

Abstract
The molecular mechanisms underlying the behavioral effects of glucocorticoids are poorly understood. We report here that hippocampal neuronal nitric oxide synthase (nNOS) is a crucial mediator. Chronic mild stress and glucocorticoids exposures caused hippocampal nNOS overexpression via activating mineralocorticoid receptor. In turn, hippocampal nNOS-derived nitric oxide (NO) significantly downregulated local glucocorticoid receptor expression through both soluble guanylate cyclase (sGC)/cGMP and peroxynitrite (ONOO−)/extracellular signal-regulated kinase signal pathways, and therefore elevated hypothalamic corticotrophin-releasing factor, a peptide that governs the hypothalamic-pituitary-adrenal axis. More importantly, nNOS deletion or intrahippocampal nNOS inhibition and NO-cGMP signaling blockade (using NO scavenger or sGC inhibitor) prevented the corticosterone-induced behavioral modifications, suggesting that hippocampal nNOS is necessary for the role of glucocorticoids in mediating depressive behaviors. In addition, directly delivering ONOO− donor into hippocampus caused depressive-like behaviors. Our findings reveal a role of hippocampal nNOS in regulating the behavioral effects of glucocorticoids.
Introduction
Major depressive disorder (MDD) is a severe, life-threatening, and highly prevalent psychiatric disorder (Bartolomucci and Leopardi, 2009). Stressful life events have a substantial causal association with depression and stress paradigms have long been used to model the disease (Martinowich et al., 2007; Bartolomucci and Leopardi, 2009). Glucocorticoids secreted by the adrenal gland, via activating glucocorticoids receptors, well characterized ligand-activated transcription factors (Krishnan and Nestler, 2008), play an important role in mediating the behavioral consequences of stress (Revest et al., 2005). The release of glucocorticoids is controlled by the hypothalamic–pituitary–adrenal (HPA) axis. Feedback regulation of the HPA axis by glucocorticoids is mediated through activation of two distinct intracellular receptor subtypes referred to as mineralocorticoid receptor (MR), a high affinity receptor, and glucocorticoid receptor (GR), a lower affinity receptor for endogenous corticosteroids. The GR is believed to be more important in the regulation of the response to stress when endogenous levels of glucocorticoids are high. This has led to a prevailing view that impaired hippocampal GR function is correlated with the hyperactivity of the HPA axis, and could be an etiologic factor in depression (Boyle et al., 2005). Although copious information is available on the implication of hippocampal GR in the stress-related behavioral effects of glucocorticoids (de Kloet et al., 2005; Revest et al., 2005; Berton and Nestler, 2006; Krishnan and Nestler, 2008), the underlying molecular mechanisms are unknown.
Neuronal nitric oxide synthase (nNOS)-derived nitric oxide (NO), a free radical with many physiological and pathological functions, including neurogenesis, hippocampal long-term potentiation, synaptogenesis, neurodegeneration, neuronal survival, and differentiation (Cho et al., 2009; Zhou and Zhu, 2009), may be an important mediator between glucocorticoids and hippocampal GR. nNOS is enriched throughout the limbic system (Bredt et al., 1991), an area important in emotional behaviors. A number of studies have shown that nNOS has significant implications for the psychiatric disorders characterized by depression (Zhou and Zhu, 2009), anxiety (Zhang et al., 2010), schizophrenia (Shinkai et al., 2002; Reif et al., 2006; O'Donovan et al., 2008), and aggressiveness (Nelson et al., 1995; Chiavegatto et al., 2001). Here, we report that nNOS-mediated GR downregulation in the adult hippocampus underlies the stress-related behavioral effects of glucocorticoids.
Materials and Methods
Animals.
Young adult (6- to 7-week-old) male homozygous nNOS-deficient mice (B6;129S4-Nos1tm1Plh, KO, stock number: 002633) and their wild-type controls of similar genetic background (B6129SF2, WT) (both from Jackson Laboratories; maintained at Model Animal Research Center of Nanjing University, Nanjing, China), young adult (6- to 7-week-old) ICR mice, and newborn (postnatal days 0–1) ICR mice, newborn nNOS KO and WT mice (from Model Animal Research Center of Nanjing University, Nanjing, China), and Sprague Dawley rat pups (10-d-old) (Shanghai Silaike Experimental Animal) were used in this study. Animals were housed in an air-conditioned room (20 ± 2°C), with food and water ad libitum, except when specified otherwise. All procedures involving the use of animals were approved by the Institutional Animal Care and Use Committee of Nanjing Medical University.
Drugs.
Corticosterone (40 mg/kg/d) and metyrapone (100 mg/kg/d) were subcutaneously injected, and 7-nitroindazole (7-NI; 30 mg/kg/d) was intraperitoneally injected. Stereotaxic surgery was used to deliver 10 μm corticosterone, 1 mm SIN-1, 10 μm 7-NI, 20 μm CPTIO, 10 μm ODQ, or 10 μm U0126 into the hippocampus (coordinates: 2.3 mm posterior to bregma, 1.3 mm lateral to the midline, and 2.0 mm below dura) or to deliver 10 μm corticosterone into the paraventricular nucleus (PVN) of the hypothalamus (coordinates: 0.85 mm caudal to bregma, 0.15 mm lateral to the midline, and 5.5 mm below the surface of the skull). The designated drug solution in 2 μl of volume was microinjected (0.2 μl/min). All these drugs were purchased from Sigma-Aldrich. For stereotaxic surgery, adult mice were anesthetized with 0.07 ml of a mixture of ketamine (90.9 mg/ml) and xylazine (9.1 mg/ml).
Chronic mild stress and behavioral measures.
The procedure of chronic mild stress (CMS) was designed as described previously (Zhou et al., 2007). Stress-induced modifications in mice were assessed using body weight gain, immobility time in the tail suspension test (TST) and forced swimming test (FST), open-field test (OFT), and sucrose preference test (SPT). The TST, FST, and OFT were measured as described previously (Zhou et al., 2007). The duration of immobility in the TST and FST was recorded using the Hamilton kinder TS100 on PC computer and MotorMonitor System SF16R, respectively. For the OFT, the test arena was constructed of a plastic plate (56.13 × 56.13 cm); lines drawn on the plate divided it into 256 squares. It was surrounded by a 35.18-cm-high plastic wall. Each mouse was placed on a corner square of the arena, facing the corner, and allowed to freely explore the open field for 5 min per trial. After each trial, the plate was cleaned with 70% EtOH. No stressor was applied to the animals for at least 12 h before the test. Mobility was scored when an animal crossed a sector border with both its hindlimbs. Parameters assessed were the number of square crossings (horizontal) and the times of standing (vertical) during the 5 min test.
The SPT was performed as described previously (Grippo et al., 2005). In brief, an SPT consisted of first removing the food and water from each mouse's cage for a period of 20 h. Water and 1% sucrose were then placed in the cages in preweighed plastic bottles and animals were allowed to consume the fluids for a period of 10 h. The bottles were then removed and weighed. Two baseline fluid intake tests were performed, separated by at least 5 d, and the results were averaged. Fluid intake was calculated on an absolute basis (sucrose and water intake separately) and as a preference score (sucrose preference, relative to total fluid intake).
Culture of hippocampal neurons.
Primary hippocampal neurons were isolated and cultured as reported (Zhang et al., 2010), with minor modifications. In brief, the hippocampus of embryonic day 18 mouse was freshly isolated and enzymatically dissociated in calcium- and magnesium-free-HBSS containing 0.125% trypsin at 37°C for 10 min. Then the digestion was terminated with DMEM/F12 (1:1) containing 10% FBS and tissues were triturated with a fire-polished glass pipet. Dissociated cells were centrifuged, resuspended in Neurobasal medium (Invitrogen) containing 2% B27 supplement, 0.5 mm l-glutamine, 5 IU penicillin, and 5 μg/ml streptomycin, and plated on 10 μg/ml polyornithine-coated dishes (diameter, 3.5 cm) at 5 × 104 cells/cm2. Half of the medium was replaced with fresh Neurobasal/B27 medium every 4–5 d.
NO production in living cells.
The production of NO in living cells was estimated using the NO-sensitive fluorescent dye (Nagano, 1999), which reacts with NO in the presence of oxygen to form highly fluorescent triazolofluorescein. We used a membrane-permeable form of the dye, 3-amino,4-aminomethyl-2′7′difluorescein diacetate (DAF-FM DA) (Calbiochem), which can be taken up by cells and hydrolyzed by cellular esterases to form membrane-impermeable compound DAF-FM. The neurons cultured for 11 d were treated with a selective endothelial nitric oxide synthase (eNOS) inhibitor N5-(1-iminoethyl)-l-ornithine (10 μm; Biomol Research Laboratories) for 1 h, and loaded with 10 μm DAF-FM DA in the dark incubator. At 30 and 60 min after incubation, fluorescent signals of NO produced by cells were captured with a confocal laser-scanning microscope (LSM510; Zeiss).
Preparation and maintenance of organotypic hippocampal slice cultures.
Organotypic hippocampal slice cultures (OHSCs) were prepared as described previously (Stoppini et al., 1991) with some modification. Briefly, the hippocampus was dissected from decapitated, 10-d-old Sprague Dawley rat pups, and cut transversely into 400-μm-thick slices with a McIlwain tissue chopper. The slices were placed on the top of Millicell-CM tissue culture inserts (0.4 μm pore; Millipore) in six-well plates and maintained at 35°C in a humidified incubator with 5% CO2 for 10–14 d in a culture medium, pH 7.3, composed of 50% MEM, 25% heat-inactivated horse serum, 25% HBSS, and 25 mm HEPES. The medium was changed on the first day after culture and every 3–4 d thereafter.
Immunohistochemistry.
Serial hippocampal sections (40 μm) were made on an oscillating tissue slicer. For nNOS immunofluorescence, the sections were incubated with rabbit anti-nNOS antibody (1:500; Zymed Laboratories) in 0.1 m PBS with 3% goat serum and 0.3% Triton X-100, and binding was visualized with a Cy3-conjugated secondary antibody (1:200; Millipore Bioscience Research Reagents). Images of immunostained neurons in all groups were captured with a Zeiss Axio Cam MRC 5(D) camera mounted on Carl Zeiss Axio Observer A1 microscope under same conditions. An experimenter coded all slides from the experiments before quantitative analysis. All nNOS-positive cells in the hippocampus were counted in each section by another experimenter blinded to the study code.
Western blot analysis.
Western bolt analysis of samples from cultured hippocampal neurons, OHSCs, and hippocampal or hypothalamic tissues of animals was performed as described previously (Zhang et al., 2010). The primary antibodies were as follows: mouse anti-inducible NOS (iNOS; 1:2000; Santa Cruz Biotechnology), rabbit anti-nNOS (1:1000; Millipore Bioscience Research Reagents), mouse anti-eNOS (1:2000; Millipore Bioscience Research Reagents), rabbit anti-extracellular signal-regulated kinase (ERK; 1:500; Bioworld Technology), rabbit antiphospho-ERK (1:500; Bioworld Technology), rabbit anti-GR (1:200; Santa Cruz Biotechnology), rabbit anti-MR (1:3000; Santa Cruz Biotechnology), mouse anti-nitrotyrosine (1:3000; Millipore Bioscience Research Reagents), and rabbit anti-corticotrophin-releasing factor (anti-CRF; 1:500; Santa Cruz Biotechnology). Appropriate horseradish peroxidase-linked secondary antibodies were used for detection by enhanced chemiluminescence (Pierce).
NOS activity assay.
The hippocampus was homogenized in ice-cold PBS, pH 7.4, and centrifuged at 10,000 g for 20 min at 4°C. The supernatant was ultracentrifuged at 100,000 g for 15 min at 4°C using a 300 kDa molecular weight cutoff filter by centrifugation. NOS activity in the filtrates was measured using a commercially available kit (Jiancheng). Inducible NOS activity was measured by adding EGTA at 3 mmol/L to chelate-free Ca2+ from the reaction mixture. To measure nNOS activity, 1 mmol/L l-nomega-iminoethyl-l-ornithine (Sigma-Aldrich), a selective eNOS inhibitor, was added into the reaction mixture. Neuronal NOS activity was computed by subtracting the iNOS activity from the total NOS activity with the inhibited fraction of eNOS. Endothelial NOS activity was computed by subtracting the iNOS and nNOS activity from the total NOS activity. NOS activities were expressed as unit (U). One unit was defined as nanomoles of NO formed in 1 min by 1 mg of protein.
Corticosterone measurement.
For basal corticosterone level measurement, mice were decapitated between 9:00 and 10:00 A.M. Blood from angulus oculi vessels was collected in heparinized tubes, and corticosterone in plasma was measured with a corticosterone ELISA kit according to the instructions of the manufacturer (Cayman Chemical).
Statistical analysis.
Data are presented as means ± SEM. Comparisons among multiple groups were made with one-way ANOVA (one factor) or two-way ANOVA (two factors), followed by Scheffe's post hoc test. Comparisons between two groups were made with two-tail Student's t test. Differences were considered significant when p < 0.05.
Results
Glucocorticoids upregulate hippocampal nNOS expression via MR
Our previous study showed that CMS exposure upregulated hippocampal nNOS expression in mice (Zhou et al., 2007). To examine whether glucocorticoids account for the CMS-induced nNOS increase, we measured nNOS and NO metabolite peroxynitrite (ONOO−, measured by nitrotyrosine) levels in the hippocampus of adult mice exposed to CMS for 21 d with or without metyrapone (100 mg/kg/d s.c.), a corticosteroids synthesis inhibitor, and found that metyrapone abolished the CMS-induced ONOO− production (F(2,9) = 9.98, p = 0.010) and nNOS expression revealed by protein level (F(2,6) = 20.39, p = 0.006) (Fig. 1A) and the number of nNOS-positive neurons in the hippocampus (Fig. 1B).
Figure 1.

Glucocorticoids upregulate hippocampal nNOS expression. A, Immunoblots showing hippocampal nNOS and nitrotyrosine levels in mice (n = 3–4). B, Representative sample of nNOS+ neurons from the hippocampal CA3 and DG areas (top) and number of nNOS+ neurons in the hippocampus in mice (bottom; n = 4–5). A, B, Mice were exposed to CMS for 21 d with or without metyrapone (100 mg/kg/d, s.c.). C, Immunoblots showing hippocampal nNOS and nitrotyrosine levels of the mice treated with corticosterone (40 mg/kg/d s.c.) for 0, 7, 14, or 21 d (n = 3). D, Immunoblots showing nNOS levels in the cultured hippocampal neurons incubated with 10 μm corticosterone or its vehicle for 0.5, 2, 6, 12, or 24 h (n = 3). E, Immunoblots showing nNOS levels in the OHSCs incubated with corticosterone for 24 h at the concentrations indicated (n = 3). F, Representative imaging of NO synthesis revealed by DAF-FM staining (top) and histograms showing the fluorescence intensities (bottom) in the hippocampal neurons exposed to 10 μm corticosterone for 24 h (n = 4). Error bars denote SEM; *p < 0.05, **p < 0.01, one-way ANOVA. NT, Nitrotyrosine; Met, metyrapone.
To further confirm the effect of glucocorticoids on hippocampal nNOS, we treated adult mice with corticosterone (40 mg/kg/d s.c.) for 0, 7, 14, or 21 d. It significantly increased hippocampal nNOS after treatment for 7, 14, and 21 d (F(3,8) = 19.76, p = 0.013 for 7 d; p = 0.002 for 14 d; p = 0.001 for 21 d), and ONOO− level after treatment for 14 and 21 d (F(3,8) = 6.86, p = 0.023 for 14 d; p = 0.036 for 21 d) (Fig. 1C). Three isoforms of NOS exist, including nNOS, eNOS, and iNOS (Zhou and Zhu, 2009). Accordingly, we measured iNOS and eNOS expressions and enzymatic activities in the hippocampus of adult mice treated with corticosterone for 7 d. The drug did not change iNOS or eNOS expressions (iNOS/β-actin: 0.323 ± 0.016 vs 0.393 ± 0.033, n = 3, p = 0.131; eNOS/β-actin: 0.527 ± 0.007 vs 0.638 ± 0.078, n = 3, p = 0.233) or activities (iNOS: 0.314 ± 0.079 U vs 0.302 ± 0.017 U, n = 5–6, F(5,27) = 99.86, p = 1.000; eNOS: 0.913 ± 0.047 U vs 1.003 ± 0.087 U, n = 5–6, F(5,27) = 99.86, p = 0.990), although it increased nNOS enzymatic activity (1.929 ± 0.137 U vs 2.381 ± 0.104 U, n = 5–6, F(5,27) = 99.86, p = 0.040) compared with vehicle.
However, the subcutaneous injection of corticosterone may cause systemic effects. We therefore examined nNOS expression in the cultured hippocampal neurons exposed to 10 μm corticosterone for 0.5, 2, 6, 12, or 24 h. The drug significantly increased nNOS level after exposure for 24 h (F(1,4) = 8.80, p = 0.041, 1.82-fold relative to vehicle) (Fig. 1D). OHSCs are advantageous for examining hippocampal function for a number of weeks in a well controlled in vitro environment (Becker et al., 2008). We thus used the OHSCs to assess the effects of corticosterone on nNOS expression. Rat OHSCs were cultured in vitro for 10 d and exposed to 0.1, 1.0, or 10 μm corticosterone for 24 h. The drug increased nNOS expression concentration dependently (1.55-, 1.82-, and 3.38-fold for 0.1, 1.0, and 10 μm corticosterone, respectively, relative to vehicle) (Fig. 1E). Additionally, we measured NO synthesis in the neurons exposed to 10 μm corticosterone for 24 h using fluorescent dye DAF-FM (NO probe) (Chvanov et al., 2006). The drug significantly increased DAF-FM fluorescent signal compared with vehicle (100 ± 3.62 vs 146.8 ± 10.15, n = 4, p = 0.005, t test) (Fig. 1F), suggesting an increased NO production by corticosterone.
To investigate how glucocorticoids regulate nNOS expression, we incubated hippocampal neurons and the OHSCs with 10 μm corticosterone for 24 h in the presence of 50 μm mifepristone (a GR-selective antagonist) and/or 50 μm spironolactone (a MR-selective antagonist) (de Kloet et al., 1998). Interestingly, spironolactone but not mifepristone abolished the effect of corticosterone on nNOS expressions in the hippocampal neurons (F(4,10) = 35.34, p < 0.001 for spironolactone; p = 0.629 for mifepristone) (Fig. 2A) and in the OHSCs (F(4,15) = 6.84, p = 0.047 for spironolactone; p = 0.702 for mifepristone) (Fig. 2B). As spironolactone did not alter nNOS expression in the corticosterone-free hippocampal neurons (F(3,8) = 0.36, p = 0.819) (Fig. 2C), it is unlikely that unspecific pharmacological effect accounts for the antagonism of spironolactone. Our findings suggest that MR mediates the effect of glucocorticoids on hippocampal nNOS expression.
Figure 2.

MR mediates glucocorticoid-induced nNOS expression. A, B, Immunoblots showing nNOS levels in the cultured hippocampal neurons (A, n = 3) or OHSCs (B, n = 4) exposed to 10 μm corticosterone for 24 h with 50 μm mifepristone and/or 50 μm spironolactone. C, Immunoblots showing nNOS levels in the cultured hippocampal neurons exposed to 50 μm mifepristone or 50 μm spironolactone for 24 h (n = 3). Error bars denote SEM; *p < 0.05, **p < 0.01, ***p < 0.001, one-way ANOVA.
Requirement of nNOS for the behavioral effects of glucocorticoids
We used the CMS model of depression to characterize despair behavior in the TST and FST, and sensitivity to reward in SPT in the adult mice (Berton and Nestler, 2006). To determine whether nNOS is implicated in depressive behaviors of glucocorticoids, we treated adult mice with corticosterone (40 mg/kg/d s.c.) for 21 d with or without 7-NI (30 mg/kg/d i.p.), a selective nNOS inhibitor. Treatment with 7-NI abolished the corticosterone-induced immobility in the TST (F(3,40) = 7.00, p = 0.020) and FST (F(3,40) = 4.52, p = 0.029) (Fig. 3A) and reduction in sucrose preference (F(3,39) = 17.40, p < 0.001) (Fig. 3B) compared with vehicle. The immobility time changes cannot be explained by nonspecific effects of treatments on activity levels, because these treatments did not alter locomotor activities of mice (data not shown).
Figure 3.

nNOS is required for the behavioral effects of glucocorticoids. A, B, Immobility time in the TST and FST (A, n = 11) and sucrose preference (B, n = 10–11) of the adult mice treated with corticosterone alone (40 mg/kg/d s.c.) or in combination with 7-NI (30 mg/kg/d i.p.) for 21 d. C, D, Immobility time in the TST and FST (C) and sucrose preference (D) of the adult mice treated with corticosterone alone (40 mg/kg s.c.) or in combination with 10 μm 7-NI (hippocampal microinjection, 2 μl; n = 10–11). Corticosterone was administrated for 21 d and started at day 4 after 7-NI microinjection. E–G, Immobility time in the TST and FST (E), sucrose preference (F), and body weight gain (G) of the adult mice exposed to CMS for 21 d with or without 10 μm 7-NI (hippocampal microinjection, 2 μl; n = 11–12). CMS was started at day 4 after hippocampal 7-NI microinjection. H, I, Immobility time in the TST (H) and FST (I) of the nNOS KO (n = 9–10) and WT (n = 14) mice treated with 10 μm corticosterone (hippocampal microinjection, 2 μl). The TST and FST were performed at day 21 after hippocampal microinjection. Error bars denote SEM; *p < 0.05, **p < 0.01, ***p < 0.001, one-way ANOVA (A–G), two-way ANOVA (H–I). Cort, Corticosterone.
Next, we delivered 7-NI (2 μl, 10 μm/L) into bilateral hippocampi of mice by microinjection to inhibit local nNOS activity. We exposed mice to corticosterone (40 mg/kg/d s.c.) or CMS for 21 d. The microinjection of 7-NI was performed at day 4 before CMS exposure or after starting corticosterone treatment. The intrahippocampal 7-NI use abolished the effects of systemic corticosterone on immobility in the TST (F(2,29) = 5.23, p = 0.029) and FST (F(2,29) = 5.21, p = 0.029) (Fig. 3C) and on sucrose preference (F(2,29) = 15.94, p = 0.001) (Fig. 3D), and prevented the CMS-induced immobility in the TST (F(2,33) = 5.23, p < 0.001) and FST (F(2,33) = 5.23, p = 0.035) (Fig. 3E), sucrose preference reduction (F(2,32) = 15.28, p < 0.001) (Fig. 3F), and body weight loss (F(2,33) = 38.33, p = 0.027) (Fig. 3G). These treatments did not affect locomotor activities of mice (data not shown). Thus, hippocampal nNOS is required for the stress-related depressive behaviors of glucocorticoids.
To further address this notion, we delivered corticosterone (2 μl, 10 μm/L) into bilateral hippocampi of nNOS KO and WT mice and performed behavioral tests at day 21 after microinjection. As expected, intrahippocampal corticosterone use significantly increased immobility in the TST (F(1,43) = 10.34, p = 0.003) (Fig. 3H) and FST (F(1,43) = 5.72, p = 0.021) (Fig. 3I) in WT mice but was ineffective in KO mice. Furthermore, vehicle-treated KO mice displayed a lower latency in the TST (F(1,43) = 24.00, p < 0.001) and FST (F(1,43) = 4.14, p = 0.048) than vehicle-treated WT mice did (Fig. 3H,I). Intrahippocampal corticosterone use did not change locomotor activities in WT or KO mice (data not shown).
nNOS mediates GR regulation of glucocorticoids
Changes of GR level in forebrain are implicated in depressive behaviors (Pariante and Miller, 2001; Boyle et al., 2005). To investigate stress-related GR alteration by glucocorticoids, we measured GR levels in the hippocampus of mice exposed to CMS for 21 d with or without metyrapone (100 mg/kg/d s.c.) and found that the drug reversed CMS-induced GR reduction (F(2,6) = 21.62, p = 0.005) (Fig. 4A). Next, we incubated the OHSCs with 10 μm corticosterone for 24, 48, or 72 h and found that GR downregulation by corticosterone occurred at 72 h after treatment (F(1,4) = 191.93, p < 0.001) (Fig. 4B), whereas nNOS upregulation by corticosterone in the hippocampal neurons appeared at 24 h after treatment (Fig. 1D). Similarly, in the hippocampus of mice treated with corticosterone (40 mg/kg/d s.c.) for 0, 7, 14, or 21 d, GR expression reduction occurred at 14 d after treatment (F(3,8) = 12.05, p = 0.041) (Fig. 4C), which is dramatically later than hippocampal nNOS expression increase by the drug (Fig. 1B). These results raise a possibility that nNOS may be an upstream signal of GR expression. Indeed, 10 μm 7-NI reversed the corticosterone-induced GR reduction in the OHSCs incubated with these drugs for 72 h (F(2,6) = 13.13, p = 0.029) (Fig. 4D). Intrahippocampal 7-NI microinjection (2 μl, 10 μm/L) prevented the GR reductions that resulted from CMS exposure (F(2,6) = 11.00, p = 0.012) (Fig. 4E) or corticosterone administration (40 mg/kg/d s.c.) for 21 d (F(2,6) = 8.19, p = 0.034) (Fig. 4F). Furthermore, although systemic corticosterone (40 mg/kg/d s.c. for 21 d) significantly decreased GR levels in the hippocampus in WT mice (F(1,8) = 35.51, p < 0.001), it was ineffective in nNOS KO mice (Fig. 4G). To further confirm the effect of nNOS on GR expression, we measured hippocampal GR levels in the mice treated with 7-NI (30 mg/kg/d i.p. for 7 d) or in the nNOS KO mice. As expected, nNOS inhibition or deletion significantly increased hippocampal GR levels (7-NI vs vehicle, 1.0 ± 0.033 vs 2.08 ± 0.126, n = 3, p = 0.001; WT vs KO, 1.0 ± 0.173 vs 1.77 ± 0.133, n = 4, p = 0.012, t test). More importantly, the upregulation of nNOS activity by 50 mm KCl exposure (Fig. 4H) (Sasaki et al., 2000) resulted in a decrease in GR expression in the cultured hippocampal neurons (vehicle vs KCl, 1.0 ± 0.08 vs 0.61 ± 0.113, n = 3, p = 0.047, t test). Thus, these data collectively indicate that glucocorticoids regulate GR via nNOS.
Figure 4.
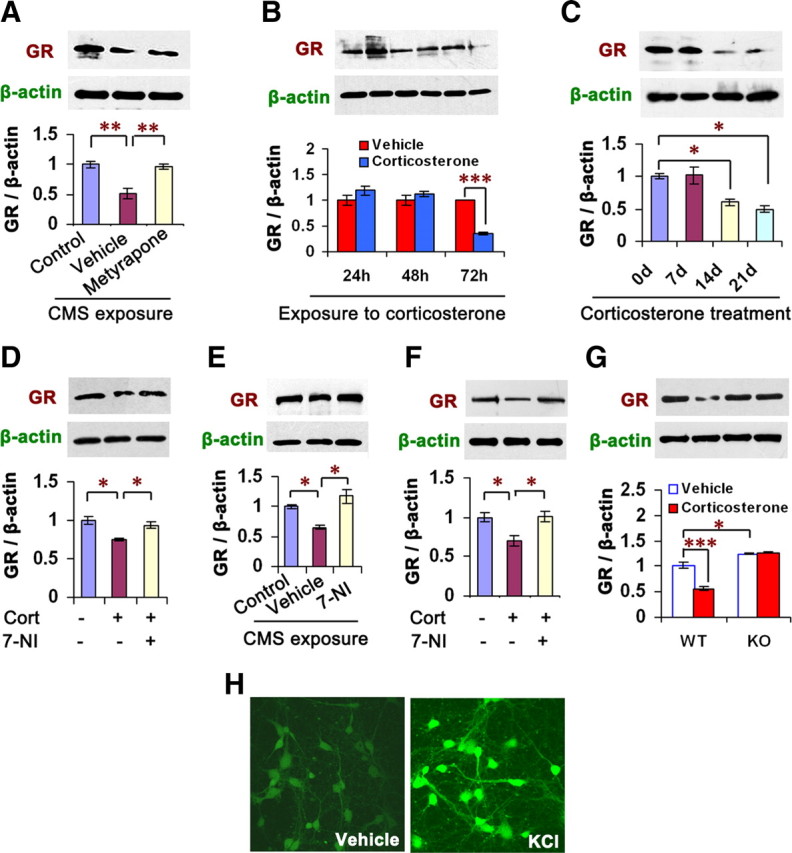
nNOS mediates GR downregulation of glucocorticoids. A, Immunoblots showing hippocampal GR levels of the mice exposed to CMS with or without metyrapone (100 mg/kg/d s.c.) for 21 d (n = 3). B, Immunoblots showing GR levels in the OHSCs incubated with 10 μm corticosterone for 24, 48, or 72 h (n = 3). C, Immunoblots showing hippocampal GR levels of the mice treated with corticosterone (40 mg/kg/d s.c.) for 0, 7, 14, or 21 d (n = 3). D, Immunoblots showing GR levels in the OHSCs incubated with 10 μm corticosterone with or without 7-NI for 72 h (n = 3). E–F, Immunoblots showing hippocampal GR levels of the mice exposed to CMS (E) or treated with corticosterone (F; 40 mg/kg/d s.c.) for 21 d with or without 10 μm 7-NI (hippocampal microinjection, 2 μl; n = 3). CMS and corticosterone injection were performed at day 4 after 7-NI microinjection. G, Immunoblots showing hippocampal GR levels of nNOS KO and WT mice treated with corticosterone (40 mg/kg/d s.c.) for 21 d (n = 3). H, Representative imaging of NO synthesis revealed by DAF-FM staining in the hippocampal neurons exposed to 50 mm KCl for 24 h. Error bars denote SEM; *p < 0.05, **p < 0.01, ***p < 0.001; two-way ANOVA (G), one-way ANOVA (A–F, H). Cort, Corticosterone.
To examine whether glucocorticoids affect MR expression, we measured MR levels in the hippocampus of adult mice exposed to CMS for 21 d with or without metyrapone (100 mg/kg/d s.c.) and found that these treatments had no effect on MR expression (n = 3, p > 0.05, between groups) (data not shown). Consistent with this result, the mice treated with corticosterone (40 mg/kg/d s.c.) for 0, 7, 14, or 21 d exhibited basal MR level in the hippocampus (n = 3, p > 0.05, between groups) (data not shown). To investigate the effect of glucocorticoids on MR expression with nNOS deletion, we examined hippocampal MR levels at day 7 after intrahippocampal corticosterone microinjection (2 μl) in nNOS KO and WT mice. The treatment had no effect on MR expression in either WT or KO mice (n = 3, p > 0.05, between groups) (data not shown). Moreover, in the OHSCs, 10 μm corticosterone alone or in combination with 7-NI (exposure for 72 h) did not affect MR expression (n = 3, p > 0.05, between groups) (data not shown).
To link nNOS-derived NO and GR regulation of glucocorticoids, we measured nNOS, ONOO−, and GR levels at day 21 after intrahippocampal corticosterone microinjection (2 μl) in nNOS KO and WT mice. The treatment significantly increased nNOS (F(1,12) = 44.00, p < 0.001) and ONOO− (F(1,12) = 18.38, p = 0.001) levels, decreased GR levels (F(1,8) = 97.57, p < 0.001) in WT mice, but was ineffective in KO mice (Fig. 5A). Next, we incubated the cultured hippocampal neurons with 100 μm DETA/NONOate, a NO donor, for 24 h. The drug significantly increased ONOO− (F(2,6) = 40.40, p = 0.001) and decreased GR (F(2,6) = 62.65, p < 0.001) levels. CPTIO (10 μm), a cell-impermeable NO scavenger, abolished the effects of DETA/NONOate on both ONOO− (F(2,6) = 40.40, p = 0.001) and GR (F(2,6) = 62.65, p < 0.001) (Fig. 5B). More importantly, DETA/NONOate inhibited GR expression both in nNOS KO neurons (F(1,8) = 15.84, p = 0.004) and WT neurons (F(1,8) = 22.07, p = 0.002) (Fig. 5C). Together, these data suggest that nNOS-derived NO may be responsible for the GR regulation of glucocorticoids.
Figure 5.

NO downregulates GR via cGMP-dependent and cGMP-independent mechanisms. A, Immunoblots showing hippocampal nNOS, nitrotyrosine, and GR levels of nNOS KO and WT mice treated with 10 μm corticosterone (hippocampal microinjection, 2 μl) at day 21 after the drug microinjection (n = 4). B, Immunoblots showing nitrotyrosine and GR levels in the cultured hippocampal neurons exposed to 100 μm DETA/NONOate with or without 10 μm CPTIO for 24 h (n = 3). C, Immunoblots showing GR levels in the nNOS KO and WT hippocampal neurons exposed to 100 μm DETA/NONOate for 24 h (n = 3). D, Immunoblots showing GR levels in the OHSCs exposed to 100 μm DETA/NONOate with or without 10 μm ODQ or 1.0 mm UA for 24 h (n = 3). E, Immunoblots showing nitrotyrosine and GR levels in the cultured hippocampal neurons exposed to SIN-1 at the concentrations indicated with or without 1.0 mm UA for 24 h (n = 3). F, G, Immobility time in the TST and FST (F) and body weight gain (G) of the adult mice treated with corticosterone alone (40 mg/kg/d × 21 d s.c.) or in combination with 20 μm CPTIO or 10 μm ODQ (intrahippocampal microinjection, 2 μl; n = 10–12). Error bars denote SEM; *p < 0.05, **p < 0.01, ***p < 0.001 (E, vs 0 mm); #p < 0.05, ##p < 0.01 (E, vs 1.0 mm); two-way ANOVA (C), one-way ANOVA (A, B, D–G). Cort, Corticosterone; NT, nitrotyrosine; UR, uric acid.
The NO-mediated actions are mostly due to cGMP formation, but cGMP-independent mechanisms, such as peroxynitrite-produced tyrosine nitration, have been suggested as potential signaling pathways modulating the NO-induced responses (Marcondes et al., 2006). Accordingly, we exposed the OHSCs to 100 μm DETA/NONOate for 24 h with or without 10 μm ODQ, a soluble guanylate cyclase inhibitor, and 1.0 mm uric acid (UA), a natural scavenger of ONOO− (Hooper et al., 1998). Pretreatment of the OHSCs with ODQ abolished the effect of DETA/NONOate on GR expression (F(5,12) = 47.24, p < 0.001), suggesting a cGMP-dependent mechanism. However, UA abolished the effect of DETA/NONOate on GR expression also (F(5,12) = 47.24, p < 0.001), implying a cGMP-independent mechanism (Fig. 5D).
To further address this hypothesis, we incubated the cultured hippocampal neurons with SIN-1, an ONOO− donor (Lipton et al., 1993), for 24 h. As expected, SIN-1 produced abundant ONOO− (1.504-, 2.82-, and 3.44-fold of control for 0.1, 0.5, and 1.0 mm SIN-1, respectively) and inhibited GR expression concentration dependently (88.4, 59.3, and 32.0% of control for 0.1, 0.5, and 1.0 mm SIN-1, respectively). Treatment with 1.0 mm UA abolished ONOO− production (F(4,10) = 10.55, p = 0.018) and reversed the effect of SIN-1 on GR (F(4,10) = 18.23, p = 0.002) (Fig. 5E).
To further determine whether hippocampal NO–cGMP pathway is implicated in the role of glucocorticoids in depressive behaviors, we treated mice with corticosterone (40 mg/kg/d s.c.) for 21 d. We delivered CPTIO (2 μl, 20 μm/L) or ODQ (2 μl, 10 μm/L) into bilateral hippocampi by microinjection at day 4 and performed behavioral tests at day 22 after starting corticosterone treatment. Both CPTIO and ODQ abolished the corticosterone-induced prolongation of immobility time in the TST (F(3,41) = 6.48, p = 0.029 for CPTIO, p = 0.020 for ODQ) and FST (F(3,41) = 4.92, p = 0.039 for CPTIO, p = 0.029 for ODQ) (Fig. 5F), and relieved the corticosterone-induced body weight loss (F(3,41) = 47.34, p = 0.042 for CPTIO, p = 0.026 for ODQ) (Fig. 5G) compared with vehicle.
To determine whether NO influences MR expression, we incubated the cultured hippocampal neurons with 100 μm DETA/NONOate alone or in combination with 10 μm CPTIO and found that these treatments had no effect on MR level (n = 3, p > 0.05, between groups) (data not shown).
ERK activation is critical for the GR regulation and behavioral effects of ONOO−
Experimental evidence suggests that mitogen-activated protein kinase (MAPK) signaling pathway is implicated in the stress-related behavioral effects of glucocorticoids (Revest et al., 2005). Consistently, mice exposed to CMS for 21 d displayed an elevated phosphorylated extracellular signal-regulated kinase (pERK) level in the hippocampus; treatment with metyrapone reversed the CMS-induced ERK activation (pERK/ERK: 0.867 ± 0.008, 1.609 ± 0.214, or 0.954 ± 0.029 for control, vehicle-treated CMS, or metyrapone-treated CMS, respectively; F(2,6) = 10.60, p = 0.016, control vs vehicle; p = 0.028, vehicle vs metyrapone), suggesting an ERK activation by glucocorticoids. To further address this finding, we incubated the OHSCs with 10 μm corticosterone for 24 h. This treatment significantly increased pERK level (pERK1/ERK1: F(4,10) = 70.32, p < 0.001; pERK2/ERK2: F(4,10) = 19.22, p = 0.001). Pretreatment with 50 μm spironolactone (pERK1/ERK1: F(4,10) = 70.32, p = 0.001; pERK2/ERK2: F(4,10) = 19.22, p = 0.019) but not with 50 μm mifepristone (pERK1/ERK1: F(4,10) = 70.32, p = 1.000; pERK2/ERK2: F(4,10) = 19.22, p = 0.996) significantly inhibited the corticosterone-induced ERK activation, suggesting that glucocorticoids regulate ERK phosphorylation via MR (Fig. 6A). To determine whether nNOS contributes to the glucocorticoid-induced ERK activation, we incubated the OHSCs with 10 μm corticosterone for 24 h with or without 10 μm 7-NI and found that 7-NI abolished the corticosterone-induced ERK activation (pERK1/ERK1: F(2,6) = 13.74, p = 0.012; pERK2/ERK2: F(2,6) = 13.67, p = 0.012) (Fig. 6B). Furthermore, intrahippocampal corticosterone microinjection (2 μl, 10 μm/l) significantly increased local pERK level in WT mice (pERK1/ERK1: F(1,8) = 98.78, p < 0.001; pERK2/ERK2: F(1,8) = 50.74, p < 0.001) but was ineffective in KO mice (pERK1/ERK1: F(1,8) = 0.14, p = 0.718; pERK2/ERK2: F(1,8) = 1.89, p = 0.207) (Fig. 6C). Thus, nNOS is required for the effect of glucocorticoids on ERK activation.
Figure 6.

ERK activation is essential for the GR regulation and behavioral effects of ONOO−. A, Immunoblots showing pERK/ERK levels in the OHSCs exposed to 10 μm corticosterone with 50 μm mifepristone and/or 50 μm spironolactone for 24 h (n = 3). B, Immunoblots showing pERK/ERK levels in the OHSCs exposed to 10 μm corticosterone with or without 10 μm 7-NI for 24 h (n = 3). C, Immunoblots showing hippocampal pERK/ERK levels of nNOS KO and WT mice at day 21 after intrahippocampal corticosterone microinjection (2 μl, 10 μm/L; n = 3). D, Immunoblots showing pERK/ERK levels in the hippocampal neurons exposed to SIN-1 (0–1.0 mm) with or without 1.0 mm UA for 24 h (n = 3). E, Immunoblots showing pERK/ERK and GR levels in the OHSCs exposed to 10 μm corticosterone with 1.0 mm UA or 10 μm U0126 for 72 h (n = 3). F, Immunoblots showing nNOS levels in the OHSCs exposed to 10 μm corticosterone with 10 μm U0126 for 24 h (n = 4). G, Immunoblots showing pERK/ERK and GR levels in the OHSCs exposed to 1.0 mm SIN-1 with or without 10 μm U0126 for 24 h (n = 3). H, I, Immobility in the TST and FST (H) and sucrose preference (I) of mice at day 20 after intrahippocampal microinjection of SIN-1 (1 μl, 1.0 mm/L) alone or in combination with U0126 (1 μl, 10 μm/L; n = 11–13). J, Immunoblots showing pERK/ERK levels in the OHSCs exposed to 100 μm DETA/NONOate with or without 10 μm ODQ or 1.0 mm UA for 24 h (n = 3). Error bars denote SEM; *p < 0.05, **p < 0.01, ***p < 0.001; two-way ANOVA (C), one-way ANOVA (A, B, D–J). Cort, Corticosterone; Mif, mifepristone; Spir, spironolactone.
To investigate the effect of ONOO− on ERK activation, we incubated the cultured hippocampal neurons with 0, 0.1, 0.5, or 1.0 mm SIN-1 with or without UA for 24 h. As shown in Fiugre 6D, 1.0 mm SIN-1 significantly increased pERK level (pERK1/ERK1: F(4,10) = 14.92, p = 0.001; pERK2/ERK2: F(4,10) = 12.12, p = 0.005), indicating ERK activation by ONOO−, which is consistent with previous reports (Zhang et al., 2000; Csibi et al., 2010). UA abolished the effect of 1.0 mm SIN-1 on ERK activation. Next, we incubated the OHSCs with 10 μm corticosterone for 72 h with or without UA or U0126, an ERK phosphorylation inhibitor. Both UA and U0126 abolished the effects of corticosterone on GR expression (F(3,8) = 8.22, p = 0.024 for U0126; p = 0.028 for UA) and ERK phosphorylation (pERK1/ERK1: F(3,8) = 14.64, p = 0.004 for both U0126 and UA; pERK2/ERK2: F(3,8) = 8.47, p = 0.024 for U0126, p = 0.014 for UA) (Fig. 6E). Because U0126 did not change the corticosterone-induced nNOS expression in the OHSCs (F(2,9) = 15.09, p = 0.836) (Fig. 6F), nNOS may constitute an upstream signaling of ERK activation and GR regulation of glucocorticoids. Interestingly, U0126 abolished SIN-1-induced GR reductions and ERK phosphorylation in the OHSCs (GR: F(2,6) = 26.96, p = 0.002; pERK1/ERK1: F(2,6) = 34.25, p = 0.001; pERK2/ERK2: F(2,6) = 19.93, p = 0.005) (Fig. 6G). Together, these data indicate that ERK activation is critical for the peroxynitrite-induced GR downregulation.
To determine whether hippocampal ERK activation is essential for the effect of ONOO− on depressive behaviors, we delivered 1.0 mm SIN-1 (1 μl) alone or in combination with 10 μm U0126 (1 μl; 30 min interval between two injections) into bilateral hippocampi of adult mice and performed behavioral examinations at day 10 or 20 after treatments. At day 20 after microinjection, the mice exposed to SIN-1 had significantly prolonged immobility time in the TST (F(2,33) = 8.02, p = 0.002) and FST (F(2,33) = 5.02, p = 0.048) and reduced sucrose preference (F(2,33) = 7.00, p = 0.011) compared with vehicle-treated mice, exhibiting a depressive phenotype. U0126 reversed the effects of SIN-1 on immobility in the TST (F(2,33) = 8.02, p = 0.035) and FST (F(2,33) = 5.02, p = 0.028) (Fig. 6H) and on sucrose preference (F(2,33) = 7.00, p = 0.010) (Fig. 6I). No altered behavioral phenotype was observed at day 10 after the treatment (data not shown).
To determine whether NO-mediated ERK activation is cGMP-dependent, we exposed the OHSCs to 100 μm DETA/NONOate for 24 h with or without 10 μm ODQ or 1.0 mm UA. As shown in Figure 6J, UA but not ODQ abolished the effect of DETA/NONOate on ERK phosphorylation (pERK1/ERK1: F(5,12) = 15.48, p = 0.022 for UA; p = 0.909 for ODQ; pERK2/ERK2: F(5,12) = 12.30, p = 0.018 for UA; p = 1.000 for ODQ), indicating that NO activates ERK pathway via cGMP-independent mechanism.
Hippocampal nNOS mediates CRF regulation of glucocorticoids
Hyperactivity of HPA axis has been identified as a characteristic feature of depression (Berton and Nestler, 2006). To explore the role of nNOS in glucocorticoid-induced HPA axis hyperactivity, we delivered 10 μm 7-NI or its vehicle (2 μl) into bilateral hippocampi of mice by microinjection. Four days later, the mice were exposed to CMS for 21 d. Interestingly, intrahippocampal 7-NI use abolished the CMS-induced elevations of corticosterone (F(2,18) = 23.48, p < 0.001) and hypothalamic CRF, a peptide that governs the HPA axis (F(2,12) = 13.09, p = 0.003) (de Kloet et al., 2005), compared with vehicle (Fig. 7A,B).
Figure 7.

Hippocampal NO upregulates hypothalamic CRF. A, Plasma corticosterone levels of the adult mice exposed to CMS for 21 d with or without 10 μm 7-NI (2 μl, hippocampal microinjection 4 d before CMS; n = 7). B, Immunoblots showing hypothalamic CRF levels of the adult mice exposed to CMS for 21 d with or without 10 μm 7-NI (2 μl, hippocampal microinjection 4 d before CMS; n = 5). C, Immunoblots showing hypothalamic CRF levels of the adult mice treated with corticosterone (40 mg/kg/d s.c.) for 21 d with or without 10 μm 7-NI (hippocampal microinjection, 2 μl). Corticosterone injection was started at day 4 after 7-NI microinjection (n = 3). D, Immunoblots showing hypothalamic CRF levels of nNOS KO and WT mice treated with 10 μm corticosterone (hippocampal microinjection, 2 μl) at day 21 after the drug microinjection (n = 3). E, F, Immunoblots showing hippocampal GR levels (E) and hypothalamic CRF and GR levels (F) of adult mice treated with 1.0 mm SIN-1 alone or in combination with 10 μm U0126 (hippocampal microinjection, 1 μl) at day 7 after microinjections (n = 3). Error bars denote SEM; *p < 0.05, **p < 0.01, ***p < 0.001; two-way ANOVA (D), one-way ANOVA (A–C, E, F).
Next, we treated mice with corticosterone (40 mg/kg/d delivered s.c.) for 21 d; 4 d later, we delivered 7-NI (2 μl, 10 μm/l) or its vehicle into bilateral hippocampi by microinjection. Although acute treatment with corticosterone caused a decrease in CRF expression (CRF/β-actin: 0.668 ± 0.037 vs 0.411 ± 0.076, vehicle vs corticosterone, n = 3, p = 0.038), the long-term treatment with corticosterone significantly elevated the CRF level (F(2,6) = 9.67, p = 0.022). Hippocampal 7-NI microinjection abolished the corticosterone-induced CRF upregulation (F(2,6) = 9.67, p = 0.029) (Fig. 7C). To further confirm the role of nNOS, we delivered 10 μm corticosterone (2 μl, 10 μm/L) into the bilateral hippocampi of nNOS KO or WT mice and then examined CRF levels at 21 d after microinjection. As shown in Figure 7D, intrahippocampal corticosterone microinjection significantly increased CRF level in WT mice (F(1,12) = 22.38, p < 0.001) but was ineffective in KO mice (F(1,12) = 0.22, p = 0.65). Although nNOS exists in the hypothalamus, the glucocorticoid-induced CRF expression cannot be explained by hypothalamic nNOS alteration, because intrahippocampal corticosterone microinjection did not alter hypothalamic nNOS expression (nNOS/β-actin: 0.5999 ± 0.034 vs 0.661 ± 0.035, vehicle vs corticosterone, n = 4, p = 0.255).
GR density is the highest in the PVN of the hypothalamus (de Kloet et al., 2005). To exclude the possibility that hippocampal NO mediates CRF regulation of glucocorticoids via hypothalamic GR, we delivered SIN-1 (1 μl, 1.0 mm/L) alone or in combination with U0126 (1 μl, 10 μm/L) into bilateral hippocampi and measured hypothalamic CRF and GR levels and hippocampal GR levels at day 7 microinjections. The intrahippocampal SIN-1 significantly inhibited hippocampal GR expression (F(2,6) = 10.45, p = 0.021) and increased hypothalamic CRF level (F(2,6) = 18.55, p = 0.003), and U0126 abolished the effects of SIN-1 on CRF (F(2,6) = 18.55, p = 0.047) and hippocampal GR (F(2,6) = 10.45, p = 0.022). These treatments did not affect hypothalamic GR level (F(2,6) = 1.77, p = 0.800) (Fig. 7E,F). Furthermore, intrahippocampal corticosterone microinjection did not alter hypothalamic GR expression (GR/β-actin: 0.487 ± 0.034 vs 0.529 ± 0.06, vehicle vs corticosterone, n = 3, p = 0.572). Thus, hypothalamic GR does not account for the effect of hippocampal NO on HPA axis.
Discussion
Despite a strong relationship between hippocampal GR and stress-related HPA axis hyperacitivity and behavioral effects of glucocorticoids, the molecular mechanisms by which glucocorticoids regulate the hippocampal GR and, in turn, produce behavioral modifications, are not clear. In the present study, we have identified a signaling cascade whereby glucocorticoid-induced hippocampal nNOS upregulation mediates GR reduction and depressive behaviors (Fig. 8). Glucocorticoids upregulate nNOS expression in the hippocampus by activating MR and, in turn, lead to hippocampal GR downregulation by cGMP-dependent and cGMP-independent mechanisms. This modification causes HPA axis hyperactivity, thereby modulating stress-related depressive behaviors.
Figure 8.
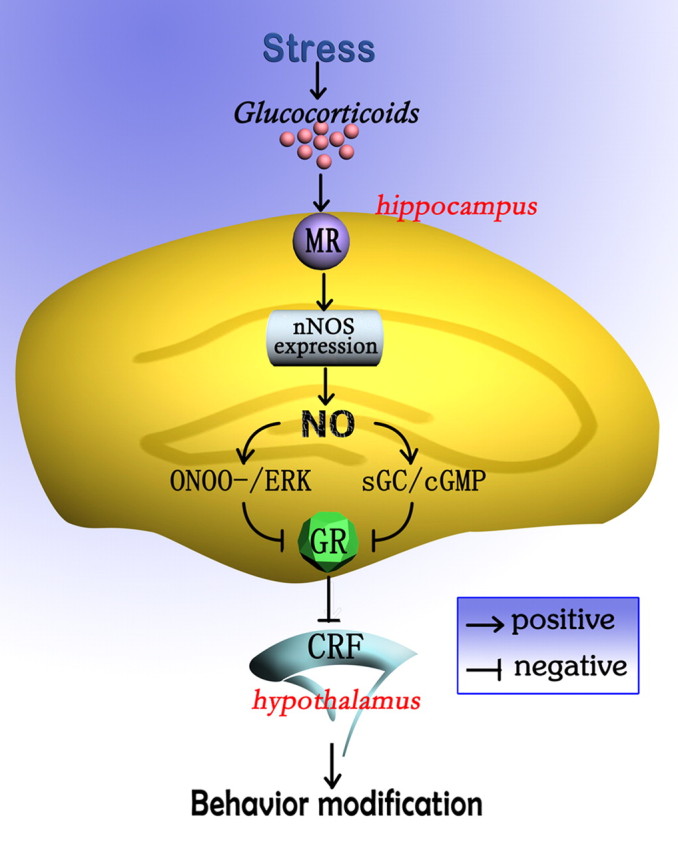
A model of signaling pathway whereby hippocampal nNOS mediates stress-related depressive behaviors of glucocorticoids. Stress-induced glucocorticoids release, acting via MR, stimulates hippocampal nNOS expression and, in turn, leads to GR downregulation by cGMP-dependent and cGMP-independent mechanisms. This modification causes CRF upregulation in the hypothalamus, thereby modulating stress-related depressive behaviors.
It has been demonstrated that nNOS, mainly responsible for NO production in the nervous system (Bredt and Snyder, 1994), is implicated in depression (Zhou and Zhu, 2009) and anxiety (Zhang et al., 2010). Although nNOS is richly expressed throughout the limbic system (Bredt et al., 1991), the hippocampal nNOS may be the most important for the stress-related depression, not only because both CMS and corticosterone exposures caused hippocampal nNOS overexpression, but also because the inhibition of hippocampal nNOS almost abolished both CMS- and corticosterone-induced depressive behaviors. GR and MR colocalization is found in the hippocampus of almost all species (de Kloet et al., 2005). However, our data indicate that the hippocampal nNOS upregulation by glucocorticoids depends on MR activation. The MR selectivity of nNOS expression may result because MR binds corticosterone with ∼10-fold higher affinity than colocalized GR does (de Kloet et al., 1998). GR is widely expressed throughout the brain, whereas most MR in the brain is located in the hippocampus where they may be coexpressed with GR by many, but not all, neurons in rodents (Rozeboom et al., 2007). The selective hippocampal distribution of MR may account for why neither CMS exposure nor hippocampal corticosterone microinjections changed hypothalamic nNOS expression. Therefore, hypothalamic CRF elevation by hippocampal corticosterone microinjection is not due to hypothalamic nNOS alteration, although NO is implicated in the hypothalamic regulation of stress (Calabrese et al., 2007). Additionally, only nNOS-derived NO is related to the glucocorticoid-mediated behavioral modifications because corticosterone exposure had no effect on iNOS or eNOS expressions or enzymatic activities, even though nNOS, eNOS, and iNOS are isoenzymes (Zhou and Zhu, 2009).
How does the glucocorticoid-induced hippocampal nNOS overexpression lead to stress-related behavioral modifications? Hippocampal GR downregulation could be a crucial step. The formation of MR and GR homodimers and heterodimers, as well as monomers, allows receptor responsivity over a wide concentration range. MR is implicated in the appraisal process and the onset of the stress response, whereas GR terminates stress reactions and mobilizes the energy resources required for this purpose and facilitates recovery (de Kloet et al., 2005). GR thus plays an important role in the negative feedback regulation of the HPA axis and adaptation to stress (Pariante and Miller, 2001). GR sensitivity has been shown to be reduced in MDD in both in vitro and in vivo studies. Transgenic animals with partial impairment of GR function show behavioral changes consistent with MDD (Ridder et al., 2005). Moreover, reduced expression of GR has been reported in the distinct regions of the cortex and hippocampus of MDD bipolar disorder brains (Matsubara et al., 2006). Deletion of limbic GR (except in the hypothalamic PVN) induces a robust depression-like phenotype in mice (Boyle et al., 2005). Our data indicate that CMS exposure reduced hippocampal GR level and corticosteroid synthesis inhibitor abolished the effect of CMS. Identically, corticosterone exposures inhibited hippocampal GR expression in vitro and in vivo. The reduction in GR expression can be explained by the glucocorticoid-induced hippocampal nNOS overexpression, because it was abolished by nNOS inhibition. More importantly, nNOS overexpression caused an impaired GR repression. Because hippocampal nNOS inhibition and NO donor had no effect on MR expression, the nNOS-mediated GR reduction may change the GR:MR ratio, an index important for HPA system homeostasis (Rozeboom et al., 2007).
It is well accepted that NO-mediated actions are due to activation of the soluble guanylate cyclase (sGC) leading to an increase of cGMP, but cGMP-independent mechanisms have been suggested as potential signaling pathways modulating the NO-induced responses (Marcondes et al., 2006). NO can act by directly modifying cysteine residues in target proteins (Stamler et al., 2001; Hess et al., 2005). Cysteine nitrosylation (S-nitrosylation) is now regarded as a selective and specific signal controlled by cellular NO (Stamler et al., 2001). NO can regulate gene expression via S-nitrosylation of transcription factors in the cytoplasm (Marshall et al., 2004) or via S-nitrosylation of nuclear proteins (Nott et al., 2008). Moreover, ONOO−, the product of the diffusion-controlled reaction of NO with superoxide O2·− radical, can nitrate tyrosine residues and consequently modify the functional properties of several proteins (Csibi et al., 2010). Importantly, ONOO− is able to affect other signal-transduction pathways, including PKC, MAPK, and Src tyrosine kinases (Szabó et al., 2007). In this study, both NO and ONOO− donors inhibited hippocampal GR expression, and both selective sGC inhibitor and ONOO− scavenger abolished the effect of NO donor on GR, suggesting that hippocampal nNOS-derived NO represses GR expression via both cGMP-dependent and cGMP-independent mechanisms.
Hippocampal MAPK pathway is involved in stress-related behavioral effects of glucocorticoids (Revest et al., 2005). Our data show that glucocorticoids promote ERK phosphorylation via upregulating nNOS expression, and that ONOO− caused a substantial ERK activation. In fact, both NO–cGMP signaling pathway and ONOO− are implicated in ERK activation (Zhang et al., 2000; Rangaswami et al., 2009). In this study, the glucocorticoid-mediated ERK activation led to hippocampal GR downregulation. More importantly, ERK phosphorylation inhibitor not only abolished SIN-1-induced GR reduction, but also reversed depressive behaviors of hippocampal SIN-1 microinjection, indicating an nNOS–ERK–GR signaling pathway. Given that CRF is essential for HPA axis homeostasis (de Kloet et al., 2005), hypothalamic CRF alteration may account for the role of hippocampal nNOS–ERK–GR signaling pathway in stress-related behaviors, because, as we show here, nNOS in the hippocampus is critical for hypothalamic CRF expression. We thus conclude that hippocampal nNOS overexpression, and thereby ERK activation and GR reduction, are responsible for the stress-related depressive behaviors of glucocorticoids, although more studies are needed to exclude the effects of other molecules that are associated with NO and emotion, such as histone deacetylase and 5-HT1A receptor (de Kloet et al., 2005; Krishnan and Nestler, 2008; Nott et al., 2008; Zhang et al., 2010).
Footnotes
This work was supported by grants from National Natural Science Foundation of China (30870900, 30971021, and 81030023 to D.Y.Z. and 81000586 to Q.G.Z.), Natural Science Foundation of Jiangsu Province (BK2007728 to D.Y.Z.), and Specialized Research Fund for the Doctoral Program of Higher Education (200803120008 to D.Y.Z.). We thank Y. Hu, W. X. Sun, and H. B. Xu for technical assistance.
References
- Bartolomucci A, Leopardi R. Stress and depression: preclinical research and clinical implications. PLoS One. 2009;4:e4265. doi: 10.1371/journal.pone.0004265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker N, Wierenga CJ, Fonseca R, Bonhoeffer T, Nägerl UV. LTD induction causes morphological changes of presynaptic boutons and reduces their contacts with spines. Neuron. 2008;60:590–597. doi: 10.1016/j.neuron.2008.09.018. [DOI] [PubMed] [Google Scholar]
- Berton O, Nestler EJ. New approaches to antidepressant drug discovery: beyond monoamine. Nat Rev Neurosci. 2006;7:137–151. doi: 10.1038/nrn1846. [DOI] [PubMed] [Google Scholar]
- Boyle MP, Brewer JA, Funatsu M, Wozniak DF, Tsien JZ, Izumi Y, Muglia LJ. Acquired deficit of forebrain glucocorticoid receptor produces depression-like changes in adrenal axis regulation and behavior. Proc Natl Acad Sci U S A. 2005;102:473–478. doi: 10.1073/pnas.0406458102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bredt DS, Snyder SH. Transient nitric oxide synthase neurons in embryonic cerebral cortical plate, sensory ganglia, and olfactory epithelium. Neuron. 1994;13:301–313. doi: 10.1016/0896-6273(94)90348-4. [DOI] [PubMed] [Google Scholar]
- Bredt DS, Glatt CE, Hwang PM, Fotuhi M, Dawson TM, Snyder SH. Nitric oxide synthase protein and mRNA are discretely localized in neuronal populations of the mammalian CNS together with NADPH diaphorase. Neuron. 1991;7:615–624. doi: 10.1016/0896-6273(91)90374-9. [DOI] [PubMed] [Google Scholar]
- Calabrese V, Mancuso C, Calvani M, Rizzarelli E, Butterfield DA, Stella AM. Nitric oxide in the central nervous system: neuroprotection versus neurotoxicity. Nat Rev Neurosci. 2007;8:766–775. doi: 10.1038/nrn2214. [DOI] [PubMed] [Google Scholar]
- Chiavegatto S, Dawson VL, Mamounas LA, Koliatsos VE, Dawson TM, Nelson RJ. Brain serotonin dysfunction accounts for aggression in male mice lacking neuronal nitric oxide synthase. Proc Natl Acad Sci U S A. 2001;98:1277–1281. doi: 10.1073/pnas.031487198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho DH, Nakamura T, Fang J, Cieplak P, Godzik A, Gu Z, Lipton SA. S-nitrosylation of Drp1 mediates beta-amyloid-ralated mitochondrial fission and neuronal injury. Science. 2009;324:102–105. doi: 10.1126/science.1171091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chvanov M, Gerasimenko OV, Petersen OH, Tepikin AV. Calcium-dependent release of NO from intracellular S-nitrosothiols. EMBO J. 2006;25:3024–3032. doi: 10.1038/sj.emboj.7601207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Csibi A, Communi D, Müller N, Bottari SP. Angiotensin II inhibits insulin-stimulated GLUT4 translocation and Akt activation through tyrosine nitration-dependent mechanisms. PLoS One. 2010;5:e10070. doi: 10.1371/journal.pone.0010070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Kloet ER, Vreugdenhil E, Oitzl MS, Joëls M. Brain corticosteroid receptor balance in health and disease. Endocr Rev. 1998;19:269–301. doi: 10.1210/edrv.19.3.0331. [DOI] [PubMed] [Google Scholar]
- de Kloet ER, Joëls M, Holsboer F. Stress and the brain: from adaptation to disease. Nat Rev Neurosci. 2005;6:463–475. doi: 10.1038/nrn1683. [DOI] [PubMed] [Google Scholar]
- Grippo AJ, Sullivan NR, Damjanoska KJ, Crane JW, Carrasco GA, Shi J, Chen Z, Garcia F, Muma NA, Van de Kar LD. Chronic mild stress induces behavioral and physiological changes, and may alter serotonin 1A receptor function, in male and cycling female rats. Psychopharmacology. 2005;179:769–780. doi: 10.1007/s00213-004-2103-4. [DOI] [PubMed] [Google Scholar]
- Hess DT, Matsumoto A, Kim SO, Marshall HE, Stamler JS. Protein S-nitrosylation: purview and parameters. Nat Rev Mol Cell Biol. 2005;6:150–166. doi: 10.1038/nrm1569. [DOI] [PubMed] [Google Scholar]
- Hooper DC, Spitsin S, Kean RB, Champion JM, Dickson GM, Chaudhry I, Koprowski H. Uric acid, a natural scavenger of peroxynitrite, in experimental allergic encephalomyelitis and multiple sclerosis. Proc Natl Acad Sci U S A. 1998;95:675–680. doi: 10.1073/pnas.95.2.675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnan V, Nestler EJ. The molecular neurobiology of depression. Nature. 2008;455:894–902. doi: 10.1038/nature07455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipton SA, Choi YB, Pan ZH, Lei SZ, Chen HS, Sucher NJ, Loscalzo J, Singel DJ, Stamler JS. A redox-based mechanism for the neuroprotective and neurodestructive effects of nitric oxide and related nitroso-compounds. Nature. 1993;364:626–632. doi: 10.1038/364626a0. [DOI] [PubMed] [Google Scholar]
- Marcondes S, Cardoso MH, Morganti RP, Thomazzi SM, Lilla S, Murad F, De Nucci G, Antunes E. Cyclic GMP-independent mechanisms contribute to the inhibition of platelet adhesion by nitric oxide donor: a role for alpha-actinin nitration. Proc Natl Acad Sci U S A. 2006;103:3434–3439. doi: 10.1073/pnas.0509397103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall HE, Hess DT, Stamler JS. S-nitrosylation: physiological regulation of NF-κB. Proc Natl Acad Sci U S A. 2004;101:8841–8842. doi: 10.1073/pnas.0403034101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinowich K, Manji H, Lu B. Mew insights into BDNF function in depression and anxiety. Nat Neurosci. 2007;10:1089–1093. doi: 10.1038/nn1971. [DOI] [PubMed] [Google Scholar]
- Matsubara T, Funato H, Kobayashi A, Nobumoto M, Watanabe Y. Reduced glucocorticoid receptor alpha expression in mood disorder patients and first-degree relatives. Biol Psychiatry. 2006;59:689–695. doi: 10.1016/j.biopsych.2005.09.026. [DOI] [PubMed] [Google Scholar]
- Nagano T. Practical methods for detection of nitric oxide. Luminescence. 1999;14:283–290. doi: 10.1002/(SICI)1522-7243(199911/12)14:6<283::AID-BIO572>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- Nelson RJ, Demas GE, Huang PL, Fishman MC, Dawson VL, Dawson TM, Snyder SH. Behavioural abnormalities in male mice lacking neuronal nitric oxide synthase. Nature. 1995;378:383–386. doi: 10.1038/378383a0. [DOI] [PubMed] [Google Scholar]
- Nott A, Watson PM, Robinson JD, Crepaldi L, Riccio A. S-nitrosylation of histone deacetylase 2 induces chromatin remodeling in neurons. Nature. 2008;455:411–415. doi: 10.1038/nature07238. [DOI] [PubMed] [Google Scholar]
- O'Donovan MC, Craddock N, Norton N, Williams H, Peirce T, Moskvina V, Nikolov I, Hamshere M, Carroll L, Georgieva L, Dwyer S, Holmans P, Marchini JL, Spencer CC, Howie B, Leung HT, Hartmann AM, Möller HJ, Morris DW, Shi Y, et al. Identification of loci associated with schizophrenia by genome-wide association and follow-up. Nat Genet. 2008;40:1053–1055. doi: 10.1038/ng.201. [DOI] [PubMed] [Google Scholar]
- Pariante CM, Miller AH. Glucocorticoid receptors in major depression: relevance to pathophysiology and treatment. Biol Psychiatry. 2001;49:391–404. doi: 10.1016/s0006-3223(00)01088-x. [DOI] [PubMed] [Google Scholar]
- Rangaswami H, Marathe N, Zhuang S, Chen Y, Yeh JC, Frangos JA, Boss GR, Pilz RB. Type II cGMP-dependent protein kinase mediates osteoblast mechanotransduction. J Biol Chem. 2009;284:14796–14808. doi: 10.1074/jbc.M806486200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reif A, Herterich S, Strobel A, Ehlis AC, Saur D, Jacob CP, Wienker T, Töpner T, Fritzen S, Walter U, Schmitt A, Fallgatter AJ, Lesch KP. A neuronal nitric oxide synthase (NOS-I) haplotype associated with schizophrenia modifies prefrontal cortex function. Mol Psychiatry. 2006;11:286–300. doi: 10.1038/sj.mp.4001779. [DOI] [PubMed] [Google Scholar]
- Revest JM, Di Blasi F, Kitchener P, Rougé-Pont F, Desmedt A, Turiault M, Tronche F, Piazza PV. The MAPK pathway and Egr-1 mediate stress-related behavioral effects of glucocorticoids. Nat Neurosci. 2005;8:664–672. doi: 10.1038/nn1441. [DOI] [PubMed] [Google Scholar]
- Ridder S, Chourbaji S, Hellweg R, Urani A, Zacher C, Schmid W, Zink M, Hörtnagl H, Flor H, Henn FA, Schütz G, Gass P. Mice with genetically altered glucocorticoid receptor expression show altered sensitivity for stress-induced depressive reactions. J Neurosci. 2005;25:6243–6250. doi: 10.1523/JNEUROSCI.0736-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozeboom AM, Akil H, Seasholtz AF. Mineralocorticoid receptor overexpression in forebrain decreases anxiety-like behavior and alters the stress response in mice. Proc Natl Acad Sci U S A. 2007;104:4688–4693. doi: 10.1073/pnas.0606067104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki M, Gonzalez-Zulueta M, Huang H, Herring WJ, Ahn S, Ginty DD, Dawson VL, Dawson TM. Dynamic regulation of neuronal NO synthase transcription by calcium influx through a CREB family transcription factor-dependent mechanism. Proc Natl Acad Sci U S A. 2000;97:8617–8622. doi: 10.1073/pnas.97.15.8617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shinkai T, Ohmori O, Hori H, Nakamura J. Allelic association of the neuronal nitric oxide synthase (NOS1) gene with schizophrenia. Mol Psychiatry. 2002;7:560–563. doi: 10.1038/sj.mp.4001041. [DOI] [PubMed] [Google Scholar]
- Stamler JS, Lamas S, Fang FC. Nitrosylation: the prototypic redox-based signaling mechanism. Cell. 2001;106:675–683. doi: 10.1016/s0092-8674(01)00495-0. [DOI] [PubMed] [Google Scholar]
- Stoppini L, Buchs PA, Muller D. A simple method for organotypic cultures of nervous tissue. J Neurosci Methods. 1991;37:173–182. doi: 10.1016/0165-0270(91)90128-m. [DOI] [PubMed] [Google Scholar]
- Szabó C, Ischiropoulos H, Radi R. Peroxynitrite: biochemistry, pathophysiology and development of therapeutics. Nat Rev Drug Discov. 2007;8:662–680. doi: 10.1038/nrd2222. [DOI] [PubMed] [Google Scholar]
- Zhang J, Huang XY, Ye ML, Luo CX, Wu HY, Hu Y, Zhou QG, Wu DL, Zhu LJ, Zhu DY. Neuronal nitric oxide synthase alteration accounts for the role of 5-HT1A-receptor in modulating anxiety-related behaviors. J Neurosci. 2010;30:2433–2441. doi: 10.1523/JNEUROSCI.5880-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang P, Wang YZ, Kagan E, Bonner JC. Peroxynitrite targets the epidermal growth factor receptor, Raf-1, and MEK independently to activate MAPK. J Biol Chem. 2000;275:22479–22486. doi: 10.1074/jbc.M910425199. [DOI] [PubMed] [Google Scholar]
- Zhou L, Zhu DY. Neuronal nitric oxide synthase: structure, subcellular localization, regulation, and clinical implications. Nitric Oxide. 2009;20:223–230. doi: 10.1016/j.niox.2009.03.001. [DOI] [PubMed] [Google Scholar]
- Zhou QG, Hu Y, Hua Y, Hu M, Luo CX, Han X, Zhu XJ, Wang B, Xu JS, Zhu DY. Neuronal nitric oxide synthase contributes to chronic stress-induced depression in mice. J Neurochem. 2007;103:1843–1854. doi: 10.1111/j.1471-4159.2007.04914.x. [DOI] [PubMed] [Google Scholar]