

Abstract
Mutations in leucine-rich repeat kinase 2 (LRRK2) are the single most common cause of inherited Parkinson's disease. Little is known about its involvement in the pathogenesis of Parkinson's disease mainly because of the lack of knowledge about the physiological role of LRRK2. To determine the function of LRRK2, we studied the impact of short hairpin RNA-mediated silencing of LRRK2 expression in cortical neurons. Paired recording indicated that LRRK2 silencing affects evoked postsynaptic currents. Furthermore, LRRK2 silencing induces at the presynaptic site a redistribution of vesicles within the bouton, altered recycling dynamics, and increased vesicle kinetics. Accordingly, LRRK2 protein is present in the synaptosomal compartment of cortical neurons in which it interacts with several proteins involved in vesicular recycling. Our results suggest that LRRK2 modulates synaptic vesicle trafficking and distribution in neurons and in consequence participates in regulating the dynamics between vesicle pools inside the presynaptic bouton.
Introduction
Parkinson's disease (PD) is a common neurodegenerative disease clinically characterized by bradykinesia, rigidity, and resting tremor. A hallmark of the disease is the progressive loss of dopaminergic (DA) neurons in the substantia nigra and the formation of Lewy bodies (Moore et al., 2005; Hardy et al., 2006). Although the majority of cases are sporadic, mutations in the leucine-rich repeat kinase 2 (LRRK2) gene (PARK8; Online Mendelian Inheritance in Men database identification number 609007) are linked to late-onset autosomal dominant PD, accounting for up to 13% of familial PD cases compatible with dominant inheritance (Paisán-Ruíz et al., 2004; Zimprich et al., 2004) and 1–2% of sporadic PD patients, thus suggesting this protein as the most significant player in PD pathogenesis identified to date (Aasly et al., 2005; Berg et al., 2005; Taylor et al., 2006). The LRRK2 protein has a molecular weight (MW) of ∼280 kDa and contains several domains, including a Ras/GTPase like (Roc), a C-terminal of Roc (COR), a kinase (similar to mitogen-activated protein kinase kinase kinases), and a WD40 domain (Bosgraaf and Van Haastert, 2003; Guo et al., 2006). Several single nucleotide alterations have been identified in LRRK2 (Brice, 2005), covering all functional domains, but only five missense mutations clearly segregate with PD in large family studies (Goldwurm et al., 2005; Bonifati, 2006). Some of these mutations cause increased kinase activity (West et al., 2005; Gloeckner et al., 2006, 2009; Greggio et al., 2006).
Despite its predominance in PD, the physiological function of LRRK2 is not known, and therefore its precise role in the etiology of PD is far from being understood, although in vivo studies of mutant animals suggested an involvement in neurotransmitter release (Tong et al., 2009). Perturbations of LRRK2 expression have been shown to influence neurite extension (MacLeod et al., 2006; Plowey et al., 2008; Wang et al., 2008; Gillardon, 2009) and vesicle endocytosis (Shin et al., 2008).
To elucidate the physiological role of LRRK2, we determined the presynaptic and postsynaptic properties of cortical neurons in which LRRK2 had been silenced by short hairpin (sh)-mediated RNA interference (Bauer et al., 2009). Electrophysiological analysis of such neurons revealed that LRRK2 silencing alters synaptic transmission. LRRK2 silencing perturbs vesicle dynamics and distribution within the recycling pool. On the molecular level, LRRK2 interacts with a number of proteins involved in synaptic vesicle (SV) trafficking. Together, our data suggest that LRRK2 participates in a protein network regulating synaptic vesicle trafficking in the presynaptic bouton.
Materials and Methods
Lentiviral vector constructs, virus production, and plasmids.
Lrrk2 and Dlk1 target sequences were identified using Ambion web-based oligo-search software, and two sequences for LRRK2 (miB3, AAGTTGATAGTCAGGCTGAAT; miB4, AGTGCTCCGGTATCAGATG) and one for Dlk1 (miB5, AATGGAGTCTGCAAGGAAC) were selected, synthesized, and cloned into green fluorescent protein (GFP)-expressing pLVTH as described previously (Bauer et al., 2008, 2009). In brief, oligonucleotides (oligo-nt) coding for a 5′-pseudoBglII site, a sense oligo-nt loop antisense oligo-nt, a transcription termination site, and a 3′pseudo-XbaI restriction site were purchased from Metabion. Sense and antisense oligos were annealed and subsequently phosphorylated. The fragments were cloned 3′ to the H1 promoter of pBC KS+(ClaI)–H1, resulting in pBC KS+(ClaI)–sh. The H1sh cassettes were isolated with ClaI, blunted, and cloned into the blunted ClaI/BamH1 site of pLV transfer vector. pLV is a modified plasmid transfer vector derived from original pLVTH (Wiznerowicz and Trono, 2003), in which the BamH1–tetO–H1–ClaI fragment was excised. All recombinant lentiviruses were produced by transient transfection of HEK293T cells according to standard protocols (Wiznerowicz and Trono, 2003). Primary cortical cultures were transduced with LV vectors at multiplicity of infection 1. Empty virus (LVTH) has been used as control. For pull-down experiments, several hLRRK2 domains were subcloned into pDEST15 [glutathione S-transferase (N-terminal fusion GST) tag] using Gateway system (Invitrogen): N terminus (N-term) (amino acids 1–397), ankyrin repeats (ANK) (amino acids 394–995), leucine-rich repeats (LRRs) (amino acids 921–1356), Roc–COR–kinase domain (R-C-K) (amino acids 1340–2142), and WD40 (amino acids 2124–2527).
Cortical neuron cultures, fractionation, and chemicals.
Cortical neuron cultures were prepared from embryonic day 17.5–18.5 mouse cortexes (CD1). High-density (750–1000 cells/mm2) and medium-density (150–200 cells/mm2) neuron cultures were plated and grown as described on six-well plastic tissue culture plates (Iwaki; Bibby Sterilin) or on 12-mm-diameter coverslips put into 24-well plastic tissue culture plates (Iwaki; Bibby Sterilin) (Romorini et al., 2004). Cells were infected with viruses at 10 d in vitro (DIV10) and assayed as described throughout text at DIV16–DIV18. Subcellular fractions have been prepared as described (Dodd et al., 1981). Briefly, high-density cultures were collected in HEPES-buffered sucrose (0.32 m sucrose and 4 mm HEPES, pH 7.4) and spun at 600 × g at 4°C for 5 min to pellet the nuclear fraction (P1). The resulting supernatant was centrifuged at 10,000 × g at 4°C for 15 min to obtain a cytosolic supernatant (S2) and a crude synaptosomal pellet that was lysed by hypoosmotic shock in 4 mm HEPES, pH 7.4. A final centrifugation step (at 25,000 × g at 4°C for 20 min) generated a supernatant (S3, crude synaptic vesicle fraction) and a pellet (P3, lysed synaptosomal membrane fraction). An equal amount of protein, as determined by Bradford assay (Bio-Rad), was then separated on SDS-PAGE. Unless otherwise stated, all of the other chemicals came from Applichem.
Pull-down, immunoprecipitation, and antibodies.
LRRK2 GST fusion domains were expressed in Escherichia coli BL21 strain (Invitrogen), purified as described (Frangioni and Neel, 1993). Ten micrograms of each GST fusion protein was loaded on glutathione–Sepharose resin (GE Healthcare) and coincubated with adult mouse brain lysate (1 mg of total protein). In immunoprecipitation assays, 10 mg of the indicated antibodies (Abs) were incubated with 2 mg of adult mouse brain lysate and loaded on protein G–Sepharose resin (GE Healthcare). In both procedures, resins were extensively washed in Tris–EDTA buffer (10 mm Tris-HCl, pH 8.0, 1 mm EDTA,150 mm NaCl, and 0.2% Triton -X100) and sample eluted with Laemli's buffer. For protein identification, samples were loaded onto 6–12% SDS-PAGE gels; the proteins were transferred onto nitrocellulose membrane (Sigma) at 80 V for 120 min at 4°C. The primary antibodies were applied overnight in blocking buffer (20 mm Tris, pH 7.4, 150 mm NaCl, 0.1% Tween 20, and 5% nonfat dry milk) and included the following (source in parentheses): rat monoclonal anti-LRRK2 1E11 at 1:1000 (developed by Dr. E. Kremmer, Helmholtz Zentrum München, Munich, Germany); rabbit anti-postsynaptic density 95 (PSD-95) at 1:1000 (gift from E. Kim, Korea Advanced Institute of Science and Technology, Daejeon, South Korea); mouse anti-synaptosome-associated protein of 25 kDa (SNAP-25) at 1:1000 and rabbit anti-glutamate receptor subunit 2 (GluR2) at 1:250 (Millipore Bioscience Research Reagents); rabbit anti-N-ethylmaleimide sensitive factor (NSF) at 1:1000 and rabbit anti-heat shock protein 90 (HSP90) (Cell Signaling Technology); rabbit anti-vesicle-associated membrane protein 1 (Vamp1) at 1:1000 (Synaptic Systems); and mouse anti-syntaxin 1A at 1:1000, mouse anti-synaptophysin at 1:1000, and mouse anti-α-tubulin at 1:1000 (Sigma). The secondary antibodies (HRP-conjugated anti-mouse, anti-rabbit, or anti-rat) (Jackson ImmunoResearch) were used in a ratio of 1:2000. The signal was detected using an ECL detection system (GE Healthcare) and quantified by means of NIH ImageJ software.
Tandem mass spectrometry identification.
Liquid chromatography tandem mass spectrometry (LC-MS/MS) analysis was performed on an Ultimate3000 nano HPLC system (Dionex) online coupled to a LTQ OrbitrapXL (Thermo Fisher Scientific) mass spectrometer by a nano spray ion source. The system was equipped with a nano trap column (100 μm inner diameter × 2 cm, packed with Acclaim PepMap100 C18, 5 μm, 100 Å; LC Packings) and an analytical column (75 μm inner diameter × 15 cm, Acclaim PepMap100 C18, 3 μm, 100 Å; LC Packings). Samples originating from prefractionation experiments were dissolved in 50 μl of 2% acetonitrile and 0.5% trifluoroacetic acid by incubation for 15 min at 4°C under agitation. The samples were automatically injected and loaded onto the trap column at a flow rate of 30 μl/min in 5% buffer B (80% acetonitrile and 0.1% formic acid (FA) in HPLC-grade water) and 95% buffer A (5% acetonitrile and 0.1% FA in HPLC-grade water). After 5 min, the peptides were eluted and separated on the analytical column by a 140 min gradient from 5 to 40% of buffer B at 300 nl/min flow rate. Remaining peptides were eluted by a short gradient from 50 to 100% buffer B in 5 min. The eluting peptides were ionized by nano spray ionization and the ionized peptides analyzed in the Orbitrap. From the MS prescan, the 10 most intense peptide ions were selected for fragment analysis in the linear ion trap if they exceeded an intensity of at least 200 counts and if they were at least doubly charged. The normalized collision energy for collision-induced dissociation was set to a value of 35, and the resulting fragments were detected with normal resolution in the linear ion trap. While the fragment analysis took place, a high-resolution (60,000 full-width half maximum) MS spectrum was acquired in the Orbitrap with a mass range from 200 to 1500 Da. The lock mass option was activated, and a background signal with a mass of 445.120020 was used as lock mass (Olsen et al., 2005). Every ion selected for fragmentation was excluded for 30 s by dynamic exclusion. The acquired spectra were processed and analyzed by using the Bioworks Browser software and the SEQUEST algorithm (Eng et al., 1994) for database searching. Monoisotopic masses and full tryptic cleavage were selected. The peptide tolerance was set to 10 ppm and the fragment ions tolerance to 1 Da. Only Y and Z ions were considered for the identification. The threshold for peak detection was set to 100 counts and the molecular weight range to 380–4500 Da. No fixed modifications and methionine oxidation, serine, threonine and tyrosine phosphorylation, and cystein carbamidomethylation were allowed as variable modifications with a maximum of three modifications per peptide allowed. The database used was a mouse subset of the Uniref100 database (version 14.4, released on Nov. 11, 2008).
Sequest result files were analyzed by the Scaffold software. Scaffold (version Scaffold_2_02_03; Proteome Software) was used to validate MS/MS-based peptide and protein identifications. Peptide identifications were accepted if they could be established at >95% probability as specified by the Peptide Prophet algorithm (Keller et al., 2002). Protein identifications were accepted if they could be established at >99% probability and contained at least two identified unique peptides. Protein probabilities were assigned by the Protein Prophet algorithm (Nesvizhskii and Aebersold, 2004). Proteins that contained similar peptides and could not be differentiated based on MS/MS analysis alone were grouped to satisfy the principles of parsimony. Only hits confirmed by more than two independent experiments and absent in the GST control sample were taken in consideration.
Synaptotagmin antibodies uptake assay.
The assay to monitor synapses recycling was performed using rabbit polyclonal antibodies directed against the intravesicular domain of synaptotagmin 1 (Synaptic Systems), applied for 5 min if not indicated otherwise at room temperature on the cultures, as described previously (Matteoli et al., 1992). Incubations with the antibody (1:400) were performed in Tyrode's solution containing 124 mm NaCl, 5 mm KCl, 2 mm MgCl2, 30 mm glucose, 25 mm HEPES, pH 7.4, and 2 mm CaCl2 if not indicated otherwise or in Tyrode's solution containing 50 mm KCl or 2 μm TTX or 0.5 m sucrose. After fixation and permeabilization, a synaptophysin counterstaining visualized the totality of synapses. Acquired images were processed and quantitatively analyzed with NIH ImageJ software as described previously (Verderio et al., 1999). Briefly, GFP-positive (GFP+) processes were manually tracked, and the number of synaptotagmin and synaptophysin-positive clusters doubled present in the region of interest (ROI) was automatically counted. To track synaptic vesicle movements, the neurons were incubated for 5 min with cyanine 3 (Cy3)-coupled anti-synaptotagmin 1 antibody (Synaptic Systems) in Tyrode's solution at room temperature. The neurons were then washed, transferred to the microscope, and imaged. Movies were acquired 2–5 min after labeling using a Carl Zeiss LSM 510 confocal microscope equipped with a Carl Zeiss 63× objective at 0.5 or 2 Hz sample frequency. Resulting images were optimized and processed on NIH ImageJ. Single particle has been manually tracked using an algorithm implementation published by Sbalzarini and Koumoutsakos (2005). Briefly, images have been thresholded, and circular ROIs were automatically generated around clusters. The position of the center of each ROI along the time has been annotated. The diffusion coefficient D was calculated using the equation MSD(Δt) = 4DΔt, where MSD(Δt) indicates the mean square displacement of the single vesicle (Saxton, 1994). Only movies captured at 2 Hz sampling frequency have been included in the analysis, and only particle tracks longer than 10 frames whose MSD(Δt) were consistent with the dynamic of spatial confined particles (Kusumi et al., 1993; Saxton, 1994) have been considered. At least three vesicles were analyzed for each experiment.
Electron microscopy.
Specimens from siLRRK2 and control infected neurons have been prepared for electron microscopy as described previously (Rudelius et al., 2006). Briefly, neuron cultures were fixed in cacodylate-buffered 3% glutaraldehyde for 12 h and subsequently Epon embedded. Ultrathin sections were cut, mounted on copper grids, contrasted with uranyl acetate and lead citrate, and observed with an electron microscope (Carl Zeiss EM10 at 60 kV). EM images have been processed on NIH ImageJ before performing the analysis on LoClust tool (Nikonenko and Skibo, 2004). Single vesicle has been manually annotated; only intact presynaptic boutons with similar size (longer than 300 nm and including more than 40 vesicles) have been included in the analysis. The n values throughout the text refer to presynaptic boutons measured.
Electrophysiology.
Paired recordings were obtained from low-density cultures of DIV14– DIV16 neurons transfected at DIV10 bathed in an external solution (Krebs'–Ringer's–HEPES) of the following composition: 125 mm NaCl, 5 mm KCl, 1.2 mm MgSO4, 1.2 mm KH2PO4, 2 mm CaCl2, 6 mm glucose, and 25 mm HEPES, pH 7.4. Transfected pyramidal neurons identified via positive GFP fluorescence were selected as the presynaptic neuron with an adjacent nontransfected pyramidal neuron serving as its postsynaptic target. Both neurons were voltage clamped at −70 mV in the whole-cell configuration using a MultiClamp 700A amplifier interfaced to a personal computer via a Digidata 1320 (Molecular Devices) before depolarizing the presynaptic neuron with a single pulse (1 ms, 100 mV pulse) up to 30 mV while recording the response in the postsynaptic neuron as an EPSC. Currents were sampled at 5–10 kHz and filtered at 5 kHz, and data were acquired and analyzed offline using Clampfit 10 (Molecular Devices). Average EPSCs were obtained by measuring consecutive EPSCs (n = 20–40) for each neuronal pair in which events had to exceed a threshold of two to three times the SD of the baseline noise. Synaptic latency was calculated as the time spanning between the stimulus artifact and the 5% of EPSC amplitude (Markram et al., 1997; Bardoni et al., 2004). Spontaneous synaptic responses [miniature EPSCs (mEPSCs)] were monitored in medium-density neuron cultures in the presence of 1 μm TTX. mEPSC amplitudes and frequencies were determined using Clampfit 10 software. Detection threshold for mEPSCs was set at 7–10 pA, depending on the amplitude of the membrane noise observed. Recording pipettes were fabricated from capillary glass using a two stage puller (Narishige) and had tip resistances of 3–5 MΩ when filled with the intracellular solution of the following composition (in mm): 130 K-gluconate, 10 KCl, 1 EGTA, 10 HEPES, 2 MgCl2, 4 MgATP, and 0.3 Tris-GTP. Contaminating GABAA-mediated responses were avoided by voltage clamping neurons near the calculated equilibrium potential for Cl− (−63 mV) under these conditions.
Statistical analysis.
All data are expressed as mean ± SE. Data were analyzed with an unpaired Student's t test (two classes) or ANOVA followed by Tukey's post hoc test (more than two classes). The indication of number of experiments (n) and level of significance (p) are indicated throughout text.
Supplemental Figures S1–S4 (available at www.jneurosci.org as supplemental material) provide additional confirmations for the evidences reported in the manuscript. Supplemental Movies 1–8 (available at www.jneurosci.org as supplemental material) report single vesicle dynamic in control and siLRRK2 neurons. Supplemental Table 1 (available at www.jneurosci.org as supplemental material) lists additional information related to protein identification.
Results
LRRK2 is positively associated to neuron maturation
LRRK2 is widely expressed in neurons throughout the CNS, including cortex, striatum, and other brain areas (Melrose et al., 2006; Higashi et al., 2007). As LRRK2 expression increases during postnatal development correlating with synaptic maturation (Biskup et al., 2007), we investigated LRRK2 protein expression in cortical neurons in vitro by Western blotting at five different time points, DIV4, DIV8, DIV12, DIV16, and DIV20, that can be considered representative for neuron maturation (Lee and Sheng, 2000; Ziv and Garner, 2001). We detected LRRK2 by Western blot using a custom-made rat monoclonal antibody (1E11) specific for LRRK2 (Bauer et al., 2009) (Fig. 1A, top). Because the predicted MW of LRRK2 is ∼286 kDa, we considered and analyzed the band at ∼280 kDa as endogenous full-length LRRK2. Interestingly, LRRK2 levels were positively associated with synapse maturation, as indicated by the increasing level of synaptophysin and PSD-95 proteins (Verderio et al., 1999; Buckby et al., 2004; Mielke et al., 2005) (Fig. 1A,B). In contrast, no significant increase in tubulin was detected. This finding suggests a physiological role of LRRK2 in mature neurons.
Figure 1.

LRRK2 expression increases during in vitro neuron development. A, Neurons were cultured and solubilized at the indicated DIV. Equal amounts of protein were loaded on SDS-PAGE gel and stained with the indicated antibodies. LRRK2 expression increases along in vitro synapse maturation, as demonstrated by the parallel increment of PSD-95 and synaptophysin amount. Two representative independent experiments are shown. B, Mean ± SE protein levels expressed as optical density (in arbitrary units) normalized against total protein amount. n = 5, *p < 0.05 versus DIV4. C, Cortical neurons were left untreated (mock) or infected at DIV10 with control (LVTH) or two LRRK2-silencing (miB3 and miB4) viruses, solubilized at DIV18 and analyzed for the expression of the indicated protein. D, LRRK2 protein levels expressed as percentage over control; mean ± SE; **p < 0.01, n = 5, ANOVA followed by Tukey's post hoc test. s-physin, Synaptophysin.
LRRK2 silencing alters evoked postsynaptic currents
A recent publication proposed an involvement for LRRK2 in neurotransmitter release (Tong et al., 2009). Therefore, we tested whether LRRK2 may modulate synaptic function. For these experiments and subsequent ones, we impaired cellular LRRK2 expression via short hairpin-mediated RNA interference. We infected cortical neurons with lentiviruses expressing the silencing constructs on DIV10 and performed specific experimental procedures on DIV18. To test the efficiency of LRRK2 silencing, we infected mouse cortical neurons or NIH3T3 cells with control virus (LVTH) or with a virus expressing either the short hairpin construct miB3 or miB4, both of them specifically silencing LRRK2 (Bauer et al., 2009). miB3 small interfering RNA (siRNA)-mediated silencing led to an efficient reduction of the signal detected by the 1E11 antibody in cortical neurons (Fig. 1C, miB3 lane) as well as in NIH3T3 cells (supplemental Fig. 1A, available at www.jneurosci.org as supplemental material). The same result was obtained when a second independent silencing construct (miB4) was transduced in cortical neurons (Fig. 1C, miB4 lane). Because both miB3 and miB4 constructs reduced LRRK2 protein expression to a similar extent [Fig. 1D; LRRK2 protein levels, % of expression over control (mean ± SE): LVTH, 100 ± 15.7; miB3, 8.93 ± 1.84; miB4, 30.99 ± 9.95; n = 5; p < 0.01], we mainly used miB3 for additional experiments (hereafter siLRRK2, whereas control refers to the LVTH virus). For the electrophysiological recordings, instead of infecting the cells with viruses, we transfected cortical neurons with a lentiviral plasmid construct. Because the successful transfection of all constructs can be tracked via coexpressed GFP, we were able to identify transfected neurons as GFP-positive cells. First, we asked whether LRRK2 might directly modulate neuron intrinsic excitability. To this purpose, we selected and patched nontransfected or control or siLRRK2-transfected neurons surrounded by nontransfected cells. When we recorded resting membrane potential and spontaneous miniature currents, we did not observe any obvious phenotype in siLRRK2 neurons (supplemental Fig. 2A–C, available at www.jneurosci.org as supplemental material). Next we tested whether LRRK2 silencing might affect presynaptic mechanism by measuring evoked EPSCs in pairs of synaptically connected pyramidal cortical neurons. Therefore, two adjacent neurons were chosen such that the presynaptic neuron was either a lentiviral construct control or an siLRRK2-transfected neuron, whereas the postsynaptic neuron was always a nontransfected cell (supplemental Fig. 2D, available at www.jneurosci.org as supplemental material). The two neurons were whole-cell voltage clamped at −70 mV, and the presynaptic one was stimulated by a brief depolarization up to +30 mV generating an EPSC in the connected postsynaptic neuron. We first measured EPSCs after a single presynaptic stimulation (100 mV, 1 ms) in paired neurons (Fig. 2A shows representative traces from control and siLRRK2 couples). We found that EPSC amplitude in postsynaptic neurons connected to siLRRK2 silenced presynaptic neurons (now referred to as siLRRK2 pairs) increased more than twofold compared with that measured in control pairs [Fig. 2C, first peak; EPSC amplitude in nA (mean ± SE): control, −0.09 ± 0.01; siLRRK2, −0.23 ± 0.02; n = 8; p < 0.001]. Furthermore, siLRRK2 pairs showed a higher probability to generate an EPSC over the baseline after a presynaptic trigger [% of successful events (mean ± SE): control, 84.63 ± 2.66; siLRRK2, 100 ± 2; n = 8; p < 0.001]. However, LRRK2 silencing did not significantly affect postsynaptic current latency and decay (Fig. 2D, first peak) (supplemental Fig. 2E, available at www.jneurosci.org as supplemental material). We next tested the impact of LRRK2 silencing after paired-pulse stimulation (Matveev et al., 2002; Zucker and Regehr, 2002). Pairs of stimuli (100 mV, 1 ms) were elicited in the presynaptic cell at an interpulse interval of 50 ms (Fig. 2B shows representative traces from control and siLRRK2 couples). In control pairs, the amplitude of the second EPSC was slightly increased, whereas LRRK2 silencing was associated with a robust impairment of the second EPSC [Fig. 2C, supplemental Fig. 2F (available at www.jneurosci.org as supplemental material), % EPSC2/EPSC1 (mean ± SE): control, 115.77 ± 5.79; siLRRK2, 55.44 ± 2.39; n = 5; p < 0.01]. Additionally, an increase in the latency of the second current was observed [Fig. 2D, second peak; postsynaptic current latency in ms (mean ± SE): control, 1.9 ± 0.13; siLRRK2, 2.8 ± 0.05; n = 5; p < 0.01]. When we tried to rescue LRRK2 levels in siLRRK2 neurons by the means of LRRK2 overexpression, we observed cell death and we failed to record EPSC (data not shown). This might be attributable to the fact that LRRK2 overexpression in vitro is toxic (Greggio et al., 2006; Iaccarino et al., 2007; Li et al., 2010). Notwithstanding this, we believe these results suggest a role for LRRK2 in modulating presynaptic vesicle release.
Figure 2.

LRRK2 silencing modifies synaptic transmission. Paired recordings were performed between a control or siLRRK2-transfected neuron (presynaptic) and an adjacent nontransfected neuron. A, Representative EPSC traces from control and siLRRK2 pairs after a single depolarizing stimulus (100 mV, 1 ms). B, Representative traces from control and siLRRK2 pairs after a paired-pulse stimulation protocol (100 mV, 1 ms, 50 ms interpulse interval). C, LRRK2 silencing significantly increased evoked EPSC amplitude after a single-pulse stimulation but enhanced paired-pulse depression. D, LRRK2 silencing increased the latency of the second EPSC. Data are expressed as mean ± SE. **p < 0.01, ***p < 0.001, Student's t test; n = 5.
LRRK2 silencing alters synaptic vesicle recycling
Given the electrophysiological changes associated with LRRK2 silencing, we asked whether LRRK2 might be involved in presynaptic processes. We thus looked at the ratio of recycling synapses by exposing control and siLRRK2-infected cortical neuron cultures to anti-synaptotagmin antibody as described previously (Matteoli et al., 1992; Bacci et al., 2001). These antibodies are internalized in the lumen of synaptic vesicles after their exo-endocytosis (Matteoli et al., 1992; Kraszewski et al., 1995; Malgaroli et al., 1995; Verderio et al., 1999), and their uptake closely reflects levels of vesicle recycling. Neurons were fixed after the given treatment, and then putative synapses were labeled after fixation with synaptophysin antibodies. The synapses appeared as synaptotagmin and synaptophysin-positive (i.e., recycling synapses) or only synaptophysin-positive (i.e., not recycling synapses) clusters along GFP-positive neuronal processes (Fig. 3A) (supplemental Fig. 3A, available at www.jneurosci.org as supplemental material). The analysis showed that, under basal conditions (Fig. 3B, untreated), LRRK2 silencing induced a significant increase in the number of synaptotagmin and synaptophysin-positive clusters [Fig. 3C; % of synaptotagmin+ synaptophysin+ clusters (mean ± SE): untreated control, 34.17 ± 4.15; siLRRK2, 55.87 ± 6.21; n = 22; p < 0.01]. Neurons infected with control virus behaved as non-infected neurons, whereas an increase of ratio of recycling synapses was measured when we silenced LRRK2 using miB4 virus [supplemental Fig. 3B,C (available at www.jneurosci.org as supplemental material); % of synaptotagmin+ synaptophysin+ clusters (mean ± SE): non-infected, 30.90 ± 1.72; control, 33.82 ± 1.98; miB4, 55.39 ± 7.89; n = 12; p < 0.05]. To test the specificity of the phenotype observed in siLRRK2 neurons, we infected the cells with a virus (miB5) bearing a sequence designed to silence Dlk1, a protein involved in DA neuron differentiation and not expressed at detectable levels in cortical neurons (Bauer et al., 2008). Interestingly, we did not report any increase of the ratio of recycling synapses in miB5-infected neurons [supplemental Fig. 3B,C (available at www.jneurosci.org as supplemental material); % of synaptotagmin+ synaptophysin+ clusters (mean ± SE): miB5, 34.59 ± 2.33; n = 12; p > 0.05]. We then exposed cultures to anti-synaptotagmin Abs in the presence of 50 mm KCl (Fig. 3B, KCl). In control neurons, the stimulation increased the ratio of recycling synapses as expected (Bacci et al., 2001). However, the siLRRK2 neurons did not react to KCl depolarization [Fig. 3C, KCl; % of synaptotagmin+ synaptophysin+ clusters (mean ± SE): control, 79.15 ± 3.22; siLRRK2, 60.68 ± 5.47; n = 22; p < 0.01]. Finally, once we blocked action potential propagation with 2 μm TTX treatment (Fig. 3B, TTX), the ratio of recycling synapses was impaired in both control and siLRRK2 neurons [Fig. 3C, TTX; % of synaptotagmin+ synaptophysin+ clusters (mean ± SE): control, 21.67 ± 4.16; siLRRK2, 27.39 ± 5.63; n = 16; p > 0.05]. The total number of synaptic contacts, however, remained unaltered despite pharmacological treatment and viral transduction [Fig. 3D, supplemental Fig. 3D (available at www.jneurosci.org as supplemental material); number of synaptophysin+ clusters/10 μm (mean ± SE): n = 15; p > 0.05]. The observation that LRRK2 silencing increases EPSC and the ratio of recycling synapses might imply that LRRK2 alters the Ca2+ affinity of release. To test this hypothesis, we measured the ratio of recycling synapses in the presence of different concentration of extracellular Ca2+ (Yamasaki et al., 2006; Chang and Südhof, 2009) (Fig. 4A). We noticed that overall, siLRRK2 neurons, when compared with control cells, were characterized by a higher ratio of recycling synapses, except when 10 mm extracellular Ca2+ concentration was applied [Fig. 4B; % of synaptotagmin+ synaptophysin+ clusters (mean ± SE): 0 mm Ca2+ control, 11.63 ± 1.23; siLRRK2, 16.35 ± 2.87; 2 mm Ca2+ control, 27.02 ± 1.67; siLRRK2, 45.70 ± 3.32; 5 mm Ca2+ control, 42.15 ± 6.36; siLRRK2, 60.26 ± 4.27; 10 mm Ca2+ control, 62.09 ± 4.94; siLRRK2, 63.63 ± 5.19 n = 14, p < 0.05]. The total number of synaptic contacts, however, remained unaltered [Fig. 4C; number of synaptophysin+ clusters/10 μm (mean ± SE); n = 14; p > 0.05]. These data might indicate that LRRK2 deletion partially alters the Ca2+ affinity of release. Another alternative explanation for this phenotype might come from a direct role of LRRK2 in controlling SV trafficking mechanisms. To further test this hypothesis, we analyzed vesicle movement over time. Control, siLRRK2 and miB5 (data not shown) infected neurons were loaded with anti-synaptotagmin Abs coupled to Cy3 fluorochrome. Synaptotagmin-positive clusters within GFP-positive processes were then tracked in basal conditions under laser-assisted confocal microscopy (Fig. 5A). siRNA-mediated LRRK2 knockdown increased clusters relative mobility, as demonstrated by analysis of cluster motion over time (Fig. 5B shows representative cluster path) (supplemental Movies 1–8, available at www.jneurosci.org as supplemental material). To compare cluster motion in control, miB5- and siLRRK2-infected cultures, we calculated the diffusion coefficient D from the equation MSD(Δt) = 4DΔt, where MSD(Δt) indicate the mean square displacement of the cluster (Saxton, 1982; Kusumi et al., 1993). The coefficient D increased by a log factor in siLRRK2 neurons when compared with control and miB5-infected neurons [Fig. 5C,D; in μm2/s (mean ± SE): control, 0.009 ± 0.0006; miB5, 0.011 ± 0.001; siLRRK2, 0.152 ± 0.0146; n = 9; p < 0.001]. Together, these results suggest that LRRK2 modulates vesicle motility inside the presynaptic bouton.
Figure 3.

LRRK2 silencing modifies synaptic vesicle recycling. The synaptotagmin uptake assay was performed on cortical neurons at DIV18, infected at DIV10 with control or siLRRK2 virus. A, Synaptotagmin (s-tagmin)-positive spots colocalized with synaptophysin (s-physin) clusters along neuron processes. B, Neurons were left untreated or treated with KCl (50 mm, 5 min) or TTX (30 min before labeling, 2 μm) and then assayed for exo-endocytosis. Scale bar, 5 μm. CTRL, Control. C, The percentage of synaptotagmin and synaptophysin-positive clusters within the totality of synaptophysin-positive clusters reflects the pool of recycling synapses. LRRK2-silenced neurons showed an increased ratio of recycling synapses in basal condition but a reduced activity after KCl stimulation. Instead, LRRK2 knockdown did not modify the number of recycling synapses during TTX repression. NT, Nontreated. D, Active zone number, monitored as synaptophysin-positive dots along neuronal processes, was not modified by LRRK2 silencing. Data are expressed as mean ± SE. *p < 0.05, **p < 0.01, same treatment, Student's t test, #p < 0.01 same infection, ANOVA, Tukey's post hoc test, n = 25.
Figure 4.
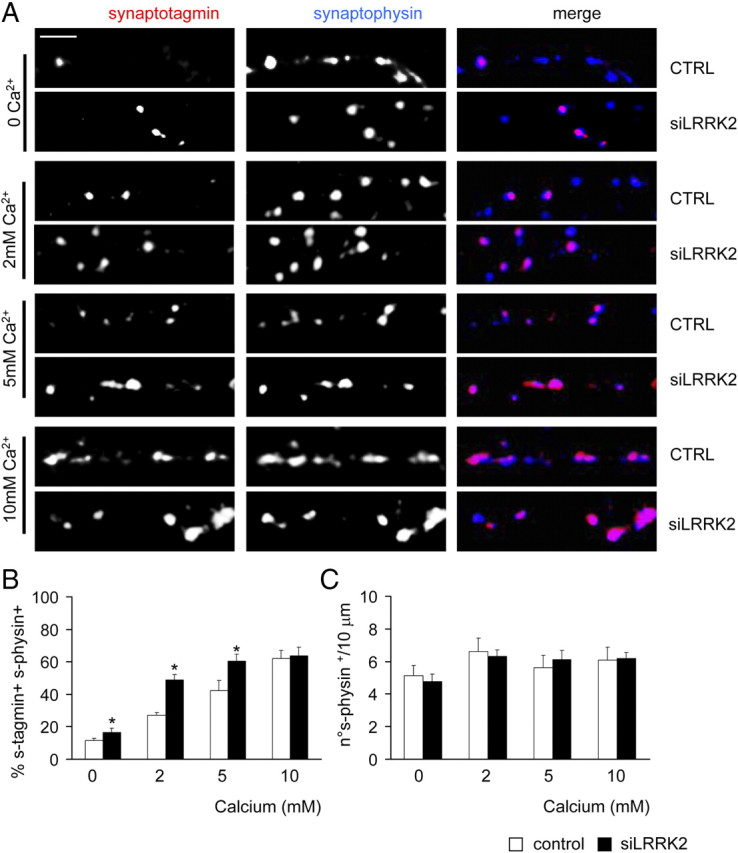
LRRK2 silencing affects Ca2+ sensitivity. A, The ratio of recycling synapses was assayed in neurons in presence of increasing external Ca2+ concentration. Scale bar, 5 μm. CTRL, Control. B, The ratio of recycling synapses rose to a higher extent in siLRRK2 neurons in response to increasing Ca2+ concentration. C, Active zone number was not modified by LRRK2 silencing or Ca2+ concentration. Data are expressed as mean ± SE. *p < 0.05 versus control, Student's t test, n = 14.
Figure 5.

LRRK2 silencing increases vesicle motility. A, Control and siLRRK2-infected neurons were exposed to anti-synaptotagmin antibody coupled to a fluorochrome. Synaptotagmin (s-tagmin)-positive clusters within GFP-positive processes were tracked in basal condition under laser confocal microscopy. Clusters in siLRRK2 neurons showed an increased motility. Yellow arrows indicate the position of cluster at t = 0 s. White arrows report instead the position of the clusters relative to the previous time point. Scale bar, 5 μm. B, The diagram reports representative path of single cluster from control or siLRRK2 neuron. Scale bar, 1 μm. C, Quantification of diffusion coefficient D, where MSD(Δt) = 4Dt; mean ± SE; ***p < 0.001, Student's t test, n = 9. CTRL, Control.
LRRK2 interacts with presynaptic proteins
Because synaptic vesicle mobility and distribution is orchestrated by an array of presynaptic proteins (Südhof, 2004; Montecucco et al., 2005; Jahn and Scheller, 2006), we tested first whether LRRK2 silencing affects synaptic protein expression in cortical neurons. However, we found that the expression of major presynaptic proteins such as NSF, syntaxin 1A, synaptotagmin 1, and synaptophysin as well as actin was not altered by LRRK2 silencing. Also, the levels of postsynaptic proteins such as PSD-95 and GluR2 were not changed (supplemental Fig. 1B, available at www.jneurosci.org as supplemental material). The function of these proteins is, however, primarily conferred via specific protein interactions (Rizo and Rosenmund, 2008; Südhof and Rothman, 2009). We therefore asked whether LRRK2 in cortical neurons specifically interacts with presynaptic proteins testing for NSF, a vesicle-fusing ATPase and a key player in vesicular endocytosis (Otto et al., 1997; Littleton et al., 1998, 2001; Kawasaki and Ordway, 2009). NSF was specifically coprecipitated together with endogenous LRRK2 from adult mouse brain lysate using anti LRRK2 1E11 monoclonal antibody (Fig. 6A, top). To further prove this finding, we performed a reciprocal immunoprecipitation using a rabbit anti-NSF antibody (Fig. 6A, bottom). In fact, NSF coprecipitated endogenous LRRK2 as well as syntaxin 1A, a well known interactor of NSF (Hanson et al., 1995). To identify additional LRRK2 interactors from adult mouse brain lysate in a domain-specific fashion, we expressed five different proteins containing specific domains of LRRK2 as GST fusion proteins: GST–N-term, GST–ANK, GST–LRR, GST–Roc–COR–kinase (GST–R-C-K), and GST–WD40 (supplemental Fig. 1C, available at www.jneurosci.org as supplemental material). Pulled proteins were eluted and tryptically digested, and the resulting peptides were identified by LC-MS/MS (Table 1). The analysis revealed 13 putative interactors specifically binding to GST–LRRK2 domains but not to GST alone. Notably, the interactors found were mainly proteins involved in presynaptic vesicular trafficking, including AP-2 complex subunits, synapsin 1, synaptic vesicle glycoprotein 2A (SV2A), and, as expected, NSF. To confirm the results obtained by mass spectrometry, all pull-downs were tested by Western blotting (Fig. 6B). Western blotting confirmed LRRK2 interaction with NSF, syntaxin 1A, and actin mainly through its WD40 domain. Furthermore, SNAP-25 and synaptophysin were not found to bind GST–LRRK2 domains (supplemental Fig. 1D, available at www.jneurosci.org as supplemental material). Precedent studies showed that LRRK2 is mainly associated with mitochondria but also with multiple vesicles structure, including synaptic vesicles (Biskup et al., 2006). To verify whether LRRK2 might localize within the cell in a cellular compartment in which its putative interactors are present, we performed a subcellular fractionation of dissociated neuronal cortical cultures (Dodd et al., 1981). LRRK2 showed a partial enrichment in a fraction containing its putative interactors, NSF, syntaxin1A, and actin (Fig. 6C). Interestingly, also the presynaptic markers Vamp1 and synaptotagmin and the postsynaptic marker PSD-95, but not the cytosolic marker HSP90, were found enriched in the same fraction (Fig. 6D).These data suggest that LRRK2 interacts with a subset of the vesicle fusion protein complex, putting it perfectly in place to modulate presynaptic vesicle trafficking and distribution.
Figure 6.

LRRK2 interacts with presynaptic protein. A, Immunoprecipitation (IP) of endogenous LRRK2 (top) and NSF (bottom) from adult brain lysate shows that LRRK2 and NSF interact physiologically. NSF antibody precipitates equally syntaxin1 and LRRK2. B, LRRK2 interacts with presynaptic proteins. GST–LRRK2 domains have been used to pull down putative LRRK2 interactors from adult mouse brain. LRRK2 WD40 domain precipitates presynaptic proteins as NSF and syntaxin 1A and actin. C, D, LRRK2 is present in a subcellular fraction (P3) in which its putative interactors and presynaptic and postsynaptic markers are found. s-tagmin, Synaptotagmin.
Table 1.
LRRK2 interactors
| Identified proteins | Accession number | Molecular weight | n peptides | Domain | Function |
|---|---|---|---|---|---|
| α-Actin-2 | P62737 | 42 kDa | 10 | WD40 | Component of cytoskeleton |
| AP-1 complex subunit β-1 | O35643 | 104 kDa | 8 | WD40 | Required for vesicle sorting during endocytosis |
| AP-2 complex subunit α-1 | P17426 | 108 kDa | 5 | WD40 | AP-2 complex is a heterotetramer; it mediates the recruitment of clathrin to membranes and plays a role in the recycling of synaptic vesicle membranes from the presynaptic surface |
| AP-2 complex subunit α-2 | P17427 | 104 kDa | 7 | ANK | |
| AP-2 complex subunit β-1 | Q9DBG3 | 105 kDa | 5 | WD40 | |
| Calcium-dependent secretion activator 1 | Q80TJ1 | 153 kDa | 3 | WD40 | Calcium-binding protein involved in the exocytosis of synaptic vesicles |
| Clathrin coat assembly protein AP180 | Q61548 | 92 kDa | 6 | WD40 | Components of the adapter complex that links clathrin to coated vesicles |
| Clathrin heavy chain 1 | Q68FD5 | 192 kDa | 52 | WD40 | Main coat of coated pits and vesicles |
| Dynamin-1 | P39053 | 97 kDa | 27 | WD40 | Microtubule-associated protein involved in endocytosis of vesicles |
| Synapsin-1 | O88935 | 74 kDa | 4 | WD40 | Neuronal phosphoprotein that coats synaptic vesicles |
| Synaptic vesicle glycoprotein 2A | Q9JIS5 | 83 kDa | 5 | WD40 | Regulates vesicle fusion by maintaining the readily releasable pool |
| Syntaxin 1B | P61264 | 33 kDa | 5 | WD40 | Involved in docking of synaptic vesicles at presynaptic active zones |
| Vesicle-fusing ATPase | P46460 | 83 kDa | 21 | WD40 | Required for vesicle-mediated transport |
A domain-based GST pull-down approach was performed to explore LRRK2 interactome. GST fusion proteins covering full-length LRRK2 and mimicking its functional domain (GST–N-term, GST–ANK, GST–LRR, GST–R-C-K, and GST–WD40) was used to retain interactors from adult mouse brain lysate. The nature of the pulled proteins including putative interactors was identified by LC-MS/MS. Peptide identifications were accepted if they could be established at greater than 95%, whereas protein identifications were accepted if they could be established at greater than 99% probability and contained at least two identified unique peptides. Only hits confirmed by more than two independent experiments and absent in the GST control sample were taken into consideration. The table reports protein name, UniProtKB/Swiss-Prot accession number, protein molecular weight, number of unique peptides identifying the indicated protein, GST fusion domain bound, and protein putative function as annotated in UniProt database. Supplemental Table 1 reports additional peptides information (available at www.jneurosci.org as supplemental material).
LRRK2 silencing alters synaptic vesicle distribution
Synaptic vesicles within the presynaptic compartment are distributed in pools distinguished by the relative distance to the presynaptic membrane (Schikorski and Stevens, 2001; Rizzoli and Betz, 2004). Given the data reported above, we asked whether SV number, distribution, and clustering might be influenced by LRRK2 silencing. To test this, we analyzed the presynaptic boutons in control and siLRRK2-infected neurons by electron microscopy (Fig. 7A). Electron microscopic analysis revealed that LRRK2 knockdown did not cause obvious abnormalities in presynaptic terminals with respect to control neurons. LRRK2 silencing did not affect total pool size, because the average number of SVs per presynaptic terminal was equal in both conditions (supplemental Fig. 4A, available at www.jneurosci.org as supplemental material). Also, the analysis of SV dimension, reported by the measurement of major axis length, did not show any differences between the two groups (supplemental Fig. 4B, available at www.jneurosci.org as supplemental material). To clarify whether LRRK2 might influence synaptic vesicle organization, we analyzed SV distribution in terms of shortest distance to the active zone (AZD) (Nikonenko and Skibo, 2004). We observed differences in the spatial distribution of SV in terms of relative abundance of vesicles located in two specific SV pools, identified by their AZD (supplemental Fig. 4D, available at www.jneurosci.org as supplemental material). siLRRK2 synapses were characterized by a significant reduction in the number of docked SVs, i.e., vesicles in physical contact with the presynaptic membrane (Schikorski and Stevens, 2001) [Fig. 7C, untreated; docked SV in vesicles/μm (mean ± SE): control, 17 ± 1.32; siLRRK2, 11.29 ± 0.53; n = 35; p < 0.01]. LRRK2 silencing also caused a significant increase of vesicles located distally to the presynaptic membrane [Fig. 7D, untreated; fraction of vesicles within a range of 75–150 nm to the presynaptic membrane (%) (mean ± SE): control, 21.11 ± 1.76; siLRRK2, 26.45 ± 1.58; n = 35; p < 0.05]. We then depolarized cortical neurons with 50 mm KCl before fixation (Fig. 7B). LRRK2 silencing did not affect total pool size (supplemental Fig. 4C, available at www.jneurosci.org as supplemental material) but altered SV distribution (supplemental Fig. 4E, available at www.jneurosci.org as supplemental material). In LRRK2-silenced synapses, the amount of docked vesicles was unaltered compared with controls [Fig. 7C, KCl; docked SV, vesicle/μm (mean ± SE): control, 10.74 ± 0.51; siLRRK2, 10.92 ± 0.48; n = 35; p > 0.5], but in these neurons, the pool within the 75–150 nm range was reduced [Fig. 7D, KCl; fraction of vesicles within a range of 75–150 nm to the presynaptic membrane (%) (mean ± SE): control, 25.28 ± 1.74; siLRRK2, 18.88 ± 1.56; n = 35; p < 0.01]. To determine whether LRRK2 silencing might affect the size of the readily releasable pool (RRP), we applied a hypertonic sucrose pulse (0.5 m sucrose, 45 s) to cultured neurons while performing the exo-endocytic assay (Fig. 8A). Hypertonic sucrose is thought to stimulate the release of the entire RRP in neuron in culture, thereby allowing the estimation of the RRP itself (Rosenmund and Stevens, 1997; Chang and Südhof, 2009). The hypertonic pulse improved the ratio of recycling synapses in control neurons as expected (Pyle et al., 2000); instead, we did not observe any additional increase in the ratio of recycling synapses in siLRRK2 neurons. Interestingly, control and siLRRK2 neurons were demonstrated to uptake synaptotagmin antibody in a similar extent after stimulation [Fig. 8B; % of synaptotagmin+ synaptophysin+ clusters (mean ± SE): control untreated, 13.58 ± 2.07; 0.5 m sucrose, 27.84 ± 1.91, n = 15, p < 0.01; siLRRK2 untreated, 24.85 ± 1.66; 0.5 m sucrose, 26.18 ± 1.51, n = 15, p > 0.05]. The total number of synaptic contacts remained unaltered despite the treatments (Fig. 8C; number of synaptophysin+ clusters/10 μm (mean ± SE); n = 15; p > 0.05). Together, these findings suggest an involvement of LRRK2 in controlling synaptic vesicle distribution within the presynaptic bouton without affecting RRP size.
Figure 7.
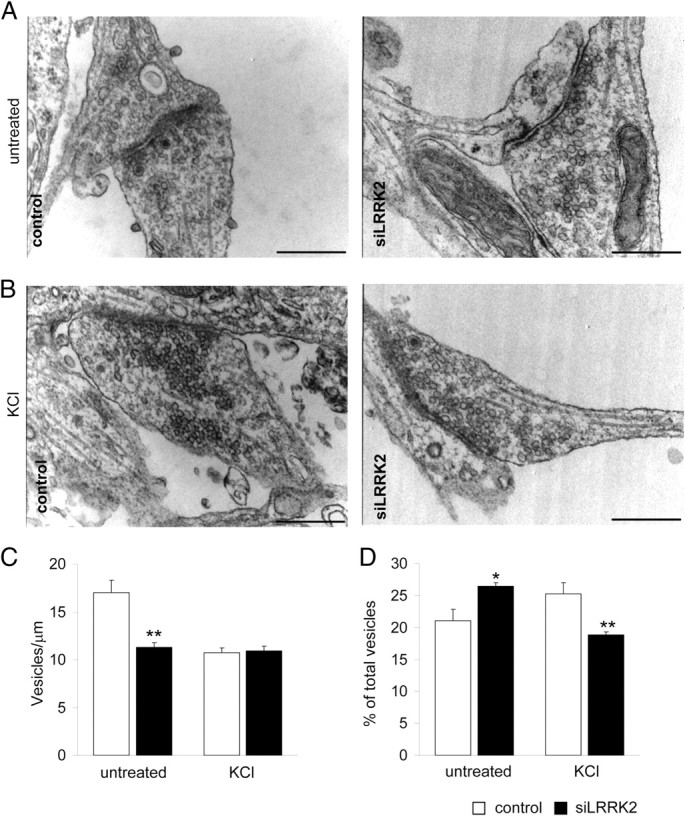
LRRK2 silencing perturbs vesicle distribution inside the presynaptic bouton. Neurons were infected at DIV10 as described and processed for electron microscopy at DIV18. A, B, High-magnification (40,000×) images from control or LRRK2-silenced neurons kept in basal condition (A) or depolarized with 50 mm KCl 5 min (B). C, The number of docked vesicle (vesicle/μm presynaptic membrane) was reduced in siLRRK2 neurons kept in basal condition. D, LRRK2 silencing affected the fraction of total vesicle within the 75–150 nm range from the presynaptic membrane in untreated and depolarized neurons. Data are expressed as mean ± SE. *p < 0.05, **p < 0.01, Student's t test; n = 35. Scale bar, 500 nm.
Figure 8.

LRRK2 silencing does not affect RRP size. A, The ratio of recycling synapses was assayed in neurons in presence of a hypertonic sucrose solution (0.5 m sucrose, 45 s). Scale bar, 5 μm. B, The ratio of recycling synapses was not significantly different between control and siLRRK2 neurons after sucrose application. C, Active zone number was not modified by LRRK2 silencing or sucrose application. Data are expressed as mean ± SE. *p < 0.05 versus control, #p < 0.05 versus untreated, ANOVA, Tukey's post hoc test; n = 15. s-physin, Synaptophysin; s-tagmin, synaptotagmin.
Discussion
In light of the presented data, we suggest that LRRK2 is part of a functional protein network that controls SV trafficking within the recycling pool by interacting with a subset of presynaptic proteins. A role of LRRK2 in vesicle trafficking involving Rab5b had already been suggested (Shin et al., 2008), but we can now show for the first time that electrophysiological properties as well as vesicular trafficking in the presynaptic pool depend on the presence of LRRK2 as an integral part of presynaptic protein complex. We have in fact identified presynaptic proteins—NSF, AP-2 complex subunits, SV2A, synapsing, and syntaxin 1—as well as actin as putative LRRK2 interactors. These proteins have been described previously as key elements of synaptic vesicle trafficking (Takamori et al., 2006). NSF catalyzes the release of the SNARE complex (SNAP-25, syntaxin 1, and Vamp) and allows the first step of the endocytic cycle (Littleton et al., 1998, 2001). The clathrin complex [clathrin, AP-2 adaptor complex, and accessories protein as dynamin and AP180 (Jung and Haucke, 2007)] constitutes one of the major pathways for SV recycling from the membrane to the resting pool (RP) (Murthy and De Camilli, 2003; Granseth et al., 2006). The control of storage and mobilization of SV in the RRP depends instead on the synaptic vesicle glycoproteins SV2A and SV2B (Xu and Bajjalieh, 2001; Chang and Südhof, 2009), whereas synapsins are thought to immobilize SV in the reserve pool (RP) by crosslinking vesicles to the actin cytoskeleton (Greengard et al., 1993; Hilfiker et al., 1999). RRP and RP constitute together the recycling pool [for a more comprehensive review, see (Südhof, 2000, 2004)]. Ultrastructural analysis of presynaptic boutons revealed that LRRK2 silencing affected the size of a distal pool of SVs and the number of docked ones. According to the distance from the active zone, SVs within a range of 75–150 nm can be considered as belonging to the recycling pool (Schikorski and Stevens, 2001; Genoud et al., 2004). These data suggest that LRRK2 is implicated in the mobilization of the recycling pool. The fact that, in siLRRK2 neuron synapses, we described a similar amount of total SVs but a decreased number of docked ones seems to contradict the synaptotagmin uptake and electrophysiology results. However, even if it has generally been assumed that the vesicles closest to release sites and the docked ones represent the RRP and thus are recruited first during synapse activity (Schikorski and Stevens, 2001), studies have shown that the RRP vesicles are distributed without a specific localization within the bouton in a range of 100 nm from the active zone (Rizzoli and Betz, 2004). Furthermore, other studies have described modifications of synaptic activity not correlated to ultrastructural changes (Augustin et al., 1999; Rosenmund et al., 2002; Moulder et al., 2006). A mechanism other than docking, such as SV recruiting, priming, or release efficacy, might be involved in synaptic adaptation to activity (Morales et al., 2000; Moulder et al., 2006). Given that we did not discriminate between resting SV and recycling SV in our ultrastructural analysis, we cannot draw here a direct link between distribution/number of vesicle and fusion events. Notwithstanding this, in our hands, control neurons exhibited a reduction of docked vesicle after KCl depolarization, an effect that might be related to massive vesicle release. Similarly, we described here a reduced amount of docked SVs in basal condition after LRRK2 knockdown. This outcome might arise from a reduction of RRP size or from an alteration of firing rate or/and vesicle fusion probability. Given that the total RRP size, as estimated by hypertonic sucrose application, was not altered in siLRRK2 neurons, we propose that the lack of LRRK2 affects SV release. Accordingly, the ratio of recycling synapses, monitored by anti-synaptotagmin antibody uptake, was increased in siLRRK2 neurons under basal conditions. Interestingly, we observed in siLRRK2-silenced neurons an inability to further increase the ratio of recycling synapses after long-lasting depolarization. This lack of response may reflect the depletion of the RRP as a result of a perturbation of the recycling machinery. In fact, it has been shown that sustained repetitive activity beyond the first presynaptic release depends not only on the RRP but on SV mobilization from the RP to overcome the rapid depletion of RRP (Dobrunz and Stevens, 1997; Murthy and Stevens, 1998). Indeed, the distribution of SV after long-lasting depolarization was altered by LRRK2 silencing. Given that the blockage of action potential propagation through TTX treatment impaired the ratio of recycling synapses in both control and siLRRK2 neurons, we suggest that LRRK2 participates mainly in the trafficking of SVs driven by evoked activity. Our findings that the presynaptic lack of LRRK2 alters EPSC further support the idea of an involvement of LRRK2 in presynaptic mechanisms. EPSC amplitude depends on the presynaptic factors of vesicle number (N), probability of successful fusion (P), together with the postsynaptic quantal response (Q), EPSC = NPQ (for review, see Schneggenburger et al., 2002). Indeed, LRRK2 silencing in the presynaptic neurons was associated with a reduction in failure rate and with an increase of EPSC amplitude after a single stimulation. Furthermore, LRRK2 silencing induced a significant paired-pulse depression (PPD) and an increase of the second current latency. In a simple model, PPD can be explained as the consequence of the depletion of RRP after multiple triggers (Zucker and Regehr, 2002), but experimental evidences have highlighted how other mechanisms might influence short-term plasticity (Waldeck et al., 2000; Xu and Wu, 2005; Sullivan, 2007). PPD has been associated with an increased latency and a reduction of P (Waldeck et al., 2000; Boudkkazi et al., 2007); additionally, manipulation that modifies P significantly affects synaptic latency (Boudkkazi et al., 2007). Variations in P might derive from local modifications of presynaptic Ca2+ concentrations, from modulation of the Ca2+ sensitivity of the release machinery, and finally from the availability and the correct spatial organization of the components of the fusion machinery (Paisán-Ruíz et al., 2004; Wadel et al., 2007; Kawasaki and Ordway, 2009; Matz et al., 2010). The analysis of the ratio of recycling synapses at different Ca2+ concentrations might suggest that LRRK2 silencing affects Ca2+ sensitivity. Another explanation for the effect on evoked currents we registered during single or repetitive stimulation might arise from a disorganization of SV storage and/or mobilization. FRAP measurements of SV mobility have concluded that recycling vesicles have poor mobility in cultured hippocampal neurons (D = ∼0.003 μm2/s) (Gaffield et al., 2006). The tight confinement of SV depends on molecular anchors such as synapsins and actin and is important for the correct SV trafficking (for a comprehensive review, see Rizzoli and Betz, 2005), but interestingly, neither synapsins knock-out nor actin filament disruption has been shown to increase severely SV mobility (Gaffield et al., 2006; Gaffield and Betz, 2007). Thus, synapsins and actin are not uniquely required for the regulation of SV dynamics, but they might act in cooperation with other molecules. We propose that LRRK2 executes its main function at the presynaptic site; given its relative position as an integral part of a presynaptic protein network, LRRK2 may serve as a molecular hub coordinating both the storage and the mobilization of SVs driven by activity. Accordingly, the analysis of vesicle motion showed that SVs in siLRRK2 neurons were characterized by an increased spatial freedom, with a measured diffusion coefficient approaching that of vesicles in free solution (D = ∼0.15 μm2/s) (Luby-Phelps et al., 1987). Therefore, if in basal condition the lack of LRRK2 might confer to vesicles a higher probability to contact the membrane and fuse, it might affect the organization of the presynaptic machinery as well, thus impairing SV mobilization required during high activity. An intriguing possibility yet to be explored is that the impact of LRRK2 on SV trafficking mainly interests silent synapses (Moulder et al., 2004, 2006, 2008) or the pool of reluctant vesicles described in glutamatergic terminals (Sun and Wu, 2001; Moulder and Mennerick, 2005). In fact, interestingly, LRRK2 knockdown and hypertonic sucrose stimulation were associated with a comparable increase in the ratio of recycling synapses. This might suggest that LRRK2 controls a pool of SV larger than the RRP that is not involved during basal activity but instead can be released by hypertonic sucrose stimulation (Moulder and Mennerick, 2005). Even if a recent study (Andres-Mateos et al., 2009) has reported that LRRK2 knock-out in mice does not result in any observable phenotype, it has been shown that LRRK2 mutation affects activity-dependent DA neurotransmission and catecholamine release (Tong et al., 2009). Furthermore, considering the complexity and the importance of the fusion machinery and the expression in neurons of a homologous LRRK2, LRRK1 (Biskup et al., 2007; Westerlund et al., 2008), compensatory mechanisms during embryonic development of LRRK2 knock-out mice cannot be excluded. Data obtained from our domain-based interaction studies finally suggest that LRRK2 interacts with presynaptic partners mainly through its WD40 C-terminal domain. This domain is required for both LRRK2 toxic and physiological role (Jorgensen et al., 2009; Sheng et al., 2010), and it harbors the mutation G2385R, considered as the main risk factor for Parkinson's disease in Chinese Han population (Mata et al., 2005). Future studies are now needed to determine whether perturbed regulation of vesicle trafficking may contribute to Parkinson's disease associated with this gene variant. Given the correlation described recently between LRRK2 and α-synuclein (Lin et al., 2009; Carballo-Carbajal et al., 2010) and the impact of α-synuclein overexpression on synaptic vesicle recycling (Nemani et al., 2010), the regulation of neurotransmitter release might arise as one the main biological pathway compromised during neuropathology onset.
Footnotes
This work was supported by the National Genome Research Framework programs NGFN-Plus FKZ01GS08140 and NGFN2 SMP Proteomics FKZ01GR0449 and the Helmholtz Alliance for Mental Health in an Aging Society. The research leading to these results has received funding from the European Community's Seventh Framework Programme FP7/2009 under grant agreement numbers 241955, SYSCILIA, and 241481, AFFINOMICS. C.S. was supported by Telethon-Italy Grant GGP09196, Fondazione CARIPLO Project 2009-264, RSTL-CNR, and Regione Lombardia Project SAL-50-16983 TERDISMENTAL. M.M was supported by Fondazione CARIPLO Project 2008-3184 and European Union 7th Framework Program Grant HEALTH F2-2009-241498 (EUROSPIN Project). S.B.C. was supported by Fondazione Cariplo “Brain on Chip” Grant 2006-0948. We thank Elisabeth Kremmer for producing monoclonal anti LRRK2 antibodies. We are grateful to Elöd Kortvély and Matteo Gorza for scientific discussions and critical reading of this manuscript and Luise Jennen for performing the electron microscopy. We thank Silke Becker and Sandra Helm for technical assistance on MS analysis and Martin Distel for his support on confocal microscopy.
References
- Aasly JO, Toft M, Fernandez-Mata I, Kachergus J, Hulihan M, White LR, Farrer M. Clinical features of LRRK2-associated Parkinson's disease in central Norway. Ann Neurol. 2005;57:762–765. doi: 10.1002/ana.20456. [DOI] [PubMed] [Google Scholar]
- Andres-Mateos E, Mejias R, Sasaki M, Li X, Lin BM, Biskup S, Zhang L, Banerjee R, Thomas B, Yang L, Liu G, Beal MF, Huso DL, Dawson TM, Dawson VL. Unexpected lack of hypersensitivity in LRRK2 knock-out mice to MPTP (1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine) J Neurosci. 2009;29:15846–15850. doi: 10.1523/JNEUROSCI.4357-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Augustin I, Rosenmund C, Südhof TC, Brose N. Munc13-1 is essential for fusion competence of glutamatergic synaptic vesicles. Nature. 1999;400:457–461. doi: 10.1038/22768. [DOI] [PubMed] [Google Scholar]
- Bacci A, Coco S, Pravettoni E, Schenk U, Armano S, Frassoni C, Verderio C, De Camilli P, Matteoli M. Chronic blockade of glutamate receptors enhances presynaptic release and downregulates the interaction between synaptophysin-synaptobrevin-vesicle-associated membrane protein 2. J Neurosci. 2001;21:6588–6596. doi: 10.1523/JNEUROSCI.21-17-06588.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bardoni R, Torsney C, Tong CK, Prandini M, MacDermott AB. Presynaptic NMDA receptors modulate glutamate release from primary sensory neurons in rat spinal cord dorsal horn. J Neurosci. 2004;24:2774–2781. doi: 10.1523/JNEUROSCI.4637-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauer M, Szulc J, Meyer M, Jensen CH, Terki TA, Meixner A, Kinkl N, Gasser T, Aebischer P, Ueffing M. Delta-like 1 participates in the specification of ventral midbrain progenitor derived dopaminergic neurons. J Neurochem. 2008;104:1101–1115. doi: 10.1111/j.1471-4159.2007.05037.x. [DOI] [PubMed] [Google Scholar]
- Bauer M, Kinkl N, Meixner A, Kremmer E, Riemenschneider M, Förstl H, Gasser T, Ueffing M. Prevention of interferon-stimulated gene expression using microRNA-designed hairpins. Gene Ther. 2009;16:142–147. doi: 10.1038/gt.2008.123. [DOI] [PubMed] [Google Scholar]
- Berg D, Schweitzer K, Leitner P, Zimprich A, Lichtner P, Belcredi P, Brussel T, Schulte C, Maass S, Nagele T. Type and frequency of mutations in the LRRK2 gene in familial and sporadic Parkinson's disease. Brain. 2005;128:3000–3011. doi: 10.1093/brain/awh666. [DOI] [PubMed] [Google Scholar]
- Biskup S, Moore DJ, Celsi F, Higashi S, West AB, Andrabi SA, Kurkinen K, Yu SW, Savitt JM, Waldvogel HJ, Faull RL, Emson PC, Torp R, Ottersen OP, Dawson TM, Dawson VL. Localization of LRRK2 to membranous and vesicular structures in mammalian brain. Ann Neurol. 2006;60:557–569. doi: 10.1002/ana.21019. [DOI] [PubMed] [Google Scholar]
- Biskup S, Moore DJ, Rea A, Lorenz-Deperieux B, Coombes CE, Dawson VL, Dawson TM, West AB. Dynamic and redundant regulation of LRRK2 and LRRK1 expression. BMC Neurosci. 2007;8:102. doi: 10.1186/1471-2202-8-102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonifati V. Parkinson's disease: the LRRK2–G2019S mutation: opening a novel era in Parkinson's disease genetics. Eur J Hum Genet. 2006;14:1061–1062. doi: 10.1038/sj.ejhg.5201695. [DOI] [PubMed] [Google Scholar]
- Bosgraaf L, Van Haastert PJ. Roc, a Ras/GTPase domain in complex proteins. Biochim Biophys Acta. 2003;1643:5–10. doi: 10.1016/j.bbamcr.2003.08.008. [DOI] [PubMed] [Google Scholar]
- Boudkkazi S, Carlier E, Ankri N, Caillard O, Giraud P, Fronzaroli-Molinieres L, Debanne D. Release-dependent variations in synaptic latency: a putative code for short- and long-term synaptic dynamics. Neuron. 2007;56:1048–1060. doi: 10.1016/j.neuron.2007.10.037. [DOI] [PubMed] [Google Scholar]
- Brice A. Genetics of Parkinson's disease: LRRK2 on the rise. Brain. 2005;128:2760–2762. doi: 10.1093/brain/awh676. [DOI] [PubMed] [Google Scholar]
- Buckby LE, Mummery R, Crompton MR, Beesley PW, Empson RM. Comparison of neuroplastin and synaptic marker protein expression in acute and cultured organotypic hippocampal slices from rat. Brain Res Dev Brain Res. 2004;150:1–7. doi: 10.1016/j.devbrainres.2004.01.006. [DOI] [PubMed] [Google Scholar]
- Carballo-Carbajal I, Weber-Endress S, Rovelli G, Chan D, Wolozin B, Klein CL, Patenge N, Gasser T, Kahle PJ. Leucine-rich repeat kinase 2 induces alpha-synuclein expression via the extracellular signal-regulated kinase pathway. Cell Signal. 2010;22:821–827. doi: 10.1016/j.cellsig.2010.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang WP, Südhof TC. SV2 renders primed synaptic vesicles competent for Ca2+-induced exocytosis. J Neurosci. 2009;29:883–897. doi: 10.1523/JNEUROSCI.4521-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobrunz LE, Stevens CF. Heterogeneity of release probability, facilitation, and depletion at central synapses. Neuron. 1997;18:995–1008. doi: 10.1016/s0896-6273(00)80338-4. [DOI] [PubMed] [Google Scholar]
- Dodd PR, Hardy JA, Oakley AE, Edwardson JA, Perry EK, Delaunoy JP. A rapid method for preparing synaptosomes: comparison, with alternative procedures. Brain Res. 1981;226:107–118. doi: 10.1016/0006-8993(81)91086-6. [DOI] [PubMed] [Google Scholar]
- Eng JK, McCormack AL, Yates JR., 3rd An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. J Am Soc Mass Spectrometry. 1994;5:976–989. doi: 10.1016/1044-0305(94)80016-2. [DOI] [PubMed] [Google Scholar]
- Frangioni JV, Neel BG. Solubilization and purification of enzymatically active glutathione S-transferase (pGEX) fusion proteins. Anal Biochem. 1993;210:179–187. doi: 10.1006/abio.1993.1170. [DOI] [PubMed] [Google Scholar]
- Gaffield MA, Betz WJ. Synaptic vesicle mobility in mouse motor nerve terminals with and without synapsin. J Neurosci. 2007;27:13691–13700. doi: 10.1523/JNEUROSCI.3910-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaffield MA, Rizzoli SO, Betz WJ. Mobility of synaptic vesicles in different pools in resting and stimulated frog motor nerve terminals. Neuron. 2006;51:317–325. doi: 10.1016/j.neuron.2006.06.031. [DOI] [PubMed] [Google Scholar]
- Genoud C, Knott GW, Sakata K, Lu B, Welker E. Altered synapse formation in the adult somatosensory cortex of brain-derived neurotrophic factor heterozygote mice. J Neurosci. 2004;24:2394–2400. doi: 10.1523/JNEUROSCI.4040-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillardon F. Leucine-rich repeat kinase 2 phosphorylates brain tubulin-beta isoforms and modulates microtubule stability—a point of convergence in Parkinsonian neurodegeneration? J Neurochem. 2009;110:1514–1522. doi: 10.1111/j.1471-4159.2009.06235.x. [DOI] [PubMed] [Google Scholar]
- Gloeckner CJ, Kinkl N, Schumacher A, Braun RJ, O'Neill E, Meitinger T, Kolch W, Prokisch H, Ueffing M. The Parkinson disease causing LRRK2 mutation I2020T is associated with increased kinase activity. Hum Mol Genet. 2006;15:223–232. doi: 10.1093/hmg/ddi439. [DOI] [PubMed] [Google Scholar]
- Gloeckner CJ, Schumacher A, Boldt K, Ueffing M. The Parkinson disease-associated protein kinase LRRK2 exhibits MAPKKK activity and phosphorylates MKK3/6 and MKK4/7, in vitro. J Neurochem. 2009;109:959–968. doi: 10.1111/j.1471-4159.2009.06024.x. [DOI] [PubMed] [Google Scholar]
- Goldwurm S, Di Fonzo A, Simons EJ, Rohé CF, Zini M, Canesi M, Tesei S, Zecchinelli A, Antonini A, Mariani C, Meucci N, Sacilotto G, Sironi F, Salani G, Ferreira J, Chien HF, Fabrizio E, Vanacore N, Dalla Libera A, Stocchi F, et al. The G6055A (G2019S) mutation in LRRK2 is frequent in both early and late onset Parkinson's disease and originates from a common ancestor. J Med Genet. 2005;42:e65. doi: 10.1136/jmg.2005.035568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granseth B, Odermatt B, Royle SJ, Lagnado L. Clathrin-mediated endocytosis is the dominant mechanism of vesicle retrieval at hippocampal synapses. Neuron. 2006;51:773–786. doi: 10.1016/j.neuron.2006.08.029. [DOI] [PubMed] [Google Scholar]
- Greengard P, Valtorta F, Czernik AJ, Benfenati F. Synaptic vesicle phosphoproteins and regulation of synaptic function. Science. 1993;259:780–785. doi: 10.1126/science.8430330. [DOI] [PubMed] [Google Scholar]
- Greggio E, Jain S, Kingsbury A, Bandopadhyay R, Lewis P, Kaganovich A, van der Brug MP, Beilina A, Blackinton J, Thomas KJ, Ahmad R, Miller DW, Kesavapany S, Singleton A, Lees A, Harvey RJ, Harvey K, Cookson MR. Kinase activity is required for the toxic effects of mutant LRRK2/dardarin. Neurobiol Dis. 2006;23:329–341. doi: 10.1016/j.nbd.2006.04.001. [DOI] [PubMed] [Google Scholar]
- Guo L, Wang W, Chen SG. Leucine-rich repeat kinase 2: relevance to Parkinson's disease. Int J Biochem Cell Biol. 2006;38:1469–1475. doi: 10.1016/j.biocel.2006.02.009. [DOI] [PubMed] [Google Scholar]
- Hanson PI, Otto H, Barton N, Jahn R. The N-ethylmaleimide-sensitive fusion protein and alpha-SNAP induce a conformational change in syntaxin. J Biol Chem. 1995;270:16955–16961. doi: 10.1074/jbc.270.28.16955. [DOI] [PubMed] [Google Scholar]
- Hardy J, Cai H, Cookson MR, Gwinn-Hardy K, Singleton A. Genetics of Parkinson's disease and parkinsonism. Ann Neurol. 2006;60:389–398. doi: 10.1002/ana.21022. [DOI] [PubMed] [Google Scholar]
- Higashi S, Moore DJ, Colebrooke RE, Biskup S, Dawson VL, Arai H, Dawson TM, Emson PC. Expression and localization of Parkinson's disease-associated leucine-rich repeat kinase 2 in the mouse brain. J Neurochem. 2007;100:368–381. doi: 10.1111/j.1471-4159.2006.04246.x. [DOI] [PubMed] [Google Scholar]
- Hilfiker S, Pieribone VA, Czernik AJ, Kao HT, Augustine GJ, Greengard P. Synapsins as regulators of neurotransmitter release. Philos Trans R Soc Lond B Biol Sci. 1999;354:269–279. doi: 10.1098/rstb.1999.0378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iaccarino C, Crosio C, Vitale C, Sanna G, Carrì MT, Barone P. Apoptotic mechanisms in mutant LRRK2-mediated cell death. Hum Mol Genet. 2007;16:1319–1326. doi: 10.1093/hmg/ddm080. [DOI] [PubMed] [Google Scholar]
- Jahn R, Scheller RH. SNAREs: engines for membrane fusion. Nat Rev Mol Cell Biol. 2006;7:631–643. doi: 10.1038/nrm2002. [DOI] [PubMed] [Google Scholar]
- Jorgensen ND, Peng Y, Ho CC, Rideout HJ, Petrey D, Liu P, Dauer WT. The WD40 domain is required for LRRK2 neurotoxicity. PLoS One. 2009;4:e8463. doi: 10.1371/journal.pone.0008463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung N, Haucke V. Clathrin-mediated endocytosis at synapses. Traffic. 2007;8:1129–1136. doi: 10.1111/j.1600-0854.2007.00595.x. [DOI] [PubMed] [Google Scholar]
- Kawasaki F, Ordway RW. Molecular mechanisms determining conserved properties of short-term synaptic depression revealed in NSF and SNAP-25 conditional mutants. Proc Natl Acad Sci U S A. 2009;106:14658–14663. doi: 10.1073/pnas.0907144106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller A, Nesvizhskii AI, Kolker E, Aebersold R. Empirical statistical model to estimate the accuracy of peptide identifications made by MS/MS and database search. Anal Chem. 2002;74:5383–5392. doi: 10.1021/ac025747h. [DOI] [PubMed] [Google Scholar]
- Kraszewski K, Mundigl O, Daniell L, Verderio C, Matteoli M, De Camilli P. Synaptic vesicle dynamics in living cultured hippocampal neurons visualized with CY3-conjugated antibodies directed against the lumenal domain of synaptotagmin. J Neurosci. 1995;15:4328–4342. doi: 10.1523/JNEUROSCI.15-06-04328.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kusumi A, Sako Y, Yamamoto M. Confined lateral diffusion of membrane receptors as studied by single particle tracking (nanovid microscopy). Effects of calcium-induced differentiation in cultured epithelial cells. Biophys J. 1993;65:2021–2040. doi: 10.1016/S0006-3495(93)81253-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SH, Sheng M. Development of neuron-neuron synapses. Curr Opin Neurobiol. 2000;10:125–131. doi: 10.1016/s0959-4388(99)00046-x. [DOI] [PubMed] [Google Scholar]
- Li X, Moore DJ, Xiong Y, Dawson TM, Dawson VL. Reevaluation of phosphorylation sites in the Parkinson's disease associated leucine rich repeat kinase-2. J Biol Chem. 2010;285:29569–29576. doi: 10.1074/jbc.M110.127639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin X, Parisiadou L, Gu XL, Wang L, Shim H, Sun L, Xie C, Long CX, Yang WJ, Ding J, Chen ZZ, Gallant PE, Tao-Cheng JH, Rudow G, Troncoso JC, Liu Z, Li Z, Cai H. Leucine-rich repeat kinase 2 regulates the progression of neuropathology induced by Parkinson's-disease-related mutant alpha-synuclein. Neuron. 2009;64:807–827. doi: 10.1016/j.neuron.2009.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Littleton JT, Chapman ER, Kreber R, Garment MB, Carlson SD, Ganetzky B. Temperature-sensitive paralytic mutations demonstrate that synaptic exocytosis requires SNARE complex assembly and disassembly. Neuron. 1998;21:401–413. doi: 10.1016/s0896-6273(00)80549-8. [DOI] [PubMed] [Google Scholar]
- Littleton JT, Barnard RJ, Titus SA, Slind J, Chapman ER, Ganetzky B. SNARE-complex disassembly by NSF follows synaptic-vesicle fusion. Proc Natl Acad Sci U S A. 2001;98:12233–12238. doi: 10.1073/pnas.221450198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luby-Phelps K, Castle PE, Taylor DL, Lanni F. Hindered diffusion of inert tracer particles in the cytoplasm of mouse 3T3 cells. Proc Natl Acad Sci U S A. 1987;84:4910–4913. doi: 10.1073/pnas.84.14.4910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacLeod D, Dowman J, Hammond R, Leete T, Inoue K, Abeliovich A. The familial Parkinsonism gene LRRK2 regulates neurite process morphology. Neuron. 2006;52:587–593. doi: 10.1016/j.neuron.2006.10.008. [DOI] [PubMed] [Google Scholar]
- Malgaroli A, Ting AE, Wendland B, Bergamaschi A, Villa A, Tsien RW, Scheller RH. Presynaptic component of long-term potentiation visualized at individual hippocampal synapses. Science. 1995;268:1624–1628. doi: 10.1126/science.7777862. [DOI] [PubMed] [Google Scholar]
- Markram H, Lübke J, Frotscher M, Roth A, Sakmann B. Physiology and anatomy of synaptic connections between thick tufted pyramidal neurones in the developing rat neocortex. J Physiol. 1997;500:409–440. doi: 10.1113/jphysiol.1997.sp022031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mata IF, Kachergus JM, Taylor JP, Lincoln S, Aasly J, Lynch T, Hulihan MM, Cobb SA, Wu RM, Lu CS, Lahoz C, Wszolek ZK, Farrer MJ. Lrrk2 pathogenic substitutions in Parkinson's disease. Neurogenetics. 2005;6:171–177. doi: 10.1007/s10048-005-0005-1. [DOI] [PubMed] [Google Scholar]
- Matteoli M, Takei K, Perin MS, Südhof TC, De Camilli P. Exo-endocytotic recycling of synaptic vesicles in developing processes of cultured hippocampal neurons. J Cell Biol. 1992;117:849–861. doi: 10.1083/jcb.117.4.849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matveev V, Sherman A, Zucker RS. New and corrected simulations of synaptic facilitation. Biophys J. 2002;83:1368–1373. doi: 10.1016/S0006-3495(02)73907-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matz J, Gilyan A, Kolar A, McCarvill T, Krueger SR. Rapid structural alterations of the active zone lead to sustained changes in neurotransmitter release. Proc Natl Acad Sci U S A. 2010;107:8836–8841. doi: 10.1073/pnas.0906087107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melrose H, Lincoln S, Tyndall G, Dickson D, Farrer M. Anatomical localization of leucine-rich repeat kinase 2 in mouse brain. Neuroscience. 2006;139:791–794. doi: 10.1016/j.neuroscience.2006.01.017. [DOI] [PubMed] [Google Scholar]
- Mielke JG, Comas T, Woulfe J, Monette R, Chakravarthy B, Mealing GA. Cytoskeletal, synaptic, and nuclear protein changes associated with rat interface organotypic hippocampal slice culture development. Brain Res Dev Brain Res. 2005;160:275–286. doi: 10.1016/j.devbrainres.2005.09.009. [DOI] [PubMed] [Google Scholar]
- Montecucco C, Schiavo G, Pantano S. SNARE complexes and neuroexocytosis: how many, how close? Trends Biochem Sci. 2005;30:367–372. doi: 10.1016/j.tibs.2005.05.002. [DOI] [PubMed] [Google Scholar]
- Moore DJ, West AB, Dawson VL, Dawson TM. Molecular pathophysiology of Parkinson's disease. Annu Rev Neurosci. 2005;28:57–87. doi: 10.1146/annurev.neuro.28.061604.135718. [DOI] [PubMed] [Google Scholar]
- Morales M, Colicos MA, Goda Y. Actin-dependent regulation of neurotransmitter release at central synapses. Neuron. 2000;27:539–550. doi: 10.1016/s0896-6273(00)00064-7. [DOI] [PubMed] [Google Scholar]
- Moulder KL, Mennerick S. Reluctant vesicles contribute to the total readily releasable pool in glutamatergic hippocampal neurons. J Neurosci. 2005;25:3842–3850. doi: 10.1523/JNEUROSCI.5231-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moulder KL, Meeks JP, Shute AA, Hamilton CK, de Erausquin G, Mennerick S. Plastic elimination of functional glutamate release sites by depolarization. Neuron. 2004;42:423–435. doi: 10.1016/s0896-6273(04)00184-9. [DOI] [PubMed] [Google Scholar]
- Moulder KL, Jiang X, Taylor AA, Olney JW, Mennerick S. Physiological activity depresses synaptic function through an effect on vesicle priming. J Neurosci. 2006;26:6618–6626. doi: 10.1523/JNEUROSCI.5498-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moulder KL, Jiang X, Chang C, Taylor AA, Benz AM, Conti AC, Muglia LJ, Mennerick S. A specific role for Ca2+-dependent adenylyl cyclases in recovery from adaptive presynaptic silencing. J Neurosci. 2008;28:5159–5168. doi: 10.1523/JNEUROSCI.5317-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murthy VN, De Camilli P. Cell biology of the presynaptic terminal. Annu Rev Neurosci. 2003;26:701–728. doi: 10.1146/annurev.neuro.26.041002.131445. [DOI] [PubMed] [Google Scholar]
- Murthy VN, Stevens CF. Synaptic vesicles retain their identity through the endocytic cycle. Nature. 1998;392:497–501. doi: 10.1038/33152. [DOI] [PubMed] [Google Scholar]
- Nemani VM, Lu W, Berge V, Nakamura K, Onoa B, Lee MK, Chaudhry FA, Nicoll RA, Edwards RH. Increased expression of alpha-synuclein reduces neurotransmitter release by inhibiting synaptic vesicle reclustering after endocytosis. Neuron. 2010;65:66–79. doi: 10.1016/j.neuron.2009.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nesvizhskii AI, Aebersold R. Analysis, statistical validation and dissemination of large-scale proteomics datasets generated by tandem MS. Drug Discov Today. 2004;9:173–181. doi: 10.1016/S1359-6446(03)02978-7. [DOI] [PubMed] [Google Scholar]
- Nikonenko AG, Skibo GG. Technique to quantify local clustering of synaptic vesicles using single section data. Microsc Res Tech. 2004;65:287–291. doi: 10.1002/jemt.20134. [DOI] [PubMed] [Google Scholar]
- Olsen JV, de Godoy LM, Li G, Macek B, Mortensen P, Pesch R, Makarov A, Lange O, Horning S, Mann M. Parts per million mass accuracy on an Orbitrap mass spectrometer via lock mass injection into a C-trap. Mol Cell Proteomics. 2005;4:2010–2021. doi: 10.1074/mcp.T500030-MCP200. [DOI] [PubMed] [Google Scholar]
- Otto H, Hanson PI, Jahn R. Assembly and disassembly of a ternary complex of synaptobrevin, syntaxin, and SNAP-25 in the membrane of synaptic vesicles. Proc Natl Acad Sci U S A. 1997;94:6197–6201. doi: 10.1073/pnas.94.12.6197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paisán-Ruíz C, Jain S, Evans EW, Gilks WP, Simón J, van der Brug M, López de Munain A, Aparicio S, Gil AM, Khan N, Johnson J, Martinez JR, Nicholl D, Carrera IM, Pena AS, de Silva R, Lees A, Martí-Massó JF, Pérez-Tur J, Wood NW, Singleton AB. Cloning of the gene containing mutations that cause PARK8-linked Parkinson's disease. Neuron. 2004;44:595–600. doi: 10.1016/j.neuron.2004.10.023. [DOI] [PubMed] [Google Scholar]
- Plowey ED, Cherra SJ, 3rd, Liu YJ, Chu CT. Role of autophagy in G2019S-LRRK2-associated neurite shortening in differentiated SH-SY5Y cells. J Neurochem. 2008;105:1048–1056. doi: 10.1111/j.1471-4159.2008.05217.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pyle JL, Kavalali ET, Piedras-Rentería ES, Tsien RW. Rapid reuse of readily releasable pool vesicles at hippocampal synapses. Neuron. 2000;28:221–231. doi: 10.1016/s0896-6273(00)00098-2. [DOI] [PubMed] [Google Scholar]
- Rizo J, Rosenmund C. Synaptic vesicle fusion. Nat Struct Mol Biol. 2008;15:665–674. doi: 10.1038/nsmb.1450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizzoli SO, Betz WJ. The structural organization of the readily releasable pool of synaptic vesicles. Science. 2004;303:2037–2039. doi: 10.1126/science.1094682. [DOI] [PubMed] [Google Scholar]
- Rizzoli SO, Betz WJ. Synaptic vesicle pools. Nat Rev Neurosci. 2005;6:57–69. doi: 10.1038/nrn1583. [DOI] [PubMed] [Google Scholar]
- Romorini S, Piccoli G, Jiang M, Grossano P, Tonna N, Passafaro M, Zhang M, Sala C. A functional role of postsynaptic density-95-guanylate kinase-associated protein complex in regulating Shank assembly and stability to synapses. J Neurosci. 2004;24:9391–9404. doi: 10.1523/JNEUROSCI.3314-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenmund C, Stevens CF. The rate of aldehyde fixation of the exocytotic machinery in cultured hippocampal synapses. J Neurosci Methods. 1997;76:1–5. doi: 10.1016/s0165-0270(97)00061-7. [DOI] [PubMed] [Google Scholar]
- Rosenmund C, Sigler A, Augustin I, Reim K, Brose N, Rhee JS. Differential control of vesicle priming and short-term plasticity by Munc13 isoforms. Neuron. 2002;33:411–424. doi: 10.1016/s0896-6273(02)00568-8. [DOI] [PubMed] [Google Scholar]
- Rudelius M, Osanger A, Kohlmann S, Augustin M, Piontek G, Heinzmann U, Jennen G, Russ A, Matiasek K, Stumm G, Schlegel J. A missense mutation in the WD40 domain of murine Lyst is linked to severe progressive Purkinje cell degeneration. Acta Neuropathol. 2006;112:267–276. doi: 10.1007/s00401-006-0092-6. [DOI] [PubMed] [Google Scholar]
- Saxton MJ. Lateral diffusion in an archipelago. Effects of impermeable patches on diffusion in a cell membrane. Biophys J. 1982;39:165–173. doi: 10.1016/S0006-3495(82)84504-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saxton MJ. Single-particle tracking: models of directed transport. Biophys J. 1994;67:2110–2119. doi: 10.1016/S0006-3495(94)80694-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sbalzarini IF, Koumoutsakos P. Feature point tracking and trajectory analysis for video imaging in cell biology. J Struct Biol. 2005;151:182–195. doi: 10.1016/j.jsb.2005.06.002. [DOI] [PubMed] [Google Scholar]
- Schikorski T, Stevens CF. Morphological correlates of functionally defined synaptic vesicle populations. Nat Neurosci. 2001;4:391–395. doi: 10.1038/86042. [DOI] [PubMed] [Google Scholar]
- Schneggenburger R, Sakaba T, Neher E. Vesicle pools and short-term synaptic depression: lessons from a large synapse. Trends Neurosci. 2002;25:206–212. doi: 10.1016/s0166-2236(02)02139-2. [DOI] [PubMed] [Google Scholar]
- Sheng D, Qu D, Kwok KH, Ng SS, Lim AY, Aw SS, Lee CW, Sung WK, Tan EK, Lufkin T, Jesuthasan S, Sinnakaruppan M, Liu J. Deletion of the WD40 domain of LRRK2 in zebrafish causes Parkinsonism-like loss of neurons and locomotive defect. PLoS Genet. 2010;6:e1000914. doi: 10.1371/journal.pgen.1000914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin N, Jeong H, Kwon J, Heo HY, Kwon JJ, Yun HJ, Kim CH, Han BS, Tong Y, Shen J, Hatano T, Hattori N, Kim KS, Chang S, Seol W. LRRK2 regulates synaptic vesicle endocytosis. Exp Cell Res. 2008;314:2055–2065. doi: 10.1016/j.yexcr.2008.02.015. [DOI] [PubMed] [Google Scholar]
- Südhof TC. The synaptic vesicle cycle revisited. Neuron. 2000;28:317–320. doi: 10.1016/s0896-6273(00)00109-4. [DOI] [PubMed] [Google Scholar]
- Südhof TC. The synaptic vesicle cycle. Annu Rev Neurosci. 2004;27:509–547. doi: 10.1146/annurev.neuro.26.041002.131412. [DOI] [PubMed] [Google Scholar]
- Südhof TC, Rothman JE. Membrane fusion: grappling with SNARE and SM proteins. Science. 2009;323:474–477. doi: 10.1126/science.1161748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan JM. A simple depletion model of the readily releasable pool of synaptic vesicles cannot account for paired-pulse depression. J Neurophysiol. 2007;97:948–950. doi: 10.1152/jn.00554.2006. [DOI] [PubMed] [Google Scholar]
- Sun JY, Wu LG. Fast kinetics of exocytosis revealed by simultaneous measurements of presynaptic capacitance and postsynaptic currents at a central synapse. Neuron. 2001;30:171–182. doi: 10.1016/s0896-6273(01)00271-9. [DOI] [PubMed] [Google Scholar]
- Takamori S, Holt M, Stenius K, Lemke EA, Grønborg M, Riedel D, Urlaub H, Schenck S, Brügger B, Ringler P, Müller SA, Rammner B, Gräter F, Hub JS, De Groot BL, Mieskes G, Moriyama Y, Klingauf J, Grubmüller H, Heuser J, Wieland F, Jahn R. Molecular anatomy of a trafficking organelle. Cell. 2006;127:831–846. doi: 10.1016/j.cell.2006.10.030. [DOI] [PubMed] [Google Scholar]
- Taylor JP, Mata IF, Farrer MJ. LRRK2: a common pathway for parkinsonism, pathogenesis and prevention? Trends Mol Med. 2006;12:76–82. doi: 10.1016/j.molmed.2005.12.004. [DOI] [PubMed] [Google Scholar]
- Tong Y, Pisani A, Martella G, Karouani M, Yamaguchi H, Pothos EN, Shen J. R1441C mutation in LRRK2 impairs dopaminergic neurotransmission in mice. Proc Natl Acad Sci U S A. 2009;106:14622–14627. doi: 10.1073/pnas.0906334106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verderio C, Coco S, Bacci A, Rossetto O, De Camilli P, Montecucco C, Matteoli M. Tetanus toxin blocks the exocytosis of synaptic vesicles clustered at synapses but not of synaptic vesicles in isolated axons. J Neurosci. 1999;19:6723–6732. doi: 10.1523/JNEUROSCI.19-16-06723.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wadel K, Neher E, Sakaba T. The coupling between synaptic vesicles and Ca2+ channels determines fast neurotransmitter release. Neuron. 2007;53:563–575. doi: 10.1016/j.neuron.2007.01.021. [DOI] [PubMed] [Google Scholar]
- Waldeck RF, Pereda A, Faber DS. Properties and plasticity of paired-pulse depression at a central synapse. J Neurosci. 2000;20:5312–5320. doi: 10.1523/JNEUROSCI.20-14-05312.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Xie C, Greggio E, Parisiadou L, Shim H, Sun L, Chandran J, Lin X, Lai C, Yang WJ, Moore DJ, Dawson TM, Dawson VL, Chiosis G, Cookson MR, Cai H. The chaperone activity of heat shock protein 90 is critical for maintaining the stability of leucine-rich repeat kinase 2. J Neurosci. 2008;28:3384–3391. doi: 10.1523/JNEUROSCI.0185-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West AB, Moore DJ, Biskup S, Bugayenko A, Smith WW, Ross CA, Dawson VL, Dawson TM. Parkinson's disease-associated mutations in leucine-rich repeat kinase 2 augment kinase activity. Proc Natl Acad Sci U S A. 2005;102:16842–16847. doi: 10.1073/pnas.0507360102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westerlund M, Belin AC, Anvret A, Bickford P, Olson L, Galter D. Developmental regulation of leucine-rich repeat kinase 1 and 2 expression in the brain and other rodent and human organs: implications for Parkinson's disease. Neuroscience. 2008;152:429–436. doi: 10.1016/j.neuroscience.2007.10.062. [DOI] [PubMed] [Google Scholar]
- Wiznerowicz M, Trono D. Conditional suppression of cellular genes: lentivirus vector-mediated drug-inducible RNA interference. J Virol. 2003;77:8957–8961. doi: 10.1128/JVI.77.16.8957-8961.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Wu LG. The decrease in the presynaptic calcium current is a major cause of short-term depression at a calyx-type synapse. Neuron. 2005;46:633–645. doi: 10.1016/j.neuron.2005.03.024. [DOI] [PubMed] [Google Scholar]
- Xu T, Bajjalieh SM. SV2 modulates the size of the readily releasable pool of secretory vesicles. Nat Cell Biol. 2001;3:691–698. doi: 10.1038/35087000. [DOI] [PubMed] [Google Scholar]
- Yamasaki M, Hashimoto K, Kano M. Miniature synaptic events elicited by presynaptic Ca2+ rise are selectively suppressed by cannabinoid receptor activation in cerebellar Purkinje cells. J Neurosci. 2006;26:86–95. doi: 10.1523/JNEUROSCI.2258-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimprich A, Müller-Myhsok B, Farrer M, Leitner P, Sharma M, Hulihan M, Lockhart P, Strongosky A, Kachergus J, Calne DB, Stoessl J, Uitti RJ, Pfeiffer RF, Trenkwalder C, Homann N, Ott E, Wenzel K, Asmus F, Hardy J, Wszolek Z, Gasser T. The PARK8 locus in autosomal dominant parkinsonism: confirmation of linkage and further delineation of the disease-containing interval. Am J Hum Genet. 2004;74:11–19. doi: 10.1086/380647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziv NE, Garner CC. Principles of glutamatergic synapse formation: seeing the forest for the trees. Curr Opin Neurobiol. 2001;11:536–543. doi: 10.1016/s0959-4388(00)00246-4. [DOI] [PubMed] [Google Scholar]
- Zucker RS, Regehr WG. Short-term synaptic plasticity. Annu Rev Physiol. 2002;64:355–405. doi: 10.1146/annurev.physiol.64.092501.114547. [DOI] [PubMed] [Google Scholar]