

Abstract
Multiple cortical areas are involved in pain processing, including the primary somatosensory cortex (S1) and the anterior cingulate cortex (ACC). Although accumulations of evidence suggest that the S1 activity increases under chronic pain conditions, whether plastic change occurs or not within the S1, and whether and how the plastic change contributes to chronic pain behavior, is unknown. Here, we provide the first evidence that intra-regional remodeling within the mouse S1 accelerates chronic pain behavior by modulating neuronal activity in the ACC, one of the important cortical areas for chronic pain. Using two-photon Ca2+ imaging, we found that the spontaneous activity of layer 2/3 neurons in the S1 and then response to sensory and layer 4 stimulations increased under chronic pain conditions. In addition, pharmacological attenuation and facilitation of S1 activity attenuated and facilitated the chronic pain behavior, respectively. Furthermore, electrical response of the ACC to peripheral stimulation successfully correlated with S1 neuronal activity, and inhibition of ACC activity alleviated the mechanical allodynia. The present results will provide development of efficient therapeutic strategies against chronic pain by focusing on the S1 and ACC.
Introduction
Chronic pain is a major health issue all over the world, and is caused by tissue or nerve injury under different disease conditions. Treatment of chronic pain is a clinical challenge because the mechanisms are less well known. In addition to the spinal cord (Woolf and Salter, 2000), multiple brain areas, such as the primary somatosensory cortex (S1) and anterior cingulate cortex (ACC), are involved in acute pain processing (Price, 2000; Vogt, 2005; Zhuo, 2008). These areas also contribute to chronic pain (Moisset and Bouhassira, 2007). The S1 has a role in the sensory aspects of pain, such as the location and duration of pain (Bushnell et al., 1999). Although several brain imaging experiments show increase in the S1 activity under chronic pain conditions (Peyron et al., 1998; Petrovic et al., 1999; Endo et al., 2008), little is known about the plastic change of neuronal activity in the S1 and its contribution to the pathological behavior under the chronic pain condition.
Since pain-related areas may interact with each other in processing pain information (Moisset and Bouhassira, 2007), we focused on layer 2/3 (L2/3) excitatory neurons of the S1, which integrate sensory information originating from peripheral nerves and transmit the signals to other pain-related cortical areas (Ferezou et al., 2007; Kamatani et al., 2007). Here, we found facilitation of the L2/3 excitatory neuronal activity in the S1 using in vivo two-photon Ca2+ imaging, which exaggerated pain behavior by facilitation of ACC activity.
Materials and Methods
Animal preparation.
C57BL/6 male mice (4 weeks old) and vesicular GABA transporter (VGAT)-Venus male mice, which express Venus in the inhibitory neurons (Wang et al., 2009), were used. To induce chronic inflammatory pain, 40 μl of Complete Freund's adjuvant (CFA) (Sigma) was injected subcutaneously into the hindpaw 1 week before experiments. Control mice were injected with 40 μl of saline only. All relevant experimental protocols were approved by the Animal Research Committee of the National Institutes of Natural Sciences, Japan.
Surgery.
Mice were anesthetized with urethane (1.7 g/kg body weight, i.p.) and atropine (0.4 mg/kg body weight, i.p.). The skull was exposed and cleaned. A 3-mm-wide cranial window was made above the S1. The exposed dura was covered with a glass coverslip and the space was filled with agarose (Wako Pure Chemical Industries, 2% in ACSF).
Optical imaging of intrinsic signal.
For imaging of intrinsic signals, the cortical surface was illuminated by blue light-emitting diodes (490 nm) and signal was detected as a decrease in reflective light intensity in response to the hindpaw stimulation using mechanical manipulator (1 s, 4.0 g/cm2) (Shibuki et al., 2003).
Labeling procedure and in vivo Ca2+ imaging.
Multicell bolus loading of L2/3 neurons and astrocytes with membrane-permeable Ca2+ indicator Oregon Green 488 BAPTA-1 (OGB-1) AM (Invitrogen) or fura-2 AM (Invitrogen) was performed as previously reported (Kerr et al., 2005; Kameyama et al., 2010).
Ca2+ indicators (OGB-1 or fura-2) were excited at 800 nm, and Venus and sulforhodamine 101(SR101) were excited at 950 nm. A water-immersion objective (20×, 0.95 numerical aperture, 40×, 0.8 numerical aperture, all from Olympus) was used to acquire images.
Fluorescence data acquisition and analysis.
Images (256 × 256 pixels, 0.428 s per frame) was analyzed with Olympus Fluoview software. Hindpaw stimulation was repeatedly applied at each plane. The baseline intensity, F0, was obtained by averaging the intensity values during the prestimulus period (5 s). Cells were considered responsive if the fluorescence intensity change was >10% of baseline value, because we can clearly distinguish responses from baseline. Amplitude from OGB-1, fura-2 signals were calculated as ΔF/F0 (ΔF = F − F0), −ΔF/F0, respectively.
Intra-regional electrical stimulation.
In some experiments, we placed a bipolar electrode (Inter Medical) at the depth of 500–550 μm below the cortical surface. Electrical pulses were generated using a stimulator (SEN-7203, Nihon Kohden) attached to an isolator unit (SS-403J, Nihon Kohden). Ten pulses of 200 μs constant current (10–100 μA, interval of 1 min, 20 Hz) were applied in each trial.
Slice preparation and whole-cell patch-clamp recording.
Brain slices were made as previously reported (Eto et al., 2007). Electrical recordings were made using the conventional whole-cell patch-clamp recording technique at a holding potential of −60 mV (Axopatch 700B, Molecular Devices). Patch pipettes were filled with the internal solution, which contained (in mm:) 130 cesium methanesulfonate, 4 CsCl, 2 EGTA, 4 Mg-ATP, and 10 HEPES, 5 QX314, pH 7.3 (7–9 MΩ). Evoked EPSCs were recorded from the L2/3 pyramidal neurons in the S1 hindpaw area in the presence of 50 μm d-(−)-2-amino-5-phosphonopentanoic acid (AP-5), an NMDA receptor antagonist, and elicited by applying voltage pulses (100 μs, 0.05 Hz) to the S1 layer 4. Data were acquired using pCLAMP 10 software, filtered at 2 kHz, digitized at 10 kHz.
In vivo electrophysiology.
A craniotomy was made in the region overlying the ACC and S1. Field EPSPs (fEPSPs) in the ACC (1.3–1.4 mm from the surface) were recorded using a DiI-coated tungsten electrode. After amplification (ER-91, NeuroData) and bandpass filtering (10–1 kHz), the signal was sent to an analog-to-digital converter (PowerLab/4ST). The drug [AMPA receptor antagonist 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX), or SR95531 (2-(3-carboxyl)-3-amino-6-(4-methoxyphenyl)-pyridazinium bromide) with 0.1% fluorescein isothiocyanate (FITC)] was applied to the L2/3 region of the S1 hindpaw area, and drug expansion was assessed by the fluorescence from FITC.
Mechanical allodynia test and drug application.
The mechanical allodynia tests were performed before injection of saline or CFA into the hindpaw, 7 d after the injection, and after drug injection using the von Frey test (Chaplan et al., 1994). A small hole (1 mm) was made on the skull above the S1, and drugs (CNQX, vehicle containing 1% DMSO, SR95531) were injected using a glass pipette at the L2/3 region under brief anesthesia, using isoflurane (1–3% as needed) inhalation with 30% oxygen balanced with nitrogen. In control mice, injection of a higher concentration (3 mm) of CNQX increased pain threshold (n = 4). Thus, we used injection of 1 mm CNQX in this study. For drug application to the ACC, small holes were made on the skull above the bilateral ACC, and CNQX (1 mm, 0.5 μl) was injected into the bilateral ACC.
Statistical analysis.
All values are expressed as mean ± SEM. Statistical comparisons were performed using ANOVA followed by a Bonferroni test or unpaired Student's t test. The difference between cumulative distributions was assessed using the Kolmogorov–Smirnov test. The difference was considered significant at p < 0.05.
Results
CFA was injected into the hindpaw of a wild-type mouse or a VGAT-Venus mouse (see Material and Methods) to generate a chronic inflammatory pain model. Before Ca2+ imaging, the area of S1 topographically corresponding to the contralateral hindpaw area was identified by optical imaging of intrinsic signals (Fig. 1a, right), which were evoked by pressure stimulation applied to the hindpaw of anesthetized mice with either saline (control) or CFA injection (Fig. 1a, left).
Figure 1.
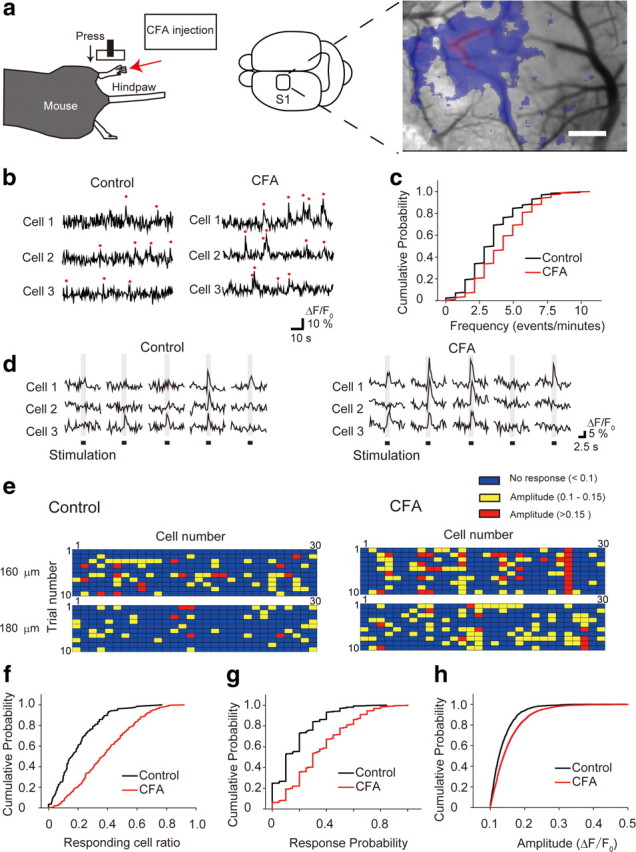
Increased spontaneous activity and sensory-evoked response of L2/3 neurons in the S1 under chronic pain conditions. a, Schematic diagrams of pressure stimulation of the hindpaw (left) and a brain showing the imaged area of the S1 as a square area (middle), and the intrinsic optical response in the S1 elicited by stimulation of the hindpaw in control mice (right). Scale bar, 0.5 mm. b, Typical traces of spontaneous Ca2+ transients (red dots) were obtained from different L2/3 neurons. c, Cumulative probability graph of spontaneous Ca2+ transient frequency. d, Typical traces of Ca2+ transients in L2/3 neurons evoked by hindpaw stimulation (indicated by black bars). e, Representative distribution of Ca2+ responses to each hindpaw stimulation (10 successive stimulations). Each box represents either no response (blue), or small (from 0.1 to 0.15) (yellow) or large amplitude of ΔF/F0 (>0.15) (red) in the Ca2+ imaging. f–h, Cumulative probability histograms of responding cell ratio (f), response probability per cell (g), and amplitude of Ca2+ transients (h). The responding cell ratio and response probability were determined as the number of responsive cells divided by total imaged cells at each plane, and as the number of responsive trials divided by the total number of trials, respectively. In each analysis, a significant difference between the control and CFA-injected group was detected (Kolmogorov–Smirnov test, p < 0.05).
For the measurement of L2/3 neuronal activity, the Ca2+ indicator OGB-1 AM was used (Kerr et al., 2005). The frequency of spontaneous Ca2+ transients was increased in the CFA-injected mice (4.26 ± 0.12 transients/min, 172 cells, 2 animals, p < 0.05) when compared with the control mice (3.42 ± 1.8 transients/min, 130 cells, 2 animals). Cumulative probability histograms were significantly different between the control and CFA-injected mice (Fig. 1b,c). Response pattern of Ca2+ transients to a train of hindpaw stimulation was valid among neurons of control and CFA-injected mice (Fig. 1d,e). The L2/3 neuronal activity of the CFA-injected mice increased in responding cell ratio (38.0 ± 1.06%, 350 trials in 5 animals, p < 0.05), response probability per cell (0.35 ± 0.01, 886 cells in 5 animals, p < 0.05), and amplitude (0.154 ± 0.078, 4660 responses in 5 animals, p < 0.05) compared with the control mice, which displayed values of 19.4 ± 1.00% (209 trials in 4 animals), 0.17 ± 0.01 (534 cells in 5 animals), and 0.137 ± 0.099 (1420 responses in 4 animals), respectively (Fig. 1f–h). Considering that the amplitude of evoked Ca2+ transient reflects the number of action potentials (Kerr et al., 2005), this result suggests that spontaneous activity and excitability of L2/3 neurons in response to peripheral stimulation increased under chronic pain condition.
To clearly distinguish the activity of L2/3 excitatory neurons, which integrate and transmit sensory information to other cortical areas, the L2/3 excitatory neuronal activity elicited in response to hindpaw pressure stimulation was examined by fura-2 Ca2+ imaging (Kameyama et al., 2010) (Fig. 2a,b). The L2/3 excitatory neuronal activity of the CFA-injected mice in response to sensory stimulation increased in responding cell ratio (22.2 ± 0.01%, 400 trials in 4 animals, p < 0.05), response probability per cell (0.219 ± 0.007, 450 cells in 4 animals, p < 0.05), and amplitude (0.149 ± 0.001, 1880 responses in 4 animals, p < 0.05), compared with the control mice, which displayed values of 17.3 ± 0.01% (320 trials in 3 animals), 0.136 ± 0.005 (610 cells in 3 animals), and 0.133 ± 0.001 (1651 responses in 3 animals), respectively (Fig. 2c–e). These results clearly demonstrate the hyperexcitability of L2/3 excitatory neurons in chronic pain conditions.
Figure 2.
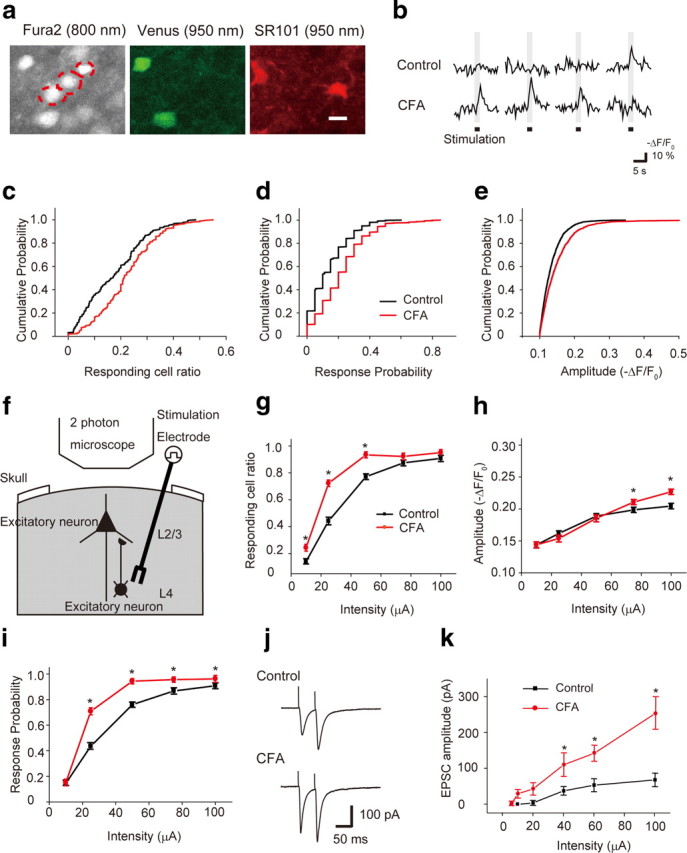
Intracortical remodeling between the L2/3 and L4 in the S1 contributes to the hypersensitivity of L2/3 excitatory neurons to sensory stimulation under chronic pain conditions. a, Visualization of L2/3 excitatory neurons (red dotted circles in the fura-2 image). Cells expressing Venus (inhibitory neurons, green) and cells stained with SR101 (astrocytes, red) were excluded from the analysis. Scale bar, 12 μm. b, Typical traces of sensory-evoked Ca2+ transients of L2/3 excitatory neurons in a control (upper traces) and CFA-injected (lower traces) mouse. c–e, Cumulative probability histograms of the responding cell ratio (c), response probability per cell (d), and amplitude of Ca2+ transients (e). In each analysis, a significant difference between control and CFA-injected mice was observed (Kolmogorov–Smirnov test, p < 0.05). f, Schematic drawing of in vivo Ca2+ imaging of L2/3 excitatory neurons in response to L4 electrical stimulation. g–i, The relationship between stimulus intensity and responding cell ratio (g), amplitude of Ca2+ transients (h), and response probability per cell (i). Excitability of L2/3 excitatory neurons of S1 evoked by electrical stimulation of the L4 region increased in the CFA-injected mice. j, Averaged traces of EPSCs evoked by paired-pulse stimulations of 60 μA in control (top) and CFA-injected (bottom) mice. k, Stimulus intensity–response curves from control (7 cells, filled squares) and CFA-injected (8 cells, filled circles) mice. *p < 0.05, significantly different from the control group by ANOVA followed by Bonferroni test. Error bars represent ±SEM.
To examine plastic changes of synaptic transmission within the S1 in CFA-injected mice, we next measured Ca2+ transients in L2/3 excitatory neurons evoked by electrical stimulation of the layer 4 (L4) region in vivo (Fig. 2f). Evoked Ca2+ transients increased with respect to the responding cell ratio, response probability per responding cell, and amplitude, compared with the respective values of control mice (control, 3 animals; CFA, 4 animals) (Fig. 2g–i), which may indicate intracortical remodeling between the L4 and L2/3 neurons under chronic pain conditions in vivo. This intracortical remodeling was further confirmed by patch-clamp recording of EPSCs from L2/3 pyramidal neurons in brain slice preparations (Fig. 2j,k). The obtained intensity–response curves revealed that the amplitudes of EPSCs evoked by electrical stimulation of the L4 region were increased in the CFA-injected mice compared with the control mice (Fig. 2j,k). In addition, the paired-pulse ratio (second EPSC/first EPSC, amplitude) was significantly smaller in CFA-injected mice (1.06 ± 0.08, 8 cells) than in control mice (1.68 ± 0.42, 7 cells, p < 0.05) (Fig. 2j). This increased synaptic input from L4 to L2/3 pyramidal neurons also suggests that the intra-regional network from L4 to L2/3 excitatory neurons is remodeled in chronic pain conditions.
To directly examine the role of hyperactivities of S1 neurons in pain behavior under a chronic inflammatory pain condition, CNQX (1 mm, 1 μl) was microinjected into the surface area of the S1 in CFA-injected mice. Injection of CNQX increased the paw-withdrawal threshold (Fig. 3a). In contrast, microinjection of CNQX at the same dose to the S1 did not affect the paw-withdrawal threshold in control mice (Fig. 3a) (see Material and Methods). However, an injection of a GABAA receptor antagonist, SR95531, decreased the paw-withdrawal threshold in control mice (Fig. 3b). Thus, this result suggests that S1 neuronal hyperactivity contributes to the facilitation of mechanical allodynia under chronic pain conditions.
Figure 3.

The S1 neuronal activity modulated mechanical allodynia. a, Microinjection of AMPA receptor antagonist CNQX into the L2/3 region attenuated mechanical allodynia in CFA-injected mice (closed circles, 5 mice), but did not result in any observable changes in control mice (open circles, 3 mice). b, Microinjection of SR95531 (3 μm) into the S1 significantly reduced the paw-withdrawal thresholds in control mice (6 mice). *p < 0.05, significantly different from the value just before drug application by ANOVA, followed by Bonferroni test. Error bars represent ±SEM.
Since L2/3 excitatory neurons project to other cortical areas (Ferezou et al., 2007; Kamatani et al., 2007), the plastic change may modulate activity of other pain-related cortical areas, which may contribute to expression of mechanical allodynia. To clarify the influence of increased outputs from the S1 on the ACC, one of the important areas for chronic pain (Zhuo, 2007; Xu et al., 2008), we recorded fEPSPs in the ACC evoked by sensory stimulation with the suppression or disinhibition of the neuronal activity in the S1 (Fig. 4). Under normal conditions (control mice), local CNQX (1 mm, 1 μl) injection into the S1 did not alter the slope of fEPSP of the ACC in response to electrical stimulation of the hindpaw (Fig. 4b,c). However, in chronic pain conditions (CFA-injected mice), CNQX injection into the S1 decreased the slope of the fEPSP of the ACC in response to electrical stimulation of the hindpaw (Fig. 4b,c). In contrast, disinhibition of S1 activity by local application of SR95531 (3 μm, 1 μl) into the S1 facilitated sensory-evoked fEPSP in the ACC in control mice (Fig. 4d,e). To clarify the role of the ACC activity in chronic pain behavior, we applied CNQX into the ACC, which increased paw-withdrawal thresholds (Fig. 4f). This result suggests that the facilitated outputs from the S1 enhanced ACC activity, which leads to acceleration of mechanical allodynia under chronic pain conditions.
Figure 4.
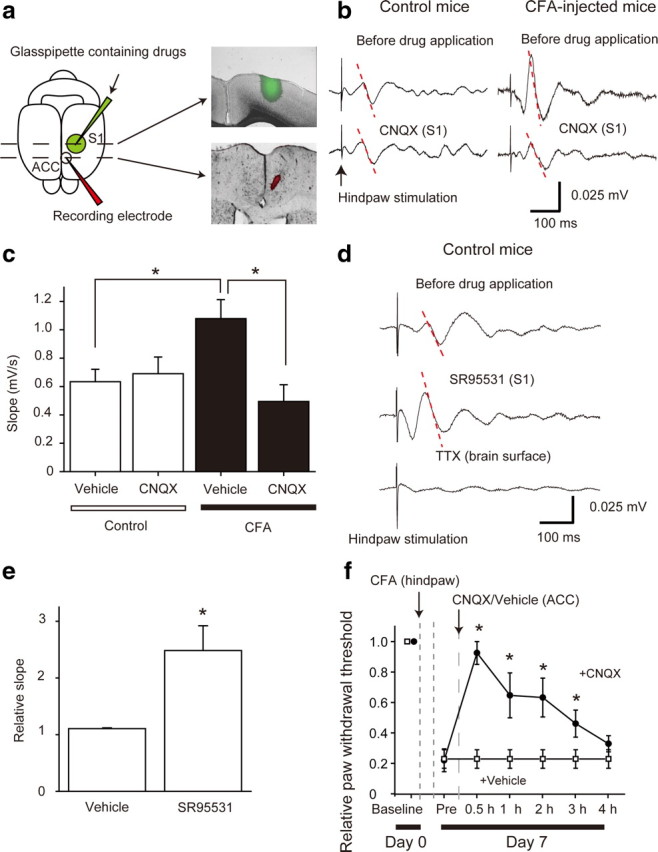
The S1 neuronal activity modulated ACC activity. a, Schematic drawing of the experimental configuration. DiI-coated recording electrode was placed in the ACC (red) and a glass pipette filled with a drug including FITC (green) in the L2/3 region of the S1, respectively (left). Coronal sectional images of the S1 and ACC show an area of drug diffusion (green, FITC) (right top) and the location of the recording electrode (red, DiI) (right bottom). In the slice containing the ACC, fluorescence from FITC was not detected. b, Averaged traces of fEPSPs evoked by hindpaw stimulations with (bottom) and without (top) CNQX applied to the S1 in control (left) and CFA-injected (right) mice. The slope (dotted line) was then analyzed. c, Mean slopes of the fEPSPs with vehicle and CNQX in the S1 in control and CFA-injected mice. d, Averaged traces of the fEPSPs evoked by hindpaw stimulation with (middle) and without (top) the GABAA receptor antagonist SR95531. TTX broadly applied to the brain surface completely abolished the response (bottom). Microinjection of SR95531 (3 μm) to the S1 enhanced the electrical response to hindpaw stimulation in the ACC of control mice. e, Mean slopes of the fEPSPs with vehicle (4 animals) and SR95531 (4 animals) in the S1 in control mice. Data were subjected to ANOVA followed by Bonferroni test. f, Microinjection of CNQX into the ACC attenuated mechanical allodynia in CFA-injected mice (closed circles, 4 animals), but injection of vehicle did not alter the thresholds (open square, 4 animals). *p < 0.05, significantly different from the control group by ANOVA followed by Bonferroni test. Error bars represent ±SEM.
Discussion
Cortical plasticity of the S1 has been discussed in chronic pain as a maladaptive memory formation (Vartiainen et al., 2009). Recently, brain imaging studies revealed the involvement of the S1 in the processing of allodynia in both humans and animals (Peyron et al., 1998; Petrovic et al., 1999; Endo et al., 2008). In the present study, in vivo imaging with single-cell resolution further demonstrated the increased excitability of L2/3 excitatory neurons. In the S1, neurons in the S1 code intensity and location of touch and pain stimulations (Kenshalo and Isensee, 1983). Since L2/3 neurons project to neurons in the same layer (Cheetham et al., 2007), increased outputs from the L2/3 neurons could enhance neuronal activity in the same layer, which may lead to recognition of touch sensation as exaggerated pain sensation.
In addition, we observed plastic changes of local cortical circuits from L4 to L2/3 in the S1 (Fig. 2). The S1 reorganization occurs constantly in response to the inputs from peripheral afferent neurons to the L2/3 neurons, which exhibit activity-dependent plasticity, such as long-term potentiation (Walcott and Langdon, 2002; Ismailov et al., 2004) and long-term depression (Allen et al., 2003). Since hyperexcitability in peripheral neurons enhances nociceptive inputs to supraspinal sites in chronic pain conditions (Urban and Gebhart, 1999; Woolf and Salter, 2000), such increased inputs may have caused the observed activity-dependent plastic changes in the afferents to L2/3 neurons within the S1, as seen in spinal circuits (Woolf and Salter, 2000). In pain models with exaggerated pain sensation, functional plastic change occurs within the ACC (Wei and Zhuo, 2001) and structural change of layer 5 neurons occurs in the S1 (Kim and Nabekura, 2011). Thus, the functional and structural plastic change within the pain-related cortical areas may have a critical role in chronic pain syndrome.
We demonstrated that pharmacological inhibition of excitatory synaptic transmission in L2/3 of the S1 successfully attenuated the mechanical allodynia and hyperexcitable activity of the ACC, while disinhibition of the transmission facilitated the pain behavior (Fig. 3) and ACC activity (Fig. 4). Excessive sensory information from the L2/3 excitatory neurons in the S1 to the ACC may increase the activity of the ACC. The experience of pain is a result of both sensory and emotional aspects, which are independently processed via different pathways involving the S1 and ACC, respectively (Price, 2000; Moisset and Bouhassira, 2007), and both types of cortical areas have been expected to interact with each other (Rainville et al., 1997). Since modulation of S1 activity altered electrical responses of the ACC to sensory stimulation (Fig. 4), plastic change of a sensory-related area could modulate the neuronal activity of a brain area related to emotional aspects of pain. In the S1 cortex, L2/3 neurons project to several cortical areas, including motor cortex (Ferezou et al., 2007), and the S2 (Kamatani et al., 2007). Since S2 projects to the insular cortex (Friedman and Murray, 1986), which has interaction between the ACC (Price, 2000), facilitated outputs from L2/3 excitatory neurons may modulate the ACC neuronal activity via indirect pathways, and the possibility of the direct pathways still exists. The ACC is involved in chronic pain behavior via activation of the descending pain facilitatory system (Calejesan et al., 2000; Xu et al., 2008). Thus, the facilitation of the ACC activity by the S1 inputs via a direct or indirect pathway could enhance chronic pain behavior. Actually, in the present study, inhibition of ACC neuronal activity attenuated the chronic pain behavior (Fig. 4). Thus, the hyperactivity of the L2/3 neurons in the S1 have a critical role in the expression of chronic pain behavior. However, it could not be excluded that the modulation of S1 activity directly manipulates pain behavior, which in turn may alter the ACC activity evoked by sensory stimulation, since neurons in the S1 code intensity and location of pain stimulations (Kenshalo and Isensee, 1983). This study may shed light on the central mechanisms of chronic pain and provide a new efficient therapeutic strategy against chronic pain by focusing on the interaction between the S1 and ACC.
Footnotes
This study was supported by Grants-in-Aid from the Ministry of Education, Culture, Sports, Science and Technology, Japan (to J.N.). We thank M. Yoshitomo and T. Ohba (National Institute for Physiological Sciences) for technical assistance, Dr. A. Miyawaki for providing pCS2-Venus, Dr. K. Ohki and Dr. K. Sohya for technical advice, and Dr. C.S. Yin for comments on the manuscript.
The authors declare no competing financial interests.
References
- Allen CB, Celikel T, Feldman DE. Long-term depression induced by sensory deprivation during cortical map plasticity in vivo. Nat Neurosci. 2003;6:291–299. doi: 10.1038/nn1012. [DOI] [PubMed] [Google Scholar]
- Bushnell MC, Duncan GH, Hofbauer RK, Ha B, Chen JI, Carrier B. Pain perception: is there a role for primary somatosensory cortex? Proc Natl Acad Sci U S A. 1999;96:7705–7709. doi: 10.1073/pnas.96.14.7705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calejesan AA, Kim SJ, Zhuo M. Descending facilitatory modulation of a behavioral nociceptive response by stimulation in the adult rat anterior cingulate cortex. Eur J Pain. 2000;4:83–96. doi: 10.1053/eujp.1999.0158. [DOI] [PubMed] [Google Scholar]
- Chaplan SR, Bach FW, Pogrel JW, Chung JM, Yaksh TL. Quantitative assessment of tactile allodynia in the rat paw. J Neurosci Methods. 1994;53:55–63. doi: 10.1016/0165-0270(94)90144-9. [DOI] [PubMed] [Google Scholar]
- Cheetham CE, Hammond MS, Edwards CE, Finnerty GT. Sensory experience alters cortical connectivity and synaptic function site specifically. J Neurosci. 2007;27:3456–3465. doi: 10.1523/JNEUROSCI.5143-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Endo T, Spenger C, Hao J, Tominaga T, Wiesenfeld-Hallin Z, Olson L, Xu XJ. Functional MRI of the brain detects neuropathic pain in experimental spinal cord injury. Pain. 2008;138:292–300. doi: 10.1016/j.pain.2007.12.017. [DOI] [PubMed] [Google Scholar]
- Eto K, Arimura Y, Nabekura J, Noda M, Ishibashi H. The effect of zinc on glycinergic inhibitory postsynaptic currents in rat spinal dorsal horn neurons. Brain Res. 2007;1161:11–20. doi: 10.1016/j.brainres.2007.05.060. [DOI] [PubMed] [Google Scholar]
- Ferezou I, Haiss F, Gentet LJ, Aronoff R, Weber B, Petersen CC. Spatiotemporal dynamics of cortical sensorimotor integration in behaving mice. Neuron. 2007;56:907–923. doi: 10.1016/j.neuron.2007.10.007. [DOI] [PubMed] [Google Scholar]
- Friedman DP, Murray EA. Thalamic connectivity of the second somatosensory area and neighboring somatosensory fields of the lateral sulcus of the macaque. J Comp Neurol. 1986;252:348–373. doi: 10.1002/cne.902520305. [DOI] [PubMed] [Google Scholar]
- Ismailov I, Kalikulov D, Inoue T, Friedlander MJ. The kinetic profile of intracellular calcium predicts long-term potentiation and long-term depression. J Neurosci. 2004;24:9847–9861. doi: 10.1523/JNEUROSCI.0738-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamatani D, Hishida R, Kudoh M, Shibuki K. Experience-dependent formation of activity propagation patterns at the somatosensory S1 and S2 boundary in rat cortical slices. Neuroimage. 2007;35:47–57. doi: 10.1016/j.neuroimage.2006.08.049. [DOI] [PubMed] [Google Scholar]
- Kameyama K, Sohya K, Ebina T, Fukuda A, Yanagawa Y, Tsumoto T. Difference in binocularity and ocular dominance plasticity between GABAergic and excitatory cortical neurons. J Neurosci. 2010;30:1551–1559. doi: 10.1523/JNEUROSCI.5025-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenshalo DR, Jr, Isensee O. Responses of primate SI cortical neurons to noxious stimuli. J Neurophysiol. 1983;50:1479–1496. doi: 10.1152/jn.1983.50.6.1479. [DOI] [PubMed] [Google Scholar]
- Kerr JN, Greenberg D, Helmchen F. Imaging input and output of neocortical networks in vivo. Proc Natl Acad Sci U S A. 2005;102:14063–14068. doi: 10.1073/pnas.0506029102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SK, Nabekura J. Rapid synaptic remodeling in the adult somatosensory cortex following peripheral nerve injury and its association with neuropathic pain. J Neurosci. 2011;31:5477–5482. doi: 10.1523/JNEUROSCI.0328-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moisset X, Bouhassira D. Brain imaging of neuropathic pain. Neuroimage. 2007;37(Suppl 1):S80–S88. doi: 10.1016/j.neuroimage.2007.03.054. [DOI] [PubMed] [Google Scholar]
- Petrovic P, Ingvar M, Stone-Elander S, Petersson KM, Hansson P. A PET activation study of dynamic mechanical allodynia in patients with mononeuropathy. Pain. 1999;83:459–470. doi: 10.1016/S0304-3959(99)00150-5. [DOI] [PubMed] [Google Scholar]
- Peyron R, García-Larrea L, Grégoire MC, Convers P, Lavenne F, Veyre L, Froment JC, Mauguière F, Michel D, Laurent B. Allodynia after lateral-medullary (Wallenberg) infarct. A PET study. Brain. 1998;121:345–356. doi: 10.1093/brain/121.2.345. [DOI] [PubMed] [Google Scholar]
- Price DD. Psychological and neural mechanisms of the affective dimension of pain. Science. 2000;288:1769–1772. doi: 10.1126/science.288.5472.1769. [DOI] [PubMed] [Google Scholar]
- Rainville P, Duncan GH, Price DD, Carrier B, Bushnell MC. Pain affect encoded in human anterior cingulate but not somatosensory cortex. Science. 1997;277:968–971. doi: 10.1126/science.277.5328.968. [DOI] [PubMed] [Google Scholar]
- Shibuki K, Hishida R, Murakami H, Kudoh M, Kawaguchi T, Watanabe M, Watanabe S, Kouuchi T, Tanaka R. Dynamic imaging of somatosensory cortical activity in the rat visualized by flavoprotein autofluorescence. J Physiol. 2003;549:919–927. doi: 10.1113/jphysiol.2003.040709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urban MO, Gebhart GF. Supraspinal contributions to hyperalgesia. Proc Natl Acad Sci U S A. 1999;96:7687–7692. doi: 10.1073/pnas.96.14.7687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vartiainen N, Kirveskari E, Kallio-Laine K, Kalso E, Forss N. Cortical reorganization in primary somatosensory cortex in patients with unilateral chronic pain. J Pain. 2009;10:854–859. doi: 10.1016/j.jpain.2009.02.006. [DOI] [PubMed] [Google Scholar]
- Vogt BA. Pain and emotion interactions in subregions of the cingulate gyrus. Nat Rev Neurosci. 2005;6:533–544. doi: 10.1038/nrn1704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walcott EC, Langdon RB. Synaptically driven spikes and long-term potentiation in neocortical layer 2/3. Neuroscience. 2002;112:815–826. doi: 10.1016/s0306-4522(02)00131-8. [DOI] [PubMed] [Google Scholar]
- Wang Y, Kakizaki T, Sakagami H, Saito K, Ebihara S, Kato M, Hirabayashi M, Saito Y, Furuya N, Yanagawa Y. Fluorescent labeling of both GABAergic and glycinergic neurons in vesicular GABA transporter (VGAT)-venus transgenic mouse. Neuroscience. 2009;164:1031–1043. doi: 10.1016/j.neuroscience.2009.09.010. [DOI] [PubMed] [Google Scholar]
- Woolf CJ, Salter MW. Neuronal plasticity: increasing the gain in pain. Science. 2000;288:1765–1769. doi: 10.1126/science.288.5472.1765. [DOI] [PubMed] [Google Scholar]
- Xu H, Wu LJ, Wang H, Zhang X, Vadakkan KI, Kim SS, Steenland HW, Zhuo M. Presynaptic and postsynaptic amplifications of neuropathic pain in the anterior cingulate cortex. J Neurosci. 2008;28:7445–7453. doi: 10.1523/JNEUROSCI.1812-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhuo M. A synaptic model for pain: long-term potentiation in the anterior cingulate cortex. Mol Cells. 2007;23:259–271. [PubMed] [Google Scholar]
- Zhuo M. Cortical excitation and chronic pain. Trends Neurosci. 2008;31:199–207. doi: 10.1016/j.tins.2008.01.003. [DOI] [PubMed] [Google Scholar]