

Abstract
Kinesin-1 is a microtubule plus-end-directed motor that transports various cargos along the axon. Previous studies have elucidated the physical and genetic interactions between kinesin-1 and cytoplasmic dynein, a microtubule minus-end-directed motor, in neuronal cells. However, the physiological importance of kinesin-1 in the dynein-dependent retrograde transport of cargo molecules remains obscure. Here, we show that Caenorhabditis elegans kinesin-1 forms a complex with dynein via its interaction with UNC-16, which binds to the dynein light intermediate (DLI) chain. Both kinesin-1 and UNC-16 are required for localization of DLI-1 at the plus ends of nerve process microtubules. In addition, retrograde transport of APL-1 depends on kinesin-1, UNC-16, and dynein. These results suggest that kinesin-1 mediates the anterograde transport of dynein using UNC-16 as a scaffold and that dynein in turn mediates the retrograde transport of cargo molecules in vivo. Thus, UNC-16 functions as an adaptor for kinesin-1-mediated transport of dynein.
Introduction
The general mechanistic principles of axonal transport center involve an interplay among cytoskeletal filaments and force-generating motor proteins. Axonal cargo is transported after being linked to a protein motor, which pulls it stepwise along a filament track. Long-distance transport in axons is accomplished by members of the kinesin and dynein families, which use microtubules as tracks (Vale, 2003; Hirokawa and Takemura, 2005). Microtubules in axons are organized with their plus end oriented toward the axonal terminal and their minus ends oriented toward the cell body. Conventional kinesin is a plus-end-directed motor and is responsible for anterograde axonal transport. Cytoplasmic dynein is minus end directed and is the primary motor for retrograde axonal transport (Vale, 2003). The importance of these processes is highlighted by the observation that mutation of the motors or other transport machinery components can cause neurodegenerative diseases in humans and analogous phenotypes in model organisms (Guzik and Goldstein, 2004).
In Drosophila, mutations in kinesin-1 and cytoplasmic dynein components exhibit defects in transport wherein synaptic vesicle (SV) components and endosomal cargo become clogged in motor neuron axons (Martin et al., 1999; Bowman et al., 2000). Interestingly, mutants in anterograde kinesin pathways also exhibit similar phenotypes (Hurd and Saxton, 1996; Gindhart et al., 1998, 2003). Experimental evidence from animal cells indicates that kinesin-1 directly binds to the dynein complex (Ligon et al., 2004). Such physical interaction could indicate that one motor is a cargo of the other. This notion is supported by recent results showing that fungal dynein, which accumulates at the plus ends of microtubules, requires kinesin-1 for targeting to microtubule plus ends in Aspergillus nidulans (Han et al., 2001; Lee et al., 2003; Sheeman et al., 2003; Zhang et al., 2003).
One key question is how do cargos become linked to particular motors. Kinesin–vesicle linkages appear to be mediated by scaffolding proteins. Kinesin-1 is a heterotetramer composed of two heavy (KHC) and two light (KLC) chains (Vale, 2003). Recently, KLC has been shown to bind members of the c-Jun N-terminal kinase (JNK)-interacting protein (JIP) group, including JIP1, JIP2, and JIP3, which function as molecular tethers and cargos for kinesin-1 (Bowman et al., 2000; Verhey et al., 2001). Localization of JIPs in cultured cells requires kinesin-1 function, demonstrating that JIPs are kinesin-1 cargos (Verhey et al., 2001). JIP3 is expressed exclusively in neurons and likely mediates the transport of synaptic components. Jip3 deletion mutant mice die shortly after birth and exhibit severe morphological defects in the telencephalon (Kelkar et al., 2003). The Drosophila homolog of JIP3, Sunday Driver, was identified genetically in screens for mutants exhibiting larval sluggishness and tail flip phenotypes, similar to the phenotypes observed in kinesin heavy chain (khc) mutants. In sunday driver mutants, axonal cargo misaccumulates in the axons (Bowman et al., 2000). These results implicate Drosophila JIP3 in kinesin-dependent axonal vesicular transport.
To better understand the relationship between kinesin-1 and dynein, we have taken a genetic approach using the nematode Caenorhabditis elegans as a model system. The C. elegans JIP3 homolog is encoded by the unc-16 gene (Byrd et al., 2001). Previously, we have shown that loss-of-function mutations in unc-16 result in the improper localization of SV markers in multiple classes of neurons. Mutations in the KHC gene, unc-116, cause similar defects, and UNC-16 localization is altered in unc-116 mutants. Genetic analysis with double mutants supports the conclusion that UNC-16/JIP3 functions as a cargo adaptor for kinesin-1. JIP3 function in vesicular cargo trafficking seems to be mediated by an interaction of JIP3 with KLC. We have also shown that cytoplasmic dynein is required for retrograde transport of SV proteins from nerve endings in touch sensory neurons (Koushika et al., 2004).
In this study, we show that kinesin-1 forms a complex with dynein via its interaction with UNC-16, which binds to the dynein light intermediate (DLI) chain. Localization of DLI-1 at plus ends of nerve process microtubules depends on both kinesin-1 and UNC-16. We also found that kinesin-1, UNC-16, and dynein are required for the retrograde transport of an amyloid precursor protein (APP) homolog APL-1. These results suggest that kinesin-1 mediates the anterograde transport of dynein via UNC-16, which functions as an adaptor to link kinesin-1 with dynein.
Materials and Methods
Strains.
The following strains were used: Bristol N2, wild type; CB109, unc-16(e109) III; FF41, unc-116(e2281) III; KU301, kmEx301; KU306, kmEx306; KU308, kmEx308; KU310, dhc-1(or195ts) I; kmEx301; KU314, kmEx314; KU324, unc-16 (e109) III; kmEx301; KU326, unc-16 (e109) III; kmEx306; KU327, unc-116(e2281) III; kmEx301; KU330, unc-116(e2281) III; kmEx306; KU331, unc-116(rh24) III; kmEx314; KU332, kmEx311; KU334, kmEx312; KU336, unc-116(e2281) III; kmEx313; KU337, jsIs37[mec-7p::SNB-1::GFP] V; KU338, unc-116(e2281) III; jsIs37 V; KU339, unc-16(e109) III; jsIs37 V; KU347, unc-116(e2281) III; kmEx312; KU348, dli-1(ku266) IV/nT1[qIs51] (IV, V); kmEx301; KU349, kmEx315; KU350, kmEx316; KU351, unc-16 (e109) III; kmEx315; KU352, unc-16 (e109) III; kmEx316; KU353, unc-116(e2281) III; kmEx315; KU354, unc-116(e2281) III; kmEx316; KU355, kmEx317; KU356, unc-116(e2281) III; kmEx317; KU357, unc-16 (e109) III; kmEx317; KU358, dhc-1(js121) I/hT2 [qIs48](I, III); kmEx317; KU359, dhc-1(js121) I/hT2 [qIs48](I, III); unc-116(e2281) III; jsIs37 V; KU360, unc-116(e2281) III; kmEx301; kmEx318; NM1489, dhc-1(js319) I; jsIs37 V; NM2040, dhc-1(js121) I/hT2 [qIs48](I, III); jsIs37 V.
Plasmid constructions.
The ebp-1 cDNA was amplified by PCR from a C. elegans cDNA library (Kawasaki et al., 1999). The dli-1 and klp-16 cDNAs were amplified by PCR using yk1141b6 and yk1497d4 as templates, respectively. The amplified fragments were subcloned into the pCR2.1-TOPO vector (Invitrogen) and sequenced. The dli-1 cDNA was subcloned into a vector carrying green fluorescent protein (GFP) and the jkk-1 promoter to generate the jkk-1p::gfp::dli-1 plasmid. The apl-1::yfp, apl-1::cfp, apl-1::mcherry, and jkk-1p::yfp::dli-1 plasmids were generated by replacing the GFP fragment with yellow fluorescent protein (YFP), cyan fluorescent protein (CFP), and mCHERRY, respectively. The mec-7p::yfp::dli-1 plasmid was made by inserting the yfp::dli-1 gene into the pPD52.102 vector. The apl-1p::cfp and apl-1p::mcherry plasmids were generated by truncating the coding region of the apl-1::cfp and apl-1::mcherry plasmids, respectively (Hornsten et al., 2007). The apl-1p::cfp::ebp-1 and apl-1p::cfp::klp-16 plasmids were made by inserting the respective cDNAs into the apl-1p::cfp plasmid. The mec-7p::cfp::ebp-1 and mec-7p::cfp::klp-16 plasmids were made by inserting the respective cDNAs into the mec-7p::cfp plasmid. The mec-7p::gfp::snt-1 plasmid was made by inserting both a genomic fragment of SNT-1 and a GFP fragment into the pPD52.102 vector. The ofm-1p::dsRed monomer and myo-2p::dsRed monomer plasmids were made by inserting dsRed monomer-N1 (Clontech) into plasmids carrying the ofm-1 promoter and pPD30.69, respectively. The mammalian expression constructs for T7-UNC-16 and HA-KLC-2 plasmids were described previously (Sakamoto et al., 2005). The FLAG-DLI-1 plasmid was generated by inserting the dli-1 cDNA into the pFlag-CMV vector. The yeast expression plasmids, pBTM116-UNC-16F, pBTM116-UNC-16N, pBTM116-UNC-16M, and pACT2-KLC-2, were described previously (Sakamoto et al., 2005). To make an unc-116 genomic construct, a 5.5 kb unc-116 genomic DNA was amplified by PCR and subcloned into pCR2.1 vector.
Microinjection experiments.
Germ line transformation was performed following standard procedures as previously described (Kawasaki et al., 1999). The rol-6d pRF4 plasmid (50 μg/ml), ofm-1p::gfp (50 μg/ml), or ofm-1p::dsRed monomer (50 μg/ml) was used as a coinjection marker. Transgenic arrays were generated using different concentrations as follows: apl-1::gfp (5 μg/ml) was used in kmEx301[apl-1::gfp + rol-6d] and kmEx314[apl-1::gfp + ofm-1p::gfp]; apl-1p::mcherry (50 μg/ml) and jkk-1p::gfp::dli-1 (20 μg/ml) were used in kmEx306[apl-1p::mcherry + jkk-1p::gfp::dli-1 + rol-6d]; apl-1p::cfp::ebp-1 (20 μg/ml) and jkk-1p::yfp::dli-1 (20 μg/ml) were used in kmEx308[apl-1p::cfp::ebp-1 + jkk-1p::yfp::dli-1 + rol-6d]; apl-1p::cfp::ebp-1 (20 μg/ml) and apl-1::yfp (5 μg/ml) were used in kmEx311[apl-1p::cfp::ebp-1 + apl-1::yfp + rol-6d]; apl-1p::cfp::klp-16 (50 μg/ml) and apl-1::yfp (5 μg/ml) were used in kmEx312[apl-1p::cfp::klp-16 + apl-1::yfp + rol-6d]; apl-1p::cfp::klp-16 (50 μg/ml) and jkk-1p::yfp::dli-1 (20 μg/ml) were used in kmEx313[apl-1p::cfp::klp-16 + jkk-1p::yfp::dli-1 + rol-6d]; mec-7p::cfp::ebp-1 (50 μg/ml) and mec-7p::yfp::dli-1 (20 μg/ml) were used in kmEx315[mec-7p::cfp::ebp-1 + mec-7p::yfp::dli-1 + ofm-1p::dsRed monomer]; mec-7p::cfp::klp-16 (20 μg/ml) and mec-7p::yfp::dli-1 (20 μg/ml) were used in kmEx316[mec-7p::cfp::klp-16 + mec-7p::yfp::dli-1 + ofm-1p::dsRed monomer]; mec-7p::gfp::snt-1 (25 μg/ml) was used in kmEx317[mec-7p::gfp::snt-1 + myo-2p::dsRed monomer]; an unc-116 genomic construct (25 μg/ml) was used in kmEx318[unc-116 genomic fragment + myo-2p::dsRed monomer].
Animals carrying these extrachromosomal arrays were crossed into the appropriate mutants for additional analysis. Transgenic animals were selected by their rolling behaviors or fluorescence.
Microscopy.
Standard fluorescent images of transgenic worms were observed under a Zeiss Plan-Apochromat 63× objective of a Zeiss Axioplan II fluorescent microscope and photographed with a Hamamatsu 3CCD camera. Size measurements of misaccumulated SNB-1::GFP at nerve endings were performed as described previously (Koushika et al., 2004). Confocal fluorescent images of fluorescent proteins and time-lapse images of APL-1::GFP were taken on an Olympus FV500 or FV1000 confocal laser-scanning microscope with a 100, 60, or 40× objective. For fluorescent images, nematodes were immobilized using 0.5% 1-phenoxy-2-propanol in 2% agarose. For time-lapse images, nematodes were anesthetized with levamisole (7.5 mm) in 2% agarose. The sublateral neurons of nematodes were bleached for 17 s and then imaged to observe vesicle movement using an Olympus FV1000 confocal laser-scanning microscope. Time-lapse images were captured every 0.2 s. Kymographs were generated using Olympus FluoView software for the FV1000 confocal microscope. Velocities of APL-1::GFP movements were calculated from the diagonal streaks on the kymographs. Movement of >87 APL-1::GFP specimens in each mutant and wild-type animal were used to generate histograms. For quantification of APL-1::GFP, the fluorescence intensities along lateral nerve processes were measured using an Olympus FV500 confocal laser microscope system.
Yeast two-hybrid analysis.
Screening and interaction assays by yeast two-hybrid system were performed as previously described (Sakamoto et al., 2005).
Immunoprecipitation, immunoblotting, and immunostaining.
Monkey COS-7 kidney cells were maintained in DMEM supplemented with 10% fetal calf serum. Mammalian expression vectors were transfected into COS-7 cells using FuGENE 6 transfection reagent according to the manufacturer's instruction (Roche). After 48 h, cells were harvested for additional analysis. Preparations of cell lysates, immunoprecipitation, and Western blotting procedures have been described previously (Kawasaki et al., 1999). For Western blotting of C. elegans lysates, nematodes were resolved by SDS-PAGE and subjected to immunoblotting using anti-APL-1EXT rabbit polyclonal antiserum (Hornsten et al., 2007) and anti-tubulin mouse monoclonal antibody (TAT1; a kind gift from Prof. K. Gull, University of Oxford, Oxford, UK), respectively. Intensities of the detected bands were quantified from scanned immunoblots using Epson GT-X900 scanner and Adobe Photoshop CS software. Immunostaining using anti-GFP and anti-SNT-1 antiserum were performed as described previously (Koushika et al., 2004).
Results
UNC-16 associates with a component of cytoplasmic dynein
The C. elegans JIP3 homolog UNC-16 is involved in the kinesin-1-dependent transport of vesicle cargo in neurons (Byrd et al., 2001). We have previously reported that UNC-16 associates with the middle region of KLC-2, the light chain of kinesin-1 (Fig. 1A) (Sakamoto et al., 2005). To identify additional components of the UNC-16–kinesin-1 complex involved in axonal transport, we performed a yeast two-hybrid screen using a bait construct encoding an N-terminal fragment of UNC-16 (1–240). From this screen, we identified DLI-1, cytoplasmic dynein light intermediate chain (Fig. 1A). However, we did not observe an interaction between full-length UNC-16 and DLI-1 in the yeast two-hybrid assay (Fig. 1A), suggesting that the full-length UNC-16 forms a closed conformation that might block DLI-1 binding in the absence of other factors.
Figure 1.

Interaction among UNC-16, DLI-1, and KLC-2. A, Interactions by yeast two-hybrid assay. Yeasts carrying the indicated plasmids were grown on a selective plate lacking histidine for 5 d. UNC-16 constructs used for this assay are shown below. The black and hatched boxes represent coiled-coil and leucine zipper domains, respectively. B, Interactions by coimmunoprecipitation in mammalian cells. COS-7 cells were transfected with control vector, T7-UNC-16, FLAG-DLI-1, and/or HA-KLC-2 as indicated. Cell lysates were immunoprecipitated (IP) with anti-FLAG antibodies. Immunoprecipitates were immunoblotted (IB) with anti-T7, anti-HA, and anti-FLAG antibodies. Whole-cell extracts (WCE) were immunoblotted with anti-T7 and anti-HA antibodies.
To determine whether UNC-16 can physically associate with DLI-1 protein, we performed immunoprecipitation assays. When T7-tagged UNC-16 and FLAG-tagged DLI-1 were coexpressed in COS-7 cells, we observed no coimmunoprecipitation of T7-UNC-16 with FLAG-DLI-1 (Fig. 1B, lane 3). Since UNC-16 binds to KLC-2 via a domain distinct from the DLI-1 binding domain (Fig. 1A), we imagined that binding of KLC-2 could convert UNC-16 to a conformation accessible by DLI-1. Consistent with this possibility, we observed that T7-UNC-16 coimmunoprecipitated with FLAG-DLI-1 when HA-tagged KLC-2 was also coexpressed in the cell (Fig. 1B, lane 4). Interestingly, HA-KLC-2 weakly associated with FLAG-DLI-1 and this interaction was enhanced in the presence of UNC-16 (Fig. 1B, lanes 2 and 4). These results raise the possibility that UNC-16 acts as a scaffold protein linking kinesin-1 and dynein.
Localization of dynein at the microtubule plus end is determined by the UNC-16–kinesin-1 complex
Several studies have shown that cytoplasmic dynein is present in cargos that are transported by kinesin (Caviston and Holzbaur, 2006; Colin et al., 2008; Yamada et al., 2008, 2010). Our results showed that UNC-16 has the capacity to bind both kinesin-1 and dynein motors and thereby serve as a scaffold to link them. Based on these results, we speculated that kinesin-1 may deliver the dynein complex to the microtubule plus ends via UNC-16. To address this possibility, we used the DLI-1 protein fused to YFP or GFP to examine whether the localization of dynein is affected by mutations of kinesin-1 and unc-16. Expression of YFP::DLI-1 in neurons by the pan-neuronal promoter was able to rescue the uncoordinated defect observed in dli-1(ku266) mutants (data not shown), suggesting that the subcellular localization of the reporter very likely reflects that of the endogenous protein.
We examined localization of dynein in ALM, a pair of lateral mechanosensory neurons. In ALM neurons, the main neuronal process extends to the nose region of the worm, and a branch generated from this process extends into the nerve ring (Fig. 2A) (Chalfie, 1993). We first determined the polarity of microtubules in ALM neurons using CFP fused to EBP-1 and KLP-16, a nematode homolog of microtubule plus end marker protein EB1 and an NCD/KAR1-like minus-end-directed C-terminal kinesin, respectively (Robin et al., 2005; Motegi et al., 2006). We observed that CFP::EBP-1 weakly localized along nerve processes in ALM neurons and apparently accumulated at nerve ends around the nose and synaptic regions in the nerve ring (Fig. 2B). In contrast, CFP::KLP-16 localized exclusively in cell bodies but did not accumulate at nerve ends or synaptic regions (Fig. 2B). These results indicate that microtubules orient and extend their plus ends from the cell bodies to the nerve processes.
Figure 2.

Localization of DLI-1 in ALM touch neurons. A, Schematic image of ALM touch neuron. Presynapses are indicated by green. B, Localization of CFP::EBP-1 and CFP::KLP-16 in ALM neurons. The yellow arrowhead indicates the location of CFP::EBP-1 at the nerve ending of ALM neuron. The red arrowhead indicates the accumulation of CFP::KLP-16 in the cell body. C, Localization of YFP::DLI-1 and CFP::EBP-1 in ALM neurons in wild-type, unc-116(e2281), and unc-16(e109) mutant worms. The top and bottom panels show the processes and nerve rings of ALM neurons, respectively. The arrows indicate the positions of colocalized YFP::DLI-1 and CFP::EBP-1 at the ends of ALM neuron of processes and nerve rings. The yellow arrowheads indicate the locations at the nerve endings. The merged images are shown in right panels. Scale bars, 10 μm.
We observed that in wild-type worms, YFP::DLI-1 was expressed in a weakly diffused pattern along nerve processes and accumulated exclusively at the nerve endings and the nerve ring of ALM neurons (Fig. 2C). The accumulated YFP::DLI-1 colocalized with CFP::EBP-1, suggesting that DLI-1 accumulates at the plus ends of microtubules in ALM neurons. However, in unc-116(e2281) mutants, localization of GFP::DLI-1 was greatly decreased or absent at the nerve endings and the nerve ring (Fig. 2C). Similar phenotypes were also observed in unc-16(e109) mutant worms (Fig. 2C). Since localization of CFP::EBP-1 at nerve ends and the nerve ring was still observed in unc-116 and unc-16 mutants (Fig. 2C), this means that mutations of the kinesin-1 and unc-16 have no effect on microtubule structures or nerve process polarities. These results suggest that dynein is transported to the microtubule plus end by an UNC-16–kinesin-1 complex.
Kinesin-1 and UNC-16 regulate localization of SV proteins in touch neurons
Our previous study revealed that cytoplasmic dynein components are required for the retrograde transport of SV proteins in touch neurons (Koushika et al., 2004). We therefore examined whether the kinesin-1–UNC-16–dynein complex is involved in the axonal transport of SV proteins. In C. elegans, SV proteins such as VAMP2/synaptobrevin SNB-1 are transported from the neuronal cell body by a plus-end-directed motor UNC-104, a kinesin-3 homolog, and they are enriched at synapses along the neuronal branch (Hall and Hedgecock, 1991; Otsuka et al., 1991). To visualize the localization of SNB-1 in live animals, we used a fluorescently tagged SNB-1 protein fused to GFP (SNB-1::GFP) under the control of the mec-7 promoter, which drives expression in the six C. elegans mechanosensory neurons (Koushika et al., 2004). In wild-type animals, SNB-1::GFP fluorescence was expressed in a punctate pattern in touch neurons and was primarily concentrated at the synaptic regions in the nerve ring, but not at nerve ends of ALM neurons (Fig. 3A) (Nonet, 1999). As observed previously (Koushika et al., 2004), in dhc-1(js121) mutants defective in the dynein heavy chain, SNB-1::GFP localized at the ends of ALM neuronal processes and near nerve ring synapses (Fig. 3A, Table 1). In unc-116(e2281) and unc-16(e109) mutants, SNB-1::GFP also accumulated in touch neurons at the nerve ends and in the region of the nerve ring, a phenotype similar to, but less pronounced than that observed in dhc-1 mutants (Fig. 3A, Table 1). In double dhc-1(js121) and unc-116(e2281) mutants, these misaccumulations were not enhanced (Table 1), suggesting that kinesin-1 and dynein function in the same pathway. Previous studies have shown that SV proteins accumulate in the cell bodies of ALM neurons in unc-104 mutants (Nonet, 1999; Koushika et al., 2004). In contrast, SNB-1::GFP did not accumulate in cell bodies in unc-116 mutants (data not shown), consistent with the previous finding that kinesin-1 is not the major motor for anterograde transport of SNB-1 (Hall et al., 1991). Misaccumulation of SNB-1 near the tips of the mechanosensory neuronal processes in mutants defective in the dynein complex is caused by a defect in SNB-1 retrograde transport (Koushika et al., 2004). These results suggest that the UNC-16–kinesin-1 complex is required for the retrograde transport of SNB-1::GFP in touch neurons.
Figure 3.
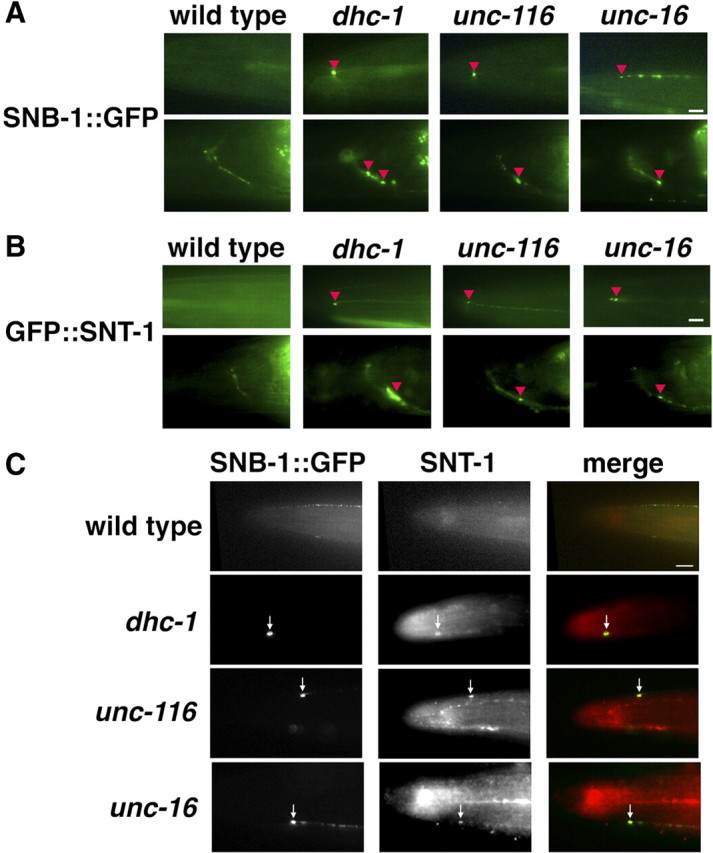
Localization of SV proteins in ALM touch neurons. A, B, Localization of SNB-1::GFP (A) and GFP::SNT-1 (B) in the head region of ALM neurons. The top and bottom panels show the processes and nerve rings of ALM neurons, respectively. The red arrowheads indicate the positions of accumulated SNB-1::GFP (A) and GFP::SNT-1 (B) proteins at the ends of ALM neuronal processes and near nerve ring synapses. C, Colocalization of SNB-1::GFP and endogenous SNT-1 in the head region of ALM neurons. The arrows indicate the positions of accumulated SNB-1::GFP and SNT-1 proteins at the tip of ALM neurons. The merged images are shown in the bottom panels. Scale bars, 10 μm.
Table 1.
Area of SNB-1::GFP misaccumulations
| Genotype | Animals with misaccumulation of SNB-1::GFP | Size of SNB-1::GFP misaccumulations (μm2) (SEM) |
|---|---|---|
| Wild type | 0/20 | |
| dhc-1(js121) | 20/20 | 3.5 (0.6) |
| unc-116(e2281) | 32/40 | 1.6 (0.7) |
| unc-16(e109) | 37/40 | 1.5 (0.6) |
| dhc-1(js121); unc-116(e2281) | 11/11 | 3.6 (0.8) |
The C. elegans synaptotagmin protein SNT-1 misaccumulates at nerve ends of ALM neurons in mutants defective in the dynein complex (Koushika et al., 2004). We therefore determined the localization of SNT-1 using GFP-tagged SNT-1 under the control of the mec-7 promoter. In wild-type worms, GFP::SNT-1 fluorescence was expressed at the nerve ring but did not accumulate at the nerve ends of ALM neurons (Fig. 3B). In contrast, GFP::SNT-1 misaccumulated in ALM neurons at the nerve ends and in the region of the nerve ring in dhc-1(js121), unc-116(e2281), and unc-16(e109) mutants (Fig. 3B). We further determined the localization of endogenous SNT-1 in mutant animals by immunostaining with specific antibodies. In wild-type worms, SNT-1 proteins did not accumulate at nerve ends (Fig. 3C). Consistent with a previous report (Koushika et al., 2004), we observed that SNT-1 and SNB-1::GFP misaccumulated at the ends of the neuronal processes in dhc-1 mutants (Fig. 3C). Similar to the dhc-1 phenotype, SNT-1 misaccumulated together with SNB-1::GFP at the ends of ALM neuronal processes in unc-116(e2281) and unc-16(e109) mutants (Fig. 3C). Together, these results suggest that the kinesin-1–UNC-16 complex is required for the retrograde transport of dynein cargos.
Effects of kinesin-1 and UNC-16 on localization and axonal transport of APL-1 in sublateral neurons
Mutation of the motors or other transport machinery components can cause neurodegenerative diseases in humans (Guzik and Goldstein 2004). APP has been implicated in the pathogenesis of early-onset Alzheimer's disease (AD) in humans. It is known that APP is inserted into the membranes of vesicles that are transported along microtubules. The anterograde movement of APP reflects the kinesin-dependent transport of a subset of vesicles in axons (Muresan and Muresan, 2005; Araki et al., 2007). Furthermore, knockdown of dynein results in the accumulation of APP at the neurite tips, where the microtubule plus ends are localized (Kimura et al., 2007). We therefore examined whether the UNC-16–kinesin-1 complex is involved in dynein-dependent retrograde transport of C. elegans APL-1, a protein related to the human APP (Daigle and Li, 1993). Because of poor detection of the endogenous APL-1 proteins by anti-APL-1 antibody staining, we explored localization using APL-1::GFP. The APL-1::GFP fusion protein was shown to be functional, as it rescued the phenotypes of an apl-1 deletion (Hornsten et al., 2007). In addition, since overexpression of APL-1 has toxic effects in animals (Hornsten et al., 2007), we used animals expressing APL-1::GFP at lower levels than the endogenous APL-1 protein (Fig. 4A). As observed previously (Hornsten et al., 2007), in wild-type worms, APL-1::GFP was expressed in a subset of head, ventral, and sublateral neurons (Fig. 4B). In particular, along the nerve processes of sublateral neurons, APL-1::GFP localized in a punctate pattern (Fig. 4B), suggesting that APL-1::GFP is located in the vesicular compartments of the cell.
Figure 4.

Expression and localization of APL-1::GFP. A, Immunoblotting of APL-1 proteins. Whole extracts were prepared from worms at mixed stages. The top panels show immunoblotting with anti-APL-1 antibody. Positions of APL-1::GFP and endogenous APL-1 are indicated by arrows. The asterisks indicate nonspecific bands. The bottom panels show immunoblotting with anti-tubulin antibody as a loading control. B, Image of fluorescent APL-1::GFP protein in the heads of L4 larva. The red open boxes indicate the cell body and nerve processes of the sublateral motor neurons. The yellow line with an asterisk indicates gut fluorescent granules. Images are from a confocal Z series projected onto a single plane. Scale bar, 5 μm.
Sublateral motor neurons, such as SIB, SID, SMB, and SMD, consist of a cell body located at the head region of worms and a nerve process that extends to the body region (Fig. 5A). These neurons are thought to form synapses with muscles (Duerr et al., 2008). We first examined the polarity of microtubules along the processes and the effect of the UNC-16–kinesin-1 complex on dynein localization in sublateral neurons. We observed that CFP::EBP-1 preferentially localized in a punctate pattern along the sublateral nerve processes (Fig. 5B), whereas CFP::KLP-16 localized in cell bodies but not along the nerve processes (Fig. 6B). These results indicate that microtubules orient and extend their plus ends from the cell bodies to the nerve processes. Coexpression of YFP::DLI-1 with CFP::EBP-1 revealed that DLI-1 puncta colocalized with EBP-1 (Fig. 5B). However, in unc-116(e2281) and unc-16(e109) mutants, GFP::DLI-1 did not localize in the lateral processes, but rather accumulated in neuronal cell bodies (Fig. 5C). When YFP::DLI-1 and CFP::KLP-16 were coexpressed in unc-116 mutants, DLI-1 was found to colocalize with KLP-16 in cell bodies (Fig. 5D). These results suggest that the UNC-16–kinesin-1 complex regulates localization of dynein in sublateral neurons as well as touch sensory neurons.
Figure 5.

Localization of DLI-1 in sublateral motor neurons. A, Schematic image of sublateral motor neurons. SIB, SID, SMB, and SMD neurons are indicated by green lines. B, Localization of YFP::DLI-1 and CFP::EBP-1 in the nerve processes of sublateral neurons. The merged image is also indicated. The arrowheads indicate the merged sites. Scale bar, 5 μm. C, Localization of GFP::DLI-1 in the sublateral neurons. The left and right panels show cell bodies and nerve processes, respectively. The arrowheads indicate the accumulated positions of GFP::DLI-1 in cell bodies. Scale bar, 5 μm. D, Localization of YFP::DLI-1 and CFP::KLP-16 in the cell bodies of sublateral neurons in unc-116(e2281) mutants. The arrowheads indicate the merged sites.
Figure 6.

Localization of APL-1 in sublateral motor neurons. A, C, Localization of APL-1::YFP and CFP::EBP-1 in the sublateral neurons of wild-type (A) and unc-116(e2281) (C) worms. The merged images in each animal are also indicated. The arrowheads indicate the merged sites. The right panels show magnified images of merged sites in nerve processes. Anterior is to the left in panels. Scale bars, 1 μm. B, Localization of APL-1::YFP and CFP::KLP-16 in sublateral neurons of wild-type worms.
APL-1::YFP showed a localization pattern in sublateral neurons similar to that of APL-1::GFP (Fig. 6A). We could detect partial colocalization or close apposition of APL-1::YFP with CFP::EBP-1, but not with CFP::KLP-16, along the nerve processes of sublateral neurons (Fig. 6A,B). This suggests that APL-1 is transported to and accumulates around the microtubule plus ends. We tested the effect of kinesin-1 mutations on localization of APL-1 in sublateral neurons. In unc-116(rh24) mutants defective in the heavy chain of kinesin-1, the fluorescent intensity of APL-1::GFP puncta was significantly decreased compared with that of wild-type animals (Fig. 7A), whereas the numbers of puncta per length were almost the same (Fig. 7B). This is consistent with the possibility that kinesin-1 is required for anterograde transport of APL-1. However, in unc-116(e2281) mutants carrying a weak allele generated by a transposon insertion at the C-terminal domain of UNC-116, APL-1::YFP localized as puncta along nerve processes and partially colocalized with CFP::EBP-1, similar to that in the wild-type animals (Fig. 6C).
Figure 7.

Effects of the unc-116 mutation on APL-1. A, Relative fluorescent intensities of APL-1::GFP per micrometer along the lateral processes in wild-type and unc-116(rh24) mutant worms. B, Numbers of accumulated spots of APL-1::GFP per micrometer along the lateral processes in wild-type and unc-116(rh24) mutant worms.
To confirm that the unc-116(e2281) mutation does not affect APL-1 anterograde transport, we examined the transport of APL-1::GFP in sublateral neurons by real-time fluorescent imaging. In wild-type worms, vesicles carrying APL-1::GFP were observed as small dots that transported from the cell body to the distal nerve process (anterograde transport) as well as from the distal nerve process to the cell body (retrograde transport) (Fig. 8A,B; supplemental Movie S1, available at www.jneurosci.org as supplemental material). Approximately 52% of the moving vesicles were transported in the anterograde direction, whereas 48% were transported in the retrograde direction (Fig. 8C). The average velocity of anterograde transport was 1.1 μm/s and that of retrograde transport was 1.6 μm/s. In unc-116(e2281) mutants, anterograde transport of APL-1::GFP vesicle appeared normal, with an average velocity of 1.2 μm/s. Unexpectedly, however, retrograde transport was never observed in unc-116 mutants (Fig. 8A–C; supplemental Movie S2, available at www.jneurosci.org as supplemental material). Introduction of the unc-116 gene as an extrachromosomal array significantly rescued the defect in retrograde transport of APL-1::GFP observed in unc-116(e2281) mutants (Fig. 8A). Thus, C. elegans kinesin-1 is required for both anterograde and retrograde transport of APL-1 along the nerve processes.
Figure 8.

Movement of APL-1 in sublateral motor neurons. A, Time-lapse images of APL-1::GFP fluorescence along the sublateral neurons of worms. Each photograph was taken at 0.8 s intervals. The red and pale blue arrowheads indicate APL-1::GFP vesicles moving toward neurite endings (anterograde) and cell bodies (retrograde), respectively. Scale bar, 2.5 μm. B, Kymographs for APL-1::GFP movements along the lateral processes. The horizontal arrow represents 10 μm. The vertical arrow represents 8.3 s. Schematic diagrams are shown below. The red and blue lines represent anterograde and retrograde movements, respectively. C, Velocity profiles of moving vesicles containing APL-1::GFP in wild-type, unc-116(e2281), unc-16(e109), dli-1(ku266), and dhc-1(or195ts) worms. Moving structures were tracked, and the velocities were calculated as described in Materials and Methods.
We examined whether dynein is involved in the axonal transport of APL-1. In dli-1(ku266) mutants defective in dynein light intermediate chain, retrograde transport of APL-1::GFP was greatly impaired, whereas its anterograde transport was normal (Fig. 8C). We studied the phenotype of APL-1::GFP transport in mutants defective in the dhc-1 gene encoding the dynein heavy chain. We found that the temperature-sensitive dhc-1(or195ts) mutation affected retrograde but not anterograde transport of APL-1 when animals were cultured at the semipermissive temperature (Fig. 8C). Thus, cytoplasmic dynein mediates the retrograde transport of APL-1.
The above results showed that kinesin-1 mediates the anterograde transport of dynein via its interaction with UNC-16. This raised the possibility that kinesin-1 is indirectly required for APL-1 retrograde transport by controlling anterograde transport of dynein, which in turn mediates APL-1 retrograde transport. Consistent with this possibility, we found that unc-16(e109) mutants were defective in retrograde but not anterograde transport of APL-1::GFP (Fig. 8C). These results support the possibility that UNC-16 acts as an adaptor that links kinesin-1 and dynein for anterograde transport of dynein, which mediates APL-1 retrograde transport.
Discussion
Recent studies have begun to elucidate how molecular motors are attached to the cargos they transport (Vale, 2003; Hirokawa and Takemura, 2005). A general picture is emerging showing that kinesin family members are typically linked to their cargos by adaptor proteins. In the case of kinesin-1, the kinesin light chain KLC subunit binds directly to JIP3. As an adaptor/scaffold protein, JIP3 protein functions to link kinesin-1 to vesicular cargos. Our genetic analysis presented here suggests that kinesin-1 and UNC-16/JIP3 are required for retrograde transport of various axonal proteins in C. elegans. We observe that different domains of UNC-16 are responsible for its association with KLC-2 kinesin light chain and DLI-1 dynein light intermediate chain. KLC-2 by itself can form a weak complex with DLI-1, but this complex is enhanced in the presence of UNC-16. Our results also demonstrate that kinesin-1 and UNC-16 are required for the localization of cytoplasmic dynein to microtubule plus ends. These results suggest that the interaction between UNC-16 and KLC-2 may trigger the loading of the dynein complex to the kinesin-1 motor at microtubule minus ends in cell bodies. Thus, the UNC-16–kinesin-1 complex acts as a motor for anterograde transport of the dynein complex in neurons. We also found accumulations of DLI-1 at microtubule plus ends in neurons, consistent with previous studies that microtubule plus ends provide a reservoir of inactive dynein motors for retrograde endosome transport in other organisms (Han et al., 2001; Lee et al., 2003; Sheeman et al., 2003; Zhang et al., 2003).
Based on the data presented here, we propose the following model showing how kinesin-1, UNC-16, and dynein regulate the axonal transport of SV proteins such as SNB-1. In this model, UNC-104 transports SNB-1 in the anterograde direction and kinesin-1 delivers the dynein complex to microtubule plus ends, using UNC-16 as a scaffold. Once at the microtubule plus ends, SNB-1 is loaded onto dynein for retrograde transport. Thus, the kinesin-1–UNC-16 complex is required for dynein-dependent retrograde transport of SNB-1. KLC-2 might regulate the dynein-mediated retrograde transport of various axonal and synaptic cargos by recruiting adaptor proteins such as UNC-16.
In Drosophila, mutations in kinesin-1, dynein, and sunday driver all exhibit a similar posterior-paralysis phenotype caused by defects in trafficking (Martin et al., 1999; Bowman et al., 2000). In addition, trans-heterozygous mutants of kinesin-1 and dynein exhibit a similar phenotype, suggesting that they function cooperatively in the transport processes (Martin et al., 1999). Therefore, it is likely that the relationship among kinesin, dynein, and UNC-16/JIP3 may be conserved beyond this species. UNC-16/JIP3 also functions as a scaffold protein for a JNK signaling module (Byrd et al., 2001; Verhey et al., 2001). Drosophila Sunday Driver/JIP3 interacts with kinesin-1 and the dynactin complex and retrogradely transports activated JNK from the site of nerve injury (Cavalli et al., 2005). However, C. elegans JNK does not play a major role in UNC-16-regulated retrograde transport of SNB-1 (M. Arimoto, K. Matsumoto, and N. Hisamoto, unpublished results). In PC12 cells, JIP3/JSAP1 transport to the growth cone neurites is apparently independent of JNK signaling (Sato et al., 2004). Thus, JIP3 plays roles both in the JNK MAP (mitogen-activated protein) kinase signaling pathway and independent of JNK.
In mammals, there are several observations indicating an association between dynein and kinesin-1 in axonal transport (Ligon et al., 2004; Colin et al., 2008; Yamada et al., 2008). Most recently, Yamada et al. (2010) demonstrated that, in cultured neurons, kinesin-1 binds and transports “transportable microtubules” (tMTs) via mammalian nuclear distribution gene C homolog (mNUDC). These tMTs act as carriers to transport cytoplasmic dynein. Thus, kinesin-1 transports the tMTs–dynein multimeric complex, with mNUDC functioning as an adaptor between kinesin-1 and the tMTs–dynein complex (Yamada et al., 2010). Although not shown in their study, it is possible that kinesin-1 delivers dynein to the microtubule plus ends where dynein is used for retrograde transport of other cargos. These results suggest that neurons may use one of two alternative systems depending on the circumstances for dynein transport: (1) when the axon requires large amounts of microtubules to grow, dynein transport with tMTs is preferentially used; (2) in mature neurons with established structure, UNC-16-dependent transport of dynein is preferentially used.
Our results also show that kinesin-1, UNC-16, and dynein are required for retrograde transport of APL-1. A recent genetic study in humans has revealed that duplication of the APP gene locus causes early-onset AD (Rovelet-Lecrux et al., 2006), suggesting that the amount of APP is a critical factor for the pathogenesis of AD. In addition, dynein-dependent retrograde transport of APP is an important process in the control of APP protein levels in mammalian neurons (Kimura et al., 2007). Based on these observations, it is tempting to speculate that human JIP3/UNC-16 and kinesin also mediate retrograde transport required for the control of APP amounts. Additional studies should contribute to our understanding of the function of these molecules in the control of APP metabolism.
Footnotes
This work was supported by grants from Ministry of Education, Culture and Science of Japan, The Sumitomo Foundation, and The Takeda Foundation (K.M., N.H.). B.C.C. is supported by a fellowship from the Council of Scientific and Industrial Research, and research in the laboratory of S.P.K. is supported by the Department of Biotechnology, Ministry of Science and Technology. We thank A. Fire, Y. Kohara, K. Gull, I. Mori, and Caenorhabditis Genetic Center for materials.
References
- Araki Y, Kawano T, Taru H, Saito Y, Wada S, Miyamoto K, Kobayashi H, Ishikawa HO, Ohsugi Y, Yamamoto T, Matsuno K, Kinjo M, Suzuki T. The novel cargo Alcadein induces vesicle association of kinesin-1 motor components and activates axonal transport. EMBO J. 2007;26:1475–1486. doi: 10.1038/sj.emboj.7601609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowman AB, Kamal A, Ritchings BW, Philp AV, McGrail M, Gindhart JG, Goldstein LS. Kinesin-dependent axonal transport is mediated by the sunday driver (SYD) protein. Cell. 2000;103:583–594. doi: 10.1016/s0092-8674(00)00162-8. [DOI] [PubMed] [Google Scholar]
- Byrd DT, Kawasaki M, Walcoff M, Hisamoto N, Matsumoto K, Jin Y. UNC-16, a JNK-signaling scaffold protein, regulates vesicle transport in C. elegans. Neuron. 2001;32:787–800. doi: 10.1016/s0896-6273(01)00532-3. [DOI] [PubMed] [Google Scholar]
- Cavalli V, Kujala P, Klumperman J, Goldstein LS. Sunday Driver links axonal transport to damage signaling. J Cell Biol. 2005;168:775–787. doi: 10.1083/jcb.200410136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caviston JP, Holzbaur EL. Microtubule motors at the intersection of trafficking and transport. Trends Cell Biol. 2006;16:530–537. doi: 10.1016/j.tcb.2006.08.002. [DOI] [PubMed] [Google Scholar]
- Chalfie M. Touch receptor development and function in Caenorhabditis elegans. J Neurobiol. 1993;24:1433–1441. doi: 10.1002/neu.480241013. [DOI] [PubMed] [Google Scholar]
- Colin E, Zala D, Liot G, Rangone H, Borrell-Pagès M, Li XJ, Saudou F, Humbert S. Huntingtin phosphorylation acts as a molecular switch for anterograde/retrograde transport in neurons. EMBO J. 2008;27:2124–2134. doi: 10.1038/emboj.2008.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daigle I, Li C. apl-1, a Caenorhabditis elegans gene encoding a protein related to the human β-amyloid protein precursor. Proc Natl Acad Sci U S A. 1993;90:12045–12049. doi: 10.1073/pnas.90.24.12045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duerr JS, Han HP, Fields SD, Rand JB. Identification of major classes of cholinergic neurons in the nematode Caenorhabditis elegans. J Comp Neurol. 2008;506:398–408. doi: 10.1002/cne.21551. [DOI] [PubMed] [Google Scholar]
- Gindhart JG, Jr, Desai CJ, Beushausen S, Zinn K, Goldstein LS. Kinesin light chains are essential for axonal transport in Drosophila. J Cell Biol. 1998;141:443–454. doi: 10.1083/jcb.141.2.443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gindhart JG, Chen J, Faulkner M, Gandhi R, Doerner K, Wisniewski T, Nandlestadt A. The kinesin-associated protein UNC-76 is required for axonal transport in the Drosophila nervous system. Mol Biol Cell. 2003;14:3356–3365. doi: 10.1091/mbc.E02-12-0800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guzik BW, Goldstein LS. Microtubule-dependent transport in neurons: steps towards an understanding of regulation, function and dysfunction. Curr Opin Cell Biol. 2004;16:443–450. doi: 10.1016/j.ceb.2004.06.002. [DOI] [PubMed] [Google Scholar]
- Hall DH, Hedgecock EM. Kinesin-related gene unc-104 is required for axonal transport of synaptic vesicles in C. elegans. Cell. 1991;65:837–847. doi: 10.1016/0092-8674(91)90391-b. [DOI] [PubMed] [Google Scholar]
- Hall DH, Plenefisch J, Hedgecock EM. Ultrastructural abnormalities of kinesin mutant unc-116. J Cell Biol. 1991;115:389A. [Google Scholar]
- Han G, Liu B, Zhang J, Zuo W, Morris NR, Xiang X. The Aspergillus cytoplasmic dynein heavy chain and NUDF localize to microtubule ends and affect microtubule dynamics. Curr Biol. 2001;11:719–724. doi: 10.1016/s0960-9822(01)00200-7. [DOI] [PubMed] [Google Scholar]
- Hirokawa N, Takemura R. Molecular motors and mechanisms of directional transport in neurons. Nat Rev Neurosci. 2005;6:201–214. doi: 10.1038/nrn1624. [DOI] [PubMed] [Google Scholar]
- Hornsten A, Lieberthal J, Fadia S, Malins R, Ha L, Xu X, Daigle I, Markowitz M, O'Connor G, Plasterk R, Li C. APL-1, a Caenorhabditis elegans protein related to the human β-amyloid precursor protein, is essential for viability. Proc Natl Acad Sci U S A. 2007;104:1971–1976. doi: 10.1073/pnas.0603997104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurd DD, Saxton WM. Kinesin mutations cause motor neuron disease phenotypes by disrupting fast axonal transport in Drosophila. Genetics. 1996;144:1075–1085. doi: 10.1093/genetics/144.3.1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawasaki M, Hisamoto N, Iino Y, Yamamoto M, Ninomiya-Tsuji J, Matsumoto K. A Caenorhabditis elegans JNK signal transduction pathway regulates coordinated movement via type-D GABAergic motor neurons. EMBO J. 1999;18:3604–3615. doi: 10.1093/emboj/18.13.3604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelkar N, Delmotte MH, Weston CR, Barrett T, Sheppard BJ, Flavell RA, Davis RJ. Morphogenesis of the telencephalic commissure requires scaffold protein JNK-interacting protein 3 (JIP3) Proc Natl Acad Sci U S A. 2003;100:9843–9848. doi: 10.1073/pnas.1733944100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura N, Imamura O, Ono F, Terao K. Aging attenuates dynactin-dynein interaction: down-regulation of dynein causes accumulation of endogenous tau and amyloid precursor protein in human neuroblastoma cells. J Neurosci Res. 2007;85:2909–2916. doi: 10.1002/jnr.21408. [DOI] [PubMed] [Google Scholar]
- Koushika SP, Schaefer AM, Vincent R, Willis JH, Bowerman B, Nonet ML. Mutations in Caenorhabditis elegans cytoplasmic dynein components reveal specificity of neuronal retrograde cargo. J Neurosci. 2004;24:3907–3916. doi: 10.1523/JNEUROSCI.5039-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee WL, Oberle JR, Cooper JA. The role of the lissencephaly protein Pac1 during nuclear migration in budding yeast. J Cell Biol. 2003;160:355–364. doi: 10.1083/jcb.200209022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ligon LA, Tokito M, Finklestein JM, Grossman FE, Holzbaur EL. A direct interaction between cytoplasmic dynein and kinesin I may coordinate motor activity. J Biol Chem. 2004;279:19201–19208. doi: 10.1074/jbc.M313472200. [DOI] [PubMed] [Google Scholar]
- Martin M, Iyadurai SJ, Gassman A, Gindhart JG, Jr, Hays TS, Saxton WM. Cytoplasmic dynein, the dynactin complex, and kinesin are interdependent and essential for fast axonal transport. Mol Biol Cell. 1999;10:3717–3728. doi: 10.1091/mbc.10.11.3717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motegi F, Velarde NV, Piano F, Sugimoto A. Two phases of astral microtubule activity during cytokinesis in C. elegans embryos. Dev Cell. 2006;10:509–520. doi: 10.1016/j.devcel.2006.03.001. [DOI] [PubMed] [Google Scholar]
- Muresan Z, Muresan V. Coordinated transport of phosphorylated amyloid-beta precursor protein and c-Jun NH2-terminal kinase-interacting protein-1. J Cell Biol. 2005;171:615–625. doi: 10.1083/jcb.200502043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nonet ML. Visualization of presynaptic terminal specializations in live C. elegans using synaptic vesicle protein-GFP fusions. J Neurosci Methods. 1999;89:33–40. doi: 10.1016/s0165-0270(99)00031-x. [DOI] [PubMed] [Google Scholar]
- Otsuka AJ, Jeyaprakash A, García-Añoveros J, Tang LZ, Fisk G, Hartshorne T, Franco R, Born T. The C. elegans unc-104 gene encodes a putative kinesin heavy chain-like protein. Neuron. 1991;6:113–122. doi: 10.1016/0896-6273(91)90126-k. [DOI] [PubMed] [Google Scholar]
- Robin G, DeBonis S, Dornier A, Cappello G, Ebel C, Wade RH, Thierry-Mieg D, Kozielski F. Essential kinesins: characterization of Caenorhabditis elegans KLP-15. Biochemistry. 2005;44:6526–6536. doi: 10.1021/bi048157h. [DOI] [PubMed] [Google Scholar]
- Rovelet-Lecrux A, Hannequin D, Raux G, Le Meur N, Laquerrière A, Vital A, Dumanchin C, Feuillette S, Brice A, Vercelletto M, Dubas F, Frebourg T, Campion D. APP locus duplication causes autosomal dominant early-onset Alzheimer disease with cerebral amyloid angiopathy. Nat Genet. 2006;38:24–26. doi: 10.1038/ng1718. [DOI] [PubMed] [Google Scholar]
- Sakamoto R, Byrd DT, Brown HM, Hisamoto N, Matsumoto K, Jin Y. The Caenorhabditis elegans UNC-14 RUN domain protein binds to the kinesin-1 and UNC-16 complex and regulates synaptic vesicle localization. Mol Biol Cell. 2005;16:483–496. doi: 10.1091/mbc.E04-07-0553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato S, Ito M, Ito T, Yoshioka K. Scaffold protein JSAP1 is transported to growth cones of neurites independent of JNK signaling pathways in PC12h cells. Gene. 2004;329:51–60. doi: 10.1016/j.gene.2003.12.034. [DOI] [PubMed] [Google Scholar]
- Sheeman B, Carvalho P, Sagot I, Geiser J, Kho D, Hoyt MA, Pellman D. Determinants of S. cerevisiae dynein localization and activation. Implications for the mechanism of spindle positioning. Curr Biol. 2003;13:364–372. doi: 10.1016/s0960-9822(03)00013-7. [DOI] [PubMed] [Google Scholar]
- Vale RD. The molecular motor toolbox for intracellular transport. Cell. 2003;112:467–480. doi: 10.1016/s0092-8674(03)00111-9. [DOI] [PubMed] [Google Scholar]
- Verhey KJ, Meyer D, Deehan R, Blenis J, Schnapp BJ, Rapoport TA, Margolis B. Cargo of kinesin identified as JIP scaffolding proteins and associated signaling molecules. J Cell Biol. 2001;152:959–970. doi: 10.1083/jcb.152.5.959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada M, Toba S, Yoshida Y, Haratani K, Mori D, Yano Y, Mimori-Kiyosue Y, Nakamura T, Itoh K, Fushiki S, Setou M, Wynshaw-Boris A, Torisawa T, Toyoshima YY, Hirotsune S. LIS1 and NDEL1 coordinate the plus-end-directed transport of cytoplasmic dynein. EMBO J. 2008;27:2471–2483. doi: 10.1038/emboj.2008.182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada M, Toba S, Takitoh T, Yoshida Y, Mori D, Nakamura T, Iwane AH, Yanagida T, Imai H, Yu-Lee LY, Schroer T, Wynshaw-Boris A, Hirotsune S. mNUDC is required for plus-end-directed transport of cytoplasmic dynein and dynactins by kinesin-1. EMBO J. 2010;29:517–531. doi: 10.1038/emboj.2009.378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Li S, Fischer R, Xiang X. Accumulation of cytoplasmic dynein and dynactin at microtubule plus ends in Aspergillus nidulans is kinesin dependent. Mol Biol Cell. 2003;14:1479–1488. doi: 10.1091/mbc.E02-08-0516. [DOI] [PMC free article] [PubMed] [Google Scholar]