

Abstract
Presynaptic kainate receptors (KARs) exert a modulatory action on transmitter release. This effect can be switched from facilitation to inhibition by an increased concentration of KAR agonists. We here report that activation of presynaptic GluK1-containing KARs facilitates GABA release on oxytocin and vasopressin neurons in the supraoptic nucleus of the hypothalamus. Increase in ambient levels of glutamate associated with the physiological reduction of astrocytic coverage of oxytocin neurons in lactating rats switches this KAR-mediated facilitation to inhibition of GABAergic transmission. This effect was reproduced in both oxytocin and vasopressin neurons of virgin rats when glutamate transporters were blocked pharmacologically, thereby establishing that enhanced levels of extracellular glutamate induce the switch in KAR-mediated action. The facilitation of GABA release was inhibited with philanthotoxin, a Ca2+-permeable KAR antagonist, suggesting that this effect was associated with an ionotropic mode of action. Conversely, KAR-mediated inhibition was compromised in the presence of U73122, a phospholipase C inhibitor, in agreement with the involvement of a metabotropic pathway. We thus reveal that physiological astrocytic plasticity modifies the mode of action of presynaptic KARs, thereby inversing their coupling with GABA release.
Introduction
Kainate receptors (KARs) are ubiquitous ionotropic glutamate receptors in the CNS (Wisden and Seeburg, 1993). They are encoded by five genes producing GluK1, GluK2, GluK3, GluK4 and GluK5 subunits (novel nomenclature) (Collingridge et al., 2009) and assemble in tetrameric stoichiometry (Bettler and Mulle, 1995; Chittajallu et al., 1999). GluK1–GluK3 can form functional homomeric or heteromeric receptors whereas GluK4 and GluK5 participate only in heteromeric receptors, partnering with any of the GluK1-3 subunits. Activation of presynaptic KARs can regulate the release of glutamate or GABA in a positive or negative manner, thereby playing a key role in modulating the excitability of neuronal networks (Pinheiro and Mulle, 2006). Moreover, a bidirectional modulation of transmitter release by KAR activation has been described in the hippocampus (Jiang et al., 2001; Schmitz et al., 2001), cerebellum (Delaney and Jahr, 2002), amygdala (Braga et al., 2003), and spinal cord (Kerchner et al., 2001; Youn and Randic, 2004). In these areas, an experimental increase in the concentration of KAR agonist resulted in a switch from facilitation to inhibition of transmitter release. Nevertheless, the mechanisms underlying such facilitation and inhibition and their physiological and/or pathological relevance remain elusive. To address these issues, we examined the supraoptic nucleus (SON) of the hypothalamus in which perisynaptic glutamate levels change significantly under physiological conditions.
The SON is part of the hypothalamo-neurohypophysial system (HNS) composed of magnocellular neurons synthesizing either oxytocin (OT) or vasopressin (VP), two neuropeptides essential to the central and peripheral regulation of reproductive functions and body fluid homeostasis, respectively. During stimulated conditions of neurosecretion such as lactation, parturition, and chronic dehydration, the HNS undergoes a remarkable anatomical remodeling that is notably characterized by a pronounced reduction of the astrocytic coverage of OT neurons (Theodosis, 2002). One consequence of such a glial withdrawal is a deficiency in glutamate clearance at perisynaptic sites resulting in increased levels of extracellular glutamate (Oliet et al., 2001; Boudaba et al., 2003).
Magnocellular neuron activity controls the release in the blood stream of OT and VP from axon terminals located in the neurohypophysis (Poulain and Wakerley, 1982). This activity itself depends on excitatory and inhibitory synaptic inputs originating from different brain regions (Leng et al., 1999). Among these, GABA inputs are of particular importance since they represent the main synaptic input to SON neurons (Gies and Theodosis, 1994). Moreover, numerous electrophysiological studies have highlighted an important role of the inhibitory control on the activity of magnocellular neurons (Armstrong, 1995). However, although the release of GABA can be modulated by a variety of presynaptic receptors in the SON (Shibuya et al., 2000), the presence and eventual roles of KARs has not been yet investigated.
In the present study, we first established the presence of functional KARs on GABAergic terminals in the SON. We then found that physiological enhancement of extracellular levels of glutamate resulting from glial withdrawal could lead to a switch in KAR activity. Finally, we identified the underlying mechanisms for KAR facilitation and inhibition.
Materials and Methods
Slice preparation.
Experiments were performed on acute hypothalamic slices obtained from two groups of Wistar rats, one composed of 2–3 month-old unstimulated rats [normally hydrated males (n = 80) and virgin females (n = 25)] and the other of 4 month-old females on day 8–15 of lactation (n = 29). The rats were anesthetized with isoflurane and decapitated. The brain was then quickly removed and placed in ice-cold artificial CSF (ACSF) saturated with 95% O2 and 5% CO2. Hypothalamic slices (300 μm) including the SON were obtained and allowed to recover for at least 1 h at 33°C in a submerged chamber containing ACSF before recording. After 30 min recovery at room temperature, one slice was transferred and submerged in a recording chamber where it was continuously perfused (1–2 ml/min) with ACSF composed of (in mm): NaCl, 123; KCl, 2.5; Na2HPO4, 1; NaHCO3, 26.2; MgCl2, 1.3; CaCl2, 2.5; and glucose, 10 (pH 7.4; 295–300 mOsm · kg−1). All experiments were conducted with respect to European and French directives on animal experimentation (authorization no. 33 0004).
Patch-clamp recording.
Magnocellular neurons in the SON were identified visually using infrared differential interference contrast microscopy (Olympus BX50). The patch-clamp recording pipettes (3–5 MOhm) were filled with a solution containing (in mm): CsCl, 130; NaCl, 10; HEPES, 10; QX-314, 5; EGTA, 1; and CaCl2, 0.1 (adjusted to pH 7.1–7.3 with CsOH; 292–296 mOsm · kg−1). Biocytin (0.2%) was included in the pipette solution for post hoc identification of the recorded cells. Membrane currents were recorded using an Axopatch-1D amplifier (Molecular Devices); signals were filtered at 2 kHz and digitized at 5 kHz via a DigiData 1440A interface (Molecular Devices). Series resistance (6–15 MOhm) was monitored online and cells were excluded from data analysis if a >20% change occurred during the course of the experiment. All cells were held at −70 mV in voltage-clamp mode. Spontaneous unitary synaptic currents (miniatures) obtained in the presence of tetrodotoxin (TTX, 0.5 μm) were stored on videotape via a pulse-code modulator (Neurodata), detected, and analyzed off-line using Axograph (Molecular Devices). At least 200 events were analyzed for each cell in each condition. To evoke IPSCs, a glass-stimulating electrode connected to an isolated stimulator (AMPI) was placed in the hypothalamic region dorsomedial to the SON (Kombian et al., 1996). Synaptic responses were evoked at 0.05 Hz, using square pulses of 0.1 ms duration, and analyzed online using a data acquisition and analysis software (Molecular Devices). To study the paired-pulse facilitation ratio (PPR), two synaptic responses (P1 and P2) were evoked by two stimuli given at 50–60 ms intervals. PPR was expressed as the ratio of the amplitude of the second synaptic response over the first synaptic response (P2/P1).
The data, reported as means ± SE, were compared statistically with the paired or unpaired Student's t test. Amplitude and frequency distributions of the miniatures were compared using the non-parametric Kolmogorov–Smirnov test. Significance was assessed at p < 0.05.
Drugs.
All drugs were bath-applied. Appropriate stock solutions were made and diluted with ACSF just before application. QX-314 chloride, a voltage-gated sodium channel blocker (Alomone Labs) was diluted directly in the patch solution. The drugs used were the following: biocytin, glutamic-pyruvic transaminase (GPT), Philanthotoxin 433 Tris (trifluoroacetate) salt (PhTx), 2,3-dihydroxy-6-nitro-7 sulfonyl-benzo[f] quinoxaline (NBQX), 6,7-dinitroquinoxaline-2,3-dione (DNQX), TTX, 2-amino-2-methyl-4-phosphonobutanoic acid (MAP4), (RS)-2-amino-3-(3-hydroxy-5-tert-butylisoxazol-4-yl) propanoic acid (ATPA), (S)-1-(2-amino-2-carboxyethyl)-3-(2-carboxybenzyl) pyrimidine-2,4-dione (UBP302), d,l-threo-b-benzyloxyaspartic acid (d,l-TBOA), 1-[6-[[(17b)-3- methoxyestra-1,3,5(10)-trien-17-yl] amino]hexyl]-1H-pyrrole-2,5-dione (U73122), 3-(4-[4-chlorophenyl]piperazin-1-yl)-methyl-1H-pyrrolo[2,3- b]pyridine trihydrochloride (L-745,870 trihydrochloride), [S-(R*,R*)][-[3-[[1-(3,4-dichlorophenyl)ethyl]amino]-2-hydroxypropyl](cyclohexylmethyl) phosphinic acid (CGP 54626 hydrochloride), 17a-hydroxyyohimban-16a-carboxylic acid methyl ester hydrochloride (Yohimbine hydrochloride), N,2,3,3-tetramethylbicyclo[2.2.1]heptan-2-amine hydrochloride (Mecamylamine hydrochloride), N-(piperidin-1-yl)-5-(4- iodophenyl)-1-(2,4-dichlorophenyl)-4-methyl-1H-pyrazole-3-carboxamide (AM 251), d-(-)-2-amino-5-phosphonopentanoic acid (d-AP5), 2S)-2-amino-2-[(1S,2S)-2-carboxycycloprop-1-yl]-3-(xan th-9-yl) propanoic acid (LY 341495), 8-cyclopentyl-1,3-dipropylxanthine (DPCPX). Apart from biocytin, PhTx, and GPT, which were obtained from Sigma; all the drugs were obtained from Tocris Bioscience. As described previously (Min et al., 1998) GPT (from porcine heart 115 kDa dimer) was dialyzed for 3 h with a 10 kDa cutoff membrane (Slide-A-Lyzer, Pierce Chemical) before the experiments.
Neuron identification.
After recording, magnocellular neurons filled with biocytin (0.2%) were identified further by immunocytochemistry. At the end of the recording period, slices were fixed by immersion in 4% paraformaldehyde and 0.15% picric acid (2 h, room temperature). The biocytin was visualized after incubation (overnight at 4°C) with streptavidin-conjugated 7-amino-4-methyl-coumarin-3-acetic acid (AMCA; AbCys S.A.) diluted to 1:200. Sections containing biocytin-positive neurons were then incubated for 7 d (at 4°C) in a mixture of a monoclonal mouse antibody raised against VP [diluted to 1:20,000; gift from V. Geenen, University of Liege, Liege, Belgium; for production and specificity, see the study by Moll et al. (1988)] and a polyclonal rabbit serum raised against OT [diluted to 1:4000; gift from T. Higuchi, University of Fukui, Fukui, Japan; for production and specificity, see the study by Higuchi et al., (1983)]. Immunoreactivities were then revealed by incubation in a mixture with goat anti-mouse immunoglobulins conjugated to fluorescein isothiocyanate (FITC; AbCys S.A.) (diluted to 1:1600) and goat anti-rabbit immunoglobulins conjugated to Texas Red (AbCys S.A.) (diluted to 1:500, overnight at 4°C). All antibodies were diluted in a solution of TBS containing 0.25% BSA and 2% Triton X-100. Controls including omission of primary antibodies and incubation with inappropriate secondary antibodies yielded no specific immunolabeling. Slices were mounted in Vectashield (AbCys S.A.) and examined with a confocal microscope (Leica DMR TCS SP2 AOBS on an upright stand, using objective HCX Plan Apo CS 63× numerical aperture 1.40). The lasers used were Argon (488 nm), Green Helium-Neon (543 nm), and a diode laser (405 nm). Images were acquired using Leica TCS software with a sequential mode to avoid interference between each channel. All confocal images are projections of 20 consecutive optical sections (0.5 μm). Contrast/brightness enhancement was done in parallel on obtained stacks using Adobe Photoshop 8.0.
Results
KARs are positively coupled to GABA release in the SON
Earlier studies described the action of various presynaptic receptors on GABA terminals in the SON (Kabashima et al., 1997; Schrader and Tasker, 1997; Wang et al., 1998; Oliet and Poulain, 1999; Li et al., 2001; Stern and Ludwig, 2001; Azdad et al., 2003) but no particular attention was paid to that of KARs. For this purpose, we performed recordings on miniature IPSCs (mIPSCs) in slices from animals under basal, nonstimulated conditions of neurosecretion, i.e., normally hydrated male and virgin female rats. Results between male and virgin female rats were statistically similar and were therefore pooled together. The mean amplitude and frequency of mIPSCs varied a lot from one cell to another (from 0.25 to 6.51 Hz and from 115 to 338 pA, respectively). Part of this variability was due to differences between OT and VP neurons as previously reported (Li et al., 2007) as well as differences associated with the physiological state of the animals (Brussaard et al., 1999).
The recordings were made in the presence of TTX and NBQX (1 μm) to block action potentials and AMPA receptor-mediated currents, respectively. Application of kainate (KA, 3 μm) induced a reversible increase in mIPSC activity (Fig. 1A) that was associated with a significant enhancement in the frequency (+38.31 ± 8.88%; n = 9; p < 0.05;) but not the amplitude (+2.57 ± 6.29%; n = 9; p > 0.05) of these unitary events (Fig. 1B). That only the frequency of mIPSCs was affected indicated that the KA-mediated action on GABAergic transmission occurred at a presynaptic locus.
Figure 1.
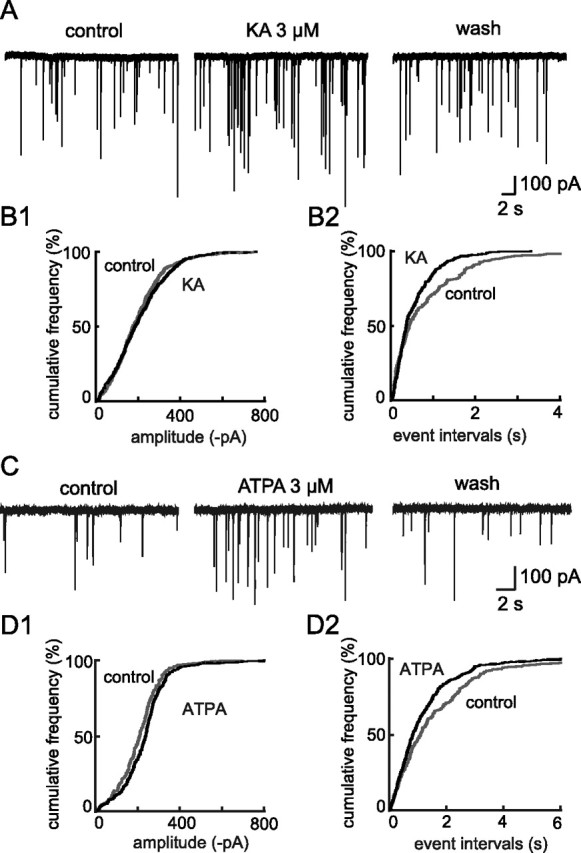
Presynaptic kainate receptors (KARs) are positively coupled to GABA release in the SON. A, C, Traces showing mIPSC activity recorded before (control), during and after (wash) bath application of the agonists KA (A) and ATPA, a selective agonist for GluK1-containing KARs (C). B, D, Cumulative plots of amplitude (B1 and D1) and event interval (B2 and D2) distributions before (gray thin line) and during (black thick line) applications of KA or ATPA on cells shown in A and C, respectively. Neither KA nor ATPA affected mIPSC amplitude, but each induced a leftward shift of the event interval distribution, reflecting an increase in mIPSC frequency and therefore a presynaptic locus of action.
To identify the KAR subtype(s) involved in this effect, we applied the GluK1-containing KARs selective agonist ATPA (3 μm) (Clarke et al., 1997; Bortolotto et al., 1999). ATPA induced an increase in mIPSC activity similar to that induced by KA (n = 9) (Fig. 1C), specifically increasing the frequency (+22.94 ± 2.19%; p < 0.05) but not the amplitude (−1.36 ± 6.67%; p > 0.05) (Fig. 1C,D). In the presence of UBP302 (10 μm), a selective antagonist of GluK1-containing KARs (More et al., 2004), the effect of ATPA on mIPSC frequency was completely abolished (−2.48 ± 3.75%; p > 0.05; n = 10) (Fig. 2A) whereas their amplitude remained unaffected (−7.05 ± 5.54%; n = 10; p > 0.05) (Fig. 2A). This confirmed that ATPA selectively activated GluK1-containing KARs and demonstrated the presence of functional presynaptic GluK1-containing KARs on GABAergic terminals.
Figure 2.
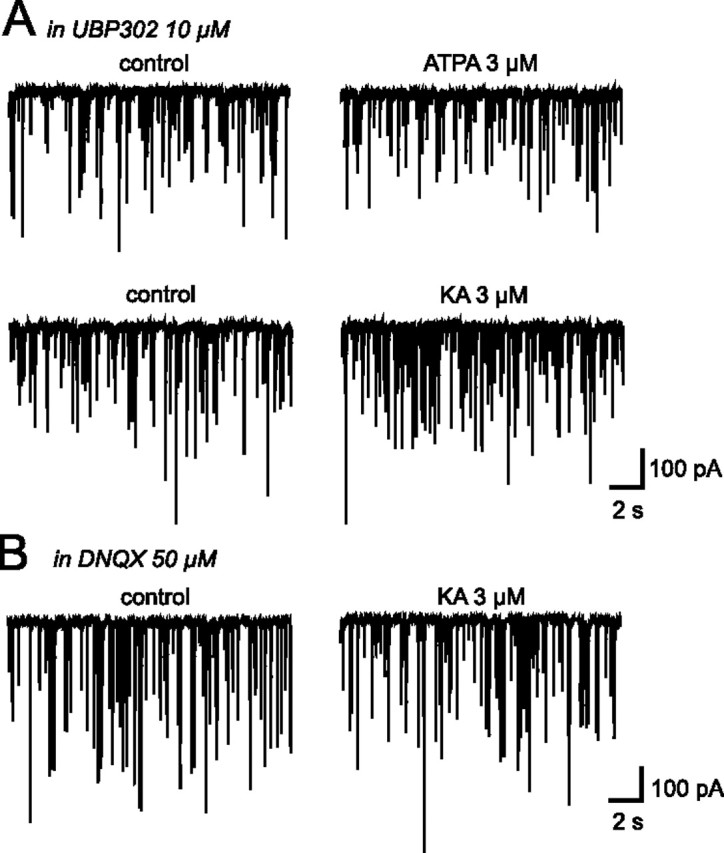
Existence of two different types of KARs on GABAergic synapses in the SON. A, Traces showing mIPSC activity recorded in the presence of UBP302, a selective GluK1-containing KARs antagonist, before and during bath application of ATPA and KA. B, Traces showing mIPSC activity recorded in the presence of a broad spectrum antagonist for AMPA/KA receptors, DNQX, before and during bath application of KA.
Interestingly, ATPA was less efficient than KA in increasing mIPSC frequency, which may reveal the presence of at least one other type of KARs on GABAergic terminals, in addition to GluK1-containing receptors. To test this possibility, we applied KA (3 μm) in the presence of UBP302 (10 μm). The increase in mIPSC frequency triggered by KA was only partially abolished (+21.30 ± 1.39%; p < 0.05; n = 6) (Fig. 2A), which made it highly probable that an additional KAR subtype is present on these inhibitory terminals. If this was the case, the remaining KA-induced response should have been completely blocked by a broad-spectrum AMPA/KA receptor inhibitor such as DNQX (50 μm). This was the case, since, in the presence of this antagonist (Fig. 2B), KA (3 μm) no longer affected mIPSC activity (−3.59 ± 5.57%; p > 0.05 for mIPSC frequency and −2.05 ± 3.22%; p > 0.05 for mIPSC amplitude; n = 12). Two different types of KARs are thus present on GABAergic terminals impinging on magnocellular neurons in the SON.
Ambient glutamate tonically activates presynaptic GluK1-containing KARs
The above experiments also revealed that UBP302 by itself had an effect on mIPSC activity. This effect was characterized by a reversible reduction in mIPSC frequency (−27.05 ± 2.41%; n = 13; p < 0.05, Fig. 3A) whereas mIPSC amplitude remained unaffected (−5.08 ± 3.55%; n = 13; p > 0.05). This result argues in favor of a tonic activation of presynaptic GluK1-containing KARs by ambient glutamate, activation responsible for a positive tone on GABA release. On the other hand, DNQX (50 μm) did not induce any change in mIPSC activity in the presence of UBP302 (+6.49 ± 8.67%; n = 12; p > 0.05 for mIPSC frequency and −7.97 ± 5.60%; n = 12; p > 0.05 for mIPSC amplitude) (Fig. 3B), indicating that only GluK1-containing KARs are activated by ambient glutamate.
Figure 3.
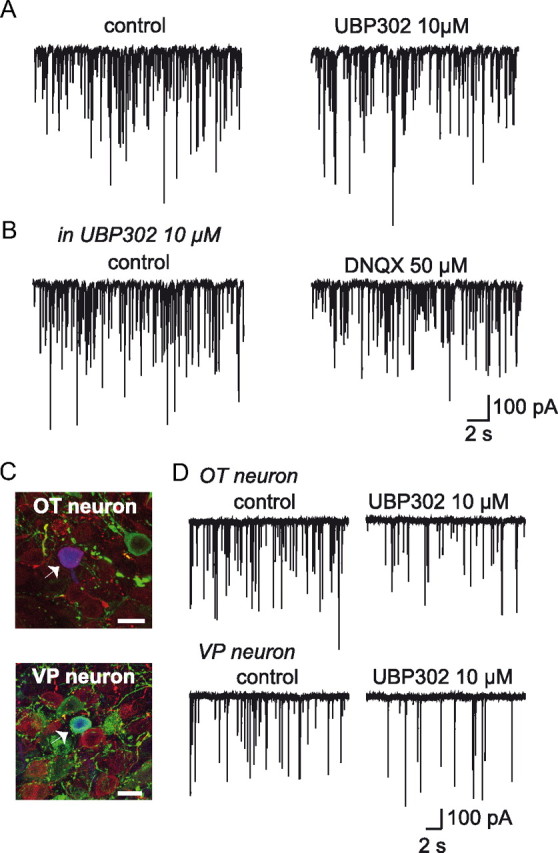
Activation of GluK1-containing KARs by ambient glutamate facilitates GABA release on OT and VP neurons. A, Traces showing mIPSC activity recorded before and during bath application of UBP302, a selective GluK1-containing KARs antagonist. B, Traces showing mIPSC activity recorded in the presence of UBP302 before and during bath application of DNQX. C, Recorded neurons (white arrowheads) displayed fluorescence for biocytin (blue) and immunoreactivity for either oxytocin (OT, red) (top) or vasopressin (VP, green) (bottom). All confocal images are projections of 20 consecutive optical sections (0.5 μm). Scale bar, 20 μm. D, Traces showing mIPSC activity in an OT and a VP neuron before and during bath application of UPB302.
The rat SON contains both OT and VP cells, in about equal proportions (Rhodes et al., 1981). To see whether UBP302 differentially affected OT and VP neurons, recorded neurons were identified post hoc using immunofluorescence with specific antibodies for either of the neuropeptides (Fig. 3C). As shown in Figure 3D, inhibition of GluK1-containing KARs induced a reversible reduction in mIPSC frequency in both OT (−21.63 ± 2.79%; n = 5; p < 0.05) and VP (−31.02 ± 3.84%; n = 6; p < 0.05) neurons. As expected, mIPSC amplitude remained unchanged in either type of neuron (−9.82 ± 4.69%; n = 5; p > 0.05 and −1.35 ± 6.33%; n = 6; p > 0.05 for OT and for VP neurons, respectively) (Fig. 3D). These results indicate that under unstimulated conditions of neurosecretion, GluK1-containing KARs regulate GABA release in a similar manner on both OT and VP neurons in the SON.
Lactation-induced switch in GluK1 activation on OT neurons
KAR-mediated effects on synaptic transmission can switch from facilitation to inhibition when the concentration of KAR-agonist is increased (Jiang et al., 2001; Kerchner et al., 2001; Schmitz et al., 2001; Delaney and Jahr, 2002; Braga et al., 2003, Youn and Randic, 2004). Then, a physiological modification in glutamate levels could modify KAR-mediated activity. Indeed, because glutamate clearance is ensured essentially by surrounding astrocyte transporters, a significant increased in ambient glutamate occurs in the SON of lactating animals resulting from a reduction of astrocytic coverage of OT neurons and their synapses (Oliet et al., 2001; Theodosis, 2002). We, thus, wanted to test whether such a physiological variation in extracellular levels of glutamate in lactating rats could modify the activity of KARs on GABAergic inputs of OT neurons. However, increased levels of ambient glutamate have been shown to affect presynaptic group III mGluRs that are coupled to neurotransmitter release (Piet et al., 2003, 2004), we thus blocked these receptors with MAP4 (250 μm) to get rid of this possible contaminating effect.
Surprisingly, in lactating rats, UBP302 increased mIPSC frequency in OT neurons (+32.62 ± 7.09%; n = 8; p < 0.05) (Fig. 4A), an effect opposite not only to that observed in unstimulated animals but also to the inhibitory effect on mIPSC frequency of VP neurons from lactating rats (−25.38 ± 2.02%; n = 5; p < 0.05) (Fig. 4A). In lactating rats, therefore, presynaptic GluK1-containing KARs are differentially coupled to GABA release according to the cell contacted. This differential effect is likely to result from the anatomical neuron-glia remodeling that affects specifically the OT system in lactating animals (Theodosis, 2002).
Figure 4.

Lactation-induced switch in GluK1 effect on OT neurons. A, Traces showing mIPSC activity recorded in an OT and a VP neuron in lactating rats before, during and after bath application of UPB302. B, Traces showing mIPSC activity recorded in an OT and a VP neuron in lactating rats before, during, and after bath application of ATPA.
Under conditions where the coupling of KARs with GABA release switches from facilitation to inhibition, stimulation of GluK1-containing receptors should reduce GABAergic transmission. Surprisingly, ATPA did not affect mIPSC frequency (−1.4 ± 2.76%; n = 9; p > 0.05) (Fig. 4B) in OT SON neurons of lactating rats, whereas it facilitated GABA transmission to the same extent in VP neurons recorded in lactating (+ 23.73 ± 4.62%; n = 4; p < 0.05) (Fig. 4B) and unstimulated rats (Fig. 1C). One possible explanation could be that GluK1-containing KARs on GABA terminals are fully saturated by ambient glutamate in these animals, resulting in an occlusion of the ATPA effect. To test this hypothesis, we incubated slices obtained from lactating rats with an enzymatic glutamate scavenger [glutamic-pyruvic transaminase (GPT) 5U/ml] to reduce ambient levels of the excitatory amino acid (Min et al., 1998). In the presence of GPT and 2 mm pyruvate, ATPA now caused a significant change mIPSC frequency that was either increased (+28.02 ± 2.41%; n = 6; p < 0.05) (Fig. 5A,B) or decreased (−10.41 ± 4.70%; n = 6; p < 0.05) (Fig. 5A,B) in OT neurons, respectively. As expected, in the absence of pyruvate that is required for the enzymatic action of GPT, ATPA did not affect mIPSC frequency in OT neurons (−0.32 ± 1.47%; n = 3; p > 0.05; data not shown). Furthermore, ATPA-mediated facilitation of GABA release on VP neurons was unaffected by the presence of GPT and pyruvate (+25.72 ± 3.23% of mIPSC frequency; n = 7; p < 0.05) (Fig. 5A,B). That we observed two opposite effects on OT neurons could reflect variations in the reduction of ambient glutamate induced by the scavenger. In some experiments, this reduction might be sufficient to unsaturate the GluK1 receptors without affecting their coupling to GABA release, while in others, the reduction might be so important that it also switched back the coupling of these receptors from inhibitory to facilitatory.
Figure 5.

Reducing glutamate levels affects mIPSC activity in OT neurons of lactating rats. A, Traces showing mIPSC activity recorded in two OT and one VP neurons in lactating rats before, during and after bath application of ATPA in the presence of GPT and pyruvate. ATPA had two opposite actions on OT neurons. It either increased (upper traces) or decreased (middle traces) mIPSC activity. B, Summary histogram illustrating the change in mIPSC mean frequency induced by ATPA in the presence of GPT and pyruvate in lactating rats. In 6 OT neurons (empty bars; left), the agonist increased mIPSC frequency whereas in 6 others OT cells (empty bars, middle), it reduced it. In contrast, the effect of ATPA in VP neurons (black bars; n = 7) was similar to that recorded in the absence of the glutamate scavenger (see Fig. 4). *p < 0.05.
Increasing glutamate concentration switches GluK1-containing KAR activity
If the opposite effect of GluK1-containing KARs on GABA transmission observed in OT neurons of lactating rats results from increased levels of ambient glutamate due to the lack of intervening astrocytic processes, then it should be possible to mimic this effect in unstimulated rats by an experimentally induced increase in extracellular glutamate. To this end, we decreased glutamate clearance using the broad spectrum inhibitor of glutamate transporters d,l-TBOA (25 μm). Once again, experiments were performed in the presence of MAP4 to avoid any mGluRIII-mediated side effects.
Under these conditions, UBP302 (10 μm) induced a reversible increase in mIPSC activity (Fig. 6A) sustained by a significant increase in the frequency (+ 21.70 ± 2.91%; n = 8; p < 0.05) but not in the amplitude (−6.07 ± 5.66%; n = 8; p > 0.05) of miniature events. In this condition, both OT and VP neurons should be affected by the d,l-TBOA-induced increased glutamate concentration. Indeed, among those 8 cells, we were able to identify 2 VP and 2 OT neurons thereby indicating that increasing extracellular glutamate concentration in the SON reverses the coupling effect of GluK1-containing receptors on GABA release in both types of neurons. Therefore, increasing glutamate concentration with d,l-TBOA in unstimulated rats switched the effect of GluK1-containing KARs activation from facilitator into inhibitor, thereby mimicking the responses recorded in OT neurons in lactating animals.
Figure 6.

An increased extracellular glutamate concentration in unstimulated rats switches the activity of GluK1-containing KARs. A, Traces showing mIPSC activity before, during and after bath application of UBP302 in the presence of a selective inhibitor of glutamate transporters, d,l-TBOA. B, Traces showing mIPSC activity before, during, and after bath application of ATPA in the presence of d,l-TBOA.
In agreement with such a switch in KAR activity, ATPA effect on mIPSC frequency in the presence of d,l-TBOA (25 μm) was reversed from a facilitation into an inhibition (−18.35 ± 1.81%; n = 7; p < 0.05) (Fig. 6B). The fact that ATPA mediates an inhibitory effect in slices from unstimulated rats treated with d,l-TBOA whereas it has no effect on OT neurons from lactating rats, suggests that in the latter, the increased ambient glutamate levels fully saturated GluK1-containing KARs. If this is true, then we should be able to reproduce this observation in unstimulated rats by causing a massive increase in extracellular glutamate levels. To this end, we increased d,l-TBOA concentrations from 25 to 100 μm to block most glutamate transporters and to enhance ambient glutamate to levels that are likely to saturate GluK1-containing KARs. In agreement with this hypothesis, in the presence of 100 μm d,l-TBOA, ATPA no longer affected mIPSC frequency (−0.65 ± 3.24%; n = 8; p > 0.05) (data not shown). This experiment demonstrates that the lack of effect of ATPA in OT neurons of lactating rats is due to the saturation of GluK1-containing KARs by ambient glutamate in these animals.
The opposite KAR-mediated regulation of GABA transmission in SON neurons as a function of extracellular glutamate levels was also observed when monitoring IPSCs evoked by afferent input stimulation. UBP302 reversibly decreased the amplitude of evoked-IPSPs in hypothalamic slices obtained from unstimulated animals (−26.69 ± 4.85%, n = 6; p < 0.05) (Fig. 7A). This effect was accompanied by an elevated paired-pulse ratio (PPR; from 1.27 ± 0.24 to 1.43 ± 0.23; p < 0.05) (Fig. 7B), confirming the presynaptic location of GluK1-containing KARs. In slices incubated with d,l-TBOA (25 μm), in the presence of MAP4 and d-AP5 to prevent mGluRIII- and NMDAR-mediated effects, UBP302 had an opposite action, increasing the amplitude of evoked-IPSCs (+ 17.86 ± 2.47%, n = 6; p < 0.05) (Fig. 7A) while decreasing PPR (from 1.49 ± 0.24 to 1.13 ± 0.20; p < 0.05) (Fig. 7B).
Figure 7.

Bidirectional modulation of evoked-IPSCs by GluK1-containing KARs. A, Superimposed traces illustrating IPSCs evoked by a pair of stimulations obtained under control conditions (black traces) and in the presence of UBP302 (10 μm) (gray traces) in slices untreated (top) or treated (bottom) with d,l-TBOA (25 μm). B, Traces illustrated in (A) were scaled to the amplitude of the first IPSC to illustrate the change in PPR. Whereas UBP302 increased PPR in untreated slices, it decreased it in d,l-TBOA-treated slices.
Mechanism underlying KAR-mediated facilitation of GABA release
To investigate the mechanism accounting for the switch in the coupling between KARs and GABA release, we first focused on the process underlying the facilitating effect of GluK1 activation on GABA transmission as observed in unstimulated condition of neurosecretion. Several mechanisms of facilitation of transmitter release by KARs have been described in the literature, including membrane depolarization of synaptic terminals leading to Ca2+ influx through voltage-gated calcium channels (Kamiya et al., 2002) and a direct Ca2+ influx through the receptor itself (Lauri et al., 2003; Pinheiro et al., 2007; Mathew and Hablitz, 2008; Sun et al., 2009). Such rises in intraterminal Ca2+ increase the probability of transmitter release. To determine whether a similar mechanism is responsible for GABA release facilitation in our experiments, we used philantotoxin-433 (PhTx, 3 μm), a blocker of Ca2+-permeable AMPA/Kainate receptors (Fletcher and Lodge, 1996). In its presence, UBP302 (10 μm) no longer induced a reduction of mIPSC activity. Most interestingly, under these conditions UBP302 caused a significant and reversible increase in mIPSC frequency (+34.63 ± 4.69%; n = 7; p < 0.05) (Fig. 8A,B), similar to that observed when extracellular glutamate levels are increased. As expected, not only the facilitating effect of ATPA disappeared in the presence of PhTx, but this effect was switched into an inhibition of mIPSC frequency (−16.35 ± 1.30%; n = 7; p < 0.05) (Fig. 8A,B). These findings indicate that the GluK1-containing KARs responsible for facilitating GABAergic transmission are permeable to Ca2+, a property that could account for the observed increase in the probability of transmitter release.
Figure 8.

Facilitation of GABA release by GluK1-containing-KARs is mediated by an ionotropic mode of action. A, Traces showing mIPSC activity before and during bath application of either UBP302 or ATPA in the presence of an inhibitor of Ca2+-permeable AMPA/Kainate receptors, Philanthotoxin-433 (PhTx). B, Summary histogram illustrating the percentage of change in mIPSC frequency induced either by UBP302 (left part of the histogram; n = 7) or by ATPA (right part; n = 7) in the presence of PhTx (black bars). For comparison purpose, the effect induced by these two compounds in the absence of PhTx is also illustrated (dotted bars). PhTx blocked the facilitatory coupling between GluK1 and GABA release. This result is in agreement with a ionotropic mode of action of GluK1. *p < 0.05.
Mechanism underlying KAR-mediated inhibition of GABA release
All experiments aimed at determining the mechanism underlying the inhibitory action of GluK1-containing KARs have been realized in unstimulated rats in the presence of d,l-TBOA (25 μm). The inhibitory action of KAR activation on GABA release could result from direct or indirect effects. We first investigated indirect effects, including the activation of GABA-B autoreceptors (Kerchner et al., 2001) and other types of presynaptic receptors whose stimulation may take place in a retrograde (Chergui et al., 2000) or heterosynaptic manner following KAR activation. For instance, activation of KARs on SON neurons or on dopaminergic afferent inputs could trigger the release of endocannabinoids or dopamine, which in turn could activate presynaptic CB1 and D4 receptors, thereby inhibiting GABA release (Azdad et al., 2003; Oliet et al., 2007). To test for this possibility we added a mixture of antagonists in the bathing solution for receptors that could be involved in such an indirect KAR-mediated effect, including GABA-B receptors (CPG5426, 10 μm), adenosine A1 receptors (DPCPX, 100 nm), mGluRs (LY341495, 100 μm), NMDA receptors (d-AP5, 50 μm), CB1 receptors (AM251, 5 μm), nicotinic receptors (mecamylamine, 10 μm), dopamine D4 receptors (L745,870, 50 μm), and α-2-adrenergic receptor (yohimbine, 100 μm). Such a mixture did not affect the inhibitory action of GluK1-containing KARs on GABA release. UBP302 (10 μm) increased mIPSCs frequency (+ 24.59 ± 3.37%; n = 6; p < 0.05; data not shown) to an extent similar to that observed in the absence of the receptor blockers (p > 0.05). This result argues against an indirect mechanism mediating the inhibitory effect of GluK1 activation on GABA release.
We then investigated the possibility that the inhibitory action of GluK1 activation on GABA release depends on a direct coupling between KARs and transmitter release through a metabotropic G-protein pathway, a mechanism already described (Rodríguez-Moreno and Lerma, 1998; Cunha et al., 2000; Frerking et al., 2001; Lauri et al., 2005, 2006; Negrete-Diaz et al., 2006; Jin and Smith, 2007). To test for a similar mechanism in our recordings, we first tried to incubate hypothalamic slices for at least 5 h in the presence of pertussis toxin to block G-proteins. However, this procedure consistently caused a deterioration of the slices, making stable and prolonged recordings impossible. Because most reported KAR-mediated metabotropic effects are associated with the activation of the phospholipase C (PLC) pathway downstream of G-protein stimulation (Rodríguez-Moreno and Lerma, 1998; Lauri et al., 2005; Jin and Smith, 2007), we tried to disrupt this intracellular cascade using the PLC inhibitor U73122 (10 μm). In its presence, the facilitation induced by blocking the tonic activation of KARs with UBP302 was no longer observed. Instead, a consistent inhibition of mIPSC frequency occurred (−21.17 ± 2.91%; n = 6) (Fig. 9A,B). Similarly, direct activation of GluK1-containing KARs with the agonist ATPA no longer had an inhibitory action. Instead, ATPA induced a facilitation of mIPSC frequency (+19.02 ± 2.02; n = 10; p < 0.05) (Fig. 9A,B) Together, these findings indicate that a PLC-dependent metabotropic pathway mediates the inhibitory effect of GluK1 on GABA release.
Figure 9.

Inhibition of GABA release by GluK1-containing-KARs involves a PLC-dependent metabotropic pathway. A, Traces showing mIPSC activity before and during bath application of either UBP302 or ATPA in the presence of an inhibitor of glutamate transporters, d,l-TBOA and a selective inhibitor of Phospholipase C (PLC), U73122. B, Summary histogram illustrating the percentage of change in mIPSC frequency induced either by UBP302 (left part of the histogram; n = 6) or by ATPA (right part; n = 10) in the presence of U73122 (black bars). For comparison purpose, the effect induced by these two compounds in the absence of U73122 is also illustrated (dotted bars; from Fig. 6). The inhibitory effect of GluK1 activation on GABA release was no longer present when PLC pathway was inhibited. This is in agreement with a metabotropic mode of action of GluK1 in high extracellular glutamate levels. *p < 0.05.
Coexistence of KAR-mediated ionotropic and metabotropic pathways
Our results obtained with PhTx and U73122 support the idea that the two modes of action of KARs coexist on the same GABAergic terminal. If this is true, the inhibitory effect of KARs unmasked by PhTx in unstimulated conditions of neurosecretion (Fig. 8), should be sensitive to PLC inhibition. Conversely, the GluK1-mediated facilitation of GABA transmission revealed by U73122 under conditions of enhanced ambient glutamate levels (Fig. 9) should be compromised in PhTx. In agreement with this hypothesis, UBP302 (10 μm) no longer affected mIPSC activity in the presence of both PhTx and U73122 in either untreated (−5.20 ± 3.13%; n = 6; p > 0.05, Fig. 10A,B) or d,l-TBOA-treated slices (+0.23 ± 2.32%; n = 6; p > 0.05) (Fig. 10C,D).
Figure 10.

Coexistence of KAR-mediated ionotropic and metabotropic pathways. A, Traces showing mIPSC activity before and during bath application of UBP302 in untreated slices from unstimulated rats, either in the presence of both U73122 and PhTx or only U73122. B, Summary histogram illustrating the percentage of change in mIPSC frequency induced by UBP302 in the presence of both U73122 and PhTx (gray bars; n = 6) or only U73122 (black bars; n = 10). For comparison purpose, the action of UBP302 under control conditions is illustrated (dotted bars). Inhibiting the PLC pathway in control conditions did not affect GluK1-mediated facilitatory effect. C, Traces showing mIPSC activity before and during bath application of UBP302 in TBOA-treated slices from unstimulated rats either in the presence of both PhTx and U73122 or only PhTx. D, Summary histogram illustrating in TBOA-treated slices the percentage of change in mIPSC frequency induced by UBP302 in the presence of both PhTx and U73122 (gray bars; n = 6) or only PhTx (black bars; n = 5). For comparison purpose, the effect of UBP302 under control conditions is illustrated (dotted bars, from Fig. 6A). Inhibiting Ca2+ entry through GluK1 in high extracellular glutamate levels, did not affect KAR-mediated inhibitory effect. Blocking the ionotropic and the metabotropic pathways in untreated and TBOA-treated slices completely inhibited GluK1 effect on GABA release, suggesting that these two modes of action coexist on the same GABAergic terminals. *p < 0.05.
We next tested whether the two modes of action are concomitantly engaged when GluK1-containing KARs are activated. If this is the case, the overall effect of KAR activation on GABA transmission is then the result of both a facilitation and an inhibition of transmitter release, with a dominant facilitating effect in unstimulated conditions and a dominant inhibitory action in conditions of high extracellular glutamate levels. Accordingly, blocking the nondominant effect should increase the dominant action of KARs. If this hypothesis is true, blocking the metabotropic pathway should enhance the facilitatory KAR-mediated effects in control animals. To this end, we tested the effect of UPB302 in the presence of the PLC inhibitor under control conditions. Interestingly, we observed that this manipulation did not affect the change in mIPSC frequency (p > 0.05) resulting from GluK1-containing KAR blockade (−28.06 ± 3.90%; n = 10; p < 0.05) (Fig. 10A,B). Vice versa, inhibiting the ionotropic pathway under conditions of high ambient glutamate should augment the inhibitory action of KARs on GABA release. We thus tested UBP302 in the presence of PhTx in TBOA-treated slices. Its facilitating effect was similar to that obtained in the absence of PhTx (p > 0.05) (+29.06 ± 8.49%; n = 5; p < 0.05) (Fig. 10C,D). Together, these results suggest that the two pathways are not activated concomitantly.
Discussion
We here report for the first time the presence of functional KARs on GABAergic inputs impinging on OT and VP neurons of the SON. Activation of these presynaptic KARs increased GABA release under unstimulated conditions of neurosecretion. Interestingly, a physiological increase in extracellular levels of ambient glutamate mediated by astrocytic process plasticity completely switches this KAR-mediated facilitation of GABA release into an inhibition. Moreover, we demonstrated that the facilitation of GABA release by KARs is mediated through an ionotropic mode of action, whereas its inhibition involves a PLC-dependent metabotropic pathway.
Glia-dependent switch in GluK1 activity in lactating rats
The major finding of this study is that the facilitating effect of GluK1 activation on GABA release in the SON of unstimulated animals is switched into an inhibitory action on OT neurons of lactating rats. As reported in previous studies, activation of presynaptic KARs can have bidirectional effects on glutamatergic or GABAergic transmission. Indeed, such a switch in KAR-mediated effects from facilitation to inhibition of transmitter release has been observed in response to an increase in either agonist concentrations (Jiang et al., 2001; Kerchner et al., 2001; Schmitz et al., 2001; Delaney and Jahr, 2002; Braga et al., 2003; Youn and Randic, 2004) or glutamatergic afferent input stimulations (Schmitz et al., 2001; Delaney and Jahr, 2002). Together, these data suggest that a rise in extracellular glutamate concentration can completely reverse the effect of KAR activation on transmitter release probability.
In our experiments, the most plausible explanation to account for the difference observed in OT neurons in lactating versus unstimulated animals may be related to the astroglial-neuronal remodeling characterizing the OT system during physiological states, such as lactation, parturition, or chronic dehydration (Theodosis, 2002). The most striking feature of such an anatomical reorganization is a pronounced and specific reduction of astrocytic coverage of OT neurons and their synapses, a remodeling that does not affect the VP system (Chapman et al., 1986; Theodosis et al., 1986). Analysis of synaptic transmission in the SON of lactating and dehydrated rats demonstrated that such an anatomical modification of the glial environment of OT neurons leads to increased levels of ambient glutamate, presumably resulting from a deficient transport of the excitatory amino acid (Oliet et al., 2001; Boudaba et al., 2003). The switch in the activity of GluK1 in lactating rats is observed only in OT, which strongly argues that this effect is actually due to the increased glutamate levels resulting from glial retraction. In agreement with this hypothesis, degrading extracellular glutamate with GPT caused the reversal of this switch in about half the OT neurons recorded from lactating rats under these conditions.
Incubating hypothalamic slices from unstimulated rats with the non-transportable inhibitor of glutamate transporters, d,l-TBOA (Shimamoto et al., 1998, 2000; Jabaudon at al., 1999) induced in both OT and VP neurons a similar switch to that recorded in OT neurons of lactating animals. This observation supports the hypothesis that a pharmacological increase in extracellular glutamate concentration, if sufficient, can reverse the activity of KARs. Moreover, this experimental switch in GluK1 activity affected both OT and VP systems in unstimulated animals as expected if the increase in extracellular glutamate levels was uniform in the entire SON. The switch from facilitation to inhibition in TBOA-treated slices was observed when KARs were activated by either ambient glutamate, as revealed using the antagonist UBP302, or a selective GluK1-containing KAR agonist, ATPA.
Together, these experiments demonstrate that GluK1-containing KARs present on GABAergic terminals contacting both VP and OT neurons possess the capacity to switch their coupling with GABA release. These results establish for the first time that under physiological conditions, KAR activity can be drastically modified by the glial environment of neurons through the modulation of ambient glutamate levels.
Coexistence of ionotropic and metabotropic KAR modes of action
The mechanism responsible for this switch is presently unknown. The facilitating effect mediated by KARs on GABA transmission can be easily explained by direct Ca2+ influx through the receptor itself causing an increased probability of transmitter release (Lauri et al., 2003; Pinheiro et al., 2007). This was confirmed by the use of PhTx, a toxin inhibiting Ca2+-permeable AMPA/Kainate receptors. In its presence, the facilitation of GABA release was compromised indicating that Ca2+ influx through KARs is likely to be involved in the facilitation of inhibitory transmission.
The inhibitory action of KAR activation observed in SON neurons could be accounted for an indirect or direct action of KARs onto GABA release. An indirect action involving third-party signaling molecules, like endocannabinoids, dopamine, adenosine (Chergui et al., 2000), or glutamate, is unlikely in view of the results obtained with the mixture of antagonists for various receptors including GABA-B, CB1, A1, D4, mGluRs, NMDA, nicotinic and α-2 adrenergic receptors. A direct action of KARs on transmitter release involving a G-protein-coupled signaling pathway has already been reported in other structures (Rodríguez-Moreno and Lerma, 1998; Cunha et al., 2000; Frerking et al., 2001; Lauri et al., 2005, 2006; Negrete-Diaz et al., 2006; Jin and Smith, 2007). This seems to be also the case in our recordings as indicated by the abolition of the KAR-mediated inhibitory effect in the presence of the PLC inhibitor, U73122. This finding demonstrates that under certain circumstances, activation of presynaptic GluK1-containing KARs stimulates a metabotropic pathway leading to a decrease in GABA release probability. That facilitation and inhibition of GABA transmission involve an ionotropic and a metabotropic mechanism, respectively, has been already demonstrated in other structures (Rodríguez-Moreno and Lerma, 1998; Jin and Smith, 2007; Mathew and Hablitz, 2008). However, these observations were made separately from each other. Furthermore, it was never demonstrated that the biphasic activity of KARs due to increased agonist concentrations resulted from a switch from ionotropic to metabotropic mode of action. The present study thus provides for the first time the demonstration that at given GABAergic synapses, KAR activation can switch from a ionotropic to a metabotropic mode of action, thereby accounting for the biphasic modulation of transmitter release.
Our findings raise a lot of questions and in particular whether the ionotropic and metabotropic actions of KARs are mediated by the same or by two distinct types of GluK1-containing receptors. Although our data do not address this issue, it is clear that these two pathways coexist within the same terminals, in agreement with previous studies that have reported the presence of these two mechanisms in a single neuron (Rozas et al., 2003; Ruiz et al., 2005; Rivera et al., 2007). The inhibition of Ca2+ entry through GluK1, using PhTx, unmasked an inhibitory metabotropic pathway activated by GluK1-containing KARs in slices from unstimulated rats. Vice versa, we revealed a GluK1-mediated facilitation of GABA release in TBOA-treated slices when the PLC pathway was inhibited. It is reasonable to assume, therefore, that in the SON, the two modes of action of GluK1-containing KARs, ionotropic and metabotropic, can be engaged at individual GABAergic synapses. The question then arises as to whether both ionotropic facilitatory and metabotropic inhibitory pathways engaged whenever GluK1 KARs were activated. This does not seem to be the case since inhibiting PLC in untreated slices did not modify the extent of the KAR-mediated facilitation of GABA release, and blocking the ionotropic effect of KARs with PhTx in TBOA-treated slices did not affect the magnitude of the KAR-mediated inhibition of GABA release.
Another possibility for our results is that the ionotropic and metabotropic pathways are not engaged concomitantly. A simple explanation could be the existence of a negative cross talk between the two pathways preventing them to be recruited in parallel.
Physiological relevance of the switch
During unstimulated conditions, tonic activation of KARs will enhance the GABAergic drive on magnocellular neurons, thereby reducing their overall excitability, and in consequence, maintaining a low level of neurohormone release in steady-state conditions. In lactating rats, the inhibitory action on GABA release exerted by the tonic activation of KARs by ambient glutamate is likely to cause a disinhibition of OT neurons, thereby increasing neuronal excitability and neuropeptide secretion.
Finally, we enlighten in this study physiological conditions in which a switch in KAR activity from facilitation to inhibition can occur. This switch in KAR activity results from a switch in the mode of action of KARs on GABA release. Because this switch is caused by enhanced levels of extracellular glutamate, it is under the control of the glial environment of neurons that govern glutamate clearance and diffusion in the CNS. Such a switch may be relevant in other brain regions in which similar astrocytic changes occur under physiological conditions, as for example in the suprachiasmatic nucleus, the barrel cortex, the visual cortex, and the hippocampus (Theodosis et al., 2008). Likewise, this type of control may be an important mechanism that may participate in pathologies, such as neurodegenerative diseases, epilepsy, and ischemia, which are associated with greatly enhanced levels of extracellular glutamate. Under such conditions, the disinhibition resulting from the switch in the KAR coupling with GABA release may amplify the excitotoxic process and thus be very deleterious for the neuronal survival.
Footnotes
This work was supported by grants from Inserm, Fondation pour la Recherche Médicale and the Human Frontier Science Program. V.B. was supported by studentships from the “Ministère de l'Education Nationale de la Recherche et de la Technologie” and the “Fondation pour la Recherche Médicale.” The confocal microscopy was performed in the Bordeaux Imaging Center (BIC), Bordeaux. We thank Drs. Christophe Mulle and Paulo Pinhero for helpful discussions and comments on an earlier version of the manuscript, as well as Dr. Laurent Aniksztejn and Camille Allène for precious advise with the glutamate scavenger. We thank Philippe Legros, Laure Malicieux, and Christel Poujol for their technical assistance.
References
- Armstrong WE. Morphological and electrophysiological classification of hypothalamic supraoptic neurons. Prog Neurobiol. 1995;47:291–339. [PubMed] [Google Scholar]
- Azdad K, Piet R, Poulain DA, Oliet SH. Dopamine D4 receptor-mediated presynaptic inhibition of GABAergic transmission in the rat supraoptic nucleus. J Neurophysiol. 2003;90:559–565. doi: 10.1152/jn.00226.2003. [DOI] [PubMed] [Google Scholar]
- Bettler B, Mulle C. Review: neurotransmitter receptors.II. AMPA and kainate receptors. Neuropharmacology. 1995;34:123–139. doi: 10.1016/0028-3908(94)00141-e. [DOI] [PubMed] [Google Scholar]
- Bortolotto ZA, Clarke VR, Delany CM, Parry MC, Smolders I, Vignes M, Ho KH, Miu P, Brinton BT, Fantaske R, Ogden A, Gates M, Ornstein PL, Lodge D, Bleakman D, Collingridge GL. Kainate receptors are involved in synaptic plasticity. Nature. 1999;402:297–301. doi: 10.1038/46290. [DOI] [PubMed] [Google Scholar]
- Boudaba C, Linn DM, Halmos KC, Tasker JG. Increased tonic activation of presynaptic metabotropic glutamate receptor in the rat supraoptic nucleus following chronic dehydration. J Physiol. 2003;551:815–823. doi: 10.1113/jphysiol.2003.042739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braga MF, Aroniadou-Anderjaska V, Xie J, Li H. Bidirectional modulation of GABA release by presynaptic glutamate receptor 5 kainate receptors in the basolateral amygdala. J Neurosci. 2003;23:442–452. doi: 10.1523/JNEUROSCI.23-02-00442.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brussaard AB, Devay P, Leyting-Vermeulen JL, Kits KS. Changes in properties and neurosteroid regulation of GABAergic synapses in the supraoptic nucleus during the mammalian female reproductive cycle. J Physiol. 1999;516:513–524. doi: 10.1111/j.1469-7793.1999.0513v.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman DB, Theodosis DT, Montagnese C, Poulain DA, Morris JF. Osmotic stimulation causes structural plasticity of neurone-glia relationships of the oxytocin but not vasopressin secreting neurones in the hypothalamic supraoptic nucleus. Neuroscience. 1986;17:679–686. doi: 10.1016/0306-4522(86)90039-4. [DOI] [PubMed] [Google Scholar]
- Chergui K, Bouron A, Normand E, Mulle C. Functional GluR6 kainate receptors in the striatum: indirect dowregulation of synaptic transmission. J Neurosci. 2000;20:2175–2182. doi: 10.1523/JNEUROSCI.20-06-02175.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chittajallu R, Braithwaite SP, Clarke VR, Henley JM. Kainate receptors: subunits, synaptic localization and function. Trends Pharmacol Sci. 1999;20:26–35. doi: 10.1016/s0165-6147(98)01286-3. [DOI] [PubMed] [Google Scholar]
- Clarke VR, Ballyk BA, Hoo KH, Mandelzys A, Pellizzari A, Bath CP, Thomas J, Sharpe EF, Davies CH, Ornstein PL, Schoepp DD, Kamboj RK, Collingridge GL, Lodge D, Bleakman D. A hippocampal GluK1 kainate receptor regulating inhibitory synaptic transmission. Nature. 1997;389:599–603. doi: 10.1038/39315. [DOI] [PubMed] [Google Scholar]
- Collingridge GL, Olsen RW, Peters J, Spedding M. A nomenclature for ligand-gated ion channels. Neuropharmacology. 2009;56:2–5. doi: 10.1016/j.neuropharm.2008.06.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunha RA, Malva JO, Ribeiro JA. Pertussis toxin prevents presynaptic inhibition by kainate receptors of rat hippocampal [3H]GABA release. FEBS Lett. 2000;469:159–162. doi: 10.1016/s0014-5793(00)01272-2. [DOI] [PubMed] [Google Scholar]
- Delaney AJ, Jahr CE. Kainate receptors differentially regulate release at two parallel fiber synapses. Neuron. 2002;36:475–482. doi: 10.1016/s0896-6273(02)01008-5. [DOI] [PubMed] [Google Scholar]
- Fletcher EJ, Lodge D. New developments in the molecular pharmacology of alpha-amino-3-hydroxy-5-methyl-4-isoxazole propionate and kainate receptors. Pharmacol Ther. 1996;70:65–89. doi: 10.1016/0163-7258(96)00014-9. [DOI] [PubMed] [Google Scholar]
- Frerking M, Schmitz D, Zhou Q, Johansen J, Nicoll RA. Kainate receptors depress excitatory synaptic transmission at CA3→CA1 synapses in the hippocampus via a direct presynaptic action. J Neurosci. 2001;21:2958–2966. doi: 10.1523/JNEUROSCI.21-09-02958.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gies U, Theodosis DT. Synaptic plasticity in the rat supraoptic nucleus during lactation involves GABA innervation and oxytocin neurons: a quantitative immunocytochemical analysis. J Neurosci. 1994;14:2861–2869. doi: 10.1523/JNEUROSCI.14-05-02861.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higuchi T, Honda K, Fukuoka T, Negoro H, Hosono Y, Nishida E. Pulsatile secretion of prolactin and oxytocin during nursing in the lactating rat. Endocrinol Jpn. 1983;30:353–359. doi: 10.1507/endocrj1954.30.353. [DOI] [PubMed] [Google Scholar]
- Jabaudon D, Shimamoto K, Yasuda-Kamatani Y, Scanziani M, Gähwiler BH, Gerber U. Inhinition of uptake unmasks rapid extracellular turnover of glutamate of nonvesicular origin. Proc Natl Acad Sci U S A. 1999;96:8733–8738. doi: 10.1073/pnas.96.15.8733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang L, Xu J, Nedergaard M, Kang J. A kainate receptor increases the efficacity of GABAergic synapses. Neuron. 2001;30:503–513. doi: 10.1016/s0896-6273(01)00298-7. [DOI] [PubMed] [Google Scholar]
- Jin XT, Smith Y. Activation of presynaptic kainate receptors suppresses GABAergic synaptic transmission in the rat globus pallidus. Neuroscience. 2007;149:338–349. doi: 10.1016/j.neuroscience.2007.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabashima N, Shibuya I, Ibrahim N, Ueta Y, Yamashita H. Inhibition of spontaneous EPSCs and IPSCs by presynaptic GABAB receptors on rat supraoptic magnocellular neurons. J Physiol. 1997;504:113–126. doi: 10.1111/j.1469-7793.1997.113bf.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamiya H, Ozawa S, Manabe T. Kainate receptor-dependent short term plasticity of presynaptic Ca2+ influx at the hippocampal mossy fiber synapses. J Neurosci. 2002;22:9237–9243. doi: 10.1523/JNEUROSCI.22-21-09237.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kerchner GA, Wang GD, Qui CS, Huettner JE, Zhuo M. Direct presynaptic regulation of GABA/Glycine release by kainate receptors in the dorsal horn: an ionotropic mechanism. Neuron. 2001;32:477–488. doi: 10.1016/s0896-6273(01)00479-2. [DOI] [PubMed] [Google Scholar]
- Kombian SB, Zidichouski JA, Pittman QJ. GABAB receptors presynaptically modulate excitatory synaptic transmission in the rat supraoptic nucleus in vitro. J Neurophysiol. 1996;76:1166–1179. doi: 10.1152/jn.1996.76.2.1166. [DOI] [PubMed] [Google Scholar]
- Lauri SE, Bortolotto ZA, Nistico R, Bleakman D, Ornstein PL, Lodge D, Isaac JT, Collingridge GL. A role for Ca2+ stores in kainate receptor-dependent synaptic facilitation and LTP at mossy fiber synapses in the hippocampus. Neuron. 2003;39:327–341. doi: 10.1016/s0896-6273(03)00369-6. [DOI] [PubMed] [Google Scholar]
- Lauri SE, Segerstråle M, Vesikansa A, Maingret F, Mulle C, Collingridge GL, Isaac JT, Taira T. Endogenous activation of kainate receptors regulates glutamate release and network activity in the developing hippocampus. J Neurosci. 2005;25:4473–4484. doi: 10.1523/JNEUROSCI.4050-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauri SE, Vesikansa A, Segerstråle M, Collingridge GL, Isaac JTR, Taira T. Functional maturation of CA1 synapses involves activity-dependent loss of tonic kainate receptor-mediated inhibition of glutamate release. Neuron. 2006;50:415–429. doi: 10.1016/j.neuron.2006.03.020. [DOI] [PubMed] [Google Scholar]
- Leng G, Brown CH, Russell JA. Physiological pathways regulating the activity of magnocellular neurosecretory cells. Prog Neurobiol. 1999;57:625–655. doi: 10.1016/s0301-0082(98)00072-0. [DOI] [PubMed] [Google Scholar]
- Li C, Tripathi PK, Armstrong WE. Differences in spike train variability in rat vasopressin and oxytocin neurons and their relationship to synaptic activity. J Physiol. 2007;581:221–240. doi: 10.1113/jphysiol.2006.123810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li DP, Pan YZ, Pan HL. Acetylcholine attenuates synaptic GABA release to suparoptic neurons through presynaptic nicotinic receptors. Brain Res. 2001;920:151–158. doi: 10.1016/s0006-8993(01)03055-4. [DOI] [PubMed] [Google Scholar]
- Mathew SS, Hablitz JJ. Calcium release via activation of presynaptic IP3 receptors contributes to kainate-induced IPSC facilitation in rat neocortex. Neuropharmacology. 2008;55:106–116. doi: 10.1016/j.neuropharm.2008.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Min MY, Rusakov DA, Kullmann DM. Activation of AMPA, kainate, and metabotropic receptors at hippocampal mossy fiber synapses: role of glutamate diffusion. Neuron. 1998;21:561–570. doi: 10.1016/s0896-6273(00)80566-8. [DOI] [PubMed] [Google Scholar]
- Moll UM, Lane BL, Robert F, Geenen V, Legros JJ. Abundant occurrence of oxytocin-, vasopressin-, and neurophysin-like peptides in epithelial cells. Histochemistry. 1988;89:385–390. doi: 10.1007/BF00500641. [DOI] [PubMed] [Google Scholar]
- More JC, Nistico R, Dolman NP, Clarke VR, Alt AJ, Ogden AM, Buelens FP, Troop HM, Kelland EE, Pilato F, Bleakman D, Bortolotto ZA, Collingridge GL, Jane DE. Characterisation of UBP296: a novel, potent and selective kainate receptor antagonist. Neuropharmacology. 2004;47:46–64. doi: 10.1016/j.neuropharm.2004.03.005. [DOI] [PubMed] [Google Scholar]
- Negrete-Díaz JV, Sihra TS, Delgado-García JM, Rodríguez-Moreno A. Kainate receptor-mediated inhibition of glutamate release involves protein kinase A in the mouse hippocampus. J Neurophysiol. 2006;96:1829–1837. doi: 10.1152/jn.00280.2006. [DOI] [PubMed] [Google Scholar]
- Oliet SH, Poulain DA. Adenosine-induced presynaptic inhibition of IPSCs and EPSCs in rat hypothalamic supraoptic nucleus neurones. J Physiol. 1999;520:815–825. doi: 10.1111/j.1469-7793.1999.00815.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliet SH, Baimoukhametova DV, Piet R, Bains JS. Retrograde regulation of GABA transmission by the tonic release of oxytocin and endocannabinoids governs postsynaptic firing. J Neurosci. 2007;27:1325–1333. doi: 10.1523/JNEUROSCI.2676-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliet SHR, Piet R, Poulain DA. Control of glutamate clearance and synaptic efficacy by glial coverage of neurons. Science. 2001;292:923–926. doi: 10.1126/science.1059162. [DOI] [PubMed] [Google Scholar]
- Piet R, Bonhomme R, Theodosis DT, Poulain DA, Oliet SH. Modulation of GABAergic transmission by endogenous glutamate in the supraoptic nucleus. Eur J Neurosci. 2003;17:1777–1785. doi: 10.1046/j.1460-9568.2003.02611.x. [DOI] [PubMed] [Google Scholar]
- Piet R, Vargová L, Syková E, Poulain DA, Oliet SH. Physiological contribution of the astrocytic environment of neurons to intersynaptic crosstalk. Proc Natl Acad Sci U S A. 2004;101:2151–2155. doi: 10.1073/pnas.0308408100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinheiro P, Mulle C. Kainate receptors. Cell Tissue Res. 2006;326:457–482. doi: 10.1007/s00441-006-0265-6. [DOI] [PubMed] [Google Scholar]
- Pinheiro P, Perrais D, Coussen F, Barhanin J, Bettler B, Mann JR, Malva JO, Heinemann SF, Mulle C. GluR7 is an essential subunit of presynaptic kainate autoreceptors at hippocampal mossy fiber synapses. Proc Natl Acad Sci U S A. 2007;104:12181–12186. doi: 10.1073/pnas.0608891104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poulain DA, Wakerley JB. Electrophysiology of hypothalamic magnocellular neurones secreting oxytocin and vasopressin. Neuroscience. 1982;7:773–808. doi: 10.1016/0306-4522(82)90044-6. [DOI] [PubMed] [Google Scholar]
- Rhodes CH, Morrell JI, Pfaff DW. Immunohistochemical analysis of magnocellular elements in rat hypothalamus: distribution and numbers of cells containing neurophysin, oxytocin, and vasopressin. J Comp Neurol. 1981;198:45–64. doi: 10.1002/cne.901980106. [DOI] [PubMed] [Google Scholar]
- Rivera R, Rozas JL, Lerma J. PKC-dependent autoregulation of membrane kainate receptors. EMBO J. 2007;26:4359–4367. doi: 10.1038/sj.emboj.7601865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodríguez-Moreno A, Lerma J. Kainate receptor modulation of GABA release involves a metabotropic function. Neuron. 1998;20:1211–1218. doi: 10.1016/s0896-6273(00)80501-2. [DOI] [PubMed] [Google Scholar]
- Rozas JL, Paternain AV, Lerma J. Noncanonical signaling by ionotropic kainate receptors. Neuron. 2003;39:543–553. doi: 10.1016/s0896-6273(03)00436-7. [DOI] [PubMed] [Google Scholar]
- Ruiz A, Sachidhanandam S, Utvik JK, Coussen F, Mulle C. Distinct subunits in heteromeric kainite receptors mediate ionotropic and metabotropic function at hippocampal mossy fiber synapses. J Neurosci. 2005;25:11710–11718. doi: 10.1523/JNEUROSCI.4041-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitz D, Mellor J, Nicoll RA. Presynaptic kainate receptor mediation of frequency facilitation at hippocampal mossy fiber synapses. Science. 2001;291:1972–1976. doi: 10.1126/science.1057105. [DOI] [PubMed] [Google Scholar]
- Schrader LA, Tasker JG. Presynaptic modulation by metabotropic glutamate receptors of excitatory and inhibitory synaptic inputs to hypothalamic magnocellular neurons. J Neurophysiol. 1997;77:527–536. doi: 10.1152/jn.1997.77.2.527. [DOI] [PubMed] [Google Scholar]
- Shibuya I, Kabashima N, Ibrahim N, Setiadji SV, Ueta Y, Yamashita H. Pre- and postsynaptic modulation of the electrical activity of rat supraoptic neurones. Exp Physiol. 2000;85:S145–S151. doi: 10.1111/j.1469-445x.2000.tb00018.x. [Spec No] [DOI] [PubMed] [Google Scholar]
- Shimamoto K, Lebrun B, Yasuda-Kamatani Y, Sakaitani M, Shigeri Y, Yumoto N, Nakajima T. DL-threo-beta-Benzyloxyaspartate, a potent blocker of excitatory amino acid transporters. Mol Pharmacol. 1998;53:195–201. doi: 10.1124/mol.53.2.195. [DOI] [PubMed] [Google Scholar]
- Shimamoto K, Shigeri Y, Yasuda-Kamatani Y, Lebrun B, Yumoto N, Nakajima T. Syntheses of optically pure beta-hydroxyaspartate derivates as glutamate transporter blockers. Bioorg Med Chem Lett. 2000;10:2407–2410. doi: 10.1016/s0960-894x(00)00487-x. [DOI] [PubMed] [Google Scholar]
- Stern JE, Ludwig M. NO inhibits supraoptic oxytocin and vasopressin neurons via activation of GABAergic synaptic inputs. Am J Physiol Regul Integr Comp Physiol. 2001;280:R1815–R1822. doi: 10.1152/ajpregu.2001.280.6.R1815. [DOI] [PubMed] [Google Scholar]
- Sun HY, Bartley AF, Dobrunz LE. Calcium-permeable presynaptic kainate receptors involved in excitatory short-term facilitation onto somatostatin interneurons during natural stimulus patterns. J Neurophysiol. 2009;101:1043–1055. doi: 10.1152/jn.90286.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theodosis DT. Oxytocin-secreting neurons: a physiological model of morphological neuronal and glial plasticity in the adult hypothalamus. Front Neuroendocrinol. 2002;23:101–135. doi: 10.1006/frne.2001.0226. [DOI] [PubMed] [Google Scholar]
- Theodosis DT, Chapman DB, Montagnese C, Poulain DA, Morris JF. Structural plasticity in the hypothalamic supraoptic nucleus at lactation affects oxytocin, -but not vasopressin-secreting neurones. Neuroscience. 1986;17:661–678. doi: 10.1016/0306-4522(86)90038-2. [DOI] [PubMed] [Google Scholar]
- Theodosis DT, Poulain DA, Oliet SH. Activity-dependent structural and functional plasticity of astrocyte-neuron interactions. Physiol Rev. 2008;88:983–1008. doi: 10.1152/physrev.00036.2007. [DOI] [PubMed] [Google Scholar]
- Wang YF, Shibuya I, Kabashima N, Setiadji VS, Isse T, Ueta Y, Yamashita H. Inhibition of spontaneous inhibitory postsynaptic currents (IPSC) by noradrenaline in rat supraoptic neurons through presynaptic α2-adrenoreceptors. Brain Res. 1998;807:61–69. doi: 10.1016/s0006-8993(98)00732-x. [DOI] [PubMed] [Google Scholar]
- Wisden W, Seeburg PH. A complex mosaic of high-affinity kainate receptors in the brain. J Neurosci. 1993;13:3582–3598. doi: 10.1523/JNEUROSCI.13-08-03582.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youn DH, Randic M. Modulation of excitatory synaptic transmission in the spinal substantia gelatinosa of mice deficient in the kainate receptor GluK1 and/or GluR6 subunit. J Physiol. 2004;555:683–698. doi: 10.1113/jphysiol.2003.057570. [DOI] [PMC free article] [PubMed] [Google Scholar]