

Abstract
We studied wild-type (WT) and Cav1.3−/− mouse chromaffin cells (MCCs) with the aim to determine the isoform of L-type Ca2+ channel (LTCC) and BK channels that underlie the pacemaker current controlling spontaneous firing. Most WT-MCCs (80%) were spontaneously active (1.5 Hz) and highly sensitive to nifedipine and BayK-8644 (1,4-dihydro-2,6-dimethyl-5-nitro-4-[2-(trifluoromethyl)phenyl]-3-pyridinecarboxylic acid, methyl ester). Nifedipine blocked the firing, whereas BayK-8644 increased threefold the firing rate. The two dihydropyridines and the BK channel blocker paxilline altered the shape of action potentials (APs), suggesting close coupling of LTCCs to BK channels. WT-MCCs expressed equal fractions of functionally active Cav1.2 and Cav1.3 channels. Cav1.3 channel deficiency decreased the number of normally firing MCCs (30%; 2.0 Hz), suggesting a critical role of these channels on firing, which derived from their slow inactivation rate, sizeable activation at subthreshold potentials, and close coupling to fast inactivating BK channels as determined by using EGTA and BAPTA Ca2+ buffering. By means of the action potential clamp, in TTX-treated WT-MCCs, we found that the interpulse pacemaker current was always net inward and dominated by LTCCs. Fast inactivating and non-inactivating BK currents sustained mainly the afterhyperpolarization of the short APs (2–3 ms) and only partially the pacemaker current during the long interspike (300–500 ms). Deletion of Cav1.3 channels reduced drastically the inward Ca2+ current and the corresponding Ca2+-activated BK current during spikes. Our data highlight the role of Cav1.3, and to a minor degree of Cav1.2, as subthreshold pacemaker channels in MCCs and open new interesting features about their role in the control of firing and catecholamine secretion at rest and during sustained stimulations matching acute stress.
Introduction
In most pacemaking neurons, spontaneous subthreshold depolarizations are associated with Na+ entry through either hyperpolarization-activated cation channels or voltage-gated Na+ channels (Raman and Bean, 1999). There are, however, neurons in which Ca2+ channels also contribute to the pacemaker current (Bean, 2007). Cav1.3 L-type channels (LTCCs) are particularly suitable for pacemaking neurons given their low threshold of activation and slow inactivation time course (Platzer et al., 2000; Koschak et al., 2001; Lipscombe et al., 2004; Striessnig and Koschak, 2008). Cav1.3 channels drive the spontaneous firing of dopaminergic neurons in substantia nigra (SNc) (Chan et al., 2007) and striatal neurons (Olson et al., 2005) and are likely to contribute to the spontaneous action potential (AP) firing of suprachiasmatic nucleus (SCN) and midbrain dopamine neurons (Jackson et al., 2004; Puopolo et al., 2007).
Rodent adrenal chromaffin cells (RCCs) also fire spontaneously (Nassar-Gentina et al., 1988; Gullo et al., 2003), and their activity is associated with basal release of catecholamines (Zhou and Misler, 1995). In intact adrenal glands, basal secretion is triggered by the resting activity of the splanchnic nerve, and, in isolated cells, spontaneous firing rises from still unidentified ion channels that open at subthreshold voltages. We have shown recently that MCCs and RCCs undergo spontaneous firings that are inhibited or fully blocked by nifedipine and enhanced by compounds that upregulate LTCC gating (Marcantoni et al., 2007, 2009). LTCCs shape the APs, contribute to the pacemaker current, and sustain Ca2+-dependent exocytosis. This derives from two peculiar properties of LTCCs: they activate at low voltages and contribute to ∼45% of total Ca2+ current in rodent chromaffin cells. In RCCs, LTCCs are tightly coupled to BK channels, and AP firing is regulated by the strict interaction between these two channel types (Prakriya and Lingle, 1999). In addition, bovine and rat chromaffin cells express both Cav1.2 and Cav1.3 channels (García-Palomero et al., 2000; Baldelli et al., 2004). Thus, we wanted to study how the two LTCC isoforms contribute to the generation of AP firings and how their coupling to BK channels conditions the AP frequency. We did this by comparing the Ca2+ and K+ currents contributing to the spontaneous firing of wild-type (WT) and Cav1.3−/− knock-out (KO) mice (Platzer et al., 2000).
Here we show that WT-MCCs express comparable fractions of functional Cav1.3 and Cav1.2 channels that contribute to the pacemaker current driving MCC firing. Deletion of Cav1.3 decreases the number of normally firing MCCs (from 80 to 30%) and makes the frequency of remaining firing cells more variable. Action potential shaping and firing frequency are mainly controlled by the coupling between Cav1.3 (Cav1.2) and BK channels, which can be either fast inactivating or non-inactivating, the fast inactivating BK (BKi) channels being more closely coupled to the Cav1.3 isoform and most likely affecting the pacemaker current. Our data highlight the role of Cav1.3 and Cav1.2 as pacemaker channels in MCCs uncovering a new function of LTCCs in the modulation of catecholamine secretion during physiological and pathological stress.
Materials and Methods
Isolation and culture of WT and Cav1.3−/− mouse adrenal medulla chromaffin cells.
All experiments were performed in accordance with the guidelines established by the National Council on Animal Care and were approved by the local Animal Care Committee of Turin University. Chromaffin cells were obtained from young (1–3 months) C57BL/6N mice and Cav1.3−/− mice (Platzer et al., 2000) Animals were killed by cervical dislocation, and chromaffin cells were cultured following a slightly modified version of the method by Sørensen et al. (2003) described previously (Marcantoni et al., 2009). After removal, the adrenal glands were placed in Ca2+- and Mg2+-free Locke's buffer containing the following (in mm): 154 NaCl, 3.6 KCl, 5.6 NaHCO3, 5.6 glucose, and 10 HEPES, pH 7.2, at room temperature. The glands were decapsulated, and the medullas were precisely separated from the cortical tissue. Medulla digestion was achieved for 60 min at 37°C in a DMEM solution containing the following: 1.5 mm l-cysteine, 1 mm CaCl2, 0.5 mm EDTA, and 20 U/ml papain (Worthington Biochemicals). The cell suspension was then centrifuged for 5 min at 900 rpm and washed two times with a Locke's solution containing 1 mm CaCl2 and 10 mg/ml BSA. Cells were then resuspended in 2 ml of DMEM supplemented with 15% fetal calf serum (FCS) and plated in four-well plastic dishes treated with poly-l-ornithine (0.5 mg/ml) and laminin (10 μg/ml in L-15 carbonate) by placing a drop of concentrated cell suspension in the center of each well. After 1 h, 1.8 ml of DMEM supplemented with 15% FCS (Invitrogen), 50 IU/ml penicillin, and 50 μg/ml streptomycin (Invitrogen) was added to the wells. Cells were then incubated at 37°C in a water-saturated atmosphere with 5% CO2 and used within 2–4 d after plating.
Voltage-clamp and current-clamp recordings.
Current-clamp and voltage-clamp recordings were made in perforated-patch conditions using either an Axopatch 200-A or 200-B amplifier and pClamp 10.0 software programs (Molecular Devices). Patch pipettes were made of thin borosilicate glass (Kimax 51; Witz Scientific) and filled with different solutions depending on whether we measured Ca2+ or K+ currents. For Ca2+ currents, the pipette contained the following (in mm): 135 CsMeSO3, 8 NaCl, 2 MgCl2, and 20 HEPES, pH 7.3 with CsOH plus amphotericin B (Sigma). The external bath contained the following (in mm): 135 tetraethylammonium (TEA)-Cl, 2 CaCl2, 2 MgCl2, 10 glucose, and 10 HEPES, pH 7.4 with CsOH. For K+ currents, the pipette contained the following (in mm): 135 KAsp, 8 NaCl, 2 MgCl2, and 20 HEPES, pH 7.3 with KOH plus amphotericin B. The external bath contained the following (in mm): 137 NaCl, 4 KCl, 2 CaCl2, 1 MgCl2, 10 glucose, and 10 HEPES, pH 7.4 with NaOH. When required, a 0 Ca2+ external solution was prepared by replacing the extracellular Ca2+ (2 mm) with an equal amount of Mg2+. This abolished inward Ca2+ currents without producing a significant voltage shift to the activation–inactivation gating of voltage-dependent Ca2+ channels. Amphotericin B was dissolved in dimethylsulfoxide (DMSO) stored at −20°C in stock aliquots of 50 mg/ml and used at a final concentration of 500 μg/ml. To facilitate the sealing, the pipette was first dipped in a beaker containing the internal solution and then backfilled with the same solution containing amphotericin B. The syringe used for filling the pipettes and containing the internal solution plus amphotericin B was kept cold (0–4°C) during the experiments.
Pipettes with series resistance of 1–2 MΩ were used to form giga seals. Recording of Ca2+ and K+ currents started when the access resistance decreased below 15 MΩ, which usually happened within 10 min after sealing (Cesetti et al., 2003). Series resistance was compensated by 80% and monitored throughout the experiment. Ca2+ currents were evoked by step depolarization of 10–600 ms to a fixed potential (+10 mV), sequentially increasing test potentials, or action potential trains (APTs) (“action potential clamp”). Voltage-gated and Ca2+-activated K+ currents were elicited using a double-pulse protocol described previously (Prakriya and Lingle, 1999). The holding potential (Vh) was −70 mV throughout the experiments, except for the experiments shown in Figure 1c, where the Vh was −50 mV, mimicking the MCCs resting potential. Current traces were filtered using a low-pass Bessel filter set at 1–2 kHz and sampled at 10 kHz. Fast capacitative transients during step depolarization were minimized online by the patch-clamp analog compensation. Uncompensated capacitative currents were further reduced by subtracting the averaged currents in response to P/4 hyperpolarizing pulses. The indicated voltages were not corrected for the liquid junction potential (LJP), whose estimate is conditioned by the undetermined junction potential of the patch. LJP was 15 mV (absolute value) in current-clamp and voltage-clamp control conditions when measuring AP firing or recording K+ and Ca2+ currents. This value increased by 5 mV when adding 135 mm TEA to the bath to measure Ca2+ currents. TEA was added after seal formation, and the cell was voltage clamped. For all the other cases, when using micromolar concentrations of drugs or toxins, the LJP remained constant. All the experiments were performed at room temperature (22–24°C).
Figure 1.

Cav1.2 and Cav1.3 expression in the adrenal medulla and cortex and their DHP sensitivity in WT-MCCs and KO-MCCs. a, Quantitative comparison of different LTCC isoform transcripts in adrenal medulla and cortex of WT mice. Relative abundance of isoforms is given as percentage of the total copy numbers of all LTCC α1-subunit transcripts in each experiment (n = 3). Cav1.1 and Cav1.4 copy numbers did not exceed assay detection limits (n = 2). b, Expression of Cav1.2 α1 subunit in adrenal medulla and cortex in Cav1.3−/− mice relative to WT compared by ΔΔCt analysis using GAPDH and β-actin (bAct) as reference genes (n = 3). c, Dose–response relationship of Ca2+ current block by nifedipine (nife) recorded from WT-MCCs (n = 7) and KO-MCCs (n = 5). Percentage of block was measured using 20 ms pulses to +10 mV from Vh of −50 mV. The smooth curves represent the fit to the data with IC50 of 0.21 ± 0.07 μm (WT) and 0.48 ± 0.12 μm (KO) and Hill slopes of 1.22 ± 0.52 and 1.35 ± 0.48, respectively. d, Time course of peak Ca2+ current recorded from a WT-MCC before, during, and after sequential application of 0.01, 0.3, 1, and 3 μm nifedipine (nife). Step depolarization to +10 mV was applied every 10 s. The inset shows the current traces recorded at the time indicated by the letters. e, Time course of peak Ca2+ current recorded from a WT-MCC during sequential application of 3 μm nifedipine (nife), 3.2 μm ω-CTx-GVIA + 1 μm ω-CTx-MVIIC (GVIA+MVIIC), and 400 nm SNX-482 (SNX). Notice the partial recovery after washing that uncovers the irreversible block of N- and P/Q-type channels by ω-CTx-GVIA and ω-CTx-MVIIC. The inset shows the current traces recorded at the time indicated by the letters.
In the action potential-clamp mode, the cell was voltage clamped using a train of three action potentials recorded previously during spontaneous firing in current-clamp conditions. This allowed studying the time course of Ca2+ and K+ currents underlying an APT (Bean, 2007). Extracellular and intracellular solutions in this case were the same used for current-clamp measurements listed below. To simplify the recording procedure, we used the same train of APs in all MCCs (WT and KO). This was an almost obligatory choice because spontaneous firing in WT-MCCs and KO-MCCs was rather irregular, and selecting a representative pattern of APs required a certain amount of time that could become critical within a short recording period (5–7 min). Finally, the selected APT had sufficiently large overshoots to allow the reversal equilibrium potential for Ca2+ to be determined during current recording (Marcantoni et al., 2009).
APs were recorded in perforated-patch-clamp conditions using an intracellular solution containing the following (in mm): 135 KAsp, 8 NaCl, 20 HEPES, 2 MgCl2, and 5 EGTA. The external bath contained the following (in mm): 137 NaCl, 4 KCl, 2 CaCl2, 1 MgCl2, 10 glucose, and 10 HEPES, pH 7.4 with NaOH. Spontaneous action potentials could be recorded in current-clamp mode at resting conditions without injecting any current. For measuring the input resistance and to determine the threshold of action potential firing, the cells were first hyperpolarized at Vh of −70 mV by passing negative current (−3 to −15 pA) to stop spontaneous firing. Positive current step injections of variable duration (5–100 ms) and amplitude (5–30 pA) were then delivered until single or multiple action potentials were elicited. APs were recorded in the “normal” current-clamp mode with no corrections for the slow patch-clamp responses to “fast” current injections (Magistretti et al., 1996). This introduces partial distortion to the rising and falling phase of APs but did not bias the present analysis based on the “comparative effects” of drugs on spontaneously occurring APs. The shape of APs reported here is rather similar to that recorded from RCCs in the fast current-clamp mode (Gullo et al., 2003).
Solutions.
External solutions were exchanged as reported previously (Carabelli et al., 2007). Nifedipine (3 μm), BayK-8644 (1,4-dihydro-2,6-dimethyl-5-nitro-4-[2-(trifluoromethyl)phenyl]-3-pyridinecarboxylic acid, methyl ester) (1 μm), paxilline (1 μm), and apamine (200 nm) were purchased from Sigma, and tetrodotoxin citrate (TTX) (300 nm) from Tocris Bioscience. ω-Agatoxin-IVA (ω-Aga-IVA), ω-conotoxin-GVIA (ω-CTx-GVIA), ω-conotoxin-MVIIC (ω-CTx-MVIIC), and SNX-482 were purchased from Peptide Institute (Osaka, Japan) and used for blocking P/Q-, N-, and R-type channels. Cells pretreated with the toxins were bathed for 20 min in a control solution containing 2 μm ω-Aga-IVA and 3.2 μm ω-CTx-GVIA and then used in free-toxin solutions. BAPTA and EGTA (Invitrogen) were dissolved in DMSO and introduced by incubating the cells for 30–40 min in 20 μm of the AM-ester form dissolved in normal DMEM as described above (Prakriya and Lingle, 2000).
Data are given as mean ± SEM for n numbers of cells. Statistical significance was calculated by either using Student's paired t test or one-way ANOVA, followed by Bonferroni's post hoc test when multiple data comparison were required (Fig. 2a–c). Values of p ≤ 0.05 were considered significant.
Figure 2.
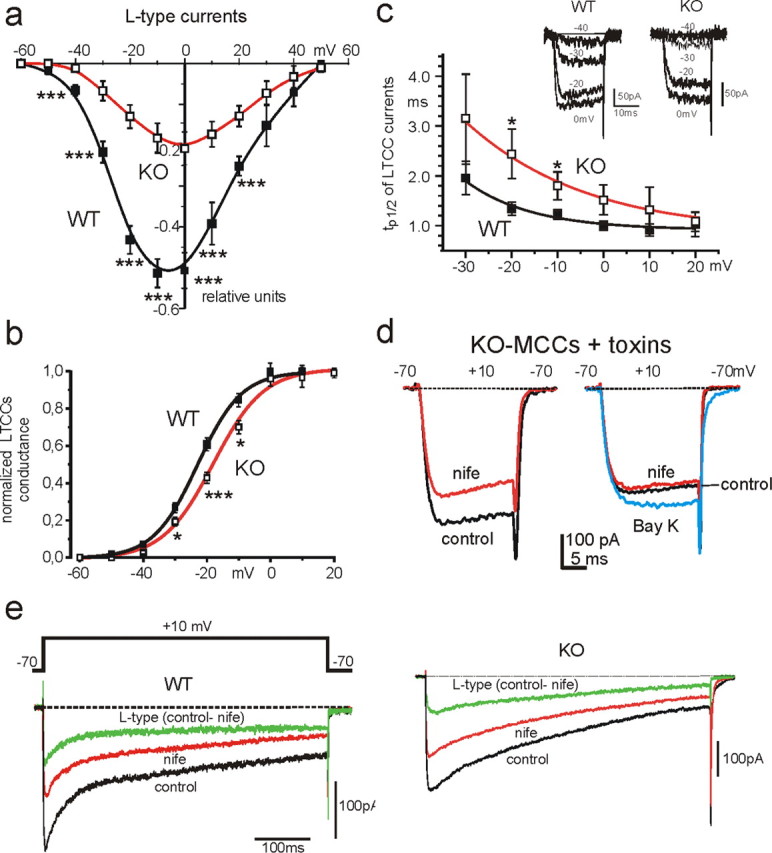
Voltage-dependent characteristics of LTCCs in WT-MCCs and KO-MCCs. a, I–V characteristics of normalized L-type currents in ω-toxin-treated WT-MCCs (filled squares; n = 14) and KO-MCCs (open squares; n = 12). Vh was −70 mV. The L-type current amplitude was determined as the difference between control and nifedipine-insensitive currents, using 3 μm nifedipine to block LTCCs (for the estimated DHP blocking potency at Vh of −70 mV, see text and supplemental Fig. S1, available at www.jneurosci.org as supplemental material). L-type currents are plotted as fractions of the total current. b, Voltage dependence of Cav1.3 and Cav1.2 channel conductance in WT-MCCs and KO-MCCs. The normalized LTCC conductance was calculated as Ipeak/(V − Vrev) with Vrev = +50 mV from n = 8 WT-MCCs and n = 5 KO-MCCs. The two continuous curves are Boltzmann functions best fitting the data points with V1/2 = −23.0 mV and slope factor k = 6.9 mV for WT-MCCs and V1/2 = −18.1 mV and k = 7.9 mV for KO-MCCs. c, Voltage dependence of tp1/2 taken as an index of LTCC activation in ω-toxin-treated WT-MCCs and KO-MCCs. tp1/2 was smaller in WT-MCCs and significantly prolonged at −20 and −10 mV in KO-MCCs (*p < 0.05). Inset, Time course of two families of L-type currents recorded from a WT-MCC and a KO-MCC during brief step depolarization to −40, −30, −20, and 0 mV from −70 mV Vh. d, KO-MCCs displayed heterogeneous distribution of Cav1.2 channel expression. To the left are shown the blocking effect of nifedipine (nife) on Ca2+ currents in a ω-toxin-treated KO-MCC responding normally to the DHP. To the right are shown the effects of nifedipine and BayK-8644 (Bay K) on a KO-MCC characterized by a weak response to the DHPs. e, Cav1.3 currents inactivate less than Cav1.2 during prolonged membrane depolarization. L-type currents were elicited using pulses of 600 ms to +10 mV and calculated by subtracting nifedipine (nife)-insensitive from control currents. In the WT-MCC (left), the resulting L-type current after an initial fast inactivation of ∼100 ms reached a steady-state value that did not change further for the remaining 500 ms. In the KO-MCC (right), inactivation persists for the entire duration of the pulse reaching a steady-state value after 600 ms. The datasets were compared using one-way ANOVA, followed by Bonferroni's post hoc test (*p < 0.05; ***p < 0.001).
RNA extraction, reverse transcription, and quantitative PCR.
Total RNA was prepared from tissue pools of intact glands from 1-month-old male C57BL/6N mice (Carabelli et al., 2007; Sinnegger-Brauns et al., 2009). Reverse transcription (RT) was performed as published previously (Sinnegger-Brauns et al., 2009), using RNAqueous-4PCR kit (Ambion).
Quantitative PCR was performed with Taqman Gene Expression assays (Applied Biosystems) as published previously (Sinnegger-Brauns et al., 2009). Assay GenBank accession numbers were as follows: Cav1.2, Mm00437917_m1; Cav1.3-42a (recognizing the short C-terminal Cav1.3 splice variant), custom designed (forward primer, GGAAGTACCCTGCGAAGAACAC; reverse primer, CTCAGGCAGAGAACTCTAAAGCAT; probe, TTGCCCTACAGATGCTTG); Cav1.3-49 (recognizing full-length Cav1.3 transcripts), Mm 01209927_g1; Cav1.4, Mm00490443_m1; Cav1.1, Mm00489257_m1; Cav2.3, Mm01284736_m1 and Mm0494444_m1; glyceraldehyde-3-phosphate dehydrogenase (GAPDH), Mm99999915_g1; and β-actin, Mm00607939_s1. Data in Figure 1a were obtained from two independent RNA preparations, each from pools of 25 dissected glands.
Comparative expression in WT and KO glands was investigated with ΔΔCt analysis using GAPDH and β-actin as reference genes. Total RNA was obtained from tissue pools (30 adult mice) in parallel. Three independent, parallel reverse transcriptions were performed for comparative analysis of Cav1.2, Cav1.1, Cav1.4, and Cav2.3 expression, respectively (see above). Relative standard curves using mouse whole-brain cDNA (0.0005–50 ng RNA equivalents) as a template served to compare the assays used. Because of insignificant slope variations, mean slope values (−3.52 for Cav1.2, −3.61 for Cav1.4, and −3.67 for Cav2.3 analysis) could be used for ΔΔCt calculations. Statistical analysis (linear regression for standard curves and one-sample t test for KO/WT expression ratios) was performed using GraphPad Prism 5.00 software (GraphPad Software). All experimental data points were obtained as triplicates.
Results
Expression of Cav1.3 and Cav1.2 channels in WT-MCCs and KO-MCCs
Cav1.2 and Cav1.3 α1 subunits are effectively expressed in MCCs. Using quantitative RT-PCR, we found that, in the medulla, Cav1.3 (long- and short-length α1-subunit) contributes to 80% of the total LTCC α1 transcripts extracted from 25 mouse adrenal glands (Fig. 1a, left). The Cav1.2 α1 contributes to 20%. The number of Cav1.1 and Cav1.4 transcripts was below the detection limit of the assays used (Sinnegger-Brauns et al., 2009). This differed sharply from the distribution of Cav1.2 and Cav1.3 subunits in the adrenal cortex in which the contribution of the LTCC subunits was nearly reversed (Fig. 1a, right). This suggests that, in Cav1.3−/− KO mice, the only functioning LTCC is Cav1.2, whereas in WT-MCCs, both Cav1.3 and Cav1.2 contribute to the total Ca2+ current. Cav1.3-42a channels (short C-terminal splice variant), which exhibit more rapid Ca2+-induced inactivation (Singh et al., 2008), contribute only to a small fraction of Cav1.3 in medulla and cortex, suggesting that most of the Cav1.3 current is mediated by the long splice variant (Cav1.3-49). As an additional control, we tested for changes of Cav1.2 expression in KO mice. As shown in Figure 1b, the ratio of Cav1.2 mRNA expression between KO and WT was not significantly different from unity in the adrenal medulla when using GAPDH and β-actin as reference genes, and it was only minimally decreased in the adrenal cortex (0.86 and 0.73). Also, Cav1.1 and Cav1.4 mRNA expression was below detection levels in the medulla and cortex of KO adrenal glands.
Dihydropyridine sensitivity of Cav1.3 and Cav1.2 channels in WT-MCCs
To assay the contribution of Cav1.2 and Cav1.3 to Ca2+ currents and AP firings, we first tested the sensitivity of LTCCs to nifedipine in WT-MCCs and KO-MCCs at voltages near the interpulse potential (−50 mV) at which spontaneous firing starts and at which the dihydropyridines (DHPs) have maximal blocking effect on Cav1.2 (Welling et al., 1997) and Cav1.3 (Koschak et al., 2001). Figure 1c shows that WT-MCCs were equally sensitive to nifedipine as KO-MCCs that possessed only the Cav1.2 isoform. Nifedipine blocked the total Ca2+ currents available at +10 mV in a concentration-dependent manner (Fig. 1d). Near maximal block was obtained with 3 μm nifedipine: 52.8 ± 3.4% (n = 7) in WT-MCCs and 22.4 ± 2.2% (n = 5) in KO-MCCs. Figure 1e shows also that, after blocking LTCCs with 3 μm nifedipine, the remaining non-L-type currents were fully blocked by sequential applications of ω-CTx-GVIA (3.2 μm), ω-CTx-MVIIC (10 μm), and SNX-482 (0.4 μm), which selectively block N-, P/Q-, and R-type channels. This was observed in six WT-MCCs and suggests that 3 μm nifedipine is a suitable concentration for fully blocking LTCCs in WT and KO-MCCs at Vh of −50 mV.
Interestingly, WT-MCCs and KO-MCCs had comparable total Ca2+ current amplitudes (−148.7 ± 8.4 pA, n = 29; −140.6 ± 12.3 pA, n = 26; Vh of −50 mV) and membrane capacitance (10.1 ± 0.4 pF, n = 29; 10.1 ± 0.6 pF, n = 26). This suggests that loss of the Cav1.3 isoform is compensated with an increased density of non-L-type currents.
Conductance and activation–inactivation kinetics of Cav1.3 and Cav1.2 channels in WT-MCCs and KO-MCCs
We next studied the voltage dependence of channel conductance and the activation–inactivation kinetics of the two LTCC subunits. Because MCCs express different densities of N-, L-, P/Q-, and R-type channels (Hernandez-Guijo et al., 1998), we maximally blocked N- and P/Q-type channels by pretreating MCCs with ω-CTx-GVIA (3.2 μm) and ω-Aga-IVA (2 μm). Under these conditions, the amplitude of Ca2+ currents was smaller and carried by R- and L-type channels. Figure 2a shows the I–V characteristics of LTCCs in WT-MCCs and KO-MCCs obtained by subtracting from controls the currents remaining after adding 3 μm nifedipine from Vh of −70 mV. The more negative Vh was used to determine the LTCCs available near resting potential (−50 mV). Data are given as fractional amounts of total current densities that were not significantly different in WT-MCCs and KO-MCCs (−15.1 ± 1.7 pA/pF, n = 23; −14.5 ± 1.6 pA/pF, n = 17). These values compare well with those at Vh of −50 mV and suggest that KO-MCCs compensated for the loss of Cav1.3 by mainly upregulating the density of R-type channels, although we cannot exclude partial upregulation of N- and P/Q-type channels because the test measurements were done under perfusion conditions after toxin incubation and thus a small fraction of bound toxins could have been washed out. Confirmation that R-type channels were upregulated in KO-MCCs came also from a comparative quantitative PCR analysis in which Cav2.3 α1-subunit transcripts were significantly upregulated by 37.0 ± 0.03% (n = 3) when normalized to GAPDH in adrenal medulla but not in cortex.
It is worth noticing that, at −70 mV Vh, higher doses of nifedipine are required to fully block Cav1.3 channels, with respect to −50 mV Vh (Koschak et al., 2001). In a series of experiments (supplemental Fig. S1, available at www.jneurosci.org as supplemental material), we tested this specific issue and concluded that, when using 3 μm nifedipine at −70 mV Vh, the full block of LTCCs could be underestimated by 15–20%. This partially underestimates the true size of Cav1.3 current at −70 mV Vh but does not significantly alter the data analysis of Figure 2.
In WT-MCCs, LTCCs started activating at approximately −50 mV and reached peak values between −10 and 0 mV (52% of the total). In KO-MCCs, the I–V relationship was shifted toward more positive voltages, and current amplitudes were reduced (peak current at 0 mV was 24%). The calculated Cav1.2 channel conductance was shifted by ∼5 mV toward more positive potentials than WT-MCCs containing Cav1.3 channels (Fig. 2b). An even more marked shift was evident on the activation kinetics. The half-time-to-peak (tp1/2) was larger at every potential and shifted to the right in KO-MCCs (Fig. 2c), in good agreement with the reported faster activation of Cav1.3 compared with Cav1.2 (Koschak et al., 2001; Helton et al., 2005). Finally, an interesting feature of toxin pretreated KO-MCCs was the heterogeneous blocking potency of nifedipine, which revealed a population of cells responding to the DHP (80% of the cells) (Fig. 2d, left) and cells rather insensitive to nifedipine and BayK-8644 (20%) (Fig. 2d, right).
The inactivation time course of L-type currents was studied using single step depolarizations of 600 ms to +10 mV from Vh of −70 mV. These experiments could not be tested at different potentials on the same cells given the large quantity of Ca2+ entering the cells. Figure 2, e and f, shows that, in WT-MCCs after a rapid decay lasting 100 ms (attributable to the Ca2+-dependent inactivation of LTCCs), the residual L-type current inactivates very little during the remaining 500 ms. This differs from the inactivation of Cav1.2 in KO-MCCs, which is fast and persists for the entire pulse duration. On average, the LTCC inactivated by 49 ± 5% (n = 7) after 600 ms in WT-MCCs and by 73 ± 3% in KO-MCCs (n = 8; p < 0.01).
In conclusion, despite the contribution of Cav1.2 to WT-MCCs recordings, Cav1.3 possesses kinetic properties resembling those of other cell preparations (Platzer et al., 2000; Lipscombe et al., 2004). The Cav1.3 channel of MCCs activates at more negative potentials and inactivates more slowly during prolonged depolarization than Cav1.2. Both properties favor a role of Cav1.3 in sustaining inward pacemaker currents during spontaneous firing.
Different coupling of Cav1.3 and Cav1.2 to BK channels in WT-MCCs and KO-MCCs
Ca2+-activated BK channels are highly expressed in RCCs (Neely and Lingle, 1992) and are preferentially coupled to LTCCs (Prakriya and Lingle, 1999; Marcantoni et al., 2007). We tested whether this was also the case in MCCs and whether there could be a preferential coupling between BK channels and Cav1.3 or Cav1.2. To address this, we first applied the same double-pulse protocol used for separating BK from voltage-gated K+ currents in RCCs (Herrington et al., 1995). Two pulses of 400 ms to +80 mV were delivered in a 10 s sequence to activate outward K+ currents. One pulse was applied directly from −70 to +80 mV to activate voltage-gated K+ currents (KV) that quickly reached mean amplitudes of 3.2 nA and inactivated slowly during the long pulse to +80 mV (Fig. 3a, filled circles). The second pulse was delivered after a prestep of 10 ms to +10 mV to briefly open all available voltage-gated Ca2+ channels. The pulse activated both KV and BK currents that reached mean peak amplitudes of 5.2 nA in ∼5 ms and inactivated (Fig. 3a) or deactivated (Fig. 3b) back to the KV current (open circles). BK currents were transient, with a decaying time constant of 50.9 ± 5.3 ms (n = 19) in WT and 42.3 ± 7.4 ms (n = 21) in KO-MCCs and could be fully blocked by applying 500 μm Cd2+ (Fig. 3a, left), applying the selective BK channel blocker paxilline (1 μm) (Sanchez and McManus, 1996) (Fig. 3a, right), or replacing external Ca2+ with Mg2+ to give nominally 0 external Ca2+ concentration. Interestingly, in 30% of KO-MCCs, the short prepulse was unable to produce sizeable BK currents, although Ca2+ currents were present.
Figure 3.
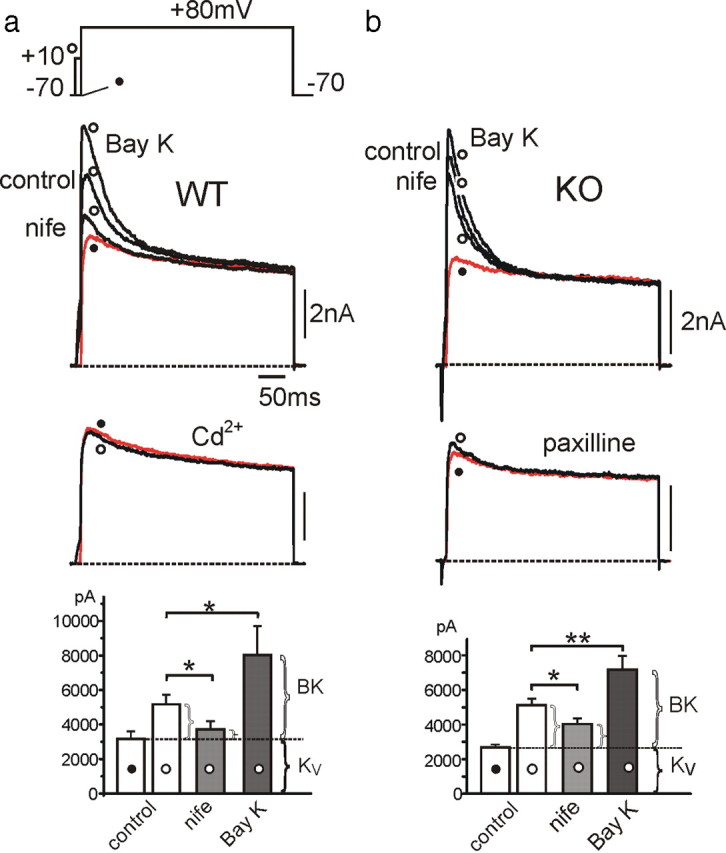
LTCC coupling to BK channels in WT-MCCs and KO-MCCs. a, Ca2+-activated and voltage-gated K+ currents recorded from a WT-MCC at control and during application of nifedipine (nife; 3 μm), BayK-8644 (Bay K; 1 μm), or Cd2+ (500 μm). The voltage command consisted of a double-pulse protocol with a test potential of 400 ms to +80 mV preceded (open circle) or not (filled circle) by a 10 ms prepulse to +10 mV to activate maximal inward Ca2+ currents. Ca2+ inward currents were not always visible because of the fast BK and KV channel activation. Below are the same recordings in the presence of 500 μm Cd2+ obtained from the same MCC. To the bottom are the peak amplitude values of K+ currents (n = 20; *p < 0.05). The horizontal dotted line indicates the mean peak amplitude of KV currents estimated at +80 mV without prepulse (filled circle). The amount of currents exceeding the dotted line represents the BK current. b, BK and KV currents recorded from a KO-MCC. The protocol was as in a except that BK currents were blocked by paxilline (1 μm). The recordings are from the same MCC. Addition of Cd2+ (500 μm) or 0 Ca2+ solutions caused similar blocking effects. To the bottom are the mean values of peak KV and BK currents as in a (n = 16; *p < 0.05; **p < 0.01).
Nifedipine markedly reduced the transient BK component in WT-MCCs (64.5%) (Fig. 3, left) and to a lesser extent in KO-MCCs (45.8%) (Fig. 3, right). Considering that LTCCs contribute to ∼50% to the total Ca2+ currents at +10 mV from −70 mV in WT-MCCs and 25% in KO-MCCs, it appears that BK channels are more coupled to L-type than non-L-type channels (Prakriya and Lingle, 1999). Strong coupling to LTCCs is also suggested by the potentiating effects of BayK-8644 on BK currents by 142% (n = 5) in WT-MCCs and by only 82% (n = 5) in KO-MCCs. We also measured the voltage-dependent deactivation kinetics of KV and BK channels in WT-MCCs and KO-MCCs after short depolarizing pulses and found no significant kinetic differences (supplemental Fig. S2, available at www.jneurosci.org as supplemental material).
To better understand the degree of coupling between BK and Ca2+ channels and the real nature of BK channel inactivation, we also measured the time course of K+ currents during prolonged Ca2+ loading steps and in the presence of intracellular Ca2+ buffers (EGTA and BAPTA). Prolongation of preloading steps (from 10 to 90 ms) caused different responses in WT-MCCs and KO-MCCs (Fig. 4a,b). The majority of WT-MCCs (90%) had BKi channels, which increased their amplitude with increasing preloading steps but remained substantially fast inactivating (n = 12) (Fig. 4a). In contrast, the majority of KO-MCCs (70%; n = 10) had fast decaying BK currents during brief presteps that became non-inactivating after prolonged Ca2+ preloading, uncovering the presence of slowly inactivating BK (BKs) channels (Fig. 4b). In this case, the fast decaying phase is indeed a “fast deactivation” because BKs channels deactivate quickly after short preloading steps, presumably in accordance with the termination of Ca2+ influx and rapid fall of submembrane Ca2+ (Prakriya et al., 1996). The remaining 30% of KO-MCCs were unable to produce sizeable BK currents even after prolonged presteps, despite Ca2+ currents were present. In all cases, addition of paxilline (1 μm) or a 0 Ca2+ solution fully blocked BKi and BKs channels (Fig. 4c).
Figure 4.

Effects of varying the duration of Ca2+ loading steps in the absence (control) or in the presence of EGTA-AM or BAPTA-AM on the time course and amplitude of BK currents in WT-MCCs (a) and KO-MCCs (b). a, Recordings from three WT-MCCs in control solution, after cell incubation with 20 μm EGTA for 40 min or 20 μm BAPTA-AM for 40 min, as indicated. Notice the increasing amplitude of fast inactivating BK currents with increasing the duration of preloading steps in control conditions, the persistence of pure fast inactivating BK currents in the presence of EGTA, and the absence of any BK current in the presence of BAPTA. On the insets are reported the values of Ip and Iss measured as indicated in d from n = 12 (control), 11 (EGTA), and 8 (BAPTA) WT-MCCs (**p < 0.01 vs control using Student's paired t test). b, Same as in a but from three different KO-MCCs. Notice how the prolonged Ca2+ preloading steps uncover a large non-inactivating BKs current and how EGTA and BAPTA are both effective in preventing this BK current. Ip and Iss were derived as in a from n = 10 (control), 13 (EGTA), and 6 (BAPTA) KO-MCCs (*p < 0.05, **p < 0.01 vs control using Student's paired t test). c, Overlapped current traces recorded from a WT-MCC in control conditions, 0 Ca2+ solution (red traces), and after adding 1 μm paxilline (blue traces). The double-pulse protocol was as in Figure 3 with a Ca2+ preloading step of 90 ms. Notice the full block of the transient BK current in the two conditions. d, Measure of Ip and Iss from a WT-MCC. The pulse protocol was as indicated in the inset: the test potential was to +80 mV, and the preloading steps of 10 and 90 ms were to +10 mV (Vh of −70 mV).
The bars in the insets indicate the percentage of BK currents relative to KV measured at the peak after a short preloading step (Ip) and at the steady state of a prolonged step (Iss) (Fig. 4d). Ip is indicative of the amount of BKi channels that open transiently after short pulses, whereas Iss is indicative of the BKs channels that remain open after long depolarizations. Iss was significantly larger in KO-MCCs with respect to WT-MCCs (p < 0.05). This indicates that KO-MCCs express mainly BKs channels, whereas WT-MCCs express a predominance of BKi and a minor fraction of BKs channels. Supplemental Figure S3 (available at www.jneurosci.org as supplemental material) shows a selection of BK currents that furnishes an overview of their differences and variability in WT-MCCs and KO-MCCs. In 8 of 12 WT-MCCs, the recordings were like those on the bottom, whereas in the remaining four cells the time courses were as shown on the top.
The responses to preloading steps changed drastically when using EGTA as intracellular Ca2+ buffer. EGTA was either applied intracellularly in whole-cell conditions (10 mm) or extracellularly in perforated patches (EGTA-AM, 20 μm for 30–40 min). EGTA completely abolished the BKs currents in both WT-MCCs (n = 11) and KO-MCCs (n = 13). All WT-MCCs exhibited only BKi currents that were maximal after the first preloading step (10 ms) and then declined progressively with longer presteps (attributable to the BKi channel inactivation developing during the prestep) (Fig. 4a, middle). On the contrary, KO-MCCs either displayed no BK currents (n = 8) (Fig. 4b, middle) or had small BKi currents (n = 5). These latter markedly increased by adding BayK-8644 (1 μm) (supplemental Fig. S4, available at www.jneurosci.org as supplemental material). This indicates that Cav1.3 is closely coupled to BKi and that BKs are rather distant from Ca2+ channels and effectively uncoupled by EGTA. In contrast to EGTA, BAPTA-AM (20 μm for 30–40 min) abolished all BK currents in both WT (n = 8) and KO-MCCs (n = 6), suggesting that, despite Ca2+ channels and BKi channels being close enough to overcome the buffering action of millimolar EGTA, they are far enough to be uncoupled by BAPTA (see Discussion).
Resting potential and firing modes in WT-MCCs and KO-MCCs
As reported previously (Marcantoni et al., 2009), the majority of WT-MCCs (80%) had a mean resting potential of −49 mV and exhibited spontaneous firing over periods of 5–10 min (Fig. 5a, left). In most cells, the firing was continuous but irregular. The frequency of APs changed greatly from cell to cell (from 0.2 to 4.9 Hz; mean, 1.5 ± 0.2 Hz; n = 41) and could change significantly while recording from the same cell. In a small fraction of WT-MCCs, the firing occurred in bursts, with periods of regular activity interrupted by brief silent periods of some seconds. These findings are in good agreement with data in MCCs from intact adrenal glands (Nassar-Gentina et al., 1988).
Figure 5.
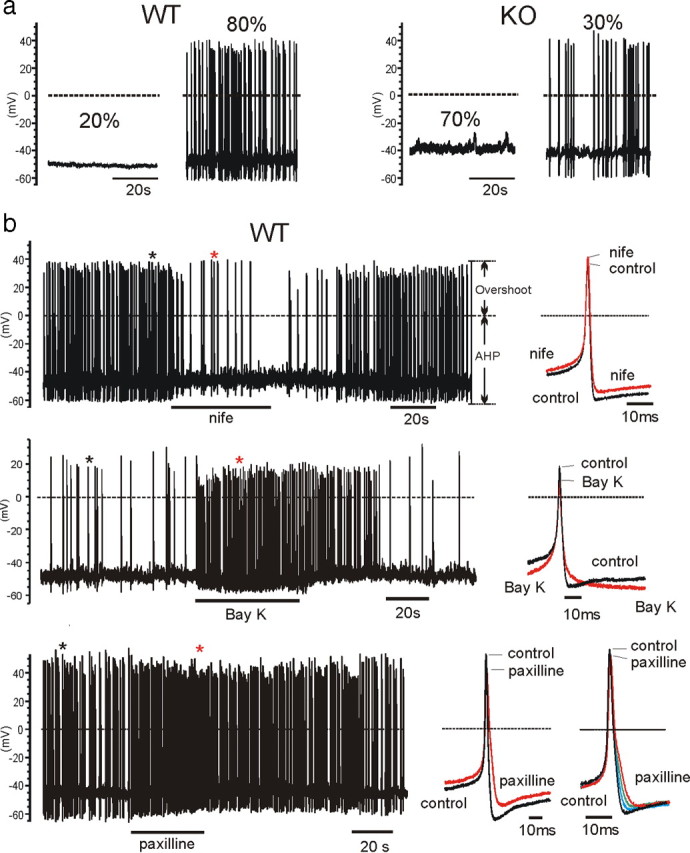
a, Representative recordings and percentages of firing and nonfiring WT-MCCs and KO-MCCs. Only 30% of KO-MCCs showed spontaneous firing, whereas the majority of WT-MCCs (80%) had spontaneous activity. b, Effects of nifedipine (nife), BayK-8644 (Bay K), and paxilline on pacemaking in WT-MCCs and KO-MCCs. The panels show the spontaneous firing of three WT-MCCs before, during, and after application of 3 μm nifedipine (top), 1 μm BayK-8644 (middle), or 1 μm paxilline (bottom). To the right are shown two overlapped action potentials on an expanded timescale corresponding to those indicated by the asterisks. Nifedipine had a full blocking action, which recovered completely after washing, whereas BayK-8644 and paxilline accelerated the firing rate. Paxilline reduced also the AHP and broadened AP width. This is more evident on the overlapped APs to the right recorded during paxilline action in a different WT-MCC. The arrows on the top show how the overshoot and the AHP were determined.
The loss of Cav1.3 in KO-MCCs abolished the firing in 70% of KO-MCCs (Fig. 5a, right). The interpulse starting potential was slightly more positive (−45 mV; p < 0.03) (Table 1), and this was most likely the reason of the increased mean frequency observed in KO-MCCs exhibiting spontaneous firing (1.9 ± 0.4 Hz; n = 30) that was, however, not statistically significant (p < 0.29) (Table 1). By comparing Ca2+ currents near resting potentials and during AP firing conditions (n = 12), we found that nonfiring KO-MCCs had usually not measurable Ca2+ currents at −40 mV despite their size at +10 mV, and the coupling with BK channels were normal.
Table 1.
Parameters defining the action potential shape in spontaneously firing KO-MCCs and WT-MCCs
| Interp. pot. (mV) |
Maximal rate of rise (ms) |
Overshoot (mV) |
Width at 0 mV (ms) |
AHP (mV) |
τAHP (ms) |
Frequency (Hz) |
||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| KO | WT | KO | WT | KO | WT | KO | WT | KO | WT | KO | WT | KO | WT | |
| Mean | −45.1 | −49.0 | 55.7 | 60.7 | 37.2 | 40.6 | 2.2 | 1.9 | −57.9 | −63.7 | 18.7 | 29.8 | 1.99 | 1.51 |
| SEM | 1.0 | 1.5 | 4.0 | 9.5 | 3.0 | 2.7 | 0.2 | 0.1 | 1.8 | 1.7 | 2.8 | 7.3 | 0.42 | 0.24 |
| t test | 0.03* | 0.56 | 0.40 | 0.11 | 0.02* | 0.12 | 0.29 | |||||||
Mean and SEM for the interpulse potential(Interp.pot.), maximal rate of rise, overshoot, width at 0 mV, AHP, and time constant on returning to the interpulse potential after the AHP (τAHP) were obtained from n = 24 KO-MCCs and n = 26 WT-MCCs. The values for the frequency were obtained from n = 30 WT-MCCs and n = 41 KO-MCCs. The only two statistically different values were the interpulse potential (*p < 0.03) and the AHP (*p < 0.02).
Despite these differences, firing WT-MCCs and KO-MCCs had both comparably high input resistance (4.1 ± 0.5 GΩ, n = 7; 3.1 ± 0.8 GΩ, n = 5, respectively) that was significantly higher than nonfiring MCCs (WT, 1.5 ± 0.8 GΩ, n = 5; KO, 1.8 ± 0.5 GΩ, n = 5; p < 0.05) (Nassar-Gentina et al., 1988). A consequence of this was that current injections of 5–8 pA were sufficient to generate AP trains from MCCs held at −70 mV, whereas nonfiring cells required 25–30 pA to induce AP trains (data not shown).
LTCC coupling to BK channels regulates the AP shape and firing frequency in WT-MCCs
In WT-MCCs, nifedipine completely blocked the firing in 20 of 28 cells, and, in the remaining cells, the frequency was decreased by 59% (from 1.7 ± 0.4 to 0.7 ± 0.3 Hz; p < 0.05). Nifedipine changed the shape of the AP and had variable effects on the interpulse potential, either increasing or decreasing it by a few millivolts. In 5 of 11 MCCs, the interpulse potential increased slightly (mean, +3.1 mV), whereas in five cells, it decreased (mean, −5.4 mV). In one cell, it remained unchanged. Altogether, the DHP produced a mean increased overshoot of 5.5 mV (p < 0.05) and a 3.5 mV reduction of the afterhyperpolarization (AHP) (Fig. 5b, top). Nifedipine produced also a slight broadening of the AP width (from 2 to 2.6 ms) that was not evident in every cell. These effects are consistent with a strong coupling between LTCCs and BK channels. The increased overshoot and reduced AHP indicate an L-type-mediated block of BK channels during spikes, whereas the variable effects on the interspike potential reflect either direct block of LTCCs (hyperpolarization) or indirect block of BK channels (depolarization). This point will be discussed later.
BayK-8644 had opposing effects to nifedipine on AP shape and firing frequency (Fig. 5b, middle). The DHP agonist decreased the overshoot by 9.6 mV (p < 0.05) and increased the AHP and the interpulse potential by 5.2 and 11.9 mV (p < 0.01). BayK-8644 also shortened the interpulse interval, and the firing mode became more regular. In the majority of BayK-8644-treated MCCs, the frequency increased approximately threefold (from 0.8 ± 0.3 to 2.1 ± 0.4 Hz; p < 0.01). The AHP was prolonged and followed by a steeper subthreshold depolarization that shortened the interspike interval (Fig. 5b, middle). This indicates that BK channels coupled to LTCCs have higher probability of opening with BayK-8644 and dominate the deeper AHP phase, whereas the increased L-type current accelerates the slow depolarization and increases the spike frequency.
The role of BK channels on setting the shape and frequency of AP spikes was also evident when testing the effects of paxilline (Fig. 5b, bottom). In 12 WT-MCCs, 1 μm paxilline caused an average increase of 86% of the firing frequency (from 1.4 to 2.6 Hz), whereas in another seven cells, it had no effects, suggesting that BK channels contribute to slow down the frequency of AP firings and that their distribution varies from cell to cell. Paxilline also reduced the interpulse potential by 2.4 mV and the AHP by 5.7 mV (n = 5) (p < 0.05), broadening the AP width by 2.0 ms at −20 mV (p < 0.05). All this indicates that BK channels contribute markedly to the repolarization phase of the APs. In the examples of Figure 5b, bottom, paxilline caused a clear reduction and prolongation of the AHP and a decreased overshoot. This latter was most likely attributable to the increased firing rate and resting depolarization. Notice that, although nifedipine and paxilline have comparable effects on AP shape, they have opposite effects on firing frequency, emphasizing the opposing roles that LTCCs and BK channels play in setting the firing frequency: LTCCs accelerate and BK channels slow down the firing.
Spontaneously firing of KO-MCCs are sensitive to nifedipine and BayK-8644
As stated above, 30% of KO-MCCs were spontaneously firing. We therefore tested how Cav1.2 channels and coupled BK channels controlled this activity. Nifedipine had a reduced effect on firing KO-MCCs. Only in 6 of 11 KO-MCCs did the DHP stop the firing, whereas in the remaining cells, the frequency was decreased by 46% (from 5.1 ± 1.9 to 2.8 ± 1.6 Hz; p < 0.02) (Fig. 6, top). Nifedipine had less evident effects on the AP shape than in WT-MCCs. The changes of the overshoots, width, and interpulse potential were not significantly different from the control values of KO-MCCs (Table 1). The only parameter that was consistently different in the KO-MCCs was the AHP, which decreased by 4.9 mV (from −58.4 to −52.5 mV; n = 5), suggesting that the strong action of BK on the AP shape observed in WT-MCCs was strongly attenuated in KO-MCCs. The blocking effects of nifedipine nevertheless uncovered a critical role of Cav1.2 channels (when sufficiently expressed) in the control of firing frequency.
Figure 6.

Effects of nifedipine and BayK-8644 on three spontaneously firing KO-MCCs. Top and middle, Same protocols and analysis as in Figure 5b. The black and red asterisks indicate the position of the overlapped potentials shown to the right. The two DHPs acted moderately on both the shape and frequency of the AP firing. Bottom, The paradoxical effect of BayK-8644 on a spontaneously firing KO-MCC. Cell firings was rather irregular and application of BayK-8644 first caused an acceleration of the firing, followed by a net depolarization to −10 mV. To the right is the recording inside the dashed rectangle on an expanded timescale. The cell returned spontaneously to the resting potential after washing BayK-8644.
BayK-8644 had clearly different effects on KO-MCCs. In 14 KO-MCCs, six responded with a moderate increase of firing frequency (79%) and a sizeable reduction of AHP (2.9 mV) (Fig. 6b, middle), with no additional significant changes to the AP shape. The remaining eight cells responded with an unexpected transient membrane depolarization, perhaps indicating the presence of a sufficient density of Cav1.2 channels and weak coupling to BK channels responsible for this paradoxical response (Fig. 6b, bottom). With BayK-8644, the amplitude of APs decreased rapidly because of the quickly repeated depolarizations, the Ca2+ entry through BayK-8644-activated Cav1.2 channels, and opening of other Ca2+ channels, contributing to the net depolarization.
Cav1.3 and Cav1.2 contribute to the pacemaker current driving WT-MCCs and KO-MCCs firing
LTCCs contribute markedly to the pacemaker Ca2+ current in WT-MCCs (Marcantoni et al., 2009). This is most evident using the action potential-clamp technique (see Materials and Methods). In the presence of TTX and TEA to block Na+ and K+ channels, Ca2+ currents possessed two well separated inward components: an early slowly activating phase (prespike) that was highly sensitive to nifedipine and a late short-lasting component (postspike), also carried by non-L type channels (Fig. 7b, inset). The prespike current contributed to both the pacemaker potential and the rapid action potential upstroke, whereas the postspike current sustained the AP repolarization. In WT-MCCs, the prespike component started soon after the end of the AHP phase and increased progressively to reach a peak during the rapid AP rise (Fig. 7a,b). In 16 MCCs, the mean prespike current measured at −45 mV was −17.4 ± 3.6 pA (n = 16), and nifedipine blocked it by 75%, suggesting a significant contribution of LTCCs (Cav1.3 and Cav1.2) (Fig. 7b and left inset in c). Notice that the prespike current reached mean peak values of −89 pA just before the spike upstroke at approximately −30 mV.
Figure 7.

Ca2+ currents in WT-MCCs and KO-MCCs during an action potential clamp recorded in current-clamp conditions from a spontaneously firing cell. K+ and Na+ currents were blocked by 135 mm TEA and 0.3 μm TTX in the bath solution. In a is illustrated the voltage-clamp command consisting of a train of three APs separated by different interpulse intervals. The cell was initially held at −70 mV and then clamped at the AP waveform (see Materials and Methods). In b are shown the overlapped Ca2+ currents recorded from a WT-MCC before, during, and after application of 3 μm nifedipine (nife). Right inset, Ca2+ current traces corresponding to the second AP (dashed rectangle) on a more expanded timescale. c, Same as in b except that the recording was from a nonfiring KO-MCC. The cell possessed a small prespike Ca2+ current that was potentiated by BayK-8644 (Bay K; n = 8). Notice the large prespike L-type current increase and how the mean amplitude at −45 mV with BayK-8644 increases to nearly the same size of WT-MCC control current (gray box in b). Left inset, Mean amplitudes of the prespike Ca2+ currents measured from WT-MCCs and KO-MCCs (n = 16) at the time when the interpulse potential reached −45 mV, before the second AP (arrow in a). Notice the strong current reduction induced by nifedipine in both WT-MCCs and KO-MCCs. **p < 0.01, ***p < 0.001 vs control using Student's paired t test.
In nonfiring KO-MCCs, the prespike current was remarkably smaller (−5.6 ± 1.1 pA at −45 mV; n = 16; p < 0.05), and nifedipine decreased its size by 79% (Fig. 7c, left inset). The small amplitude of the prespike current in KO-MCCs was expected because Cav1.2 channels contribute only to a minor fraction of the total Ca2+ current in KO-MCCs. They also have more complete inactivation during long depolarizations and lower probability of opening at −45 mV with respect to Cav1.3. In most KO-MCCs, the prespike component was barely detectable, but addition of BayK-8644 enhanced it by approximately a factor of 3 (Fig. 7c, left inset). This may explain the increased firing frequency of a fraction of BayK-8644-treated KO-MCCs (Fig. 6).
At variance with the prespike, the postspike current was also carried by non-LTCCs (N, P/Q, and R) and had different amplitudes in WT-MCCs and KO-MCCs (−320.0 ± 24.6 pA, n = 16; −535.4 ± 77.6 pA, n = 16, respectively; p < 0.05), indicating that, in KO-MCCs, the smaller prespike current was followed by a larger postspike current. A possible reason for this increase is both the reduced intracellular Ca2+ loading during the prespike that increases Ca2+ ions reversal potential (ECa) and the higher percentage of non-LTCCs expressed in KO-MCCs that contribute to the postspike current. In WT-MCCs, the larger prespike Ca2+ loading causes robust Ca2+ accumulation that lowers ECa much below +52 mV (AP overshoot). This causes a net outward current at the peak of the AP and a smaller postspike current (Fig. 7b, right inset). In KO-MCCs, Ca2+ preloading is much attenuated, ECa increases to +52 mV or more, and the postspike currents increase proportionally.
LTCC-activated K+ channels contribute critically to the pacemaker current in WT-MCCs
To quantify the contribution of BK and voltage-gated K+ channels to the spontaneous firing, we used the action potential-clamp and TEA-free control solutions plus 0.3 μm TTX to block Na+ channels. Figure 8a shows that, in WT-MCCs, K+ outward currents are visible during spikes, whereas Ca2+ currents are evident during the prespike interval. Nifedipine had a marked blocking effect on both the prespike (86%; n = 10) and the transient K+ current (51%) (Fig. 8a, left inset), suggesting that the two currents were mostly driven by Cav1.3 and Cav1.2 channels.
Figure 8.
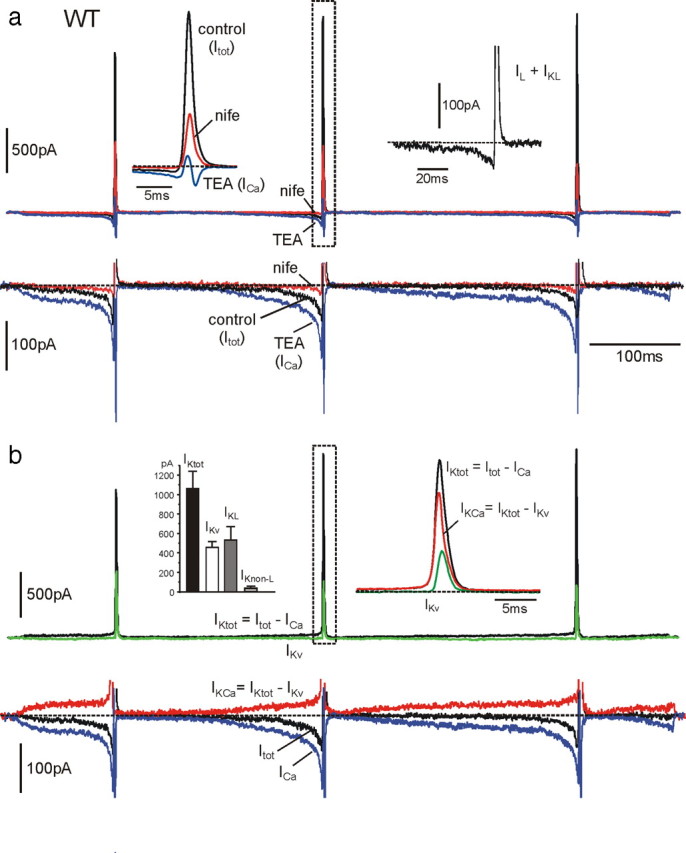
Contribution of BK and voltage-gated K+ and Ca2+ currents during an AP train clamp in a WT-MCC. a, Three overlapped current traces recorded in control conditions, in the presence of nifedipine (nife; 3 μm) and with 135 mm TEA. On the top row are evident the large transient K+ currents of increasing amplitudes during spikes and the small prespike currents preceding AP generation. As shown on a more expanded vertical scale (bottom row), the prespike control current was always net inward and increased markedly after addition of TEA (ICa). Left top inset, K+ and Ca2+ currents just before and during the second spike on a more expanded timescale corresponding to the dashed rectangle. The inward and outward control currents were strongly blocked by nifedipine. Right top inset, Sum of L-type currents (IL) and L-type-activated BK currents (IKL) obtained by subtracting nifedipine-insensitive currents from control currents. The current is net inward before the spike and turns rapidly out during the upstroke. The top part of the outward current is blanked photographically. b, Time courses of the total (IKtot) and the Ca2+-activated K+ current (IKCa) calculated from the recordings of a overlapped to the voltage-gated K+ current (IKv) measured in 0 external Ca2+. IKtot was obtained by subtracting the Ca2+ current recorded in the presence of 135 mm TEA (ICa) from the total control current (Itot), whereas IKCa was obtained by subtracting IKv from IKtot, as indicated. Right top inset, IKtot, IKCa, and IKv during the second spike on an expanded timescale. Left top inset, Mean values of IKtot, IKv, and BK currents activated by L-type (IKL) and non-L-type (IKnon-L) Ca2+ channels (n = 14), calculated as explained in Results. Notice that non-L-type-activated K+ currents are nearly absent in WT-MCCs. In the bottom row is illustrated the time course of the control current (Itot), which is net inward during the interspike intervals and the outward (IKCa) and inward (ICa) components contributing to this current. As shown, the inward Ca2+ current is always larger than IKCa during the interspike intervals.
To determine the time course of BK currents (IKCa), we first determined the time course of the total K+ current (IKtot) by subtracting Ca2+ (ICa) (in 135 mm TEA) from control currents (Itot) (Fig. 8a,b). Next, we measured the time course of voltage-gated K+ currents (IKv) by blocking IKCa in a 0 Ca2+ solution (external Ca2+ replaced by Mg2+). As shown in Figure 8b, IKtot exhibited both a large transient outward current during the APs and a small slowly rising prespike outward component, whereas IKv contributed only during the APs (Fig. 8b, right inset). IKCa was finally obtained by subtracting IKv from IKtot (IKCa = IKtot − IKv) (Fig. 8b). As shown, IKCa dominated the K+ current during APs and contributed to most of the small prespike outward current. Thus, subtraction of ICa and IKv from Itot uncovers the slowly rising BK current that counterbalances the inward Ca2+ current dominated by LTCCs. The resulting current remains net inward and sets the pacemaking of MCCs (Fig. 8b, black trace). It is important to notice that, because fast inactivating BKi channels activate at more negative potentials with respect to non-inactivating BKs channels (Sun et al., 2009), it is likely that BKi channels carry most of the slow prespike outward current. BKs channels are expected to sustain the postspike current together with BKi channels.
Given the main role of L-type currents (IL) and their associated BK currents (IKL), we analyzed the time course of their sum (IL + IKL) by subtracting nifedipine-insensitive currents from control currents (Itot) (Fig. 8a, right inset). IL + IKL was close to the baseline after the first AP and net inward for most of the interspike interval, reaching mean peaks of −69 pA (prespike) before turning outward and mean peaks of 539 pA during the second AP (Fig. 8b, left inset). Although having remarkably different time courses and amplitudes, the inward and outward component of IL + IKL carried comparable quantities of charges when integrated over an entire firing cycle, inclusive of prespike and postspike intervals. The 0 current level was accurately set at the current value measured at Vh (−70 mV). The ratio of charges carried outward to that carried inward was either <1 (outward currents < inward currents; n = 11) or >1 (n = 8). This may explain the variability of nifedipine action on the interpulse potential described above. In MCCs with net inward currents during a cycle (ratio <1), addition of nifedipine may induce hyperpolarization, whereas in MCCs with net outward currents (ratio >1), nifedipine may depolarize the cell (Fig. 5b, top).
The left inset in Figure 8b summarizes the mean amplitudes of IKL, IKnon-L, and IKv contributing to IKtot during the second spike. IKnon-L represents the non-L-type-activated BK current and was obtained by subtracting IKL from IKCa. From the figure, the dominant contribution of IKL with respect to IKnon-L in WT-MCCs is evident.
BayK-8644 and paxilline potentiate the inward pacemaker current in WT-MCCs
Depending on the coupling of LTCCs to BK channels, the sum of prespike ICa and IKCa was found to change considerably from cell to cell, perhaps explaining the extremely variable set of firing frequencies (0.2–4 Hz). In most cases, the prespike current was close to the baseline at the end of the first AP and turned net inward at approximately half the interpulse interval. Figure 9a shows an extreme case in which, in the presence of BayK-8644, ICa + IKCa was inward soon after the first APs and remained as such for most of the interpulse duration, reaching a peak of −60 pA before turning outward at the next AP. Notice that a net inward current of a few picoamperes passing through a membrane resistance of 3–4 GΩ is sufficient to raise the membrane potential to the threshold of AP firing. This implies that, in the cell of Figure 9a, the natural firing that would have occurred in the presence of BayK-8644 would have been at a much higher rate than that used to evoke the currents. The inset and the bottom traces show the near total inhibition of control and BayK-8644-potentiated outward currents by nifedipine.
Figure 9.
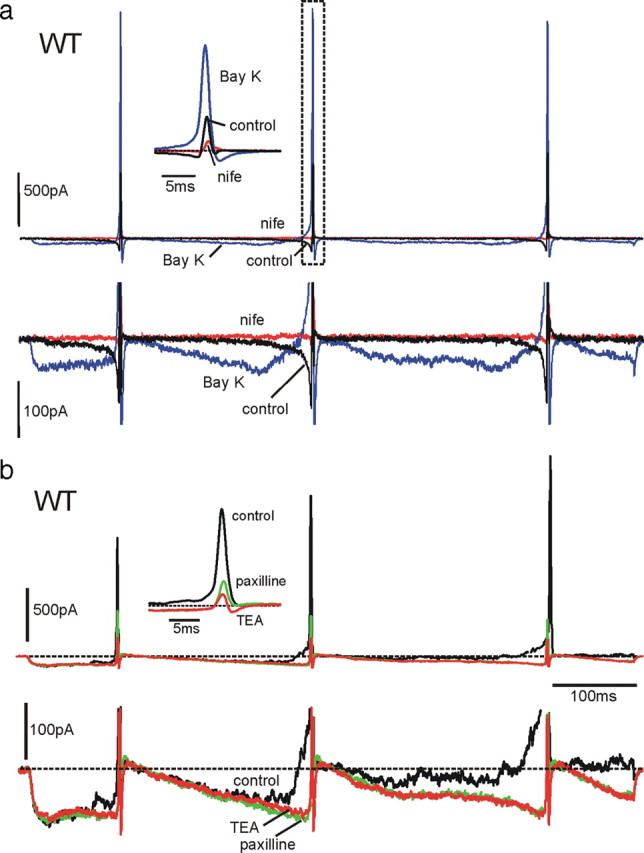
BayK-8644 and paxilline increase the net inward pacemaker current in WT-MCCs. a, In the top row are shown the effects of BayK-8644 (Bay K; 1 μm) and nifedipine (nife; 3 μm) on inward and outward currents during an action potential train clamp. As shown at more expanded vertical (bottom row) and timescales (inset), BayK-8644 increases dramatically the prespike current and generates a dominant BK current component during the three spikes. Both inward and outward currents are primarily blocked by nifedipine. Notice also the brief inward L-type current after the AHP associated with the closing of BayK-8644-modified LTCCs. This current contributed to the quick redepolarization after the increased AHP induced by BayK-8644. b, Effects of paxilline on K+ outward currents in a BayK-8644-treated WT-MCC. The DHP activator induces a large inward L-type current that activates a noisy BK outward current that rises gradually during the interpulse. Paxilline blocks the noisy current, increases the net inward prespike current, and drastically lowers the K+ outward component during the spike (inset). The residual K+ outward current is mainly associated with voltage-gated K+ channels. The block of the slow outward current by paxilline is similar to that induced by TEA, confirming that the current component blocked by paxilline is carried by Ca2+-activated BK channels.
We also assayed the contribution of BK channels to IKCa by using 1 μm paxilline. Paxilline had an effective blocking action on both the slowly activating and fast transient IKCa (Fig. 9b). In most WT-MCCs (n = 12), paxilline fully blocked the slow component and only partially the transient outward current (mean block of 60%). In the case of Figure 9b, a WT-MCC was pretreated with BayK-8644 (1 μm) to overactivate both LTCCs and BK channels. The control prespike current was inward soon after the first AP and turned outward before the second AP. The trace was noisy, indicating the presence of a robust current carried by large-conductance BK channels. Addition of paxilline removed the noise, increased the amplitude of the inward current, and drastically lowered the amplitude of the transient IK, preserving only ICa and IKv (see inset). Addition of 135 mm TEA caused complete block of IKv and uncovered the prespike Ca2+ current. Thus, the BK channels blocked by paxilline are the only K+ channels contributing to the prespike current controlling AP firings.
In a small percentage of cells (20%), the block of the prespike IKCa by paxilline was only partial (mean of 80%), whereas that of the transient component was preserved. The reason for this partial block is unclear and could be attributable to the existence of SK channels. This is supported by preliminary experiments using the SK channel blocker apamine (200 nm) and RT-PCR analysis indicating abundance of SK2 mRNA in MCCs (D. H. F. Vandael, D. Gavello, and E. Carbone, unpublished observations), which require additional work for full clarification. The possibility that other ion channels beside SK contribute to the pacemaker current is an open issue that has not been addressed here (Marcantoni et al., 2009, their Discussion).
Pacemaker currents carried by L and BK channels are strongly attenuated in KO-MCCs
As shown in Figure 7, the loss of Cav1.3 channels markedly decreases the inward Ca2+ currents between spikes in KO-MCCs. An easy expectation is that the coupling of L-type to BK channels produces proportionally lower IKL and that nifedipine has a limited action on outward K+ currents while preserving its blocking effects on the prespike current. We found that this was true in all the 19 KO-MCCs tested. Nifedipine blocked the prespike current by 86% and the outward K+ current by only 14%. Figure 10a shows an example of currents recorded from a KO-MCC in which nifedipine blocked almost completely the prespike current and by only 20% the outward K+ component. On average, KO-MCCs possessed transient K+ currents (IKtot) of smaller amplitude with respect to WT-MCCs (750 ± 107 pA, n = 19 vs 1061 ± 178 pA, n = 14; p < 0.05) (Fig. 10a, bottom left inset), whereas IKv had nearly the same size (457 ± 61 vs 505 ± 82 pA). On the contrary, there was a nearly fivefold decrease of IKL (from 538 ± 134 to 100 ± 26 pA; p < 0.01) primarily compensated by a sevenfold increase of IKnon-L (from 36 ± 21 to 268 ± 100 pA; p < 0.05). Thus, in KO-MCCs, the BK currents were primarily activated by non-L-type channels and contributed less to the total K+ currents. This could explain the reduced effects of nifedipine and paxilline on AP shape and suggests that KO-MCCs differ from WT-MCCs not only for the reduced L-type currents but also for the reduced contribution of BK currents to the spontaneous firing.
Figure 10.

Time course and contribution of K+ and Ca2+ currents to the AP firing is markedly different in KO-MCCs. Test solutions and protocols were similar to those used for WT-MCCs (see Fig. 8). a, Overlapped current traces recorded in control conditions, during nifedipine application (3 μm; nife; red trace), and with 135 mm TEA (blue trace) in a KO-MCC. The prespike Ca2+ current was particularly small compared with the postspike current. Left inset, K+ and Ca2+ currents relative to the dashed rectangle on a more expanded timescale. Nifedipine had a marked action on the inward currents but had only minor blocking effects on the outward K+ current, suggesting a small IKL component. Middle inset, Time course of IKtot, IKCa, and IKv, showing the larger contribution of IKv. IKtot and IKCa were obtained as described in Results. Right inset, Mean values of IKtot, IKL, IKnon-L, and IKv (n = 19). Notice the small contribution of IKL compared with WT-MCCs. b, K+ and Ca2+ currents recorded from a KO-MCC in which the BK currents (IKCa) were particularly small. Despite the near full block of the sizeable Cav1.2 current, nifedipine (nife; 3 μm) caused a paradoxical increase of the outward K+ current. As shown to the right, IKv was responsible for most of the outward K+ current.
Interestingly, despite the lower density of functioning Ca2+ channels in approximately half of the KO-MCCs (4 of 10), the coupling between L-type and BK channels was effective and comparable with that in WT-MCCs (Fig. 8a, right inset), whereas in the remaining six cells, it was weak or nearly absent. In these KO-MCCs, even in the presence of sizeable L-type and non-L-type currents, the coupled BK currents were either absent or very small. An example is given in Figure 10b, left, in which nifedipine blocked the prespike Cav1.2 current of approximately −60 pA without producing any block of IKtot. On the contrary, we observed an increased IKtot because of the block of L-type currents. The cell exhibited also an overall IKCa that was nearly one order of magnitude smaller than IKv (Fig. 10b, right). Thus, a fraction of KO-MCCs was weakly coupled to functioning BK channels and could account for the anomalous responses to BayK-8644 characterized by marked depolarizations in current-clamp conditions (Fig. 6, bottom).
Discussion
We provided new evidence that L-type Ca2+ channels and their coupling to BK channels play a key role in controlling the firing activity of mouse chromaffin cells. Cultured MCCs fire spontaneously at low frequencies (0.2–5 Hz) (Marcantoni et al., 2009) and thus require a set of channels that activate at relatively low voltages (−50 mV), carry sufficient inward current, and weakly inactivate during prolonged prespike subthreshold depolarizations. These properties belong to the Cav1.3 channel that is expressed at comparable densities with the Cav1.2 isoform in MCCs. Loss of Cav1.3 channels induces drastic changes to the firing modes in Cav1.3−/− MCCs. Most KO-MCCs do not fire any longer (70%), and some of those firing have either irregular activity, fire at higher frequency, and respond with prolonged depolarizations to BayK-8644. These anomalous effects cannot be simply attributed to the loss of half available LTCCs but are related to the critical features of Cav1.3 and Cav1.2 and their coupling to BK channels.
Cav1.3 and BK channels in MCCs
Our findings emphasize two main properties that are at the basis of spontaneous firings: the peculiar activation–inactivation gating of Cav1.3 channels, which activate at slightly more negative voltages than Cav1.2 and inactivate very slowly during long depolarizations (Platzer et al., 2000; Koschak et al., 2003; Lipscombe et al., 2004), and the preferential coupling of Cav1.3 to fast inactivating BKi channels (Prakriya and Lingle, 1999). The first property is most evident during the prespike subthreshold depolarizations of 300–1000 ms, in which the pacemaker Ca2+ current is fourfold larger in WT-MCCs with respect to KO-MCCs and dominated by LTCCs. This L-type current is not only responsible for the prespike inward current driving WT-MCC activity but originates the slow BK current that activates during the prespike period and most of the fast BK component during spikes. The former current is likely to be mostly carried by BKi channels that open at relatively low voltages at a given Ca2+ (Sun et al., 2009), whereas the latter is sustained by both BKi and BKs channels. Both currents are strongly attenuated in KO-MCCs. A slowly non-inactivating pacemaker Ca2+ current similar to WT-MCCs and sensitive to DHPs has been reported also in midbrain dopaminergic (Puopolo et al., 2007) and suprachiasmatic nucleus (Jackson et al., 2004) neurons that fire spontaneously at low frequencies.
The second main property concerns the coupling of BKi to Cav1.3 channels, which conditions the shape of APs and the firing frequency as highlighted by the effects of nifedipine, BayK-8644, and paxilline. The blocking action of nifedipine is similar to that of other LTCC blockers that reduce or even block the firing frequency of cardiac myocytes (Mangoni and Nargeot, 2008) and central dopaminergic neurons (Jackson et al., 2004; Puopolo et al., 2007; Guzman et al., 2009). The effects of paxilline, which blocks fast and slowly inactivating BK channels (Sanchez and McManus, 1996), also resemble those of other BK channel blockers that increase rather than decreasing the spontaneous firing in central and peripheral neurons (Nelson et al., 2003; Swensen and Bean, 2003; Zhang et al., 2003).
The persistent firing in the presence of paxilline underlines also an important issue of BK channel function in MCCs. These channels modulate the AP shape, stabilize the repolarization phase, and set the firing frequency but are not crucial for generating spontaneous APs. In fact, the MCCs of BK channel-deficient mice (BK−/−) exhibit regular spontaneous activity blocked by nifedipine (D. H. F. Vandael, A. Marcantoni, P. Ruth, and E. Carbone, unpublished observation). The block or absence of BK channels is likely to be well compensated by voltage-gated Kv channels in MCCs. These channels contribute to approximately half of the K+ current activated during spikes and can sustain MCC repolarization and repetitive firing. This may not apply to the RCCs in which the BK channels carry fivefold more current than Kv channels (Solaro et al., 1995).
Fast inactivating and non-inactivating BK channels and their coupling to LTCCs
We found that, similarly to RCCs (Solaro and Lingle, 1992), WT-MCCs express both BKi and BKs channels. BKi channels are evident during brief Ca2+ preloading depolarizations, whereas BKs channels require longer presteps to be identified. After short preloading steps, BKs channels deactivate quickly as Ca2+ falls (Prakriya et al., 1996) and can be erroneously identified as BKi channels. We found of great significance that, whereas WT-MCCs possess mainly BKi channels and smaller fractions of BKs, KO-MCCs possessed mostly BKs channels, indicating preferential coupling between BKi and Cav1.3 channels. Because BKi channel inactivation is associated with the presence of a specific BK β2 subunit that shifts channel activation toward more negative voltages at a given Ca2+ and inactivate open channels (Xia et al., 2000; Sun et al., 2009), our data suggest also that loss of Cav1.3 channels may lead to the loss of BK β2 subunits. This could also indicate that β2 is the critical element regulating the close coupling between Cav1.3 and BKi channels. In the absence of β2, the coupling between BKs and Cav1.3 (or Cav1.2) channels is weaker and less critical for cell firing.
Close coupling between Cav1.3 and fast inactivating BKi channels in WT-MCCs is supported by its persistence in the presence of EGTA and is very similar to the effects of EGTA/BAPTA Ca2+ buffering on BK channel activation in RCCs (Prakriya and Lingle, 2000). A main conclusion of this latter study is that the BK channels that are close enough to Ca2+ channels to be resistant to the buffering action of EGTA are located at a distance between 50 and 160 nm, whereas those sensitive to EGTA are more distant. If we extrapolate this analysis to MCCs, we can conclude that most BKs channels, whose activation is fully prevented by EGTA, are weakly coupled to Ca2+ channels, whereas BKi channels that persist in EGTA are closely coupled to Ca2+ channels. This is particularly valid for LTCCs, as suggested by the blocking action of nifedipine (Fig. 3). The near complete loss of coupling in KO-MCCs in the presence of EGTA (Fig. 4b) further highlights the effective coupling existing between Cav1.3 and BKi channels that is apparently the most effective channel arrangement controlling spontaneous firing in WT-MCCs. An increased contribution of BKs currents together with a lower density of LTCCs in KO-MCCs further decreases the ability of these cells to generate spontaneous firing.
A final remark concerns the existence of a group of KO-MCCs responding to BayK-8644 with a net depolarization (Fig. 6b), indicative of a loss (or a reduced density) of BK channels in these cells. This is supported by noticing that a fraction of KO-MCCs shows weak coupling to BK channels even after prolonged preloading steps, and similar responses to BayK-8644 are often observed in MCCs of BK−/− mice (Vandael, Marcantoni, Ruth, and Carbone, unpublished results) but not in WT-MCCs.
Cav1.3 as pacemaker channels in neurons and role in chromaffin cells exocytosis
The role of Cav1.3 channels in generating pacemaker currents is widely debated and convincingly shown in striatal and SNc neurons (Olson et al., 2005; Guzman et al., 2009) and sinoatrial node cardiomyocytes (Mangoni et al. 2003). Cav1.3 channels may also support the persistent inward current sustaining plateau potentials in spinal motoneurons (Perrier et al., 2002) and are likely to control the tonic firing of several central neurons (Raman and Bean, 1999; Taddese and Bean, 2002; Do and Bean, 2003), including dopamine SNc neurons in which nimodipine fully blocks spontaneous firing (Puopolo et al., 2007). SNc neurons fire at ∼1 Hz and exhibit cobalt-sensitive prespike Ca2+ currents with time course and amplitude impressively similar to the slow nifedipine-sensitive currents of WT-MCCs. MCCs differ from SNc neurons for the presence of a slow prespike BK current that contributes and partially counterbalances the inward Cav1.3 current sustaining MCCs firing. In this respect, the prespike current of MCCs resemble more the sum of Ca2+ and BK currents regulating the firing of SCN neurons (Jackson et al., 2004). The main difference is the small size of the prespike Ca2+ current in SCN neurons compared with WT-MCCs, which accounts for the weak depressing effects of nimodipine on SCN firing. Similar to brain neurons, however, BK channels contribute significantly to the K+ current during spikes and help set the fast AHP.
Regulation of electrical activity of chromaffin cells, under splanchnic nerve stimulation, has been only partially investigated. Cultured bovine (Fenwick et al., 1982; Wallace et al., 2002), rat (Brandt et al., 1976; Gullo et al., 2003), and mouse (Nassar-Gentina et al., 1988; Marcantoni et al., 2009) chromaffin cells undergo spontaneous firing, but little is known about the ion currents regulating their resting activity. AP-induced secretion in chromaffin cells occurs under basal firing conditions, set by the sympathetic tone, and varies remarkably by changes in AP firing patterns (Duan et al., 2003; Fulop et al., 2005). Our data showing the presence of net inward Cav1.3 and Cav1.2 currents during interspike intervals highlights the role that LTCCs play in regulating AP firing and secretion. This is relevant, because LTCCs can be modulated in opposing ways by cAMP/PKA (Hernandez-Guijo et al., 1999; Cesetti et al., 2003) and cGMP/PKG (Carabelli et al., 2002) cascades inducing marked changes to the inward Ca2+ currents driving spontaneous firing and catecholamine secretion (Marcantoni et al., 2009).
Footnotes
This work was supported by Marie Curie Research Training Network “CavNET” Contract MRTN-CT-2006-035367, Ministero dell'Istruzione, dell'Università, e della Ricerca Grant 2005054435, Austrian Science Fund Grant P20670, and University of Innsbruck and San Paolo Company Grant 2008.2191.
References
- Baldelli P, Hernández-Guijo JM, Carabelli V, Novara M, Cesetti T, Andrés-Mateos E, Montiel C, Carbone E. Direct and remote modulation of L-channels in chromaffin cells: distinct actions on alpha1C and alpha1D subunits? Mol Neurobiol. 2004;29:73–96. doi: 10.1385/MN:29:1:73. [DOI] [PubMed] [Google Scholar]
- Bean BP. The action potential in mammalian central neurons. Nat Rev Neurosci. 2007;8:451–465. doi: 10.1038/nrn2148. [DOI] [PubMed] [Google Scholar]
- Brandt BL, Hagiwara S, Kidokoro Y, Miyazaki S. Action potentials in the rat chromaffin cell and effects of acetylcholine. J Physiol. 1976;263:417–439. doi: 10.1113/jphysiol.1976.sp011638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carabelli V, D'Ascenzo M, Carbone E, Grassi C. Nitric oxide inhibits neuroendocrine Ca(V)1 L-channel gating via cGMP-dependent protein kinase in cell-attached patches of bovine chromaffin cells. J Physiol. 2002;541:351–366. doi: 10.1113/jphysiol.2002.017749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carabelli V, Marcantoni A, Comunanza V, de Luca A, Díaz J, Borges R, Carbone E. Chronic hypoxia up-regulates alpha1H T-type channels and low-threshold catecholamine secretion in rat chromaffin cells. J Physiol. 2007;584:149–165. doi: 10.1113/jphysiol.2007.132274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cesetti T, Hernández-Guijo JM, Baldelli P, Carabelli V, Carbone E. Opposite action of β1- and β2-adrenergic receptors on CaV1 L-channel current in rat adrenal chromaffin cells. J Neurosci. 2003;23:73–83. doi: 10.1523/JNEUROSCI.23-01-00073.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan CS, Guzman JN, Ilijic E, Mercer JN, Rick C, Tkatch T, Meredith GE, Surmeier DJ. “Rejuvenation” protects neurons in mouse models of Parkinson's disease. Nature. 2007;447:1081–1086. doi: 10.1038/nature05865. [DOI] [PubMed] [Google Scholar]
- Do MT, Bean BP. Subthreshold sodium currents and pacemaking of subthalamic neurons: modulation by slow inactivation. Neuron. 2003;39:109–120. doi: 10.1016/s0896-6273(03)00360-x. [DOI] [PubMed] [Google Scholar]
- Duan K, Yu X, Zhang C, Zhou Z. Control of secretion by temporal patterns of action potentials in adrenal chromaffin cells. J Neurosci. 2003;23:11235–11243. doi: 10.1523/JNEUROSCI.23-35-11235.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fenwick EM, Marty A, Neher E. A patch-clamp study of bovine chromaffin cells and of their sensitivity to acetylcholine. J Physiol. 1982;331:577–597. doi: 10.1113/jphysiol.1982.sp014393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fulop T, Radabaugh S, Smith C. Activity-dependent differential transmitter release in mouse adrenal chromaffin cells. J Neurosci. 2005;25:7324–7332. doi: 10.1523/JNEUROSCI.2042-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- García-Palomero E, Cuchillo-Ibáñez I, García AG, Renart J, Albillos A, Montiel C. Greater diversity than previously thought of chromaffin cell Ca2+ channels, derived from mRNA identification studies. FEBS Lett. 2000;481:235–239. doi: 10.1016/s0014-5793(00)01984-0. [DOI] [PubMed] [Google Scholar]
- Gullo F, Ales E, Rosati B, Lecchi M, Masi A, Guasti L, Cano-Abad MF, Arcangeli A, Lopez MG, Wanke E. ERG K+ channel blockade enhances firing and epinephrine secretion in rat chromaffin cells: the missing link to LQT2-related sudden death? FASEB J. 2003;17:330–332. doi: 10.1096/fj.02-0200fje. [DOI] [PubMed] [Google Scholar]
- Guzman JN, Sánchez-Padilla J, Chan CS, Surmeier DJ. Robust pacemaking in substantia nigra dopaminergic neurons. J Neurosci. 2009;29:11011–11019. doi: 10.1523/JNEUROSCI.2519-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helton TD, Xu W, Lipscombe D. Neuronal L-type calcium channels open quickly and are inhibited slowly. J Neurosci. 2005;25:10247–10251. doi: 10.1523/JNEUROSCI.1089-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernández-Guijo JM, de Pascual R, García AG, Gandía L. Separation of calcium channel current components in mouse chromaffin cells superfused with low- and high-barium solutions. Pflugers Arch. 1998;436:75–82. doi: 10.1007/s004240050606. [DOI] [PubMed] [Google Scholar]
- Hernández-Guijo JM, Carabelli V, Gandía L, García AG, Carbone E. Voltage-independent autocrine modulation of L-type channels mediated by ATP, opioids and catecholamines in rat chromaffin cells. Eur J Neurosci. 1999;11:3574–3584. doi: 10.1046/j.1460-9568.1999.00775.x. [DOI] [PubMed] [Google Scholar]
- Herrington J, Solaro CR, Neely A, Lingle CJ. The suppression of Ca2+- and voltage-dependent outward K+ current during mAChR activation in rat adrenal chromaffin cells. J Physiol. 1995;485:297–318. doi: 10.1113/jphysiol.1995.sp020731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson AC, Yao GL, Bean BP. Mechanism of spontaneous firing in dorsomedial suprachiasmatic nucleus neurons. J Neurosci. 2004;24:7985–7998. doi: 10.1523/JNEUROSCI.2146-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koschak A, Reimer D, Huber I, Grabner M, Glossmann H, Engel J, Striessnig J. alpha 1D (Cav1.3) subunits can form L-type Ca2+ channels activating at negative voltages. J Biol Chem. 2001;276:22100–22106. doi: 10.1074/jbc.M101469200. [DOI] [PubMed] [Google Scholar]
- Koschak A, Reimer D, Walter D, Hoda JC, Heinzle T, Grabner M, Striessnig J. Cav1.4 α1 subunits can form slowly inactivating dihydropyridine-sensitive L-type Ca2+ channels lacking Ca2+-dependent inactivation. J Neurosci. 2003;23:6041–6049. doi: 10.1523/JNEUROSCI.23-14-06041.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipscombe D, Helton TD, Xu W. L-type calcium channels: the low down. J Neurophysiol. 2004;92:2633–2641. doi: 10.1152/jn.00486.2004. [DOI] [PubMed] [Google Scholar]
- Magistretti J, Mantegazza M, Guatteo E, Wanke E. Action potentials recorded with patch-clamp amplifiers: are they genuine? Trends Neurosci. 1996;19:530–534. doi: 10.1016/s0166-2236(96)40004-2. [DOI] [PubMed] [Google Scholar]
- Mangoni ME, Nargeot J. Genesis and regulation of the heart automaticity. Physiol Rev. 2008;88:919–982. doi: 10.1152/physrev.00018.2007. [DOI] [PubMed] [Google Scholar]
- Mangoni ME, Couette B, Bourinet E, Platzer J, Reimer D, Striessnig J, Nargeot J. Functional role of L-type Cav1.3 Ca2+ channels in cardiac pacemaker activity. Proc Natl Acad Sci U S A. 2003;100:5543–5548. doi: 10.1073/pnas.0935295100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcantoni A, Baldelli P, Hernandez-Guijo JM, Comunanza V, Carabelli V, Carbone E. L-type calcium channels in adrenal chromaffin cells: role in pace-making and secretion. Cell Calcium. 2007;42:397–408. doi: 10.1016/j.ceca.2007.04.015. [DOI] [PubMed] [Google Scholar]
- Marcantoni A, Carabelli V, Vandael DH, Comunanza V, Carbone E. PDE type-4 inhibition increases L-type Ca2+ currents, action potential firing, and quantal size of exocytosis in mouse chromaffin cells. Pflugers Arch. 2009;457:1093–1110. doi: 10.1007/s00424-008-0584-4. [DOI] [PubMed] [Google Scholar]
- Nassar-Gentina V, Pollard HB, Rojas E. Electrical activity in chromaffin cells of intact mouse adrenal gland. Am J Physiol. 1988;254:C675–C683. doi: 10.1152/ajpcell.1988.254.5.C675. [DOI] [PubMed] [Google Scholar]
- Neely A, Lingle CJ. Two components of calcium-activated potassium current in rat adrenal chromaffin cells. J Physiol. 1992;453:97–131. doi: 10.1113/jphysiol.1992.sp019220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson AB, Krispel CM, Sekirnjak C, du Lac S. Long-lasting increases in intrinsic excitability triggered by inhibition. Neuron. 2003;40:609–620. doi: 10.1016/s0896-6273(03)00641-x. [DOI] [PubMed] [Google Scholar]
- Olson PA, Tkatch T, Hernandez-Lopez S, Ulrich S, Ilijic E, Mugnaini E, Zhang H, Bezprozvanny I, Surmeier DJ. G-protein coupled receptor modulation of striatal Cav1.3 L-type Ca2+ channels is dependent on a shank-binding domain. J Neurosci. 2005;25:1050–1062. doi: 10.1523/JNEUROSCI.3327-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perrier JF, Alaburda A, Hounsgaard J. Spinal plasticity mediated by postsynaptic L-type Ca2+ channels. Brain Res Rev. 2002;40:223–229. doi: 10.1016/s0165-0173(02)00204-7. [DOI] [PubMed] [Google Scholar]
- Platzer J, Engel J, Schrott-Fischer A, Stephan K, Bova S, Chen H, Zheng H, Striessnig J. Congenital deafness and sinoatrial node dysfunction in mice lacking class D L-type Ca2+ channels. Cell. 2000;102:89–97. doi: 10.1016/s0092-8674(00)00013-1. [DOI] [PubMed] [Google Scholar]
- Prakriya M, Lingle CJ. BK channel activation by brief depolarizations requires Ca2+ influx through L-and Q-type Ca2+ channels in rat chromaffin cells. J Neurophysiol. 1999;81:2267–2278. doi: 10.1152/jn.1999.81.5.2267. [DOI] [PubMed] [Google Scholar]
- Prakriya M, Lingle CJ. Activation of BK channels in rat chromaffin cells requires summation of Ca2+ influx from multiple Ca2+ channels. J Neurophysiol. 2000;84:1123–1135. doi: 10.1152/jn.2000.84.3.1123. [DOI] [PubMed] [Google Scholar]
- Prakriya M, Solaro CR, Lingle CJ. [Ca2+]i elevations detected by BK channels during Ca2+ influx and muscarine-mediated release of Ca2+ from intracellular stores in rat chromaffin cells. J Neurosci. 1996;16:4344–4359. doi: 10.1523/JNEUROSCI.16-14-04344.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puopolo M, Raviola E, Bean BP. Roles of subthreshold calcium current and sodium current in spontaneous firing of mouse midbrain dopamine neurons. J Neurosci. 2007;27:645–656. doi: 10.1523/JNEUROSCI.4341-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raman IM, Bean BP. Ionic currents underlying spontaneous action potentials in isolated cerebellar Purkinje neurons. J Neurosci. 1999;19:1663–1674. doi: 10.1523/JNEUROSCI.19-05-01663.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez M, McManus OB. Paxilline inhibition of the alpha-subunit of the high-conductance calcium-activated potassium channel. Neuropharmacology. 1996;35:963–968. doi: 10.1016/0028-3908(96)00137-2. [DOI] [PubMed] [Google Scholar]
- Singh A, Gebhart M, Fritsch R, Sinnegger-Brauns MJ, Poggiani C, Hoda JC, Engel J, Romanin C, Striessnig J, Koschak A. Modulation of voltage- and Ca2+-dependent gating of Cav1.3 L-type calcium channels by alternative splicing of a C-terminal regulatory domain. J Biol Chem. 2008;283:20733–20744. doi: 10.1074/jbc.M802254200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinnegger-Brauns MJ, Huber IG, Koschak A, Wild C, Obermair GJ, Einzinger U, Hoda JC, Sartori SB, Striessnig J. Expression and 1,4-dihydropyridine-binding properties of brain L-type calcium channel isoforms. Mol Pharmacol. 2009;75:407–414. doi: 10.1124/mol.108.049981. [DOI] [PubMed] [Google Scholar]
- Solaro CR, Lingle CJ. Trypsin-sensitive, rapid inactivation of a calcium-activated potassium channel. Science. 1992;257:1694–1698. doi: 10.1126/science.1529355. [DOI] [PubMed] [Google Scholar]
- Solaro CR, Prakriya M, Ding JP, Lingle CJ. Inactivating and noninactivating Ca2+- and voltage-dependent K+ current in rat adrenal chromaffin cells. J Neurosci. 1995;15:6110–6123. doi: 10.1523/JNEUROSCI.15-09-06110.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sørensen JB, Nagy G, Varoqueaux F, Nehring RB, Brose N, Wilson MC, Neher E. Differential control of the releasable vesicle pools by SNAP-25 splice variants and SNAP-23. Cell. 2003;114:75–86. doi: 10.1016/s0092-8674(03)00477-x. [DOI] [PubMed] [Google Scholar]
- Striessnig J, Koschak A. Exploring the function and pharmacotherapeutic potential of voltage-gated Ca2+ channels with gene knockout models. Channels. 2008;2:233–251. doi: 10.4161/chan.2.4.5847. [DOI] [PubMed] [Google Scholar]
- Sun L, Xiong Y, Zeng X, Wu Y, Pan N, Lingle CJ, Qu A, Ding J. Differential regulation of action potentials by inactivating and noninactivating BK channels in rat adrenal chromaffin cells. Biophys J. 2009;97:1832–1842. doi: 10.1016/j.bpj.2009.06.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swensen AM, Bean BP. Ionic mechanisms of burst firing in dissociated Purkinje neurons. J Neurosci. 2003;23:9650–9663. doi: 10.1523/JNEUROSCI.23-29-09650.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taddese A, Bean BP. Subthreshold sodium current from rapidly inactivating sodium channels drives spontaneous firing of tuberomammillary neurons. Neuron. 2002;33:587–600. doi: 10.1016/s0896-6273(02)00574-3. [DOI] [PubMed] [Google Scholar]
- Wallace DJ, Chen C, Marley PD. Histamine promotes excitability in bovine adrenal chromaffin cells by inhibiting an M-current. J Physiol. 2002;540:921–939. doi: 10.1113/jphysiol.2001.013370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welling A, Ludwig A, Zimmer S, Klugbauer N, Flockerzi V, Hofmann F. Alternatively spliced IS6 segments of the alpha 1C gene determine the tissue-specific dihydropyridine sensitivity of cardiac and vascular smooth muscle L-type Ca2+ channels. Circ Res. 1997;81:526–532. doi: 10.1161/01.res.81.4.526. [DOI] [PubMed] [Google Scholar]
- Xia XM, Ding JP, Zeng XH, Duan KL, Lingle CJ. Rectification and rapid activation at low Ca2+ of Ca2+-activated, voltage-dependent BK currents: consequences of rapid inactivation by a novel β subunit. J Neurosci. 2000;20:4890–4903. doi: 10.1523/JNEUROSCI.20-13-04890.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang XF, Gopalakrishnan M, Shieh CC. Modulation of action potential firing by iberiotoxin and NS1619 in rat dorsal root ganglion neurons. Neuroscience. 2003;122:1003–1011. doi: 10.1016/j.neuroscience.2003.08.035. [DOI] [PubMed] [Google Scholar]
- Zhou Z, Misler S. Action potential-induced quantal secretion of catecholamines from rat adrenal chromaffin cells. J Biol Chem. 1995;270:3498–3505. [PubMed] [Google Scholar]