

Abstract
The maintenance of chronic pain states requires the regulation of gene expression, which relies on an influx of calcium. Calcium influx through neuronal L-type voltage-gated calcium channels (LTCs) plays a pivotal role in excitation–transcription coupling, but the involvement of LTCs in chronic pain remains unclear. We used a peptide nucleic acid (transportan 10-PNA conjugates)-based antisense strategy to investigate the role of the LTC subtypes CaV1.2 and CaV1.3 in long-term pain sensitization in a rat model of neuropathy (spinal nerve ligation). Our results demonstrate that specific knockdown of CaV1.2 in the spinal dorsal horn reversed the neuropathy-associated mechanical hypersensitivity and the hyperexcitability and increased responsiveness of dorsal horn neurons. Intrathecal application of anti-CaV1.2 siRNAs confirmed the preceding results. We also demonstrated an upregulation of CaV1.2 mRNA and protein in neuropathic animals concomitant to specific CaV1.2-dependent phosphorylation of the cAMP response element (CRE)-binding protein (CREB) transcription factor. Moreover, spinal nerve ligation animals showed enhanced transcription of the CREB/CRE-dependent gene COX-2 (cyclooxygenase 2), which also depends strictly on CaV1.2 activation. We propose that L-type calcium channels in the spinal dorsal horn play an important role in pain processing, and that the maintenance of chronic neuropathic pain depends specifically on channels comprising CaV1.2.
Introduction
Acute pain and the associated transient period of hypersensitivity are adaptive mechanisms that warn individuals of the potential or actual danger of tissue damage. However, in pathological conditions, such as neuropathic syndromes, pain may become permanent, thereby altering the physical and emotional functioning of patients, decreasing their quality of life and impairing their ability to work (Costigan et al., 2009). Neuropathic pain is an expression of pathological operation of the nervous system, which commonly results from nerve injury (Campbell and Meyer, 2006). Most treatments have been empirically established, and their efficacy sometimes remains unsatisfactory. It is therefore necessary to pursue a deeper understanding of the cellular mechanisms maintaining chronic neuropathic pain states (Baron, 2006).
Chronic pain is associated with long-lasting changes in gene expression involving calcium channels (Woolf and Salter, 2000). One of the main sources of calcium influx mediating gene regulation is L-type voltage-gated calcium channels (LTCs) (Bading et al., 1993; Bito et al., 1996; Deisseroth et al., 1998; Dolmetsch et al., 2001; West et al., 2001). However, the involvement of LTCs in neuropathic pain processing is unclear [for review, see Vanegas and Schaible (2000) and Yaksh (2006)]. In the CNS, two isoforms of the α1 subunit (CaV1.2 and CaV1.3) define two classes of LTC (Hell et al., 1993; Koschak et al., 2001; Xu and Lipscombe, 2001; Striessnig and Koschak, 2008), and it is known that CaV1.2 is involved in gene regulation (Dolmetsch et al., 2001; Weick et al., 2003; Gomez-Ospina et al., 2006; Zhang et al., 2006). We used a peptide nucleic acid (covalent transportan 10-S-S-PNA conjugates)-based antisense strategy (Pooga et al., 1998) to investigate the role of CaV1.2 in the maintenance of long-term mechanical hypersensitivity in the “spinal nerve ligation” (SNL) model of neuropathy in the rat (Kim and Chung, 1992). We showed that specific knockdown of the CaV1.2 gene, but not of the CaV1.3 gene, totally reversed this component of the neuropathic sensitization to pain. Intrathecal application of two distinct anti-CaV1.2 siRNAs confirmed the preceding results. CaV1.2 gene knockdown also reversed the enhanced excitability and responsiveness of spinal dorsal horn neurons. We also showed that CaV1.2 transcript and protein expression was upregulated in SNL rats. We explored the intracellular signaling pathways associated with CaV1.2-mediated neuropathic pain, and demonstrated a preferential CaV1.2-mediated increase in intranuclear calcium, and a specific CaV1.2-dependent phosphorylation of the cAMP response element (CRE)-binding protein (CREB) transcription factor, concomitant with an enhanced transcription of the CREB/CRE-dependent gene cyclooxygenase 2 (COX-2). Altogether, our data demonstrate that LTCs in the spinal dorsal horn, and especially those comprising CaV1.2, are crucial factors for the maintenance of chronic neuropathic pain.
Materials and Methods
Animals
All experimental procedures followed the ethical guidelines of the International Association for the Study of Pain and were approved by the local ethics committee (agreement number AP 1/04/2005). The experiments were done on adult Wistar rats (250–300 g).
Animal model of neuropathy and behavioral tests
Forty-six adult male rats were used in the spinal nerve ligation model experiments (Kim and Chung, 1992). The right L5 and L6 spinal nerves were isolated and tightly ligated with a 7.0 silk thread. After complete hemostasis, the incision was sutured. Surgery was performed on all rats under gaseous anesthesia with a mixture of halothane (5% for induction and 2% for maintenance) and a 1:1 flow ratio of air/O2. The rats resumed normal activity within 30 min after termination of the gaseous anesthesia.
Mechanical response thresholds were monitored at 1 d before surgery (day −1, reference value for each animal), and at days 7 and 11 [24 h after the last peptide nucleic acid (PNA) injection] after surgery (Fig. 1). The rats were placed in the testing cage 1 h before the test for habituation. The withdrawal threshold of the leg on the operated side was determined in response to mechanical stimuli applied to the plantar surface of the foot. The limb withdrawal threshold was measured by an electronic device (Bioseb) derived from von Frey filaments. Nonoperated rats were used as controls.
Figure 1.

Experimental procedure. Spinal nerve ligations (branches 5 and 6 of the sciatic nerve; SNL model of neuropathy) were performed at day 0. The mechanical threshold was measured 1 d before the surgery (−1) and at 7 and 11 d after surgery. TP10-PNA constructs were injected daily from day 7 to day 10.
Intrathecal drug administration
Rats were subjected to gaseous anesthesia with halothane or isoflurane (5% for induction and 2% for maintenance). For nicardipine chronic administration, an Alzet osmotic pump (1007D, DURECT) was filled with 100 mm nicardipine, placed subcutaneously, and connected to a catheter (PE-10) placed intrathecally above the lumbar part of the spinal cord. Nicardipine was released with a 0.5 μl/h (26 μg/h) flow during 5 d. For PNA chronic injections, the same catheter (PE-10) was also introduced above the lumbar part of the spinal cord, and the external portion of the catheter ran under the skin up to the neck (Jasmin and Ohara, 2001). The incision was closed using 3-0 surgical sutures. The drugs were injected using a Hamilton syringe connected to the catheter and flushed with 10 μl of saline (dead volume of the catheter).
PNA design and application
PNA sequences (15-mers) were designed to target regions upstream to the translation start site of CaV1.2 and CaV1.3 mRNA. Care was taken to minimize the stability of the hairpin secondary structures under physiological conditions: anti-CaV1.2 PNA: Cys-CGTGAGATTGTAATG; anti-CaV1.3 PNA: Cys-GTTACTGATAGGTAG; mismatch PNA: Cys-CGTGAATTAGTTAGG.
To improve their intracellular delivery, cysteine-extended PNAs were coupled to the cell-penetrating peptide transportan TP10 [AGYLLGKINLK(C)ALAALAKKIL-amide] (Soomets et al., 2000) through a disulfide bond. After reduction of the bond in the intracellular medium, the transportan 10-PNA constructs were dissociated, and the PNAs could either associate with target CaV1.2 or CaV1.3 mRNA in the cytosol or translocate into the nucleus, thereby interacting with the corresponding DNA sequences.
The PNAs were synthesized automatically on an Applied Biosystems 433A peptide synthesizer using the t-Boc strategy (Egholm et al., 1993). The carrier peptide, transportan 10 (TP10), was synthesized on the Applied Biosystems 431A peptide synthesizer and purified before conjugation with PNA on a reverse-phase HPLC C18 column. The masses of the purified peptide and of the PNA were verified by MALDI-TOF (ABI Voyager-DE STR) mass spectrometry. Conjugation was performed in a DMSO/dimethylformamide/0.1 m acetic buffer, pH 5.5, 1/1/3 mixture overnight, and the products were separated on a C18 HPLC column. The correct conjugate was determined via an absorbance profile created by a multiwavelength detector of HPLC and by MALDI-TOF mass spectrometry. To assess the penetration into tissues, biotin-coupled transportan 10-PNA constructs were also synthesized. Transportan 10-PNA constructs were injected daily for 4 d, a time interval consistent with calcium channel turnover (Dobremez et al., 2005). Behavioral and electrophysiological experiments were performed 24 h after the last injection.
RNA interference
We used siRNA targeting several splice variants of CaV1.2 (“Silencer Select Pre-designed and Validated siRNA”, Ambion). They consisted of a pool of two 21 nt duplex. siRNAs were selected to target two distinct CaV1.2 mRNA regions to enhance silencing. The antisense sequences were as follows: UCUAUUGUCAUAUCGCAGG and UAUCCGAACAGGUAUAGAG.
In contrast to PNA, these siRNAs targeted the 5′-coding region. Mismatch siRNA was a nontargeting 21 nt duplex designed as a negative control. The siRNAs (2 μg) were solubilized in 10 μl of i-Fect reagent (Neuromics) following Neuromics instructions and published protocol (Luo et al., 2005), and applied intrathecally according to the same protocol as for the PNA.
Immunohistochemistry
Control, sham, SNL, and PNA-injected rats were perfusion fixed through the ascending aorta with 4% paraformaldehyde in phosphate buffer (0.1 m, pH 7.4). Immunohistochemistry was performed on young rats after intrathecal injection of either anti-CaV1.2, anti-CaV1.3, or mismatch PNAs (n = 4 for each condition). Double immunodetection of CaV1.2 or CaV1.3, and microtubule-associated protein 2 (MAP2), a somatodendritic neuronal marker, was performed in spinal cord cryostat sections (30 μm thickness; Microm). Briefly, sections were incubated overnight at 4°C with mouse anti-MAP2 (1:1000; Sigma) and rabbit anti-CaV1.2 or anti-CaV1.3 (1:20; Alomone) antibodies. After rinsing, the sections were incubated with biotinylated goat anti-rabbit antibody (1:200; Vector Laboratories) and next with Alexa 488-conjugated streptavidin (1:500) and Alexa 568-conjugated goat anti-mouse (1:500) antibodies (Molecular Probes/Invitrogen). We also used a mouse anti-CaV1.2 antibody (1:200; NeuroMab Facility, Antibodies Incorporated). After rinsing, the sections were incubated with biotinylated horse anti-mouse antibody (1:200; Vector Laboratories) and next with Alexa 488-conjugated streptavidin (1:500) (Molecular Probes/Invitrogen). The sections were then mounted and viewed with an epifluorescence microscope (Zeiss Axiophot 2 imaging). Specificity of antibodies was determined as previously described (Dobremez et al., 2005).
For immunofluorescence quantification, CaV1.2-immunolabeled sections were viewed with a confocal microscope (Leica SP2, Leica Microsystems). Images to be compared were collected during the same session using identical scanning settings. They were then imported into “ImageJ” free software (version 1.42q) for quantitative analysis. Background was subtracted by thresholding, and the mean gray level corresponding to fluorescence intensity was measured.
Biotin-coupled constructs were injected intrathecally as described above, and their tissular penetration was assessed in perfusion-fixed spinal sections. Briefly, the sections were incubated with peroxidase-conjugated streptavidin (1:500; Vector Laboratories). After rinsing, the peroxidase activity was revealed with diaminobenzidine. The sections were finally dehydrated, mounted and observed with bright-field illumination.
Spinal cord cell cultures and calcium imaging
Dissociated cultures were made from the lumbar spinal cord of E18 rat embryos. The embryos were delivered by cesarean section from deeply anesthetized pregnant females and killed by decapitation.
Slices of the lumbar region of the spinal cord, without dorsal root ganglia, were exposed to a 1× trypsin solution for 25 min at 37°C. Slices were then mechanically dissociated by forcing them through fine-tipped pipettes. Cells were plated at a density of 100,000/100 μl on glass coverslips previously coated with polylysine (0.1 mg/ml) and laminin (0.05 mg/ml). The cultures were kept for 1 week in serum-free Neurobasal medium (Invitrogen) that was supplemented with B27 and GlutaMax (both Invitrogen), and then were incubated in a 5% CO2/95% O2 atmosphere at 36.5°C. Half of the medium was changed twice a week. After 1 week, the cultures were treated daily with PNA (50 μm) for 4 d.
Spinal cells were filled with the Ca2+ indicator fluo4/AM (10 μm), and changes in [Ca2+]i were measured using a CCD camera and observed using a 60× objective lens. The cells were continuously superfused with artificial CSF (ACSF) containing the following (in mm): 130 NaCl, 3 KCl, 2.5 CaCl2, 1.3 MgSO4, 0.58 NaH2PO4, 25 NaHCO3, and 10 glucose. The ACSF was adjusted to pH 7.4 with HCl, and equilibrated with 95% CO2–5% O2 at room temperature (25–26°C). Tetrodotoxin (0.1 μm) was added to the saline to decrease spontaneous activity. Increases of [Ca2+]i induced by 7 min bath applications of 30 mm K+ were acquired by recording a series of 300 frames (1 frame/3 s, exposure time: 20–250 ms) with IPLab software (Scanalytics). For the global Ca2+ transient measurements, changes in fluorescence (FT) were calculated for each cell relative to the averaged baseline fluorescence before stimulation (Frest) and expressed as %ΔF/F = [(FT − Frest)/Frest] × 100. To evaluate the nucleus/cytoplasm fluorescence ratio, analysis was performed at the maximum of the Ca2+ response in individual cells presenting a visible nucleus. A rectangle region of interest (ROI) was defined, including a part of the cytoplasm and the nucleus. The intensities were averaged in each pixel column of the ROI, and plotted along the x-axis. We extracted from these plots the minimal and maximal fluorescence intensities, which were attributed to the cytoplasm and nucleus, respectively.
Immunoautoradiography
Control, sham, SNL, and PNA-injected rats were perfusion fixed through the ascending aorta with 4% paraformaldehyde in phosphate buffer (0.1 m, pH 7.4). Immunoautoradiography was performed on adult rat spinal cord cryostat sections (14 μm) that were thaw mounted onto Super-Frost Gold slides (CML). Sections corresponding to different experimental models [sham (n = 6), SNL (n = 6), SNL + anti-CaV1.2 (n = 5) and SNL + anti-CaV1.3 (n = 5) injected rats] were mounted together on the same slides and treated as one batch. The sections were first preincubated for 10 min in PBS-NaI-BSA buffer [0.15 g/L NaI, 30 g/L bovine serum albumin (Sigma) in PBS 0.01 m, pH 7.4], and then incubated at 4°C for 24 h with one of the following primary antibodies (1:500 in PBS-NaI-BSA): anti-phosphoP44/42 mitogen-activated protein kinase (MAPK) antibodies (Cell Signaling Technology); anti-CREB or anti-ser133 phosphoCREB antibodies (PhosphoPlus CREB antibody kit); anti-phospho p38 MAPK antibodies (Cell Signaling Technology); anti-phospho-CaMKII T286 antibodies (AbCam). The sections were rinsed in PBS-NaI-BSA at room temperature. They were then incubated with a biotinylated anti-rabbit or anti-mouse secondary antibody (Vector Laboratories) (1:100 in PBS-NaI-BSA) for 30 min at room temperature. Sections were rinsed in PBS-NaI-BSA and then incubated with radiolabeled (35S) streptavidin (GE Healthcare Biosciences) (1:100 in PBS-NaI-BSA) for 30 min at room temperature. The sections were rinsed twice in PBS-NaI-BSA, and for 10 min in distilled water to remove the excess salt. After being air dried, the sections were processed for radioactive imaging and quantitative analysis.
The images were acquired with a β-sensitive radioimager (Micro-imager, Biospace Mesures), allowing real-time acquisition with a 15 μm spatial resolution, and quantification (Laniece et al., 1998). The signal obtained was a linear function of the radioactivity.
A thin foil of scintillating paper was placed in contact with the slides. β-Particles emitted by the radioactive streptavidin were identified by acquisition of the light spot emissions for 6 h by a charge-coupled device (CCD), coupled to an image intensifier. Image analysis was done with β vision+ software (Biospace Mesures). The average surfacic activity (SA) (cpm/mm2) was calculated for superficial and deep laminae. Areas of interest that were ipsilateral to the ligation (SAi) were compared to the corresponding areas on the contralateral side (SAc). The variations (ΔSA) were expressed as percentages of the contralateral side according to ΔSA = [(SAi − SAc)/SAc] × 100 (mean ± SEM).
Electrophysiology
Extracellular recordings were made as previously described (Fossat et al., 2007). Briefly, rats were placed in a stereotaxic frame to ensure stability during electrophysiological recordings. Recordings of wide dynamic range (WDR) dorsal horn neurons (DHNs) were made with borosilicate glass capillaries (2 MΩ, filled with 4% NaCl) (Harvard Apparatus). The depth of the neurons from the surface of the dorsal horn of the spinal cord was monitored. The criterion for neuron selection was a depth ranging from 500 to 1000 μm and the presence of an Aβ-fiber-evoked response followed by a C-fiber-evoked response to electrical stimulation of the ipsilateral paw. The response of the neuron to various natural innocuous (brush, pressure) and noxious (pinch) stimulations of the most responsive part of the receptive field was characterized. To quantify natural mechanical stimuli, we used an electronic device derived from von Frey filaments (Bioseb). Four calibrated pressures were tested (5 g, 10 g, 25 g, and 50 g).
Data were acquired by means of a CED 1401 interface (Cambridge Electronic Design) and analyzed with the Spike 2 software (Cambridge Electronic Design). The data were plotted with the Sigma-Plot software (version 6.00, Systat Software).
Western blots
Membrane preparation.
Ipsilateral lumbar spinal cord tissue from sham, SNL, anti-CaV1.2 siRNA-injected, and anti-CaV1.2 PNA-injected animals were homogenized in ice-cold buffer (20 mm HEPES, 0.15 mm EDTA, and 10 mm KCl, pH 8) containing protease inhibitors (Roche) and centrifuged for 5 min at 2500 rpm. The supernatant was centrifuged for 30 min at 14,000 rpm. The pellet was homogenized in the same buffer adjusted at 15% sucrose. The homogenate was centrifuged for 5 min at 2000 rpm to remove genomic DNA. The supernatant containing the membranes was centrifuged again for 30 min at 14,000 rpm. Spinal cord membranes were solubilized in a medium containing 20 mm HEPES, 1% Triton X-100, 150 mm NaCl, and 5 mm EDTA, pH 8, and then incubated for 45 min. The sample was centrifuged for 45 min at 14,000 rpm. All steps were performed at 4°C. Protein concentration was evaluated with a Bio-Rad protein assay.
Western blotting.
Equal amounts of proteins were subjected to SDS-PAGE electrophoresis on 7% polyacrylamide gels. Western blots were reprobed for GAPDH to verify the amount of proteins loaded on each lane. Membrane was incubated overnight at 4°C with mouse anti-CaV1.2 calcium channel antibody (1/2000, clone L57/46) (NeuroMab Facility, Antibodies Incorporated). Immunoreactivity was detected using polyclonal goat anti-mouse immunoglobulins-HRP (DakoCytomation) and visualized using an ECL detection system (GE Healthcare). Pictures of the blots were taken with a ChemiGenius 2XE apparatus under the control of GeneSnap program (Syngene). Quantitative analysis of Western blots was performed by densitometry with the corresponding “Gene tools” analysis program (Syngene).
Quantitative RT-PCR
Total RNA was purified with the TRI Reagent (Sigma-Aldrich) according to the manufacturer's instructions. cDNA was synthesized from 1 μg of total RNA using the SuperScript III first strand synthesis kit (Invitrogen) and oligo-dT primers. PCR amplification was performed on a DNA Engine Opticon2 fluorescence detection System (MJResearch/Bio-Rad) with primer pairs designed to span exon boundaries and to generate amplicons of ∼70 bp. Primer sets for CaV1.2, CaV1.3, and COX-2 were tested by quantitative reverse transcription (qRT)-PCR and gel electrophoresis for the absence of primer-dimer artifacts and multiple products. Triplicate qRT-PCRs were done twice for each sample, using transcript-specific primers (600 nm) and cDNA (10 ng) in a final volume of 10 μl. The DyNAmo SYBR Green qPCR kit (Finnzymes) was used with the following PCR amplification cycles: initial denaturation, 95°C for 15 min; followed by 40 cycles with denaturation, 95°C for 20 s and annealing-extension, 61°C for 35 s. A dissociation curve was generated at the end of the 40th cycle to verify that a single product was amplified. The Ct value of each gene was normalized against that of succinate dehydrogenase complex subunit A (SDHA). The relative level of expression was calculated using the comparative (2ΔΔCT) method (Livak and Schmittgen, 2001).
Statistical analysis
Values are expressed as mean ± SEM. For intracellular signaling pathways, statistical analyses were conducted using GraphPad InStat (version 3.0, GraphPad Software). Ipsilateral/contralateral radioactivity ratios were compared between groups using an unpaired t test followed by a Welch's corrected test. For electrophysiology, the analysis was performed using SigmaStat (version 3.5, Systat Software). Experimental groups were compared using a paired Student's t test. For morphological, behavioral, Western blots, and qRT-PCR quantifications, the groups were compared using a Mann–Whitney test. In all cases, the criterion for statistical significance was p < 0.05.
Results
Reversal of neuropathic tactile allodynia by knockdown of the CaV1.2 gene
In the first step, we tested for the implication of LTCs in long-term sensitization associated with neuropathic pain. This chronic pain state may be characterized by painful hypersensitivity to innocuous stimuli, a phenomenon known as tactile allodynia. In the SNL model of neuropathy, we monitored, 7 d after the spinal nerve ligatures, the mechanical threshold for withdrawal of the ipsilateral foot in response to stimulation of the paw with von Frey hairs (Fig. 1, day 7), and compared it to the threshold measured in the same animal just before the surgery (day −1, reference value 100 ± 3.1%). The threshold was lowered to 49.5 ± 4.5% of the control in the operated animals, but remained unchanged in sham animals (106.5 ± 8.9%). Then, over 5 d, a group of SNL animals received a chronic intrathecal administration of nicardipine, a dihydropyridine that blocks LTCs. The LTC antagonist was administered in the lumbar region of the spinal cord, via a catheter connected to a subcutaneous osmotic pump (26 μg/h). A second group of SNL animals was injected with the vehicle (artificial CSF). At day 11, the nicardipine-injected animals showed a significantly higher mechanical threshold, compared to the preinjection day 7 (68.4 ± 4.2% and 49.5 ± 4.5% of control, respectively, p = 0.03 SNL + nicardipine vs SNL), whereas the threshold did not change in the CSF group (53.5 ± 2.5% of control, p = 0.61 SNL + CSF vs SNL). This indicates that LTCs are involved in the SNL-induced mechanical allodynia.
Because neuronal access of intrathecally applied dihydropyridines is limited, and because these antagonists do not distinguish between the two LTC isoforms, in the next step, we used an antisense-based strategy to knock down each variant in SNL rats. We considered the effects of intrathecal injections of PNA antisense molecules specifically targeting the expression of one of the CaV1.2 or CaV1.3 isoforms. We first checked that biotinylated PNAs coupled to TP10, actually penetrated in the lumbar dorsal horn when applied intrathecally in the lumbar region of the spinal cord (Fig. 2Aa, star). Interestingly, this penetration did not extend to the thoracic (Fig. 2Ab) or the sacral regions (data not shown), or to the ipsilateral DRG (Fig. 2Ad). As a confirmation, the expression of the CaV1.2 transcript evaluated by qRT-PCR in the ipsilateral DRGs of SNL rats (85.1 ± 11.9% of sham, n = 4) did not differ significantly from sham animals (100 ± 5.5%, n = 4, p = 0.34), or SNL rats injected with anti-CaV1.2 PNA (91.6 ± 6.5%, n = 3, p = 0.86) or with mismatch PNA (108.5 ± 17.3%, n = 3, p = 0.23) (supplemental Fig. S1A, available at www.jneurosci.org as supplemental material). We then verified the specific extinction of immunoreactivity for the LTC subtypes targeted by the antisense molecules, i.e., anti-CaV1.2 PNA (Fig. 3A–C; see also Fig. 7Ac), and anti-CaV1.3 PNA (Fig. 3D–F) (see quantifications in supplemental Fig. S2, A and B, respectively, available at www.jneurosci.org as supplemental material). Again, these extinctions were restricted to the lumbar region of the spinal cord (Fig. 3G,H,J,K), whereas the thoracic (Fig. 3I,L) and sacral (data not shown) segments, and ipsilateral DRG (Fig. 2B; see quantification in supplemental Fig. S1B, available at www.jneurosci.org as supplemental material) remained unaffected. Finally, qRT-PCR analysis confirmed that the PNA-induced decrease in protein expression was actually due to knocking down the corresponding LTC mRNAs in the lumbar region of the spinal cord (see below, Fig. 6A,B).
Figure 2.
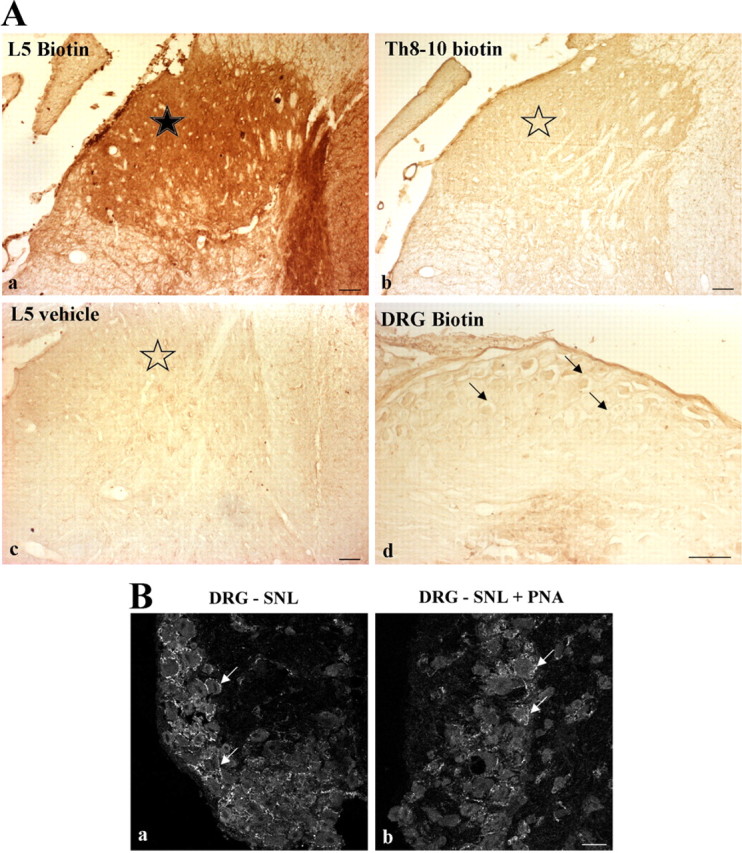
Penetration of TP10-PNAs restricted to the lumbar spinal dorsal horn. A, Naive animals received an intrathecal injection of either biotin-coupled transportan 10-anti-CaV1.2 PNA construct (Aa, Ab, Ad), or vehicle (Ac). Then, peroxidase activity was revealed in the dorsal horn of the L5 lumbar (Aa, Ac) and Th8-10 thoracic (Ab) spinal cord, and in the L5 DRG (Ad). A positive signal was seen only in L5 dorsal horn after injection of biotinylated TP10-PNA constructs (black star in Aa). No signal was observed in thoracic segments (open star in Ab), or after vehicle injection (open star in Ac). Sensory neurons in the L5 DRG also remained unlabeled (arrows in Ad). Sections were observed from three animals in each condition (biotinylated PNA- or vehicle-injected animals). B, Confocal views of ipsilateral DRG sections from SNL rats, before (Ba) or after (Bb) intrathecal application of TP10-anti-CaV1.2 PNA construct. Immunofluorescence of individual neurons is similar in both conditions (arrows, see quantification in supplemental Fig. S1B, available at www.jneurosci.org as supplemental material). Scale bars: 50 μm in A, 20 μm in B.
Figure 3.
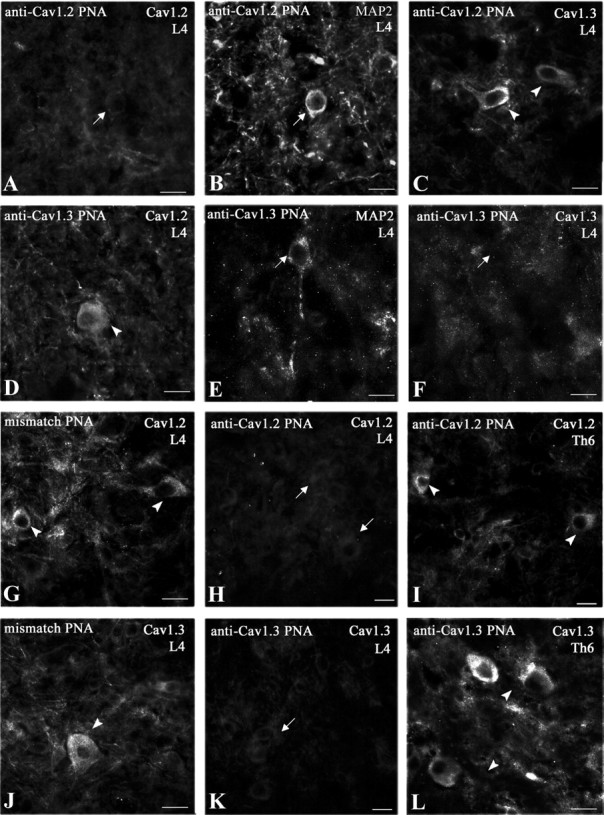
Selective suppression of LTC proteins after intrathecal TP10-PNA application. Immunohistochemical detection of CaV1.2 (A, D, G–I), CaV1.3 (C, F, J–L), and MAP2 (B, E) in lamina V of rat spinal cords after intrathecal injection of anti-CaV1.2 (A–C, H, I), anti-CaV1.3 (D–F, K, L), or mismatch (G, J) TP10-PNA. TP10-PNA injection selectively inhibited the expression of the related α1 subunit in MAP2-positive neurons (arrows in A, B, E, and F) without affecting the expression of the other subtypes (arrowheads in C and D). Mismatch TP10-PNA did not block CaV1.2 (arrowheads in G) or CaV1.3 (arrowheads in J) synthesis. Intrathecally injected TP10-PNA inhibited protein synthesis in the lumbar region (L4, arrows in H and K), but did not diffuse to the upper spinal segments. It had no effects in the thoracic region (Th6, arrowheads in I and L). Observations were done from two to five sections in each region, from three animals in each condition (anti-CaV1.2, anti-CaV1.3, and mismatch PNAs). The sections in H and I and in K and L were taken from the same animals, respectively. Scale bars: 20 μm.
Figure 7.

Dorsal horn-specific increase of CaV1.2 in neuropathic rats. A, Immunofluorescence micrographs showing detection of CaV1.2 protein in the superficial dorsal horn (DH, Aa, Ab, arrows) and lamina IX of the ventral horn (VH, Ad, Ae; arrows indicate large presumptive motoneuronal somata), in lumbar spinal cord of control (sham, Aa, Ad) and neuropathic (SNL, Ab, Ae) rats. Absence of CaV1.2 immunolabeling after intrathecal application of anti-CaV1.2 TP10-PNA in both dorsal (Ac) and ventral horns (Af). Scale bar, 20 μm. B, Quantification of CaV1.2 immunofluorescence. In neuropathic animals, CaV1.2 immunolabeling significantly increased in the dorsal horn (B1, black bar) but not in the ventral horn (B2, black bar). After anti-CaV1.2 TP10-PNA application, CaV1.2 immunolabeling significantly decreased in DH and VH (B1, B2, white bars), whereas application of anti-CaV1.3 TP10-PNA had no effect (gray bars). n, The first and second numbers indicate the number of quantified sections and of animals, respectively; two stars, p < 0.01; NS, nonsignificant.
Figure 6.

SNL-induced overexpression of CaV1.2. Efficient knockdown after intrathecal anti-CaV1.2 antisenses application. A, Expression of CaV1.2 mRNA (qRT-PCR) increased by more than twofold in SNL rats compared to sham animals (black bar). CaV1.2 mRNA levels were significantly reduced after intrathecal injection of anti-CaV1.2 TP10-PNA (white bar) or siRNA (dotted bar). Application of either anti-CaV1.3 TP10-PNA (gray bar) or mismatch PNA or mismatch siRNA (middle and right hatched bars, respectively) had no effect. B, Expression of CaV1.3 mRNA (qRT-PCR) showed a significant reduction in SNL rats compared to sham animals (black bar), and was further decreased after intrathecal injection of anti-CaV1.3 TP10-PNA (gray bar). Application of either anti-CaV1.2 TP10-PNA (white bar) or mismatch PNA (hatched bar) had no effect. C, Western blot analysis of the CaV1.2 expression from ipsilateral lumbar spinal cord of sham, SNL, siRNA-injected, and PNA-injected animals (C1). The protein level increased in the spinal cord of SNL animals (black bar). Application of anti-CaV1.2 PNA or siRNA (white and dotted bars, respectively) suppressed CaV1.2 overexpression (C2). n, Number of animals; one star, p < 0.05; two stars, p < 0.01; three stars, p < 0.001; NS, nonsignificant; stars above black bars are statistics versus sham.
In this series of experiments, 7 d after the ligation, the mechanical threshold for withdrawal of the ipsilateral paw was again lowered by ∼50% (Fig. 4A, SNL, day 7) in all the operated animals, whereas it remained unchanged in the sham animals (data not shown). Then, for 4 d, animals received daily intrathecal injections of either anti-CaV1.2 PNA or anti-CaV1.3 PNA (see Materials and Methods). Vehicle or mismatch PNA injections were used as controls. The threshold was further measured 1 d after the last injection (Fig. 1, day 11). As shown in Figure 4A, the specific knockdown of CaV1.2 expression brought the threshold from 41.5 ± 2.3% (day 7, n = 8) back to 95.6 ± 4.1% of the reference value (day 11, n = 8; p < 0.0001 vs day 7 value, p = 0.47 vs reference value). In contrast, anti-CaV1.3 PNA had no effects (Fig. 4A, bar group 3). No changes were observed in the untreated animals (SNL) (Fig. 4A, bar group 1) or after the injection of either the mismatch PNA (Fig. 4A, bar group 4) or the vehicle (Fig. 4A, bar group 5). These results, therefore, demonstrate that only the knockdown of the CaV1.2 LTC isoform was able to reverse the neuropathy-induced sensitization of the withdrawal response and restore a normal level of mechanical sensitivity. On the contrary, in intact animals, injection of either one of the two PNAs had no effect on the mechanical threshold (p = 0.7 for CaV1.2 and p = 0.6 for CaV1.3, Student's t test, data not shown). Together, our results indicate that CaV1.2 is specifically involved in the SNL-induced long-term sensitization to pain, but not in the normal response. Daily testing of the paw withdrawal threshold revealed the progressive recovery of a normal sensitivity level during the period of PNA injection, and the progressive reinstallation of the sensitization after the last application (Fig. 4B), indicating that CaV1.2 LTCs are required for the maintenance of the sensitization. To confirm the implication of CaV1.2-comprising LTCs in tactile allodynia, we also coapplied intrathecally, according to the previous protocol, two siRNAs targeting distinct nucleotide sequences of CaV1.2 (see Materials and Methods). The mechanical threshold for withdrawal of the ipsilateral paw 7 d after the nerve ligation was 43.5 ± 2.1% (n = 6 animals, data not shown) of the threshold measured at control day −1 before surgery (100 ± 1.9%; p < 0.002, SNL/control). One day after the last of 4 daily intrathecal siRNA applications, the withdrawal threshold recovered to 81.1 ± 2.1% of the control (n = 6 animals, p < 0.002, day 11/day 7). Therefore, CaV1.2 knockdown by PNAs or siRNAs produced similar reversal of tactile allodynia in neuropathic rats.
Figure 4.

Suppression of long-term mechanical allodynia by blockade of CaV1.2 expression. A, SNL induced a lowering of the mechanical threshold at day 7 (46.1 ± 1.0% of the reference value, von Frey test), which persisted at day 11 (49.7 ± 2.3%). In a second group of rats, the specific knockdown of CaV1.2 channels by intrathecal injection of the corresponding TP10-PNA antisense restored the threshold from 41.5 ± 2.3% (day 7) to 95.6 ± 4.1% of the reference value (day 11, p < 0.01 vs day 7 value, p = 0.47 vs reference value). No reversal occurred after anti-CaV1.3 TP10-PNA (p = 0.54 vs day 7 value), mismatch TP10-PNA (p = 0.31 vs day 7 value), or vehicle (p = 0.24 vs day 7 value) injections. n, Number of animals; two stars, p < 0.01; NS, nonsignificant. B, Progressive recovery of a normal mechanical threshold during a period of daily injection of anti-CaV1.2 TP10-PNA (arrows) in SNL animals. The allodynia resumed progressively after the last TP10-PNA injection. CaV1.2 is necessary for the maintenance of the neuropathic pain sensitization. n = 4 animals; one star, p < 0.05; two stars, p < 0.01.
Ventral horn neurons and especially plateau potential-generating motor neurons also express CaV1.2 LTCs (Westenbroek et al., 1998; Anelli et al., 2007) that are knocked down by anti-CaV1.2 PNAs (see Fig. 7Ae,Af). In spinal cord, motor responses to sensory stimulation can be perturbed by LTC blockade (Martinez-Gomez and Lopez-Garcia, 2007). Because tactile allodynia is usually monitored by means of a flexion reflex including such a sensorimotor response, we checked whether the increase of the withdrawal threshold back to control could be partly due to a PNA-mediated degradation of motor performance. We monitored the latency to fall from a rotating beam (Rotarod) (see supplemental method and Fig. S3, available at www.jneurosci.org as supplemental material). In the SNL model, two of the three segmental ventral branches of the sciatic nerve are also ligatured, and, accordingly, SNL animals showed a significant decrease in the latency to fall (supplemental Fig. S3, available at www.jneurosci.org as supplemental material) (23.9 ± 5.6% of control day −1; n = 10, p = 0.001, SNL/control). However, subsequent intrathecal PNA applications did not change significantly this latency (supplemental Fig. S3, available at www.jneurosci.org as supplemental material) (24.6 ± 8.0% of control; n = 5, p = 0.69, CaV1.2 PNA/SNL; 13.9 ± 5.5% of control; n = 5, p = 0.84, mismatch PNA/SNL). In summary, it does not appear that application of anti-CaV1.2 PNA affected the motor performance of SNL rats.
Reversal of dorsal horn neurons hyperexcitability by knockdown of CaV1.2 gene
Given the involvement of CaV1.2 in the sensitization of the behavioral response to mechanical stimuli, we next tested the effects of blocking the expression of the LTC subtypes on the excitability and responsiveness of deep DHNs. By means of extracellular recordings, we selected WDR neurons identified in the dorsal horn as neurons responding to both innocuous (brush and pressure) and noxious (pinch) stimulations of their receptive field on the paw (Fig. 5A). As shown in Figure 5B, the spontaneous discharge frequency of WDRs in the SNL animals was substantially higher than that in the sham animals (Fig. 5B) (SNL 1.92 ± 0.36 Hz vs sham 0.39 ± 0.3 Hz, p < 0.05). Again, the increase in excitability was selectively reversed after intrathecal application of PNA targeted against CaV1.2 (Fig. 5B) (0.28 ± 0.1 Hz). However, anti-CaV1.3 PNA or mismatch PNA again had no effect.
Figure 5.

Reversal of the SNL-induced increase in neuronal excitability and responsiveness by blockade of CaV1.2 expression. A, In vivo extracellular recording of the discharge pattern (lower trace) and firing frequency (upper trace) of a deep WDR neuron, in response to physiological innocuous (brush, pressure) and noxious (pinch) stimulation of its receptive field on the foot-paw (control animal). B, Mean spontaneous discharge frequency in sham and SNL animals and in rats intrathecally injected with anti-CaV1.2, anti-CaV1.3, and mismatch TP10-PNAs. In the SNL animals, spontaneous activity increased substantially, and was restored to normal by the specific blockade of CaV1.2 expression. Stars, p < 0.05. C, Mean neuronal responses to mechanical stimulation of the paw with von Frey hairs of increasing forces (5 and 10 g, innocuous; 25 and 50 g, noxious) in sham (filled symbols) and SNL animals (open symbols). Neuronal responses to natural stimulation were increased in SNL animals whatever the force. Three stars, p < 0.001, sham versus SNL. D, In SNL animals, normal responsiveness was restored by the specific blockade of CaV1.2 expression (filled circles), whereas the sensitization remained unchanged after application of anti-CaV1.3 (open circles) or mismatch TP10-PNAs (filled triangles). One star, p < 0.05; two stars, p < 0.01; three stars, p < 0.001 (“SNL + anti-CaV1.2” vs SNL). After anti-CaV1.2 TP10-PNA application, the responses were equivalent to the sham responses (p > 0.05). n, Number of recorded neurons. B–D were obtained from 8 sham, 26 SNL, 7 anti-CaV1.2 PNA-injected, 4 anti-CaV1.3 PNA-injected, and 6 mismatch PNA-injected rats.
We further considered the responses of the same neurons to mechanical stimulation of their receptive field on the ipsilateral paw, with von Frey hairs of increasing rigidity. In the SNL animals, WDRs presented much stronger responses than in the sham animals, for both innocuous (Fig. 5C, 5–10 g) and noxious (Fig. 5C, 25–50 g) stimulations. This increase in WDR sensitivity was totally reversed after intrathecal application of anti-CaV1.2 PNA (Fig. 5D, filled circles), whereas anti-CaV1.3 PNA (Fig. 5D, open circles) or mismatch PNA (Fig. 5D, triangles) had no effects. Our results therefore indicate that specific calcium influx through CaV1.2 LTC is a critical factor of DHN sensitization in the SNL model of neuropathic pain.
Upregulation of CaV1.2 LTCs
This specific involvement of CaV1.2 LTCs in DHN sensitization shown by neuropathic rats, fits with an increase in the expression of CaV1.2 reported in other models of nerve injury (Dobremez et al., 2005). We therefore evaluated the expression of CaV1.2 mRNA in neuropathic animals by quantitative RT-PCR. As shown in Figure 6A, the expression of CaV1.2 mRNA increased by more than twofold in the SNL rats compared to the sham animals (215% of sham, p < 0.01) (Fig. 6A, black bar). As expected, this overexpression was strongly reduced after anti-CaV1.2 PNA intrathecal application (40.1% of sham, equivalent to 19% of SNL, p = 0.002) (Fig. 6A, white bar). Intrathecal application of anti-CaV1.2 siRNA also strongly reduced the overexpression of CaV1.2 transcripts (88.6% of sham, equivalent to 41% of SNL, p < 0.01) (Fig. 6A, dotted bar), whereas application of either anti-CaV1.3 PNA (Fig. 6A, gray bar) or mismatch PNA or mismatch siRNA (Fig. 6A, middle and right hatched bars, respectively) had no effect on CaV1.2 mRNA expression (230.3 ± 29.3%, 196.5 ± 25.3%, and 237.3 ± 33.4% of sham, respectively, p = 0.93, 0.59, and 0.38 vs SNL). Unlike CaV1.2, the expression of CaV1.3 transcripts showed a significant decrease in the SNL rats (58.3% ± 7.4% of sham, p < 0.01) (Fig. 6B, black bar). As predicted, this expression was further reduced after intrathecal application of anti-CaV1.3 PNA (22.6% of sham, p < 0.05) (Fig. 6B, gray bar). Importantly, however, intrathecal application of anti-CaV1.2 PNA in SNL animals had no effect on CaV1.3 mRNA expression (50.1% of sham, equivalent to 86% of SNL, p = 0.62) (Fig. 6B, white bar), like the application of mismatch PNA (55% of sham, equivalent to 94% of SNL, p = 0.85) (Fig. 6B, hatched bar). This indicates that reduction of the SNL-induced allodynia by anti-CaV1.2 PNA cannot be mediated by the alteration of CaV1.3 mRNA level.
CaV1.2 upregulation in SNL rats was confirmed by Western blots. Expression of CaV1.2 protein was examined by Western blotting in total extracts of ipsilateral lumbar spinal cord. The protein level increased in the spinal cord of SNL animals (Fig. 6C1,C2, black bar) (154.3 ± 5.2% of sham, p < 0.01). This overexpression was suppressed by intrathecal application of anti-CaV1.2 PNA or siRNA (Fig. 6C2, white and dotted bars, respectively) (103.3 ± 1.5% of sham, equivalent to 66.94 ± 0.9% of SNL, p < 0.001, PNA vs SNL, and 113.2 ± 1.5% of sham, equivalent to 73.37 ± 0.9% of SNL, p < 0.001, siRNA vs SNL).
As reported previously (Westenbroek et al., 1998; Dobremez et al., 2005), CaV1.2 immunostaining can be observed throughout the whole dorsal horn of the lumbar spinal cord. The intensity of CaV1.2 immunofluorescence significantly increased in neuropathic rats (Fig. 7Aa,Ab; Fig. 7B1, black bar) (129.6% of sham, p = 0.002). CaV1.2 immunofluorescence also appeared in the ventral horn, especially on motor neurons (Fig. 7Ad,Ae, arrows), but did not significantly increased in SNL animals (Fig. 7B2, black bar) (112.9% of sham, p = 0.08), unlike what occurs in chronic spinal cord injury (Anelli et al., 2007). In both dorsal and ventral horns, immunolabeling of CaV1.2 strongly decreased after intrathecal applications of anti-CaV1.2 PNA (Fig. 7Ac,Af; Fig. 7B, white bars) (62.4 ± 6.2% and 61.7 ± 6.8 of sham, respectively, p < 0.01). Conversely, application of anti-CaV1.3 had no effect on CaV1.2 immunolabeling (Fig. 7B, gray bars) (136.5 ± 8.9% and 109.9 ± 7.8 of sham in DH and VH, respectively, p = 0.26 and 0.45 anti-CaV1.3 vs SNL).
Together, our results showed that, in neuropathic pain conditions, CaV1.2 LTCs were upregulated in dorsal horn neurons.
LTC-dependent calcium rise
In long-term sensitization, calcium influx leads to gene regulation (Ji et al., 2003). Thus, we investigated the intracellular mechanisms activated by an LTC-mediated calcium rise in dorsal horn neurons. We first conducted calcium-imaging experiments on dissociated spinal cell cultures to test for subtype-specific calcium entry in the somatic compartments (Fig. 8A). Stimulation of the cultures with K+ application induced a long-lasting rise in [Ca2+]i (Fig. 8B) yielding to a %ΔF/F (see Materials and Methods) of 35.85 ± 2.49 (mean ± SEM, n = 84 cells in 11 cultures). This global Ca2+ transient was significantly decreased in cultures treated with anti-CaV1.2 or anti-CaV1.3 PNA, although the decrease was much more pronounced with the anti-CaV1.2 PNA (%ΔF/F = 16.34 ± 1.85 and 30.00 ± 2.84, respectively). After PNA treatments, the reduction of Ca2+ transients occurred in both the cytoplasmic and nuclear compartments (see Materials and Methods). However, the ratio between nucleus and cytoplasmic fluorescence decreased significantly in the presence of anti-CaV1.2 PNA, whereas the change remained nonsignificant in the presence of anti-CaV1.3 or mismatch PNA (Fig. 8C), indicating that CaV1.2 channels contribute more prominently to the calcium rise in the nucleus.
Figure 8.
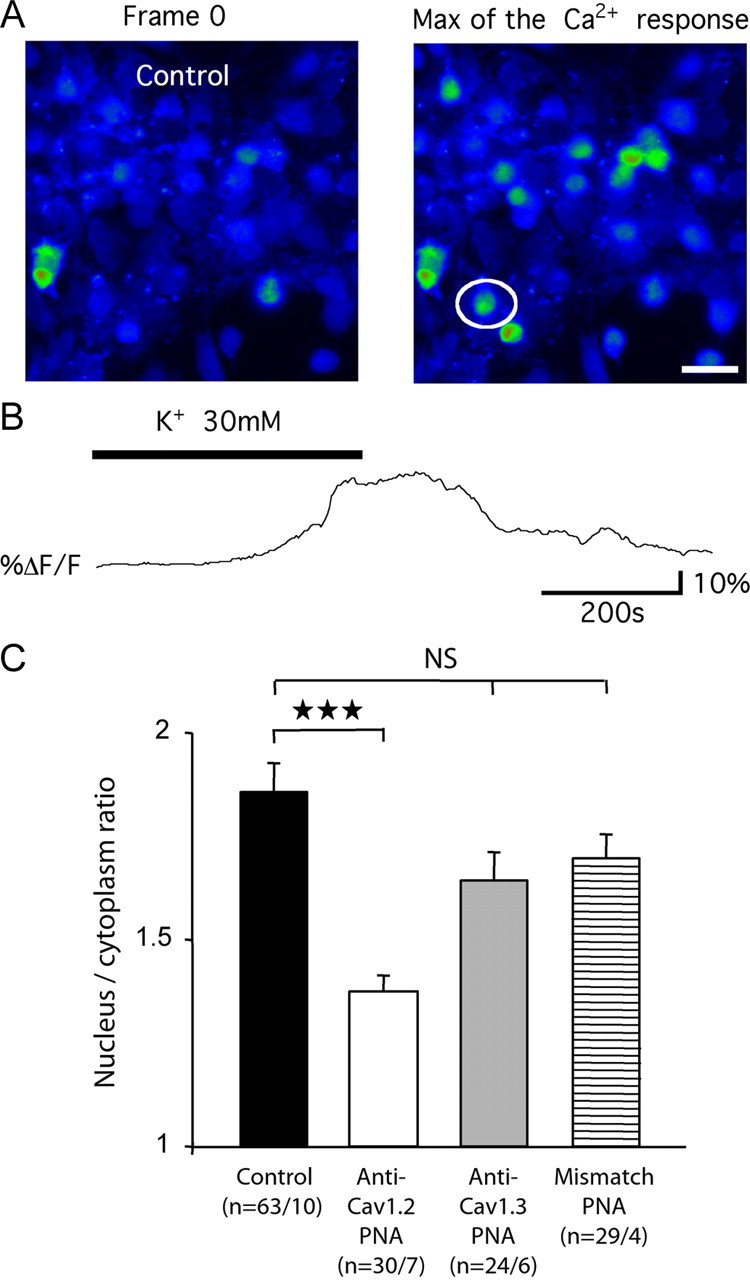
Intracellular Ca2+ dynamic and L-type channels. A, Fluorescent image of dissociated spinal neurons loaded with 10 μm Fluo4/AM, in response to K+ application. Images were taken at the beginning of the acquisition (frame 0) and at the maximum of the Ca2+ response, in nontransfected cultures. White circle, cell recorded in B. Scale bar, 30 μm. B, Example trace of change in global intracellular Ca2+ concentration in response to bath application of 30 mm K+. C, Ratio of the nucleic versus cytoplasmic fluorescence responses obtained in control nontransfected cultures, and in cultures transfected with anti-CaV1.2, anti-CaV1.3, or mismatch TP10-PNAs. n, The first and second numbers indicate the number of tested cells, and of tested cultures, respectively. Three stars, p < 0.001; NS, nonsignificant.
The stronger CaV1.2-dependent calcium rise in the nucleus, and the fact that LTCs have long been known to induce regulation of gene expression (Bading et al., 1993), suggest that the effect of this L-channel subtype in SNL-derived sensitization involves gene activation.
CaV1.2-dependent gene activation
To test for this possibility, we investigated the phosphorylation of CREB, a transcription factor activated by LTC-mediated calcium influx (Bading et al., 1993; Deisseroth et al., 1998; Dolmetsch et al., 2001). To do so, we used immunoautoradiography and Micro-imager analysis to compare, quantitatively, the amount of phosphorylated, active forms of CREB in the following experimental conditions: sham-operated animals, SNL animals, and SNL animals with anti-CaV1.2 intrathecal application. As we showed (see above) that CaV1.3-mediated calcium influx is not involved in the SNL-induced sensitization, we also used anti-CaV1.3 PNA application as a negative control. The study was performed separately in more superficial (I–II) and deeper laminae (V–VI), the former of which receive most of the nociceptive inputs, whereas the latter are part of the nociceptive output stage.
As in other models of neuropathy (Ma and Quirion, 2001; Miletic et al., 2002; Song et al., 2005; Crown et al., 2006), the expression of the phosphorylated form of the CREB transcription factor significantly increased in the ipsilateral dorsal horn of SNL animals (Fig. 9, SNL, black bars), both in superficial and deep laminae (respectively +21.0 ± 2.8% and +21.4 ± 2.0% compared to contralateral dorsal horn). Intrathecal application of the anti-CaV1.2 PNA in neuropathic rats restored phospho-CREB (pCREB) amounts almost to the sham values (+4.0 ± 2.0% vs +0.7 ± 2.0% in sham, p = 0.55, in superficial laminae; +5.7 ± 1.1% vs +0.6 ± 1.0% in sham, p = 0.11, in deep laminae) (Fig. 9, white vs dotted bars). In contrast, anti-CaV1.3 PNA application did not reverse the SNL-associated increase in pCREB levels (Fig. 9, gray bars). In addition, our data showed that the immunoreactivity for total CREB (phosphorylated plus nonphosphorylated forms) did not change after CaV1.2 PNA injection (+22.8 ± 3.1% vs +26.5 ± 4.3% in SNL, p = 0.73, in superficial laminae; +18.8 ± 5.9% vs +10.3 ± 2.6% in SNL, p = 0.20, in deep laminae) (data not shown). This further confirms that, in SNL animals, calcium influx through the CaV1.2 subtype preferentially induces CREB phosphorylation rather than modifying CREB expression. This specific CaV1.2-dependent phosphorylation of CREB is consistent with the specific role of this calcium channel subtype in pain-associated mechanical hypersensitivity that is also shown in the present study (Fig. 4).
Figure 9.
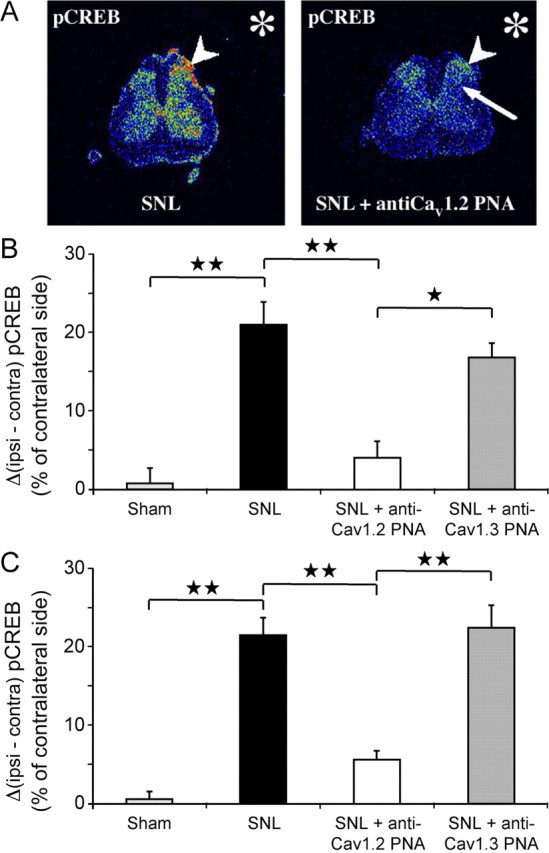
SNL-induced upregulation of pCREB. Reversal after intrathecal anti-CaV1.2 TP10-PNA application. A, Radioactive immunolabeling of pCREB in the spinal dorsal horn (transverse section in the lumbar spinal cord). B, Quantification in superficial dorsal horn. The values represent the difference in surfacic activity between ipsilateral and contralateral sides expressed in percentage of the contralateral side (ΔSA = [(SAi − SAc)/SAc] × 100; see Materials and Methods). C, Quantification in deep dorsal horn. In SNL animals (A, left), pCREB expression increased (arrowhead) ipsilateral to the ligature (asterisk), both in superficial (black bar in B) and deep (black bar in C) laminae. Intrathecal application of anti-CaV1.2 TP10-PNA (A, right) suppressed the ipsilateral increase in pCREB, both in the superficial (arrowhead in A, right, white bar in B) and deep laminae (arrow in A, right, white bar in C). In contrast, CaV1.3 knockdown did not modify the SNL-induced increase in pCREB expression, either in the superficial (gray bar in B) or in the deep (gray bar in C) laminae. One star, p < 0.05; two stars, p < 0.01.
Next, we investigated the classical MAPK [extracellular signal-regulated kinase 1 and 2 (ERK1/2) and p38] and CaM kinase II (CaMKII) intracellular signaling pathways involved in CREB activation (West et al., 2001), and elicited by chronic pain (Woolf and Salter, 2000). The expression of the phosphorylated forms of the three kinases (ERK1/2, p38 MAPK, and CaMKII) significantly increased in the ipsilateral dorsal horn of neuropathic animals (supplemental Fig. S4, available at www.jneurosci.org as supplemental material, SNL, black bars). However, the intrathecal application of either anti-CaV1.2 or anti-CaV1.3 PNA did not reverse these kinase upregulations (supplemental Fig. S4, white and gray bars, available at www.jneurosci.org as supplemental material). The preceding data therefore indicate that the CaV1.2-specific phosphorylation of CREB in SNL animals is not mediated by intracellular signaling pathways involving ERK1/2, p38 MAPK, or CaMKII.
The CaV1.2-dependent upregulation of P-CREB should lead to an enhanced level of transcription of CRE-dependent genes. To test for this possibility, we monitored the expression of COX-2, a key enzyme regulating the production of prostaglandins, central mediators of inflammation, whose expression may be regulated by pCREB (Pham et al., 2006; Tsatsanis et al., 2006). Using qRT-PCR, we compared the expression of COX-2 mRNA in sham and SNL rats, before and after the intrathecal application of PNA targeting the two LTC α-1 subunits. The expression of COX-2 mRNA increased by more than threefold in the SNL rats compared to the sham animals (316% of sham, n = 7, p < 0.01) (Fig. 10, black bar). This overexpression was totally reversed to the sham value after anti-CaV1.2 PNA intrathecal application (103% of sham, n = 4, p = 0.79) (Fig. 10, white bar). Again, application of anti-CaV1.3 PNA (297% of sham, n = 3, p < 0.02) (Fig. 10, gray bar) or mismatch PNA (252% of sham, n = 3, p < 0.05) (Fig. 10, hatched bar) did not reverse the SNL-associated increase in COX-2 mRNA expression. Therefore, we conclude that it was the CaV1.2-dependent increase in pCREB expression in SNL animals that led to the enhancement of the CRE-dependent COX-2 gene transcription.
Figure 10.

SNL-induced CaV1.2-dependent upregulation of the CRE-dependent COX-2 gene. qRT-PCR analyses showed that COX-2 mRNA increased by more than threefold in SNL rats compared to sham animals (black bar). Anti-CaV1.2 TP10-PNA intrathecal injections restored COX-2 mRNA expression to sham animal levels (white bar). Anti-CaV1.3 (gray bar) or mismatch TP10-PNA (hatched bar) intrathecal applications had no effect on COX-2 expression in SNL rats. n, Number of animals; one star, p < 0.05; two stars, p < 0.01; NS, nonsignificant; all values versus sham.
Discussion
Two types of L calcium channels, which contain either the CaV1.2 or the CaV1.3 α1 subunit, are expressed in the CNS (Ertel et al., 2000). By using an antisense-based strategy, we demonstrated here that the specific knockdown of CaV1.2, but not of CaV1.3 channel expression, totally reversed the long-term sensitization associated with chronic neuropathic pain. We showed that CaV1.2 is upregulated in the SNL model of neuropathic pain, and that this upregulation is associated with an increase in pCREB expression, leading to the transcription enhancement of the CRE-dependent COX-2 gene.
The CaV1.2 LTC maintains long-term neuropathic sensitization
The use of antisense that specifically targeted either CaV1.2 or CaV1.3 is particularly relevant because, except for a difference in dose dependence for dihydropyridines (Xu and Lipscombe, 2001), all the available L-type voltage-gated calcium channel (VGCC) antagonists do not discriminate between the two LTC subtypes. Our antisense strategy used PNA conjugated to TP10, a shorter analog of the cell-penetrating peptide transportan (Soomets et al., 2000). Moreover, the intrathecal procedure used (chronic intrathecal catheter) allowed us to target the lumbar spinal cord while limiting the diffusion of biologically active constructs. Actually, with biotinylated PNAs, we checked that they reached neither the DRGs, nor the thoracic or sacral spinal cord segments. qRT-PCR and quantification of immunolabeling confirmed that expression of both CaV1.2 mRNA and protein was not altered in DRGs.
In accordance with the original study of Kim and Chung (1992), the SNL procedure led to long-term sensitization, marked by a robust tactile allodynia. Focal and selective knockdown of CaV1.2 by PNAs resulted in the restoration of the normal threshold for mechanical sensitivity. The intrathecal application of siRNAs targeting two distinct regions of CaV1.2 produced similar effects. Moreover, this knockdown reversed the hyperexcitability of deep DHNs, which restored normal background activity and responsiveness to physiological stimuli. Interestingly, these functional modifications were concomitant with an upregulation of CaV1.2 mRNA and protein in SNL animals, consistent with the increase in CaV1.2 channels reported in other models of nerve injury (Dobremez et al., 2005). Altogether, our data strongly suggest that the upregulated CaV1.2 calcium channel subtype actually plays a prominent role in the maintenance of long-term neuropathic sensitization.
CaV1.2 gene activation
Calcium influx through neuronal LTCs plays a pivotal role in excitation–transcription coupling, triggering changes in gene expression that result in long-term CNS plasticity, especially via a potent activation of the CRE/CREB-dependent transcription (Bading et al., 1993; Bito et al., 1996; Deisseroth et al., 1998; Dolmetsch et al., 2001; West et al., 2001). Pathological changes in neuronal sensitization associated with chronic pain, involve such multiple gene regulation following neuronal hyperactivity (Woolf and Salter, 2000), and activation of the CREB transcription factor contributes to long-term modifications in the processing of nociceptive information in the spinal cord (Ma and Quirion, 2001; Miletic et al., 2002; Wu et al., 2002; Song et al., 2005; Crown et al., 2006). The present study shows a similar activation of CREB signaling in the SNL model of neuropathy, and demonstrates the essential and specific contribution of the CaV1.2 channel in this process.
Downstream of LTCs, CREB activation may be controlled by distinct cytoplasmic and nuclear calcium signaling pathways (Hardingham et al., 1997; West et al., 2001). Classical cascades result from the phosphorylation of the ERK1/2 MAP kinases, the p38 MAPK, or the CaM kinase II. Activation of these signaling pathways in the dorsal horn participates in pain hypersensitivity (Ji et al., 1999, 2003; Kawasaki et al., 2004; Song et al., 2005; Zhuang et al., 2005; Crown et al., 2006). Our quantitative analysis confirmed the overexpression of the three phosphokinases in the ipsilateral dorsal horn of SNL rats, but also showed that the knockdown of either of the two LTCs had no effect on this overexpression.
In addition to cytoplasmic kinases, nuclear kinases such as the calmodulin-activated kinase IV (CaMKIV) may phosphorylate CREB (Bito et al., 1996). This may be achieved by nuclear translocation from the cytoplasm of a calcium-activated calmodulin (Deisseroth et al., 1998, 2003). But alternatively, CREB may function as a nuclear calcium-responsive transcription factor (Hardingham et al., 1997), whose phosphorylation by CaMKIV and induction of transcriptional activity requires a calcium rise in the nucleus (Chawla et al., 1998; Hardingham et al., 2001). As evidenced by our calcium imaging experiments, CaV1.2 is an important source of nuclear calcium in spinal neurons. This suggests that CREB-activating nuclear calcium signals downstream of the CaV1.2 channels in spinal neurons may control the long-term plasticity associated with neuropathic pain. The involvement of CaMKIV in this CaV1.2-dependent sensitization requires further investigation.
We also report here a downregulation of CaV1.3 mRNA. But although the CaV1.3 LTC-mediated Ca2+ influx may also activate pCREB signaling (H. Zhang et al., 2005), our selective knockdown of CaV1.3 in the dorsal horn of SNL animals showed no evidence that these channels promote CREB phosphorylation. Accordingly, the knockdown of CaV1.3 had no effects on both allodynia and DHN hyperexcitability. This is consistent with data showing that mice lacking the CaV1.3 subunit present unmodified mechanical and thermal nociceptive sensitivity (Clark et al., 2003). This absence of effect is noteworthy in light of our previous observation that both CaV1.2 and CaV1.3 are strictly necessary for the expression of windup (Fossat et al., 2004), a form of short-term sensitization to pain dependent on LTCs (Fossat et al., 2007), but not requiring gene activation.
The phosphorylation of CREB is not by itself sufficient to induce its transcription activity (Chawla et al., 1998; X. Zhang et al., 2005). Therefore, we verified that the CaV1.2-sensitive increase in p-CREB that occurred in SNL animals actually enhanced the downstream transcription of CRE-dependent genes. We chose to monitor the expression of COX-2 because (1) it is an inducible enzyme strongly upregulated in the spinal cord by proinflammatory stimuli, (2) it is implicated in chronic pain states [for review, see Bingham et al. (2006), Tsatsanis et al. (2006), and Zeilhofer (2007)], and (3) it may be controlled by several transcription factors including p-CREB (Pham et al., 2006; Tsatsanis et al., 2006). We demonstrated a clear increase in COX-2 transcription in the dorsal horn ipsilateral to the spinal nerve ligation, and a recovery after specific knockdown of CaV1.2 LTCs in the dorsal horn neurons. We may therefore conclude that the two LTC subtypes expressed in the dorsal horn have differential effects, and that the specific calcium influx through CaV1.2 maintains long-term central sensitization associated with chronic neuropathic pain, acting via regulation of gene expression.
The CaV1.2 LTC in neuropathic pain
There is increasing evidence that N-type VGCCs (CaV2.1) in DRG and the spinal cord, and T-type channels (CaV3.2) in DRG play an important role in acute nociception and sensitization to pain (Bourinet et al., 2005) [for review, see Vanegas and Schaible (2000), Yaksh (2006), and Swayne and Bourinet (2008)]. The involvement of L-type channels, however, is less clear and seems to depend on the experimental model, the type of pain, the mode of application of the agonists and antagonists, and most probably the dose of these compounds (Vanegas and Schaible, 2000; Yaksh, 2006). Although L-type channel blockers decrease primary and secondary hyperalgesia and mechanical allodynia in monoarthritic rat (Neugebauer et al., 1996; Schaible et al., 2000), their effects in neuropathic pain remain controversial. It was reported that they did not significantly influence tactile allodynia in the SNL model (Chaplan et al., 1994) or in mice treated with vincristine (Fukuizumi et al., 2003). In streptozotocin-induced diabetic rats, intrathecal application of verapamil, diltiazem or nimodipine had no effect on tactile allodynia (Calcutt and Chaplan, 1997), whereas systemic nimodipine decreased thermal hyperalgesia (Gupta et al., 2003). The discrepancies between these reports might result from the low potency of intrathecally applied dihydropyridine antagonists. In the present work actually, even prolonged infusion of nicardipine with an implanted osmotic pump decreased the SNL-induced mechanical hyper-sensitivity by 40% only. This could explain why the many treatments for cardiovascular diseases with oral or systemic dihydropyridines in humans yielded no analgesic effects. According to animal studies on dihydropyridine bioavailability in brain ventricles after systemic injection (Tsukahara et al., 1989), one may approximate a difference of three orders of magnitude between intra-arterial and CSF concentrations. At therapeutic intravenous doses (Porchet et al., 1992; Cheung et al., 1999), the concentration of dihydropyridine in the CSF must be far too low to exert any antihyperalgesic effect. In any event, our antisense strategy demonstrated that knockdown of the CaV1.2 LTC gene induced a strong antiallodynic effect in the SNL model, and a reversal of dorsal horn neuron's hyperexcitability, thereby providing direct evidence for a pronociceptive role of this calcium channel in the neuropathic pain state. CaV1.2, but not CaV1.3, mediates long-term pathological sensitization through CREB phosphorylation. Altogether, our results provide a direct demonstration of the role of L-type calcium channels in supporting central nociception, and pathological sensitization.
Concluding remarks
Our data demonstrate that the LTCs in the spinal dorsal horn, especially those comprising CaV1.2, are crucial factors for the maintenance of chronic neuropathic pain. Handling LTC blockers in clinics may be complicated regarding the lack of isoform-selective antagonists. But a better understanding of the mechanisms elicited by LTC-dependent sensitization should indicate new therapeutic avenues to control neuropathic pain by targeting either LTC-auxiliary subunits, associated proteins, or downstream signaling pathways regulating gene expression.
Footnotes
This work was supported by grants from the Conseil Régional d'Aquitaine (20010301213), the Agence Nationale de la Recherche (ANR-07-Neuro-015-01, the Direction Générale des Armées (01.34.012.00470.75.01), the Swedish Research Councils for Natural and Engineering Sciences (VR-NT) and Medical Research (VR-Med), the Wallenberg Foundation, and the European Community FP5 project QLK3-CT-2002-01989. We are grateful to Drs. Nicole Barthes and Brigitte Brouillaud (INSERM U551) for technical assistance in using the Micro-imager. We thank Prof. Bernard Rayner (INSERM U383) for his help in initial PNA-construct purification.
References
- Anelli R, Sanelli L, Bennett DJ, Heckman CJ. Expression of L-type calcium channel alpha(1)-1.2 and alpha(1)-1.3 subunits on rat sacral motoneurons following chronic spinal cord injury. Neuroscience. 2007;145:751–763. doi: 10.1016/j.neuroscience.2006.12.043. [DOI] [PubMed] [Google Scholar]
- Bading H, Ginty DD, Greenberg ME. Regulation of gene expression in hippocampal neurons by distinct calcium signaling pathways. Science. 1993;260:181–186. doi: 10.1126/science.8097060. [DOI] [PubMed] [Google Scholar]
- Baron R. Mechanisms of disease: neuropathic pain—a clinical perspective. Nat Clin Pract Neurol. 2006;2:95–106. doi: 10.1038/ncpneuro0113. [DOI] [PubMed] [Google Scholar]
- Bingham S, Beswick PJ, Blum DE, Gray NM, Chessell IP. The role of the cylooxygenase pathway in nociception and pain. Semin Cell Dev Biol. 2006;17:544–554. doi: 10.1016/j.semcdb.2006.09.001. [DOI] [PubMed] [Google Scholar]
- Bito H, Deisseroth K, Tsien RW. CREB phosphorylation and dephosphorylation: a Ca(2+)- and stimulus duration-dependent switch for hippocampal gene expression. Cell. 1996;87:1203–1214. doi: 10.1016/s0092-8674(00)81816-4. [DOI] [PubMed] [Google Scholar]
- Bourinet E, Alloui A, Monteil A, Barrère C, Couette B, Poirot O, Pages A, McRory J, Snutch TP, Eschalier A, Nargeot J. Silencing of the Cav3.2 T-type calcium channel gene in sensory neurons demonstrates its major role in nociception. EMBO J. 2005;24:315–324. doi: 10.1038/sj.emboj.7600515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calcutt NA, Chaplan SR. Spinal pharmacology of tactile allodynia in diabetic rats. Br J Pharmacol. 1997;122:1478–1482. doi: 10.1038/sj.bjp.0701538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell JN, Meyer RA. Mechanisms of neuropathic pain. Neuron. 2006;52:77–92. doi: 10.1016/j.neuron.2006.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaplan SR, Pogrel JW, Yaksh TL. Role of voltage-dependent calcium channel subtypes in experimental tactile allodynia. J Pharmacol Exp Ther. 1994;269:1117–1123. [PubMed] [Google Scholar]
- Chawla S, Hardingham GE, Quinn DR, Bading H. CBP: a signal-regulated transcriptional coactivator controlled by nuclear calcium and CaM kinase IV. Science. 1998;281:1505–1509. doi: 10.1126/science.281.5382.1505. [DOI] [PubMed] [Google Scholar]
- Cheung AT, Guvakov DV, Weiss SJ, Savino JS, Salgo IS, Meng QC. Nicardipine intravenous bolus dosing for acutely decreasing arterial blood pressure during general anesthesia for cardiac operations: pharmacokinetics, pharmacodynamics, and associated effects on left ventricular function. Anesth Analg. 1999;89:1116–1123. [PubMed] [Google Scholar]
- Clark NC, Nagano N, Kuenzi FM, Jarolimek W, Huber I, Walter D, Wietzorrek G, Boyce S, Kullmann DM, Striessnig J, Seabrook GR. Neurological phenotype and synaptic function in mice lacking the CaV1.3 alpha subunit of neuronal L-type voltage-dependent Ca2+ channels. Neuroscience. 2003;120:435–442. doi: 10.1016/s0306-4522(03)00329-4. [DOI] [PubMed] [Google Scholar]
- Costigan M, Scholz J, Woolf CJ. Neuropathic pain: a maladaptive response of the nervous system to damage. Annu Rev Neurosci. 2009;32:1–32. doi: 10.1146/annurev.neuro.051508.135531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crown ED, Ye Z, Johnson KM, Xu GY, McAdoo DJ, Hulsebosch CE. Increases in the activated forms of ERK 1/2, p38 MAPK, and CREB are correlated with the expression of at-level mechanical allodynia following spinal cord injury. Exp Neurol. 2006;199:397–407. doi: 10.1016/j.expneurol.2006.01.003. [DOI] [PubMed] [Google Scholar]
- Deisseroth K, Heist EK, Tsien RW. Translocation of calmodulin to the nucleus supports CREB phosphorylation in hippocampal neurons. Nature. 1998;392:198–202. doi: 10.1038/32448. [DOI] [PubMed] [Google Scholar]
- Deisseroth K, Mermelstein PG, Xia H, Tsien RW. Signaling from synapse to nucleus: the logic behind the mechanisms. Curr Opin Neurobiol. 2003;13:354–365. doi: 10.1016/s0959-4388(03)00076-x. [DOI] [PubMed] [Google Scholar]
- Dobremez E, Bouali-Benazzouz R, Fossat P, Monteils L, Dulluc J, Nagy F, Landry M. Distribution and regulation of L-type calcium channels in deep dorsal horn neurons after sciatic nerve injury in rats. Eur J Neurosci. 2005;21:3321–3333. doi: 10.1111/j.1460-9568.2005.04177.x. [DOI] [PubMed] [Google Scholar]
- Dolmetsch RE, Pajvani U, Fife K, Spotts JM, Greenberg ME. Signaling to the nucleus by an L-type calcium channel-calmodulin complex through the MAP kinase pathway. Science. 2001;294:333–339. doi: 10.1126/science.1063395. [DOI] [PubMed] [Google Scholar]
- Egholm M, Buchardt O, Christensen L, Behrens C, Freier SM, Driver DA, Berg RH, Kim SK, Norden B, Nielsen PE. PNA hybridizes to complementary oligonucleotides obeying the Watson-Crick hydrogen-bonding rules. Nature. 1993;365:566–568. doi: 10.1038/365566a0. [DOI] [PubMed] [Google Scholar]
- Ertel EA, Campbell KP, Harpold MM, Hofmann F, Mori Y, Perez-Reyes E, Schwartz A, Snutch TP, Tanabe T, Birnbaumer L, Tsien RW, Catterall WA. Nomenclature of voltage-gated calcium channels. Neuron. 2000;25:533–535. doi: 10.1016/s0896-6273(00)81057-0. [DOI] [PubMed] [Google Scholar]
- Fossat P, Dobremez E, Landry M, Sibon I, Bouali-Benazzouz R, Langel Ü, Kilk K, Le Masson G, Nagy F. NMDA receptors and L-type calcium channels cooperate in short term sensitization to pain in the rat spinal cord. Soc Neurosci Abstr. 2004;30:644–613. [Google Scholar]
- Fossat P, Sibon I, Le Masson G, Landry M, Nagy F. L-type calcium channels and NMDA receptors: a determinant duo for short-term nociceptive plasticity. Eur J Neurosci. 2007;25:127–135. doi: 10.1111/j.1460-9568.2006.05256.x. [DOI] [PubMed] [Google Scholar]
- Fukuizumi T, Ohkubo T, Kitamura K. Spinally delivered N-, P/Q- and L-type Ca2+-channel blockers potentiate morphine analgesia in mice. Life Sci. 2003;73:2873–2881. doi: 10.1016/s0024-3205(03)00700-8. [DOI] [PubMed] [Google Scholar]
- Gomez-Ospina N, Tsuruta F, Barreto-Chang O, Hu L, Dolmetsch R. The C terminus of the L-type voltage-gated calcium channel Ca(V)1.2 encodes a transcription factor. Cell. 2006;127:591–606. doi: 10.1016/j.cell.2006.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta M, Singh J, Sood S, Arora B. Mechanism of antinociceptive effect of nimodipine in experimental diabetic neuropathic pain. Methods Find Exp Clin Pharmacol. 2003;25:49–52. doi: 10.1358/mf.2003.25.1.772547. [DOI] [PubMed] [Google Scholar]
- Hardingham GE, Chawla S, Johnson CM, Bading H. Distinct functions of nuclear and cytoplasmic calcium in the control of gene expression. Nature. 1997;385:260–265. doi: 10.1038/385260a0. [DOI] [PubMed] [Google Scholar]
- Hardingham GE, Arnold FJ, Bading H. Nuclear calcium signaling controls CREB-mediated gene expression triggered by synaptic activity. Nat Neurosci. 2001;4:261–267. doi: 10.1038/85109. [DOI] [PubMed] [Google Scholar]
- Hell JW, Westenbroek RE, Warner C, Ahlijanian MK, Prystay W, Gilbert MM, Snutch TP, Catterall WA. Identification and differential subcellular localization of the neuronal class C and class D L-type calcium channel alpha 1 subunits. J Cell Biol. 1993;123:949–962. doi: 10.1083/jcb.123.4.949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jasmin L, Ohara PT. Long-term intrathecal catheterization in the rat. J Neurosci Methods. 2001;110:81–89. doi: 10.1016/s0165-0270(01)00420-4. [DOI] [PubMed] [Google Scholar]
- Ji RR, Baba H, Brenner GJ, Woolf CJ. Nociceptive-specific activation of ERK in spinal neurons contributes to pain hypersensitivity. Nat Neurosci. 1999;2:1114–1119. doi: 10.1038/16040. [DOI] [PubMed] [Google Scholar]
- Ji RR, Kohno T, Moore KA, Woolf CJ. Central sensitization and LTP: do pain and memory share similar mechanisms? Trends Neurosci. 2003;26:696–705. doi: 10.1016/j.tins.2003.09.017. [DOI] [PubMed] [Google Scholar]
- Kawasaki Y, Kohno T, Zhuang ZY, Brenner GJ, Wang H, Van Der Meer C, Befort K, Woolf CJ, Ji RR. Ionotropic and metabotropic receptors, protein kinase A, protein kinase C, and Src contribute to C-fiber-induced ERK activation and cAMP response element-binding protein phosphorylation in dorsal horn neurons, leading to central sensitization. J Neurosci. 2004;24:8310–8321. doi: 10.1523/JNEUROSCI.2396-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SH, Chung JM. An experimental model for peripheral neuropathy produced by segmental spinal nerve ligation in the rat. Pain. 1992;50:355–363. doi: 10.1016/0304-3959(92)90041-9. [DOI] [PubMed] [Google Scholar]
- Koschak A, Reimer D, Huber I, Grabner M, Glossmann H, Engel J, Striessnig J. α1D (Cav1.3) subunits can form L-type Ca2+ channels activating at negative voltages. J Biol Chem. 2001;276:22100–22106. doi: 10.1074/jbc.M101469200. [DOI] [PubMed] [Google Scholar]
- Laniece P, Charon Y, Cardona A, Pinot L, Maitrejean S, Mastrippolito R, Sandkamp B, Valentin L. A new high resolution radioimager for the quantitative analysis of radiolabelled molecules in tissue section. J Neurosci Methods. 1998;86:1–5. doi: 10.1016/s0165-0270(98)00128-9. [DOI] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- Luo MC, Zhang DQ, Ma SW, Huang YY, Shuster SJ, Porreca F, Lai J. An efficient intrathecal delivery of small interfering RNA to the spinal cord and peripheral neurons. Mol Pain. 2005;1:29. doi: 10.1186/1744-8069-1-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma W, Quirion R. Increased phosphorylation of cyclic AMP response element-binding protein (CREB) in the superficial dorsal horn neurons following partial sciatic nerve ligation. Pain. 2001;93:295–301. doi: 10.1016/S0304-3959(01)00335-9. [DOI] [PubMed] [Google Scholar]
- Martinez-Gomez J, Lopez-Garcia JA. Simultaneous assessment of the effects of L-type current modulators on sensory and motor pathways of the mouse spinal cord in vitro. Neuropharmacology. 2007;53:464–471. doi: 10.1016/j.neuropharm.2007.06.007. [DOI] [PubMed] [Google Scholar]
- Miletic G, Pankratz MT, Miletic V. Increases in the phosphorylation of cyclic AMP response element binding protein (CREB) and decreases in the content of calcineurin accompany thermal hyperalgesia following chronic constriction injury in rats. Pain. 2002;99:493–500. doi: 10.1016/S0304-3959(02)00242-7. [DOI] [PubMed] [Google Scholar]
- Neugebauer V, Vanegas H, Nebe J, Rümenapp P, Schaible HG. Effects of N- and L-type calcium channel antagonists on the responses of nociceptive spinal cord neurons to mechanical stimulation of the normal and the inflamed knee joint. J Neurophysiol. 1996;76:3740–3749. doi: 10.1152/jn.1996.76.6.3740. [DOI] [PubMed] [Google Scholar]
- Pham H, Shafer LM, Slice LW. CREB-dependent cyclooxygenase-2 and microsomal prostaglandin E synthase-1 expression is mediated by protein kinase C and calcium. J Cell Biochem. 2006;98:1653–1666. doi: 10.1002/jcb.20899. [DOI] [PubMed] [Google Scholar]
- Pooga M, Soomets U, Hällbrink M, Valkna A, Saar K, Rezaei K, Kahl U, Hao JX, Xu XJ, Wiesenfeld-Hallin Z, Hökfelt T, Bartfai T, Langel U. Cell penetrating PNA constructs regulate galanin receptor levels and modify pain transmission in vivo. Nat Biotechnol. 1998;16:857–861. doi: 10.1038/nbt0998-857. [DOI] [PubMed] [Google Scholar]
- Porchet HC, Loew F, Gauthey L, Dayer P. Serum concentration-effect relationship of (+/-)-nicardipine and nifedipine in elderly hypertensive patients. Eur J Clin Pharmacol. 1992;43:551–553. doi: 10.1007/BF02285101. [DOI] [PubMed] [Google Scholar]
- Schaible HG, Nebe J, Neugebauer V, Ebersberger A, Vanegas H. The role of high-threshold calcium channels in spinal neuron hyperexcitability induced by knee inflammation. Prog Brain Res. 2000;129:173–190. doi: 10.1016/S0079-6123(00)29013-4. [DOI] [PubMed] [Google Scholar]
- Song XS, Cao JL, Xu YB, He JH, Zhang LC, Zeng YM. Activation of ERK/CREB pathway in spinal cord contributes to chronic constrictive injury-induced neuropathic pain in rats. Acta Pharmacol Sin. 2005;26:789–798. doi: 10.1111/j.1745-7254.2005.00123.x. [DOI] [PubMed] [Google Scholar]
- Soomets U, Lindgren M, Gallet X, Hällbrink M, Elmquist A, Balaspiri L, Zorko M, Pooga M, Brasseur R, Langel U. Deletion analogues of transportan. Biochim Biophys Acta. 2000;1467:165–176. doi: 10.1016/s0005-2736(00)00216-9. [DOI] [PubMed] [Google Scholar]
- Striessnig J, Koschak A. Exploring the function and pharmacotherapeutic potential of voltage-gated Ca2+ channels with gene knockout models. Channels (Austin) 2008;2:233–251. doi: 10.4161/chan.2.4.5847. [DOI] [PubMed] [Google Scholar]
- Swayne LA, Bourinet E. Voltage-gated calcium channels in chronic pain: emerging role of alternative splicing. Pflugers Arch. 2008;456:459–466. doi: 10.1007/s00424-007-0390-4. [DOI] [PubMed] [Google Scholar]
- Tsatsanis C, Androulidaki A, Venihaki M, Margioris AN. Signalling networks regulating cyclooxygenase-2. Int J Biochem Cell Biol. 2006;38:1654–1661. doi: 10.1016/j.biocel.2006.03.021. [DOI] [PubMed] [Google Scholar]
- Tsukahara T, Arista A, Kassell NF. The distribution of intravenous nicardipine in rat brain after subarachnoid hemorrhage. Surg Neurol. 1989;32:188–194. doi: 10.1016/0090-3019(89)90177-8. [DOI] [PubMed] [Google Scholar]
- Vanegas H, Schaible H-G. Effects of antagonists to high-threshold calcium channels upon spinal mechanisms of pain, hyperalgesia and allodynia. Pain. 2000;85:9–18. doi: 10.1016/s0304-3959(99)00241-9. [DOI] [PubMed] [Google Scholar]
- Weick JP, Groth RD, Isaksen AL, Mermelstein PG. Interactions with PDZ proteins are required for L-type calcium channels to activate cAMP response element-binding protein-dependent gene expression. J Neurosci. 2003;23:3446–3456. doi: 10.1523/JNEUROSCI.23-08-03446.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West AE, Chen WG, Dalva MB, Dolmetsch RE, Kornhauser JM, Shaywitz AJ, Takasu MA, Tao X, Greenberg ME. Calcium regulation of neuronal gene expression. Proc Natl Acad Sci U S A. 2001;98:11024–11031. doi: 10.1073/pnas.191352298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westenbroek RE, Hoskins L, Catterall WA. Localization of Ca2+ channel subtypes on rat spinal motor neurons, interneurons, and nerve terminals. J Neurosci. 1998;18:6319–6330. doi: 10.1523/JNEUROSCI.18-16-06319.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woolf CJ, Salter MW. Neuronal plasticity: increasing the gain in pain. Science. 2000;288:1765–1769. doi: 10.1126/science.288.5472.1765. [DOI] [PubMed] [Google Scholar]
- Wu J, Fang L, Lin Q, Willis WD. The role of nitric oxide in the phosphorylation of cyclic adenosine monophosphate-responsive element-binding protein in the spinal cord after intradermal injection of capsaicin. J Pain. 2002;3:190–198. doi: 10.1054/jpai.2002.123653. [DOI] [PubMed] [Google Scholar]
- Xu W, Lipscombe D. Neuronal CaV1.3α1 L-type channels activate at relatively hyperpolarized membrane potentials and are incompletely inhibited by dihydropyridines. J Neurosci. 2001;21:5944–5951. doi: 10.1523/JNEUROSCI.21-16-05944.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yaksh TL. Calcium channels as therapeutic targets in neuropathic pain. J Pain. 2006;7:S13–S30. doi: 10.1016/j.jpain.2005.09.007. [DOI] [PubMed] [Google Scholar]
- Zeilhofer HU. Prostanoids in nociception and pain. Biochem Pharmacol. 2007;73:165–174. doi: 10.1016/j.bcp.2006.07.037. [DOI] [PubMed] [Google Scholar]
- Zhang H, Maximov A, Fu Y, Xu F, Tang TS, Tkatch T, Surmeier DJ, Bezprozvanny I. Association of CaV1.3 L-type calcium channels with Shank. J Neurosci. 2005;25:1037–1049. doi: 10.1523/JNEUROSCI.4554-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Fu Y, Altier C, Platzer J, Surmeier DJ, Bezprozvanny I. Ca1.2 and CaV1.3 neuronal L-type calcium channels: differential targeting and signaling to pCREB. Eur J Neurosci. 2006;23:2297–2310. doi: 10.1111/j.1460-9568.2006.04734.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Odom DT, Koo SH, Conkright MD, Canettieri G, Best J, Chen H, Jenner R, Herbolsheimer E, Jacobsen E, Kadam S, Ecker JR, Emerson B, Hogenesch JB, Unterman T, Young RA, Montminy M. Genome-wide analysis of cAMP-response element binding protein occupancy, phosphorylation, and target gene activation in human tissues. Proc Natl Acad Sci U S A. 2005;102:4459–4464. doi: 10.1073/pnas.0501076102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhuang ZY, Gerner P, Woolf CJ, Ji RR. ERK is sequentially activated in neurons, microglia, and astrocytes by spinal nerve ligation and contributes to mechanical allodynia in this neuropathic pain model. Pain. 2005;114:149–159. doi: 10.1016/j.pain.2004.12.022. [DOI] [PubMed] [Google Scholar]