

Abstract
Mutations of the myosin Va gene cause the neurological diseases Griscelli syndrome type 1 and Elejalde syndrome in humans and dilute phenotypes in rodents. To understand the pathophysiological mechanisms underlying the neurological disorders in myosin Va diseases, we conducted an integrated analysis at the molecular, cellular, electrophysiological, and behavioral levels using the dilute-neurological (d-n) mouse mutant. These mice manifest an ataxic gait and clonic seizures during postnatal development, but the neurological disorders are ameliorated in adulthood. We found that smooth endoplasmic reticulum (SER) rarely extended into the dendritic spines of Purkinje cells (PCs) of young d-n mice, and there were few, if any, IP3 receptors. Moreover, long-term depression (LTD) at parallel fiber–PC synapses was abolished, consistent with our previous observations in juvenile lethal dilute mutants. Young d-n mice exhibited severe impairment of cerebellum-dependent motor learning. In contrast, adult d-n mice showed restoration of motor learning and LTD, and these neurological changes were associated with accumulation of SER and IP3 receptors in some PC spines and the expression of myosin Va proteins in the PCs. RNA interference-mediated repression of myosin Va caused a reduction in the number of IP3 receptor-positive spines in cultured PCs. These findings indicate that myosin Va function is critical for subsequent processes in localization of SER and IP3 receptors in PC spines, LTD, and motor learning. Interestingly, d-n mice had defects of motor coordination from young to adult ages, suggesting that the role of myosin Va in PC spines is not sufficient for motor coordination.
Introduction
An actin-based molecular motor myosin Va is predominantly expressed in the brain (Espreafico et al., 1992; Langford and Molyneaux, 1998; Reck-Peterson et al., 2000; Desnos et al., 2007), and mutation of the myosin Va gene causes the human hereditary disease Griscelli syndrome type 1 (GS1) [Online Mendelian Inheritance in Man (OMIM) 214450] (Pastural et al., 2000) and the dilute phenotypes of mice and rats. Elejalde syndrome (ES) (OMIM 256710) (Ivanovich et al., 2001) in humans is also thought to be the same disease entity as GS1. GS1 and ES patients manifest hypopigmentation of the hair and skin and neurological dysfunctions such as marked motor development delay, hypotonia, ataxia, and mental retardation. Pathological examinations of the nervous systems of GS1 and ES patients have not been reported. However, studies of dilute mutants have demonstrated various abnormalities in the nervous system caused by myosin Va deficiency, suggesting a role for myosin Va in myelination (Sloane and Vartanian, 2007), synaptic plasticity at neuromuscular junctions (Röder et al., 2008), axonal transport of neurofilaments (Alami et al., 2009), and hippocampal long-term potentiation (Correia et al., 2008).
We also demonstrated that smooth endoplasmic reticulum (SER) and associated IP3 receptors are depleted in the dendritic spines of Purkinje cells (PCs) and that cerebellar long-term depression (LTD), a cellular basis of motor learning, is abolished in the myosin Va-null mutants, dilute-lethal (d-l) mice and dilute-opisthotonus (dop) rats (Dekker-Ohno et al., 1996; Takagishi et al., 1996; Miyata et al., 2000; Takagishi and Murata, 2006). Our results implied that myosin Va is important for the extension of SER into PC spines and is involved in synaptic plasticity of the cerebellum. This finding might partially account for the disease background of GS1 and ES patients who show ataxic cerebellar movement and cerebellar atrophy (Sanal et al., 2000, 2002; Ivanovich et al., 2001). However, they are still insufficient to understand all of the pathological processes of this neurological defect. Since homozygosity for the d-l and dop mutations is a juvenile lethal condition in mice and rats, these alleles are relatively poor candidates for a study aimed at elucidating behavior tasks. Therefore, we exploited the viable myosin Va mutant dilute-neurological (d-n) that has a missense mutation in the myosin Va gene (Huang et al., 1998). The d-n mice exhibit neurological abnormalities at 2–4 weeks of age, similar to those of the juvenile lethal mutants, but have a normal life span because of an amelioration of the neurological disorders as the mice age (Huang et al., 1998). Here, we tested cerebellum-dependent motor learning and motor coordination in d-n mice, and also investigated myosin Va function in PC spines and synaptic plasticity at PC synapses in juvenile and adult mice. We found that there was a link among the absence of SER and IP3 receptors in the PC spines, LTD deficits, and impaired motor learning. The present findings provide an important platform that accounts for the cerebellar disorder observed in dilute mutants and in GS1 and ES patients.
Materials and Methods
Animals
Dilute-neurological (gene symbol, Myo5ad-n) mice were provided by N. A. Jenkins (National Cancer Institute, Frederick Cancer Research and Development Center, Frederick, MD) (Huang et al., 1998). Homozygous (d-n/d-n) mice were obtained from the mating of a homozygous male and a heterozygous (d-n/+) female. Mutant mice can be distinguished from normal mice by 4∼5 d after birth by their diluted coat color. Homozygous mice, and heterozygous littermates or wild-type (C57BL/6J) mice as controls, were used from postnatal day 10 (P10) to 2 months of age for all experiments in this study. The mice were housed in a room with controlled humidity and temperature, and a 12 h light/dark cycle. The animals were provided with standard laboratory chow and water ad libitum. All experiments were conducted in accordance with the guideline of the Committee for Animal Experiments of Nagoya University.
Morphological analysis
Preparation of tissue sections for immunohistochemistry.
Developing [postnatal day 10 (P10)∼P22] and adult (2-month-old) mice were used for this study. The animals were perfused intracardially with 4% paraformaldehyde (PFA) in PBS for 10 min; the cerebellum was removed and bisected in the sagittal plane. The cerebellar pieces were immersed immediately in the same fixative at 4°C overnight. They were cryoprotected with graded sucrose (10∼30%) in PBS and then frozen in liquid N2. Cryostat sections were prepared for calbindin D-28K (CD-28k) or myosin Va immunohistochemistry. For IP3 receptor immunostaining, we used electron-microscopic cryomicrotomy, because thin (1.0 μm) sections were required to detect localization of IP3 receptors in PC spines. In brief, small tissue blocks were infused with 2.3 m sucrose in PBS, placed on the specimen stubs, and frozen in liquid N2. One micrometer sections were cut at −110°C with a Reichert ultramicrotome Ultracut E equipped with cryosectioning system FC-4 (Reichert).
Immunofluorescence.
The following antibodies were used: (1) anti-CD-28K antibody (Millipore Bioscience Research Reagents); (2) anti-myosin Va antibodies raised against the head domain of chicken myosin Va [gifts from S. Ohashi (Nihon University, Funahashi, Japan) and R. Larson (Universidade de São Paulo, São Paulo, Brazil)] (Ohashi et al., 2002); (3) anti-IP3 receptor antibody [a gift from K. Mikoshiba (University of Tokyo, Tokyo, Japan)]. After incubation with primary antibodies at 4°C overnight, the sections were incubated with Alexa 488- or 568-labeled species-specific secondary antibodies (diluted to 1:250∼500; Jackson ImmunoResearch Laboratories) for 1 h. The immunostained sections were examined with a Zeiss LSM 510 confocal laser-scanning microscope (Carl Zeiss MicroImaging).
Electron microscopy.
Young (P17∼P19) and adult (P40∼P64) mice were intracardially perfused with 2% PFA and 2.5% glutaraldehyde in 0.1 m phosphate buffer (PB). The cerebellum was dissected out and the tissues were cut into small pieces. They were postfixed with 1% OsO4 in PB for 1 h. After dehydration with graded alcohols, the tissues were embedded in epoxy resin. Semithin (1.0∼1.5 μm) sections were cut and stained with toluidine blue for light microscopy. Ultrathin sections were examined with a JEOL 1210 electron microscope (JEOL).
The proportion of PC spines containing SER was estimated using randomly selected EM negatives (5000× magnification) of the molecular layer. PC spines in synaptic contacts with parallel fibers (PFs) were counted and those containing SER identified.
Western blot analysis
Total proteins were extracted from the cerebella of d-n and normal littermate mice at P10 and 2 months. Protein concentrations were estimated by the BCA assay (Pierce). Aliquots of 30 μg of protein were separated on 4–12% SDS-PAGE gels, and then transferred to a PVDF (polyvinylidene fluoride) membrane (Millipore). The membranes were immersed in 5% nonfat milk in 0.1% Tween–Tris-buffered saline (T-TBS) for 1 h, and then were incubated with anti-myosin Va antibodies (gifts from S. Ohashi and R. Larson) and anti-β-actin antibodies (Cell Signaling Technology) at 4°C overnight. After washing with T-TBS, the membranes were incubated with HRP-conjugated antibodies (Jackson ImmunoResearch Laboratories) and developed using SuperSignal West Pico ECL Substrate (Pierce).
Electrophysiological acquisition and data analysis
Young (P29∼P34) and adult (P56∼P67) d-n mice and control age-matched C57BL/6 Cr mice were deeply anesthetized with halothane and decapitated, and their cerebella were dissected rapidly. Sagittal or parasagittal cerebellar slices (250 μm) were cut in ice-cold artificial CSF (ACSF) with a vibratome (Leica; VT1000S) and kept in a submerged chamber for >1 h with 95% O2/5% CO2-saturated ACSF (125 mm NaCl, 2.5 mm KCl, 1.25 mm NaH2PO4, 1 mm MgSO4, 2 mm CaCl2, 26 mm NaHCO3, and 20 mm glucose) at 30∼32°C. The ACSF was equilibrated with 95% O2/5% CO2, pH 7.3, and infused at a rate of 3.0 ml/min. During recordings, ACSF containing 10 μm (−)-bicuculline methochloride (Tocris Bioscience) was used for blocking GABAA receptors. All experiments were performed at 30∼32°C.
PCs were visualized using a BX50WI microscope (Olympus) with an infrared differential interference contrast video system (C2400-79H; Hamamatsu Photonics). Whole-cell recordings from PCs in current-clamp configurations were made with 2–4.5 MΩ recording pipettes containing 120 mm K-gluconate, 10 mm HEPES, 1.0 mm EGTA, 2 mm MgCl2, 0.1 mm CaCl2, 10 mm NaCl, 2 mm Na2ATP, and 0.5 mm Na2GTP, pH 7.3 with KOH, 295–310 mOsm. Axoclamp 2B (Molecular Devices/MDS Analytical Technologies) or EPC9 (HEKA) patch-clamp amplifiers were used for whole-cell current-clamp recordings. Synaptic responses were evoked by a concentric electrode (tip diameter, 25 μm; Inter Medical) placed on the edge of the molecular layer for recording EPSPs of PFs to a PC (PF-EPSPs) and on the white matter for recording the electrophysiological response of a climbing fiber (CF) to PC synapses (CF response) as previously reported (Miyata et al., 1999, 2000). The stimulus consisted of a 100 μs duration bipolar pulse of constant current steps (<100 μA) by a biphasic isolator (BAK Electronics).
LTD was estimated from the 10–90% slopes of the PF-EPSPs. LTD experiments were accepted if the resting potential and membrane resistance of the recorded cells remained constant during the recording. Data were acquired using the PULSE program (HEKA, version 8.54). PULSE FIT (HEKA, version 8.54) and Igor Pro (Wavemetrics) were used to analyze the obtained data.
To record the PF-mediated EPSCs (PF-EPSCs), PCs were recorded using an internal solution composed of 60 mm CsCl, 10 mm Cs d-gluconate, 20 mm TEA (tetraethylammonium)-Cl, 20 mm BAPTA, 4 mm MgCl2, 4 mm ATP, and 30 mm HEPES, pH 7.3, adjusted with CsOH. PCs were held at −80 mV, and PFs were stimulated at 0.5 Hz using stimulating electrodes filled with ACSF.
Eyeblink classical conditioning
Surgery.
C57BL/J mice (CLEA Japan) at P17∼P19, P28∼P29, and 2 months of age were used to assess eyeblink conditioning during normal development and for comparison with young (P28∼P29) and adult (P60) d-n mice and their normal littermates; surgery was performed as previously described (Kishimoto et al., 1997, 2001a). Mice were anesthetized with ketamine (80 mg/kg, i.p.; Sankyo) and xylazine (20 mg/kg, i.p.; Bayer), and four Teflon-coated stainless-steel wires (100 μm in diameter; A-M Systems) were subcutaneously implanted under the left eyelid. Two of the wires were used to deliver the unconditioned stimulus (US) and the remaining two to record an electromyogram (EMG) from the musculus orbicularis oculi (MOO), which is responsible for eyelid closure. To avoid picking up other facial muscle responses, small slits were made at the outer and inner edges of the left eye.
Conditioning procedures.
The conventional experimental design used for eyeblink conditioning in adult mice is not suitable for juvenile mice because their skulls are soft and the electrodes could not be maintained for a prolonged experimental period. We, therefore, applied a novel protocol for the conditioning over a reduced experimental period; the conditioning procedure was modified from a previous protocol (Stanton et al., 1992; Kishimoto et al., 1997). At least 24 h were allotted for recovery and 1 h for acclimation (with no stimulus) to the conditioning chamber after the surgery. A 352 ms tone (1 kHz, 80 dB) was used as a conditioned stimulus (CS) and 100 ms electrical shock (0.2 mA, 100 Hz square pulses) as the US. In the delayed eyeblink conditioning paradigm, the US was given to overlap the CS in time such that the two stimuli terminated simultaneously. Mice received six sessions within 2 d at 4 h intervals (three sessions per day). Each session consisted of 100 trials grouped in 10 blocks, with 10 CS only (every 10th trial) and 90 CS–US paired trials. The intertrial interval was randomized between 20 and 40 s with a mean of 30 s. The spontaneous eyeblink frequency was measured by 100 “no stimulus” trials during acclimation before the conditioning experiment began, and the startle response to a tone was measured during the first 100 trials of the first session of conditioning. Experiments were performed during the light phase of the light/dark cycle in a container (10 cm in diameter) placed in a sound- and light-attenuating chamber. All experiments including surgery were performed by an operator who was blind to the genotype of the mice. The EMG was measured and analyzed as described previously (Kishimoto and Kano, 2006).
Rota-rod and fixed-bar test
Normal and d-n mice were screened at young (P26∼P29) and adult (P60∼P63) stages. The rota-rod and fixed-bar tests were basically performed as described previously (Kadotani et al., 1996). The rota-rod (Muromachi Kikai) consisted of a gritted metal roller (3 cm in diameter). A mouse was placed on a roller rotating at 10 rpm, and the time it remained on the rotating roller was measured. A maximum of 120 s was allowed per animal. In the fixed-bar test, the time the animal remained on a wooden bar (5 mm in width and 40 cm above the ground) was measured. A maximum of 60 s was allowed per animal.
Statistical analysis
Data were statistically analyzed by a two-tailed Student t test with the Microsoft Excel program, or by repeated-measures ANOVA after post hoc Sheffé test with the SPSS 6.1 program. Differences were considered significant when p < 0.05.
Purkinje cell culture, short interfering RNA transfection, and immunostaining
A primary culture of PCs was performed as described previously (Tanaka et al., 2009). Briefly, cerebella were dissected from C57BL/6 mice at P0 or P1 and dissociated in PBS containing 8 U/ml papain (Worthington Biochemicals), 20 U/ml DNase I (Sigma-Aldrich), and 5 mm cysteine-HCl for 30 min at 37°C. After trituration with a pipette tip, cells were resuspended in a serum-free culture medium composed of Eagle's basal medium with Earle's salts supplemented with 1 mg/ml bovine serum albumin, 5.6 g/L d-glucose, 3 mm l-glutamine, 5 μg/ml bovine insulin, 5 μg/ml human transferrin, 30 nm sodium selenite, 20 nm progesterone, and 1 mm sodium pyruvate. Then, the cell suspension was plated on a plastic-bottom dish (μ-Dish; ibidi, Nippon Genetics) coated with poly-l-lysine (molecular weight, >300,000; Sigma-Aldrich) at a density of 1.5∼1.8 × 105 cells per square centimeter. After 1.5 h incubation, 0.5 ml of the Sumitomo Nerve-Cell Culture Medium (Sumitomo Bakelite) was added to each dish, and the cultures were maintained at 37°C in 5% CO2/95% air.
PCs at 11 d in vitro (DIV) were transfected with short interfering RNAs (siRNAs) by single-cell electroporation (Tanaka et al., 2009). The myosin Va siRNA (target sequence, GCUGAAGAAACUCAAAAUU) and the negative control siRNA were purchased from Ambion/Applied Biosystems. Micropipettes were pulled on a micropipette puller (PB-7; Narishige) using 1.2 mm outer diameter filament glass capillaries (GD-1.2; Narishige). The siRNAs were diluted to 1 μm in a solution containing 150 mm potassium methanesulfonate and 5 mm HEPES, pH 7.2. Alexa Fluor 594 (AF594) (Invitrogen) was added at 0.25 mm to the siRNA solution to monitor the electroporation and visualize the electroporated cells. Micropipette tips (∼0.5 μm in diameter and 30–40 MΩ resistance) were filled with 3 μl of siRNA/AF594. A silver wire electrode (cathode) was placed in the micropipette and a ground electrode (anode) was placed in the culture medium. The culture dish containing the dissociated PCs was placed on the stage of an inverted microscope equipped with epifluorescence (Axiovert; Carl Zeiss MicroImaging). PCs were identified morphologically by phase contrast microscopy. The tip of a micropipette controlled by a micromanipulator (5170; Eppendorf) was touched to a PC, and square electric pulses (−4 V, 1 ms width, 100 Hz, 2 s duration) generated with an electroporator (Axoporator 800A; Molecular Devices) were applied to transfer siRNAs and AF594 into the cell.
Ten days after transfection (at 21 DIV), PCs were fixed with 4% PFA in PBS for 40 min and then double immunofluorescence was performed using anti-IP3 receptor antibody [1:50; a gift from N. Maeda (Tokyo Metropolitan Institute for Neuroscience, Tokyo, Japan)] and anti-CD-28K antibody (1:2000; Swant) or anti-myosin Va antibody (1:200; Sigma-Aldrich). Immunoreactivities were visualized using the appropriate Cy2-, Cy3-, or Cy5-conjugated secondary antibodies (Jackson ImmunoResearch). Images were acquired using a Zeiss LSM 510 META confocal laser-scanning microscope (Carl Zeiss MicroImaging). For quantification of the spine density, protrusions of 0.4∼1.5 μm in length were identified as spines. The lengths of spines and dendrites were measured using Zeiss LSM software, version 4.0.
Results
Neurological phenotypes of d-n mice
The d-n mice exhibit neurological disorders during development similar to those of the myosin Va-null mutants, d-l mice, and dop rats. In brief, they begin to display abnormalities in gait at P10∼P11; later, they suffer clonic seizures (convulsion and opisthotonus with forelimbs and hindlimbs beating up and down) (Fig. 1A). The abnormalities are most severe at 2∼4 weeks of age. Surprisingly, after 1 month of age, the mice cease to suffer from clonic seizures, so that superficially they appear normal (Fig. 1A). Before the onset of the neurological abnormalities, the growth rate of d-n mice is similar to that of normal mice; however, after this time, they show poor weight gain, even as adults (data not shown). The cerebellar weights of the mutant mice were slightly lower than those of normal mice, and these weights scarcely increased from 3 weeks of age to adulthood (data not shown). We showed previously that the cerebellum of d-n mice had a normal foliation pattern, a typical layer organization of the cortex, and normal monolayer alignment of PCs (Fig. 1B) (Takagishi et al., 2007). Likewise, the PCs formed well developed and branched dendritic trees (Fig. 1C). Thus, no gross alteration of cerebellar architecture was found in d-n mice during postnatal development or adulthood. Nevertheless, the smaller size of the cerebellum in these mice is consistent with that of GS1 and ES patients (Sanal et al., 2000, 2002).
Figure 1.

d-n mice and morphological analysis of the cerebellum. A, Homozygous mutant (d-n) mice at young (P21) and adult (P60) ages. The young mouse manifests convulsions, whereas the adult mouse has recovered from clonic seizures. B, The cerebellar cortices of normal and d-n mice at P22; plastic sections, toluidine blue staining. PCs, Purkinje cell layer; ML, molecular layer; GL, granular layer. Scale bars, 50 μm. C, Purkinje cells immunostained with anti-CD-28K antibodies in normal and d-n mice at P60. PCs, Purkinje cells; PCD, Purkinje cell dendrites. Scale bars, 50 μm.
A previous study reported that the amount of myosin Va protein was dramatically reduced (<1% of normal levels) in the d-n mouse brain (Huang et al., 1998). We examined whether myosin Va was expressed in PCs of d-n mice by immunohistochemistry. For this study, we used anti-myosin Va antibodies that were raised against the head domain of myosin Va protein (Ohashi et al., 2002). In normal mice at P10 and adulthood, PCs and granule cells in the internal granular layer were positively stained (Fig. 2Aa,c). In particular, the PCs strongly expressed myosin Va throughout the soma to dendrites (Fig. 2Ae). The molecular layer was well stained, and thus it appeared that myosin Va was expressed in PC spines as well as PC dendrites and was also present in PFs. In contrast, positive staining was hardly detected in d-n cerebella including PCs at P10 (Fig. 2Ab). Interestingly, in adult d-n cerebella, staining was found in the PCs (Fig. 2Ad); the somata of many PCs were clearly stained and the PC dendrites showed weak staining (Fig. 2Af, arrows). The staining was not evident in the internal granular layer in d-n cerebella. These observations suggested that myosin Va proteins showed an increase in expression levels or an increased rate of accumulation within PCs with age.
Figure 2.
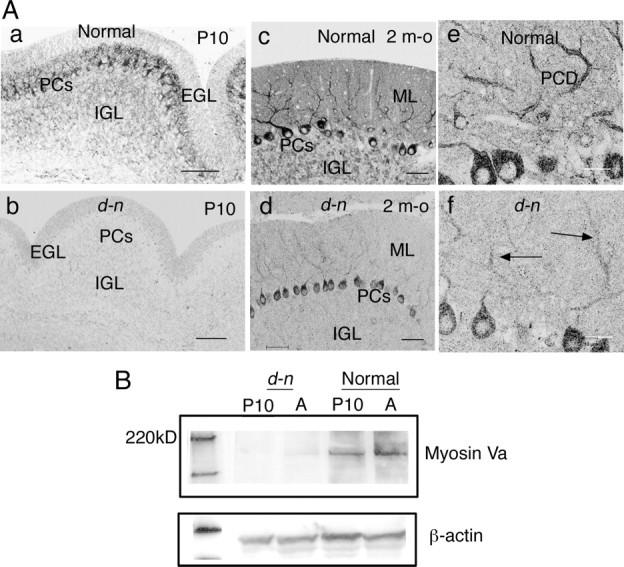
Myosin Va expression in Purkinje cells in d-n mice. A, Myosin Va immunostaining of cerebellar cortices at P10 (a, b) and at 2 months old (m-o) (c–f). At P10, PCs are well stained in the normal mouse (a), whereas no staining is found in the d-n mouse (b). In normal mice, the PCs are strongly stained throughout the soma to dendritic branches at 2 m-o (c, e). The staining is also found in ML and IGL (c). In d-n mice at 2 m-o, the positive staining is found in the PC soma and dendrites (d; f, arrows), although the staining is fairly weak compared with that in normal mice. EGL, External granular layer; ML, molecular layer. PCD, Purkinje cell dendrites. Scale bars: a–d, 50 μm; e, f, 20 μm. B, Representative Western blot of the cerebellar lysates from normal and d-n mice at P10 and 2 m-o (A). The bands of mutant mice are faint at both P10 and 2 m-o; however, the band at 2 m-o is slightly easier to detect than at P10.
Next, we performed a Western blot analysis to confirm the expression of myosin Va in young and adult d-n mouse cerebella. As expected, the band at 190∼200 kDa that represents the myosin Va protein was extremely faint in mutant cerebellar lysates at P10 and in adults (Fig. 2B). Although a quantitative analysis for mutant proteins was not feasible, it was clear that myosin Va protein was expressed in mutant cerebella.
These findings reveal that, during postnatal development, the d-n mice show very similar neurological phenotypes, including cerebellar structures and myosin Va expression in PCs, to those of the myosin Va-null mutants, d-l mice, and dop rats. First, therefore, we performed the same analysis as in d-l mice and dop rats (Dekker-Ohno et al., 1996; Takagishi et al., 1996; Miyata et al., 2000) and then tested motor learning in young d-n mice. A similar analysis was also performed in adult d-n mice because of their intriguing phenotype in which they recover from their severe neurological impairment and their PCs express the myosin Va protein.
Young d-n mice show a lack of SER and IP3 receptors in PC spines
First, we determined whether SER and IP3 receptors were absent from the PC spines of d-n mice at 2–3 weeks of age when they begin to exhibit severe neurological disorders. In an EM analysis, the SER is a distinct structure frequently seen in PC spines in normal mice (Fig. 3A). The SER extended into the spines from the dendrites and, in spines that had been sectioned in the longitudinal plane, clearly reached the spine head (Fig. 3Aa, arrows). In transverse sections, the spines could be seen to contain several SER cisternae (Fig. 3Ab, arrows). These observations indicate that the SER is ramified and forms complicated tubular systems within the spines, as previously described (Martone et al., 1993; Levine and Rabouille, 2005). In young d-n mice, SER was absent from all but a few PC spines (Figs. 3B, 6B). Similarly to myosin Va-null mutants, SER was present in the dendrites and soma of the PCs. Thus, it appeared that SER tubular networks were formed normally, except within the spine, in the PCs of d-n mice.
Figure 3.
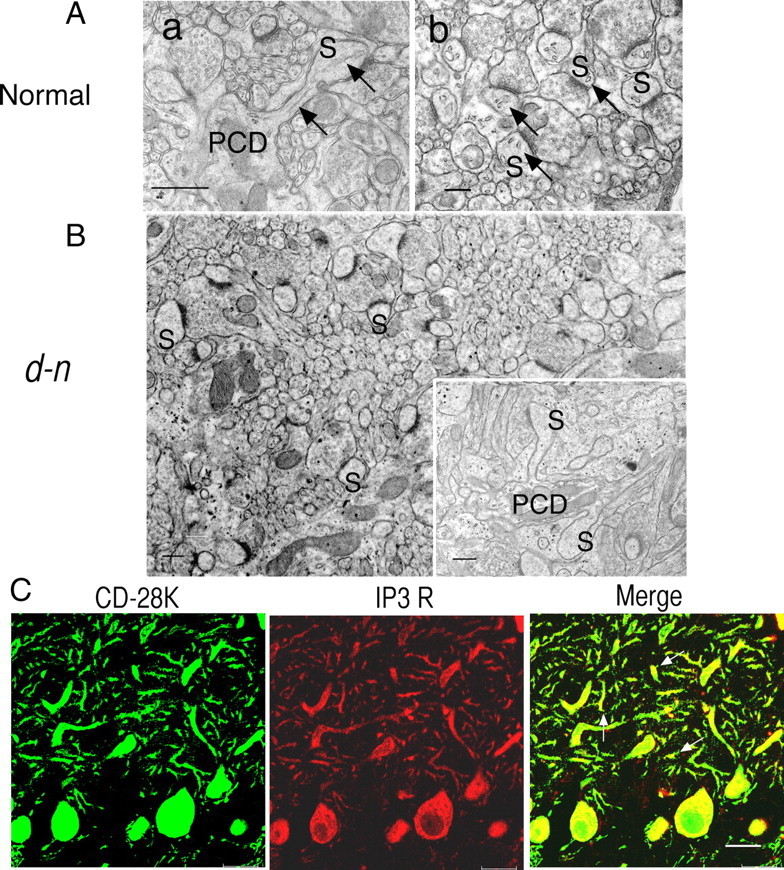
Absence of SER and IP3 receptors in PC spines of young mice. A, B, EM analysis in normal (A) and d-n (B) young mice. The arrows in A indicate SER in PC spines (S). In d-n mice (B), notice the absence of SER in the cross-sectioned plan and longitudinal-sectioned plane (inset) of PC spines (S). PCD, Purkinje cell dendrites. Scale bars, 500 nm. C, Immunohistochemical detection of IP3 receptors in PC spines. Double labeling of CD-28K and IP3 receptors in PC cells of young d-n mice. Note that IP3 receptor-positive PC spine are missing (middle), whereas dot-like profiles stained with anti-CD-28K antibodies are abundant (left). The arrows indicate CD-28K-positive and IP3 receptor-negative PC spines (right, a merged image). Scale bar, 20 μm.
Figure 6.
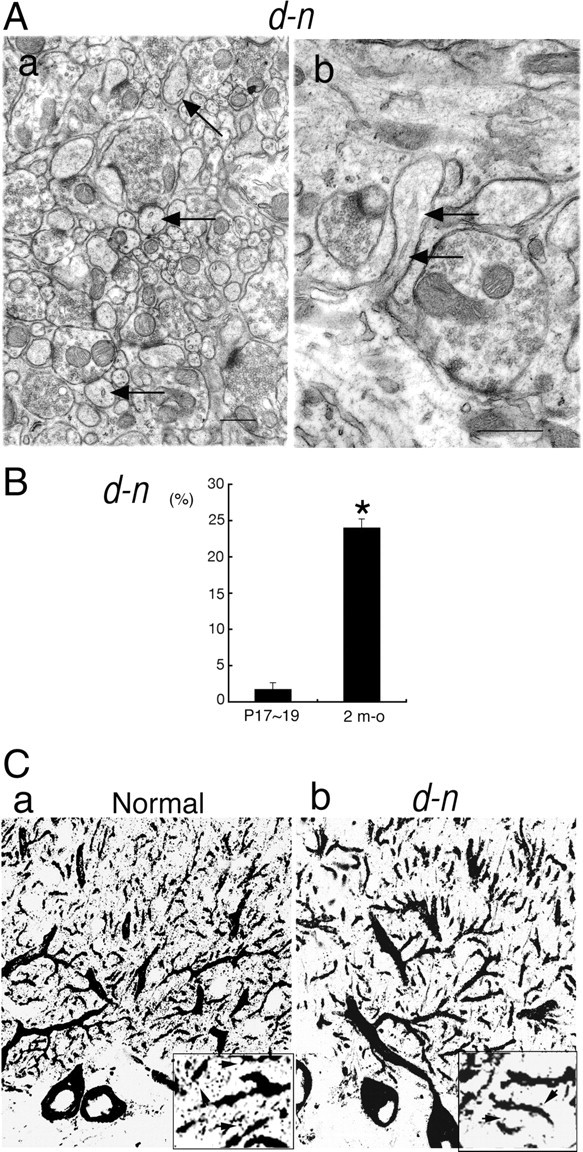
Presence of SER and IP3 receptors in PC spines of adult d-n mice. A, EM analysis of SER in PC spines of adult d-n mice. The arrows indicate SER in PC spines. a, Cross-sectioned profiles of PC spines. b, Longitudinal plan of a PC spine. B, Quantitative comparison of PC spines containing SER between young (P17∼P19) and adult [2-month-old (m-o)] d-n mice. Error bars represent SEM. *p < 0.0001, Welch's t test. C, IP3 receptor immunostaining in PC spines from normal (a) and d-n (b) adult mice. The arrows (a, b, insets) indicate IP3 receptor-positive spines.
Immunohistochemical analysis using double labeling of IP3 receptors and calbindin (CD-28K) showed that IP3 receptors were very infrequent in PC spines in young d-n mice (Fig. 3C, middle), whereas CD-28K-positive spines were abundant (Fig. 3C, left). As the two proteins colocalized in the dendrites and soma of the PCs (Fig. 3C, right), our results indicate that IP3 receptors were lost only in PC spines (Fig. 3C, arrows in the merged image). This is consistent with our EM observations described above.
LTD is impaired in young d-n mice
We first investigated the basic electrophysiological properties of synaptic transmission, resting membrane potential, and input resistance in d-n and normal mice. Previously, we reported that CF extension into PCs is reduced in the cerebella of d-n mice compared with normal mice (Takagishi et al., 2007). In d-n mice, however, CF stimulation reliably induced typically complex spikes under the current-clamp conditions, similarly to normal mice (Figs. 4Aa,Ba, 7Aa,Ba). The 10–90% rise time and the decay time constant of the PF-EPSCs were normal in d-n mice (Table 1). Our previous analysis indicates that paired-pulse facilitation of PF-EPSCs is also normal in d-n mice (Takagishi et al., 2007). These lines of evidence suggest that the excitatory synaptic transmissions to PCs are almost normal in d-n mice. In addition, resting membrane potential and input resistance in d-n mice showed no significant differences to those of normal animals. Resting membrane potentials of −64.8 ± 3.5 and −65.3 ± 2.3 mV were recorded in d-n and normal mice, respectively (n = 7 for each group; p > 0.05). Input resistances of 212 ± 10.4 and 218 ± 8.6 MΩ were obtained in d-n and normal mice, respectively (n = 7 for each group; p > 0.05).
Figure 4.

LTD is impaired in young d-n mice. Normal (A) and d-n (B) mice at young ages (P29∼P34). a, Trace for CF response. b, Traces for PF-EPSPs 5 min before and 30 min after the initiation of conjunctive stimulation. Each trace is an average of five successive responses. C, The graph shows pooled data plotted against time [mean ± SEM; n = 8 and 10 for normal (open circle) and d-n (closed circle) mice, respectively]. The data were normalized against the mean initial slope value during the control period for the first 10 min.
Figure 7.

LTD and eyeblink conditioning are restored in adult d-n mice. A, B, LTD is normal in adult d-n mice: normal mice (A); d-n mice (B). a, Trace for CF response. b, Traces for PF-EPSPs 5 min before and 30 min after the initiation of conjunctive stimulation. The graphs show pooled data plotted against time. The traces and graphs are illustrated in a same manner as in Figure 4 (n = 10 and 14 for normal and d-n mice, respectively). C–E, Eyeblink conditioning is normal in adult d-n mice. C, Development of CR% during six sessions of eyeblink conditioning in normal (open circle) and d-n mice (closed circle) (n = 7 for each). D, The averaged EMG traces on the sixth session in normal (solid line) and d-n (dashed line) mice. E, Frequency histogram of the CR peak latencies in adult d-n mice. The CR peaks on sixth session were binned into the four time windows (50–100, 100–150, 150–200, and 200–250 ms measured from the CS onset) in the normal (open column) and d-n (solid column) mice. The columns and error bars represent mean ± SEM.
Table 1.
Kinetics of the parallel fiber-mediated EPSCs
| Age | Normal | d-n | p |
|---|---|---|---|
| P11–P15 | |||
| 10–90% rise time (ms) | 1.1 ± 0.3 (n = 8) | 1.0 ± 0.2 (n = 9) | 0.413 |
| Decay time constant (ms) | 7.0 ± 2.1 (n = 8) | 7.9 ± 1.9 (n = 9) | 0.361 |
| 2 months | |||
| 10–90% rise time (ms) | 0.9 ± 0.3 (n = 10) | 0.8 ± 0.2 (n = 11) | 0.549 |
| Decay time constant (ms) | 7.5 ± 2.6 (n = 10) | 6.2 ± 2.9 (n = 11) | 0.342 |
Measurements are expressed as mean ± SD.
There was no significant difference between normal and d-n mice pairs (Mann–Whitney U test).
Next, we performed an electrophysiological analysis to determine whether the defect in the PC spines of young d-n mice was accompanied by LTD deficiency. LTD was readily induced by the conjunctive stimulation of PF and CF synapses at 1 Hz for 5 min, a protocol that is optimal for inducing LTD (Miyata et al., 1999, 2000) in normal mice (P29∼P34) (Fig. 4Ab). The mean value of the EPSP slope was significantly reduced to 54.5 ± 9.1% of the control at 30 min after conjunctive stimulation in normal mice (Fig. 4C, open circle) (n = 8; p < 0.01). In contrast, the same conjunctive stimulation failed to induce LTD in d-n mice (P29∼P34) (Fig. 4Bb). Only one cell of those recorded showed induction of LTD; the other cells showed no evidence of induced LTD but rather displayed long-term potentiation (LTP). The mean value of the EPSP slope was 133.6 ± 14.0% of the control at 30 min after conjunctive stimulation (Fig. 4C, closed circle) (p < 0.01; n = 10).
These findings indicate that LTD was deficient in young d-n mice, which also show a lack of SER and IP3 receptors in nearly all PC spines. We, therefore, next addressed the question of whether the PC spine defect causes motor learning deficiencies in young d-n mice.
Eyeblink conditioning is impaired in young d-n mice
To test cerebellum-dependent motor learning, we used the classical eyeblink conditioning assay since its conditioning paradigm has contributed greatly to the understanding of the neurological and developmental bases underlying learning and memory (Thompson and Krupa, 1994; Thompson et al., 1997; Thompson and Steinmetz, 2009). The timing of emergence of eyeblink conditioning during development has been investigated mainly in rats (Stanton et al., 1992, 1998; Freeman et al., 2003), and, as yet, no studies on mice have been reported. Here, we first tested the classical eyeblink conditioning in developing mice. To confirm the validity of the conditioning procedure and to determine the critical period for successful acquirement of the eyeblink conditioned response (CR), normal C57BL/6J mice were trained at three different ages: P17∼P19, P28∼P29, and P60 (at the first day of each test) with a delay conditioning procedure, the same protocol as used for developing rats (Stanton et al., 1992). The rate of CR at P28∼P29 and P60 progressively increased to >60% during the six sessions. In contrast, at P17∼P19, the CR increased to a nonsignificant extent (Fig. 5A). An ANOVA revealed no significant effects in the interaction between sessions and groups (F(12,126) = 1.305; p = 0.237) but did identify a significant effect of the group (F(2,21) = 9.536; p = 0.0011). Post hoc analysis revealed that no difference was detected between P28∼P29 and P60; however, the CR at P17∼P19 was significantly lower than the other two groups. Thus, eyeblink conditioning was clearly present at P28∼P29, but was difficult to detect at P17∼P19 in the wild-type mice during development. These results are consistent with the ontogenetic emergence of eyeblink conditioning in rats; rat pups trained at P17 show very little acquisition of eyeblink conditioning, whereas rats trained at P24 show adult-like acquisition of the CR (Stanton et al., 1992; Hunt et al., 2007).
Figure 5.

Eyeblink conditioning is impaired in young d-n mice. A, Normal developmental effect on mice eyeblink conditioning. Percentage of conditioned response (CR%) was evaluated in C57BL/6J mice at three ages [P17∼P19, P28∼P29, and 2 months old (M-O)]. Both P28∼P29 (n = 8) and 2 M-O (n = 8) groups successfully gained CRs during the six sessions. In contrast, the C57BL/6J mice at P17∼P19 could not acquire sufficient CRs, indicating that CR could not have been formed and/or analyzed before P17∼P19 even in wild-type mice. B, Development of CR% during eyeblink conditioning in young normal (open circle) and d-n (closed circle) mice at P28∼P29 (n = 8 for each). C, The averaged EMG amplitudes on the sixth session in normal (solid line) and d-n (dotted line) young mice. All of the EMG amplitudes obtained in one session (100 trials) were summed, representing the overall response pattern. D, Frequency histogram of the CR peak latencies in d-n mice. The CR peaks on sixth session were binned into the four time windows (50–100, 100–150, 150–200, and 200–250 ms measured from the CS onset) in the normal (open column) and d-n (solid column) young mice. Columns and error bars represent mean ± SEM.
We then performed an eyeblink conditioning analysis with young d-n mice (P28∼P29). The rate of the CR in d-n mice increased only to ∼40%, compared with >60% in normal mice as described above (Fig. 5B). An ANOVA revealed no significant interaction effects (F(5,70) = 0.909; p = 0.480) but did identify significant differences between genotypes (F(1,14) = 5.804; p = 0.030). Thus, eyeblink conditioning was significantly impaired in young d-n mice when the mice still exhibited clonic seizures.
Adult d-n mice show accumulation of SER and IP3 receptors in PC spines
We investigated whether SER and IP3 receptors were present in the PC spines of adult d-n mice. EM analysis showed that the SER was present in some PC spines in d-n mice (Fig. 6Aa,b, arrows). The SER tubules in longitudinal sections could be seen to insert into the neck of the PC spines, occasionally reaching into the head (Fig. 6Ab, arrows). However, the SER had a small vesicle-like profile and, in transverse sections, mostly had only a few stacks (Fig. 6Aa). Thus, it appeared that tubular SER networks formed poorly within the PC spines of d-n mice. We analyzed the frequency of PC spines containing SER and found that, in adult d-n mice, 23.9 ± 1.3% (mean ± SEM; n = 1113 PC spines in 57 EM micrographs from three mice) of PC spines contained SER, compared with 1.65 ± 1.0% (n = 283 PC spines in 21 EM micrographs from two mice) of PC spines from young d-n mice (Fig. 6B). The difference in frequency between the two age groups was significant (p < 0.0001, Welch's t test). We found that >97% of the PC spines of both young and adult normal mice contained SER: 97.3 ± 0.6% (mean ± SEM; n = 718 spines from three mice) at P19∼P20 and 97.7 ± 0.5% (n = 939 spines from three mice) at 2 months of age. Some of this information on EM studies also appeared in our short report (Takagishi and Murata, 2001).
Immunohistochemistry also showed that IP3 receptor-positive spines were occasionally present in adult d-n mice (Fig. 6Cb, inset, arrows). In contrast, in normal mice they were abundant and could be visualized as tiny spots around the tips and distal parts of PC dendrites (Fig. 6Ca; inset, arrows).
LTD and motor learning is restored in adult d-n mice
We tested the LTD in adult d-n mice and found that most Purkinje cells showed restoration of the LTD. Although only 1 of the 14 cells used for recording displayed potentiation of the slope of PF-EPSPs (120.8% of control), the other cells showed clear induction of the LTD. The mean value of the slope was depressed to 66.4 ± 6.3% (n = 14; p < 0.01) at 30 min after conjunctive stimulation (Fig. 7B). The value of the LTD was not significantly different from that of normal mice (61.5 ± 7.6%; n = 10; p > 0.05 vs normal mice) (Fig. 7A). Thus, the LTD is certainly restored in d-n mice as they age.
Next, we tested eyeblink conditioning in adult d-n mice. Interestingly, the rate of CR increased to >60% during the six sessions as in normal mice (Fig. 7C). An ANOVA revealed no significant interaction effects (F(5,56) = 0.631; p = 0.677) and no significant differences between genotypes (F(1,14) = 0.024; p = 0.880). Thus, adult d-n mice had acquired a CR similar to that of normal mice (Fig. 1A), indicating that the learning deficit of young mice was restored by adulthood.
The averaged EMG amplitudes obtained at the sixth session for normal and mutant mice are shown in Figures 5C and 7D, respectively. In young mice, the averaged EMG amplitude for d-n mice was clearly lower than that of normal mice because of a lower rate of CR (Fig. 5C). In contrast, no differences were observed between the amplitudes of normal and d-n adult mice (Fig. 7D). The latency of peak CR on the sixth session in normal and d-n mice at young and adult ages is shown in Figures 5D and 7E. The data are shown as histograms of the CR temporal components in the time ranges 50∼100, 100∼150, 150∼200, and 200∼250 ms from CS onset. In both age groups, there were no significant differences between normal and d-n mice. Thus, the temporal pattern of the CR is normal in d-n mice.
The motor output for eye blinking and the sensitivity to stimuli were tested in young and adult d-n mice. The frequencies of spontaneous eyeblink and startle response to the tone CS were compared; there were no significant differences in either trait between normal and d-n mice, regardless of age differences (data not shown). These results indicate that the age-dependent improvement of eyeblink conditioning in d-n mice cannot be explained by the change in ability to respond to or in sensitivity to the CS. We further tested pseudoconditioning with pseudorandomized presentations of CS and US in normal and d-n mice at young and adult ages (data not shown). Analysis of the data indicated that there were no significant interaction effects (F(15,60) = 0.335; p = 0.989) or significant genotypic effects (F(3,12) = 0.057; p = 0.981). Thus, our results confirmed that the age differences in d-n mice had no effects on nonassociated responses.
Overall, our present study in young and adult d-n mice demonstrated that defects in SER and IP3 receptors in PC spines caused the deficiency of LTD and motor learning, whereas restoration of LTD and motor learning were accompanied by accumulation of SER and IP3 receptors in PC spines.
Motor coordination is affected during development and adulthood in d-n mice
Adult d-n mice superficially appeared normal since they did not suffer from clonic seizures. However, they still displayed somewhat abnormal movements such as occasionally lifting the tail (Fig. 1A) and a tendency to waddle. Such gait abnormalities in adult d-n mice were shown most clearly in the footprint patterns made by mice after ink staining their forepaws and hindpaws. The footprint patterns of d-n mice were wider and showed a more irregular stride pattern compared with normal mice (Fig. 8A,B). We then performed a rota-rod test to determine whether motor coordination was affected in young and adult d-n mice. When the rota-rod was running, normal mice at both young and adult stages exhibited a clear improvement in performance on successive days of the experiment (Fig. 8C,D). In contrast, d-n mice at both age groups failed to stay on the rota-rod and this performance was not improved even by training for 7 d (Fig. 8C,D). An ANOVA revealed no significant interaction effects in the two groups (F(6,48) = 1.454, p = 0.214 for the young stage; F(6,90) = 1.305, p = 0.263 for the adult stage) but did identify significant differences between the genotypes (F(1,8) = 133.92, p < 0.00001 for the young stage; F(1,15) = 679.74, p < 0.000001 for the adult stage). To further assess motor coordination and balance, we performed a fixed-bar test using a narrow wooden bar (Fig. 8E,F). We found that d-n mice from both age groups were unable to stay on the bar at all. In contrast, young and adult normal mice could stay on the bar. These differences were statistically significant (p = 0.018 for the young stage; p < 0.000001 for the adult stage). Thus, severe motor incoordination still remained in adult d-n mice.
Figure 8.

Motor coordination is impaired during development through the adult age of d-n mice. A, B, Representative foot track patterns of young (P22) and adult (P60) mice. C, D, Rota-rod tests in young (P26∼P29) and adult (P60∼P63) mice. Mean times in which mice of different genotypes remained on the rota-rod (rotating at 10 rpm) during a 7 d training session. Young (C) and adult (D) d-n mice (closed circle) spent significantly less time on the rod than normal mice (open circle). A maximum of 120 s was allowed for each animal per trial. n = 5 for each normal and d-n young mice and 7 and 10 for normal and d-n adult mice. E, F, Fixed-bar test in young and adult mice. The time an animal remained on a bar of 5 mm width was measured for a maximum of 60 s. d-n mice (solid bar) spent significantly less time on the bar than the normal mice (open bar) at both young (E) and adult (F) stages. Columns and error bars represent mean ± SEM. n = 5 for each for normal and d-n young mice and 7 and 10 for normal and d-n adult mice.
Myosin Va is necessary for localization of IP3 receptors in PC spines
Finally, to confirm the role of myosin Va in the localization of IP3 receptors in PC spines, we performed RNA interference analysis in cultured PCs. Cultured PCs at 11 DIV were transfected with siRNA against mouse myosin Va mRNA by single-cell electroporation (Tanaka et al., 2009) and were maintained for 10 d after transfection (to 21 DIV). PCs at 21 DIV developed numerous dendritic spines that were immunopositive for both CD-28K and IP3 receptors (Fig. 9A). Although transfection with the negative control siRNA did not affect myosin Va expression (Fig. 9B, arrow in b1), myosin Va siRNAs caused downregulated expression of endogenous myosin Va in PCs (Fig. 9C, arrow in c1). In PCs with myosin Va knockdown, IP3 receptor-positive PC spines appeared to be decreased in number (Fig. 9Cc3). In contrast, IP3 receptor-positive PC spines were as abundant on the dendrites in PCs transfected with the negative control siRNA (Fig. 9Bb3) as in nontransfected PCs (Fig. 9Aa3). We quantified the density of IP3 receptor-positive spines in PCs at 21 DIV and found that IP3 receptor-positive PC spines were significantly reduced in number (3.1 ± 0.5 per 25 μm of dendritic surface; mean ± SEM) in PCs transfected with myosin Va siRNA compared with those with the negative control siRNA (6.7 ± 0.9 per 25 μm of dendritic surface) or untransfected (6.8 ± 0.5 per 25 μm of dendritic surface) (Fig. 9D). These results showed that reduction of myosin Va expression in PCs affected the localization of IP3 receptors in PC spines, indicating that myosin Va is involved in translocation of IP3 receptors into PC spines.
Figure 9.
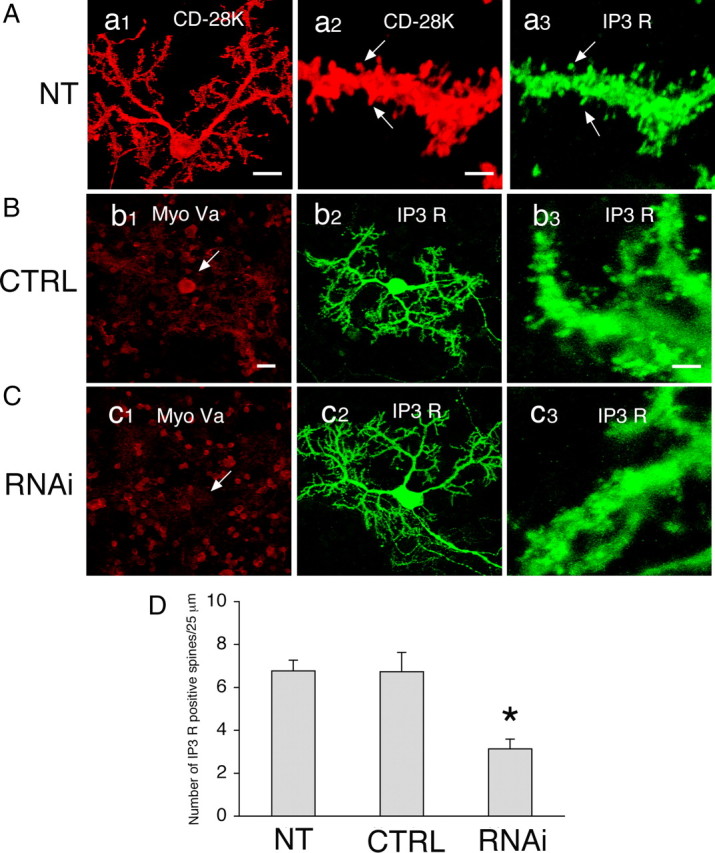
Impairment of IP3 receptor localization in PC spines by RNA interference of myosin Va. A, Cultured PCs at 21 DIV. Cells were not electroporated [nontransfected (NT)] and were double labeled for CD-28K (a1, a2) and IP3 receptors (a3). Spines (a2, a3, arrows) are immunopositive for both CD-28K and IP3 receptors. Scale bars: a1, 20 μm; a2, a3, 2 μm. B, C, Transfection of siRNAs in cultured PCs. Cells at 11 DIV were transfected with negative control siRNA (CTRL) or myosin Va siRNA (RNAi). Ten days after transfection (at 21 DIV), the cells were double labeled for myosin Va (Bb1, Cc1) and IP3 receptors (Bb2,b3, Cc2,c3). Expression of endogenous myosin Va was reduced by RNAi treatment (Cc1, arrow) but not in the control (Bb1, arrow). IP3 receptor-positive spines appeared to be decreased in number in Cc3 but not in Bb3. Scale bars: bi, bz, c1, c2, 20 μm; b3, c3, 2 μm. D, Quantification of the density of IP3 receptor-positive spines in nontransfected cells (NT), in cells transfected with negative control siRNA (CTRL), and in cells transfected with myosin Va siRNA (RNAi). The number of IP3 receptor-positive spines per 25 μm of dendritic surface was counted. n = 30, 15, and 22 for NT, CTRL, and RNAi, respectively. Error bars represent SEM. *p < 0.0005 versus CTRL, Student's t test.
Discussion
Myosin Va is necessary for extension and localization of SER and IP3 receptors into PC spines
It is clear from our previous work on myosin Va-null mutants that myosin Va is crucial for the localization of SER and IP3 receptors in PC spines (Dekker-Ohno et al., 1996; Takagishi et al., 1996; Takagishi and Murata, 2006). In the present study, we provide additional evidence for this role with our RNA interference experiment. Reduction in the expression levels of endogenous myosin Va after transfection of cultured PCs with myosin Va siRNA resulted in a decrease in the number of IP3 receptor-positive PC spines (Fig. 9C,D). [The recent paper by Wagner et al. (2011) has described the direct function of myosin Va in SER transport into PC spines.]
We suspect that differences in SER localization in PC spines between juvenile and adult d-n mice are related to the dendritic outgrowth of PC cells during development. Young d-n mice have an extremely low level of expression of myosin Va protein (Huang et al., 1998) (Fig. 2B), and this is insufficient to function in SER transport into PC spines while the PCs are undergoing rapid and extensive development of dendrites. However, in adult d-n mice, PCs do not undergo dendritic outgrowth (Berry and Bradley, 1976) and there is sufficient myosin Va protein to promote some SER transport into PC spines. We assume that, as the mice age, the increased expression of myosin Va protein or the accumulation of the protein in Purkinje cells, including dendrites, might contribute to SER transport into PC spines. Our immunohistochemistry showed that myosin Va protein was expressed in adult PCs, even though the staining was weaker than in normal PCs (Fig. 2Ad,f). In addition, our Western blot of normal cerebella showed that the level of myosin Va was higher in adults than young mice (an ∼1.8-fold increase) (our unpublished data), leading to the expectation that the mutant protein levels might also increase with age in d-n cerebella.
The mouse neurological mutant flailer (flr) expresses a hybrid gene derived from Myo5a and guanine nucleotides binding protein β 5 (Gnb5) and produces a protein with a dominant-negative effect on myosin Va function (Jones et al., 2000). Homozygous flr mice exhibit juvenile neurological disorders and juvenile flr PC spines are devoid of SER. The flr mice show improvement of their neurological impairment with age, suggesting that SER has gradually accumulated in the PC spines of older mice. Jones et al. (2000) postulated that the hybrid protein was insufficient to compete with normal protein in older mice but did act as an effective competitor during the critical period of brain maturation between 2 and 4 weeks of age.
SER in PC spines is important for the induction of cerebellar LTD and motor learning
We have previously reported that IP3-mediated Ca2+ release from SER in PC spines is essential for the induction of LTD at PF–PC synapses (Miyata et al., 2000). Consistent with this earlier investigation, the present study found that PCs in young d-n mice did not display LTD, as a consequence of a lack of SER in the PC spines, but rather showed LTP. By contrast, the induction of LTD was restored in adult d-n mice, concurrently with the reestablishment of SER in some PC spines. Our observations might be in line with previous reports that demonstrate the inverse calcium threshold model (i.e., the induction of both LTD and LTP at PF–PC synapses depends on the intracellular Ca2+ threshold; however, the threshold for LTD induction is higher than that for LTP induction) (Reynolds and Hartell, 2000; Coesmans et al., 2004). Thus, the level of Ca2+ signal in PC spines that contain almost no SER in young d-n mice might be sufficient for the induction of LTP, but not LTD, and the reestablishment of SER in PC spines might elevate the Ca2+ signal in PC spines to reach the required Ca2+ threshold for the induction of LTD in adult d-n mice.
There is evidence that SER in dendritic spines plays an important role in synaptic plasticity. In hippocampal neurons, absence of stacked SER, termed the spine apparatus, reduces the magnitude of LTP in synaptopodin knock-out (KO) mice (Deller et al., 2003, 2007). The lack of synaptopodin and spine apparatus also leads to behavioral changes, such as impairment in spatial learning in a radial arm maze, which is linked to the LTP (Deller et al., 2003, 2007). Therefore, it appears that the SER in dendritic spines is a vital element in the hippocampal LTP as well as in the induction of cerebellar LTD.
Over the past decade, a large number of studies have investigated the correlation between cerebellar LTD and delayed eyeblink conditioning. Impairments in both cerebellar LTD and eyeblink conditioning has been reported in most cases from genetically manipulated mice (Aiba et al., 1994; Ichise et al., 2000; Kishimoto et al., 2001a,b; Miyata et al., 2001; Koekkoek et al., 2003; Kishimoto and Kano, 2006). Our model of d-n mice also clearly showed that LTD and eyeblink conditioning are interrelated; impaired LTD caused deficits in the acquisition of conditioned eyeblink responses in young mutant mice (Figs. 4B, 5B), and restored LTD was associated with the rescue of the conditioned eyeblink responses in adult mutant mice (Fig. 7B,C). The poor acquisition of the CR in young d-n mice could be attributed to impairments in associative learning ability, since they exhibited normal sensitivity to the stimuli and normal motor output (data not shown). Furthermore, d-n mice exhibited normal CR timing during development until adulthood (Figs. 5D, 7E).
There is still controversy over the role of the cerebellar cortex in CR timing and CR acquisition during eyeblink conditioning. It has been postulated that CR timing is determined by the integrity of cerebellar circuits (Raymond et al., 1996). Indeed, the evidence from studies using cellular recording or lesion techniques suggests that the cerebellar cortex is critical for modulating CR timing (Perrett et al., 1993; Green and Steinmetz, 2005). Nevertheless, many different types of mutant mice that have a cerebellar LTD deficit show normal CR timing (Kishimoto et al., 2001a; Miyata et al., 2001; Kishimoto and Kano, 2006). This is true of the d-n mice, despite the slight possibility that the preserved timing of CR results from insufficient sensitivity in the detection of mouse eyelid closure by the EMG method (Koekkoek et al., 2003). In this study, we modified the conventional surgery of the EMG method and improved the sensitivity for detecting MOO activities. Our present results imply that a cerebellar function other than PF–PC synaptic plasticity contributes to CR timing. Overall, our present findings suggest that myosin Va functions in SER localization in PC spines and regulates the induction of cerebellar LTD and acquisition of motor memory.
Eyeblink conditioning deficits have been reported in patients with nonmotor psychiatric diseases such as schizophrenia (Bolbecker et al., 2009b), bipolar disorder (Bolbecker et al., 2009a), and attention deficit/hyperactivity disorder (Frings et al., 2010), as well as in a large number of human cases with cerebellar lesions (Gerwig et al., 2007; Kronenbuerger et al., 2007). Most of these studies demonstrate that progressive memory deficits occur over time. It is quite intriguing that the memory deficit of young d-n mice completely disappears in adult d-n mice without any artificial intervention.
Motor coordination is not restored in adult d-n mice
In contrast to the acquisition of motor learning ability, motor coordination (Fig. 8) did not show recovery in the adult d-n mice. Circumstantial evidence supports our finding that the recovery of LTD did not rescue the motor coordination defect of adult d-n mice. Protein kinase Cγ (PKCγ) KO mice display intact eyeblink conditioning and normal cerebellar LTD, whereas they have impaired motor coordination that is associated with impaired elimination of multiple CF innervations on PCs (Chen et al., 1995; Kano et al., 1995). The Tc1 mouse model for human Down syndrome also exhibits impairment in motor coordination but has normal LTD at PF–PC synapses (Galante et al., 2009). By contrast, mice lacking cGK1 (cGMP-dependent protein kinase type 1) in PCs show defective LTD and impaired adaptation of vestibular-ocular reflex (VOR), another form of cerebellum-dependent motor learning, but they show normal motor coordination (De Zeeuw et al., 1998). Likewise, transgenic mice specifically expressing a PKCI (PKC inhibitor) in PCs show impairment of LTD and no motor learning in the VOR; however, motor coordination is not affected (Feil et al., 2003). These studies suggest that cerebellar LTD might not be involved in motor coordination.
The neural basis of the motor coordination deficit is not clear in d-n mice but may be related to the malfunction of myosin Va during development of the nervous system in either cerebellar or noncerebellar structures. Hypomyelination of nerves in the brain and spinal cord was observed in d-l (myosin Va-null) mice (Sloane and Vartanian, 2007). Likewise, in d-l mice, malformation of the neuromuscular junctions with regressed postsynaptic sites occurs progressively with the increasing severity of neurological phenotypes (Röder et al., 2008). We suggest that, in d-n mice, a delay in myelination occurs during development of the nervous system and shows little subsequent recovery. Such myosin Va defects might contribute to motor incoordination in d-n mice. This possibility will be addressed in a subsequent study.
Concluding remarks
GS1 and ES are rare autosomal recessive genetic disorders and develop severe neurological impairment in addition to a defect of pigmentation in the hair and skin during infancy (Pastural et al., 2000; Ivanovich et al., 2001; Sanal et al., 2002). Although abnormal distribution of pigment granules in melanocytes has been shown, no pathophysiological examination of the nervous system has been made in GS1 and ES patients. Likewise, the pigmentation defects of dilute myosin Va mutants and their underlying mechanisms are well characterized (Reck-Peterson et al., 2000; Desnos et al., 2007); in contrast, the mechanisms responsible for the neurological deficits remain unclear. In our studies using dilute mutants having both neurological and coat color defects among the hundreds of dilute mutation so far identified, the data described here and in our previous study (Miyata et al., 2000) showed a consequent process of neuronal defects at the molecular, cellular, synaptic, and behavioral levels in myosin Va-deficient cerebella, and are suggestive of a mechanism partially underlying neurological disorders in myosin Va mutants. Our findings do not exclude other mechanisms for myosin Va disease, but they do provide an important platform for understanding pathophysiological aspects, some of the neurological phenotypes, of GS1 and ES patients.
Footnotes
This work was supported by Grants-in-Aid for Scientific Research 0760708 and 20500370 (Y.T.), 20790084 (Y.K.), and 21220006 (M.K.), Takeda Science Foundation (Y.K., M.M., M.T.), the Strategic Research Program for Brain Sciences (Development of Biomarker Candidates for Social Behavior) from Ministry of Education, Culture, Sports, Science, and Technology (M.K.), and Nagoya University Foundation (Y.T.). This work was also supported by Cooperative Study Program of the National Institute for Physiological Sciences. We thank R. E. Larson and S. Ohashi for the myosin Va antibodies and N. Maeda for the IP3 receptor antibody. We thank N. A. Jenkins for providing dilute-neurological mice and her discussion of this work, and K. Imoto for his encouragement of this work.
References
- Aiba A, Kano M, Chen C, Stanton ME, Fox GD, Herrup K, Zwingman TA, Tonegawa S. Deficient cerebellar long-term depression and impaired motor learning in mGluR1 mutant mice. Cell. 1994;79:377–388. [PubMed] [Google Scholar]
- Alami NH, Jung P, Brown A. Myosin Va increases the efficiency of neurofilament transport by decreasing the duration of long-term pauses. J Neurosci. 2009;29:6625–6634. doi: 10.1523/JNEUROSCI.3829-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berry M, Bradley P. The growth of the dendritic trees of Purkinje cells in irradiated agranular cerebellar cortex. Brain Res. 1976;116:361–387. doi: 10.1016/0006-8993(76)90487-x. [DOI] [PubMed] [Google Scholar]
- Bolbecker AR, Mehta C, Johannesen JK, Edwards CR, O'Donnell BF, Shekhar A, Nurnberger JI, Steinmetz JE, Hetrick WP. Eyeblink conditioning anomalies in bipolar disorder suggest cerebellar dysfunction. Bipolar Disord. 2009a;11:19–32. doi: 10.1111/j.1399-5618.2008.00642.x. [DOI] [PubMed] [Google Scholar]
- Bolbecker AR, Mehta CS, Edwards CR, Steinmetz JE, O'Donnell BF, Hetrick WP. Eye-blink conditioning deficits indicate temporal processing abnormalities in schizophrenia. Schizophr Res. 2009b;111:182–191. doi: 10.1016/j.schres.2009.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C, Kano M, Abeliovich A, Chen L, Bao S, Kim JJ, Hashimoto K, Thompson RF, Tonegawa S. Impaired motor coordination correlates with persistent multiple climbing fiber innervation in PKC gamma mutant mice. Cell. 1995;83:1233–1242. doi: 10.1016/0092-8674(95)90148-5. [DOI] [PubMed] [Google Scholar]
- Coesmans M, Weber JT, De Zeeuw CI, Hansel C. Bidirectional parallel fiber plasticity in the cerebellum under climbing fiber control. Neuron. 2004;44:691–700. doi: 10.1016/j.neuron.2004.10.031. [DOI] [PubMed] [Google Scholar]
- Correia SS, Bassani S, Brown TC, Lisé MF, Backos DS, El-Husseini A, Passafaro M, Esteban JA. Motor protein-dependent transport of AMPA receptors into spines during long-term potentiation. Nat Neurosci. 2008;11:457–466. doi: 10.1038/nn2063. [DOI] [PubMed] [Google Scholar]
- Dekker-Ohno K, Hayasaka S, Takagishi Y, Oda S, Wakasugi N, Mikoshiba K, Inouye M, Yamamura H. Endoplasmic reticulum is missing in dendritic spines of Purkinje cells of the ataxic mutant rat. Brain Res. 1996;714:226–230. doi: 10.1016/0006-8993(95)01560-4. [DOI] [PubMed] [Google Scholar]
- Deller T, Korte M, Chabanis S, Drakew A, Schwegler H, Stefani GG, Zuniga A, Schwarz K, Bonhoeffer T, Zeller R, Frotscher M, Mundel P. Synaptopodin-deficient mice lack a spine apparatus and show deficits in synaptic plasticity. Proc Natl Acad Sci U S A. 2003;100:10494–10499. doi: 10.1073/pnas.1832384100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deller T, Orth CB, Del Turco D, Vlachos A, Burbach GJ, Drakew A, Chabanis S, Korte M, Schwegler H, Haas CA, Frotscher M. A role for synaptopodin and the spine apparatus in hippocampal synaptic plasticity. Ann Anat. 2007;189:5–16. doi: 10.1016/j.aanat.2006.06.013. [DOI] [PubMed] [Google Scholar]
- Desnos C, Huet S, Darchen F. “Should I stay or should I go?”: myosin V function in organelle trafficking. Biol Cell. 2007;99:411–423. doi: 10.1042/BC20070021. [DOI] [PubMed] [Google Scholar]
- De Zeeuw CI, Hansel C, Bian F, Koekkoek SK, van Alphen AM, Linden DJ, Oberdick J. Expression of a protein kinase C inhibitor in Purkinje cells blocks cerebellar LTD and adaptation of the vestibulo-ocular reflex. Neuron. 1998;20:495–508. doi: 10.1016/s0896-6273(00)80990-3. [DOI] [PubMed] [Google Scholar]
- Espreafico EM, Cheney RE, Matteoli M, Nascimento AA, De Camilli PV, Larson RE, Mooseker MS. Primary structure and cellular localization of chicken brain myosin-V (p190), an unconventional myosin with calmodulin light chains. J Cell Biol. 1992;119:1541–1557. doi: 10.1083/jcb.119.6.1541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feil R, Hartmann J, Luo C, Wolfsgruber W, Schilling K, Feil S, Barski JJ, Meyer M, Konnerth A, De Zeeuw CI, Hofmann F. Impairment of LTD and cerebellar learning by Purkinje cell-specific ablation of cGMP-dependent protein kinase I. J Cell Biol. 2003;163:295–302. doi: 10.1083/jcb.200306148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freeman JH, Jr, Nicholson DA, Muckler AS, Rabinak CA, DiPietro NT. Ontogeny of eyeblink conditioned response timing in rats. Behav Neurosci. 2003;117:283–291. doi: 10.1037/0735-7044.117.2.283. [DOI] [PubMed] [Google Scholar]
- Frings M, Gaertner K, Buderath P, Gerwig M, Christiansen H, Schoch B, Gizewski ER, Hebebrand J, Timmann D. Timing of conditioned eyeblink responses is impaired in children with attention-deficit/hyperactivity disorder. Exp Brain Res. 2010;201:167–176. doi: 10.1007/s00221-009-2020-1. [DOI] [PubMed] [Google Scholar]
- Galante M, Jani H, Vanes L, Daniel H, Fisher EM, Tybulewicz VL, Bliss TV, Morice E. Impairments in motor coordination without major changes in cerebellar plasticity in the Tc1 mouse model of Down syndrome. Hum Mol Genet. 2009;18:1449–1463. doi: 10.1093/hmg/ddp055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerwig M, Kolb FP, Timmann D. The involvement of the human cerebellum in eyeblink conditioning. Cerebellum. 2007;6:38–57. doi: 10.1080/14734220701225904. [DOI] [PubMed] [Google Scholar]
- Green JT, Steinmetz JE. Purkinje cell activity in the cerebellar anterior lobe after rabbit eyeblink conditioning. Learn Mem. 2005;12:260–269. doi: 10.1101/lm.89505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang JD, Mermall V, Strobel MC, Russell LB, Mooseker MS, Copeland NG, Jenkins NA. Molecular genetic dissection of mouse unconventional myosin-VA: tail region mutations. Genetics. 1998;148:1963–1972. doi: 10.1093/genetics/148.4.1963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunt PS, Fanselow MS, Richardson R, Mauk MD, Freeman JH, Jr, Stanton ME. Synapses, circuits, and the ontogeny of learning. Dev Psychobiol. 2007;49:649–663. doi: 10.1002/dev.20250. [DOI] [PubMed] [Google Scholar]
- Ichise T, Kano M, Hashimoto K, Yanagihara D, Nakao K, Shigemoto R, Katsuki M, Aiba A. mGluR1 in cerebellar Purkinje cells essential for long-term depression, synapse elimination, and motor coordination. Science. 2000;288:1832–1835. doi: 10.1126/science.288.5472.1832. [DOI] [PubMed] [Google Scholar]
- Ivanovich J, Mallory S, Storer T, Ciske D, Hing A. 12-year-old male with Elejalde syndrome (neuroectodermal melanolysosomal disease) Am J Med Genet. 2001;98:313–316. doi: 10.1002/1096-8628(20010201)98:4<313::aid-ajmg1098>3.0.co;2-p. [DOI] [PubMed] [Google Scholar]
- Jones JM, Huang JD, Mermall V, Hamilton BA, Mooseker MS, Escayg A, Copeland NG, Jenkins NA, Meisler MH. The mouse neurological mutant flailer expresses a novel hybrid gene derived by exon shuffling between Gnb5 and Myo5a. Hum Mol Genet. 2000;9:821–828. doi: 10.1093/hmg/9.5.821. [DOI] [PubMed] [Google Scholar]
- Kadotani H, Hirano T, Masugi M, Nakamura K, Nakao K, Katsuki M, Nakanishi S. Motor discoordination results from combined gene disruption of the NMDA receptor NR2A and NR2C subunits, but not from single disruption of the NR2A or NR2C subunit. J Neurosci. 1996;16:7859–7867. doi: 10.1523/JNEUROSCI.16-24-07859.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kano M, Hashimoto K, Chen C, Abeliovich A, Aiba A, Kurihara H, Watanabe M, Inoue Y, Tonegawa S. Impaired synapse elimination during cerebellar development in PKC gamma mutant mice. Cell. 1995;83:1223–1231. doi: 10.1016/0092-8674(95)90147-7. [DOI] [PubMed] [Google Scholar]
- Kishimoto Y, Kano M. Endogenous cannabinoid signaling through the CB1 receptor is essential for cerebellum-dependent discrete motor learning. J Neurosci. 2006;26:8829–8837. doi: 10.1523/JNEUROSCI.1236-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kishimoto Y, Kawahara S, Kirino Y, Kadotani H, Nakamura Y, Ikeda M, Yoshioka T. Conditioned eyeblink response is impaired in mutant mice lacking NMDA receptor subunit NR2A. Neuroreport. 1997;8:3717–3721. doi: 10.1097/00001756-199712010-00012. [DOI] [PubMed] [Google Scholar]
- Kishimoto Y, Kawahara S, Suzuki M, Mori H, Mishina M, Kirino Y. Classical eyeblink conditioning in glutamate receptor subunit delta 2 mutant mice is impaired in the delay paradigm but not in the trace paradigm. Eur J Neurosci. 2001a;13:1249–1253. doi: 10.1046/j.0953-816x.2001.01488.x. [DOI] [PubMed] [Google Scholar]
- Kishimoto Y, Hirono M, Sugiyama T, Kawahara S, Nakao K, Kishio M, Katsuki M, Yoshioka T, Kirino Y. Impaired delay but normal trace eyeblink conditioning in PLCbeta4 mutant mice. Neuroreport. 2001b;12:2919–2922. doi: 10.1097/00001756-200109170-00033. [DOI] [PubMed] [Google Scholar]
- Koekkoek SK, Hulscher HC, Dortland BR, Hensbroek RA, Elgersma Y, Ruigrok TJ, De Zeeuw CI. Cerebellar LTD and learning-dependent timing of conditioned eyelid responses. Science. 2003;301:1736–1739. doi: 10.1126/science.1088383. [DOI] [PubMed] [Google Scholar]
- Kronenbuerger M, Gerwig M, Brol B, Block F, Timmann D. Eyeblink conditioning is impaired in subjects with essential tremor. Brain. 2007;130:1538–1551. doi: 10.1093/brain/awm081. [DOI] [PubMed] [Google Scholar]
- Langford GM, Molyneaux BJ. Myosin V in the brain: mutations lead to neurological defects. Brain Res Brain Res Rev. 1998;28:1–8. doi: 10.1016/s0165-0173(98)00020-4. [DOI] [PubMed] [Google Scholar]
- Levine T, Rabouille C. Endoplasmic reticulum: one continuous network compartmentalized by extrinsic cues. Curr Opin Cell Biol. 2005;17:362–368. doi: 10.1016/j.ceb.2005.06.005. [DOI] [PubMed] [Google Scholar]
- Martone ME, Zhang Y, Simpliciano VM, Carragher BO, Ellisman MH. Three-dimensional visualization of the smooth endoplasmic reticulum in Purkinje cell dendrites. J Neurosci. 1993;13:4636–4646. doi: 10.1523/JNEUROSCI.13-11-04636.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyata M, Okada D, Hashimoto K, Kano M, Ito M. Corticotropin-releasing factor plays a permissive role in cerebellar long-term depression. Neuron. 1999;22:763–775. doi: 10.1016/s0896-6273(00)80735-7. [DOI] [PubMed] [Google Scholar]
- Miyata M, Finch EA, Khiroug L, Hashimoto K, Hayasaka S, Oda SI, Inouye M, Takagishi Y, Augustine GJ, Kano M. Local calcium release in dendritic spines required for long-term synaptic depression. Neuron. 2000;28:233–244. doi: 10.1016/s0896-6273(00)00099-4. [DOI] [PubMed] [Google Scholar]
- Miyata M, Kim HT, Hashimoto K, Lee TK, Cho SY, Jiang H, Wu Y, Jun K, Wu D, Kano M, Shin HS. Deficient long-term synaptic depression in the rostral cerebellum correlated with impaired motor learning in phospholipase C beta4 mutant mice. Eur J Neurosci. 2001;13:1945–1954. doi: 10.1046/j.0953-816x.2001.01570.x. [DOI] [PubMed] [Google Scholar]
- Ohashi S, Koike K, Omori A, Ichinose S, Ohara S, Kobayashi S, Sato TA, Anzai K. Identification of mRNA/protein (mRNP) complexes containing Puralpha, mStaufen, fragile X protein, and myosin Va and their association with rough endoplasmic reticulum equipped with a kinesin motor. J Biol Chem. 2002;277:37804–37810. doi: 10.1074/jbc.M203608200. [DOI] [PubMed] [Google Scholar]
- Pastural E, Ersoy F, Yalman N, Wulffraat N, Grillo E, Ozkinay F, Tezcan I, Gediköglu G, Philippe N, Fischer A, de Saint Basile G. Two genes are responsible for Griscelli syndrome at the same 15q21 locus. Genomics. 2000;63:299–306. doi: 10.1006/geno.1999.6081. [DOI] [PubMed] [Google Scholar]
- Perrett SP, Ruiz BP, Mauk MD. Cerebellar cortex lesions disrupt learning-dependent timing of conditioned eyelid responses. J Neurosci. 1993;13:1708–1718. doi: 10.1523/JNEUROSCI.13-04-01708.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raymond JL, Lisberger SG, Mauk MD. The cerebellum: a neuronal learning machine? Science. 1996;272:1126–1131. doi: 10.1126/science.272.5265.1126. [DOI] [PubMed] [Google Scholar]
- Reck-Peterson SL, Provance DW, Jr, Mooseker MS, Mercer JA. Class V myosins. Biochim Biophys Acta. 2000;1496:36–51. doi: 10.1016/s0167-4889(00)00007-0. [DOI] [PubMed] [Google Scholar]
- Reynolds T, Hartell NA. An evaluation of the synapse specificity of long-term depression induced in rat cerebellar slices. J Physiol. 2000;527:563–577. doi: 10.1111/j.1469-7793.2000.00563.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Röder IV, Petersen Y, Choi KR, Witzemann V, Hammer JA, 3rd, Rudolf R. Role of myosin Va in the plasticity of the vertebrate neuromuscular junction in vivo. PLoS One. 2008;3:e3871. doi: 10.1371/journal.pone.0003871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanal O, Yel L, Kucukali T, Gilbert-Barnes E, Tardieu M, Texcan I, Ersoy F, Metin A, de Saint Basile G. An allelic variant of Griscelli disease: presentation with severe hypotonia, mental-motor retardation, and hypopigmentation consistent with Elejalde syndrome (neuroectodermal melanolysosomal disorder) J Neurol. 2000;247:570–572. doi: 10.1007/s004150070162. [DOI] [PubMed] [Google Scholar]
- Sanal O, Ersoy F, Tezcan I, Metin A, Yel L, Ménasché G, Gürgey A, Berkel I, de Saint Basile G. Griscelli disease: genotype-phenotype correlation in an array of clinical heterogeneity. J Clin Immunol. 2002;22:237–243. doi: 10.1023/a:1016045026204. [DOI] [PubMed] [Google Scholar]
- Sloane JA, Vartanian TK. Myosin Va controls oligodendrocyte morphogenesis and myelination. J Neurosci. 2007;27:11366–11375. doi: 10.1523/JNEUROSCI.2326-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanton ME, Freeman JH, Jr, Skelton RW. Eyeblink conditioning in the developing rat. Behav Neurosci. 1992;106:657–665. doi: 10.1037//0735-7044.106.4.657. [DOI] [PubMed] [Google Scholar]
- Stanton ME, Fox GD, Carter CS. Ontogeny of the conditioned eyeblink response in rats: acquisition or expression? Neuropharmacology. 1998;37:623–632. doi: 10.1016/s0028-3908(98)00072-0. [DOI] [PubMed] [Google Scholar]
- Takagishi Y, Murata Y. Electron microscopic observation of smooth endoplasmic reticulum in the dendritic spine of Purkinje cells in dilute-neurological mice. Environ Med. 2001;45:80–82. [Google Scholar]
- Takagishi Y, Murata Y. Myosin Va mutation in rats is an animal model for the human hereditary neurological disease, Griscelli syndrome type 1. Ann N Y Acad Sci. 2006;1086:66–80. doi: 10.1196/annals.1377.006. [DOI] [PubMed] [Google Scholar]
- Takagishi Y, Oda S, Hayasaka S, Dekker-Ohno K, Shikata T, Inouye M, Yamamura H. The dilute-lethal (dl) gene attacks a Ca2+ store in the dendritic spine of Purkinje cells in mice. Neurosci Lett. 1996;215:169–172. doi: 10.1016/0304-3940(96)12967-0. [DOI] [PubMed] [Google Scholar]
- Takagishi Y, Hashimoto K, Kayahara T, Watanabe M, Otsuka H, Mizoguchi A, Kano M, Murata Y. Diminished climbing fiber innervation of Purkinje cells in the cerebellum of myosin Va mutant mice and rats. Dev Neurobiol. 2007;67:909–923. doi: 10.1002/dneu.20375. [DOI] [PubMed] [Google Scholar]
- Tanaka M, Yanagawa Y, Hirashima N. Transfer of small interfering RNA by single-cell electroporation in cerebellar cell cultures. J Neurosci Methods. 2009;178:80–86. doi: 10.1016/j.jneumeth.2008.11.025. [DOI] [PubMed] [Google Scholar]
- Thompson RF, Krupa DJ. Organization of memory traces in the mammalian brain. Annu Rev Neurosci. 1994;17:519–549. doi: 10.1146/annurev.ne.17.030194.002511. [DOI] [PubMed] [Google Scholar]
- Thompson RF, Steinmetz JE. The role of the cerebellum in classical conditioning of discrete behavioral responses. Neuroscience. 2009;162:732–755. doi: 10.1016/j.neuroscience.2009.01.041. [DOI] [PubMed] [Google Scholar]
- Thompson R, Bas S, Chen L, Ciproano B, Grethe J, Kim J, Thompson J, Tracy J, Weninger M, Krupa D. Associative learning. San Diego: Academic; 1997. [DOI] [PubMed] [Google Scholar]
- Wagner W, Brenowitz SD, Hammer JA., 3rd Myosin-Va transports the endoplasmic reticulum into the dendritic spines of Purkinje neurons. Nat Cell Biol. 2011;13:40–48. doi: 10.1038/ncb2132. [DOI] [PMC free article] [PubMed] [Google Scholar]