mm EGTA effectively decreased PS1 KO mEPSC frequency to levels observed in WT neurons (WT = 5.9 ± 1.3 Hz, n = 11; KO = 12.9 ± 2.0 Hz, n = 30; KO + EGTA = 6.4 ± 1.4 Hz, n = 28; ANOVA, p < 0.008) (Fig. 3A). The frequency of mEPSCs observed in WT neurons was only modestly decreased by the presence of 10 mm EGTA in the internal recording solution (WT + EGTA = 4.3 ± 2.0 Hz, n = 9). Therefore, calcium appears to play an enhanced role in controlling mEPSC frequency in PS1 KO neurons compared with WT neurons. This could be due to an increase in entry of calcium from outside the cell and/or efflux of calcium from internal stores.
Figure 3.

The high frequency of spontaneous mEPSCs in PS1 KO neurons is reduced to WT levels by buffering intracellular Ca2+. Including 10 mm EGTA in the intracellular recording solution significantly decreased mEPSC frequency in PS1 KOs to WT levels (ANOVA, p < 0.008; *Tukey's multiple comparison test, p < 0.05 between PS1 KO control and PS1 KO with intracellular EGTA). The number of neurons in each group is indicated and values represent the mean ± SEM.
To determine the influence of external calcium on the high mEPSC frequency in PS1 KO neurons, mEPSCs were recorded from WT and PS1 KO neurons after washing in calcium-free external solution. We found that, similar to buffering internal calcium, excluding external calcium significantly reduced PS1 KO mEPSC frequency to WT levels but had a smaller effect on the frequency of WT mEPSCs (WT = 3.4 ± 0.9 Hz, n = 8; PS1 KO = 4.7 ± 0.9 Hz, n = 16). These experiments, together with the EGTA results, not only demonstrate the calcium dependence of the high mEPSC frequency phenotype, but also effectively rule out an increase in functional synapse number as an underlying mechanism.
Next, we set out to further define the dependence of vesicle release on external calcium in PS1 KO neurons. It has been previously shown that cultured cortical PS1 KO neurons exhibit enhanced calcium influx through L- and P-type VGCCs (Cook et al., 2005). To test whether calcium entry through L-, P-, or any other type of VGCC contributes to the high mEPSC frequency in PS1 KO neurons, we performed recordings in which mEPSCs were first recorded in normal external solution and then switched to an external solution containing the general VGCC blocker cadmium (0.5 mm). If the calcium contributing to the high mEPSC frequency in PS1 KOs was coming in through any type of VGCC, then blocking them with cadmium should inhibit mEPSC frequency as effectively as calcium-free external did. On the contrary, we found that blocking VGCCs with cadmium did not reduce the frequency of mEPSCs in KO neurons (PS1 KO control = 9.6 ± 2.7 Hz; PS1 KO + Cd2+ = 13.3 ± 3.6 Hz, n = 12). Because T-type calcium channels are less sensitive to cadmium blockade than other VGCCs (Fox et al., 1987), we also tested the effect of the efficient T-type calcium channel blocker nickel (Fox et al., 1987). Nickel (100 μm) had no significant effect on mEPSC frequency in PS1 KOs (data not shown). These data suggest that calcium entry through VGCCs is not responsible for the calcium-dependent enhancement of vesicle release observed in PS1 KO neurons.
Presynaptic NMDARs have been found to modulate spontaneous neurotransmitter release in several brain regions (Corlew et al., 2008). To determine whether calcium entry through presynaptic NMDA receptors could be triggering enhanced spontaneous release, mEPSC frequency was measured in PS1 KO neurons before and after washing in the NMDA antagonist APV (50 μm). We found that PS1 KO mEPSC frequencies were not significantly reduced in the presence of APV (PS1 KO control = 27.2 ± 6.3 Hz; PS1 KO + APV = 22.6 ± 4.9 Hz, n = 8) (Parent et al., 2005), ruling out the involvement of either presynaptic or postsynaptic NMDARs.
In summary, although the enhanced release of presynaptic vesicles observed in PS1 KO neurons was dependent upon the presence of external calcium, this synaptic phenotype did not appear to involve calcium entry through either VGCC or presynaptic NMDARs. One interpretation of these findings is that normal levels of calcium entry from outside the neuron could be triggering exaggerated, abnormal release of calcium from internal stores in PS1 KO neurons. In other words, the enhanced mEPSC frequency in PS1 KO neurons could be the result of enhanced calcium-induced calcium release, which has been reported to modulate spontaneous neurotransmission in hippocampal pyramidal cells (Emptage et al., 2001). This notion seems especially appealing given that PS1 has been implicated in release of calcium from internal stores via interactions with the InsP3 receptor (Cheung et al., 2008), the ryanodine receptor (Rybalchenko et al., 2008), and/or by forming a calcium leak channel in the endoplasmic reticulum (Tu et al., 2006).
To test the contribution from ryanodine receptor-mediated calcium efflux from internal stores on spontaneous release, ryanodine receptors were blocked by inclusion of ryanodine (15 μm) in the intracellular recording solution. We found that blocking ryanodine receptors failed to significantly decrease the high mEPSC frequency exhibited by PS1 KO neurons (PS1 KO control = 11.3 ± 1.3 Hz, n = 52; PS1 KO + ryanodine = 9.4 ± 2.3 Hz, n = 17) (Fig. 4).
Figure 4.

Blocking Ca2+ release from internal stores does not significantly reduce the high frequency of mEPSCs in PS1 KO neurons. Blocking ryanodine receptors by including 15 μm ryanodine (Ryan) in the intracellular recording solution, or blocking InsP3 receptors with 3–10 μm xestospongin C (Xest; applied either inside or outside the cell), or depleting internal Ca2+ stores by pretreating cells for 30 min with 1 μm thapsigargin (Thaps; and recording in the continued presence of thapsigargin) had no significant effect on mEPSC frequency in PS1 KO neurons. The number of neurons in each group is indicated and plotted values represent the mean ± SEM.
Next, the role of InsP3 receptors was tested by blocking them with xestospongin C. Whether applied inside or outside the cell, the presence of xestospongin C (3–10 μm) had no significant effect on the high mEPSC frequency phenotype in PS1 KO neurons (PS1 KO control = 11.3 ± 1.3 Hz, n = 52; PS1 KO + xestospongin C = 13.7 ± 2.5 Hz, n = 20) (Fig. 4).
To test the possibility that other receptors/channels in the endoplasmic reticulum (ER) could be the locus of exaggerated calcium efflux, we took the alternative approach of depleting calcium from internal stores by pretreating neurons with the SERCA pump inhibitor thapsigargin. Thapsigargin blocks the uptake of calcium from the cytosol into the ER, thereby leading to the depletion of calcium from the ER. If internal stores are the source of calcium that is triggering the high rate of spontaneous vesicle release, depleting internal stores of calcium should reduce mEPSC frequency in PS1 KO neurons. We found that pretreatment for 30 min with thapsigargin (1 μm) did not significantly attenuate the high mEPSC frequency phenotype in PS1 KO neurons recorded in the continued presence of thapsigargin (PS1 KO control = 11.3 ± 1.33 Hz, n = 52; PS1 KO + thapsigargin = 10.9 ± 2.1 Hz, n = 14) (Fig. 4). Together, the ryanodine, xestospongin C, and thapsigargin experiments suggest that the endoplasmic reticulum is not a source of calcium contributing to the high mEPSC frequency in PS1 KO neurons.
A concern about the thapsigargin experiments is that depletion of ER stores could be activating capacitative calcium entry (CCE) through store-operated channels (SOCs). In fact, it has been reported that neurons lacking PS1 exhibit exaggerated CCE (Yoo et al., 2000; Ris et al., 2003). Unfortunately, attempts to determine the role of enhanced CCE in the high spontaneous mEPSC frequency in PS1 KO neurons were thwarted by clear off-target effects in both WT and PS1 KO neurons on both evoked and spontaneous neurotransmission elicited by the drugs known to block SOCs (10–100 μm 2-APB and 10 μm SKF 96365). Although there is no evidence that SOCs are active when internal stores are not depleted, it is possible, though unlikely, that enhanced calcium influx through these channels is mediating the high mEPSCs frequency in PS1 KO neurons regardless of the levels of calcium in the internal stores.
Expression of WT PS1 and familial AD-linked M146V PS1 reduces mEPSC frequency in PS1 KOs, but expression of catalytically inactive D257A PS1 does not
The results described above demonstrate that loss of PS1 significantly increases mEPSC frequency. To determine whether reintroduction of PS1 is sufficient to restore mEPSC frequency to wild-type levels, we investigated the effects of expressing WT PS1 protein in PS1 KO neurons. To gain further insight into the mechanisms by which PS1 function alters mEPSC frequency, we also investigated the effects of expressing two functionally distinct PS1 variants: (1) M146V, a familial AD-linked PS1 mutation that retains an aberrant γ-secretase activity favoring production of β-amyloid 42 and elicits exaggerated intracellular calcium signaling in neurons (LaFerla, 2002; Stutzmann, 2005; Bezprozvanny and Mattson, 2008); and (2) D257A, a PS1 mutation that blocks γ-secretase activity (Wolfe et al., 1999). WT PS1, M146V PS1, and D257A PS1 were expressed in PS1 KO neurons, along with EGFP (as a separate protein), using lentiviral vectors. Lentivirus directing expression of EGFP alone served as a control.
Expression of WT PS1 in PS1 KO neurons significantly reduced high mEPSC frequency (PS1 KO = 14.7 ± 1.9, n = 31 Hz; PS1 KO + EGFP = 15.4 ± 2.6 Hz, n = 25; PS1 KO + WT PS1 = 7.9 ± 1.1 Hz, n = 27; ANOVA, p < 0.0001) (Fig. 5A). Expression of the familial AD-linked M146V mutant also restored mEPSC frequency to WT levels (PS1 KO + M146V PS1 = 5.2 ± 0.8 Hz, n = 11) (Fig. 5A). The D257A mutant, however, was unable to reverse the high PS1 KO mEPSC frequency (PS1 KO + D257A PS1 = 16.0 ± 2.3 Hz, n = 17) (Fig. 5A). In keeping with these findings, Western blot analysis (Fig. 5B) confirmed that the lentiviral vectors restored properly processed exogenously expressed WT and M146V PS1 protein levels in PS1 KO neurons that were approximately comparable to endogenously expressed PS1 in WT neurons (Fig. 5B, far left lane; compare lentivirus WT PS1 and M146V PS1 with equivalent uninfected WT cultured neurons). Consistent with previous results (Thinakaran et al., 1996), PS1 overexpression resulted in the accumulation of unprocessed full-length PS1. Also in keeping with previous findings (Wolfe et al., 1999), the D257A PS1 mutant did not mature into a cleaved N-/C-terminal form, but was clearly expressed in its full-length form, thus ruling out the possibility that the failure of the D257A PS1 lentivirus to rescue the wild-type mEPSC phenotype was due to inadequate protein expression.
Figure 5.

γ-Secretase activity is required for reversal of high mEPSC frequency in PS1 KO neurons infected with lentivirus directing expression of WT or mutant PS1. A, Lentivirus-mediated expression of WT PS1 or familial Alzheimer's disease-linked M146V PS1—but not the catalytically inactive D257A PS1—significantly reduced the high mEPSC frequency in PS1 KO neurons. The number of neurons in each group is indicated and plotted values represent the mean ± SEM (ANOVA, p < 0.0001; *Tukey's multiple comparison test, p < 0.05 between PS1 KO neurons expressing WT PS1 or M146V PS1 and control PS1 KO neurons expressing EGFP alone). Inset, Treating WT neurons for 48–72 h with γ-secretase inhibitor L685,458 (5 μm) mimicked the effect of knocking out PS1, leading to a significant increase in the frequency of spontaneous mEPSCs (*unpaired Student's t test, p < 0.05). The number of neurons in each group is indicated and plotted values represent the mean ± SEM. B, Western blot shows expression of endogenous and exogenous full-length PS1 holoprotein (PS1fl) and processed N-terminal PS1 fragments (PS1nt), as well as actin levels, in lysates from uninfected and lentivirus-infected neuronal cultures, and from whole brain. Migration positions of molecular mass markers are on the left. Uninf, Uninfected.
These data suggest that loss of γ-secretase activity underlies the enhanced release of vesicles in PS1 KO neurons. To test this idea further, we pharmacologically inhibited γ-secretase activity in WT neurons by treating them for 48–72 h with the γ-secretase inhibitor L685,458 (5 μm). Inhibiting γ-secretase activity in WT neurons mimicked the effects of knocking out PS1 and led to a significant increase in mEPSC frequency (WT = 6.5 ± 2.1 Hz, n = 13; WT + L685,458 = 14.5 ± 3.3 Hz, n = 12; t test, p < 0.05) (Fig. 5A, inset) without significantly affecting EPSC size (WT = 6361 ± 969 pA, n = 11; WT + L685,458 = 4194 ± 1015 pA). Together, these results suggest that γ-secretase activity selectively modulates spontaneous vesicle release.
Discussion
Only a handful of studies to date have investigated the normal role of WT PS1 in synaptic function by examining the effects of selectively eliminating PS1 (Feng et al., 2001; Yu et al., 2001; Parent et al., 2005; Dewachter et al., 2008; Zhang et al., 2009, their supplemental information). To extend our knowledge about PS1 in normal neuronal function, we performed a detailed analysis of the consequences of losing PS1 on a variety of features of synaptic transmission. We found that PS1 loss selectively enhanced action potential-independent neurotransmitter release while having no effect on evoked synaptic transmission. EPSC amplitude, short-term synaptic plasticity, AMPAR/NMDAR, ratio and the apparent calcium-sensitivity of release were all unchanged in PS1 KO neurons (Fig. 1). The frequency of spontaneous mEPSCs in PS1 KO neurons was, however, more than double the rate in WT neurons, without any change in amplitude (Fig. 2). Buffering intracellular calcium or removing extracellular calcium reduced mEPSC frequency in PS1 KO neurons to WT levels, but blocking calcium entry through VGCCs or NMDARs had no effect (Fig. 3). Surprisingly, blocking release of calcium from internal stores also failed to alter mEPSC frequency in PS1 KO neurons (Fig. 4). Lentivirus-mediated expression of WT or M146V PS1 in PS1 KO neurons restored mEPSCs frequency to WT levels, but expression of the catalytically inactive (and improperly processed) D257A PS1 had no effect (Fig. 5). Consistent with these data, treatment of WT neurons with γ-secretase inhibitor increased mEPSC frequency to KO levels (Fig. 5). Together, these results indicate that γ-secretase normally suppresses presynaptic calcium influx that selectively modulates spontaneous neurotransmission, independent of any changes in spine density or neurite outgrowth.
The lack of effect on EPSC size is consistent with most previous studies that used extracellular field potential recordings from acute hippocampal slices to investigate evoked synaptic transmission in PS1 KO neurons (Feng et al., 2001; Yu et al., 2001; Zhang et al., 2009; but see Dewachter et al., 2008) and PS1 HET neurons (Morton et al., 2002). Although both loss of PS1 and inhibition of γ-secretase were found to increase immunocytochemically identified axodendritic contacts in cortical cultures (Parent et al., 2005), the ineffectiveness of either of these manipulations in altering evoked EPSC amplitudes suggests that the additional contacts may not form functional synapses. An alternative explanation is that loss of PS1 or inhibition of γ-secretase increases synapse number but reduces the probability of neurotransmitter release in response to an action potential; the absence of any change in the PPR, which typically correlates with release probability (Zucker and Regehr, 2002), or in the apparent calcium-sensitivity of release in PS1 KO neurons, argues against a decrease in release probability counteracting the effect of an increase in synapse number. The rapidity with which introduction of intracellular EGTA or removal of extracellular calcium reduced mEPSC frequency (≤6 min after breakthrough and ≤3 min after solution exchange, respectively) also supports the notion that loss of PS1 does not lead to an increase in functional synapse number.
Like EPSC size, short-term plasticity in PS1 KO neurons was not different from WT neurons, again consistent with the majority of previous studies (Feng et al., 2001; Yu et al., 2001; Zhang et al., 2009; but see Dewachter et al., 2008). Given the multiple roles that PS1 has been implicated in playing to control calcium release from internal stores, it was somewhat surprising that neither PPR nor depression during trains were altered, because two studies have reported that calcium release from internal stores contributes to short-term plasticity in hippocampal pyramidal neurons (Emptage et al., 2001; Zhang et al., 2009), although this remains controversial (Carter et al., 2002; Chakroborty et al., 2009). In any case, the lack of effect on short-term plasticity in PS1 KO neurons further supports the conclusion that PS1 does not influence evoked transmission.
The rate at which synaptic vesicles were spontaneously released was clearly changed in PS1 KO neurons. Although mEPSC frequency varied between batches of neurons, the average rate in PS1 KO neurons was consistently approximately twice the rate in batch-matched WT neurons, and in individual neurons was often much higher—indeed, over 1 s long stretches, mEPSC frequencies >40 Hz were not uncommon in PS1 KO neurons but rarely observed in WT neurons. This enhancement appears to depend on an elevated influx of calcium, because it was eliminated by removal of extracellular calcium or by buffering of intracellular calcium, manipulations that had much smaller effects on mEPSC frequency in WT neurons. Although it might seem surprising that increased calcium influx could selectively enhance spontaneous release without affecting evoked release, we note that elevating intracellular calcium concentration from its normal resting level of ∼25 nm up to ∼80 nm leads to a tripling of mEPSC frequency at the calyx of Held (Lou et al., 2005), suggesting that, in this range of very low intracellular calcium concentrations, an increase of <100 nm could be responsible for the increase in mEPSC frequency we observed in PS1 KO neurons. This amount of extra calcium would not be expected to have a significant effect on action potential-evoked release, which is triggered by intracellular calcium concentrations in the range of ∼10–25 μm (Bollmann et al., 2000; Schneggenburger and Neher, 2000).
The source of the calcium responsible for elevated spontaneous release in PS1 KO neurons remains an enigma. Although we initially suspected that enhanced calcium-induced calcium release from internal stores would play a role in the high-frequency phenotype in PS1 KO neurons, blocking or eliminating release from internal stores left mEPSC frequency unaltered in these cells. VGCCs, whose activity is enhanced in cortical neurons derived from PS1 KO mice (Cook et al., 2005), do not appear to be involved, nor do presynaptic calcium-permeable NMDARs. We were unable to determine whether SOCs might be aberrantly active when internal stores were not depleted, because of off-target effects of SOC antagonists on evoked and spontaneous neurotransmission in both PS1 KO and WT neurons. We note that presynaptic Gq-coupled receptors (such as group 1 metabotropic glutamate receptors) would be expected to act through release from internal stores, so are unlikely to be involved. In addition, in the absence of action potential-dependent loading of presynaptic terminals with sodium, the sodium/calcium exchanger should strongly favor calcium efflux, making this a highly improbable source for calcium entry in PS1 KO neurons. Similarly, the plasma membrane calcium-dependent ATPase controls intracellular calcium levels through calcium extrusion, and would not be expected to reverse its direction. Calcium-permeable transient receptor potential channels remain candidates for future studies of the mechanism underlying high mEPSC frequency in PS1 KO neurons, along with other as-yet-unidentified pathways for calcium influx.
An unexpected role for γ-secretase in the control of spontaneous neurotransmission was revealed by rescue experiments that investigated the effects of lentivirus-mediated expression of WT and mutant PS1 in PS1 KO neurons. Whereas expression of either WT or M146V PS1 significantly reduced mEPSC frequency, expression of D257A PS1 did not. Western blot data showed that in infected neuronal cultures, full-length D257A PS1 was abundant, but not properly processed, confirming the requirement for this residue in PS1 endoproteolysis as well as γ-secretase activity (Wolfe et al., 1999). Treatment of WT neurons with γ-secretase inhibitor phenocopied the loss of PS1, leading to an increase in mEPSC frequency (Kamenetz et al., 2003; Parent et al., 2005) without any change in evoked synaptic responses. Together, these results implicate a novel role for γ-secretase in the control of low-level tonic calcium influx into presynaptic axon terminals that selectively influences spontaneous release. Because γ-secretase is involved in the processing of so many different types of proteins, to hypothesize here about a specific pathway by which γ-secretase regulates spontaneous release is beyond the scope of this study. In recent years, important physiological functions have been identified for spontaneous excitatory neurotransmission (McKinney et al., 1999; Sharma and Vijayaraghavan, 2003; Sutton et al., 2006; Espinosa and Kavalali, 2009). The current study thus adds to the growing list of potential deleterious consequences of using γ-secretase inhibitors to treat AD (Panza et al., 2010).
Footnotes
This work was supported by American Health Assistance Foundation and National Institutes of Health/National Institute of Neurological Disorders and Stroke (J.M.S.), and the Veteran's Affairs Office of Research and Development Medical Research Service (D.G.C.). We thank Mike Ahlquist and Ning Li for excellent technical assistance. The original WT human PS1 plasmid used to generate the lentivirus constructs was generously provided by Dr. Mark Bothwell (University of Washington, Seattle, WA).
References
- Barry SC, Harder B, Brzezinski M, Flint LY, Seppen J, Osborne WR. Lentivirus vectors encoding both central polypurine tract and posttranscriptional regulatory element provide enhanced transduction and transgene expression. Hum Gene Ther. 2001;12:1103–1108. doi: 10.1089/104303401750214311. [DOI] [PubMed] [Google Scholar]
- Bezprozvanny I, Mattson MP. Neuronal calcium mishandling and the pathogenesis of Alzheimer's disease. Trends Neurosci. 2008;31:454–463. doi: 10.1016/j.tins.2008.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bollmann JH, Sakmann B, Borst JG. Calcium sensitivity of glutamate release in a calyx-type terminal. Science. 2000;289:953–957. doi: 10.1126/science.289.5481.953. [DOI] [PubMed] [Google Scholar]
- Carter AG, Vogt KE, Foster KA, Regehr WG. Assessing the role of calcium-induced calcium release in short-term presynaptic plasticity at excitatory central synapses. J Neurosci. 2002;22:21–28. doi: 10.1523/JNEUROSCI.22-01-00021.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakroborty S, Goussakov I, Miller MB, Stutzmann GE. Deviant ryanodine receptor-mediated calcium release resets synaptic homeostasis in presymptomatic 3xTg-AD mice. J Neurosci. 2009;29:9458–9470. doi: 10.1523/JNEUROSCI.2047-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung KH, Shineman D, Müller M, Cárdenas C, Mei L, Yang J, Tomita T, Iwatsubo T, Lee VM, Foskett JK. Mechanism of Ca2+ disruption in Alzheimer's disease by presenilin regulation of InsP3 receptor channel gating. Neuron. 2008;58:871–883. doi: 10.1016/j.neuron.2008.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook DG, Li X, Cherry SD, Cantrell AR. Presenilin 1 deficiency alters the activity of voltage-gated Ca2+ channels in cultured cortical neurons. J Neurophysiol. 2005;94:4421–4429. doi: 10.1152/jn.00745.2005. [DOI] [PubMed] [Google Scholar]
- Corlew R, Brasier DJ, Feldman DE, Philpot BD. Presynaptic NMDA receptors: newly appreciated roles in cortical synaptic function and plasticity. Neuroscientist. 2008;14:609–625. doi: 10.1177/1073858408322675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Strooper B. Aph-1, Pen-2, and nicastrin with presenilin generate an active gamma-secretase complex. Neuron. 2003;38:9–12. doi: 10.1016/s0896-6273(03)00205-8. [DOI] [PubMed] [Google Scholar]
- De Strooper B, Saftig P, Craessaerts K, Vanderstichele H, Guhde G, Annaert W, Von Figura K, Van Leuven F. Deficiency of presenilin-1 inhibits the normal cleavage of amyloid precursor protein. Nature. 1998;391:387–390. doi: 10.1038/34910. [DOI] [PubMed] [Google Scholar]
- Dewachter I, Ris L, Croes S, Borghgraef P, Devijver H, Voets T, Nilius B, Godaux E, Van Leuven F. Modulation of synaptic plasticity and Tau phosphorylation by wild-type and mutant presenilin1. Neurobiol Aging. 2008;29:639–652. doi: 10.1016/j.neurobiolaging.2006.11.019. [DOI] [PubMed] [Google Scholar]
- Emptage NJ, Reid CA, Fine A. Calcium stores in hippocampal synaptic boutons mediate short-term plasticity, store-operated Ca2+ entry, and spontaneous transmitter release. Neuron. 2001;29:197–208. doi: 10.1016/s0896-6273(01)00190-8. [DOI] [PubMed] [Google Scholar]
- Espinosa F, Kavalali ET. NMDA receptor activation by spontaneous glutamatergic neurotransmission. J Neurophysiol. 2009;101:2290–2296. doi: 10.1152/jn.90754.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng R, Rampon C, Tang YP, Shrom D, Jin J, Kyin M, Sopher B, Miller MW, Ware CB, Martin GM, Kim SH, Langdon RB, Sisodia SS, Tsien JZ. Deficient neurogenesis in forebrain-specific presenilin-1 knockout mice is associated with reduced clearance of hippocampal memory traces. Neuron. 2001;32:911–926. doi: 10.1016/s0896-6273(01)00523-2. [DOI] [PubMed] [Google Scholar]
- Fox AP, Nowycky MC, Tsien RW. Kinetic and pharmacological properties distinguishing three types of calcium currents in chick sensory neurones. J Physiol. 1987;394:149–172. doi: 10.1113/jphysiol.1987.sp016864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamenetz F, Tomita T, Hsieh H, Seabrook G, Borchelt D, Iwatsubo T, Sisodia S, Malinow R. APP processing and synaptic function. Neuron. 2003;37:925–937. doi: 10.1016/s0896-6273(03)00124-7. [DOI] [PubMed] [Google Scholar]
- LaFerla FM. Calcium dyshomeostasis and intracellular signalling in Alzheimer's disease. Nat Rev Neurosci. 2002;3:862–872. doi: 10.1038/nrn960. [DOI] [PubMed] [Google Scholar]
- Lou X, Scheuss V, Schneggenburger R. Allosteric modulation of the presynaptic Ca2+ sensor for vesicle fusion. Nature. 2005;435:497–501. doi: 10.1038/nature03568. [DOI] [PubMed] [Google Scholar]
- McKinney RA, Capogna M, Dürr R, Gähwiler BH, Thompson SM. Miniature synaptic events maintain dendritic spines via AMPA receptor activation. Nat Neurosci. 1999;2:44–49. doi: 10.1038/4548. [DOI] [PubMed] [Google Scholar]
- Morton RA, Kuenzi FM, Fitzjohn SM, Rosahl TW, Smith D, Zheng H, Shearman M, Collingridge GL, Seabrook GR. Impairment in hippocampal long-term potentiation in mice under-expressing the Alzheimer's disease related gene presenilin-1. Neurosci Lett. 2002;319:37–40. doi: 10.1016/s0304-3940(01)02512-5. [DOI] [PubMed] [Google Scholar]
- Panza F, Frisardi V, Imbimbo BP, Capurso C, Logroscino G, Sancarlo D, Seripa D, Vendemiale G, Pilotto A, Solfrizzi V. Review: gamma-secretase inhibitors for the treatment of Alzheimer's disease: the current state. CNS Neurosci Ther. 2010;16:272–284. doi: 10.1111/j.1755-5949.2010.00164.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parent AT, Barnes NY, Taniguchi Y, Thinakaran G, Sisodia SS. Presenilin attenuates receptor-mediated signaling and synaptic function. J Neurosci. 2005;25:1540–1549. doi: 10.1523/JNEUROSCI.3850-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ris L, Dewachter I, Reversé D, Godaux E, Van Leuven F. Capacitative calcium entry induces hippocampal long term potentiation in the absence of presenilin-1. J Biol Chem. 2003;278:44393–44399. doi: 10.1074/jbc.M300971200. [DOI] [PubMed] [Google Scholar]
- Rybalchenko V, Hwang SY, Rybalchenko N, Koulen P. The cytosolic N-terminus of presenilin-1 potentiates mouse ryanodine receptor single channel activity. Int J Biochem Cell Biol. 2008;40:84–97. doi: 10.1016/j.biocel.2007.06.023. [DOI] [PubMed] [Google Scholar]
- Schneggenburger R, Neher E. Intracellular calcium dependence of transmitter release rates at a fast central synapse. Nature. 2000;406:889–893. doi: 10.1038/35022702. [DOI] [PubMed] [Google Scholar]
- Sharma G, Vijayaraghavan S. Modulation of presynaptic store calcium induces release of glutamate and postsynaptic firing. Neuron. 2003;38:929–939. doi: 10.1016/s0896-6273(03)00322-2. [DOI] [PubMed] [Google Scholar]
- Shen J, Bronson RT, Chen DF, Xia W, Selkoe DJ, Tonegawa S. Skeletal and CNS defects in presenilin-1-deficient mice. Cell. 1997;89:629–639. doi: 10.1016/s0092-8674(00)80244-5. [DOI] [PubMed] [Google Scholar]
- Stevens CF, Sullivan JM. The synaptotagmin C2A domain is part of the calcium sensor controlling fast synaptic transmission. Neuron. 2003;39:299–308. doi: 10.1016/s0896-6273(03)00432-x. [DOI] [PubMed] [Google Scholar]
- Stutzmann GE. Calcium dysregulation, IP3 signaling, and Alzheimer's disease. Neuroscientist. 2005;11:110–115. doi: 10.1177/1073858404270899. [DOI] [PubMed] [Google Scholar]
- Sutton MA, Ito HT, Cressy P, Kempf C, Woo JC, Schuman EM. Miniature neurotransmission stabilizes synaptic function via tonic suppression of local dendritic protein synthesis. Cell. 2006;125:785–799. doi: 10.1016/j.cell.2006.03.040. [DOI] [PubMed] [Google Scholar]
- Tanzi RE, Bertram L. New frontiers in Alzheimer's disease genetics. Neuron. 2001;32:181–184. doi: 10.1016/s0896-6273(01)00476-7. [DOI] [PubMed] [Google Scholar]
- Thinakaran G, Borchelt DR, Lee MK, Slunt HH, Spitzer L, Kim G, Ratovitsky T, Davenport F, Nordstedt C, Seeger M, Hardy J, Levey AI, Gandy SE, Jenkins NA, Copeland NG, Price DL, Sisodia SS. Endoproteolysis of presenilin 1 and accumulation of processed derivatives in vivo. Neuron. 1996;17:181–190. doi: 10.1016/s0896-6273(00)80291-3. [DOI] [PubMed] [Google Scholar]
- Tu H, Nelson O, Bezprozvanny A, Wang Z, Lee SF, Hao YH, Serneels L, De Strooper B, Yu G, Bezprozvanny I. Presenilins form ER Ca(2+) leak channels, a function disrupted by familial Alzheimer's disease-linked mutations. Cell. 2006;126:981–993. doi: 10.1016/j.cell.2006.06.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolfe MS, Xia W, Ostaszewski BL, Diehl TS, Kimberly WT, Selkoe DJ. Two transmembrane aspartates in presenilin-1 required for presenilin endoproteolysis and gamma-secretase activity. Nature. 1999;398:513–517. doi: 10.1038/19077. [DOI] [PubMed] [Google Scholar]
- Yang Y, Cook DG. Presenilin-1 deficiency impairs glutamate-evoked intracellular calcium responses in neurons. Neuroscience. 2004;124:501–505. doi: 10.1016/j.neuroscience.2003.11.029. [DOI] [PubMed] [Google Scholar]
- Yoo AS, Cheng I, Chung S, Grenfell TZ, Lee H, Pack-Chung E, Handler M, Shen J, Xia W, Tesco G, Saunders AJ, Ding K, Frosch MP, Tanzi RE, Kim TW. Presenilin-mediated modulation of capacitative calcium entry. Neuron. 2000;27:561–572. doi: 10.1016/s0896-6273(00)00066-0. [DOI] [PubMed] [Google Scholar]
- Yu H, Saura CA, Choi SY, Sun LD, Yang X, Handler M, Kawarabayashi T, Younkin L, Fedeles B, Wilson MA, Younkin S, Kandel ER, Kirkwood A, Shen J. APP processing and synaptic plasticity in presenilin-1 conditional knockout mice. Neuron. 2001;31:713–726. doi: 10.1016/s0896-6273(01)00417-2. [DOI] [PubMed] [Google Scholar]
- Zhang C, Wu B, Beglopoulos V, Wines-Samuelson M, Zhang D, Dragatsis I, Südhof TC, Shen J. Presenilins are essential for regulating neurotransmitter release. Nature. 2009;460:632–636. doi: 10.1038/nature08177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zucker RS, Regehr WG. Short-term synaptic plasticity. Annu Rev Physiol. 2002;64:355–405. doi: 10.1146/annurev.physiol.64.092501.114547. [DOI] [PubMed] [Google Scholar]