

Abstract
The GluA2 (GluR2) subunit is critical for the regulation of AMPA receptor properties and synaptic plasticity, but the underlying mechanisms remain unclear. Here, we demonstrate that GluA2 regulates metabotropic glutamate receptor-dependent long-term depression (mGluR-LTD) through a previously unknown mechanism involving N-cadherin-dependent and cofilin-mediated actin reorganization. We show that GluA2 is indispensable for mGluR-LTD in the hippocampus, and surprisingly this action of GluA2 is mediated by its extracellular domain interaction with N-cadherin. Accordingly, we show that the function of N-cadherin is regulated by and required for mGluR-LTD. Furthermore, we show that the regulatory effect of GluA2/N-cadherin is mediated through activation of Rho GTPase Rac1 and its downstream actin regulator cofilin, and, importantly, the requirement for GluA2/N-cadherin can be overcome by manipulating cofilin. These results provide compelling evidence that the extracellular domain of GluA2 regulates long-lasting synaptic plasticity through a signaling mechanism that is distinct from those used by the other domains of the receptor subunit.
Introduction
AMPA subtype glutamate receptors (AMPARs) are the principle mediators of fast excitatory synaptic transmission and are important for the expression of various forms of long-lasting synaptic plasticity, including long-term potentiation (LTP) and long-term depression (LTD), widely studied cellular models for learning and memory (Malenka and Bear, 2004; Lüscher and Huber, 2010). In hippocampal CA1 pyramidal neurons, most AMPARs are heteromeric complexes assembled from GluA1 and GluA2 (also referred to as GluR1 and GluR2) (Hollmann and Heinemann, 1994; Collingridge et al., 2009; Lu et al., 2009). The GluA2 subunit is particularly interesting because it determines a number of important properties of AMPARs, including Ca2+ permeability, single-channel conductance, and receptor trafficking at the synapse (Isaac et al., 2007). The role of GluA2 in AMPAR trafficking has attracted special attention because it provides a key mechanism for regulating the number of synaptic receptors and plasticity (Malinow and Malenka, 2002; Bredt and Nicoll, 2003; Collingridge et al., 2004; Shepherd and Huganir, 2007; Kerchner and Nicoll, 2008). Thus, genetic disruptions of GluA2 function have been shown to cause dramatic changes in AMPAR functionality and synaptic properties (Jia et al., 1996; Gerlai et al., 1998; Yan et al., 2002; Shimshek et al., 2006a,b; Panicker et al., 2008).
The exact mechanisms by which GluA2 regulates AMPAR trafficking and synaptic plasticity are not clear. However, it is known that GluA2, via its N-terminal domain (NTD), transmembrane domain (TM), and C-terminal domain (CTD), can interact with several subunit- and domain-specific proteins, and it is thought through these specific interactions that GluA2 exerts its unique regulatory effects on AMPAR properties (Isaac et al., 2007). In particular, GluA2 CTD protein interactions with protein interacting with C kinase-1 (PICK1), N-ethylmaleimide-sensitive factor (NSF), and AMPAR-binding protein (ABP)/glutamate receptor interacting protein (GRIP) have attracted the most attention because of their demonstrated importance in regulating AMPAR internalization and the expression of several forms of LTD (Isaac et al., 2007; Lüscher and Huber, 2010). The GluA2 NTD is known to interact with the cell adhesion molecule N-cadherin to regulate spine properties, but the functional significance of this interaction in synaptic plasticity remains unknown (Passafaro et al., 2003; Saglietti et al., 2007).
In this study, we investigated the role and underlying mechanisms of the NTD of GluA2 in the regulation of synaptic plasticity at hippocampal CA1 synapses. We show that the GluA2 NTD/N-cadherin interaction is critically required for metabotropic glutamate receptor-dependent LTD (mGluR-LTD), and that this action is mediated by cofilin-dependent actin reorganization. Thus, regulation of actin-based process by the GluA2/N-cadherin interaction provides an additional mechanism by which AMPARs regulate long-lasting synaptic plasticity at the central synapse.
Materials and Methods
GluA2 and LIM kinase-1 knock-out mice.
The generation and initial characterization of GluA2 knock-out (KO), GluA3 KO, and LIM kinase-1 (LIMK-1) KO mice were described previously (Jia et al., 1996; Meng et al., 2002, 2003). To generate LIMK-1/GluA2 double KO mice, we crossed GluA2+/− with LIMK-1+/− to obtain the double heterozygotes GluA2+/−/LIMK-1+/−, which were subsequently interbred to yield GluA2−/−/LIMK-1−/− double KO mice. The double KO mice were viable and showed a similar phenotype as the GluA2 KO mice in growth, viability, and home-cage behavior. Whenever possible, the investigators were blind to the mouse genotypes used for the experiments.
Electrophysiology.
The mouse brains from various genotypes were quickly removed and sagittal 300–400 μm hippocampal slices prepared in ice-cold artificial CSF (ACSF) saturated with 95% O2/5% CO2. ACSF contained the following (in mm): 120 NaCl, 3.0 KCl, 1.2 MgSO4, 1.0 NaH2PO4, 26 NaHCO3, 2.0 CaCl2, and 11 d-glucose. The slices were recovered at room temperature for at least 2 h before a single slice was transferred to a submersion chamber perfused with 95% O2/5% CO2 saturated ACSF with (for whole-cell recordings) or without (for field recordings) 100 μm picrotoxin. Hippocampal CA1 neurons were visualized using an infrared differential interference contrast microscope (Axioscope 2, Zeiss). Synaptic transmission was evoked by stimulation (at 0.05 Hz for field recordings or 0.1 Hz for whole-cell recordings) of Schaffer collaterals and was recorded with glass pipettes (3–4 MΩ) filled with either ACSF (for field responses) or with an intracellular solution (for whole-cell response) containing the following (in mm): 130 CsMeSO4, 5 NaCl, 1 MgCl2, 0.05 EGTA, 10 HEPES, 3 Mg-ATP, 0.3 Na3GTP, and 5 QX-314, pH 7.25 (280–300 mOsm). Two protocols were used to induce mGluR-LTD: bath application of 100 μm (RS)-3.5-DHPG (3,5-dihydroxyphenylglycine) (DHPG-induced LTD) for 10 min or paired-pulse low-frequency stimulation consisting of 900 pairs of stimuli with 50 ms interstimulus interval delivered at 1 Hz (PP-LFS-induced LTD) in the presence of 100 μm NMDA receptor antagonist dl-APV. LTD was calculated and statistically evaluated by comparing the mean values of the last 10 min of the recording and the mean values of the entire baseline. Whole-cell series resistance was monitored throughout LTD experiments by applying a −3 mV step at the end of each response, and depending on whether it changed by >20% the experiment was excluded from analysis. For whole-cell mGluR-LTD experiments, cells were clamped at −65 mV throughout the experiment. For peptide and antibody infusion experiments, the lack of the effect of each drug on basal synaptic responses was independently tested by conducting baseline recordings in the presence of the drug for at least 1 h without LTD induction. For current/voltage (I/V) relation and rectification index experiments, AMPAR EPSCs (10 responses) were recorded at various holding potential (−80, −60, −40, −20, 0, +20, +40, and +60 mV) with the above intracellular solution plus 100 μm spermine and the extracellular ACSF containing 100 μm dl-APV. EPSC amplitudes were normalized to those recorded at −60 mV. Rectification indices were calculated by the ratios of EPSC amplitude at +40/−60 mV. For whole-cell recordings of cultured hippocampal neurons, the cells were washed and perfused with extracellular fluid (ECF) containing the following (in mm): 120 NaCl, 1.2 MgCl2, 3.0 KCl, 2.0 CaCl2, 25 HEPES, 25 d-glucose, 0.0005 tetrodotoxin, and 0.05 picrotoxin, pH 7.35 280–300 mOsm), and recorded under the same condition as for the acute hippocampal slices. mGluR-LTD in cultured neurons was induced by bath application of 100 μm DHPG for 5 min and recorded at −65 mV holding potential. All data acquisition and analysis were done using pCLAMP 8 (Molecular Devices) and MiniAnalysis program (Synaptosoft). When average data were plotted, data were normalized to the average of the baseline responses unless indicated otherwise. In all electrophysiological experiments, n represents the number of neurons or slices, and normally only one or two slices or neurons per animal were used.
Neuronal culture, immunostaining, and image analysis.
Hippocampal neuronal cultures were prepared from postnatal day 1 (P1) pups as previously described (Meng et al., 2002). Briefly, pups were killed and hippocampal CA1 regions were dissected in ice-cold PBS. Tissues were trypsinized (0.25%) at 37°C for 15 min, dissociated by trituration, and plated onto poly-d-lysine (50 μg/ml) coated glass coverslips (60,000 cells/ml for immunostaining experiments and 90,000 cells/ml for electrophysiological experiments). The cultures were maintained by replacing half of the medium with fresh medium every 3–5 d. The maintenance medium contained Neurobasal A, 0.5 mm GlutaMax, B27 and 5 ng/ml β-FGF. Whenever possible, the KO cultures were prepared together along with the WT littermates on the same 24-well plates for better comparison. For immunostaining (17–18 DIV), culture medium was replaced with ECF and treated with 100 μm DHPG for 5 min (same procedure used for electrophysiology in cultured neurons). Immediately following the treatment cells were then fixed with ice-cold 4% paraformaldehyde + 4% sucrose for 20 min and permeabilized (except for GluA1 surface staining) with 0.25% Triton X-100 for additional 20 min. Cells were then blocked with 3% donkey serum and 3% BSA in PBS for 1 h, incubated with primary antibodies overnight at 4°C followed by rhodamine-conjugated phalloidin and appropriate secondary antibodies (Invitrogen, Jackson ImmunoResearch) for 1 h at room temperature. After washing with PBS, coverslips were mounted using DAKO mounting medium for image collections. The primary antibodies used included: anti-GluA1 (Calbiochem), anti-Synapsin I (Santa Cruz Biotechnology), anti-MAP2 (microtubule-associated protein 2) (Millipore), anti-cofilin (Cytoskeleton), anti-β-catenin (Cell Signaling Technology), and anti-PSD95 (Millipore). Confocal images were obtained on a Zeiss LSM 510 laser scanning system at 2048 × 2048 pixels using a Zeiss 63× (numerical aperture, 1.4) objective with the same settings and configurations within each experiment. Spines were defined as any dendritic protrusions 0.3–4 μm in length. The spine length was measured from the base of the spine (defined with MAP2 staining) to the tip of the spine head. For analysis of synaptic proteins, the fluorescence puncta (with an area of >0.1 μm2) were automatically selected and counted, and manually verified. For each genotype or treatment, ∼20–35 neurons from at least three independent cultures and a total of 100–150 μm linear dendrites per neuron were randomly selected, measured, and averaged. All measurements were performed using ImageJ (National Institutes of Health) and Volocity 5.2 (Improvision) software.
Slice treatment and biochemical assays.
Hippocampal slices used for immunoprecipitation and Western immunoblotting experiments were prepared according to the same procedure as for electrophysiological recordings. Slices were recovered for 3–4 h at room temperature in ACSF saturated with 95% O2/5%CO2 and transferred to a treatment chamber for additional 30 min recovery before DHPG treatment. The treatment was initiated by applying 100 μm DHPG for 5 min. Some slices were removed immediately before DHPG application (0 min) and used as untreated controls. For the INP peptide preincubation, 50 μm INP or equivalent DMSO vehicle was added during the 30 min recovery period in the treatment chamber. At the end of DHPG treatment, slices were rapidly frozen in dry ice/ethanol slurry and stored at −20°C. To prepare protein lysate, slices samples were lysed for 45 min in ice-cold cell lysis buffer containing the following (in mm): 20 Tris pH 7.5, 150 NaCl, 1 EDTA, 1 EGTA, 1% Triton X-100, 2.5 sodium pyrophosphate, 1 β-glycerophosphate, 1 Na3VO4, 20 NaF, 1 μg/ml leupeptin, 1 phenylmethylsulfonyl fluoride, and 0.5% protease inhibitor mixture (Calbiochem). Debris was removed by centrifugation at 12,000 rpm (4°C) for 10 min. The total protein concentration of each sample was estimated using BCA assay (GE Healthcare) and equalized with loading buffer. Proteins were separated on 10% SDS-PAGE polyacrylamide gel and electrotransferred to a nitrocellulose filter. Filters were then blocked with 2.5% dry milk/2.5% normal fetal bovine serum in TBS-T (20 mm Tris base, 9% NaCl, 0.1% Tween-20, pH 7.6) and incubated overnight at 4°C with appropriate primary antibodies in TBS-T. Following washing and incubation with appropriate secondary antibodies, filters were developed using enhanced chemiluminescence (GE Healthcare) method of detection and analyzed using the AlphaEaseFC software as per manufacturer's instruction. Protein loading was further controlled by normalizing each tested protein with tubulin immunoreactivity on the same blot. For immunoprecipitation experiments, 300 μl of the protein lysate (200–300 μg, normally pooled from two to three hippocampal slices) was incubated with appropriate primary antibodies at 4°C with constant gentle rocking for 3–5 h followed by the addition of 35 μl of protein A (for rabbit antibodies) or G (for mouse antibodies) agarose bead slurry (Santa Cruz Biotechnology) and further incubation of 3 h at 4°C. The samples were microcentrifuged for 1 min at 12,000 rpm, the supernatant was carefully removed, and the beads were washed thoroughly five times with the lysis buffer and resuspended in loading buffer for Western blot analysis. For Rac1 (Ras-related C3 botulinum toxin substrate 1)/RhoA (Ras homolog gene family, member A) activation assay, a procedure recommended by the manufacturer (Biochem Kit, Cytoskeleton) was followed. Primary antibodies used included the following: anti-cofilin and anti-Rac1 (Cytoskeleton), anti-pCofilin (Santa Cruz Biotechnology), anti-ERK1/2 (extracellular signal-regulated kinase 1/2), anti-pERK1/2, anti-N-cadherin, anti-β-catenin, and anti-pTyrosine (Cell Signaling Technology). n in the summary data represents the number of independent experiments.
Viral vector construction, virus purification, and in vivo injection.
The basic principles for using helper phage-dependent adenovirus-based vectors have been described previously (Sandig et al., 2000). We used the shuttle vector pC4HSU because much of its genomic sequence can be replaced by foreign DNA (e.g., GluA2 cDNA) without perturbing virus packaging as long as the resultant construct size ranges from 27 to 38 kb. To make GluA2 viral vectors, various forms of GluA2 cDNA and enhanced green fluorescent protein (EGFP) were first subcloned respectively into unique restriction sites XhoI and SpeI of an intermediate vector modified from k18EpiSEAP. Both GluA2 and EGFP cassettes contained their own human CMV (cytomegalovirus) immediate early promoter and SV40 (simian virus 40) early mRNA polyA (polyadenylation) signal; therefore, they were transcribed and translated independently as two separate proteins. The cDNAs of GluA2 used included the following: wild-type (WT) hemagglutinin (HA)-tagged full-length GluA2, HA-tagged GluA2 Δ92, Myc-tagged GluA1-NTD87-GluA2, and Myc-tagged GluA2-NTD92-GluA1. The pC4HSU vector was then cut with AscI and NotI, blunted, and relegated to remove a 4.5 kb fragment to harbor the GluA2/EGFP cassettes. And finally, the GluA2/EGFP was released from the modified k18EpiSEAP vector with MluI and inserted into the unique MluI site of pC4HSU. The total size of the final viral constructs used in this study ranged from 30 to 35 kb.
To make a large-scale virus preparation, 15 μg of viral construct DNA was linearized by PmeI digestion to remove the plasmid backbone, and the viral DNA fragment (10 μg) was purified and transfected into the Cre-expressing packaging cell line 293Cre66 using standard calcium phosphate method at 75% confluency (CellPhect Transfection Kit, GE Healthcare). 293Cre66 cells were cultured in full growth medium containing Eagle's minimal essential medium (MEM), 10% FBS, 50 μg/ml hygromycin B (Roche), and 1× antibiotic-antimycotic (Wisent). Helper virus NG163 was added to the culture medium 12 h after transfection, and when cytopathic effect (CPE; cells became rounded and detached from the plate) occurred (usually between 48 and 72 h postinfection), cells and medium [crude viral lysate (CVL)] were transferred to a cryotube and rapidly frozen in dry ice-ethanol mixture. Twenty percent of the collected CVL was then used to infect fresh 293Cre66 cells (with the NG163 helper virus added 12 h later to induce CPE), and CVL was collected for the subsequent infection cycle. The cycle was repeated three more times to accumulate a sufficient amount of CVL for final virus production, where a spinner suspension culture system containing 3 L of calcium-free Joklik MEM (Sigma-Aldrich) was used to prevent cell aggregation.
To harvest the viral particles, cells in suspension culture were collected by centrifugation at 2000 rpm, 4°C for 10 min, resuspended in 15 ml of dilution buffer (10 mm Tris-Cl, pH 8.0), and stored at −80°C for later purification. To purify the virus, cells were lysed through multiple freezing/thawing cycles (freezing in a dry ice/ethanol bath and thawing at 37°C) followed by incubation in the detergent sodium deoxycholate (0.5%) for 30 min at room temperature with occasional shaking. The lysate was then treated with 1000 units of Benzonase Nuclease (Sigma-Aldrich) for 20 min to reduce viscosity, and the debris was removed by centrifugation for 10 min at 5000 × g and 4°C. The clear supernatant containing the viral particles were then further purified by two cycles of ultracentrifugation in CsCl density gradients. The first cycle density gradient was prepared in a thin-wall Ultra-Clear tube (catalog #344059, Beckman Coulter) containing 3 ml of low-density CsCl gradient solution (1.25 g/ml), 3 ml of medium-density CsCl gradient solution (1.35 g/ml), and 1 ml of high-density CsCl gradient solution (1.5 g/ml). Six milliliters of the viral supernatant was carefully overlaid on the top of the density gradient and centrifuged in a SW 41 Ti swinging bucket rotor for 1 h at 35,000 × g and 4°C. The opalescent band at the interface between low- and medium-density CsCl gradients was then collected and subjected to the second cycle of ultracentrifugation (for 24 h at 35,000 × g and 4°C) in a continuous medium-density gradient of CsCl (1.35 g/ml). Finally, the opalescent band containing the virus was recovered and dialyzed extensively in the dialysis buffer containing 10 mm Tris-Cl, pH 8.0, and 1 mm MgCl2 (Slide-A-Lyzer cassette, Thermo Scientific Pierce). The viral stock was then stored in 10% glycerol at −80°C before injections.
For in vivo viral injections, we used a mixture of ketamine and xylazine (80 mg/10 mg, i.p.) for anesthesia. The animal was mounted on a stereotaxic apparatus. When both bregma and lambda were set at the same dorsoventral level, a midline incision was made with no. 15 surgical scalpels. Then the subcutaneous tissue was cleaned using small surgical forceps. Stereotaxic targeting was done by lowering a glass micropipette fixed to the stereotaxic holder. The coordinates for injecting dorsal hippocampal CA1 region were lateral 2.0 mm, ventral 1.4 mm, and caudal 2.1 mm. At the skull position with the above coordinates, a micro-drill with a 0.7 mm spinning drill bit was used to thin the skull until blood vessels in the dura became clearly visible. The thinned bone was then removed and cleaned with PBS. Glass micropipettes back-filled with viral solutions (1 × 107 particles per microliter in 10 mm Tris-Cl) were slowly lowered to the desired z-coordinate of CA1. For one hippocampus, 1 μl of the viral solution was gradually injected over a period of 20 min using a 10 μl Hamilton syringe. Once the injection was completed, the wounds were sutured and cleaned, and mice were returned to their home cages. All the animal procedures used in this study were approved by the Hospital for Sick Children Animal Use Committee. All the averaged data in this study were reported as the mean ± SEM and statistically evaluated by Student's t test. A p value <0.05 was considered to be significant and is indicated with the * symbol in the summary graphs in all figures.
Results
GluA2 is indispensable for mGluR-LTD in the hippocampus
At CA1 synapses, at least two forms of LTD coexist: NMDA receptor (NMDAR)-dependent LTD (NMDAR-LTD) and mGluR-dependent LTD (mGluR-LTD) (Malenka and Bear, 2004; Lüscher and Huber, 2010). Although GluA2 has been extensively investigated in the context of NMDAR-LTD (Isaac et al., 2007), its role in mGluR-LTD remains unknown. We therefore focus this study on mGluR-LTD. We induced mGluR-LTD using two separate protocols: bath application of the group I mGluR agonist DHPG; or PP-LFS in the presence of NMDA receptor antagonist APV (Kemp and Bashir, 1999; Huber et al., 2000). Since the expression mechanism of mGluR-LTD is subject to developmental regulation (Nosyreva and Huber, 2005), we therefore focused our studies on young adults (7–9 weeks), at which time both the induction and expression mechanisms are dependent on postsynaptic changes and local protein synthesis (Huber et al., 2000; Nosyreva and Huber, 2005; Waung and Huber, 2009; Lüscher and Huber, 2010). As shown in Figure 1A, the application of DHPG in both WT and GluA3 (GluR3) KO animals induced a decrease in field EPSPs (fEPSPs) that persisted during the entire recording period, but this DHPG-induced LTD was completely abolished in GluA2 KO mice (WT = 77.03 ± 1.83%; GluA3 KO = 81.37 ± 4.59%; GluA2 KO = 109.71 ± 5.98%; p < 0.001 between WT and GluA2 KO). Similarly, application of PP-LFS induced a persistent decrease in fEPSPs in both WT and GluA3 KO mice, but this PP-LFS-induced LTD was completely absent in GluA2 KO mice (Fig. 1B: WT = 77.89 ± 3.20%; GluA3 KO = 83.14 ± 3.11%; GluA2 KO = 109.21 ± 4.86%; p < 0.001 between WT and GluA2 KO). The DHPG-induced LTD was blocked by the group I mGluR antagonist MPEP (supplemental Fig. 1A, available at www.jneurosci.org as supplemental material), confirming that this DHPG-induced LTD is mGluR-dependent. The DHPG- and PP-LFS-induced LTD was also completely abolished by the protein synthesis inhibitor cycloheximide (Fig. 1C: control = 84.42 ± 3.43; cycloheximide = 100.40 ± 2.28; p < 0.001) (Fig. 1D: control = 77.30 ± 4.75%; cycloheximide = 105.14 ± 2.62; p < 0.001), but without changes in the rectification index (supplemental Fig. 1B,C, available at www.jneurosci.org as supplemental material) or activation of AMPARs (supplemental Fig. 1D, available at www.jneurosci.org as supplemental material). These results indicate that GluA2 is indispensable for mGluR-LTD at CA1 synapses.
Figure 1.
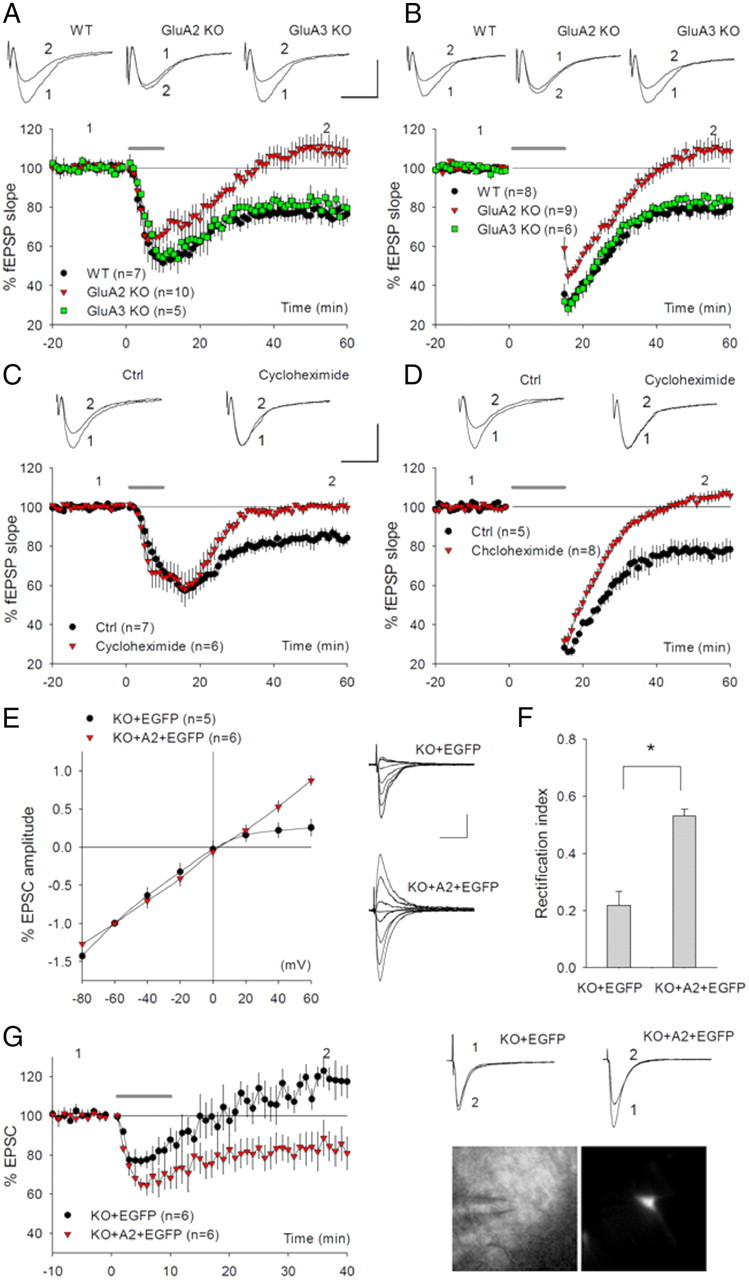
Requirement for GluA2 in hippocampal mGluR-LTD. A, Application of 100 μm DHPG for 10 min (solid line) induces LTD in WT and GluA3 KO mice, but not in GluA2 KO mice. The representative traces in this and all other graphs of electrophysiological recording experiments are averages of five successive responses at indicated time points. B, PP-LFS (solid line) in the presence of 100 μm APV induces LTD in WT and GluA3 KO mice, but not in GluA2 KO mice. C, D, Perfusion of the protein synthesis inhibitor cycloheximide (60 μm) blocks both DHPG-induced (C) and PP-LFS-induced LTD (D) in WT mice. E, I/V curves of AMPAR-mediated EPSCs and representative traces recorded at various holding potentials from GluA2 KO CA1 neurons infected with either EGFP alone (EGFP) or full-length WT GluA2 plus EGFP (A2+EGFP) HDAv viruses. Note the strong rectification and smaller EPSCs in EGFP-infected neurons, which are identical to uninfected GluA2 KO neurons, both of which are rescued in A2+EGFP-infected KO neurons. F, Averaged rectification indices (AMPAR-mediated EPSC amplitude at +40/−60 mV holding potentials) showing significant difference between EGFP- and A2+EGFP-infected GluA2 KO CA1 neurons. G, Whole-cell recordings showing the restoration of DHPG-induced LTD in GluA2 KO CA1 neurons infected with A2+EGFP, but not with EGFP viruses. Images below the traces show a CA1 neuron expressing EGFP (right) and placement of the recording electrode (left). Calibration: 0.5 mV/20 ms for slice field recordings; 50 pA/50 ms for slice whole-cell recordings in this and all other figures.
One potential problem of the above experiments was that the LTD deficit in GluA2 KO mice might be related to the chronic effect caused by GluA2 deletion. To rule out this possibility, we developed helper phage-dependent adenoviral (HDAv) vectors expressing the EGFP alone or EGFP plus HA-tagged full-length GluA2 protein (A2+EGFP) and performed rescue experiments by injecting these viruses into the adult hippocampus of GluA2 KO mice. We chose this expression system because the vector can harbor a large amount of exogenous DNA sequences to allow the expression of multiple genes in a single construct (Barcia et al., 2007). This was particularly important for this study because we preferred to express GluA2 and EGFP as two separate proteins (rather than a fusion protein) to avoid any perturbations of GluA2 function that might be caused by the fusion (supplemental Fig. 1E, available at www.jneurosci.org as supplemental material). Infection experiments using the viruses containing both GluA2 and EGFP in HEK 293 cells and cultured neurons showed that all EGFP-positive cells express GluA2, indicating that EGFP is a reliable indicator for GluA2 protein expression (data not shown). Three to five days following hippocampal viral injections, slices were prepared and EPSCs were recorded from infected CA1 neurons. As expected, the expression of EGFP alone had no effect on either I/V relation (i.e., inward rectification of AMPAR-mediated synaptic response characteristic of GluA2-lacking AMPARs) (Fig. 1E,F) or the impairment of DHPG-induced LTD (Fig. 1G) in GluA2 KO mice. However, the expression of A2+EGFP completely rescued the rectification (Fig. 1E,F: EGFP = 0.22 ± 0.05; A2+EGFP = 0.53 ± 0.03; p < 0.001), indicating that the exogenously expressed GluA2 was functional at the synapse. Importantly, DHPG-induced LTD was also completely restored in neurons expressing A2+EGFP (EGFP = 111.15 ± 5.54%; A2+EGFP = 79.76 ± 7.00%; p < 0.01). The expression of A2+EGFP had no effect on basal synaptic response, DHPG-induced LTD, or I/V relation in WT neurons (supplemental Fig. 1F–H, available at www.jneurosci.org as supplemental material). These results confirmed that the absence of mGluR-LTD is not due to developmental effects of GluA2 ablation and that GluA2 is directly involved in the regulation of the mGluR-LTD process.
GluA2 NTD interaction with N-cadherin is necessary for GluA2 to regulate hippocampal mGluR-LTD
The above results indicate that GluR2 is indispensable for mGluR-LTD expression. Since GluR2 CTD-dependent endocytosis of AMPARs is considered to be the key mechanism underlying several forms of LTD (e.g., cerebellar LTD and hippocampal NMDAR-LTD) (Malinow and Malenka, 2002; Collingridge et al., 2004; Isaac et al., 2007), then it is possible that the impaired mGluR-LTD in GluA2 KO mice is simply due to the lack of the GluA2 CTD-dependent processes. However, this interpretation may not apply in this case. It has been proposed that the role of the GluA2 CTD in activity-dependent AMPAR internalization is to release AMPARs from anchor proteins (e.g., GRIP/ABP). In neurons lacking GluA2 (i.e. GluR2 KO mice and some interneurons), AMPARs exist without being anchored by GRIP/ABP, and therefore, they are free to enter the endocytic process upon LTD induction (Brown et al., 2005). This may explain why GluR2 KO mice still show robust NMDAR-dependent LTD and NMDA-induced AMPAR internalization (Jia et al., 1996; Meng et al., 2003; Biou et al., 2008). Therefore, we reasoned that the lack of GluA2 CTD-dependent endocytosis is not likely to account for the impaired mGluR-LTD in GluA2 KO mice. To test this possibility and determine which domain of GluA2 is important for mGluR-LTD, we performed additional in vivo viral rescue experiments by using various GluA2 mutant constructs. We first examined the effects of expressing a deletion mutant GluA2, A2-Δ92, where the first 92 aa of GluA2 NTD were removed, and thus were no longer able to bind to N-cadherin, but still retained all other key unique properties of GluA2, including its TM/CTD protein interactions [i.e., with TARPs (transmembrane AMPAR regulatory proteins), PICK1, NSF, and ABP/GRIP) and channel rectification properties (Saglietti et al., 2007). As shown in Figure 2, although this mutant construct was able to restore the linear I/V relation of AMPAR-mediated currents (Fig. 2A,B: A2-Δ92 RI = 0.62 ± 0.05), confirming that it was functionally expressed at the synapse, it did not rescue mGluR-LTD in GluA2 KO mice (Fig. 2C,E: A2-Δ92 = 108.58 ± 6.95%). This result was extremely intriguing to us because it suggests that the GluA2-CTD is not sufficient for mGluR-LTD regulation and that the GluA2 NTD interaction with N-cadherin is required. To further confirm the importance of the GluA2 NTD interaction, we examined the effect of expressing another mutant GluA2, A1-87-A2, a chimeric construct where the NTD 92 aa of GluA2 were replaced with the corresponding 87 aa of GluA1 to avoid deletions of any potentially important amino acids. The expression of this construct had a similar effect on GluA2 KO neurons (i.e., it restored the linear I/V relation) (Fig. 2A,B: A1-87-A2 RI = 0.67 ± 0.17) but did not rescue the mGluR-LTD deficit (Fig. 2C,E: A1-87-A2 = 116.46 ± 3.05%). Finally, to determine whether the GluA2 NTD/N-cadherin interaction was sufficient for mGluR-LTD rescue, we tested the effect of expressing A2-92-A1, where the entire GluA2 sequence except for the NTD 92 aa, was replaced by the corresponding GluA1 sequence. This chimeric construct is still able to interact with N-cadherin but lacks all the other properties of GluA2, including its TM/CTD protein interactions, Ca2+ impermeability, and channel rectification. The expression of this construct did not restore the linear I/V relation or rectification index (Fig. 2A,B: A2-92-A1 RI = 0.27 ± 0.07), but remarkably, it completely restored mGluR-LTD (Fig. 2D,E: A2-92-A1 = 82.09 ± 4.46%). In fact, the magnitude of DHPG-induced LTD in GluA2 KO neurons infected by A2-92-A1 was indistinguishable from that of GluA2 KO neurons infected by wild-type full-length GluA2 (i.e., A2+EGFP) or that of WT neurons infected by EGFP alone (Fig. 2E). Together, these results confirm that the GluA2 NTD interaction with N-cadherin is both necessary and sufficient for GluA2 to rescue the mGluR-LTD deficit in GluA2 KO mice, indicating a critical importance for GluA2 NTD/N-cadherin interaction in mGluR-LTD regulation.
Figure 2.
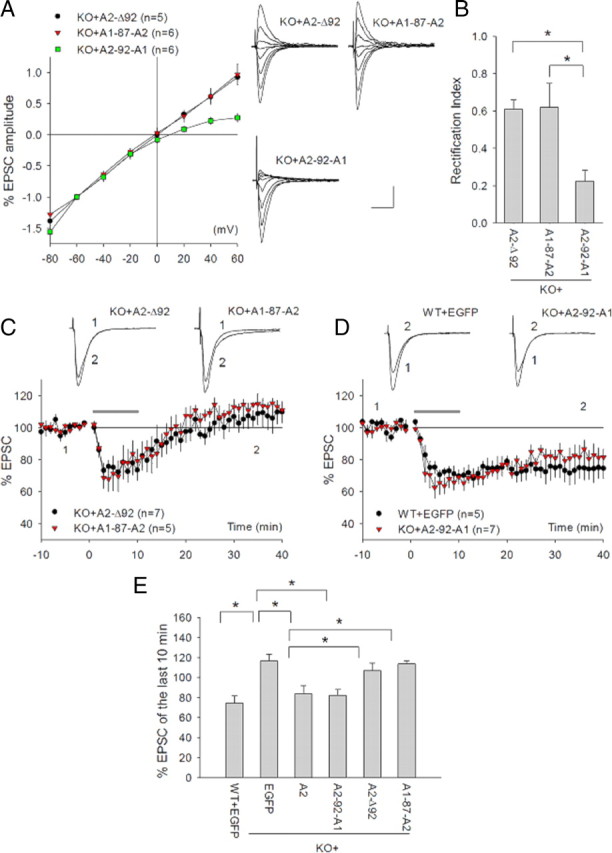
Dependence of mGluR-LTD on GluA2 NTD interaction with N-cadherin. A, I/V curves of AMPAR-mediated EPSCs and representative traces recorded at various holding potentials from GluA2 KO CA1 neurons infected with various EGFP+GluA2 mutant viruses. Note linear I/V curves in both A2-Δ92- and A1-87-A2-infected, but not in A2-92-A1-infected, KO neurons. B, Rectification indices of AMPAR-mediated EPSCs in GluA2 KO neurons infected with various GluA2 mutant viruses. C, Whole-cell recordings showing the absence of DHPG-induced LTD in GluA2 KO CA1 neurons infected with A2-Δ92 or A1-87-A2 viruses. D, Whole-cell recordings showing the rescue of DHPG-induced LTD in GluA2 KO neurons infected with A2-92-A1 viruses compared with WT neurons infected with EGFP viruses. E, Summary of the magnitudes of DHPG-induced LTD in WT and GluA2 KO neurons infected with various GluA2 viruses.
N-cadherin/catenin complex is regulated by mGluR activation and required for hippocampal mGluR LTD
Having identified the N-cadherin-binding motif on the GluA2 NTD as an essential mediator of GluA2 action in mGluR-LTD, we next examined whether N-cadherin signaling complex was also required for this form of plasticity. Earlier studies have shown that N-cadherins and their cytoplasmic partner catenins play an important role in synaptogenesis, spine structure, and LTP (Takeichi, 2007; Arikkath and Reichardt, 2008; Tai et al., 2008), but their role in mGluR-LTD has not been tested. Interestingly, the integrity and function of the N-cadherin/catenin signaling complex is critically regulated by tyrosine phosphorylation/dephosphorylation (Lilien and Balsamo, 2005), a process that is also critical for mGluR-LTD (Huang and Hsu, 2006; Moult et al., 2006; Zhang et al., 2008; Gladding et al., 2009). Therefore, we reasoned that the N-cadherin/catenin complex might be an essential signaling component for mGluR-LTD. To test this possibility, we performed the following experiments. First, we showed that the disruption of the N-cadherin/catenin complex abolished mGluR-LTD. We pretreated the hippocampal slices for 30 min with the short peptide INP (INPISGQ) or its reverse control peptide PNI (QGSIPNI) and then recorded DHPG-induced LTD in the presence of these peptides throughout the experiment. This INP peptide is mimetic of the extracellular ectodomain 1 on N-cadherin and has been shown to exert a highly specific and potent disruptive effect on N-cadherin function (Williams et al., 2000; Siu et al., 2007). As shown in Figure 3A, the peptide-treated slices did not show any appreciable amount of DHPG-induced LTD, whereas the control (PNI-treated) had a normal level of LTD (PNI = 74.20 4.01%; INP = 102.63 2.17%; p < 0.05). Neither INP nor PNI peptides had an effect in GluA2 KO mice (Fig. 3B). fEPSP recordings generated similar results (supplemental Fig. 2A, available at www.jneurosci.org as supplemental material). Application of INP peptide for up to 1 h without DHPG treatment did not have any effects on basal synaptic responses, indicating that INP treatment alone is not sufficient to induce any synaptic plasticity (supplemental Fig. 2B, available at www.jneurosci.org as supplemental material). Application of INP peptide after DHPG treatment also had no effect on DHPG-induced LTD (supplemental Fig. 2C, available at www.jneurosci.org as supplemental material). These results indicated that intact N-cadherin/catenin function is likely required for the induction phase of mGluR-LTD. Second, we performed immunoprecipitation experiments to show that DHPG was able to induce functional changes in N-cadherin/catenin complex, which was occluded by the INP peptide pretreatment. Hippocampal slices were prepared and treated with various drugs in a similar manner as for LTD recordings, but following the treatment the slices were rapidly frozen and the protein lysate was extracted for analysis of β-catenin, a major cytoplasmic binding partner and reliable functional indicator for N-cadherin. In WT slices pretreated with the control PNI peptide, DHPG treatment significantly reduced N-cadherin/β-catenin binding [Fig. 3C: DHPG (5 min) = 0.46 ± 0.13; DHPG (30 min) = 0.42 ± 0.10; p < 0.01 compared with untreated], indicative of altered N-cadherin functionality. In contrast, INP peptide pretreatment alone resulted in a significant reduction in N-cadherin/β-catenin binding, and this occluded the subsequent DHPG-induced reduction in this binding [Fig. 3C: DHPG (0 mi) = 0.61 ± 0.08; DHPG (5 min) = 0.57 ± 0.11; DHPG (30 min) = 0.55 ± 0.09; p < 0.05 compared with PNI/DHPG (0 min); p > 0.05 compared between them]. Finally, we demonstrated that DHPG-induced reduction in N-cadherin/β-catenin interaction required GluA2. As shown in Figure 3D, in GluA2 KO mice, the basal level of N-cadherin/β-catenin binding was significantly lower and DHPG treatment instead induced an increase in this binding [GluA2 KO (0 min) = 0.63 ± 0.10, p < 0.01 compared with the WT (0 min) time point; GluA2 KO/DHPG (5 min) = 1.36 ± 0.22, GluA2 KO/DHPG (30 min) = 1.20 ± 0.09, p < 0.001 compared with the WT/DHPG (5 min)]. As a further support for GluA2 requirement in N-cadherin/β-catenin signaling, we showed that both basal level and DHPG-induced increases in tyrosine-phosphorylated β-catenin, another important indicator of N-cadherin function, were also significantly altered in GluA2 KO mice (supplemental Fig. 2D,E, available at www.jneurosci.org as supplemental material). These results together indicate the following: (1) mGluR activation induces functional changes in the N-cadherin/β-catenin complex and these changes are necessary for DHPG-induced LTD; and (2) GluA2 is indispensable for the N-cadherin/β-catenin complex to perform this function. These results are consistent with the idea that GluA2 regulates mGluR-LTD through interacting with the N-cadherin/catenin complex.
Figure 3.
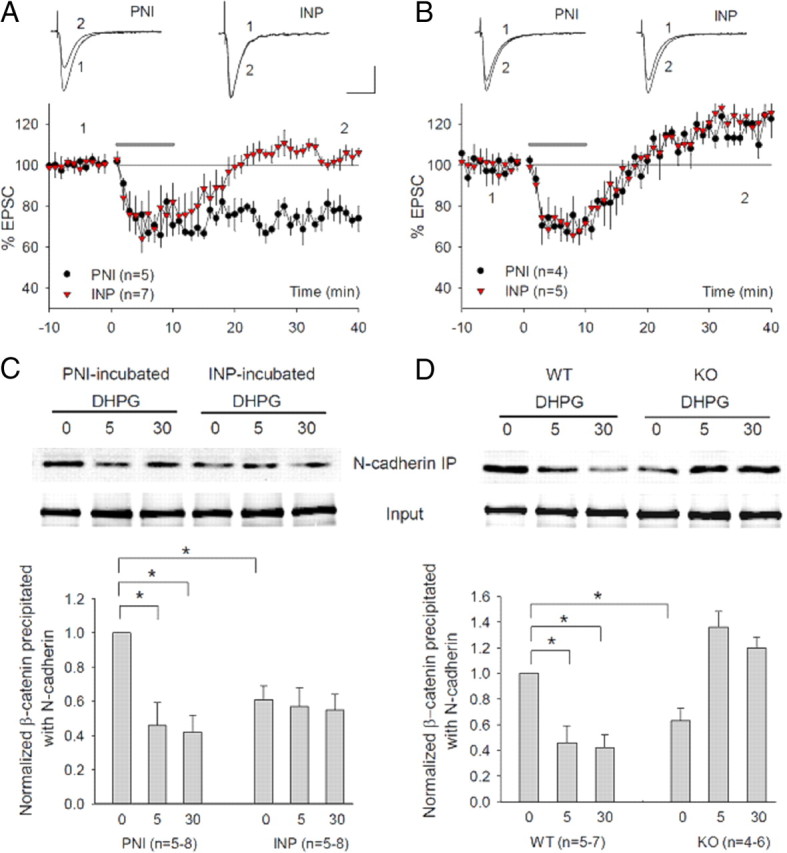
Requirement of N-cadherin/catenin in mGluR-LTD and its dependence on GluA2. A, Whole-cell recordings showing blockade of DHPG-induced LTD by pretreating the slices with the N-cadherin inhibitory peptide INP (50 μm, 30 min), but not by the control peptide PNI (50 μm, 30 min). B, Whole-cell recordings showing lack of effect for either peptide INP (50 μm, 30 min) or PNI (50 μm, 30 min) in GluA2 KO mice. C, Western blot analysis and summary graph of anti-N-cadherin immunoprecipitates using protein lysates prepared from hippocampal slices pretreated with control PNI or INP peptide (50 μm, 30 min) followed by treatment with (5, 30 min) or without (0 min) DHPG (100 μm) and probed with anti-β-catenin antibodies, showing a dramatic reduction in the basal level as well as lack of subsequent DHPG-induced reduction in β-catenin associated with the N-cadherin by the INP peptide, but not by the PNI peptide preincubation. D, Western blot analysis and summary graph of anti-N-cadherin immunoprecipitates probed with anti-β-catenin showing DHPG-induced reduction in the amount of β-catenin associated with the N-cadherin in WT samples (same as in C), but not in GluA2 KO samples. Note that the basal N-cadherin-associated β-catenin is lower, and DHPG instead induces an increase in N-cadherin/β-catenin binding in GluA2 KO slices. The total β-catenin is not altered in any treatment or in GluA2 KO mice.
Hippocampal mGluR-LTD requires cofilin-mediated actin reorganization
How then does the GluA2/N-cadherin interaction regulate mGluR-LTD? Given the prominent role of N-cadherin signaling in actin regulation (Fukata and Kaibuchi, 2001; Bamji, 2005), we reasoned that that GluA2/N-cadherin might regulate mGluR-LTD through actin reorganization. Indeed, application of F-actin perturbation agents has been shown to invariably block mGluR-LTD, but not NMDAR-LTD (Xiao et al., 2001; Zhou et al., 2004; Morishita et al., 2005; Moult et al., 2006). To address this hypothesis, we set out to identify the molecular factors important for actin regulation during mGluR-LTD. First, we showed that actin reorganization in the postsynaptic neurons was necessary for mGluR-LTD. As shown in Figure 4A, inclusion of the F-actin stabilizer phalloidin in the recording pipette to prevent F-actin depolymerization in the postsynaptic neuron completely blocked mGluR-LTD (control = 67.66 ± 4.71%; phalloidin = 98.10 ± 7.47%; p < 0.01). Surprisingly, inclusion of the actin polymerization inhibitor latrunculin A also blocked mGluR-LTD (Fig. 4B: DMSO = 70.01 ± 5.80%; latrunculin = 105.09 ± 3.36%; p < 0.01). Neither phalloidin nor latrunculin A had any effect on NMDAR-dependent LTD or basal synaptic response (supplemental Fig. 3A,B, available at www.jneurosci.org as supplemental material). These results indicated that both actin polymerization and depolymerization were required for DHPG-induced LTD. Since cofilin is a potent regulator of both processes (Bamburg, 1999; DesMarais et al., 2005), we then tested whether mGluR-LTD also depended on cofilin. We used short inhibitory peptides (active peptide M and control peptide Q) known to specifically prevent cofilin from binding to F-actin, thus inhibiting cofilin activity (Eibert et al., 2004). As shown in Figure 4C, the active peptide completely abolished DHPG-induced LTD, whereas the control peptide had no effect (M = 99.67 ± 4.83%; Q = 68.20 ± 7.21%; p < 0.005). To verify these results independently, we tested additional cofilin peptides, S3 and phospho-S3 (p-S3), which are known to enhance and block cofilin activity through inhibiting cofilin phosphorylation and dephosphorylation, respectively (Aizawa et al., 2001). As shown in Figure 4D, peptide p-S3 completely blocked DHPG-induced LTD, whereas peptide S3 had no effect (p-S3 = 100.46 ± 7.77%; S3 = 72.44 ± 4.40%; p < 0.01). Therefore, activation of cofilin through dephosphorylation is essential for DHPG-induced LTD. To identify the upstream regulators for cofilin, we tested the role of the Rho GTPases, direct substrates for N-cadherin/catenin complex and known to regulate cofilin activity (Bamburg, 1999; Fukata and Kaibuchi, 2001). Interestingly, we found that the Rac1 inhibitor (NSC23766) completely blocked DHPG-induced LTD (Fig. 4E: control = 73.96 ± 2.41%; NSC23766 = 101.03 ± 5.64%; p < 0.01), whereas the RhoA inhibitor (C3 botulinum toxin) had no inhibitory effect (C3 toxin = 54.63 ± 5.33%, p < 0.05 compared with the control). In GluA2 KO neurons infected with A2-92-A1, DHPG-induced LTD was also blocked by cycloheximide, peptide p-S3 or Rac1 inhibitor (Figs. 2, 4F: cycloheximide = 105.82 ± 7.08%; pS3 = 107.70 ± 7.56%; NSC23766 = 101.93 ± 6.19%; p < 0.05 for each compared with GluA2 KO neurons infected with WT A2 or A2-92-A1), indicating that mGluR-LTD in GluA2 KO mice share similar mechanisms when GluA2 is introduced back to these mice. These results together provide strong evidence that regulation of cofilin by Rac1 GTPase is required for mGluR-LTD in the hippocampus.
Figure 4.
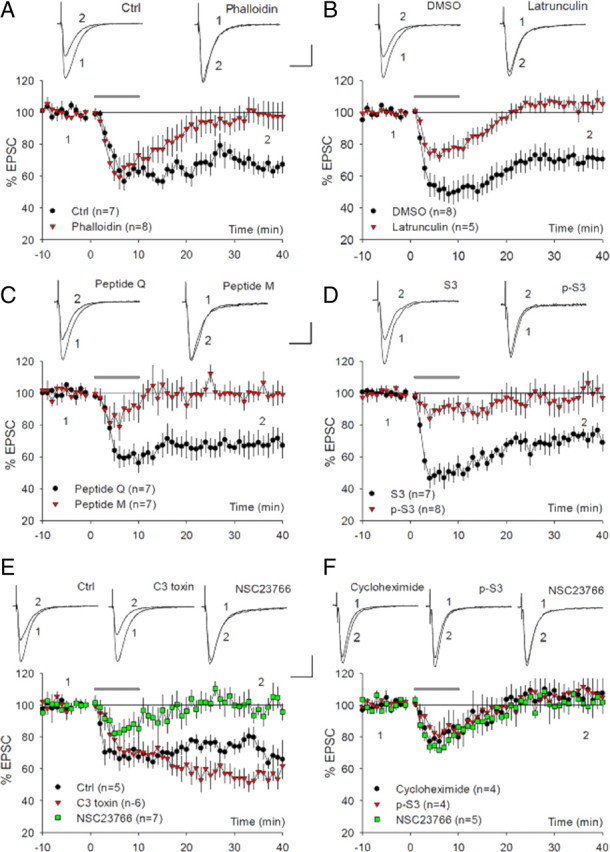
Requirement of cofilin-mediated actin reorganization for DHPG-induced LTD. A, Whole-cell recordings showing the blockade of DHPG-induced LTD in WT neurons postsynaptically infused with the F-actin stabilizer phalloidin (100 μm), but normal DHPG-induced LTD in control neurons. B, Whole-cell recordings showing the blockade of DHPG-induced LTD in WT neurons postsynaptically infused with the actin polymerization inhibitor latrunculin A (200 μm), but not in control neurons (DMSO). C, Whole-cell recordings showing the abolishment of DHPG-induced LTD in WT neurons postsynaptically infused with cofilin-inhibitory peptide (M, 200 μm), but not with control peptide (Q, 200 μm). D, Whole-cell recordings showing the abolishment of DHPG-induced LTD in WT neurons postsynaptically infused with cofilin inhibitory peptide p-S3 (200 μm), but not with cofilin-enhancing peptide S3 (200 μm). E, Whole-cell recordings showing the abolishment of DHPG-induced LTD in WT neurons infused with the Rac1-specific inhibitor NSC23766 (250 μm), but not with the RhoA-specific inhibitor C3 toxin (10 μg/ml) or control neurons. Note a small but significant enhancement of LTD in the RhoA inhibitor-treated neurons. F, Whole-cell recordings showing the blockade of DHPG-induced LTD by cycloheximide (60 μm), peptide p-S3 (200 μm), or Rac1 inhibitor NCS (250 μm) in GluA2 KO neurons infected with A2-92-A1 viruses.
GluA2/N-cadherin regulates hippocampal mGluR-LTD via cofilin
The above experiments indicated that both GluA2/N-cadherin and cofilin-mediated actin reorganization are necessary for mGluR-LTD, but the question remained whether these two processes are related. To test whether cofilin is a downstream target of GluA2/N-cadherin, we performed two sets of experiments. First, we examined the effect of mGluR activation on Rac1/cofilin and showed that DHPG induced GluA2-dependent Rac1/cofilin activation. As shown in Figure 5A,B, DHPG treatment induced rapid activation of both cofilin and Rac1 in WT slices (p-cofilin/cofilin: 0.65 ± 0.09, p < 0.05 compared with untreated; active Rac1/total Rac1: 1.50 ± 0.19, p < 0.05 compared with untreated), and this activation was absent in GluA2 KO samples. In fact, there was small but significant inhibition of both cofilin and Rac1 by the treatment in the KO sample (Fig. 5A,B) (p-cofilin/cofilin: untreated = 0.96 ± 0.04; treated = 1.23 ± 0.09; p < 0.05; active Rac1/total Rac1: untreated = 1.02 ± 0.04; treated = 0.75 ± 0.11; p = 0.05). The DHPG-induced increase (compared with a decrease in WT) in both p-cofilin and N-cadherin/β-catenin binding (Fig. 3D) in GluA2 KO mice is of interest and suggests that a GluA2-independent, opposing signaling pathway targeting N-cadherin/β-catenin and cofilin must be activated by DHPG treatment. It is possible that in the absence of GluA2, this opposing signaling pathway becomes dominant and leads to increased p-cofilin and N-cadherin/β-catenin binding. In clear contrast to Rac1, DHPG treatment induced no changes in RhoA activity in both WT and GluA2 KO samples (Fig. 5C: WT DHPG treated = 1.06 ± 0.05; GluA2 KO untreated = 0.98 ± 0.19; GluA2 KO treated = 1.02 ± 0.15; p > 0.05 compared with WT untreated).
Figure 5.
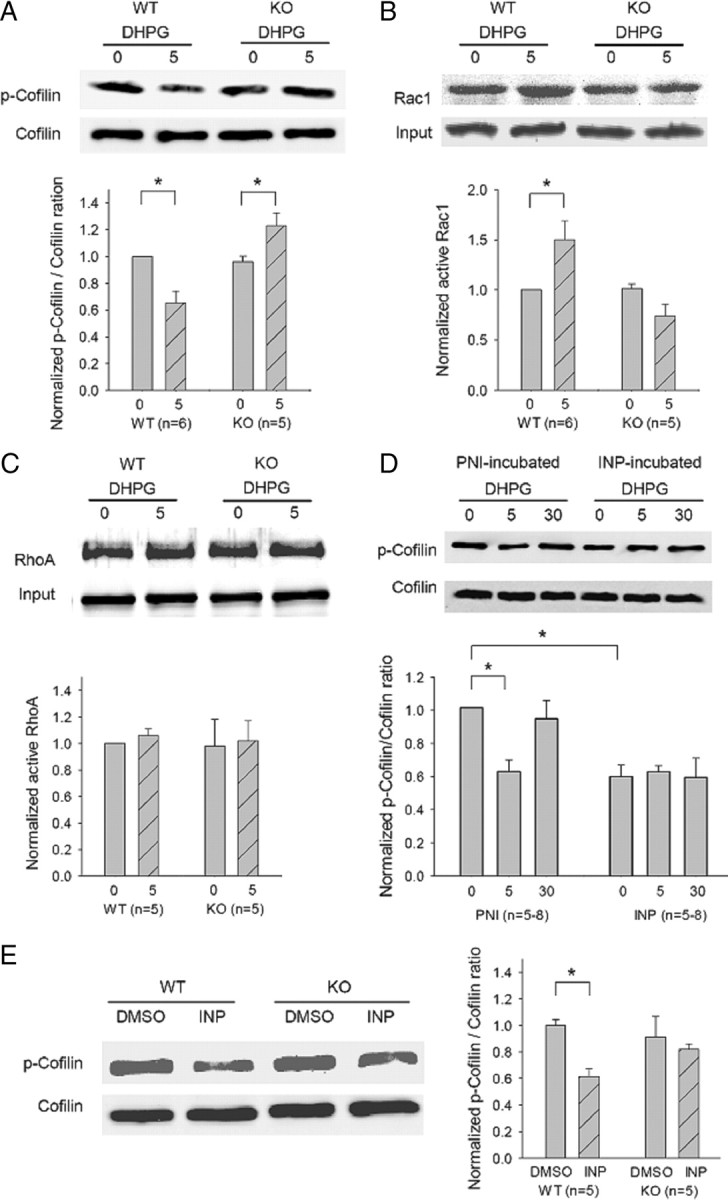
Dependence of DHPG-induced cofilin activation on GluA2 and N-cadherin. A, Western blot analysis and summary graph of cofilin activity using protein lysates prepared from hippocampal slices treated with DHPG (100 μm) for 0 min (untreated control) or for 5 min, showing a significant reduction in the amount of phosphorylated (inactive) cofilin (p-cofilin) by the treatment in WT, but not in GluA2 KO samples. The total protein level of cofilin was not affected by the treatment in either genotype. B, Western blot analysis and summary graph of Rac1 activation assay showing a significant increase in the amount of active Rac1 by DHPG treatment in WT, but not in GluA2 KO samples. The total protein level of Rac1 was not affected by the treatment in either genotype. C, Western blot analysis and summary graph of RhoA activation assay showing lack of DHPG effect in both WT and GluA2 KO samples. D, Western blot analysis and summary graph of total and phosphorylated cofilin using protein lysates prepared from hippocampal slices pretreated with control PNI or INP peptide (50 μm, 30 min) followed by treatment with (5, 30 min) or without (0 min) DHPG (100 μm) showing activation of basal cofilin and inhibition of subsequent DHPG-induced cofilin activation by the INP peptide, but not by the PNI peptide preincubation. E, Western blot analysis and summary graph of cofilin and p-cofilin using protein lysates from hippocampal slices treated with INP or PNI/DMSO peptide (50 μm, 30 min) showing cofilin activation in INP-treated slices in the WT, but not in GluA2 KO samples.
Consistent with previous studies (Gallagher et al., 2004; Mao et al., 2005), we found that an inhibitor for the mitogen-activated protein kinase ERK1/2 (control = 78.46 ± 2.11%; PD98059 = 103.14 ± 3.64%; p < 0.001) but not for the p38 kinase (DMSO = 82.55 ± 2.38%; SB239063 = 82.92 ± 5.30%) prohibited DHPG-induced LTD (supplemental Fig. 4A, available at www.jneurosci.org as supplemental material). Accordingly, DHPG treatment induced a significant increase in the active form of ERK1/2 (phosphorylated ERK1/2) in WT (supplemental Fig. 4B, available at www.jneurosci.org as supplemental material) (DHPG = 1.38 ± 0.09, p < 0.01 compared with untreated), and this effect remained intact in GluA2 KO samples (DHPG = 1.34 ± 0.08, p < 0.05 compared with untreated). These experiments indicated that GluA2 is specifically required for mGluR-dependent Rac1/cofilin activation. Second, we showed that DHPG-induced cofilin activation was also dependent on proper N-cadherin function. Again, we pretreated the slices with the INP or PNI peptide and then examined DHPG-induced cofilin activation. As shown in Figure 5D, DHPG significantly reduced the amount of phosphorylated cofilin in PNI-pretreated slices [DHPG (5 min) = 0.63 ± 0.07, p < 0.01 compared with DHPG untreated; DHPG (30 min) = 0.95 ± 0.11, p > 0.05 compared with DHPG untreated], but not in INP-pretreated slices [untreated = 0.60 ± 0.07; DHPG (5 min) = 0.63 ± 0.04; DHPG (30 min) = 0.60 ± 0.12; p < 0.05 compared with PNI/DHPG (0 min); p > 0.05 compared between them]. It is important to note that INP-pretreated slices had a significantly lower level of phosphorylated cofilin under basal conditions before DHPG treatment, indicating that INP treatment was sufficient to induce cofilin activation. In addition, cofilin activation induced by the INP treatment observed in WT slices was absent in GluA2 KO samples (Fig. 5E) (GluA2 KO/untreated = 0.91 ± 0.15; GluA2 KO/treated = 0.82 ± 0.04; p > 0.05). Since N-cadherin/catenin can also initiate signaling processes involving gene expression and protein synthesis, we tested whether DHPG-induced cofilin activation required these processes. As shown in supplemental Figure 4C (available at www.jneurosci.org as supplemental material), DHPG-induced cofilin dephosphorylation was not affected either by the transcription inhibitor actinomycin or protein synthesis inhibitor cycloheximide. These biochemical results are consistent with the idea that GluA2/N-cadherin regulates mGluR-LTD via protein synthesis-independent, cofilin-mediated actin dynamics.
mGluR activation induces GluA2-dependent β-catenin and cofilin changes at the synaptic level in hippocampal neurons
The above experiments on acute hippocampal slices have shown that GluA2/N-cadherin/cofilin signaling is activated in and required for mGluR-LTD, but whether this signaling is activated at the synaptic level remains unknown. To address this issue, we extended our studies to cultured hippocampal neurons where synaptic properties can be better analyzed. First, we established DHPG-induced LTD in the culture system and showed that it shares the same mechanistic processes as in acute slices (Fig. 6A; supplemental Fig. 5, available at www.jneurosci.org as supplemental material). Consistent with previous studies (Snyder et al., 2001), DHPG treatment caused a persistent decrease in the frequency (measured as increased interevent interval, 179.33 ± 11.39% of basal level) (Fig. 6A), but not in the amplitude (97.11 ± 4.37% of basal level) (supplemental Fig. 5A, available at www.jneurosci.org as supplemental material) of miniature EPSCs (mEPSCs). Importantly, DHPG-induced LTD in hippocampal slices was also associated with a decrease in the frequency in mEPSCs with minimal alterations in the amplitude (supplemental Fig. 5B, available at www.jneurosci.org as supplemental material). Similar to slices, DHPG-induced LTD in cultured neurons was dependent on GluA2 (Fig. 6A: WT = 179.33 ± 11.39%; KO = 115.21 ± 14.68%; p < 0.001), protein synthesis, ERK signaling, Rac1 activation, actin dynamics, and cofilin (supplemental Fig. 5C–H, available at www.jneurosci.org as supplemental material). These experiments indicated that DHPG-induced LTD in cultured neurons employs the same mechanism as that of acute hippocampal slices.
Figure 6.
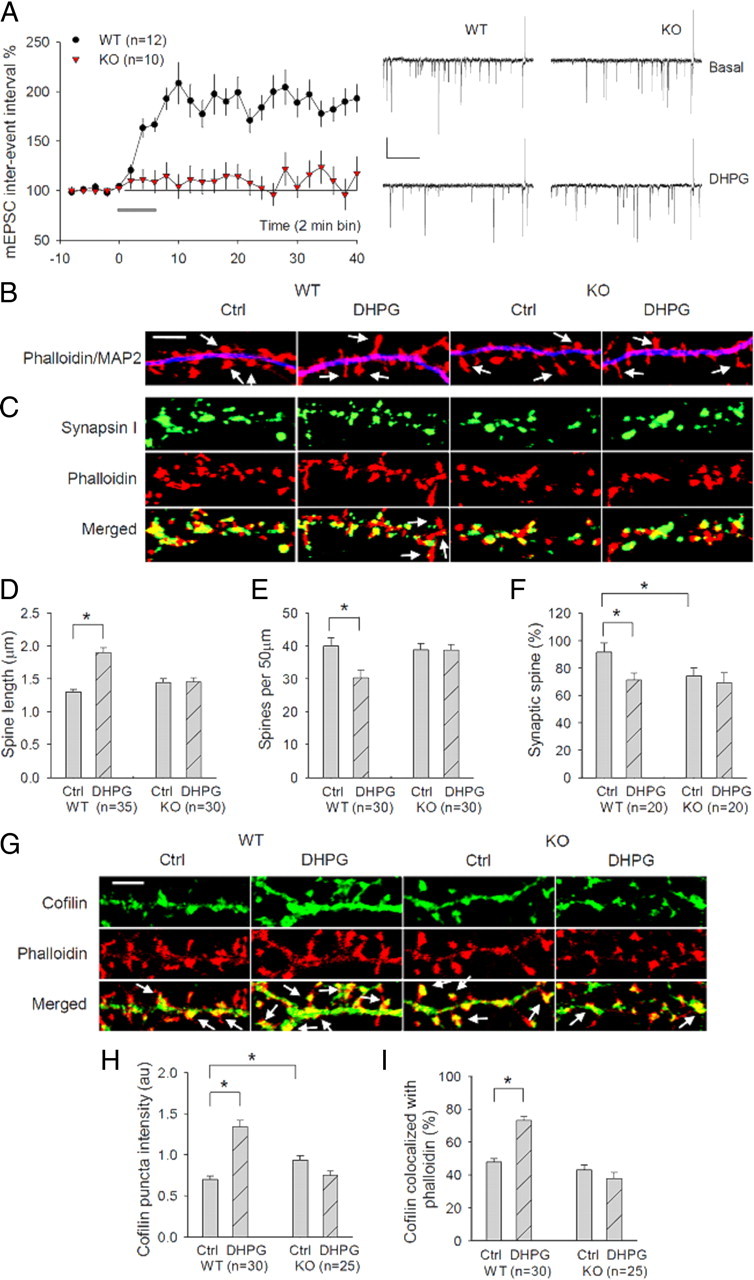
GluA2 requirement for mGluR-LTD, DHPG-induced cofilin activation and spine plasticity in cultured neurons. A, Whole-cell recordings of mEPSCs from cultured hippocampal neurons showing a persistent increase in the interevent interval (i.e., LTD) in WT neurons, but not in GluA2 KO neurons, in response to 5 min application of 100 μm DHPG added at 0 min (solid line). Calibration: 15 pA/0.5 s. B, Cultured hippocampal neurons stained with the spine marker F-actin dye phalloidin (red) and the dendritic marker MAP2 (blue) showing spine elongation and reduced spine density by DHPG treatment in WT neurons, but not in GluA2 KO neurons. Arrows indicate the dendritic spines. Scale bar, 5 μm. C, Cultured hippocampal neurons stained with the spine marker F-actin dye phalloidin (red) and the presynaptic marker synapsin showing spine elongation and decrease in synaptic spines (defined with synapsin puncta overlapping the spine head) by DHPG treatment in WT neurons, but not in GluA2 KO neurons. Arrows indicate the dendritic spines without synapsin puncta overlapping the spine head. Scale bar, 5 μm. D–F, Summary graphs showing significantly increased spine length (D), but reduced spine density (E) and synaptic spines (F), after DHPG treatment in WT neurons, but not in GluA2 KO neurons. G, Cultured hippocampal neurons costained with cofilin (green) and phalloidin (red) showing increased spine cofilin (measured by colocalization with phalloidin) after DHPG treatment in WT, but not in GluA2 KO neurons. Arrows indicate cofilin puncta colocalized with phalloidin in the spines. Also note DHPG-induced spine elongation in WT, but not in GluA2 KO neurons. Scale bar, 5 μm. H, I, Summary graphs showing a significant increase in both the mean puncta intensity (H) and spine accumulation (I) of cofilin after DHPG treatment in WT neurons, but not in GluA2 KO neurons. n represents the number of neurons from three independent cultures. au, Arbitrary unit.
Having established the in vitro system, we then examined DHPG-induced changes at the synapse by using the F-actin dye rhodamine phalloidin as a spine marker. F-actin is highly enriched in the dendritic spine and has been used widely as a useful indicator of spine and synaptic plasticity. First, we showed that DHPG-induced spine plasticity was dependent on GluA2. In WT neurons, DHPG treatment induced a significant increase in the mean spine length (Fig. 6B,D: control = 1.30 ± 0.04 μm; DHPG = 1.90 ± 0.07 μm; p < 0.001) and a significant decrease in the spine density (Fig. 6B,E: control = 39.95 ± 2.61; DHPG = 30.34 ± 2.30; p < 0.01). These results were similar to those previously reported (Vanderklish and Edelman, 2002) and were verified by using EGFP-infected neurons to visualize spines (data not shown). Because of spine elongation, the location of the presynaptic marker synaptophysin staining puncta in relation to the spine was altered; many synaptophysin puncta were found at the base of the spine after DHPG treatment as opposed to at the spine head in untreated neurons (Fig. 6C). For easy analysis, we defined the spines with synapsin or synaptophysin puncta overlapping the spine head as synaptic or normal spines (although it should be stressed here that those elongated spines with synapsin/synaptophysin puncta at the base or neck of the spine may still have functional synapses). Thus, DHPG treatment resulted in a significant reduction in normal spines (Fig. 6F: control = 91.64 ± 6.89%, DHPG = 71.27 ± 5.06%, p < 0.05; also see supplemental Fig. 6, available at www.jneurosci.org as supplemental material). However, in GluA2 KO neurons, DHPG treatment had no effect on the spine length (Fig. 6B,D: control = 1.44 ± 0.06 μm; DHPG = 1.45 ± 0.06 μm; p > 0.05), the spine density (Fig. 6E: control = 38.81 ± 2.00; DHPG = 38.75 ± 1.71; p > 0.05), or the percentage of normal spines (Fig. 6C,F: control = 74.36 ± 5.65%; DHPG = 69.20 ± 7.3%; p > 0.05). We next analyzed DHPG-induced cofilin changes in the dendritic spines. Under basal conditions, cofilin-staining puncta were partially colocalized with phalloidin puncta, indicating that only some cofilin was distributed in the spine. Following DHPG treatment, both the intensity of cofilin puncta (Fig. 6G,H: control = 1.00 ± 0.06; DHPG = 1.92 ± 0.11; p < 0.001) and their colocalization with phalloidin (Fig. 6G,I: control = 47.84 ± 2.01%; DHPG = 73.12 ± 2.43%; p < 0.001) were significantly increased in WT neurons, but these changes were not observed in GluA2 KO neurons (Fig. 6G–I) (punctum intensity: control = 1.34 ± 0.08; DHPG = 1.07 ± 0.08; p > 0.05; puncta colocalized with phalloidin: control = 43.06 ± 2.83%; DHPG = 37.61 ± 3.83%; p > 0.05).
To confirm the role of the N-cadherin/β-catenin complex and its dependence on GluA2 in the culture system, we also examined DHPG-induced changes in β-catenin distribution in the dendritic spine. In both WT and GluA2 KO neurons, β-catenin immunostaining puncta were prominent and highly colocalized with phalloidin clusters, indicating that they were predominantly localized in the spines. We therefore analyzed both the density and intensity of the β-catenin puncta pre- and post-DHPG treatment. In WT neurons, DHPG treatment significantly reduced the mean intensity of β-catenin puncta (Fig. 7A,B: basal = 1.00 ± 0.11; DHPG = 0.73 ± 0.04; p < 0.05), but this reduction was absent in GluA2 KO neurons (Fig. 7A,B: basal = 0.58 ± 0.03; DHPG = 0.50 ± 0.04; p > 0.05). Interestingly, the punctum density of β-catenin staining was not significantly altered by DHPG treatment in either genotype (Fig. 7A,C: WT basal = 55.41 ± 6.75; WT DHPG = 45.51 ± 5.03; KO basal = 51.65 ± 3.06; KO DHPG = 45.40 ± 6.95; p > 0.05 for any comparison). It is important to note that under basal conditions the mean intensity of β-catenin puncta was reduced in GluA2 KO compared with WT neurons (Fig. 7A,B). In agreement with the recording results, DHPG treatment resulted in a significant decrease in the amount of total surface and synaptic GluA1 in WT neurons (Fig. 7D,E: basal = 42.24 ± 3.41; DHPG = 29.63 ± 3.92; p < 0.05), but this DHPG-induced decrease in GluA1 was also abolished in GluA2 KO neurons (Fig. 7D,E: basal = 30.35 ± 4.96; DHPG = 29.26 ± 3.12; p > 0.05). DHPG treatment did not cause any changes in the presynaptic marker synapsin in either genotype (Fig. 7D,F) (WT: basal = 44.24 ± 3.41; DHPG = 43.07 ± 3.99; KO: basal = 40.35 ± 4.96; DHPG = 39.26 ± 3.12; p > 0.05 for any comparison). These results in cultured neurons are consistent with the idea that DHPG induces GluA2-dependent functional changes in the N-cadherin/catenin/cofilin signaling at the synapse.
Figure 7.
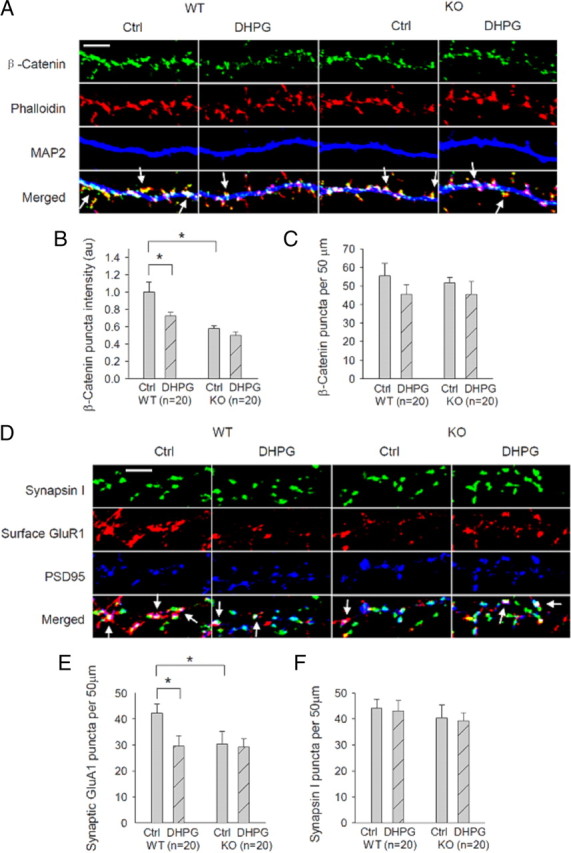
Requirement of GluA2 for DHPG-induced β-catenin changes in the dendritic spines. A, Cultured hippocampal neurons costained with the spine marker F-actin dye phalloidin (red), the dendritic marker MAP2 (blue), and anti-β-catenin (green) showing DHPG-induced decrease in the mean puncta intensity of β-catenin in WT neurons, but not in GluA2 KO neurons. Arrows indicate β-catenin immunostaining puncta colocalized with phalloidin. Scale bar, 5 μm. B, C, Summary data showing that the mean puncta intensity of β-catenin is significantly reduced by DHPG-treatment in WT but not in GluA2 KO neurons (B). Note the basal puncta intensity of β-catenin is significantly lower in GluA2 KO compared with WT neurons. The β-catenin puncta density is not significantly altered by the treatment in either genotype (C). D, Cultured hippocampal neurons costained for presynaptic marker synapsin I (green), the postsynaptic marker PSD95 (blue), and GluA1 (red), showing reduced synaptic GluA1 after DHPG treatment in WT neurons, but not in GluA2 KO neurons. Arrows indicate synaptic GluA1 puncta (i.e., surface GluA1 colocalized with both synapsin I and PSD95). Scale bar, 5 μm. E, F, Summary data showing significantly reduced synaptic GluA1 (E), but not synapsin (F), after DHPG treatment in WT neurons, but not in GluA2 KO neurons. Note the basal synaptic GluA1 is also significantly lower in GluA2 KO than in WT neurons. n represents the number of neurons from three independent cultures.
Hippocampal mGluR-LTD deficit in GluA2 KO mice is functionally rescued by manipulating cofilin activity
As a further proof that cofilin is responsible for the regulatory effect of GluA2/N-cadherin on mGluR-LTD, we performed functional rescue experiments. We reasoned that if GluA2/N-cadherin regulates mGluR-LTD through cofilin and the absence of mGluR-LTD in GluA2 KO was due to the lack of cofilin activation by mGluR, then the LTD deficit in the KO mice should be rescued or reduced by manipulating cofilin activity. We used a number of strategies to test this possibility. First, we included anti-LIMK1 antibodies or cofilin peptides in the recording pipette to enhance cofilin activity. LIMK1 is the major form of the LIMK family in the brain, and it plays the predominant role in inhibiting cofilin activity by phosphorylation (Meng et al., 2002; Bernard, 2007). We expected that by inhibiting LIMK1 in GluA2 KO neurons, cofilin activity in these cells could be increased to a level sufficient for mGluR-LTD. As shown in Figure 8A, while the anti-LIMK-1 antibody had no effect on basal synaptic responses, DHPG-induced LTD was fully restored (control IgG = 121.04 ± 7.35%; LIMK1 antibody = 66.32 ± 3.78%; p < 0.001). The control IgG had no effect on either basal transmission or DHPG-induced LTD. Neither control IgG nor anti-LIMK1 antibodies had any effects on DHPG-induced LTD in WT animals (Fig. 8B: control IgG = 75.96 ± 4.12%; LIMK1 antibody = 73.25 ± 7.17%; p > 0.05). Similarly, the inclusion of the S3 peptide, but not the p-S3 peptide, also fully rescued DHPG-induced LTD (Fig. 8C: p-S3 = 114.77 ± 6.62%; S3 = 82.12 ± 3.45%; p < 0.005). Second, we generated and analyzed double KO mice lacking both LIMK1 and GluA2. We have previously shown that cofilin activity is enhanced in the absence of LIMK1 (Meng et al., 2002), and therefore we expected that the double KO mice would have a higher cofilin activity than GluA2 KO mice, enabling mGluR-LTD induction. As shown in Figure 8D, DHPG-induced LTD was also completely restored in the double KO mice (GluA2 KO = 119.04 ± 9.28%; GluA2/LIMK1 double KO = 58.71 ± 5.30%; p < 0.001). In fact, the magnitude of DHPG-induced LTD in GluA2/LIMK1 double KO mice was similar to those in LIMK1 KO and WT animals (Fig. 8D,F) and was still sensitive to the protein synthesis inhibitor cycloheximide (supplemental Fig. 4D, available at www.jneurosci.org as supplemental material). Finally, we tested whether the blockade of mGluR-LTD by acute disruption of GluA2/N-cadherin signaling in WT mice could also be rescued by cofilin. In previous experiments, we showed that INP treatment blocked DHPG-induced LTD (Fig. 3), and if this effect was due to perturbed cofilin function, as suggested by the biochemical data (Fig. 5), then this DHPG-induced LTD deficit could also be rescued by manipulating cofilin. As shown in Figure 8E, both peptide S3 and anti-LIMK1 antibodies, but not peptide p-S3 restored DHPG-induced LTD in INP-treated slices (pS3 = 102.68 ± 2.19%; S3 = 74.16 ± 3.89%; p < 005 compared with pS3; anti-LIMK1 = 69.40 ± 2.39%; p < 0.005 compared with pS3), indicating that the effect of N-cadherin is mediated by cofilin. These rescue experiments together provided compelling functional evidence that the mGluR-LTD deficit by either genetic deletion of GluA2 or acute disruption of N-cadherin signaling is due to the lack of cofilin activation by mGluR, and together with above biochemical data indicating that cofilin is the key downstream target of GluA2/N-cadherin and that GluA2/N-cadherin controls mGluR-LTD via regulating cofilin.
Figure 8.
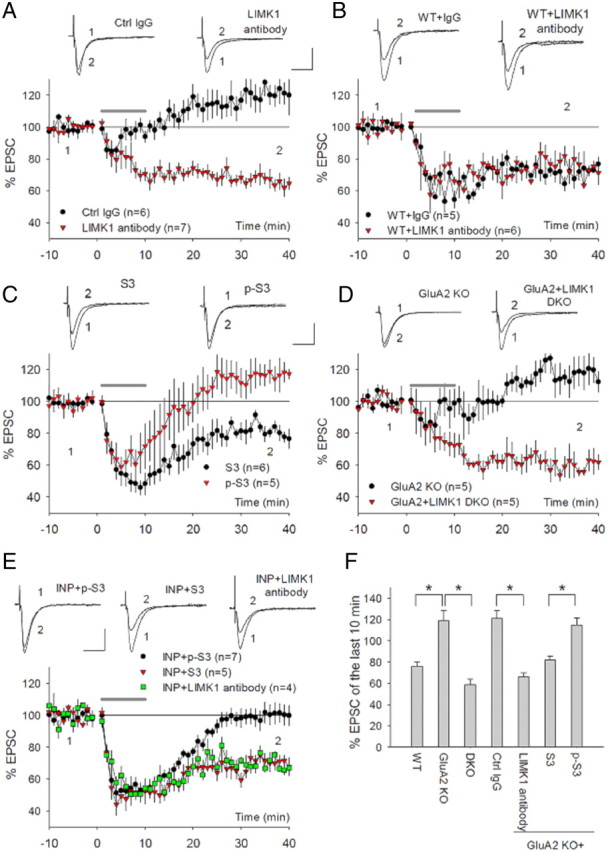
Functional rescue of mGluR-LTD deficit in GluA2 KO mice by manipulating cofilin activity. A, Whole-cell recordings showing restoration of DHPG-induced LTD in GluA2 KO neurons infused with anti-LIMK1 antibody (10 μg/ml), but not with control IgG (10 μg/ml). Neither anti-LIMK1 antibody nor control IgG had noticeable effects on basal synaptic responses before DHPG application. B, Whole-cell recordings showing lack of effect of either anti-LIMK1 antibody (10 μg/ml) or control IgG (10 μg/ml) on DHPG-induced LTP in WT neurons. C, Whole-cell recordings showing restoration of DHPG-induced LTD in GluA2 KO neurons infused with peptide S3 (200 μm), but not with peptide p-S3 (200 μm). D, Whole-cell recordings showing rescue of DHPG-induced LTD in GluA2/LIMK1 double KO mice. The DHPG-induced LTD was absent in the GluA2 KO mice, which were derived from the same breeding used for generating the double KO mice, and therefore they were similar in genetic background. E, Whole-cell recordings showing restoration of DHPG-induced LTD in INP-treated neurons (50 μm, 30 min) by infusing S3 peptide (200 μm) or anti-LIMK-1 (10 μg/ml), but not by peptide p-S3 (200 μm). F, Summary data showing the magnitudes of DHPG-induced LTD of various genotypes/manipulations used in the rescue experiments by manipulating cofilin. The DHPG-induced LTD in LIMK1 antibody-treated, peptide S3-treated, and GluA2/LIMK1 double KO neurons was similar to that of the WT neurons but was significantly different from that of untreated GluA2 KO neurons or those treated with peptide p-S3 or control IgG. DKO, GluA2/LIMK1 double KO; Ctrl IgG, GluA2 KO infused with control IgG; LIMK antibody, GluA2 KO neurons infused with anti-LIMK1 antibodies; S3, GluA2 KO neurons infused with peptide S3; p-S3, GluA2 KO neurons infused with peptide p-S3.
Discussion
Regulated protein interactions play a critical role in AMPAR properties and expression of long-lasting synaptic plasticity. However, despite the extensive studies, little is known about the significance and underlying mechanisms of the protein interactions involving the NTD of AMPARs. In this study, we demonstrate that the GluA2 NTD/N-cadherin interaction regulates hippocampal mGluR-LTD through a previously unknown actin-based mechanism that is distinct from those used by the other domains of the subunit.
In GluA2 KO mice, both DHPG- and PP-LFS-induced mGluR-LTD are absent, indicating that GluA2 is essential for mGluR-dependent plasticity in the hippocampus. The lack of mGluR-LTD in GluA2 KO mice is not likely due to a developmental effect because the deficit can be completely rescued by acutely expressing various GluA2 constructs or by manipulating cofilin in the adult hippocampal neurons (Figs. 1, 2, 8). Therefore, unlike NMDAR-dependent LTD where GluA2-independent mechanisms may also exist, GluA2 is an essential component for mGluR-LTD in the hippocampus. Although basal synaptic response is reduced in GluA2 KO mice (Jia et al., 1996; Meng et al., 2003), this change is unlikely to contribute to the LTD deficit for a number of reasons. First, the DHPG-LTD deficit of the KO mice is rescued by LIMK-1 antibodies or S3 peptide, without affecting the basal synaptic response (Fig. 8). Second, the GluA2/LIMK-1 double KO mice have similar basal synaptic responses as GluA2 KO mice (i.e., smaller than the WT mice), but DHPG-LTD can be established in the double KO mice (Fig. 8). These experiments indicate that the absence of DHPG-LTD in GluA2 KO mice is not due to smaller basal synaptic response, but rather due to a lack of cofilin activation.
GluA2 can exert its unique regulatory effects on AMPAR properties and synaptic plasticity through specific interactions with several synaptic proteins, among which the CTD interactions with NSF, PICK1, and ABP/GRIP are best studied and shown to be critically involved in AMPAR internalization and expression of LTD (Isaac et al., 2007). Thus, it is rather surprising and of special interest to us that the regulation of mGluR-LTD by GluA2 requires its NTD interaction with N-cadherin. Several pieces of evidence support this claim. First, we show that the N-cadherin binding motif on the GluA2 NTD is both necessary and sufficient for GluA2 to rescue mGluR-LTD deficit in GluA2 KO mice (Fig. 2). Second, we show that N-cadherin/catenin complex is required for mGluR-LTD and that mGluR activation induces functional changes in the N-cadherin/β-catenin complex (Fig. 3). Third, we show that GluA2 is critical for this N-cadherin/catenin function because GluA2 KO mice are impaired in both basal and DHPG-induced functional changes in the complex (Fig. 3). And finally, DHPG treatment induces changes in N-cadherin/catenin complex at the synaptic level, which are also GluA2 dependent (Fig. 7). These results together indicate a critical importance of N-cadherin in mediating the effect of GluA2 on mGluR-LTD. It is interesting to note that the extracellular domain of neuronal pentraxin receptor (NPR) has recently been shown to undergo regulated cleavage by the matrix metalloprotease tumor necrosis factor-α-converting enzyme in response to mGluR activation, and to require this process for DHPG-induced LTD in the hippocampus. It is thus proposed that the cleaved extracellular domain of NPR enables NPR to cluster and cocluster with AMPARs and thereby facilitates the rate of AMPAR internalization (Cho et al., 2008). This study, along with our present findings, supports the emerging importance of extracellular protein interactions in the regulation of long-lasting synaptic plasticity. It would be important to investigate whether NPR interacts with GluA2/N-cadherin complex, and, if so, how the interaction is regulated during mGluR-LTD.
What then are the downstream molecular substrates activated by the GluA2/N-cadherin interaction? Our study shows that the Rac1/cofilin-mediated actin is the key target. First, we demonstrate that Rac1/cofilin-mediated actin reorganization is essential for mGluR-LTD (Fig. 4; supplemental Fig. 5, available at www.jneurosci.org as supplemental material). It is interesting to note that inhibition of RhoA, a closely related family member of Rac1, has no inhibitory effect on mGluR-LTD, suggesting a specific role of Rac1-dependent cofilin regulation. Second, we show that DHPG treatment induces Rac1/cofilin activation that is dependent on both GluA2 and N-cadherin (Fig. 5). Third, the effect of the INP peptide on DHPG-induced LTD is alleviated by manipulating cofilin (Fig. 8E). And finally, the mGluR-LTD deficit in GluA2 KO mice is fully rescued by manipulating cofilin activity (Fig. 8). These results provide compelling evidence that GluA2/N-cadherin regulates hippocampal mGluR-LTD via activation of Rac1/cofilin. It is interesting to note that DHPG-induced cofilin activation is relatively short-lasting, being back to the basal level within 30 min after the treatment (Fig. 5D), as opposed to DHPG-induced reduction in N-cadherin/β-catenin binding, which remains stable for at least 30 min after the treatment (Fig. 3C,D). These results suggest that in addition to GluA2/N-cadherin, other molecules and signaling pathways must also be activated by DHPG treatment to regulate cofilin. The reason for this transient cofilin activation is unknown but may reflect the dynamic nature of actin regulation. It is conceivable that once cofilin-mediated actin reorganization has occurred, the active cofilin needs be inactivated again to stabilize the newly established actin network to maintain plasticity.
How does GluA2/N-cadherin-activated and cofilin-mediated actin reorganization regulate hippocampal mGluR-LTD? Given the well established role of the actin cytoskeleton in dictating the structural properties of the synapses and spines (Cingolani and Goda, 2008), it is likely that activation of this signaling leads to spine morphological changes and/or elimination of the synapse. Therefore, GluA2/N-cadherin interaction may provide a mechanism to regulate synaptic plasticity independent of AMPAR trafficking. However, it is also possible that actin reorganization triggered by GluA2/N-cadherin signaling may have an effect on GluA2-dependent AMPAR trafficking. Local actin turnover has been proposed to provide mechanical forces that contribute to plasma membrane curvature, vesicle scission, and propulsion of vesicles off the membrane (Kaksonen et al., 2006). It has been shown that actin reorganization and cofilin-directed vesicle trafficking are important for AMPAR internalization in hippocampal neurons and acetylcholine receptor clustering at Xenopus neuromuscular junctions, respectively (Zhou et al., 2001; Lee et al., 2009), and that GluA2/PICK1 interactions regulate NMDA-induced AMPAR internalization through their effect on actin reorganization (Rocca et al., 2008). In any case, our present study has revealed that, in addition to directly participating in AMPAR trafficking, GluA2 can also trigger a signaling process as an essential step to establish both spine and synaptic plasticity during mGluR-LTD.
It is well established that mGluR-LTD requires local protein synthesis (Lüscher and Huber, 2010). For example, rapid translation of Arc/Arg3.1 has been shown to be essential for mGluR-LTD and DHPG-induced AMPAR internalization in the hippocampus (Waung et al., 2008). In ventral tegmental area, inhibition of protein translation using GluA2-specific antisense oligos blocks mGluR-LTD, suggesting a direct requirement for GluA2 protein synthesis (Mameli et al., 2007). How then is the GluA2/N-cadherin-activated cofilin signaling that we have demonstrated in the present study related to these processes? Our data support that GluA2/N-cadherin-dependent cofilin signaling may work independently and in parallel with the protein synthesis-dependent processes (e.g., Arc/Arg3.1-dependent AMPAR trafficking). First, although DHPG induces a reduction in phosphorylated cofilin, the total protein level of cofilin is not altered by the treatment (Fig. 5). Second, GluA2/N-cadherin-mediated cofilin activation is independent of either transcription or protein synthesis (supplemental Fig. 4C, available at www.jneurosci.org as supplemental material). Third, mGluR-LTD deficit in GluA2 KO mice is fully rescued by increasing cofilin activity; therefore, the absence of mGluR-LTD in GluA2 KO mice is not likely due to the absence of GluA2 per se (e.g., GluR2 protein synthesis or its direct involvement in AMPAR internalization), but rather is due to the lack of GluA2/N-cadherin-dependent, protein synthesis-independent cofilin signaling process. Because mGluR-LTD in GluA2/LIMK-1 double KO mice still requires new protein synthesis (supplemental Fig. 4D, available at www.jneurosci.org as supplemental material), activation of cofilin alone is thus not sufficient for mGluR-LTD. This is also consistent with the result that application of INP peptide, although being capable of activating cofilin, is not sufficient to induce LTD (supplemental Fig. 2B, available at www.jneurosci.org as supplemental material). However, we cannot rule out the possibility that GluA2/N-cadherin-dependent cofilin signaling and protein synthesis may represent sequential but different aspects of LTD, with the former being important for the induction and the latter being important for a consolidation/expression mechanism. Further experiments will be needed to address these interesting questions.
Long-lasting synaptic plasticity, including mGluR-LTD, is usually associated with structural changes at the synapse (Vanderklish and Edelman, 2002; Alvarez and Sabatini, 2007; Bourne and Harris, 2008). However, how these two forms of plasticity are coordinated is poorly understood. Given the trans-synaptic nature of N-cadherin molecules and their ability to regulate the actin cytoskeleton, our demonstration that the GluA2/N-cadherin interaction is critical for mGluR-LTD and associated spine plasticity has important implications in this respect. By placing N-cadherin under the direct control of GluA2, postsynaptic responses mediated by AMPARs are coupled not only to the postsynaptic structure, but also to the presynaptic element, to achieve structural and functional coupling during long-lasting synaptic plasticity.
In summary, we have identified a previously unknown N-cadherin-dependent, cofilin-mediated signaling process by which the NTD of GluA2 regulates mGluR-LTD in the hippocampus (for a summary model, see supplemental Fig. 6, available at www.jneurosci.org as supplemental material). Further analysis of this pathway may provide a valuable venue for understanding molecular mechanisms and functional/structural coupling underlying long-lasting synaptic plasticity in the central synapse.
Footnotes
This work was supported by grants from the Canadian Institutes of Health Research (Grant MOP-42396, to Z.P.J.), Ontario Mental Health Foundation (Z.P.J.), and the National Basic Research Program (973 Program) (Grant 2005CB522501, to W.X.). We are grateful to Graham Collingridge for the suggestions on the experiments, and to Yanghong Meng, Suhail Asrar, Zarko Todorovski, Wayne Huang, Jeff Henderson, and Shouping Zhang for technical assistance and comments on the manuscript.
References
- Aizawa H, Wakatsuki S, Ishii A, Moriyama K, Sasaki Y, Ohashi K, Sekine-Aizawa Y, Sehara-Fujisawa A, Mizuno K, Goshima Y, Yahara I. Phosphorylation of cofilin by LIMkinase is necessary for semaphorin 3A-induced growth cone collapse. Nat Neurosci. 2001;4:367–373. doi: 10.1038/86011. [DOI] [PubMed] [Google Scholar]
- Alvarez VA, Sabatini BL. Anatomical and physiological plasticity of dendritic spines. Annu Rev Neurosci. 2007;30:79–97. doi: 10.1146/annurev.neuro.30.051606.094222. [DOI] [PubMed] [Google Scholar]
- Arikkath J, Reichardt LF. Cadherins and catenins at synapses: roles in synaptogenesis and synaptic plasticity. Trends Neurosci. 2008;31:487–494. doi: 10.1016/j.tins.2008.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bamburg JR. Proteins of the ADF/cofilin family: essential regulators of actin dynamics. Annu Rev Cell Dev Biol. 1999;15:185–230. doi: 10.1146/annurev.cellbio.15.1.185. [DOI] [PubMed] [Google Scholar]
- Bamji SX. Cadherins: actin with the cytoskeleton to form synapses. Neuron. 2005;47:175–178. doi: 10.1016/j.neuron.2005.06.024. [DOI] [PubMed] [Google Scholar]
- Barcia C, Jimenez-Dalmaroni M, Kroeger KM, Puntel M, Rapaport AJ, Larocque D, King GD, Johnson SA, Liu C, Xiong W, Candolfi M, Mondkar S, Ng P, Palmer D, Castro MG, Lowenstein PR. One-year expression from high-capacity adenoviral vectors in the brains of animals with pre-existing anti-adenoviral immunity: clinical implications. Mol Ther. 2007;15:2154–2163. doi: 10.1038/sj.mt.6300305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernard O. Lim kinases, regulators of actin dynamics. Int J Biochem Cell Biol. 2007;39:1071–1076. doi: 10.1016/j.biocel.2006.11.011. [DOI] [PubMed] [Google Scholar]
- Biou V, Bhattacharyya S, Malenka RC. Endocytosis and recycling of AMPA receptors lacking GluR2/3. Proc Natl Acad Sci U S A. 2008;105:1038–1043. doi: 10.1073/pnas.0711412105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourne JN, Harris KM. Balancing structure and function at hippocampal dendritic spines. Annu Rev Neurosci. 2008;31:47–67. doi: 10.1146/annurev.neuro.31.060407.125646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bredt DS, Nicoll RA. AMPA receptor trafficking at excitatory synapses. Neuron. 2003;40:361–379. doi: 10.1016/s0896-6273(03)00640-8. [DOI] [PubMed] [Google Scholar]
- Brown TC, Tran IC, Backos DS, Esteban JA. NMDA receptor-dependent activation of the small GTPase Rab5 drives the removal of synaptic AMPA receptors during hippocampal LTD. Neuron. 2005;45:81–94. doi: 10.1016/j.neuron.2004.12.023. [DOI] [PubMed] [Google Scholar]
- Cho RW, Park JM, Wolff SB, Xu D, Hopf C, Kim JA, Reddy RC, Petralia RS, Perin MS, Linden DJ, Worley PF. mGluR1/5-dependent long-term depression requires the regulated ectodomain cleavage of neuronal pentraxin NPR by TACE. Neuron. 2008;57:858–871. doi: 10.1016/j.neuron.2008.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cingolani LA, Goda Y. Actin in action: the interplay between the actin cytoskeleton and synaptic efficacy. Nat Rev Neurosci. 2008;9:344–356. doi: 10.1038/nrn2373. [DOI] [PubMed] [Google Scholar]
- Collingridge GL, Isaac JT, Wang YT. Receptor trafficking and synaptic plasticity. Nat Rev Neurosci. 2004;5:952–962. doi: 10.1038/nrn1556. [DOI] [PubMed] [Google Scholar]
- Collingridge GL, Olsen RW, Peters J, Spedding M. A nomenclature for ligand-gated ion channels. Neuropharmacology. 2009;56:2–5. doi: 10.1016/j.neuropharm.2008.06.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DesMarais V, Ghosh M, Eddy R, Condeelis J. Cofilin takes the lead. J Cell Sci. 2005;118:19–26. doi: 10.1242/jcs.01631. [DOI] [PubMed] [Google Scholar]
- Eibert SM, Lee KH, Pipkorn R, Sester U, Wabnitz GH, Giese T, Meuer SC, Samstag Y. Cofilin peptide homologs interfere with immunological synapse formation and T cell activation. Proc Natl Acad Sci U S A. 2004;101:1957–1962. doi: 10.1073/pnas.0308282100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukata M, Kaibuchi K. Rho-family GTPases in cadherin-mediated cell-cell adhesion. Nat Rev Mol Cell Biol. 2001;2:887–897. doi: 10.1038/35103068. [DOI] [PubMed] [Google Scholar]
- Gallagher SM, Daly CA, Bear MF, Huber KM. Extracellular signal-regulated protein kinase activation is required for metabotropic glutamate receptor-dependent long-term depression in hippocampal area CA1. J Neurosci. 2004;24:4859–4864. doi: 10.1523/JNEUROSCI.5407-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerlai R, Henderson JT, Roder JC, Jia Z. Multiple behavioral anomalies in GluR2 mutant mice exhibiting enhanced LTP. Behav Brain Res. 1998;95:37–45. doi: 10.1016/s0166-4328(98)00002-3. [DOI] [PubMed] [Google Scholar]
- Gladding CM, Collett VJ, Jia Z, Bashir ZI, Collingridge GL, Molnár E. Tyrosine dephosphorylation regulates AMPAR internalisation in mGluR-LTD. Mol Cell Neurosci. 2009;40:267–279. doi: 10.1016/j.mcn.2008.10.014. [DOI] [PubMed] [Google Scholar]
- Hollmann M, Heinemann S. Cloned glutamate receptors. Annu Rev Neurosci. 1994;17:31–108. doi: 10.1146/annurev.ne.17.030194.000335. [DOI] [PubMed] [Google Scholar]
- Huang CC, Hsu KS. Sustained activation of metabotropic glutamate receptor 5 and protein tyrosine phosphatases mediate the expression of (S)-3,5-dihydroxyphenylglycine-induced long-term depression in the hippocampal CA1 region. J Neurochem. 2006;96:179–194. doi: 10.1111/j.1471-4159.2005.03527.x. [DOI] [PubMed] [Google Scholar]
- Huber KM, Kayser MS, Bear MF. Role for rapid dendritic protein synthesis in hippocampal mGluR-dependent long-term depression. Science. 2000;288:1254–1257. doi: 10.1126/science.288.5469.1254. [DOI] [PubMed] [Google Scholar]
- Isaac JT, Ashby M, McBain CJ. The role of the GluR2 subunit in AMPA receptor function and synaptic plasticity. Neuron. 2007;54:859–871. doi: 10.1016/j.neuron.2007.06.001. [DOI] [PubMed] [Google Scholar]
- Jia Z, Agopyan N, Miu P, Xiong Z, Henderson J, Gerlai R, Taverna FA, Velumian A, MacDonald J, Carlen P, Abramow-Newerly W, Roder J. Enhanced LTP in mice deficient in the AMPA receptor GluR2. Neuron. 1996;17:945–956. doi: 10.1016/s0896-6273(00)80225-1. [DOI] [PubMed] [Google Scholar]
- Kaksonen M, Toret CP, Drubin DG. Harnessing actin dynamics for clathrin-mediated endocytosis. Nat Rev Mol Cell Biol. 2006;7:404–414. doi: 10.1038/nrm1940. [DOI] [PubMed] [Google Scholar]
- Kemp N, Bashir ZI. Induction of LTD in the adult hippocampus by the synaptic activation of AMPA/kainate and metabotropic glutamate receptors. Neuropharmacology. 1999;38:495–504. doi: 10.1016/s0028-3908(98)00222-6. [DOI] [PubMed] [Google Scholar]
- Kerchner GA, Nicoll RA. Silent synapses and the emergence of a postsynaptic mechanism for LTP. Nat Rev Neurosci. 2008;9:813–825. doi: 10.1038/nrn2501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee CW, Han J, Bamburg JR, Han L, Lynn R, Zheng JQ. Regulation of acetylcholine receptor clustering by ADF/cofilin-directed vesicular trafficking. Nat Neurosci. 2009;12:848–856. doi: 10.1038/nn.2322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lilien J, Balsamo J. The regulation of cadherin-mediated adhesion by tyrosine phosphorylation/dephosphorylation of beta-catenin. Curr Opin Cell Biol. 2005;17:459–465. doi: 10.1016/j.ceb.2005.08.009. [DOI] [PubMed] [Google Scholar]
- Lu W, Shi Y, Jackson AC, Bjorgan K, During MJ, Sprengel R, Seeburg PH, Nicoll RA. Subunit composition of synaptic AMPA receptors revealed by a single-cell genetic approach. Neuron. 2009;62:254–268. doi: 10.1016/j.neuron.2009.02.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lüscher C, Huber KM. Group 1 mGluR-dependent synaptic long-term depression: mechanisms and implications for circuitry and disease. Neuron. 2010;65:445–459. doi: 10.1016/j.neuron.2010.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malenka RC, Bear MF. LTP and LTD: an embarrassment of riches. Neuron. 2004;44:5–21. doi: 10.1016/j.neuron.2004.09.012. [DOI] [PubMed] [Google Scholar]
- Malinow R, Malenka RC. AMPA receptor trafficking and synaptic plasticity. Annu Rev Neurosci. 2002;25:103–126. doi: 10.1146/annurev.neuro.25.112701.142758. [DOI] [PubMed] [Google Scholar]
- Mameli M, Balland B, Luján R, Lüscher C. Rapid synthesis and synaptic insertion of GluR2 for mGluR-LTD in the ventral tegmental area. Science. 2007;317:530–533. doi: 10.1126/science.1142365. [DOI] [PubMed] [Google Scholar]
- Mao L, Yang L, Tang Q, Samdani S, Zhang G, Wang JQ. The scaffold protein Homer1b/c links metabotropic glutamate receptor 5 to extracellular signal-regulated protein kinase cascades in neurons. J Neurosci. 2005;25:2741–2752. doi: 10.1523/JNEUROSCI.4360-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng Y, Zhang Y, Tregoubov V, Janus C, Cruz L, Jackson M, Lu WY, MacDonald JF, Wang JY, Falls DL, Jia Z. Abnormal spine morphology and enhanced LTP in LIMK-1 knockout mice. Neuron. 2002;35:121–133. doi: 10.1016/s0896-6273(02)00758-4. [DOI] [PubMed] [Google Scholar]
- Meng Y, Zhang Y, Jia Z. Synaptic transmission and plasticity in the absence of AMPA glutamate receptor GluR2 and GluR3. Neuron. 2003;39:163–176. doi: 10.1016/s0896-6273(03)00368-4. [DOI] [PubMed] [Google Scholar]
- Morishita W, Marie H, Malenka RC. Distinct triggering and expression mechanisms underlie LTD of AMPA and NMDA synaptic responses. Nat Neurosci. 2005;8:1043–1050. doi: 10.1038/nn1506. [DOI] [PubMed] [Google Scholar]
- Moult PR, Gladding CM, Sanderson TM, Fitzjohn SM, Bashir ZI, Molnar E, Collingridge GL. Tyrosine phosphatases regulate AMPA receptor trafficking during metabotropic glutamate receptor-mediated long-term depression. J Neurosci. 2006;26:2544–2554. doi: 10.1523/JNEUROSCI.4322-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nosyreva ED, Huber KM. Developmental switch in synaptic mechanisms of hippocampal metabotropic glutamate receptor-dependent long-term depression. J Neurosci. 2005;25:2992–3001. doi: 10.1523/JNEUROSCI.3652-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panicker S, Brown K, Nicoll RA. Synaptic AMPA receptor subunit trafficking is independent of the C terminus in the GluR2-lacking mouse. Proc Natl Acad Sci U S A. 2008;105:1032–1037. doi: 10.1073/pnas.0711313105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Passafaro M, Nakagawa T, Sala C, Sheng M. Induction of dendritic spines by an extracellular domain of AMPA receptor subunit GluR2. Nature. 2003;424:677–681. doi: 10.1038/nature01781. [DOI] [PubMed] [Google Scholar]
- Rocca DL, Martin S, Jenkins EL, Hanley JG. Inhibition of Arp2/3-mediated actin polymerization by PICK1 regulates neuronal morphology and AMPA receptor endocytosis. Nat Cell Biol. 2008;10:259–271. doi: 10.1038/ncb1688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saglietti L, Dequidt C, Kamieniarz K, Rousset MC, Valnegri P, Thoumine O, Beretta F, Fagni L, Choquet D, Sala C, Sheng M, Passafaro M. Extracellular interactions between GluR2 and N-cadherin in spine regulation. Neuron. 2007;54:461–477. doi: 10.1016/j.neuron.2007.04.012. [DOI] [PubMed] [Google Scholar]
- Sandig V, Youil R, Bett AJ, Franlin LL, Oshima M, Maione D, Wang F, Metzker ML, Savino R, Caskey CT. Optimization of the helper-dependent adenovirus system for production and potency in vivo. Proc Natl Acad Sci U S A. 2000;97:1002–1007. doi: 10.1073/pnas.97.3.1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shepherd JD, Huganir RL. The cell biology of synaptic plasticity: AMPA receptor trafficking. Annu Rev Cell Dev Biol. 2007;23:613–643. doi: 10.1146/annurev.cellbio.23.090506.123516. [DOI] [PubMed] [Google Scholar]
- Shimshek DR, Bus T, Grinevich V, Single FN, Mack V, Sprengel R, Spergel DJ, Seeburg PH. Impaired reproductive behavior by lack of GluR-B containing AMPA receptors but not of NMDA receptors in hypothalamic and septal neurons. Mol Endocrinol. 2006a;20:219–231. doi: 10.1210/me.2005-0262. [DOI] [PubMed] [Google Scholar]
- Shimshek DR, Jensen V, Celikel T, Geng Y, Schupp B, Bus T, Mack V, Marx V, Hvalby Ø, Seeburg PH, Sprengel R. Forebrain-specific glutamate receptor B deletion impairs spatial memory but not hippocampal field long-term potentiation. J Neurosci. 2006b;26:8428–8440. doi: 10.1523/JNEUROSCI.5410-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siu R, Fladd C, Rotin D. N-Cadherin is an in vivo substrate for protein tyrosine phosphatase sigma (PTPσ) and participates in PTPσ-mediated inhibition of axon growth. Mol Cell Biol. 2007;27:208–219. doi: 10.1128/MCB.00707-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snyder EM, Philpot BD, Huber KM, Dong X, Fallon JR, Bear MF. Internalization of ionotropic glutamate receptors in response to mGluR activation. Nat Neurosci. 2001;4:1079–1085. doi: 10.1038/nn746. [DOI] [PubMed] [Google Scholar]
- Tai CY, Kim SA, Schuman EM. Cadherins and synaptic plasticity. Curr Opin Cell Biol. 2008;20:567–575. doi: 10.1016/j.ceb.2008.06.003. [DOI] [PubMed] [Google Scholar]
- Takeichi M. The cadherin superfamily in neuronal connections and interactions. Nat Rev Neurosci. 2007;8:11–20. doi: 10.1038/nrn2043. [DOI] [PubMed] [Google Scholar]
- Vanderklish PW, Edelman GM. Dendritic spines elongate after stimulation of group 1 metabotropic glutamate receptors in cultured hippocampal neurons. Proc Natl Acad Sci U S A. 2002;99:1639–1644. doi: 10.1073/pnas.032681099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waung MW, Huber KM. Protein translation in synaptic plasticity: mGluR-LTD, fragile X. Curr Opin Neurobiol. 2009;19:319–326. doi: 10.1016/j.conb.2009.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waung MW, Pfeiffer BE, Nosyreva ED, Ronesi JA, Huber KM. Rapid translation of Arc/Arg3.1 selectively mediates mGluR-dependent LTD through persistent increases in AMPAR endocytosis rate. Neuron. 2008;59:84–97. doi: 10.1016/j.neuron.2008.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams EJ, Williams G, Gour B, Blaschuk O, Doherty P. INP, a novel N-cadherin antagonist targeted to the amino acids that flank the HAV motif. Mol Cell Neurosci. 2000;15:456–464. doi: 10.1006/mcne.2000.0847. [DOI] [PubMed] [Google Scholar]
- Xiao MY, Zhou Q, Nicoll RA. Metabotropic glutamate receptor activation causes a rapid redistribution of AMPA receptors. Neuropharmacology. 2001;41:664–671. doi: 10.1016/s0028-3908(01)00134-4. [DOI] [PubMed] [Google Scholar]
- Yan J, Zhang Y, Jia Z, Taverna FA, McDonald RJ, Muller RU, Roder JC. Place-cell impairment in glutamate receptor 2 mutant mice. J Neurosci. 2002;22(RC204) doi: 10.1523/JNEUROSCI.22-03-j0002.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Venkitaramani DV, Gladding CM, Kurup P, Molnar E, Collingridge GL, Lombroso PJ. The tyrosine phosphatase STEP mediates AMPA receptor endocytosis after metabotropic glutamate receptor stimulation. J Neurosci. 2008;28:10561–10566. doi: 10.1523/JNEUROSCI.2666-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Q, Xiao M, Nicoll RA. Contribution of cytoskeleton to the internalization of AMPA receptors. Proc Natl Acad Sci U S A. 2001;98:1261–1266. doi: 10.1073/pnas.031573798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Q, Homma KJ, Poo MM. Shrinkage of dendritic spines associated with long-term depression of hippocampal synapses. Neuron. 2004;44:749–757. doi: 10.1016/j.neuron.2004.11.011. [DOI] [PubMed] [Google Scholar]