

Abstract
Type I interferons (IFN) are crucial in host defense but also are implicated as causative factors for neurological disease. Interferon regulatory factor (IRF9) is involved in type I IFN-regulated gene expression where it associates with STAT1:STAT2 heterodimers to form the transcriptional complex ISGF3. The role of IRF9 in cellular responses to type I IFN is poorly defined in vivo and hence was examined here. While transgenic mice (termed GIFN) with chronic production of low levels of IFN-α in the CNS were relatively unaffected, the same animals lacking IRF9 [GIFNxIRF9 knock-out (KO)] had cataracts, became moribund, and died prematurely. The brain of GIFNxIRF9 KO mice showed calcification with pronounced inflammation and neurodegeneration whereas inflammation and retinal degeneration affected the eyes. In addition, IFN-γ-like gene expression in the CNS in association with IFN-γ mRNA and increased phosphotyrosine-STAT1 suggested a role for IFN-γ. However, GIFNxIRF9 KO mice deficient for IFN-γ signaling developed an even more severe and accelerated disease, indicating that IFN-γ was protective. In IRF9-deficient cultured mixed glial cells, IFN-α induced prolonged activation of STAT1 and STAT2 and induced the expression of IFN-γ-like genes. We conclude that (1) type I IFN signaling and cellular responses can occur in vivo in the absence of IRF9, (2) IRF9 protects against the pathophysiological actions of type I IFN in the CNS, and (3) STAT1 and possibly STAT2 participate in alternative IRF9-independent signaling pathways activated by IFN-α in glial cells resulting in enhanced IFN-γ-like responses.
Introduction
Interferon (IFN)-α is crucial in host defense against pathogens and tumors (for review, see Sen and Ransohoff, 1993; Stark et al., 1998). This cytokine is also implicated in disease pathogenesis, including neurological disorders such as Aicardi-Goutières syndrome (AGS) (Aicardi and Goutières, 1984; Lebon et al., 1988b; Rice et al., 2007), Cree encephalitis (Black et al., 1988; Crow et al., 2003), and congenital viral encephalopathy (CVE) (Dussaix et al., 1985; Lebon et al., 1988a). AGS is a genetic disorder with increased production of IFN-α that results in neurodegeneration and characteristic cerebral calcification. Similar pathologic changes are also observed in patients with CVE (for review, see Shaw and Cohen, 1993; Kenneson and Cannon, 2007). Although the causes of AGS and CVE are unknown, a role for IFN-α is suggested by the finding that transgenic mice with astrocyte-targeted production of IFN-α (termed GIFN mice) develop similar neurodegeneration, inflammation, and cerebral calcification (Akwa et al., 1998; Campbell et al., 1999).
The biological actions of IFN-α are mediated through a well characterized signal transduction pathway (Stark et al., 1998; Schindler and Brutsaert, 1999) that triggers phosphorylation of the signal transducer and activators of transcription (STAT) 1 and STAT2 proteins. STAT1 and STAT2 then form heterodimers that translocate to the nucleus and associate with IRF9 to form the IFN-stimulated gene factor 3 (ISGF3). ISGF3 regulates the transcription of many type I IFN-regulated genes through binding to a specific DNA motif termed the IFN-stimulated response element (ISRE). Mice deficient for STAT1, STAT2, or IRF9 have impaired ISGF3-mediated responsiveness to type I IFNs and are more susceptible to microbial infection (Durbin et al., 1996; Kimura et al., 1996; Meraz et al., 1996; Park et al., 2000; Karst et al., 2003). Nevertheless, there is evidence for the existence of ISGF3-independent signal transduction pathways that are activated in the cellular response to type I IFNs (for review, see van Boxel-Dezaire et al., 2006). For example, although several studies show that IRF9-deficient cells have impaired responses to type I IFNs (John et al., 1991; Kimura et al., 1996), it is clear that such cells are not completely unresponsive (Li et al., 1996). However, the biological significance of the cellular response to type I IFN in the absence of IRF9 and its impact on the CNS is unknown. The objective of the present study was to examine this problem in the brain of GIFN mice that were sufficient or deficient for IRF9. The results showed that far from crippling the actions of IFN-α, the absence of IRF9 markedly aggravated the detrimental effects of the type I IFN in the brain. Moreover, the findings showed that some key pathologic features mediated by IFN-α in the brain that are associated with AGS and CVE can occur in an ISGF3-independent manner.
Materials and Methods
Animals.
The GIFN12 mouse line (termed GIFN) with low levels of astrocyte-directed production of IFN-α1 (Akwa et al., 1998; Campbell et al., 1999), STAT1-deficient mice [STAT1 knock-out (KO) (Durbin et al., 1996), kindly provided by Dr. Joan Durbin, The Research Institute at Nationwide Children's Hospital, Ohio State University, Columbus, OH], IRF9-deficient mice [IRF9 KO (Kimura et al., 1996), obtained from Riken Bioresource Center, Ibaraki, Japan], and IFN-γ receptor-deficient mice [IFNGR KO (Huang et al., 1993), purchased from Jackson Laboratories] have been described previously and were all on the C57BL/6 background. GIFNxSTAT1 KO, GIFNxIRF9 KO, and GIFNxIRF9 KOxIFNGR KO mice were produced by interbreeding and all genotypes were verified by PCR analysis of tail DNA. All mice were maintained under pathogen-free conditions in the animal facility of the School of Molecular and Microbial Biosciences, University of Sydney, Sydney, Australia. Ethical approval for the use of all mice in this study was obtained from the University of Sydney Animal Care and Ethics Committee.
Primary mixed glial cells and extraction of nuclear protein.
Mixed glial cell (MGC) cultures from 3-d-old mice were prepared as described by Milner and ffrench-Constant (1994) using a modified technique from McCarthy and de Vellis (Bressler et al., 1980). Briefly, meninges were removed and the forebrain finely chopped and dissociated in papain before being cultured on poly-d-lysine-coated (Sigma-Aldrich) T75 culture flasks (BD Falcon) in DMEM (Invitrogen) supplemented with 10% FBS (Thermo Scientific) and ampicillin and streptomycin (Invitrogen). The cellular composition of the MGCs was 96% astrocytes and 2–3% microglia, as determined by immunocytochemistry. For treatment with IFN-α, cells were serum starved overnight and then incubated with serum-free medium containing 1000 U/ml IFN-α (Sigma-Aldrich) for the indicated times. Cells were washed in ice-cold PBS pH 7.4, scraped from the flasks, and incubated on ice for 15 min in 500 μl of lysis buffer [10 mm HEPES pH 7.9, 1.5 mm MgCl2, 10 mm KCl, 0.5 mm DTT, 0.5 mm PMSF, protease inhibitor cocktail set III (1:200, Calbiochem) and phosphatase inhibitor cocktail set II (1:200, Calbiochem)]. Following the addition of 25 μl of 10% (v/v) NP-40, nuclei were pelleted at 4000 × g for 20 s. The pellet was lysed in 150 μl of buffer [20 mm HEPES pH 7.9, 1.5 mm MgCl2, 420 mm NaCl, 0.2 mm EDTA, 25% (v/v) glycerol, 0.5 mm PMSF, protease inhibitor cocktail set III and phosphatase inhibitor cocktail set II] for 30 min at 4°C and shaken vigorously. Nuclear extracts were recovered after centrifugation at 4°C for 10 min at 15,000 × g.
RNase protection assay.
Total RNA was isolated from snap frozen medial–ventral forebrain tissue or primary mixed glial cells using TriReagent (Sigma-Aldrich) according to the manufacturer's instructions. RNase protection assays (RPAs) were performed and analyzed as described previously (Asensio and Campbell, 1997). Multiprobe sets used for RPA included IFN-regulated genes (Asensio et al., 2001), STAT and SOCS (Maier et al., 2002), cytokines (Hobbs et al., 1993), interferons (Asensio et al., 1999), and chemokines (Asensio and Campbell, 1997; Boztug et al., 2002).
Histology and immunocytochemistry.
Mice were killed, brains removed, and one hemisphere fixed overnight in ice-cold 4% paraformaldehyde in PBS, pH 7.4. Following paraffin embedding, 5-μm-thick sections were prepared and stained with hematoxylin and eosin to determine pathological changes. For immunohistochemistry, the other hemisphere was embedded in Tissue Tek (Sakura Finetek) and snap frozen in liquid nitrogen-cooled isopentane. Immunohistochemistry was performed as described previously (Hofer et al., 2008). Dilution of the primary antibodies against CD4 and CD8 (BD Pharmingen) was 1:50; 1:50 against 7/4 (Serotec), and 1:20 against Mac1 (ATCC clone TIB 128). Immunohistochemistry for the glial fibrillary acidic protein (GFAP; 1:1000; Dako) was performed on paraffin-embedded sections. Sections incubated with secondary antibodies alone were used as controls. Stained sections were examined under a DM4000B bright field microscope (Leica) and images were captured using a Spot Flex camera and Spot V4.5 software (Diagnostic Instruments).
Immunoblot analysis.
Snap frozen medial–ventral forebrain tissue was homogenized with 50 mm Tris-HCl pH 7.4 containing 1% Nonidet-P40, 10% (v/v) glycerol, 1 mm EDTA, 1 mm DTT, protease inhibitor cocktail set III (1:200; Calbiochem) and phosphatase inhibitor cocktail set II (1:200; Calbiochem). Nuclear extracts from MGCs were obtained as described above. Proteins were fractioned by SDS/PAGE using precast gradient gels (Invitrogen) and transferred to polyvinylidene difluoride membranes. Immunoblot analysis was performed as described previously (Wang et al., 2002). All primary antibodies [phosphotyrosine (pY)-STAT1, phosphoserine (pS)-STAT1, STAT1, STAT2, pY-STAT3, STAT3, phosphothreonine and phosphotyrosine (pT/Y) ERK1/2, ERK1/2, pT/Y-P38MAPK, p38MAPK (Cell Signaling Technology), pY-STAT2 (Millipore), and TBP (Abcam)] were diluted at 1:1000 with the exception of GAPDH (Sigma-Aldrich), which was diluted 1:40,000.
Results
Lack of IRF9 in mice with CNS-targeted production of IFN-α results in severe disease
Over the time course under study, GIFN, STAT1 KO (Durbin et al., 1996), and IRF9 KO (Kimura et al., 1996) mice had survival times comparable to wild type (WT) mice (Fig. 1) and showed no obvious clinical phenotype. GIFN mice deficient for STAT1 (GIFNxSTAT1 KO) failed to thrive after birth and survived for only 1–2 months, confirming our previous finding (Wang et al., 2002). GIFN mice that lacked IRF9 (GIFNxIRF9 KO), exhibited progressive weight loss, became moribund, and showed increased lethality compared with WT mice. However, GIFNxIRF9 KO mice had a significantly longer mean survival time than GIFNxSTAT1 KO mice (72 vs 38 d, respectively) (Fig. 1). In addition, compared with WT and GIFN mice (Fig. 2A) that had normal healthy eyes, the GIFNxIRF9 KO animals developed cataracts (Fig. 2B). In summary, these observations show that in GIFN mice, loss of IRF9 results in the development of severe physical deterioration, ocular disease, and premature death.
Figure 1.

Survival of GIFN mice is reduced in the absence of STAT1 or IRF9. Survival was significantly longer in GIFNxIRF9 KO mice compared with GIFNxSTAT1 KO mice (p < 0.0001, χ2 logrank test). While IFNGR-null GIFNxIRF9 KO mice were not viable (*), IFNGR-haploinsufficiency resulted in a significantly shorter survival of these animals compared with GIFNxIRF9 KO mice (p < 0.0001, χ2 logrank test).
Figure 2.
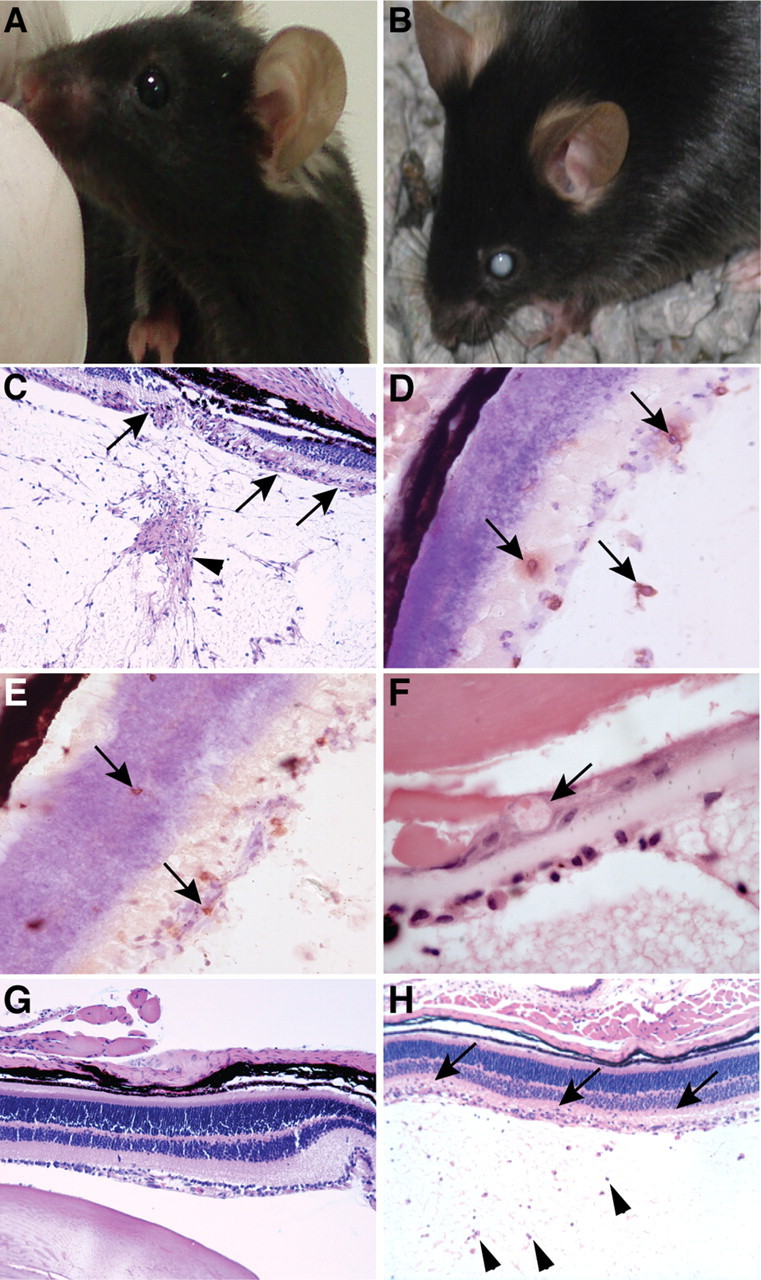
GIFN mice deficient for IRF9 develop ocular disease. A–C, In contrast to GIFN mice, whose eyes appeared normal (A), the eyes of GIFNxIRF9 KO mice showed cataracts (B). Histological examination of the eyes of GIFNxIRF9 KO mice showed destruction of the retinal architecture (C, arrows) and increased cellularity in the vitreous body (C, arrowhead). Infiltrating CD4+ (D, arrows) and CD8+ T cells (E, arrows) were present in the retina and the vitreous body, and in the lens dysplastic bladder-like fibers were present (F, arrow). No overt pathological changes were observed in the eyes of GIFN mice (G), whereas the eyes of GIFNxSTAT1 KO mice showed increased cellularity of the vitreous body (H, arrowheads) and few infiltrating cells in the retinal layers (H, arrows). C, G, H, Original magnification 50×; D, E, 400×; F, 1000×.
Inflammation and neurodegeneration in the brain and eyes of GIFN mice deficient for IRF9
The possible basis for ocular disease in GIFNxIRF9 KO mice was examined further at the cellular level. Compared with WT, no obvious histological changes were present in the eyes of GIFN mice (Fig. 2G). However, histological and immunohistochemical analysis of the eyes of GIFNxIRF9 KO mice showed epiretinal and subchoroidal edema with focal detachment of the retina from the choroid (Fig. 2C). The retinal architecture was partially destroyed with destruction of the retinal layers (Fig. 2C, arrows). Mononuclear infiltrates including CD4+ (Fig. 2D, arrows) and CD8+ T cells (Fig. 2E, arrows) were present in the retina and vitreous body. Dysplastic bladder-like fibers that are characteristic for posterior subcapsular cataracts were seen in the lenses of some GIFNxIRF9 KO mice (Fig. 2F, arrow). Moderate inflammatory changes that included increased number of cells in the vitreous body and the retina were also present in the eyes of some GIFNxSTAT1 KO mice (Fig. 2H). Yet, the overall architecture of the retinal layers was preserved in the eyes of most GIFNxSTAT1 KO mice analyzed and none of the GIFNxSTAT1 KO mice showed bladder-like fibers or developed visible cataracts. These findings indicate that GIFN mice deficient for IRF9 develop a severe inflammatory ocular disease that likely contributes to the development of cataracts in these animals.
The brain was then examined to determine the contributing underlying causes for the adverse physical signs and premature death of GIFN mice lacking either IRF9 or STAT1. No abnormal features were seen in the brain of 3-month-old WT (supplemental Fig. S1A, available at www.jneurosci.org as supplemental material), GIFN (Fig. 3A,B), STAT1 KO, or IRF9 KO mice. In contrast, reactive astrocytes with abundant eosinophilic cytoplasm, characteristic processes, loss of neurons, and infiltrates of leukocytes including lymphocytes (Fig. 3C,D, white arrowheads) and some neutrophils (Fig. 3D; inset, arrow) surrounding areas of necrosis and calcification (Fig. 3C, arrow) were seen in the medial–ventral forebrain of 2-month-old GIFNxIRF9 KO mice. Reactive astrocytosis was also confirmed by immunohistochemical staining for GFAP (Fig. 4F) while reactive astrocytosis was absent in GIFN mice (Fig. 4A). Vascular endothelial cells exhibited features of activation including hypertrophy and increased nuclear size and shape (Fig. 3D, black arrowhead). Other areas of the CNS, such as the frontal cortex, olfactory bulb, midbrain, and hindbrain, showed either no or few infiltrating leukocytes and an absence of reactive and degenerative changes. Similar pathological changes were also observed in the medial–ventral forebrain of 1-month-old GIFNxSTAT1 KO mice (Fig. 3E,F) and included marked astrocytosis (Fig. 4K). In line with the absence of a phenotype, similarly aged GIFN mice haploinsufficient for either IRF9 or STAT1 showed no apparent changes in the CNS (data not shown).
Figure 3.
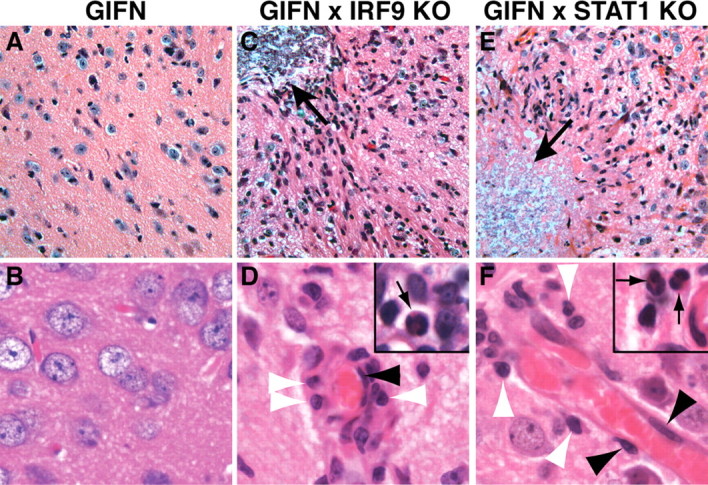
Marked pathological changes in the CNS of GIFNxSTAT1 KO and GIFNxIRF9 KO mice. A, B, The medial–ventral forebrain of 12-week-old GIFN mice showed no obvious pathological changes. C, D, In contrast, the medial–ventral forebrain of similarly aged GIFNxIRF9 KO mice showed areas of calcifying necrosis (C, arrow) with loss of neurons and leukocyte infiltrates that accumulated around blood vessels (D) but also diffusely infiltrated the parenchyma. In addition to lymphocytes (D, white arrowheads), monocytes, and macrophages/microglia, some neutrophils (D, inset, arrow) were present. Endothelial cells in the GIFNxIRF9 KO mice appeared hypertrophic and nuclei were increased and broadened in size (D, black arrowhead). E, F, Areas of calcifying necrosis (E, arrow) with neuronal loss and significant numbers of infiltrating leukocytes and microglia/macrophages were also present in the medial–ventral forebrain of GIFNxSTAT1 KO mice. Lymphocytes (F, white arrowheads) and neutrophils (F, inset, arrow) were seen and endothelial cells of blood vessels were activated (F, black arrowheads). A, C, E, Original magnification 400×; B, D, F, original magnification 750×.
Figure 4.
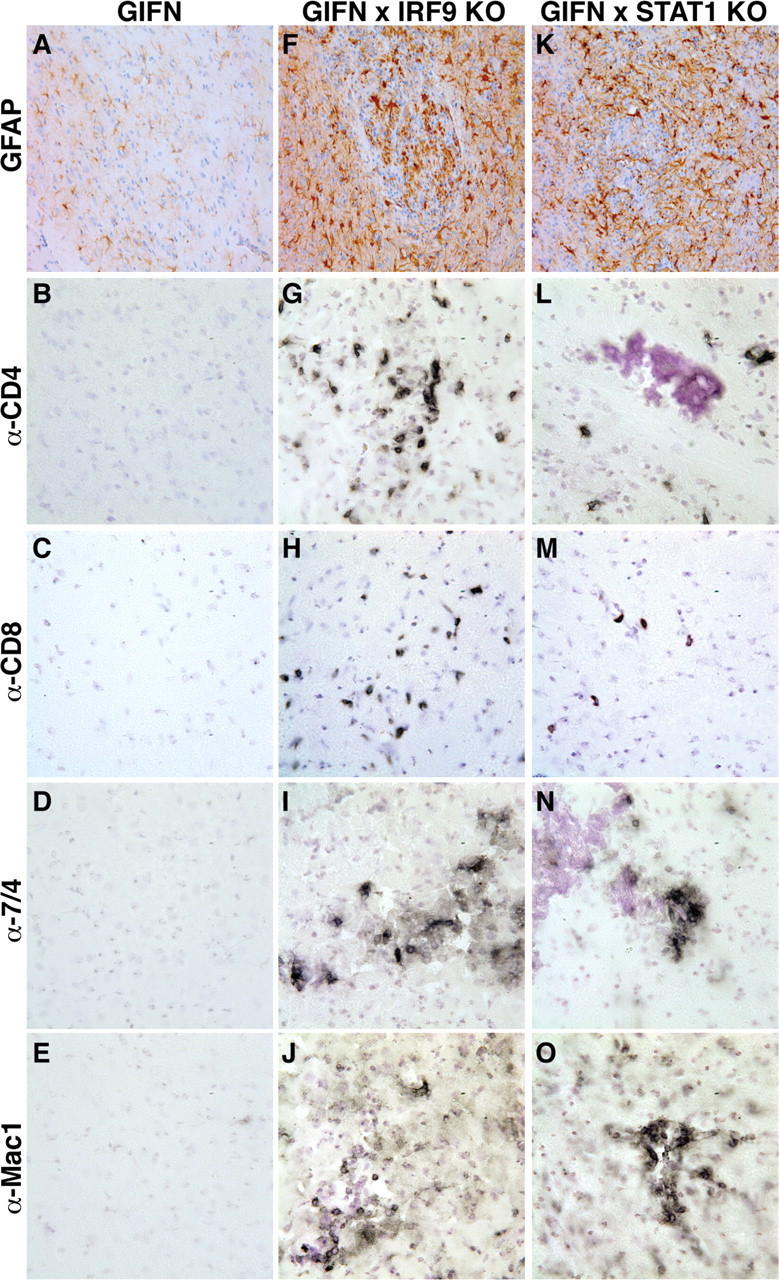
Leukocytes in the CNS of GIFNxSTAT1 KO and GIFNxIRF9 KO mice consisted of T cells, macrophages/monocytes, and neutrophils. A–E, The medial–ventral forebrain of 12-week-old GIFN mice showed no reactive astrocytosis (A) or infiltrating CD4+ (B) or CD8+ T cells (C), neutrophils (D), or macrophages (E). F–I, Pronounced reactive astrocytosis (F) was present in the medial–ventral forebrain of GIFNxIRF9 KO mice, and leukocyte infiltrates consisted of CD4+ (G) and CD8+ (H) T cells and some neutrophils (I). J, Pronounced microglia/macrophage accumulation was evident. K–O, In GIFNxSTAT1 KO mice reactive astrocytosis (K) was similar to GIFNxIRF9 KO mice. Significant numbers of infiltrating CD4+ (L) and CD8+ (M) T cells, neutrophils (N), and microglia/macrophage accumulation (O) were observed in the medial–ventral forebrain of GIFNxSTAT1 KO mice. Original magnification 400×.
Immunohistochemistry was performed to differentiate the phenotype of the infiltrating leukocytes in the medial–ventral forebrain of GIFNxIRF9 KO and GIFNxSTAT1 KO mice. Few, if any, infiltrating leukocytes were observed in WT (supplemental Fig. S1B–E, available at www.jneurosci.org as supplemental material), GIFN (Fig. 4B–E), STAT1 KO (data not shown), or IRF9 KO (data not shown) mice. In contrast, increased numbers of CD4+ (Fig. 4G) and CD8+ (Fig. 4H) T cells, 7/4 positive cells presumed to be neutrophils (Fig. 4I), and microglia/macrophages (Fig. 4J) were present in the GIFNxIRF9 KO mice. A similar pattern of infiltrating CD4+ (Fig. 4L) and CD8+ (Fig. 4M) T cells, 7/4 positive cells (Fig. 4N), and microglia/macrophages (Fig. 4O) was observed in the GIFNxSTAT1 KO mice. These results indicate that, contrary to expectation and similar to the loss of STAT1, the absence of IRF9 in GIFN mice produces an accelerated and more severe neurodegenerative disease, which is associated with an immuno-inflammatory response in the brain.
Altered cerebral expression ofIFN-regulated and cytokine genes inGIFN mice deficient for IRF9
Since IRF9 has a key role in the induction of many IFN-stimulated genes (John et al., 1991; Kimura et al., 1996), we examined the expression of a number of IFN-regulated as well as cytokine genes in the medial–ventral forebrain (Fig. 5A,B). In comparison with WT mice, PKR, 2′,5′-oligoadenylate synthetase (OAS), STAT1, STAT2, IRF9, and IRF7 mRNA transcripts were upregulated significantly in 2-month-old GIFN mice, whereas the CXCL9, T-cell specific GTPase (TGTP), IRF1, class II transactivator (CIITA), SOCS1, SOCS3, and IRF8 mRNA transcripts showed no significant changes. Consistent with the absence of clinical disease, GIFN mice showed only minor changes in TNF, IL-1α, IL-1β, and IFN-γ mRNA levels compared with levels in WT mice. In GIFNxSTAT1 KO mice, there was modest upregulation of the TGTP, OAS, SOCS1, SOCS3, STAT2, IRF7, IRF8, and IRF9 mRNA transcripts compared with WT mice. While CXCL9, TGTP, CIITA, IRF1, SOCS1, and IRF8 mRNA levels in WT mice were similar to GIFN mice, PKR, OAS, STAT1, STAT2, and IRF9 mRNA levels were significantly lower. The TNF, IL-1β, and IFN-γ mRNA levels were increased significantly in GIFNxSTAT1 KO mice compared with GIFN mice. In GIFNxIRF9 KO mice, PKR, OAS, and IRF7 mRNA levels were significantly lower than in GIFN mice, whereas STAT1 and STAT2 mRNA levels were upregulated to similar levels (Fig. 5A,B). In addition, in GIFNxIRF9 KO mice the CXCL9, TGTP, CIITA, SOCS1, SOCS3, IRF1, IRF8, TNF, IL-1α, and IFN-γ mRNA levels were significantly higher. GIFNxIRF9 KO mice also had increased IFN-α1 mRNA levels (supplemental Fig. S1K, available at www.jneurosci.org as supplemental material). This latter observation likely reflects increased GFAP-transgene expression due to reactive astrocytosis. In all, these findings indicate that in GIFNxIRF9 KO mice there is an apparent switch from a type I IFN-response to an IFN-γ-like response concomitant with increased IFN-γ gene expression.
Figure 5.

Deficiency of IRF9 in GIFN mice resulted in IFN-γ-like gene expression and increased pY-STAT1 and pY-STAT2 levels. A, RPAs with RNA from the medial–ventral forebrain showed increased expression of type I IFN-regulated genes and transcription factors in GIFN mice. In contrast to this, GIFNxIRF9 KO mice showed pronounced expression of genes associated with an IFN-γ-like response as well as of proinflammatory cytokines. Increased mRNA levels for proinflammatory cytokines were present in the CNS of GIFNxSTAT1 KO mice. GIFNxIRF9 KOxIFNGR HT mice also showed increased expression of genes characteristic of an IFN-γ-like response. B, Quantification of the autoradiographs was performed by densitometry. Values were normalized to the housekeeping gene L32 and shown as mean ± SEM. C, Immunoblot analysis of protein lysates from the medial–ventral forebrain showed increased levels for pS-STAT1 and STAT1 but not pY-STAT1 in GIFN mice compared with WT mice. In contrast, pS-STAT1, pY-STAT1, and pY-STAT2, as well as total STAT1 and STAT2, were increased in the CNS of GIFNxIRF9 KO and GIFNxIRF9 KOxIFNGR HT mice. D, Bar graphs showing quantification of immunoblot results for phosphorylated and total STAT1 and STAT2. For each genotype, three independent samples were analyzed. Protein levels were normalized to GAPDH levels and means ± SEM are shown. *p < 0.05, **p < 0.01, ***p < 0.001 compared with GIFN mice by one-way ANOVA.
IFN-γ partially protects IRF9-deficient GIFN mice from disease
The switch from type I IFN to IFN-γ-like regulated gene expression, together with the infiltration of the medial–ventral forebrain by T cells observed in GIFNxIRF9 KO mice, suggested that IFN-γ might contribute to the development of disease in these animals. To examine this possibility, we attempted to generate IFN-γ receptor (IFNGR)-deficient GIFNxIRF9 KO mice. Unexpectedly, viable GIFN mice that lacked both IRF9 and the IFNGR were not obtained (Fig. 1). Furthermore, GIFNxIRF9 KO mice haploinsufficient for the IFNGR (GIFNxIRF9 KOxIFNGR HT) developed similar but more severe disease than GIFNxIRF9 KO mice and had a mean survival time of 26 d (Fig. 1). Analysis of the brain from GIFNxIRF9 KOxIFNGR HT mice revealed marked histological changes with leukocyte infiltrates and necrosis (supplemental Fig. S1F–J, available at www.jneurosci.org as supplemental material) and the expression of IFN-γ-regulated genes and proinflammatory cytokine genes (Fig. 5A,B). The overall severity of inflammation in the medial–ventral forebrain of GIFNxIRF9 KOxIFNGR HT mice was greater than in GIFNxIRF9 KO mice. In summary, these findings demonstrate that IFN-γ signaling in the brain confers partial protection against the adverse actions of type I IFNs mediated in the absence of IRF9.
Cerebral STAT1 and STAT2 are activated in GIFNxIRF9 KO mice
Next, we investigated the possible involvement of STAT1 and STAT2 in IRF9-independent signaling in the medial–ventral forebrain of GIFNxIRF9 KO mice. The increased STAT1 and STAT2 mRNA levels observed in the GIFN and GIFNxIRF9 KO mice (Fig. 5A) were matched by increased amounts of STAT1 and STAT2 protein compared with WT mice (Fig. 5C,D). STAT2 protein levels were significantly higher in GIFNxIRF9 KO mice compared with GIFN mice but not detectable in GIFNxSTAT1 KO mice. Of note, in WT, GIFN, and IRF9 KO mice, pS727-STAT1 but not pY701-STAT1 was present and the amount of pS727-STAT1 was significantly higher in GIFN mice compared with WT or IRF9 KO mice (Fig. 5C,D). Compared with GIFN mice, GIFNxIRF9 KO mice showed a smaller increase in the level of pS-STAT1, whereas the level of pY-STAT1 was increased markedly. In addition, a high level of pY-STAT2 was detectable in the GIFNxIRF9 KO but not in the GIFN mice. The levels of total or phosphorylated ERK1/2 or p38MAPK were unaltered in GIFN, GIFNxSTAT1 KO, and GIFNxIRF9 KO mice compared with WT mice (supplemental Fig. S1L, available at www.jneurosci.org as supplemental material). In summary, these results show that in the brain of GIFN mice deficient for IRF9 but not STAT1, there was a significant activation of STAT1 and STAT2 and a shift of phosphorylation of STAT1 away from S727 to Y701.
Absence of IRF9 but not STAT1 in MGCs shifts the response to IFN-α toward an IFN-γ-like response
To determine whether IFN-α can directly activate STAT1 and STAT2 in the absence of IRF9, we analyzed nuclear extracts from primary MGCs. In nuclei from untreated WT or IRF9-deficient MGCs, low levels of STAT1 and STAT2 but not pY-STAT1 or pY-STAT2 were detected (Fig. 6). However, in nuclei from IFN-α-treated MGCs from WT and IRF9-deficient mice, the levels of phosphorylated and total STAT1 and STAT2 were increased after 0.5 h treatment, indicating that IRF9 is not required for nuclear translocation of pY-STAT1 and pY-STAT2. The levels of phosphorylated and nonphosphorylated STAT1 and STAT2 were considerably lower in the nuclei of IRF9-deficient MGCs compared with WT MGCs. Although the levels of pY-STAT1 and pY-STAT2 were increased transiently in WT MGCs, in IRF9-deficient MGCs these phosphoproteins increased over the entire 12 h period following treatment with IFN-α (Fig. 6). No STAT1 or STAT2 was detectable in the nuclei of STAT1-deficient MGCs with or without IFN-α treatment. These findings demonstrate that despite the absence of IRF9, IFN-α is able to induce, albeit at a lower level, the prolonged activation of both STAT1 and STAT2 in MGCs.
Figure 6.

IFN-α induces prolonged nuclear accumulation of pY-STAT1 and pY-STAT2 in IRF9-deficient MGCs. Immunoblot analysis of nuclear protein lysates from untreated and IFN-α-treated MGCs shows a transient accumulation of pY-STAT1 and pY-STAT2 in WT cells and prolonged accumulation in IRF9-deficient cells.
Next, we determined whether the activation of STAT1 and STAT2 mediated by IFN-α in the absence of IRF9 was associated with changes in the expression of various IFN-regulated genes in the MGCs. In untreated WT, STAT1 KO, and IRF9 KO cells, PKR, TGTP, IRF1, SOCS1, and SOCS3 mRNA levels were comparable whereas STAT1, STAT2, and IRF9 mRNA transcripts were reduced in untreated STAT1-deficient and IRF9-deficient MGCs compared with MGCs from WT mice (Fig. 7A,B). Following treatment of WT cells with IFN-α, the levels of PKR, TGTP, IRF1, STAT1, STAT2, IRF9, and SOCS1 mRNAs were significantly increased at 4 h and, with the exception of SOCS1 mRNA, remained elevated at 12 h. In IFN-α-treated STAT1-deficient cells, none of the mRNA transcripts examined were altered significantly. In IFN-α-treated IRF9-deficient MGCs, STAT1 and IRF1 mRNA levels were significantly upregulated at 4 h, whereas TGTP, IRF1, STAT1, STAT2, and SOCS1 RNA levels were significantly upregulated at 12 h. However, in contrast to WT MGCs, the expression of PKR mRNA was not altered by IFN-α treatment of MGCs that lacked IRF9. These findings show that in the absence of IRF9, IFN-α induces a subset of IFN-regulated genes that includes genes that are associated with an IFN-γ-like response.
Figure 7.

IFN-α induces IFN-γ-like gene expression in IRF9-deficient MGCs. A, RPA analysis shows a delayed expression of genes that are characteristic of an IFN-γ-like response in IRF9-deficient cells compared with WT cells. B, Quantification of the autoradiographs was performed by densitometry. Values were normalized to the housekeeping gene L32 shown as mean ± SEM. *p < 0.05, **p < 0.01, ***p < 0.001 compared with respective untreated controls by one-way ANOVA.
Discussion
Type I IFNs are critical mediators of host immune responses in the CNS and participants in neurological disease pathogenesis (for review, see Paul et al., 2007). Chronically elevated levels of IFN-α in the CNS in CVE (Dussaix et al., 1985), AGS (Lebon et al., 1988b; Rice et al., 2007), and Cree encephalitis (Black et al., 1988) are linked to neurodegeneration, inflammation, and calcification. However, the underlying pathophysiology of these disorders is not fully understood. IRF9-deficient cells are crippled in their response to type I IFNs with defective IFN-regulated ISRE-driven gene expression and increased susceptibility to viral infection (Kimura et al., 1996). Nevertheless, absence of IRF9 does not totally circumvent the ability of cells to respond to type I IFNs since U2A human fibrosarcoma cells lacking IRF9 show activation of STAT1 and STAT2 and induction of the IRF1 gene in response to IFN-α (Shuai et al., 1993). However, the significance of cellular responses to type I IFNs in the absence of IRF9 are not known and are difficult to establish in cell culture models. Here, we showed that the mild neurological phenotype caused by chronic production of low levels of IFN-α in the CNS of the GIFN mice was grossly aggravated when IRF9 was lacking, resulting in premature death. Thus, far from crippling the response to IFN-α, absence of IRF9 exacerbated the adverse CNS actions of this cytokine, causing neuropathological changes similar to those found in patients with AGS or CVE (Dussaix et al., 1985; Lebon et al., 1988b; Shaw and Cohen, 1993; Kenneson and Cannon, 2007; Rice et al., 2007).
The neurodegenerative disease in GIFNxIRF9 KO mice was associated with infiltration of T cells, microglia/macrophages, and neutrophils, calcification, and increased proinflammatory cytokine gene expression. However, cerebral expression of several ISRE-dependent genes was significantly reduced in GIFNxIRF9 KO mice compared with GIFN mice. This finding is consistent with studies in IRF9-deficient fibroblasts (Kimura et al., 1996) and U2A fibrosarcoma cells (John et al., 1991). A similar reduction in cerebral ISGF3-dependent gene expression occurred in GIFNxSTAT1-deficient mice (this study and Wang et al., 2002). Thus, disruption of canonical type I IFN signaling markedly increases the pathobiological actions of IFN-α, indicating that ISGF3-dependent signaling has an important protective function in the CNS. The molecular mechanisms responsible for neuronal loss in GIFN mice deficient for STAT1 or IRF9 are unknown. Notably, STAT1 is required for neurons to respond to IFN-α (Wang and Campbell, 2005), suggesting that indirect mechanisms are involved in GIFNxSTAT1 KO mice. It remains to be clarified whether IRF9-deficient neurons can respond to IFN-α and whether the observed neuronal loss in GIFN mice deficient for STAT1 or IRF9 occurs through comparable mechanisms.
The delayed onset of clinical symptoms and the longer survival of GIFNxIRF9 KO mice highlight differences in disease progression resulting from the loss of IRF9 versus the loss of STAT1. GIFNxIRF9 KO mice showed significant upregulation of several mRNAs, including CXCL9, TGTP, CIITA, IRF1 and SOCS1, typical of IFN-γ-regulated responses (Boehm et al., 1997; Decker et al., 1997; Shtrichman and Samuel, 2001) that were absent in GIFN and GIFNxSTAT1 KO mice. These genes are known to contain gamma interferon activation site (GAS) or GAS-like elements in the promoter that bind STAT1 homodimers or STAT1:STAT2 heterodimers (Decker et al., 1997). Accordingly, in contrast to GIFN mice that had only increased pS-STAT1 levels, there were high levels of pY-STAT1 as well as pY-STAT2 in the CNS of GIFNxIRF9 KO mice. This suggests that STAT1 homodimers and/or STAT1:STAT2 heterodimers may participate in regulating gene transcription in the brain. Thus, IRF9-deficiency might skew IFN-α signaling toward a protective IFN-γ-like response. The absence of this response in the GIFNxSTAT1 KO mice that showed similar neuropathological features as the GIFNxIRF9 KO mice suggests these GAS-regulated genes do not contribute to the core inflammatory encephalopathy in GIFN mice with disrupted canonical type I IFN signaling and may even be protective. On the other hand, the ocular pathology in GIFNxIRF9 KO mice recapitulated findings in mice with transgene-driven ocular IFN-γ production (Egwuagu et al., 1994; Geiger et al., 1994), indicating there may be an opposing effect of the IFN-γ-like response in the eyes versus the brain.
Although the cellular source of IFN-γ gene expression in the CNS of the GIFNxIRF9 KO mice was not identified, IFN-α can induce IFN-γ gene expression in murine T cells lacking either STAT1 or STAT2 via the activation of STAT4 (Nguyen et al., 2002; Wang et al., 2003). The presence of IFN-γ mRNA in the brain of the GIFNxIRF9 KO mice is therefore likely to have been induced in infiltrating T cells that were responding to IFN-α. The concurrent expression of GAS-regulated genes and IFN-γ mRNA in the brain of GIFNxIRF9 KO mice suggested that IFN-γ and not IFN-α may account for the activation of STAT1 and the observed IFN-γ-like gene expression. However, partial or complete deficiency for the IFNGR in GIFNxIRF9 KO mice resulted in a more severe disease with earlier morbidity, indicating that IFN-γ signaling was protective. A protective role for IFN-γ is observed in other immunoinflammatory diseases of the CNS, such as experimental autoimmune encephalomyelitis (EAE) (Ferber et al., 1996; Willenborg et al., 1996; Tran et al., 2000) and Borna disease virus encephalitis (Richter et al., 2009). The precise mechanism(s) for this protective function of IFN-γ remains to be resolved. In EAE there is evidence for suppression of neutrophil recruitment and accumulation via IFN-γ-mediated inhibition of neutrophil-attracting chemokines such as CXCL2 (Tran et al., 2000). However, a survey by us (S. L. Lim, M. J. Hofer, and I. L. Campbell, unpublished observation) of the expression of a number of chemokine genes, including CCL2, CXCL1, CXCL2, and CXCL10, in GIFNxIRF9 KO and GIFNxIRF9 KOxIFNGR HT mice failed to reveal significant differences, suggesting that there is not a central role for these chemokines in IFN-γ-mediated protection in our model.
Our finding that IFN-α is capable of directly activating STAT1 and STAT2 in IRF9-deficient MGCs indicates that IFN-α-signaling in the absence of IRF9 is able to induce an IFN-γ-like response. Consistent with this, the level and kinetics of expression of several GAS or GAS-like driven genes overlapped with the respective activation levels and kinetics for STAT1 and STAT2 in WT and IRF9-null MGCs. The unresponsiveness of STAT1-null MGCs to IFN-α confirms that IFN-α-stimulated GAS or GAS-like driven gene expression in MGCs lacking IRF9 is dependent on STAT1. In IRF9-deficient U2A fibrosarcoma cells, IFN-α induces STAT1 homodimers and STAT1:STAT2 heterodimers that bind to GAS-like enhancer elements in the promoter of the IRF1 gene (Haque and Williams, 1994; Li et al., 1996). Stimulation of IRF1 gene expression in these cells is abolished in the absence of STAT2, indicating that the STAT1:STAT2 heterodimer is the pivotal signaling complex. Whether STAT2 is similarly involved in the induction of IRF1 and other genes in IRF9-deficient MGCs in response to IFN-α as observed in the present study remains to be determined.
Activation of STAT1 and STAT2 in IRF9-deficient MGCs increased progressively compared with the transient activation observed in WT MGCs. A similar prolonged activation of STAT1 with increased IRF1 and induction of MHC class II occurs in STAT2-null macrophages and is due to the loss of induction of the key negative feedback regulator SOCS1 (Zhao et al., 2007). However, we found that IFN-α is capable of inducing the expression of SOCS1 mRNA in the absence of IRF9, although this effect was delayed. Therefore, unlike in STAT2 deficiency, the altered dynamics of IFN-α-mediated STAT1 activation in the absence of IRF9 is unlikely to be due to an inability to induce SOCS1.
Our findings link activation of STAT1 and STAT2 to the functional response of IRF9-deficient MGCs. The signal transduction pathway and gene expression changes in IRF9-null, IFN-α-treated MGCs overlapped remarkably with changes seen in the brain of GIFNxIRF9 KO mice, suggesting that the direct response of the glial cells to IFN-α may play a crucial role in initiating and driving the disease process. Recent findings by us indicate that the response to IFN-α in the absence of IRF9 is more extensive than expected with 100 genes upregulated in MGCs (W. Li, M. J. Hofer, and I. L. Campbell, unpublished observation). Further analysis of the promoter structure of these IFN-α-stimulated IRF9-independent genes may shed light on whether other transcription factors (e.g., IRF1) are involved in programming this response.
In summary, type I IFN-mediated, ISGF3-independent signaling due to the absence of IRF9 activates a potent immunoinflammatory response that is associated with a lethal neurological disease. A key feature of the cellular response to IFN-α in the absence of IRF9 is a shift from ISRE to a GAS or GAS-like gene expression pattern in the brain that is replicated in cultured glial cells. Yet this IFN-γ-like response clearly does not give rise to the destructive encephalopathy mediated by IFN-α in the absence of IRF9. Our findings highlight the complex molecular mechanisms that are involved in type I IFN signaling in the CNS. They demonstrate that a balanced interplay between different type I IFN signaling factors is critical for the appropriate physiological functions of IFN-α. Moreover, the results here give deeper insight into our understanding of the pathogenesis of proposed neurological “interferonopathies” such as AGS and suggest that some key neuropathological changes in these disorders may arise from noncanonical signaling mediated by IFN-α. Finally, modifications to type I IFN signaling pathways [as can occur following infection with certain viruses (Leonard and Sen, 1996; Miller et al., 1999; Palosaari et al., 2003; Zurney et al., 2009] may cause dramatic changes in the biological actions of this cytokine, including enhanced development of disease.
Footnotes
This work was supported by a National Health and Medical Research Council Grant (NH&MRC 512407) to I.L.C. M.J.H. was a postdoctoral fellow from the Deutsche Forschungsgemeinschaft (DFG HO3298/1-1). W.L. was the recipient of an Australian Postgraduate Scholarship. We thank Laura Parker for technical help. We acknowledge the help of Dr. Ilse Strempel for assistance with the histological analysis of the eye.
References
- Aicardi J, Goutières F. A progressive familial encephalopathy in infancy with calcifications of the basal ganglia and chronic cerebrospinal fluid lymphocytosis. Ann Neurol. 1984;15:49–54. doi: 10.1002/ana.410150109. [DOI] [PubMed] [Google Scholar]
- Akwa Y, Hassett DE, Eloranta ML, Sandberg K, Masliah E, Powell H, Whitton JL, Bloom FE, Campbell IL. Transgenic expression of IFN-α in the central nervous system of mice protects against lethal neurotropic viral infection but induces inflammation and neurodegeneration. J Immunol. 1998;161:5016–5026. [PubMed] [Google Scholar]
- Asensio VC, Campbell IL. Chemokine gene expression in the brains of mice with lymphocytic choriomeningitis. J Virol. 1997;71:7832–7840. doi: 10.1128/jvi.71.10.7832-7840.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asensio VC, Kincaid C, Campbell IL. Chemokines and the inflammatory response to viral infection in the central nervous system with a focus on lymphocytic choriomeningitis virus. J Neurovirol. 1999;5:65–75. doi: 10.3109/13550289909029747. [DOI] [PubMed] [Google Scholar]
- Asensio VC, Maier J, Milner R, Boztug K, Kincaid C, Moulard M, Phillipson C, Lindsley K, Krucker T, Fox HS, Campbell IL. Interferon-independent, human immunodeficiency virus type 1 gp120-mediated induction of CXCL10/IP-10 gene expression by astrocytes in vivo and in vitro. J Virol. 2001;75:7067–7077. doi: 10.1128/JVI.75.15.7067-7077.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Black DN, Watters GV, Andermann E, Dumont C, Kabay ME, Kaplan P, Meagher-Villemure K, Michaud J, O'Gorman G, Reece E, Tsoukas C, Wainberget MA. Encephalitis among Cree children in northern Quebec. Ann Neurol. 1988;24:483–489. doi: 10.1002/ana.410240402. [DOI] [PubMed] [Google Scholar]
- Boehm U, Klamp T, Groot M, Howard JC. Cellular responses to interferon-gamma. Annu Rev Immunol. 1997;15:749–795. doi: 10.1146/annurev.immunol.15.1.749. [DOI] [PubMed] [Google Scholar]
- Boztug K, Carson MJ, Pham-Mitchell N, Asensio VC, DeMartino J, Campbell IL. Leukocyte infiltration, but not neurodegeneration, in the CNS of transgenic mice with astrocyte production of the CXC chemokine ligand 10. J Immunol. 2002;169:1505–1515. doi: 10.4049/jimmunol.169.3.1505. [DOI] [PubMed] [Google Scholar]
- Bressler JP, Cole R, de Vellis J. Cell culture systems to study glial transformation. Dev Toxicol Environ Sci. 1980;8:187–192. [PubMed] [Google Scholar]
- Campbell IL, Krucker T, Steffensen S, Akwa Y, Powell HC, Lane T, Carr DJ, Gold LH, Henriksen SJ, Siggins GR. Structural and functional neuropathology in transgenic mice with CNS expression of IFN-α. Brain Res. 1999;835:46–61. doi: 10.1016/s0006-8993(99)01328-1. [DOI] [PubMed] [Google Scholar]
- Crow YJ, Black DN, Ali M, Bond J, Jackson AP, Lefson M, Michaud J, Roberts E, Stephenson JB, Woods CG, Lebon P. Cree encephalitis is allelic with Aicardi-Goutieres syndrome: implications for the pathogenesis of disorders of interferon alpha metabolism. J Med Genet. 2003;40:183–187. doi: 10.1136/jmg.40.3.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Decker T, Kovarik P, Meinke A. GAS elements: a few nucleotides with a major impact on cytokine-induced gene expression. J Interferon Cytokine Res. 1997;17:121–134. doi: 10.1089/jir.1997.17.121. [DOI] [PubMed] [Google Scholar]
- Durbin JE, Hackenmiller R, Simon MC, Levy DE. Targeted disruption of the mouse Stat1 gene results in compromised innate immunity to viral disease. Cell. 1996;84:443–450. doi: 10.1016/s0092-8674(00)81289-1. [DOI] [PubMed] [Google Scholar]
- Dussaix E, Lebon P, Ponsot G, Huault G, Tardieu M. Intrathecal synthesis of different alpha-interferons in patients with various neurological diseases. Acta Neurol Scand. 1985;71:504–509. doi: 10.1111/j.1600-0404.1985.tb03235.x. [DOI] [PubMed] [Google Scholar]
- Egwuagu CE, Sztein J, Chan CC, Reid W, Mahdi R, Nussenblatt RB, Chepelinsky AB. Ectopic expression of gamma interferon in the eyes of transgenic mice induces ocular pathology and MHC class II gene expression. Invest Ophthalmol Vis Sci. 1994;35:332–341. [PubMed] [Google Scholar]
- Ferber IA, Brocke S, Taylor-Edwards C, Ridgway W, Dinisco C, Steinman L, Dalton D, Fathman CG. Mice with a disrupted IFN-gamma gene are susceptible to the induction of experimental autoimmune encephalomyelitis (EAE) J Immunol. 1996;156:5–7. [PubMed] [Google Scholar]
- Geiger K, Howes E, Gallina M, Huang XJ, Travis GH, Sarvetnick N. Transgenic mice expressing IFN-gamma in the retina develop inflammation of the eye and photoreceptor loss. Invest Ophthalmol Vis Sci. 1994;35:2667–2681. [PubMed] [Google Scholar]
- Haque SJ, Williams BR. Identification and characterization of an interferon (IFN)-stimulated response element-IFN-stimulated gene factor 3-independent signaling pathway for IFN-alpha. J Biol Chem. 1994;269:19523–19529. [PubMed] [Google Scholar]
- Hobbs MV, Weigle WO, Noonan DJ, Torbett BE, McEvilly RJ, Koch RJ, Cardenas GJ, Ernst DN. Patterns of cytokine gene expression by CD4+ T cells from young and old mice. J Immunol. 1993;150:3602–3614. [PubMed] [Google Scholar]
- Hofer MJ, Carter SL, Muller M, Campbell IL. Unaltered neurological disease and mortality in CXCR3-deficient mice infected intracranially with lymphocytic choriomeningitis virus-Armstrong. Viral Immunol. 2008;21:425–433. doi: 10.1089/vim.2008.0057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang S, Hendriks W, Althage A, Hemmi S, Bluethmann H, Kamijo R, Vilcek J, Zinkernagel RM, Aguet M. Immune response in mice that lack the interferon-gamma receptor. Science. 1993;259:1742–1745. doi: 10.1126/science.8456301. [DOI] [PubMed] [Google Scholar]
- John J, McKendry R, Pellegrini S, Flavell D, Kerr IM, Stark GR. Isolation and characterization of a new mutant human cell line unresponsive to alpha and beta interferons. Mol Cell Biol. 1991;11:4189–4195. doi: 10.1128/mcb.11.8.4189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karst SM, Wobus CE, Lay M, Davidson J, Virgin HW., IV STAT1-dependent innate immunity to a Norwalk-like virus. Science. 2003;299:1575–1578. doi: 10.1126/science.1077905. [DOI] [PubMed] [Google Scholar]
- Kenneson A, Cannon MJ. Review and meta-analysis of the epidemiology of congenital cytomegalovirus (CMV) infection. Rev Med Virol. 2007;17:253–276. doi: 10.1002/rmv.535. [DOI] [PubMed] [Google Scholar]
- Kimura T, Kadokawa Y, Harada H, Matsumoto M, Sato M, Kashiwazaki Y, Tarutani M, Tan RS, Takasugi T, Matsuyama T, Mak TW, Noguchi S, Taniguchi T. Essential and non-redundant roles of p48 (ISGF3 gamma) and IRF-1 in both type I and type II interferon responses, as revealed by gene targeting studies. Genes Cells. 1996;1:115–124. doi: 10.1046/j.1365-2443.1996.08008.x. [DOI] [PubMed] [Google Scholar]
- Lebon P, Boutin B, Dulac O, Ponsot G, Arthuis M. Interferon gamma in acute and subacute encephalitis. Br Med J (Clin Res Ed) 1988a;296:9–11. doi: 10.1136/bmj.296.6614.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebon P, Badoual J, Ponsot G, Goutieres F, Hemeury-Cukier F, Aicardi J. Intrathecal synthesis of interferon-alpha in infants with progressive familial encephalopathy. J Neurol Sci. 1988b;84:201–208. doi: 10.1016/0022-510x(88)90125-6. [DOI] [PubMed] [Google Scholar]
- Leonard GT, Sen GC. Effects of adenovirus E1A protein on interferon-signaling. Virology. 1996;224:25–33. doi: 10.1006/viro.1996.0503. [DOI] [PubMed] [Google Scholar]
- Li X, Leung S, Qureshi S, Darnell JE, Jr, Stark GR. Formation of STAT1-STAT2 heterodimers and their role in the activation of IRF-1 gene transcription by interferon-alpha. J Biol Chem. 1996;271:5790–5794. doi: 10.1074/jbc.271.10.5790. [DOI] [PubMed] [Google Scholar]
- Maier J, Kincaid C, Pagenstecher A, Campbell IL. Regulation of signal transducer and activator of transcription and suppressor of cytokine-signaling gene expression in the brain of mice with astrocyte-targeted production of interleukin-12 or experimental autoimmune encephalomyelitis. Am J Pathol. 2002;160:271–288. doi: 10.1016/S0002-9440(10)64371-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meraz MA, White JM, Sheehan KC, Bach EA, Rodig SJ, Dighe AS, Kaplan DH, Riley JK, Greenlund AC, Campbell D, Carver-Moore K, DuBois RN, Clark R, Aguet M, Schreiber RD. Targeted disruption of the Stat1 gene in mice reveals unexpected physiologic specificity in the JAK-STAT signaling pathway. Cell. 1996;84:431–442. doi: 10.1016/s0092-8674(00)81288-x. [DOI] [PubMed] [Google Scholar]
- Miller DM, Zhang Y, Rahill BM, Waldman WJ, Sedmak DD. Human cytomegalovirus inhibits IFN-alpha-stimulated antiviral and immunoregulatory responses by blocking multiple levels of IFN-alpha signal transduction. J Immunol. 1999;162:6107–6113. [PubMed] [Google Scholar]
- Milner R, ffrench-Constant C. A developmental analysis of oligodendroglial integrins in primary cells: changes in alpha v-associated beta subunits during differentiation. Development. 1994;120:3497–3506. doi: 10.1242/dev.120.12.3497. [DOI] [PubMed] [Google Scholar]
- Nguyen KB, Watford WT, Salomon R, Hofmann SR, Pien GC, Morinobu A, Gadina M, O'Shea JJ, Biron CA. Critical role for STAT4 activation by type 1 interferons in the interferon-gamma response to viral infection. Science. 2002;297:2063–2066. doi: 10.1126/science.1074900. [DOI] [PubMed] [Google Scholar]
- Palosaari H, Parisien JP, Rodriguez JJ, Ulane CM, Horvath CM. STAT protein interference and suppression of cytokine signal transduction by measles virus V protein. J Virol. 2003;77:7635–7644. doi: 10.1128/JVI.77.13.7635-7644.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park C, Li S, Cha E, Schindler C. Immune response in Stat2 knockout mice. Immunity. 2000;13:795–804. doi: 10.1016/s1074-7613(00)00077-7. [DOI] [PubMed] [Google Scholar]
- Paul S, Ricour C, Sommereyns C, Sorgeloos F, Michiels T. Type I interferon response in the central nervous system. Biochimie. 2007;89:770–778. doi: 10.1016/j.biochi.2007.02.009. [DOI] [PubMed] [Google Scholar]
- Rice G, Patrick T, Parmar R, Taylor CF, Aeby A, Aicardi J, Artuch R, Montalto SA, Bacino CA, Barroso B, Baxter P, Benko WS, Bergmann C, Bertini E, Biancheri R, Blair EM, Blau N, Bonthron DT, Briggs T, Brueton LA, et al. Clinical and molecular phenotype of Aicardi-Goutieres syndrome. Am J Hum Genet. 2007;81:713–725. doi: 10.1086/521373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richter K, Hausmann J, Staeheli P. Interferon-gamma prevents death of bystander neurons during CD8 T cell responses in the brain. Am J Pathol. 2009;174:1799–1807. doi: 10.2353/ajpath.2009.080897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindler C, Brutsaert S. Interferons as a paradigm for cytokine signal transduction. Cell Mol Life Sci. 1999;55:1509–1522. doi: 10.1007/s000180050391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sen GC, Ransohoff RM. Interferon-induced antiviral actions and their regulation. Adv Virus Res. 1993;42:57–102. doi: 10.1016/s0065-3527(08)60083-4. [DOI] [PubMed] [Google Scholar]
- Shaw DW, Cohen WA. Viral infections of the CNS in children: imaging features. AJR Am J Roentgenol. 1993;160:125–133. doi: 10.2214/ajr.160.1.8416608. [DOI] [PubMed] [Google Scholar]
- Shtrichman R, Samuel CE. The role of gamma interferon in antimicrobial immunity. Curr Opin Microbiol. 2001;4:251–259. doi: 10.1016/s1369-5274(00)00199-5. [DOI] [PubMed] [Google Scholar]
- Shuai K, Stark GR, Kerr IM, Darnell JE., Jr A single phosphotyrosine residue of Stat91 required for gene activation by interferon-gamma. Science. 1993;261:1744–1746. doi: 10.1126/science.7690989. [DOI] [PubMed] [Google Scholar]
- Stark GR, Kerr IM, Williams BR, Silverman RH, Schreiber RD. How cells respond to interferons. Annu Rev Biochem. 1998;67:227–264. doi: 10.1146/annurev.biochem.67.1.227. [DOI] [PubMed] [Google Scholar]
- Tran EH, Prince EN, Owens T. IFN-gamma shapes immune invasion of the central nervous system via regulation of chemokines. J Immunol. 2000;164:2759–2768. doi: 10.4049/jimmunol.164.5.2759. [DOI] [PubMed] [Google Scholar]
- van Boxel-Dezaire AH, Rani MR, Stark GR. Complex modulation of cell type-specific signaling in response to type I interferons. Immunity. 2006;25:361–372. doi: 10.1016/j.immuni.2006.08.014. [DOI] [PubMed] [Google Scholar]
- Wang J, Campbell IL. Innate STAT1-dependent genomic response of neurons to the antiviral cytokine alpha interferon. J Virol. 2005;79:8295–8302. doi: 10.1128/JVI.79.13.8295-8302.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Schreiber RD, Campbell IL. STAT1 deficiency unexpectedly and markedly exacerbates the pathophysiological actions of IFN-alpha in the central nervous system. Proc Natl Acad Sci U S A. 2002;99:16209–16214. doi: 10.1073/pnas.252454799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Pham-Mitchell N, Schindler C, Campbell IL. Dysregulated Sonic hedgehog signaling and medulloblastoma consequent to IFN-alpha-stimulated STAT2-independent production of IFN-γ in the brain. J Clin Invest. 2003;112:535–543. doi: 10.1172/JCI18637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willenborg DO, Fordham S, Bernard CC, Cowden WB, Ramshaw IA. IFN-gamma plays a critical down-regulatory role in the induction and effector phase of myelin oligodendrocyte glycoprotein-induced autoimmune encephalomyelitis. J Immunol. 1996;157:3223–3227. [PubMed] [Google Scholar]
- Zhao W, Cha EN, Lee C, Park CY, Schindler C. Stat2-dependent regulation of MHC class II expression. J Immunol. 2007;179:463–471. doi: 10.4049/jimmunol.179.1.463. [DOI] [PubMed] [Google Scholar]
- Zurney J, Kobayashi T, Holm GH, Dermody TS, Sherry B. Reovirus mu2 protein inhibits interferon signaling through a novel mechanism involving nuclear accumulation of interferon regulatory factor 9. J Virol. 2009;83:2178–2187. doi: 10.1128/JVI.01787-08. [DOI] [PMC free article] [PubMed] [Google Scholar]