

Abstract
Intracellular signaling through extracellular signal-regulated kinase (ERK) is important in regulating cellular functions in a variety of tissues including the CNS. Although ERK1 and ERK2 have a very similar substrate profile and amino acid sequences, there are strikingly different phenotypes between Erk1- and Erk2-deficient mice. Thus, the question arose as to whether these two proteins are functional homologs that compensate for each other, or whether they have distinct functions. Here, we generated double knock-out mice deficient for Erk2 in the CNS, with ubiquitous homozygous deletion of Erk1, and compared the phenotypes of these mice with those of monogenic Erk2-deficient mice. Although we did obtain double knock-out newborn pups, they survived for not >1 d. These pups appeared normal just after parturition. However, they had no milk in their stomachs even 6–7 h after birth. Intracerebral hemorrhages with varying location and severity were observed. The ventricular zones and corpus callosum of the double knock-out pups did not develop adequately. Neuronal size and nuclear morphology in some brain regions were markedly aberrant in the double knock-out pups compared with controls, while deficiency in Erk2 only caused a mild phenotype. These results suggest that total ERK1/2 activity governs cellular behaviors to ensure proper brain development.
Introduction
The extracellular signal-regulated kinase 1 (ERK1) and ERK2 belong to the mitogen-activated protein kinase family of intracellular signaling molecules that are evolutionarily conserved. The ERK cascade involves sequential activation of Ras, Raf, ERK kinase (MEK), and ERK, and is activated by extracellular stimuli. ERK was originally identified as an effector of growth factor signaling. However, the ERK cascade is not restricted to growth factor signaling, but transduces a broad range of extracellular stimuli into diverse intracellular responses.
ERK1 and ERK2 share 84% amino acid identity and have a very similar substrate profile (Boulton et al., 1991). Both are activated by phosphorylation via the upstream kinases MEK1 and MEK2. However, it is not established whether ERK1 and ERK2 are functional homologs. This question has been addressed by generating Erk1- and Erk2-deficient mice to overcome the nonspecific inhibition of ERK isoforms by MEK inhibitors. Despite the broad expression patterns of ERK1 and ERK2 (Corson et al., 2003), Erk1-deficient mice displayed only subtle phenotypic effects (Pagès et al., 1999; Selcher et al., 2001), while Erk2-deficient mice were embryonically lethal (Aouadi et al., 2006). This suggests that these proteins may play different functional roles during development. However, despite the strikingly different phenotypes between Erk1- and Erk2-deficient mice, there may be another hypothesis that ERK1 and ERK2 are functional homologs and compensate for each other. Indeed, there has been a report indicating that ERK1 phosphorylation levels in Erk2-deficient mice were higher than in controls (Samuels et al., 2008). This possibility can be examined by comparing mice deficient for both Erk1 and Erk2 in the CNS with monogenic knock-out mice.
In this study, we examined the effect of abrogation of both ERK1 and ERK2 in the CNS to identify putative major ERK functions in the CNS. These mice were neonatally lethal, and the pups demonstrated histological defects in the brain. Furthermore, Erk1 deficiency enhances the abnormal neurogenesis phenotype observed in Erk2-deficient mice. These results suggest that total ERK1/2 activity governs cellular behaviors to ensure proper brain development.
Materials and Methods
Erk2 conditional knock-out (Erk2 CKO) mice (Satoh et al., 2007; Imamura et al., 2008) and Erk1 knock-out mice (Pagès et al., 1999) were generated as described previously. Both Erk1 and Erk2 mutant mice were backcrossed with C57BL/6J for >10 generations. Animals were housed on a 12 h light/dark schedule under specific pathogen-free conditions. All experiments were conducted according to the institutional ethical guidelines for animal experiments and safety guidelines for gene manipulation experiments of the National Defense Medical College, and were approved by the Committee for Animal Research at the National Defense Medical College.
Histology.
After the animals were decapitated, the skulls were opened, and the heads were immersed in 0.1 m phosphate buffer containing 4% paraformaldehyde for at least 2 d. Then, the brain was removed from the skull. Paraffin sections, 5 μm thick, were prepared and stained either with hematoxylin-eosin or by the Bodian method.
Immunohistochemical analysis was performed as described previously (Satomoto et al., 2009). Briefly, antigenic retrieval was performed by immersing mounted tissue sections in Antigen Unmasking Solution (Vector Laboratories) and heating in an autoclave (121°C) for 5 min. Deparaffinized sections were blocked for endogenous peroxidase activity followed by blocking with a nonspecific staining blocking reagent (Dako) for 1 h to reduce background staining. The sections were then incubated with primary antibodies overnight in a humidified chamber at 4°C. The primary antibodies used included those against active caspase 3 (AC3) (rabbit polyclonal, 1:100; Cell Signaling Technology), glial fibrillary acidic protein (GFAP) (mouse monoclonal, 1:50; Sigma), NeuN (mouse monoclonal, 1:100; Millipore), Ki67 antibody (rabbit polyclonal, 1:500; Novocastra), TuJ1 (mouse monoclonal, 1:500; Covance), and 2′,3′-cyclic nucleotide 3′-phosphodiesterase (CNPase) (mouse monoclonal, 1:250; Sigma). Sections were incubated with secondary antibodies (En-Vision+ system; Dako) at room temperature for 1 h, detected using 3,3′-diaminobenzidine tetrachloride (Vector Laboratories), and counterstained with hematoxylin. For fluorescent staining, secondary antibodies used were Alexa-Fluor 488-conjugated goat anti-rabbit IgG (1:500; Invitrogen) and Cy3-conjugated goat anti-mouse IgG (1:500; Jackson Immunoresearch). The fluorescent sections were examined with a fluorescent microscope (TE-2000E; Nikon) equipped with an interlined charge-coupled device camera (DS-U1; Nikon).
Terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling (TUNEL) was performed using an in situ apoptosis detection kit (ApopTag fluorescein; Millipore Bioscience Research Reagents) according to the manufacturer's protocol. Sections were counterstained with DAPI.
Western blotting.
For examination of ERK expression, preparation of protein extracts was performed as previously described (Satoh et al., 2007). Briefly, after homogenization, the homogenate was centrifuged at 15,000 × g for 30 min at 4°C. The supernatants were immunoblotted using antibodies against ERK1/2 (rabbit polyclonal; Cell Signaling Technology) and β-actin (mouse monoclonal; Sigma). The protein bands were visualized by chemiluminescence (Super Signal West Pico; Pierce). The signals on the immunoblots were analyzed using an LAS3000 digital imaging system (Fujifilm).
Cell counting.
Cell number was assessed using a StereoInvestigator system (MicroBrightField). The boundaries were drawn using StereoInvestigator, and stained cells were counted within sampling frames chosen in a systematically random manner within the areas of interest. The number of cortical cells stained for NeuN or DAPI was counted in the dorsolateral portion of the cerebral cortex, from the retrosplenial cortex medially up to the rhinal fissure ventrolaterally.
Statistical analysis.
Statistical analysis was performed using the Student's t test, as indicated in the figures. In all graphs, the error bars indicate SD.
Results
Generation of the Erk1 and Erk2 double knock-out mice
To identify putative major ERK functions that could be compensated for in monogenic Erk knock-out lines, we generated double knock-out (DKO) mice deficient for Erk2 in the CNS with ubiquitous homozygous deletion of Erk1. To abrogate Erk2 in the CNS and to overcome the embryonic lethality of the Erk2 knock-out mice, we used a conditional, region-specific genetic approach. We used a nestin promoter-driven cre transgenic mouse line, in which cre recombinant activity is confined to the CNS (Imayoshi et al., 2006). The resulting Erk2 CKO mice (Erk2ΔCNS/ΔCNS) were viable and fertile. We crossed Erk1+/− mice with Erk2+/ΔCNS mice to obtain Erk1+/−; Erk2+/ΔCNS mice (double heterozygous mice). Double heterozygotes were then intercrossed to obtain Erk1−/−; Erk2ΔCNS/ΔCNS (Erk1/2 DKO) mice. We failed to detect Erk1/2 DKO mice on postnatal day 2 (P2). We detected mice with other genotypes in a nearly Mendelian distribution.
Western blot analysis of forebrain at P0 showed that the Erk1/2 DKO mice had no detectable ERK1 or ERK2 protein (Fig. 1B). The amount of protein in mice heterozygous for the null allele was approximately half of that in the wild types.
Figure 1.
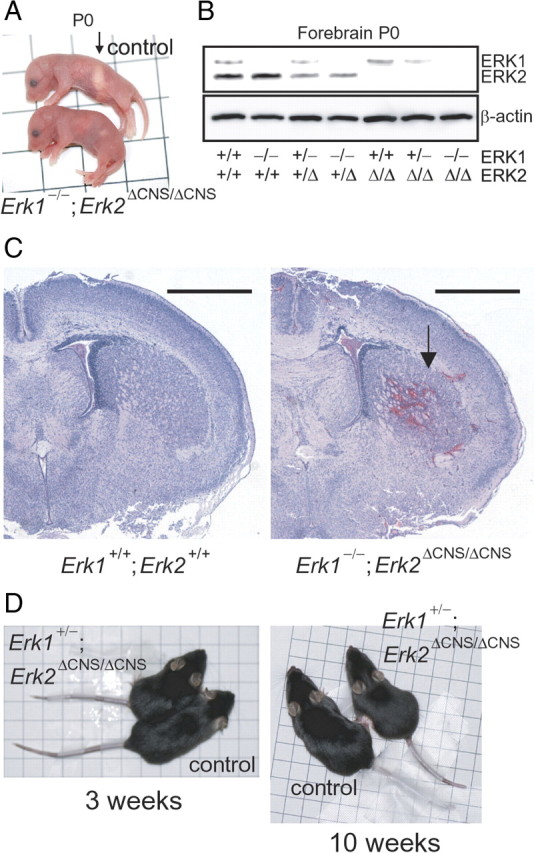
Neonatal death in Erk1/2 DKO mice. A, General appearance of mice deficient for Erk2 in the CNS with ubiquitous homozygous deletion of Erk1 (Erk1−/−; Erk2ΔCNS/ΔCNS) and wild types (control) at P0. Whereas control mice had milk in their stomachs (arrow), no milk was observed in the stomachs of the mutants. One side of each square represents 1 cm. B, Western blot analysis of ERK1 and ERK2 in the forebrain of compound heterozygous/homozygous Erk1 and Erk2 mutant mice at P0. C, Representative coronal sections from Erk1/2 DKO (Erk1−/−; Erk2ΔCNS/ΔCNS) and control (Erk1+/+; Erk2+/+) brains at P0. The arrow indicates an intracranial hemorrhage. Scale bars, 1 mm. D, General appearance of controls and mutants (Erk1+/−; Erk2ΔCNS/ΔCNS) at 3 and 10 weeks of age. Mutant mice were slightly smaller than controls. One side of each square represents 1 cm.
Neonatal death of Erk1/2 DKO mice
To further examine the Erk1−/−; Erk2ΔCNS/ΔCNS mice, various crosses (Erk1+/−; Erk2+/ΔCNS × Erk1+/−; Erk2+/f, Erk1−/−; Erk2+/ΔCNS × Erk1−/−; Erk2f/f, Erk1−/−; Erk2+/ΔCNS × Erk1−/−; Erk2+/ΔCNS, Erk1+/−; Erk2ΔCNS/ΔCNS × Erk1+/−; Erk2f/f and Erk1+/−; Erk2+/ΔCNS × Erk1−/−; Erk2+/ΔCNS) were carried out. Although newborn Erk1/2 DKO mice were obtained in all crosses, all (n = 120) died within 1 d. Erk1/2 DKO pups were reddish and appeared normal just after parturition, suggesting that the cardiovascular and respiratory systems were functional at birth. However, they had no milk in their stomachs even 6–7 h after birth, which was not observed for the other genotypes (Fig. 1A). These mice also suffered from intracerebral hemorrhages, which were not observed for the other genotypes (Fig. 1C).
When one allele of Erk2 was absent in the CNS together with ubiquitous deletion of Erk1 (Erk1−/−; Erk2+/ΔCNS), mice remained viable and fertile. Even when one allele of Erk1 was absent together with complete deletion of Erk2 in the CNS (Erk1+/−; Erk2ΔCNS/ΔCNS), mice remained viable and fertile, although they exhibited growth retardation compared with littermate controls (Fig. 1D). The Erk1+/−; Erk2ΔCNS/ΔCNS mutant mice were significantly lighter in weight than littermate controls at 3 weeks of age (controls, 8.7 ± 0.4 g; mutants, 6.9 ± 0.2 g; n = 7 for each, t test, t = 3.93, p < 0.01), and at 10 weeks of age (controls, 23.0 ± 0.7 g; mutants, 19.8 ± 0.8 g; n = 7 for each, t test, t = 2.98, p < 0.05).
The ventricular zone in Erk1/2 DKO mice is thinner than in Erk2 CKO or control mice
The ERK pathway plays critical roles in neural differentiation of cortical neural progenitor cells (NPCs) (Paquin et al., 2005). Precise regulation of progenitor cell differentiation is important for brain development because it determines cell number and laminar fate (Caviness et al., 1995; McConnell, 1998). During brain development, neurons are generated from dividing progenitors of the neocortical ventricular zone (VZ) (Tan et al., 1998; Rakic, 2002). We observed that, although the VZ was present in Erk1/2 DKO mice, it was substantially thinner than in Erk2 CKO or control mice. At embryonic day 18.5 (E18.5), we did not observe a prominent abnormality in the VZs of Erk2 CKO mice (Fig. 2A,B). However, the VZs of Erk1/2 DKO mice were markedly thinner than in Erk2 CKO or control mice. Similarly, at P0 the VZ of Erk1/2 DKO mice was substantially thinner (Fig. 2C–E) than in Erk2 CKO or control mice. Measurements of VZ thickness revealed a highly significant difference between Erk1/2 DKO and control mice at P0 (DKO vs control, n = 5 mice for each, t test, t = 11.08, p < 0.001) (Fig. 2J), suggesting a drastic reduction in the number of NPCs in the VZ of Erk1/2 DKO mice.
Figure 2.
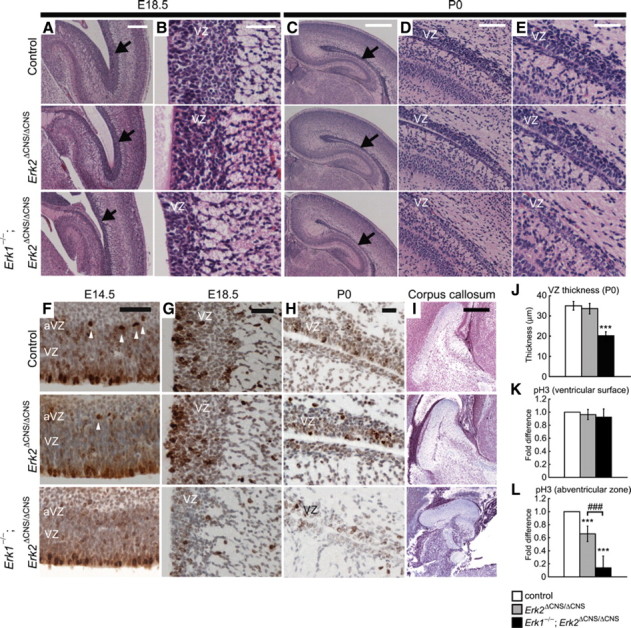
The VZ is thinner in Erk1/2 DKO mice. A, Comparison of sagittal sections at E18.5 shows that the VZ is markedly thinner in Erk1/2 DKO mice than in Erk2 CKO or control mice. Arrows indicate the VZs. B, Higher-power views of the VZs. C, Similar to at E18.5, comparison of coronal sections at P0 shows that the VZ is markedly thinner in Erk1/2 DKO than in Erk2 CKO or control mice. Arrows indicate the VZs. D, E, Higher-power views of the VZs. These sections show that the VZ was markedly thinner in Erk1/2 DKO mice. F, The majority of pH3+ cells were found on the ventricular surface in all genotypes at E14.5. In Erk2 CKO mice, the number of pH3+ cells in the abventricular zone (arrowheads) was reduced compared with controls. In Erk1/2 DKO mice, a more dramatic decrease was observed compared with controls or Erk2 CKO mice. G, At E18.5, pH3 staining was decreased in Erk1/2 DKO mice, but not in Erk2 CKO mice, compared with controls. H, At P0, staining for pH3 shows decreased proliferation in the VZ of Erk1/2 DKO mice, but not in Erk2 CKO mice, compared with controls. I, The size of the corpus callosum is reduced in Erk1/2 DKO mice compared with Erk2 CKO or control mice. J, The thickness of the VZ is reduced in Erk1/2 DKO mice compared with controls. K, L, The fold differences in the numbers of pH3+ cells in the ventricular surface and in the abventricular zone are calculated (n = 5 mice for each). J, The numbers of pH3+ cells in the ventricular surface are not different between genotypes. L, On the other hand, the numbers of pH3+ cells in the abventricular zone are significantly reduced in Erk2 CKO and Erk1/2 DKO mice compared with controls. Scale bars: C, I, 500 μm; A, 250 μm; D, 100 μm; B, E–H, 50 μm. ***p < 0.001 compared with control; ###p < 0.001.
The corpus callosum is a major fiber tract formed between ∼16 and 18 d post coitum in the mouse (Ozaki and Wahlsten, 1998), which connects the two hemispheres of the brain. We observed a reduction in the size of the corpus callosum of Erk1/2 DKO mice (Fig. 2R), compared with that of Erk2 CKO (Fig. 2Q) or control (Fig. 2I) mice at P0.
Proliferation is decreased in the VZ of Erk1/2 DKO mice
To investigate a possible cause of the reduced thickness of the VZ in Erk1/2 DKO mice, we assessed the proliferation of NPCs in the embryonic VZ. At E14.5, the peak of the neurogenic period, staining of brain for TuJ1, an early pan-neuronal marker, showed no prominent differences in the proliferative zones between the genotypes (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). We next evaluated the number of neuronal progenitors undergoing mitosis by pH3 immunostaining, which marks mitotic cells during the G2 and M phases of the cell cycle (Fig. 2F). The majority of pH3+ cells were found on the ventricular surface in all genotypes at E14.5. There was no significant difference in the number of pH3+ cells in the ventricular surface in Erk1/2 DKO mice (DKO vs control, n = 5 mice for each, t test, t = 1.37, p > 0.05) (Fig. 2K). Similarly, no prominent differences were found in the immunoreactivity for Ki67, a protein expressed in all dividing cells (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). However, the number of pH3+ cells in the abventricular zone (the basal side of the VZ) was reduced in Erk2 CKO mice compared with that in controls (CKO vs control, n = 5 mice for each, t test, t = 6.21, p < 0.001) (Fig. 2L). Furthermore, a more dramatic decrease in the number of pH3+ cells in the abventricular zone was observed in Erk1/2 DKO mice compared with controls or Erk2 CKO mice (DKO vs control, n = 5 mice for each, t test, t = 10.75, p < 0.001; DKO vs CKO, n = 5 mice for each, t test, t = 5.41, p < 0.001) (Fig. 2L). At E18.5, we found that the total number of pH3+ cells was significantly reduced in the VZ of Erk1/2 DKO mice compared with that in controls (Fig. 2G). Similarly, we found a dramatic decrease in the number of Ki67+ cells in the VZ of Erk1/2 DKO mice at E18.5 (supplemental Fig. 2, available at www.jneurosci.org as supplemental material). At P0, the number of pH3+ cells was significantly reduced in the VZ of Erk1/2 DKO mice compared with that in controls (Fig. 2H).
Erk1 deficiency enhanced the abnormal neurogenesis phenotype in Erk2-deficient mice at P0
A previous report indicated that loss of ERK2 leads to a delay in the progression of NPCs to neurons during the neurogenic period (Samuels et al., 2008). We found that cell type specification was changed in Erk2 CKO and Erk1/2 DKO mice (supplemental Figs. 3, 4, available at www.jneurosci.org as supplemental material). Furthermore, we found that neurons were generated to form each subdivision of the brain in Erk1/2 DKO mice, but that the neurons in some subdivisions were less developed than in controls and Erk2 CKO mice. These neurons were characterized by darkly stained, small, round nuclei with scant perinuclear cytoplasm. Figure 3 shows examples of the histological findings at P0. The parietal isocortex and the anterior cingulate cortex in controls exhibited laminar differentiation (Fig. 3A–C): layer V in the parietal isocortex contained typical pyramidal neurons with a triangular appearance (Fig. 3B). Their nuclei showed differentiation of hetero- and euchromatin; some of them exhibited marked nucleoli. These findings indicate that the pyramidal neurons were undergoing normal maturation processes. In Erk1/2 DKO mice, most of the cortical neurons were small and contained darkly stained, small, round nuclei that are typical of undifferentiated neurons (Fig. 3R,S). Laminar differentiation was hardly discernible except for as differences in the neuronal density. Similarly, in Erk2 CKO mice, pyramidal neurons with a triangular appearance were hardly observed and laminar differentiation was hardly discernible except for as differences in the neuronal density (Fig. 3I–K).
Figure 3.
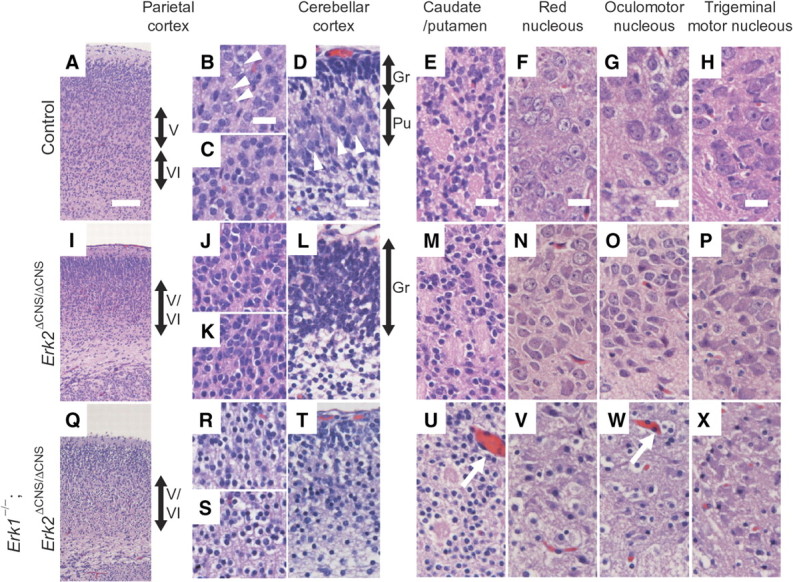
Erk1 deficiency enhanced the abnormal neurogenesis phenotype in Erk2-deficient mice at P0. A, In control mice, the parietal isocortex exhibits some laminar differentiation. B, C, While layer V of the parietal isocortex contains typical pyramidal neurons with a triangular appearance (B, arrowheads), layer VI contains small cells (C). I, Q, In Erk2 CKO (I) and Erk1/2 DKO (Q) mice, laminar differentiation is barely discernible except for as differences in the neuronal density. J, R, K, Q, Neither the outer (J, R) nor the inner (K, Q) parts of layers V and VI contains typical pyramidal neurons. D, The cerebellar cortex in controls showed large neurons representing immature Purkinje neurons (arrowheads) in the Purkinje cell layer (Pu) subjacent to the external granular layer (Gr). L, In Erk2 CKO mice, although the granular layer (Gr) is observed, the Purkinje cell layer is not. T, In Erk1/2 DKO mice, almost all the neurons are small and contain darkly stained, small, round nuclei. Only slight laminar differentiation is observed as differences in the density of neurons. E, M, Neurons in caudate/putamen are larger and contained well developed nuclei, with differentiation between heterochromatin and euchromatin in control (E) and Erk2 CKO (M) mice. U, In Erk1/2 DKO mice, some neurons are small with undifferentiated nuclei. Note that capillary telangiectasia is observed in Erk1/2 DKO mice (U, W, arrows). F–H, N–P, V–X, The brainstem nuclei show even greater differences between control and Erk1/2 DKO mice. The nuclei in controls show much larger neurons, with abundant cytoplasm and well differentiated nuclei (F–H), while those in Erk1/2 DKO mice contain smaller neurons with thinner cytoplasm and darkly stained, small, round nuclei (V–X). N–P, Neurons in Erk2 CKO mice do not exhibit significant impairment, except that some cells were smaller than those in control. Scale bars: A, 100 μm; B–H, 20 μm.
The cerebellar cortex showed the most striking differences between controls and Erk1/2 DKO mice (Fig. 3D,L,T). In controls, large neurons representing immature Purkinje neurons were observed in the Purkinje cell layer subjacent to the external granular layer (Fig. 3D). In contrast, in Erk1/2 DKO mice, almost all the neurons were small and contained darkly stained, small, round nuclei (Fig. 3T). Again, only slight laminar differentiation was observed, as a differing density of neurons (Fig. 3T). Erk2 CKO mice displayed the granular layer, but the Purkinje cell layer was not observed (Fig. 3L).
The marked differences in neuronal size and nuclear morphology in Erk1/2 DKO mice were also recognizable in the caudate nucleus (Fig. 3E,M,U). Neurons in the wild-type mice were large and contained well developed nuclei with differentiation between heterochromatin and euchromatin (Fig. 3E). However, neurons in Erk1/2 DKO mice contained small neurons with undifferentiated nuclei (Fig. 3U). Neurons in Erk2 CKO mice did not exhibit significant impairment (Fig. 3M). We also detected some capillary telangiectasia in Erk1/2 DKO mice, although the reason for this was unclear (Fig. 3U,W). The brainstem nuclei with larger, thus earlier developing, neurons showed even greater differences between wild-type and Erk1/2 DKO mice. Figure 3 depicts the red nucleus (Fig. 3F,N,V), an example of the somatic nucleus (oculomotor nucleus) (Fig. 3G,O,W), and an example of the branchial motor nucleus (trigeminal motor nucleus) (Fig. 3H,P,X). The nuclei in the wild-type animals showed much larger neurons, with abundant cytoplasm and well differentiated nuclei (Fig. 3F–H), while those in Erk1/2 DKO mice contained smaller neurons with thinner cytoplasm and darkly stained, small, round nuclei (Fig. 3V–X). Neurons in Erk2 CKO mice did not exhibit significant impairment, although some neurons in Erk2 CKO mice seemed to be smaller than those in controls (Fig. 3N–P). The neurons in the red nucleus and trigeminal motor nucleus of Erk1/2 DKO and Erk2 CKO mice were significantly smaller than those in controls (supplemental Fig. 5, available at www.jneurosci.org as supplemental material). Staining for NeuN, a marker for mature neurons showed that, in the red nucleus, large neurons with robust staining for NeuN were observed in controls (supplemental Fig. 5, available at www.jneurosci.org as supplemental material). On the other hand, the number of large neurons and robust staining for NeuN was significantly lower in Erk2 CKO mice compared with controls. Large neurons were hardly discernible, and only faint staining for NeuN was observed in Erk1/2 DKO mice.
Increased apoptosis in Erk1/2 DKO mice
As a previous study indicated that ERK signaling is implicated in promoting neuronal survival in culture (Xia et al., 1995), we investigated whether cell survival was affected in Erk1/2 DKO mice. In Erk1/2 DKO mice at P0, we observed significantly increased AC3 staining compared with that in controls (Fig. 4A,C,D,F). Figure 4, C and F, shows that the increased AC3 staining was most robust in the retrosplenial cortex and thalamus. There was no difference between Erk2 CKO and control mice (Fig. 4A,B,D,E). TUNEL in the retrosplenial cortex showed a similar pattern of apoptosis to that revealed by AC3 staining (supplemental Fig. 6, available at www.jneurosci.org as supplemental material). We next performed double labeling with AC3 and cell type-specific markers to address whether the increased apoptosis occurred specifically in the neuronal cells (supplemental Fig. 7, available at www.jneurosci.org as supplemental material). We found AC3+ cells labeled by NeuN. However, we also found AC3+ cells labeled by the astrocyte-specific marker GFAP or the oligodendrocyte maturation marker CNPase. These results suggest that apoptosis in Erk1/2 DKO mice was not cell type specific.
Figure 4.

Severe apoptosis is observed in Erk1/2 DKO mice at P0. Sections were immunohistochemically stained for activated caspase 3 (AC3). A–C, Thalamus (th) and retrosplenial cortex (rs) were heavily stained for AC3 in Erk1/2 DKO mice (C) compared with Erk2 CKO (B) or control (A) mice. D–F, Higher-magnification images of the retrosplenial cortex. Scale bars: A–C, 1 mm; D–F, 100 μm.
Although the increase in apoptosis was prominent in Erk1/2 DKO mice, the pattern of apoptosis seemed to be region specific. For instance, apoptosis was not increased in the red nucleus of Erk1/2 DKO mice compared with controls (supplemental Fig. 5, available at www.jneurosci.org as supplemental material). The numbers of AC3+ apoptotic cells in the VZ were low, and no differences were observed between genotypes at E14.5 and E18.5 (supplemental Figs. 1, 2, available at www.jneurosci.org as supplemental material). Similarly, at P0, we did not observe a prominent difference in apoptosis in the VZ between genotypes, although a prominent increase in apoptosis was observed in regions outside the VZ (supplemental Fig. 8, available at www.jneurosci.org as supplemental material). These findings suggest that inactivation of both ERK1 and ERK2 did not provoke apoptosis in the VZ throughout the neurogenic period.
Discussion
The cause of lethality in Erk1/2 DKO mice
Because of the strikingly different phenotypes between Erk1 and Erk2 mice, the question arose as to whether these two proteins are actually functional homologs. In this report, we generated double knock-out mice deficient for Erk2 in the CNS with ubiquitous homozygous deletion of Erk1, and compared these mice with monogenic Erk2-deficient mice. We found that all Erk1/2 DKO mice died within a day and that these pups had intracranial hemorrhages. The hemorrhages, however, are unlikely to represent the major cause of lethality because they were, in some cases, very mild, and were generally insufficient to cause death as a result of brain herniation. Furthermore, the hemorrhages rarely involved the vital structures of the hindbrain, and did not correlate with the impairments in neurogenesis or increased apoptosis. Indeed, severe impairments of neurogenesis in some cases occurred in the absence of hemorrhage. In addition, whereas the location of severe apoptosis was region specific, the hemorrhages occurred in random locations.
Instead, the cause of lethality might be an impairment of the sucking response, because Erk1/2 DKO mice had no milk in their stomachs even 6–7 h after birth.
Sucking behavior, such as nipple attachment and sucking movements, is regulated by complex interactions between neurons both in the sensory and motor systems, particularly related to the trigeminal and facial nuclei. At birth, the trigeminal motor nucleus of Erk1/2 DKO mice contained smaller neurons with thinner cytoplasm and darkly stained, small, round nuclei compared with those in wild types. We speculate that such a deficiency may underlie the impairment in the sucking response. Further analysis is needed to understand the cause of lethality in Erk1/2 DKO mice.
The role of ERKs in neurogenesis
A previous report has suggested that ERK2 is required for neural cell fate determination (Samuels et al., 2008). Our findings indicated that the deficits in neurogenesis were more severe in Erk1/2 DKO mice than in Erk2 CKO mice. Furthermore, our findings suggested a positive role for ERK1 in neural cell differentiation when ERK2 was abrogated, although monogenic abrogation of ERK1 has little effect (Pagès et al., 1999). Thus, it is possible that the minimal effect of ERK1 abrogation alone results from a nominal net reduction in overall ERK activity, because ERK1 is less abundant than ERK2 in neurons (Lefloch et al., 2009).
Samuels et al. (2008) reported that suppression of NPC proliferation as a consequence of ERK2 inactivation was transient because of the compensatory increase in ERK1 activity. On the other hand, inhibition of MEK1 caused NPCs to remain in an undifferentiated state, blocking neurogenesis (Paquin et al., 2005). Thus, the effect of MEK abrogation on neurogenesis seems to be more severe than the loss of ERK2. This is consistent with our findings that the deficits in Erk1/2 DKO mice were more severe than those in monogenic Erk2 CKO mice, and our findings support the hypothesis that total ERK1/2 activity governs cellular behavior (Lefloch et al., 2009).
However, it has been reported that ERK2 was selectively activated in area CA1 in hippocampal long-term potentiation (English and Sweatt, 1996), suggesting some important differences between the functions of ERK1 and ERK2. Further examination will be required to elucidate the mechanisms of redundancy and independence of ERK1/2 function.
At E14.5, a prominent decrease was observed in the number of pH3+ cells in the abventricular zone of Erk1/2 DKO mice compared with that in controls. A previous study identified pH3+ cells in the abventricular zone as intermediate progenitor cells (IPCs) (Samuels et al., 2008). IPCs are a type of transient amplifying cells that are produced from NPCs. IPCs divide at the abventricular zone and migrate to the subventricular zone (SVZ) where they produce only neurons. At E18.5, proliferation was decreased in the VZ of Erk1/2 DKO mice without apoptotic cell death. Thus, abrogation of both ERK1 and ERK2 might result in the decreased proliferation of progenitors, leading to their depletion in the VZ/SVZ. In Erk2 CKO mice, a compensatory increase in ERK1 activity might restore progenitors in the VZ.
In conclusion, our results show that abrogation of both ERK1 and ERK2 in the CNS caused impaired neurogenesis and apoptotic degeneration, leading to neonatal death. Histological analysis demonstrated that Erk1 deficiency enhances the abnormal neurogenesis phenotype in Erk2 CKO mice. The ERK pathway also plays important roles in the development of the VZ and the corpus callosum. These results strongly suggest that total ERK1/2 activity governs proper neurogenesis.
Footnotes
We thank Dr. Shogo Endo for the critical discussion regarding this work. We also thank Ms. Masako Suzuki and Kiyoko Takamiya for their excellent technical help and Dr. Kouichi Fukuda for his assistance with animal husbandry. Nestin-cre transgenic mice were a kind gift from Dr. Ryoichiro Kageyama (University of Kyoto, Kyoto, Japan).
References
- Aouadi M, Binetruy B, Caron L, Le Marchand-Brustel Y, Bost F. Role of MAPKs in development and differentiation: lessons from knockout mice. Biochimie. 2006;88:1091–1098. doi: 10.1016/j.biochi.2006.06.003. [DOI] [PubMed] [Google Scholar]
- Boulton TG, Nye SH, Robbins DJ, Ip NY, Radziejewska E, Morgenbesser SD, DePinho RA, Panayotatos N, Cobb MH, Yancopoulos GD. ERKs: a family of protein-serine/threonine kinases that are activated and tyrosine phosphorylated in response to insulin and NGF. Cell. 1991;65:663–675. doi: 10.1016/0092-8674(91)90098-j. [DOI] [PubMed] [Google Scholar]
- Caviness VS, Jr, Takahashi T, Nowakowski RS. Numbers, time and neocortical neuronogenesis: a general developmental and evolutionary model. Trends Neurosci. 1995;18:379–383. doi: 10.1016/0166-2236(95)93933-o. [DOI] [PubMed] [Google Scholar]
- Corson LB, Yamanaka Y, Lai KM, Rossant J. Spatial and temporal patterns of ERK signaling during mouse embryogenesis. Development. 2003;130:4527–4537. doi: 10.1242/dev.00669. [DOI] [PubMed] [Google Scholar]
- English JD, Sweatt JD. Activation of p42 mitogen-activated protein kinase in hippocampal long term potentiation. J Biol Chem. 1996;271:24329–24332. doi: 10.1074/jbc.271.40.24329. [DOI] [PubMed] [Google Scholar]
- Imamura O, Satoh Y, Endo S, Takishima K. Analysis of ERK2 function in neural stem/progenitor cells via nervous system-specific gene disruption. Stem Cells. 2008;26:3247–3256. doi: 10.1634/stemcells.2008-0578. [DOI] [PubMed] [Google Scholar]
- Imayoshi I, Ohtsuka T, Metzger D, Chambon P, Kageyama R. Temporal regulation of Cre recombinase activity in neural stem cells. Genesis. 2006;44:233–238. doi: 10.1002/dvg.20212. [DOI] [PubMed] [Google Scholar]
- Lefloch R, Pouysségur J, Lenormand P. Total ERK1/2 activity regulates cell proliferation. Cell Cycle. 2009;8:705–711. doi: 10.4161/cc.8.5.7734. [DOI] [PubMed] [Google Scholar]
- McConnell SK. Development and decision-making in the mammalian cerebral cortex. Brain Res Rev. 1998;13:1–23. doi: 10.1016/0165-0173(88)90002-1. [DOI] [PubMed] [Google Scholar]
- Ozaki HS, Wahlsten D. Timing and origin of the first cortical axons to project through the corpus callosum and the subsequent emergence of callosal projection cells in mouse. J Comp Neurol. 1998;400:197–206. doi: 10.1002/(sici)1096-9861(19981019)400:2<197::aid-cne3>3.0.co;2-4. [DOI] [PubMed] [Google Scholar]
- Pagès G, Guérin S, Grall D, Bonino F, Smith A, Anjuere F, Auberger P, Pouysségur J. Defective thymocyte maturation in p44 MAP kinase (Erk 1) knockout mice. Science. 1999;286:1374–1377. doi: 10.1126/science.286.5443.1374. [DOI] [PubMed] [Google Scholar]
- Paquin A, Barnabé-Heider F, Kageyama R, Miller FD. CCAAT/enhancer-binding protein phosphorylation biases cortical precursors to generate neurons rather than astrocytes in vivo. J Neurosci. 2005;25:10747–10758. doi: 10.1523/JNEUROSCI.2662-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rakic P. Neurogenesis in adult primate neocortex: an evaluation of the evidence. Nat Rev Neurosci. 2002;3:65–71. doi: 10.1038/nrn700. [DOI] [PubMed] [Google Scholar]
- Samuels IS, Karlo JC, Faruzzi AN, Pickering K, Herrup K, Sweatt JD, Saitta SC, Landreth GE. Deletion of ERK2 mitogen-activated protein kinase identifies its key roles in cortical neurogenesis and cognitive function. J Neurosci. 2008;28:6983–6995. doi: 10.1523/JNEUROSCI.0679-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satoh Y, Endo S, Ikeda T, Yamada K, Ito M, Kuroki M, Hiramoto T, Imamura O, Kobayashi Y, Watanabe Y, Itohara S, Takishima K. Extracellular signal-regulated kinase 2 (ERK2) knockdown mice show deficits in long-term memory; ERK2 has a specific function in learning and memory. J Neurosci. 2007;27:10765–10776. doi: 10.1523/JNEUROSCI.0117-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satomoto M, Satoh Y, Terui K, Miyao H, Takishima K, Ito M, Imaki J. Neonatal exposure to sevoflurane induces abnormal social behaviors and deficits in fear conditioning in mice. Anesthesiology. 2009;110:628–637. doi: 10.1097/ALN.0b013e3181974fa2. [DOI] [PubMed] [Google Scholar]
- Selcher JC, Nekrasova T, Paylor R, Landreth GE, Sweatt JD. Mice lacking the ERK1 isoform of MAP kinase are unimpaired in emotional learning. Learn Mem. 2001;8:11–19. doi: 10.1101/lm.37001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan SS, Kalloniatis M, Sturm K, Tam PP, Reese BE, Faulkner-Jones B. Separate progenitors for radial and tangential cell dispersion during development of the cerebral neocortex. Neuron. 1998;21:295–304. doi: 10.1016/s0896-6273(00)80539-5. [DOI] [PubMed] [Google Scholar]
- Xia Z, Dickens M, Raingeaud J, Davis RJ, Greenberg ME. Opposing effects of ERK and JNK-p38 MAP kinases on apoptosis. Science. 1995;270:1326–1331. doi: 10.1126/science.270.5240.1326. [DOI] [PubMed] [Google Scholar]