

Abstract
Neuroblastoma is a pediatric tumor that is thought to arise from autonomic precursors in the neural crest. Mutations in the PHOX2B gene have been observed in familial and sporadic forms of neuroblastoma and represent the first defined genetic predisposition for neuroblastoma. Here, we address the mechanisms that may underlie this predisposition, comparing the function of wild-type and mutant Phox2b proteins ectopically expressed in proliferating, embryonic sympathetic neurons. Phox2b displays a strong antiproliferative effect, which is lost in all Phox2b neuroblastoma variants analyzed. In contrast, an increase in sympathetic neuron proliferation is elicited by Phox2b variants with mutations in the homeodomain when endogenous Phox2b levels are lowered by siRNA-mediated knockdown to mimic the situation of heterozygous PHOX2B mutations in neuroblastoma. The increased proliferation is blocked by Hand2 knockdown and the antiproliferative Phox2b effects are rescued by Hand2 overexpression, implying Hand2 in Phox2b-mediated proliferation control. A Phox2b variant with a nonsense mutation in the homeodomain elicits, in addition, a decreased expression of characteristic marker genes. Together, these results suggest that PHOX2B mutations predispose to neuroblastoma by increasing proliferation and promoting dedifferentiation of cells in the sympathoadrenergic lineage.
Introduction
Neuroblastoma is a tumor of the developing autonomic nervous system that shows a variety of phenotypes, from undifferentiated tumors in patients with poor outcome to those containing differentiated cell types which, depending on the degree of differentiation, show a better prognosis (Maris et al., 2007). The correlation between the degree of differentiation and tumor growth properties suggests that different tumor stages maintain molecular mechanisms characteristic for particular stages of autonomic neuron development (Edsjö et al., 2007). A detailed knowledge of mechanisms controlling proliferation and differentiation during embryonic development may thus lead to a better understanding of neuroblastoma and eventually also to novel tumor treatment approaches.
Autonomic neuron development is controlled by a network of transcription factors which includes Phox2b, Phox2a, Ascl1, Hand2 and Gata2/3 (Goridis and Rohrer, 2002; Howard, 2005; Huber, 2006). Phox2b is essential for the initial differentiation of autonomic neurons and, together with Hand2 and Gata3, regulates noradrenergic gene expression. Hand2 additionally controls proliferation of sympathetic progenitors and immature neurons (Hendershot et al., 2008; Schmidt et al., 2009). Whereas our understanding of autonomic neuron differentiation has considerably increased in the last few years, the molecular control of neurogenesis in autonomic ganglia is not well known. In sympathetic ganglia, as opposed to parasympathetic and sensory ganglia, both progenitors and immature neurons proliferate. The major increase in sympathetic neuron number during neurogenesis is due to neuronal proliferation rather than to progenitor proliferation and subsequent differentiation (Rohrer and Thoenen, 1987; Tsarovina et al., 2008) and it is unclear how proliferation of sympathetic neurons is terminated.
The finding that Phox2b overexpression inhibits the proliferation of CNS motoneuron progenitors (Dubreuil et al., 2000) together with antiproliferative effects of Phox2a/b overexpression in several cell lines (Paris et al., 2006; Raabe et al., 2008) suggested Phox2b as a molecular link between sympathetic neuron differentiation and proliferation and a tumor-suppressor role for Phox2a/b. It is, however, unclear whether the tumor predisposition caused by mutations in the PHOX2B gene in familial (Mossé et al., 2004; Trochet et al., 2004) and sporadic (van Limpt et al., 2004; McConville et al., 2006) cases of neuroblastoma, is due to the loss of tumor-suppressor function. On the contrary, tumors in familial cases of neuroblastoma showed neither mutations of the second allele nor loss of heterozygosity, suggesting that gain-of-function or dominant-negative effects may account for the oncogenic effects (Bourdeaut et al., 2005). The absence of tumors in response to a heterozygous deletion of the PHOX2B locus (Benailly et al., 2003), also argues against a loss-of-function effect in neuroblastoma predisposition.
Here, we analyzed the effects of Phox2b neuroblastoma mutations in potential tumor founder cells in the sympathoadrenergic lineage, using cultures of immature sympathetic neurons from chick and mouse embryos. The demonstration that neuroblastoma mutations lead to gain-of-function and dominant-negative Phox2b variants that increase sympathetic neuron proliferation and dedifferentiation provides a molecular explanation for neuroblastoma predisposition.
Materials and Methods
Construction of plasmids
Phox2b mutants were based on the murine isoform of Phox2b and created via mutagenesis of pCAGGS-Phox2b (site directed mutagenesis kit, Stratagene). Primers used for the mutagenesis PCR are as follows: Phox2bR100L, 5′-CGCAAGCAGCGGCTCATCCGCACCACC-3′, 5′-GGTGGTGCGGATGAGCCGCTGCTTGCG-3′; Phox2bR141G, 5′-CCTCACAGAGGCGGGAGTCCAGGTGTGG-3′, 5′-CCACACCTGGACTCCCGCCTCTGTGAGG-3′; Phox2b676delG, 5′-GCGCTCCGGGGCGGCGGGCCC-3′, 5′-GGGCCCGCCGCCCAGGAGCGC-3′; and Phox2bK155X, 5′-GCTAGCACCACCATGTATAAAATGGAATATTCTTACCTC-3′, 5′-TTATCGATCTAGCGAAACTTAGCCCG-3′.
siRNA
The siRNA against chick Phox2b targets the following sequence: 5′-CAAGAACGGAGCGGCCGGCAA-3′ (Qiagen, siPhox2b). SiPhox2b does not target the murine isoform of Phox2b due to nucleotide mismatches. The siRNA against Hand2 binds to the sequence 5′-CACAGTTAGCAGCAGCGATAA-3′ (Qiagen, siHand2) and results in selective Hand2 knockdown (Schmidt et al., 2009). The control siRNA, siLuc, recognizes the firefly luciferase (Qiagen): 5′-CACCTAGATCCTTTTAAAT-3′.
Primary sympathetic neuron culture preparation and transfection
Chick sympathetic neuron culture.
Paravertebral lumbosacral sympathetic chain ganglia were harvested from embryonic day 7 (E7) chicken embryos. Chemical and mechanical dissociation was performed as described before (Rohrer and Thoenen, 1987; Zackenfels et al., 1995). An Amaxa Nucleofector II was used to electroporate 200,000 cells according to Amaxa's protocol for primary chicken neurons (Program G-13, transfection efficiency: ≈50%). One microgram of pCAGGS-GFP was mixed with either 2 μg of empty vector/siLuc for controls or 2 μg of the appropriate pCAGGS-plasmid containing Phox2b-variants, Hand2, p27kip1, NMYC and Id2 or 2 μg of siPhox2b and siHand2. For the Phox2b rescue experiment cells were electroporated with 0.25 μg of Phox2b and 2 μg of Hand2 pCAGGS-plasmid. Transfected cells were plated on 4-well culture dishes coated with poly-dl-ornithine (Sigma) and laminin (Invitrogen). The cells were incubated at 5% CO2 and 37°C in MEM containing 10% horse serum, 5% FCS, 1% glutamine, 1% penicillin, and streptomycin. For proliferation analysis, bromodeoxyuridine/5-ethynyl-2′-deoxyuridine (BrdU/EdU) was added to the culture medium either 24 or 3 h before fixation (1:1000, Roche). Plasmids containing chick Id2, chick Hand2, human NMYC and mouse p27kip1 were generously provided by C. Goridis (École Normale Supérieure, Paris, France), M. Howard (University of Toledo, Toledo, Spain), C. Beltinger (University Clinic Ulm, Ulm, Germany), and P. Politis (Biomedical Research Foundation, Athens, Greece), respectively.
Mouse sympathetic neuron cultures.
Conditions for culture and proliferation analysis of embryonic mouse sympathetic neuroblasts were modified from Wyatt et al. (1999). Superior cervical ganglia (SCG) and stellate ganglia were dissected from E12 mouse embryos (C57BL/6J) staged according to the method of Theiler (1989). Single cell suspensions were obtained by enzymatic (0.1% collagenase/trypsin) and mechanical dissociation. For each Phox2b construct 30.000 cells were electroporated with either Phox2b pCAGGS-plasmids (0.6 μg) and GFP pCAGGS-plasmid (0.3 μg) or an empty vector (0.6 μg) and GFP pCAGGS-plasmid (0.3 μg) as control using an Amaxa Nucleofector II (program: SCN 5). Electroporated cells were plated in a defined serum-free medium (DMEM/F12 1:1, 1% N2-Supplement (Invitrogen), 0.1% BSA, 50 U/ml penicillin, 50 μg of streptomycin and 0.2 mg/ml l-glutamine) on poly-l-ornithine/collagen-coated glass coverslips (diameter 14 mm). NGF at a concentration of 50 ng/ml was added to the medium. Cells were maintained at 37°C in a humidified atmosphere containing 5% CO2.
Protein and mRNA assays.
Dissection and dissociation were performed as described previously (Rohrer and Thoenen, 1987; Zackenfels et al., 1995). E7 chick sympathetic ganglion cells (400,000) were electroporated with 3 μg of pCAGGS-GFP/siLuc for controls or 3 μg of the appropriate plasmid/siRNA. Transfected cells were then plated on 35 mm culture dishes coated with poly-dl-ornithine and laminin. Cells were harvested by scraping and pelleted by centrifuging (150 × g, 10 min). The pellet was either used for mRNA-isolation (Qiagen, mRNEasy Mini Kit) according to Qiagen's protocol or for protein isolation. To isolate cytoplasmatic and nuclear protein, cells were lysed in cold lysis buffer (containing, in mm: 50 Tris-HCl, pH 7.4, 150 NaCl, 40 NaF, 5 EDTA, 5 EGTA, 1 Na3VO4, 1% Triton X-100, 0.1% Na-deoxycholate, 0.1% SDS, 1 PMSF, 10 μg/ml aprotinin) for 10 min on ice. After short centrifugation (10,000 ×g, 10 min, 4°C) the supernatant was collected and protein content determined with the DC protein assay (Bio-Rad).
Protein blotting
Protein lysates were analyzed by SDS-PAGE and wet blotted on a PVDF-membrane (Bio-Rad). The membrane was blocked with 5% instant milk powder in TBST (containing, in mm: 10 Tris-HCl, pH 8, 150 NaCl, 0.05% Tween 20) for 1 h before antibody incubation. Phox2b (Santa Cruz Biotechnology, SC 13224, 1:1000) and the actin antibodies (Dianova, MS-1295, 1:5000) were diluted in blocking solution and incubated overnight at 4°C. After washing 3 times with TBST, the appropriate HRP-coupled secondary antibody (1:5000 in TBST) was incubated for 1 h at room temperature and washed off twice (TBS). Blots were developed using standard ECL according to manufacturer's instructions (Pierce, SuperSignal). For quantification of protein bands the one-dimensional gel analysis software (GE Healthcare, ImageQuant TL v2005) was used.
Coimmunoprecipitation.
SY5Y neuroblastoma cells were harvested and lysed in radioimmunoprecipitation assay (RIPA) buffer (containing, in mm: 25 Tris-HCl, pH 7.6, 150 NaCl, 0.5% Triton X-100, 1% sodium deoxycholate, 0.1% SDS) with complete protease inhibitor mix (Roche). Lysates were centrifuged for 10 min at 10,000 × g and 4°C to remove cell debris and afterward precleared with 30 μl of Protein G-agarose beads (Sigma-Aldrich) for 30 min under constant rotation. Precleared SY5Y lysates were incubated with anti-Hand2 or anti-GFP antibodies (Santa Cruz Biotechnology, SC 22818, 1:1000; Invitrogen, 1:1000) overnight at 4°C under constant rotation. Protein G-agarose beads were added for 4 h at 4°C with rotation. After extensive washes in RIPA buffer, the immunoprecipitates were separated by SDS-PAGE and analyzed by Western blot using a rabbit Phox2b antibody (kind gift from Christo Goridis, Paris, France, 1:1000).
GST-pulldown.
EcoRI and XbaI restriction sites were added to the 5′ and 3′ ends, respectively, of Phox2bwt and Phox2b mutant variants and subcloned into the corresponding site in pGEX_5x_1 bacterial expression vector (GE Healthcare Biosciences). To the 5′ and 3′ ends of Hand2 sequence BamHI and HindIII restriction sites were added and subcloned into the pRSET-A bacterial expression vector (Invitrogen) which adds a C-terminal His Tag. The proteins were expressed in Escherichia coli BL21 (DE3) (Merck). Expression was induced by 0.2 mm isopropyl-1-thio-β-d-galactopyranoside at 25°C for 20 h. The molecular weight of GST-fusion proteins Phox2bwt, Phox2bR100L, Phox2bR141G and the truncated Phox2bK155X corresponds to the expected molecular weight of the proteins fused with GST, 58 kDa and 42 kDa, respectively (Trochet et al., 2009). The frameshift mutant Phox2b676delG displays a lower molecular weight, which might be due to impaired translation in the bacterial system. GST-Phox2b fusion proteins (500 μl) were incubated with 40 μl of glutathione Sepharose beads for 2 h and washed 3 times with PBS. The matrix was incubated with His-tagged Hand2 protein lysate with 0.1% Triton X-100 to prevent unspecific binding for 2 h at 4°C and washed 3 times with PBS 0.5% Triton X-100. After washing, the GST-beads were eluted twice for 5 min at 48°C with 25 mm Tris-HCl pH 7.5, 192 μm glycine, 0.1% SDS and the supernatants were analyzed by SDS-PAGE and Western blotting. GST-beads alone and GST-protein lysates without GST-beads incubated with His-tagged Hand2 protein lysates served as negative controls for specific protein interaction. GST-fusion proteins were detected by anti-GST-HRP antibodies (Abcam) (1:40.000, TBS-T with 5% low fat milk powder) and Hand2 was visualized by anti-His antibodies (Abcam) (1:5000, TBST with 5% milk powder) and anti-rabbit-HRP-coupled secondary antibodies (Dianova) (1:5.000, TBS-T with 5% milk powder). Hand2 bacterial lysate was used as positive control for the Hand2 Western blot.
Semiquanitative reverse transcription PCR
cDNA synthesis on total RNA from mRNA-isolations of sympathetic neuron cultures was performed using the SuperScript III reverse transcriptase kit (Invitrogen). The following primer pairs were used for PCR: trkA forward, 5′-CCCCTCATCATGGTCTTCGA-3′ trkA reverse, 5′-TGTGGATGTCCTGGATGCGC-3′; tyrosine hydroxylase (Th) forward, 5′-GCGGCTACAATGAGAACAACATC-3′, Th reverse, 5′-GGCTCGTATTTCACTGAGAAGGG-3′; Dbh forward, 5′-GCACAACCGAGTCTGTCAACATC-3′, Dbh reverse, 5′-CATCGTGGGAACGCAGTTTG-3′; p27kip forward, 5′-AAGGGAGGGGGAGATGTCAAAC-3′, p27kip reverse, 5′-CATCGTGGGAACGCAGTTTG-3; Tlx3 forward, 5′-CTTCCAGAAGTCGCTCAACG-3′, Tlx3 reverse, 5′-TCAGACGAGGGAGGTGAC-3′; p75 forward, 5′-TGGGGCTCCAAGGAAAAGTG-3′, p75 reverse, 5′-ACCTCGTTCTCTTCGCAGATGG-3′; and Gapdh forward, 5′-AAGGGTGGTGCTAAGCGT-3′, Gapdh reverse, 5′-GCAGGGGCTCCAACAAAG-3′.
All PCRs were performed within the linear range that had been determined empirically for each pair of primer and cell preparation. The PCR products were separated on a 1.5% agarose gel and visualized with ethidium bromide. Quantification was done with the ScionImage Software version 4.03 (Scion) and the appropriate band intensity of amplified marker cDNAs was normalized to Gapdh. All results are given as the percentage ± SEM of control transfection of at least three independent experiments and were statistically analyzed using paired two-tailed Student's t test.
Immunostaining
BrdU/EdU.
Cells were washed with PBS and fixed using 4% paraformaldehyde in 0.1 m sodium phosphate buffer for 15 min. After washing with PBS, DNA was denatured with 2N HCl in case of BrdU staining. Cells were stained with anti-BrdU or EdU labeling kit according to the manufacturer's protocol (1:100, mouse, Roche, BrdU labeling kit) (Invitrogen, Click-It EdU Imaging Kit 594). GFP-expression was analyzed by staining with anti-GFP antibody (1:500, rabbit, Invitrogen). To detect BrdU-antibodies, biotinylated anti-mouse antibodies (1:500 in PBS, 0.5% Triton X-100 and 5% FCS, Jackson ImmunoResearch) followed by Cy3-coupled Streptavidin were used (Millipore Bioscience Research Reagents). Alexa488 anti-rabbit was used as secondary antibody to anti-GFP. Nuclei were stained with DAPI (Sanofi Aventis). Stainings were covered with Aqua Polymount (Polysciences) and analyzed at room temperature with a Zeiss Axiophot 2 microscope (40× objective, 0.75 Ph2). Data were raised by analyzing GFP-positive neurons with neurites for BrdU/EdU staining in at least 20 visual fields per culture dish at a magnification of 40×. A Visitron Systems Spot RT3 camera was used for image acquisition and MetaVue (7.1.3.0) for digital image processing (adjustment of brightness and contrast). All results are given as mean ± SEM of at least three independent experiments and statistically analyzed with paired two-tailed Student's t test.
Th, phospho-histone-3, and Phox2b.
Cells were fixed and washed as described before. Primary antibodies were diluted 1:500 in staining buffer and incubated for 1 h [mouse, Th, rabbit, phospho-histone-3 (pH3); Abcam]; at room temperature or overnight (goat, Phox2b, Santa Cruz Biotechnology). After washing with PBS, appropriate secondary antibodies were used (1:500, Alexa 594, Invitrogen). Cells were washed again with PBS and mounted as described before. Transfected cells were identified by GFP-fluorescence. Stainings were analyzed as described before.
Luciferase assays.
DF1 cells were grown on 24-well plates in DMEM with 10% FCS until achieving 80% confluency and transfected with Effectene according to manufacturer's protocol (Qiagen). Fifty nanograms of the different Phox2b-constructs were combined with 300 ng of DBH promotor Luciferase reporter-plasmid (Trochet et al., 2005a) and 30 ng of pRL-CMV Renilla reporter-plasmid as internal control. After 48 h luciferase and renilla activities were assayed with the Dual-Glo Luciferase assay system (Promega) and measured with a Glomax Luminometer (Turner Biosystems). Luciferase activity results were normalized to the internal Renilla controls and analyzed with Microsoft Excel. Reporter assays were repeated for at least 4 times in duplicate.
Results
Phox2bwt displays an antiproliferative effect, which is lost in neuroblastoma variants
Mutations in PHOX2B have been linked to several congenital human diseases, including congenital central hypoventilation syndrome (CCHS), Hirschsprung's disease and neuroblastoma (Amiel et al., 2003; Weese-Mayer et al., 2003; Mossé et al., 2004; Trochet et al., 2004). Whereas CCHS is caused by heterozygous PHOX2B mutations that selectively affect the length of the polyalanine repeats (Fig. 1A) (Amiel et al., 2003; Weese-Mayer et al., 2003; Trochet et al., 2004, 2005a), there is no obvious site selectivity of mutations that predispose to neuroblastoma. Mutations in neuroblastoma are either missense alterations in highly conserved regions, or frameshift mutations that lead to an altered or truncated protein. Here we investigated the functional properties of a subset of neuroblastoma Phox2b variants with point mutations in the homeodomain (R100L, Phox2bR100L; R141G, Phox2bR141G) (Trochet et al., 2004, 2005a,b), the K155X mutation (Phox2bK155X) (Weese-Mayer et al., 2003) that results in a C-terminally truncated Phox2b protein, and a frameshift mutation (676delG, Phox2b676delG) (Mossé et al., 2004) (Fig. 1A). The analysis also included wild-type mouse Phox2b (Phox2bwt; amino acid sequence identity mouse vs human Phox2b is 100%) and a dominant-negative variant of Phox2 proteins, composed of the Phox2 homeodomain (identical in Phox2b and Phox2a) fused to the engrailed repressor domain (Phox2HDen) (Dubreuil et al., 2002) (Fig. 1A).
Figure 1.
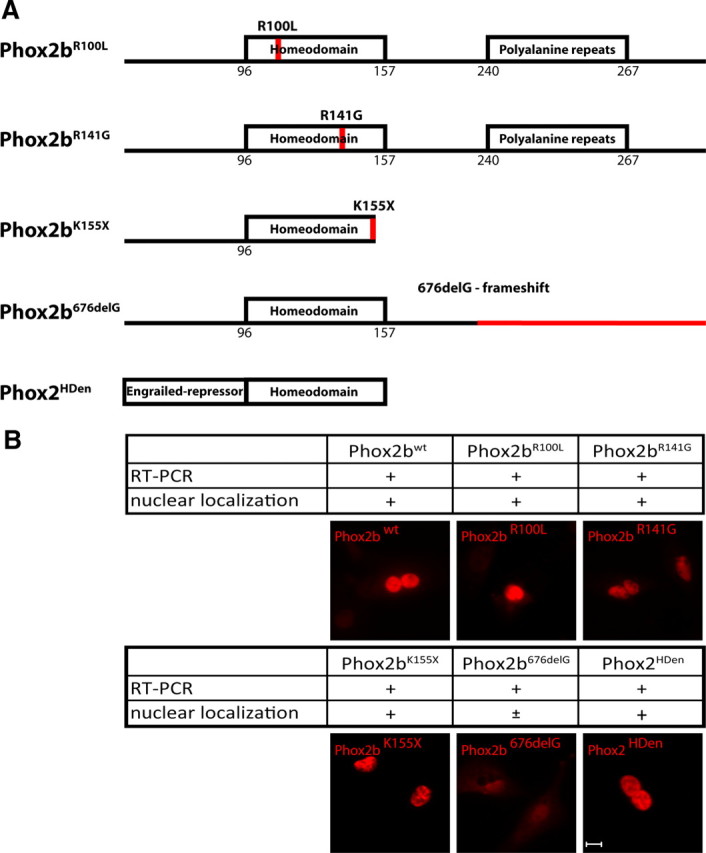
Schematic representation of Phox2bwt and mutant expression constructs (A) and cellular localization (B) of Phox2wt, Phox2bR100L, Phox2bR141G, Phox2bK155X, Phox2b676delG and Phox2HDen. A, Scheme illustrating the Phox2b structure, the location of the homeodomain and polyalanine repeats (plain boxes) and of Phox2b mutations (red). The fusion construct between the homeodomain and the engrailed repressor (Phox2HDen) is also shown schematically. B, Phox2b expression plasmids were transfected in DF1-cells and the expression of Phox2bwt and Phox2b variants was analyzed by RT-PCR and immunostaining, using an antibody directed against the Phox2 HD. All Phox2b variants were detected at RNA and protein level. Scale bar, 10 μm.
After transfection of DF-1 chick fibroblast cells, expression of Phox2bwt and all variants was observed by reverse transcription (RT)-PCR (Fig. 1B; data not shown). Immunohistological analysis of transfected fibroblasts (Fig. 1) and neurons (supplemental Fig. 1, available at www.jneurosci.org as supplemental material) revealed nuclear localization for Phox2bwt, Phox2bR100L, Phox2bR141G, Phox2bK155X and Phox2HDen. Phox2b676delG was also detected in the nucleus, albeit at lower levels (Fig. 1B).
To investigate the effects of Phox2b on proliferation in the sympathoadrenergic lineage we used sympathetic neuron cultures from E7 chick embryos. These cultures have previously been shown to contain proliferating cells that display neuronal morphology and express characteristic pan-neuronal and adrenergic markers (Rohrer and Thoenen, 1987; Zackenfels et al., 1995). They express trkB and proliferation is stimulated by BDNF (Straub et al., 2007) and forced expression of NMYC, Id2 and Hand2 (Fig. 2). TrkB signaling, NMYC and Id2 are implied in the altered proliferation control in neuroblastoma (Brodeur et al., 1984; Nakagawara et al., 1994; Jögi et al., 2002). The culture conditions closely mimic neurogenesis in vivo as neuron proliferation is controlled by Hand2 (Hendershot et al., 2008; Schmidt et al., 2009) (Fig. 2) and IGFs (Zackenfels et al., 1995). Here, the effects of Phox2bwt and Phox2b variants were analyzed by cotransfecting sympathetic ganglion cells with Phox2b and GFP expression plasmids (supplemental Fig. 1, available at www.jneurosci.org as supplemental material) and determining the proportion of BrdU-labeled GFP-positive cells (Fig. 3).
Figure 2.

Sympathetic neuron proliferation is increased by BDNF, NMYC, Id2 and Hand2 and reduced by Hand2 knockdown and p27kip1. E7 sympathetic neurons, transfected with expression vectors for GFP, or cotransfected with NMYC, Id2, Hand2, p27kip1 or siHand2 were analyzed for proportion of GFP-positive neurons labeled by BrdU as in Figure 3. BDNF was administered in a concentration of 50 μm. Data are the mean ± SEM (n = 3 independent experiments). *p < 0.05, **p < 0.01, ***p < 0.001, significantly different from control.
Figure 3.

Phox2bwt overexpression inhibits the proliferation of E7 sympathetic neurons, whereas Phox2b mutants Phox2bR100L, Phox2bR141G, Phox2bK155X, Phox2b676delG show no antiproliferative effect. Sympathetic neurons, transfected before plating with expression vectors for GFP, or cotransfected with Phox2bwt or Phox2bR100L, Phox2bR141G, Phox2bK155X and Phox2b676delG, were labeled and immunostained for BrdU (red), GFP (green) and cell nuclei (DAPI) after 2 d in culture. Expression of Phox2bwt, but not of Phox2b variants caused a decrease in the proportion of BrdU-labeled, GFP-positive transfected neurons. A, Effect of Phox2bwt and Phox2bR100L is shown. BrdU-labeled transfected neurons are indicated by arrowheads. Scale bar, 20 μm. B, The proportion of transfected GFP-positive neurons labeled by BrdU was determined after transfection with GFP only (control) or after cotransfection with Phox2bwt or the Phox2b mutants indicated. Data are the mean ± SEM (n = 3, independent experiments). ***p < 0.001, significantly reduced compared with control.
Overexpression of Phox2bwt resulted in a strong reduction in the number of BrdU-labeled cells, an effect not observed for the neuroblastoma Phox2b variants analyzed (Fig. 3A,B). Virtually identical results were obtained using cultures of embryonic (E12) mouse sympathetic neuron cultures (supplemental Fig. 2, available at www.jneurosci.org as supplemental material). The loss of antiproliferative function in Phox2b mutants is in agreement with the notion of a tumor suppressor role for Phox2b. However, the effect of ectopically expressed Phox2b variants may be restricted by the presence of the normal complement of endogenous Phox2b in sympathetic neurons, whereas in neuroblastoma, heterozygous PHOX2B mutations may be more effective due to the reduction of endogenous PHOX2Bwt. To address the question of how neuroblastoma Phox2b variants affect sympathetic neuron proliferation in cells with lower Phox2b levels, we have used chick sympathetic neuron cultures and a siRNA approach to reduce endogenous Phox2b expression.
Phox2bR100L, Phox2bR141G and Phox2bK155X stimulate sympathetic neuron proliferation after knockdown of endogenous Phox2b
Phox2b knockdown was achieved by nucleoporation of Phox2b-siRNAs and compared with Luciferase-siRNA (siLuc) as controls. Phox2b siRNA was designed to selectively interfere with endogenous chick Phox2b but not with ectopically expressed mouse Phox2b variants. Phox2b siRNA (siPhox2b) reduced the proportion of Phox2b-immunoreactive (Phox2b-IR) sympathetic neurons to 26 ± 9% compared with 100% Phox2b-IR neurons after transfection with control siRNA or pCAGGS (supplemental Fig. 3A, available at www.jneurosci.org as supplemental material). Western-blot analysis confirmed this result and showed a strong reduction (90%; n = 2) of Phox2b protein expression by siPhox2b (supplemental Fig. 3B,C, available at www.jneurosci.org as supplemental material).
For the analysis of sympathetic neuron proliferation after Phox2b knockdown, expression vectors for GFP and Phox2b variants were cotransfected with siRNAs and analyzed for BrdU-incorporation. Interestingly, the neuroblastoma Phox2b variants with mutations in the homeodomain, i.e., Phox2bR100L, Phox2bR141G, and Phox2bK155X, elicited a strong increase in sympathetic neuron proliferation when endogenous Phox2b was reduced (Fig. 4A). This is, to our knowledge, the first demonstration that neuroblastoma Phox2b mutations result in a gain-of-function phenotype. Basal proliferation of sympathetic neurons and the response to Phox2bwt and Phox2b676delG were not affected by Phox2b knockdown (compare Figs. 3B and 4A).
Figure 4.

Phox2b mutants Phox2bR100L, Phox2bR141G, Phox2bK155X increase sympathetic neuron proliferation when endogenous Phox2b levels are reduced by siRNA knockdown. A, E7 sympathetic neurons were transfected before plating with either pCAGGS-GFP (control), with pCAGGS-GFP and siPhox2b directed against endogenous chick Phox2b (Phox2b knockdown) or cotransfected with pCAGGS-GFP, siPhox2b plus pCAGGS vectors for Phox2bwt and the Phox2b mutants indicated. The proportion of BrdU-labeled transfected neurons was determined after 2 d in culture. BrdU-positive cells were reduced by Phox2bwt and increased by Phox2bR100L, Phox2bR141G, Phox2bK155X. Data are the mean ± SEM (n = 3 independent experiments). *p < 0.05, **p < 0.01, ***p < 0.001, significantly different from control. B, Effect of Phox2b mutants Phox2bR100L, Phox2bR141G, Phox2bK155X in the presence of apoptosis block and Phox2b knockdown. Phox2bR100L, Phox2bR141G, Phox2bK155X elicit increased sympathetic neuron proliferation also in the presence of the apoptosis inhibitor DEVD-CHO (5 μm). Data are the mean ± SEM (n = 3 independent experiments). *p < 0.05, **p < 0.01, significantly different from control. C, Effect of Phox2bwt, combined expression of Phox2bwt and Hand2, and Phox2b mutants Phox2bR100L, Phox2bR141G, Phox2bK155X and Phox2b676delG in the presence of apoptosis block and Phox2b/Hand2 knockdown conditions. Increased Hand2 levels rescue the antiproliferative effect of Phox2bwt. siRNA-mediated Hand2 knockdown abolishes the proliferation stimulating effect of Phox2b mutants (compare B and C). Data are the mean ± SEM (n = 4 independent experiments). ***p < 0.001, significantly different from control.
The interpretation of experiments with Phox2b knockdown are complicated by the fact that sympathetic neuron survival, determined by the number of GFP-positive transfected neurons, is impaired by reducing endogenous Phox2b levels (38% of control without siPhox2b). However, this effect is largely compensated by the coexpression of Phox2bwt and Phox2b neuroblastoma variants (Phox2bwt, Phox2bR100L, Phox2bR141G, and Phox2bK155X 87.3 ± 1.6%, 107 ± 3%, 102 ± 2%, and 107 ± 4% of control), arguing against the possibility that increased proliferation is compromised by effects on survival. To further support this notion, the effects of combined Phox2b knockdown and overexpression of Phox2b variants were analyzed under conditions where apoptotic cell death was blocked by the addition of the apoptosis inhibitor DEVD-CHO (Fig. 4B). The sympathetic neuron survival in siPhox2b-treated cultures was maintained at 91 ± 8% of controls by the addition of DEVD-CHO, compared with 38 ± 1% after Phox2b knockdown in the absence of the apoptosis inhibitor. The proliferation increase in response to Phox2bR100L, Phox2bR141G and Phox2bK155X was also observed under these conditions (Fig. 4B). The frameshift mutant Phox2b676delG showed no effect which is most likely due to the inefficient transfer to the nucleus. Together, these findings demonstrate that the increased proportion of BrdU-labeled cells in response to Phox2bR100L, Phox2bR141G and Phox2bK155X is caused by increased proliferation rather than by increased survival of proliferating cells. In agreement with this conclusion, proliferation effects by Phox2bwt, Phox2bR100L, Phox2bR141G and Phox2bK155X were also observed after short-term (3 h) BrdU-labeling (data not shown) and by phospho-histone-3 staining (supplemental Fig. 4, available at www.jneurosci.org as supplemental material), which determines the number of cells in M phase.
Proliferation effects of Phox2b and Phox2b variants involve Hand2
Hand2 is essential for the proliferation of immature sympathetic neurons in vivo (Hendershot et al., 2008; Schmidt et al., 2009) and sufficient to increase sympathetic neuron proliferation in vitro (Fig. 2). As Hand2 was previously shown to synergistically interact with Phox2a in the control of noradrenergic differentiation (Rychlik et al., 2003; Xu et al., 2003) we investigated the functional interaction of Hand2 and Phox2b in the control of sympathetic neuron proliferation by coexpression experiments. The antiproliferative effect of Phox2bwt could be completely compensated by coexpression of Hand2 (Fig. 4C) and was enforced upon siRNA-mediated Hand2 knockdown (supplemental Figs. 5, 6, available at www.jneurosci.org as supplemental material), which suggests that the Phox2bwt effect is mediated by interfering with Hand2. Interestingly, also the proliferation increase elicited by the Phox2b variants depends on Hand2, as this effect could be blocked by knockdown of Hand2 (Fig. 4C; supplemental Fig. 6, available at www.jneurosci.org as supplemental material). However, Phox2b variants display a positive functional interaction with Hand2, in contrast to Phox2bwt.
The antagonistic and stimulatory interactions between Phox2b/Phox2b variants and Hand2 could be explained by mechanisms shown to act during noradrenergic neuron specification and differentiation, i.e., effects of Phox2b/Phox2b variants on Hand2 expression (Hendershot et al., 2008) or to a direct interaction of Phox2b/Phox2b variants and Hand2 proteins. Interestingly, Phox2b overexpression in proliferating sympathetic neurons resulted in a reduction of Hand2 mRNA levels to 59.9 ± 15% of control transfections (p < 0.02), whereas siRNA-mediated Phox2b knockdown increased Hand2 mRNA levels to 172 ± 22% of controls (p < 0.05). Notably, Hand2 expression is negatively regulated by Phox2b in sympathetic neurons, in contrast to the Phox2b-dependent Hand2 expression during initial development of sympathetic progenitors (Hendershot et al., 2008). In addition, we now demonstrate the interaction between Hand2 and Phox2b by coimmunoprecipitation (Fig. 5A) and provide evidence for direct protein–protein binding between Hand2, Phox2bwt and Phox2b variants by pulldown experiments (Fig. 5C,D). Neither protein-G agarose beads coated with anti-GFP antibody (Fig. 5B) nor agarose beads alone (data not shown) showed Phox2b signal after immunoprecipitation. To study protein–protein interactions between Hand2 and Phox2b, glutathione-agarose beads containing bacterially expressed GST-Phox2bwt or GST-Phox2b variants were incubated with lysates of Hand2-His-expressing bacteria. Hand2 did not interact with the glutathione-agarose beads or GST protein alone (data not shown), but when GST-Phox2b-bound beads were added, strong signals were observed (Fig. 5D), demonstrating protein–protein interaction between Phox2bwt, Phox2b variants and Hand2. As both Phox2bwt and Phox2b variants bind to Hand2 the homeodomain mutations may elicit a change from the antagonistic function of Phox2bwt to a synergistic interaction of Phox2b variants with Hand2. Alternatively, Phox2b variants may deplete antiproliferative Phox2bwt from the Hand2 interaction partner. Together, these results suggest that the proliferation effects of Phox2b are mediated, at least in part, by Hand2 and imply both transcriptional and posttranscriptional mechanism in this interaction.
Figure 5.

Protein–protein interaction between Phox2b and Hand2. A, B, Coimmunoprecipitation of Phox2b and Hand2 from SY5Y neuroblastoma cells. Western blots with antibodies specific for Phox2b after immunoprecipitation with anti-Hand2 (A) or anti-GFP (B) antibodies. Input, Input control; SN, supernatant after agarose bead and Hand2 (A) or GFP (B) antibody incubation; W, supernatant after washing immunopellet with lysis buffer; IP, immunopellet. Anti-GFP antibody (B) and agarose beads alone (data not shown) served as negative controls. Phox2b bands with different apparent molecular weights most likely represent differentially phosphorylated forms (Adachi and Lewis, 2002). C, D, Analysis of Phox2b–Hand2 interaction by pulldown experiments. Bacterial lysates of GST-Phox2b fusion proteins were first incubated with GST-beads and subsequently with Hand2 bacterial lysate. C, Western blot analysis of GST-Phox2b fusion proteins that were used for the pulldown of Hand2 visualized by GST-HRP-coupled antibodies. D, Hand2-His was detected by antibodies against the His-tag and is present in every lane after GST-pulldown with GST-Phox2b fusion proteins. GST-beads alone and GST-Protein served as negative controls whereas Hand2 protein lysate served as positive control for specific protein–protein interaction (data not shown).
Sympathetic neuron cell cycle withdrawal is not strictly linked to p27kip1 increase
Cyclin-dependent kinase (Cdk) inhibitors (CKIs) are important regulators of cell cycle exit during development. The Cip/Kip family of inhibitors (p21cip1, p27kip1, and p57kip2) inactivate Cdk2 and Cdk1 and play a central role as negative regulators of cell cycle progression in the developing nervous system (Dyer and Cepko, 2001; Gui et al., 2007). Individual CKIs are required for cell cycle exit in specific subpopulations of cells in retina and spinal cord, suggesting that different cell populations employ a distinct molecular equipment to control cell cycle exit (Dyer and Cepko, 2001; Gui et al., 2007). A previous study implied p27kip1 downstream of Phox2a in cell cycle withdrawal of the noradrenergic CAD cell line (Paris et al., 2006). In cultured sympathetic neurons, p27kip1 overexpression results in a virtually complete proliferation block (Fig. 2). To further address the role of p27kip1 in cultured sympathetic neurons, p27kip1 levels were determined by RT-PCR under various conditions that increase or decrease sympathetic neuron proliferation. Phox2b overexpression reduced proliferation and significantly increased p27kip1 expression, whereas increased proliferation by Hand2 overexpression resulted in lower p27kip1 expression levels (supplemental Fig. 7, available at www.jneurosci.org as supplemental material). However, no correlation between sympathetic neuron proliferation and p27kip1 was observed after Phox2b and Hand2 knockdown (see Fig. 2 and supplemental Fig. 7, available at www.jneurosci.org as supplemental material). Thus, cell cycle exit of sympathetic neurons is not exclusively linked to an increase in p27kip1. Rather, additional mechanisms of action for Phox2 and Hand2 proteins in cell cycle control are implied.
In cultured proliferating sympathetic neurons Phox2a expression was not detectable by immunostaining and RT-PCR (40 cycles) (data not shown). Together with the observation that Phox2b but not Phox2a is essential for sympathetic neuron development (Coppola et al., 2005) our findings argue against an involvement of Phox2a in sympathetic neuron proliferation.
Phox2bK155X elicits a decrease in the expression of characteristic marker genes and all neuroblastoma Phox2b variants have lost the ability to induce p75 and Tlx3
One of the best understood functions of Phox2b is its role in the initial expression of Dbh and Th in sympathetic neuron progenitors (Zellmer et al., 1995; Yang et al., 1998; Pattyn et al., 1999; Adachi et al., 2000). Here, we addressed the question whether Th and Dbh expression is affected by Phox2bwt and Phox2b neuroblastoma variants in E7 chick sympathetic neurons, i.e., sympathoadrenergic cells that already have acquired noradrenergic neuron properties. In addition, effects on the neurotrophin receptors trkA, p75 and the transcription factor Tlx3 were investigated. The analysis of trkA expression and p75 was of particular interest as elevated trkA and p75 levels correlate with good prognosis in neuroblastoma patients (Kogner et al., 1993; Nakagawara et al., 1993). Effects of Phox2b on Tlx3 expression were studied since Tlx3 is the chick ortholog of Tlx2, a direct Phox2 target gene (Borghini et al., 2006). Sympathetic neurons were transfected with Phox2bwt and Phox2b mutant expression vectors and analyzed by RT-PCR for Dbh, Th, trkA, p75 and Tlx3 expression. Phox2bwt increased the expression of Tlx3 and p75 but did not affect Dbh, Th and trkA (Fig. 6). This suggests that the endogenous Phox2b levels are saturating in these cells for Dbh, Th and trkA expression, but not for Tlx3 and p75. Interestingly, Phox2bK155X elicited a significant decrease in the expression of all genes investigated. Considering the transfection efficiency of ∼50%, effects are substantially stronger if only transfected cells were taken into account. Indeed, when the effect on TH expression was analyzed by TH-immunostaining of identified transfected cells, the proportion of TH-expressing cells was reduced to 24 ± 4% (Fig. 7). The comparable decrease in Dbh, Th, trkA, p75 and Tlx3 expression in response to the Phox2bK155X and Phox2HDen (Figs. 6, 7) suggests that the K155X nonsense mutation results in a dominant-negative function of this Phox2b variant. This notion was confirmed using the DBH promotor luciferase reporter system (supplemental Fig. 8, available at www.jneurosci.org as supplemental material). Phox2bK155X and Phox2HDen completely repress the Phox2bwt-induced transcription whereas other Phox2b variants with mutations in the homeodomain were unable to antagonize the Phox2bwt effect (Phox2bR141G is shown as representative example). The transactivating function of Phox2bwt at the DBH promotor is strongly reduced in all Phox2b neuroblastoma variants analyzed using DBH luciferase reporter assays (supplemental Fig. 8, available at www.jneurosci.org as supplemental material; data not shown), which is in agreement with previous results (Trochet et al., 2005a, 2009; Raabe et al., 2008).
Figure 6.

Effects of Phox2bwt and Phox2b mutants on Dbh, trkA, Th, Tlx3 and p75 mRNA levels in E7 sympathetic neuron cultures. Sympathetic neurons were transfected before plating with pCAGGS-GFP (control) or with pCAGGS-expression vector for Phox2bR100L, Phox2bR141G, Phox2bK155X, Phox2b676delG or Phox2HDen. Cells were harvested after 2 d in culture, and expression of Dbh, trkA, Th, Tlx3 and p75 was analyzed by semiquantitative RT-PCR. Expression levels are normalized to Gapdh and referred to control transfections. Data are the mean ± SEM (n = 3 independent experiments). *p < 0.05, significantly different from control.
Figure 7.

Effects of Phox2bwt and Phox2b mutants on the expression of TH-protein in E7 sympathetic neuron cultures. Sympathetic neurons, transfected with pCAGGS-GFP (for controls), or cotransfected with Phox2bwt or Phox2b mutants were labeled and immunostained for TH (red) after 2 d in culture. A, Control GFP and Phox2HDen transfections are shown. Scale bar represents 20 μm. B, Quantitative analysis of TH-immunoreactive GFP-positive transfected neurons. Data are the mean ± SEM (n = 3 independent experiments). *p < 0.05, significantly reduced compared with control.
In conclusion, all Phox2b neuroblastoma mutations investigated abolished the transactivating properties of Phox2bwt. The K155X mutation, in addition, gives rise to a Phox2b variant with novel, dominant-negative functions that lead to dedifferentiation of sympathetic neurons.
Discussion
Heterozygous mutations in the transcription factor PHOX2B, described in familial and sporadic forms of neuroblastoma represent the first defined mutations predisposing to this childhood tumor. Here, we demonstrate that Phox2b mutations lead to both loss- and detrimental gain-of-function variants, affecting proliferation and differentiation in the sympathoadrenergic lineage. These results provide an explanation for the tumor predisposing role of PHOX2B neuroblastoma mutations.
Proliferation effects of Phox2bwt and Phox2b variants
The generation of differentiated neurons in the developing nervous system involves proliferation of progenitors, followed by cell cycle exit and onset of neuron differentiation. Phox2b coordinates cell cycle exit and neuronal differentiation in hindbrain motoneurons (Dubreuil et al., 2000, 2002). In the sympathoadrenergic lineage, Phox2b is also essential for neuron differentiation (Pattyn et al., 1999) but cell cycle exit and differentiation are not coupled in sympathoadrenal cells (Rohrer and Thoenen, 1987), and Phox2b is expressed in proliferating progenitors and proliferating sympathetic neurons. Thus, it was unclear whether cell cycle withdrawal involves Phox2b.
The present findings reveal that forced expression of Phox2b reduces sympathetic neuron proliferation and that the neuroblastoma Phox2b variants have lost their antiproliferative function. A recent comparison of the effects of Phox2b and neuroblastoma variants in neuroblastoma cell lines also showed impaired antiproliferative effects of Phox2b variants (Raabe et al., 2008). However, the importance of this loss-of-function for neuroblastoma predisposition is unclear, as Hirschsprung's disease and hypotonia but not neuroblastoma were reported in a patient with a heterozygous deletion of the entire Phox2b locus (Benailly et al., 2003). The phenotype in heterozygous Phox2b mice is even weaker and only includes atrophy of ciliary ganglia and a defective response to hypercapnia (Dauger et al., 2003; Cross et al., 2004). We now provide the first demonstration that neuroblastoma Phox2b mutations give rise to gain-of-function variants that stimulate proliferation of potential tumor founder cells of the sympathoadrenergic lineage. Thus, predisposition to tumor formation may rather rely on the acquisition of novel, proliferation-stimulating properties rather than on the loss of antiproliferative function. Increased sympathetic neuron proliferation in response to neuroblastoma variants is revealed when the levels of endogenous Phox2b are reduced by siRNA-mediated knockdown. This can be explained by the fact that the effects of ectopically expressed Phox2b variants are occluded in the presence of the normal complement of endogenous Phox2b. The Phox2b knockdown resembles the situation in neuroblastoma, where patients with PHOX2B mutations are thought to have a 50% reduction of PHOX2Bwt levels.
Why is the reduction of endogenous Phox2b levels by siRNA-mediated knockdown not sufficient to increase sympathetic neuron proliferation? This may be explained by an indirect action of Phox2b, interfering with the function of a proliferation regulating protein upon overexpression but unable to relieve its control. An alternative possibility would be that the knockdown of Phox2b does not sufficiently reduce endogenous Phox2b levels to overcome the antiproliferative effect of Phox2b. It should also be noted that the transactivation activity and protein interaction of Phox2a/b proteins is negatively regulated by phosphorylation (Adachi and Lewis, 2002) and that Phox2b overexpression may lead to an excess of active, unphosphorylated protein, whereas the knockdown effect may be compensated by dephosphorylation.
Effects of Phox2b and Phox2b variants on sympathetic neuron gene expression
The major role of Phox2b during development is the initiation of autonomic neuron differentiation. In the present study we have compared the effects of Phox2bwt and Phox2b variants on the expression of the Phox2b target genes Dbh, Th, and Tlx3 (Zellmer et al., 1995; Yang et al., 1998; Borghini et al., 2006), and trkA, p75, which are of interest in the context of neuroblastoma (Kogner et al., 1993; Nakagawara et al., 1993). Forced Phox2b expression does not affect Th, Dbh and trkA but leads to increased Tlx3 and p75 expression. This suggests that the endogenous Phox2b levels are saturating for Th, Dbh and trkA expression but not for Tlx3 and p75. All Phox2b variants analyzed have lost the ability to induce Tlx3 and p75 expression. Notably, Phox2bK155X decreased the expression of all genes investigated. As a similar decrease was observed in response to the dominant-negative Phox2HDen variant and both Phox2bK155X and Phox2HDen display a dominant-negative function at the DBH promotor, we conclude that Phox2bK155X interferes with the function of endogenous Phox2b and causes dedifferentiation. The strong dedifferentiation of sympathetic neurons observed here is in agreement with massive disturbance in the affected patients, suffering from CCHS, Hirschsprungs's disease and neuroblastoma (Weese-Mayer et al., 2003). Thus, impaired differentiation and/or dedifferentiation represent additional mechanisms underlying neuroblastoma predisposition.
Structure/function correlations of Phox2b and Phox2b variants
Phox2b and Phox2a are transcriptional activators that interact with homeodomain binding sites in the promotor region and directly stimulate the transcriptional activity of genes like Dbh (Yang et al., 1998; Adachi et al., 2000), Phox2b (Cargnin et al., 2005) and Tlx2 (Borghini et al., 2006). The effects of Phox2a/b are blocked by the dominant-negative chimera Phox2HDen (Dubreuil et al., 2000, 2002). Thus, the repression of Dbh, Th, trkA, p75 and Tlx3 by both Phox2HDen and Phox2bK155X may be explained by a dominant-negative function at homeodomain-binding promotor regions. However, as the homeodomain mutation in Phox2bK155X not only reduces promotor binding (Trochet et al., 2009) but may also affect protein–protein interaction (Kasahara et al., 2001), dominant-negative effects may be caused by heterodimerization with endogenous transcriptional regulators like Phox2b (Adachi et al., 2000) and Hand2, resulting in inactive transcription complexes. All Phox2b mutations analyzed have lost the ability to induce Tlx3 and p75 expression. This was expected from the lack of transactivation activity of Phox2bR100L, Phox2bR141G, Phox2b676delG and Phox2bK155X at the Phox2b and DBH promotor (Trochet et al., 2005a, 2009; Raabe et al., 2008). The loss-of-function is explained for the homeodomain mutations by impaired promotor interaction, for the frameshift mutation by the formation of oligomers with reduced nuclear transfer (Trochet et al., 2009).
Whereas the effects of Phox2b mutations on neuron differentiation are explained by altered functions of a transcriptional activator, the mechanisms involved in the proliferation effects seem to be more complex. Increased p27kip1 expression during cell cycle exit elicited by Phox2bwt in cultured sympathetic neurons, together with the p27kip1-induced proliferation stop suggests that p27kip1 may mediate the effect of Phox2b on the cell cycle. The correlation between increased proliferation and reduced p27kip1 expression is also observed upon Hand2 overexpression, but was not maintained upon Phox2b and Hand2 knockdown. Thus, p27kip1 levels are not strictly linked to cell cycle exit in sympathetic neurons, arguing against a general role of p27kip1 in the termination of sympathetic neuron proliferation.
An attractive mechanism would be that the proliferation effects of Phox2bwt and Phox2b variants are mediated through Hand2. This notion is supported by four kinds of observations: (1) both antiproliferative effects of Phox2bwt and proliferation stimulatory effects of Phox2b variants depend on Hand2; (2) Hand2 is essential for the proliferation of sympathetic neurons in vivo and in vitro; (3) Phox2bwt and Phox2b variants directly interact with Hand2; and (4) Phox2bwt negatively regulates the expression of Hand2 in sympathetic neurons. The proposed mechanism involving Phox2b–Hand2 interactions would imply that the homeodomain mutations lead to a change from an antagonistic to a synergistic interaction. An alternative explanation would be that Phox2b mutants displace Phox2bwt from the putative direct interaction with Hand2 and thus abrogate the antiproliferative effects of Phox2bwt by a dominant-negative mechanism. The N-terminal region of Phox2b which contains the transactivation domain of Phox2b, the CBP-interaction domain (Adachi et al., 2000) and a number of highly conserved phosphorylation sites (Trochet et al., 2009) is inferred in the proliferation stimulatory interaction of the Phox2b variants as proliferation is increased by the C-terminally truncated Phox2bK155X.
An essential role of Hand2 mediating the tumor predisposition of Phox2b mutations would also explain the restriction of the Phox2b mutation effects to sympathoadrenal cells as Hand2, in contrast to Phox2b, is not expressed in parasympathetic neurons (Müller and Rohrer, 2002).
Phox2b mutations and predisposition to neuroblastoma
The appearance of neuroblastoma in childhood or in utero indicates that early disruption of normal developmental processes plays an important role in tumor initiation. Familial forms of neuroblastoma, representing 1–2% of cases, show autosomal dominant inheritance with incomplete penetrance. Genetic analyses have identified several predisposition loci, which suggests that initiation of tumorigenesis could require multiple alterations (Maris et al., 2007). In addition to PHOX2B (Mossé et al., 2004; Trochet et al., 2004), the orphan tyrosine kinase receptor ALK has recently been identified as a second gene where mutations predispose to neuroblastoma (Chen et al., 2008; George et al., 2008; Janoueix-Lerosey et al., 2008; Mossé et al., 2008). Mutations in Phox2b and Alk increase proliferation and thus may share a similar predisposition mechanism by prolonging the period of neurogenesis and expanding the time window for tumor-initiating events, i.e., (an) additional somatic mutation(s) (Knudson, 2001). A second consequence of Phox2b mutations is dedifferentiation, reflected by a decreased expression of characteristic differentiation genes. Terminal differentiation is a critical barrier to malignant transformation (Brodeur, 2003) and the abrogation of the differentiation process by the Phox2bK155X variant may increase the susceptibility to additional tumor-initiating events. In conclusion, our data suggest that neuroblastoma Phox2b variants may predispose to this disease by increasing neurogenesis and interfering with the differentiation in the sympathoadrenal lineage.
Footnotes
This work was supported by grants from the Wilhelm Sander-Stiftung and the Schram-Stiftung. We thank J. Amiel and M. Howard for communicating unpublished results, and C. Beltinger, U. Ernsberger, C. Madry, S. Momma, and D. Schulte for critical comments on this manuscript. Thanks are due to N. Fürst for excellent technical assistance and E. Binder for the cloning of Phox2bK155X.
References
- Adachi M, Lewis EJ. The paired-like homeodomain protein, arix, mediates protein kinase A-stimulated dopamine β-hydroxylase gene transcription through its phosphorylation status. J Biol Chem. 2002;277:22915–22924. doi: 10.1074/jbc.M201695200. [DOI] [PubMed] [Google Scholar]
- Adachi M, Browne D, Lewis EJ. Paired-like homeodomain proteins Phox2a/Arix and Phox2b/NBPhox have similar genetic organization and independently regulate dopamine-β-hydroxylase gene transcription. DNA Cell Biol. 2000;19:539–554. doi: 10.1089/104454900439773. [DOI] [PubMed] [Google Scholar]
- Amiel J, Laudier B, Attié-Bitach T, Trang H, de Pontual L, Gener B, Trochet D, Etchevers H, Ray P, Simmonneau M, Vekemans M, Munnich A, Gaultier C, Lyonnet S. Polyalanine expansion and frameshift mutations of the paired-like homeobox gene PHOX2B in congenital central hypoventilation syndrome. Nat Genet. 2003;33:459–461. doi: 10.1038/ng1130. [DOI] [PubMed] [Google Scholar]
- Benailly HK, Lapierre JM, Laudier B, Amiel J, Attié T, De Blois MC, Vekemans M, Romana SP. PMX2B, a new candidate gene for Hirschsprung's disease. Clin Genet. 2003;64:204–209. doi: 10.1034/j.1399-0004.2003.00105.x. [DOI] [PubMed] [Google Scholar]
- Borghini S, Bachetti T, Fava M, Di Duca M, Cargnin F, Fornasari D, Ravazzolo R, Ceccherini I. The TLX2 homeobox gene is a transcriptional target of PHOX2B in neural-crest-derived cells. Biochem J. 2006;395:355–361. doi: 10.1042/BJ20051386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourdeaut F, Trochet D, Janoueix-Lerosey I, Ribeiro A, Deville A, Coz C, Michiels JF, Lyonnet S, Amiel J, Delattre O. Germline mutations of the paired-like homeobox 2B (PHOX2B) gene in neuroblastoma. Cancer Lett. 2005;228:51–58. doi: 10.1016/j.canlet.2005.01.055. [DOI] [PubMed] [Google Scholar]
- Brodeur GM. Neuroblastoma: biological insights into a clinical enigma. Nat Rev Cancer. 2003;3:203–216. doi: 10.1038/nrc1014. [DOI] [PubMed] [Google Scholar]
- Brodeur GM, Seeger RC, Schwab M, Varmus HE, Bishop JM. Amplification of N-myc in untreated human neuroblastomas correlates with advanced disease stage. Science. 1984;224:1121–1124. doi: 10.1126/science.6719137. [DOI] [PubMed] [Google Scholar]
- Cargnin F, Flora A, Di Lascio S, Battaglioli E, Longhi R, Clementi F, Fornasari D. PHOX2B regulates its own expression by a transcriptional auto-regulatory mechanism. J Biol Chem. 2005;280:37439–37448. doi: 10.1074/jbc.M508368200. [DOI] [PubMed] [Google Scholar]
- Chen Y, Takita J, Choi YL, Kato M, Ohira M, Sanada M, Wang L, Soda M, Kikuchi A, Igarashi T, Nakagawara A, Hayashi Y, Mano H, Ogawa S. Oncogenic mutations of ALK kinase in neuroblastoma. Nature. 2008;455:971–974. doi: 10.1038/nature07399. [DOI] [PubMed] [Google Scholar]
- Coppola E, Pattyn A, Guthrie SC, Goridis C, Studer M. Reciprocal gene replacements reveal unique functions for Phox2 paralogous homeobox genes during neural differentiation. EMBO J. 2005;24:4392–4403. doi: 10.1038/sj.emboj.7600897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cross SH, Morgan JE, Pattyn A, West K, McKie L, Hart A, Thaung C, Brunet JF, Jackson IJ. Haploinsufficiency for Phox2b in mice causes dilated pupils and atrophy of the ciliary ganglion: mechanistic insights into human congenital central hypoventilation syndrome. Hum Mol Genet. 2004;13:1433–1439. doi: 10.1093/hmg/ddh156. [DOI] [PubMed] [Google Scholar]
- Dauger S, Pattyn A, Lofaso F, Gaultier C, Goridis C, Gallego J, Brunet JF. Phox2b controls the development of peripheral chemoreceptors and afferent visceral pathways. Development. 2003;130:6635–6642. doi: 10.1242/dev.00866. [DOI] [PubMed] [Google Scholar]
- Dubreuil V, Hirsch MR, Pattyn A, Brunet JF, Goridis C. The Phox2b transcription factor coordinately regulates neuronal cell cycle exit and identity. Development. 2000;127:5191–5201. doi: 10.1242/dev.127.23.5191. [DOI] [PubMed] [Google Scholar]
- Dubreuil V, Hirsch MR, Jouve C, Brunet JF, Goridis C. The role of Phox2b in synchronizing pan-neuronal and type-specific aspects of neurogenesis. Development. 2002;129:5241–5253. doi: 10.1242/dev.129.22.5241. [DOI] [PubMed] [Google Scholar]
- Dyer MA, Cepko CL. p27Kip1 and p57Kip2 regulate proliferation in distinct retinal progenitor cell populations. J Neurosci. 2001;21:4259–4271. doi: 10.1523/JNEUROSCI.21-12-04259.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edsjö A, Holmquist L, Påhlman S. Neuroblastoma as an experimental model for neuronal differentiation and hypoxia-induced tumor cell dedifferentiation. Semin Cancer Biol. 2007;17:248–256. doi: 10.1016/j.semcancer.2006.04.005. [DOI] [PubMed] [Google Scholar]
- George RE, Sanda T, Hanna M, Fröhling S, Luther W, 2nd, Zhang J, Ahn Y, Zhou W, London WB, McGrady P, Xue L, Zozulya S, Gregor VE, Webb TR, Gray NS, Gilliland DG, Diller L, Greulich H, Morris SW, Meyerson M, et al. Activating mutations in ALK provide a therapeutic target in neuroblastoma. Nature. 2008;455:975–978. doi: 10.1038/nature07397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goridis C, Rohrer H. Specification of catecholaminergic and serotonergic neurons. Nat Rev Neurosci. 2002;3:531–541. doi: 10.1038/nrn871. [DOI] [PubMed] [Google Scholar]
- Gui H, Li S, Matise MP. A cell-autonomous requirement for Cip/Kip cyclin-kinase inhibitors in regulating neuronal cell cycle exit but not differentiation in the developing spinal cord. Dev Biol. 2007;301:14–26. doi: 10.1016/j.ydbio.2006.10.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendershot TJ, Liu H, Clouthier DE, Shepherd IT, Coppola E, Studer M, Firulli AB, Pittman DL, Howard MJ. Conditional deletion of Hand2 reveals critical functions in neurogenesis and cell type-specific gene expression for development of neural crest-derived noradrenergic sympathetic ganglion neurons. Dev Biol. 2008;319:179–191. doi: 10.1016/j.ydbio.2008.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howard MJ. Mechanisms and perspectives on differentiation of autonomic neurons. Dev Biol. 2005;277:271–286. doi: 10.1016/j.ydbio.2004.09.034. [DOI] [PubMed] [Google Scholar]
- Huber K. The sympathoadrenal cell lineage: specification, diversification, and new perspectives. Dev Biol. 2006;298:335–43. doi: 10.1016/j.ydbio.2006.07.010. [DOI] [PubMed] [Google Scholar]
- Janoueix-Lerosey I, Lequin D, Brugières L, Ribeiro A, de Pontual L, Combaret V, Raynal V, Puisieux A, Schleiermacher G, Pierron G, Valteau-Couanet D, Frebourg T, Michon J, Lyonnet S, Amiel J, Delattre O. Somatic and germline activating mutations of the ALK kinase receptor in neuroblastoma. Nature. 2008;455:967–970. doi: 10.1038/nature07398. [DOI] [PubMed] [Google Scholar]
- Jögi A, Persson P, Grynfeld A, Påhlman S, Axelson H. Modulation of basic helix-loop-helix transcription complex formation by Id proteins during neuronal differentiation. J Biol Chem. 2002;277:9118–9126. doi: 10.1074/jbc.M107713200. [DOI] [PubMed] [Google Scholar]
- Kasahara H, Usheva A, Ueyama T, Aoki H, Horikoshi N, Izumo S. Characterization of homo- and heterodimerization of cardiac Csx/Nkx2.5 homeoprotein. J Biol Chem. 2001;276:4570–4580. doi: 10.1074/jbc.M004995200. [DOI] [PubMed] [Google Scholar]
- Knudson AG. Two genetic hits (more or less) to cancer. Nat Rev Cancer. 2001;1:157–162. doi: 10.1038/35101031. [DOI] [PubMed] [Google Scholar]
- Kogner P, Barbany G, Dominici C, Castello MA, Raschellá G, Persson H. Coexpression of messenger RNA for trk protooncogene and low affinity nerve growth factor receptor in neuroblastoma with favorable prognosis. Cancer Res. 1993;53:2044–2050. [PubMed] [Google Scholar]
- Maris JM, Hogarty MD, Bagatell R, Cohn SL. Neuroblastoma. Lancet. 2007;369:2106–2120. doi: 10.1016/S0140-6736(07)60983-0. [DOI] [PubMed] [Google Scholar]
- McConville C, Reid S, Baskcomb L, Douglas J, Rahman N. PHOX2B analysis in non-syndromic neuroblastoma cases shows novel mutations and genotype-phenotype associations. Am J Med Genet A. 2006;140:1297–1301. doi: 10.1002/ajmg.a.31278. [DOI] [PubMed] [Google Scholar]
- Mossé YP, Laudenslager M, Khazi D, Carlisle AJ, Winter CL, Rappaport E, Maris JM. Germline PHOX2B mutation in hereditary neuroblastoma. Am J Hum Genet. 2004;75:727–730. doi: 10.1086/424530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mossé YP, Laudenslager M, Longo L, Cole KA, Wood A, Attiyeh EF, Laquaglia MJ, Sennett R, Lynch JE, Perri P, Laureys G, Speleman F, Kim C, Hou C, Hakonarson H, Torkamani A, Schork NJ, Brodeur GM, Tonini GP, Rappaport E, et al. Identification of ALK as a major familial neuroblastoma predisposition gene. Nature. 2008;455:930–935. doi: 10.1038/nature07261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller F, Rohrer H. Molecular control of ciliary neuron development: BMPs and downstream transcriptional control in the parasympathetic lineage. Development. 2002;129:5707–5717. doi: 10.1242/dev.00165. [DOI] [PubMed] [Google Scholar]
- Nakagawara A, Arima-Nakagawara M, Scavarda NJ, Azar CG, Cantor AB, Brodeur GM. Association between high levels of expression of the TRK gene and favourable outcome in human neuroblastoma. N Engl J Med. 1993;328:847–854. doi: 10.1056/NEJM199303253281205. [DOI] [PubMed] [Google Scholar]
- Nakagawara A, Azar CG, Scavarda NJ, Brodeur GM. Expression and function of TRK-B and BDNF in human neuroblastomas. Mol Cell Biol. 1994;14:759–767. doi: 10.1128/mcb.14.1.759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paris M, Wang WH, Shin MH, Franklin DS, Andrisani OM. Homeodomain transcription factor Phox2a, via cyclic AMP-mediated activation, induces p27Kip1 transcription, coordinating neural progenitor cell cycle exit and differentiation. Mol Cell Biol. 2006;26:8826–8839. doi: 10.1128/MCB.00575-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pattyn A, Morin X, Cremer H, Goridis C, Brunet JF. The homeobox gene Phox2b is essential for the development of all autonomic derivatives of the neural crest. Nature. 1999;399:366–370. doi: 10.1038/20700. [DOI] [PubMed] [Google Scholar]
- Raabe EH, Laudenslager M, Winter C, Wasserman N, Cole K, LaQuaglia M, Maris DJ, Mossé YP, Maris JM. Prevalence and functional consequence of PHOX2B mutations in neuroblastoma. Oncogene. 2008;27:469–476. doi: 10.1038/sj.onc.1210659. [DOI] [PubMed] [Google Scholar]
- Rohrer H, Thoenen H. Relationship between differentiation and terminal mitosis: chick sensory and ciliary neurons differentiate after terminal mitosis of precursor cells whereas sympathetic neurons continue to divide after differentiation. J Neurosci. 1987;7:3739–3748. doi: 10.1523/JNEUROSCI.07-11-03739.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rychlik JL, Gerbasi V, Lewis EJ. The interaction between dHAND and Arix at the dopamine beta-hydroxylase promoter region is independent of direct dHAND binding to DNA. J Biol Chem. 2003;278:49652–49660. doi: 10.1074/jbc.M308577200. [DOI] [PubMed] [Google Scholar]
- Schmidt M, Lin S, Pape M, Ernsberger U, Stanke M, Kobayashi K, Howard MJ, Rohrer H. The bHLH transcription factor Hand2 is essential for the maintenance of noradrenergic properties in differentiated sympathetic neurons. Dev Biol. 2009;329:191–200. doi: 10.1016/j.ydbio.2009.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Straub JA, Sholler GL, Nishi R. Embryonic sympathoblasts transiently express TrkB in vivo and proliferate in response to brain-derived neurotrophic factor in vitro. BMC Dev Biol. 2007;7:10. doi: 10.1186/1471-213X-7-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theiler K. New York: Springer; 1989. The house mouse: atlas of mouse development. [Google Scholar]
- Trochet D, Bourdeaut F, Janoueix-Lerosey I, Deville A, de Pontual L, Schleidermacher G, Coze C, Philip N, Frébourg T, Munnich A, Lyonnet S, Delattre O, Amiel J. Germline mutations of the paired-like homeobox 2B (PHOX2B) gene in neuroblastoma. Am J Hum Genet. 2004;74:761–764. doi: 10.1086/383253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trochet D, Hong SJ, Lim JK, Brunet JF, Munnich A, Kim KS, Lyonnet S, Goridis C, Amiel J. Molecular consequences of Phox2b missense, frameshift and alanine expansion mutations leading to autonomic dysfunction. Hum Mol Genet. 2005a;14:3697–3708. doi: 10.1093/hmg/ddi401. [DOI] [PubMed] [Google Scholar]
- Trochet D, O'Brien LM, Gozal D, Trang H, Nordenskjöld A, Laudier B, Svensson PJ, Uhrig S, Cole T, Niemann S, Munnich A, Gaultier C, Lyonnet S, Amiel J. PHOX2B genotype allows for prediction of tumor risk in congenital central hypoventilation syndrome. Am J Hum Genet. 2005b;76:421–426. doi: 10.1086/428366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trochet D, Mathieu Y, de Pontual L, Savarirayan R, Munnich A, Brunet JF, Lyonnet S, Goridis C, Amiel J. In vitro studies of non-poly-alanine PHOX2B mutations argue against a loss-of-function mechanism for congenital central hypoventilation. Hum Mutat. 2009;30:E421–31. doi: 10.1002/humu.20923. [DOI] [PubMed] [Google Scholar]
- Tsarovina K, Schellenberger J, Schneider C, Rohrer H. Progenitor cell maintenance and neurogenesis in sympathetic ganglia involves Notch signaling. Mol Cell Neurosci. 2008;37:20–31. doi: 10.1016/j.mcn.2007.08.010. [DOI] [PubMed] [Google Scholar]
- van Limpt V, Schramm A, van Lakeman A, Sluis P, Chan A, van Noesel M, Baas F, Caron H, Eggert A, Versteeg R. The Phox2b homeobox gene is mutated in sporadic neuroblastoma. Oncogene. 2004;23:9280–9288. doi: 10.1038/sj.onc.1208157. [DOI] [PubMed] [Google Scholar]
- Weese-Mayer DE, Berry-Kravis EM, Zhou L, Maher BS, Silvestri JM, Curran ME, Marazita ML. Idiopathic congenital central hypoventilation syndrome: analysis of genes pertinent to early autonomic nervous system embryologic development and identification of mutations in PHOX2B. Am J Med Genet. 2003;123A:267–278. doi: 10.1002/ajmg.a.20527. [DOI] [PubMed] [Google Scholar]
- Wyatt S, Andres R, Rohrer H, Davies AM. Regulation of neurotrophin receptor expression by retinoic acid in mouse sympathetic neuroblasts. J Neurosci. 1999;19:1062–1071. doi: 10.1523/JNEUROSCI.19-03-01062.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu H, Firulli AB, Zhang X, Howard MJ. HAND2 synergistically enhances transcription of dopamine-beta-hydroxylase in the presence of Phox2a. Dev Biol. 2003;262:183–193. doi: 10.1016/s0012-1606(03)00361-0. [DOI] [PubMed] [Google Scholar]
- Yang C, Kim HS, Seo H, Kim CH, Brunet JF, Kim KS. Paired-like homeodomain proteins, Phox2a and Phox2b, are responsible for noradrenergic cell-specific transcription of the dopamine β-hydroxylase gene. J Neurochem. 1998;71:1813–1826. doi: 10.1046/j.1471-4159.1998.71051813.x. [DOI] [PubMed] [Google Scholar]
- Zackenfels K, Oppenheim RW, Rohrer H. Evidence for an important role of IGF-I and IGF-II for the early development of chick sympathetic neurons. Neuron. 1995;14:731–741. doi: 10.1016/0896-6273(95)90217-1. [DOI] [PubMed] [Google Scholar]
- Zellmer E, Zhang Z, Greco D, Rhodes J, Cassel S, Lewis EJ. A homeodomain protein selectively expressed in noradrenergic tissue regulates transcription of neurotransmitter biosynthetic genes. J Neurosci. 1995;15:8109–8120. doi: 10.1523/JNEUROSCI.15-12-08109.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]