

Abstract
Morphine is one of the most potent analgesic drugs. However, the utility of morphine in the management of chronic pain is limited by its rapid development of tolerance. Morphine exerts all of its pharmacological effects via the μ-opioid receptor. In many systems, tolerance is associated with phosphorylation and desensitization of G-protein-coupled receptors (GPCRs). In case of the μ-opioid receptor, phosphorylation occurs in an agonist-selective manner. High-efficacy agonists such as [d-Ala2-MePhe4-Gly-ol]enkephalin (DAMGO), fentanyl, or etonitazene stimulate the phosphorylation of both C-terminal threonine 370 (T370) and serine 375 (S375). In contrast, morphine promotes the phosphorylation of S375 but fails to stimulate T370 phosphorylation. Here, we have assessed the contribution of S375 phosphorylation to the development of antinociceptive tolerance to high- and low-efficacy μ agonists in vivo. We show that S375 phosphorylation of the μ-opioid receptor occurs in intact mouse brain in a dose-dependent manner after administration of morphine, fentanyl, or etonitazene. In knock-in mice expressing the phosphorylation-deficient S375A mutant of the μ-opioid receptor, morphine and fentanyl exhibited greater dose-dependent antinociceptive responses than in wild-type mice. However, acute and chronic tolerance to morphine was retained in S375A mutant mice. In contrast, antinociceptive tolerance after repeated subcutaneous application of etonitazene or repeated intracerebroventricular application of DAMGO was diminished. Thus, tolerance to μ agonists with different efficacies develops through distinct pathways. Whereas tolerance induced by DAMGO or etonitazene requires agonist-driven phosphorylation of S375, the development and maintenance of antinociceptive tolerance to morphine occurs independent of S375 phosphorylation.
Introduction
The development of analgesic tolerance to morphine occurs on continued use of the drug such that the amount of drug required to elicit pain relief must be increased to compensate for diminished responsiveness. However, the molecular mechanisms of morphine tolerance are far from being understood (Nestler, 1996; Koch et al., 2005; Bailey et al., 2006; Martini and Whistler, 2007). For many clinically used drugs, tolerance has been associated with phosphorylation, desensitization and downregulation of their G-protein-coupled receptors (GPCRs). However, morphine is unique in that it activates the μ-opioid receptor without causing its rapid endocytosis (Keith et al., 1996; Schulz et al., 2004). We have recently generated phosphosite-specific antibodies for the C-terminal residues threonine 370 (T370) and serine 375 (S375), which enabled us to selectively detect either the T370-phosphorylated or the S375-phosphorylated form of the μ-opioid receptor (Doll et al., 2011). We found that full agonists such as [d-Ala2-MePhe4-Gly-ol]enkephalin (DAMGO), fentanyl, or etonitazene stimulate the phosphorylation of both T370 and S375 (Doll et al., 2011). S375 is the primary site of phosphorylation that contributes to desensitization, β-arrestin binding, and internalization of the μ-opioid receptor (El Kouhen et al., 2001; Schulz et al., 2004; McPherson et al., 2010; Kelly, 2011). In contrast, morphine promoted the phosphorylation of S375 but failed to stimulate T370 phosphorylation (Schulz et al., 2004; Doll et al., 2011). Here, we show that the morphine-mediated S375 phosphorylation also occurs in vivo shortly after administration of the drug. To directly examine the contribution of S375 phosphorylation to the development of tolerance in vivo, we generated knock-in mice expressing the phosphorylation-deficient S375A mutant of the μ-opioid receptor, and assessed opioid-mediated responses in intact animals.
Materials and Methods
Animals.
Knock-in mice expressing the S375A mutant of the μ-opioid receptor (MORS375A/S375A) were generated at Ozgene. According to Mouse Genome Informatics (MGI, The Jackson Laboratory), the official nomenclature for these mice is Oprm1tm1Shlz with accession number MGI:5000465. In this μ-opioid receptor mutant, the coding sequence for serine 375 (TCC) has been replaced using PCR with the coding sequence of alanine (GCC) (Fig. 1). Bruce 4 C57BL/6J ES cells were used to generate MORS375A/S375A mice. Mice were genotyped by PCR of genomic tail DNA using the following primers: 5′-GGC TAA TAC AGT GGA TCG AAC TAA C-3′ and 5′-TAA CTG TCT TGG CTA CAT TCC TTT C-3′. MORS375A/S375A and MOR+/+ were generated by heterozygous breeding. In behavioral experiments, male mice aged 12–16 weeks between 25 and 30 g were used. Animals were housed in a 12 h light–dark cycle and had ad libitum access to food and water. All animal experiments were performed in accordance with the Thuringian state authorities and complied with EC regulations for the care and use of laboratory animals.
Figure 1.

Generation of MORS375A/S375A mice. A, Schematic representation of targeting strategy. A genomic fragment containing the μ-opioid receptor sequence, including exons 2 and 3, was modified to contain the S375A mutation of the μ-opioid receptor. A cassette containing resistance to neomycin (PKG-Neo) and flanked by FRT sites was inserted in the intron downstream of exon 3 for selection of ES cell clones. Exon 2 and 3 are also flanked with loxP sites. B, Detection of homologous recombination in ES cells by Southern blot using the 3′ probe. C, Verification of deletion of PKG-Neo cassette by Southern blot using the 3′ probe after FlpE-mediated recombination. D, Representative example of genotyping by PCR. E, Bands in D were excised and sequenced to confirm presence of S375A mutation.
Drugs.
Morphine-HCl and naloxone-HCl were purchased from Merck, and fentanyl-citrate from Rotexmedica. DAMGO was obtained from Bachem, and etonitazene-HCl was from Novartis. Drugs were dissolved in physiological saline and injected subcutaneously in a volume of 10 ml/kg or intracerebroventricularly in a volume of 2.5 μl.
Western blot and immunoprecipitation.
Brains from μ-opioid receptor-deficient mice (MOR−/−) (provided by Dr. H. Loh, University of Minnesota, Minneapolis, MN), wild-type mice (MOR+/+), and MORS375A/S375A were quickly dissected after the indicated treatment. The cerebellum, which is devoid of μ-opioid receptors, was removed, and the remaining brain samples were immediately frozen in liquid nitrogen. In all experiments, entire brains except cerebellum were used. Samples were then transferred to ice-cold detergent buffer (50 mm Tris-HCl, pH 7.4, 150 mm NaCl, 5 mm EDTA, 10 mm NaF, 10 mm disodium pyrophosphate, 1% Nonidet P-40, 0.5% sodium deoxycholate, 0.1% SDS containing protease and phosphatase inhibitors), homogenized, and centrifuged at 16,000 × g for 30 min at 4°C. The supernatant was then immunoprecipitated with the phosphorylation-independent rabbit monoclonal anti-MOR antibody {UMB-3} bound to protein A-agarose beads for 2 h at 4°C (Lupp et al., 2011). Proteins were eluted from the beads with SDS-sample buffer for 20 min at 40°C. Samples were resolved on 8% SDS-polyacrylamide gels, and after electroblotting, membranes were incubated with guinea pig polyclonal anti-pS375 antibody {GP2} at a concentration of 0.1 μg/ml followed by detection using an enhanced chemiluminescence detection system (GE Healthcare). Blots were subsequently stripped and reprobed with phosphorylation independent guinea pig polyclonal anti-MOR antibody {GP6} at a concentration of 0.1 μg/ml to confirm equal loading of the gels. The rabbit monoclonal antibody UMB-3 was obtained from Epitomics. The phosphorylation-independent antibodies {UMB-3} and {GP6} were generated against the C-terminal tail of the mouse μ-opioid receptor. The identity of the peptide used for immunizations of the rabbits was LENLEAETAPLP, which corresponds to residues 386–398 of the mouse μ-opioid receptor. These antibodies have been extensively characterized previously (Schulz et al., 2004; Doll et al., 2011a,b; Lupp et al., 2011). The phosphosite-specific antibody for the S375-phosphorylated form of the μ-opioid receptor {GP2} was generated against the following sequence that contained a phosphorylated serine residue: REHP(pS)TANTV. This sequence corresponds to amino acids 371–380 of the mouse μ-opioid receptor. The anti-pS375 guinea pig polyclonal anti-MOR antibody {GP2} has been generated and characterized in an identical manner to that previously described for the anti-pS375 rabbit polyclonal anti-MOR antibody {2493} (Doll et al., 2011b). When indicated, lysates were sequentially incubated with WGA (wheat germ lectin agarose) beads to enrich glycoproteins after UMB-3 immunoprecipitation. These samples were then probed with an anti-transferrin receptor (TFR) antibody (Zymed) to confirm that equal amounts of lysate of each sample were used for UMB-3 immunoprecipitation.
Immunohistochemistry.
Animals were killed under ether anesthesia, and brains and spinal cords were removed, fixed in 10% buffered formaldehyde, and embedded in paraffin for immunohistochemistry. Five-micrometer sections from brain and spinal cord were cut and floated onto positively charged slides. Immunostaining was performed by an indirect peroxidase labeling method as described previously (Lupp et al., 2011). Briefly, sections were dewaxed, microwaved in 10 mm citric acid, pH 6.0, for 16 min at 600 W, and then incubated with the rabbit monoclonal anti-MOR antibody UMB-3 (dilution 1:10) overnight at 4°C. Detection of the primary antibody was performed using a biotinylated anti-rabbit IgG followed by incubation with peroxidase-conjugated avidin (Vector ABC “Elite” kit, Vector). Binding of the primary antibody was visualized using 3-amino-9-ethylcarbazole (AEC) in acetate buffer (BioGenex).
Analgesia—hot plate test.
Basal pain responses were determined at hot plate temperatures of 50°C, 52°C, 54°C, and 56°C on consecutive test days. In each antinociception assay, nociceptive latencies were assessed as the response time to the hot plate (56°C). The “response” was defined by the animal either licking or flicking his hindpaws. To avoid tissue damage, an artificial maximum time for exposure was imposed, which prevented the animal from contact with the plate for >30 s. Data are reported as the percentage maximum possible effect (% MPE), which was determined by accounting for each individual's basal response as well as the imposed maximum cutoff time using the following calculation: 100 × [(drug response time − basal response time)/(30 s − basal response time)] = % MPE. In morphine dose–response studies, withdrawal latencies were measured 30 min after a first dose of morphine (1 mg/kg, s.c.); at this time point, animals were injected with morphine (4 mg/kg, s.c.) for a cumulative dose of 5 mg/kg. Antinociception was again assessed after 30 min, and mice were again injected with morphine (5 mg/kg, s.c.) to yield a final cumulative dose of 10 mg/kg. After 30 min, antinociception was assessed once more. In fentanyl dose–response studies, withdrawal latencies were measured 30 min after a first dose of fentanyl (0.05 mg/kg, s.c.); at this time point, animals were injected with fentanyl (0.05 mg/kg, s.c.) for a cumulative dose of 0.1 mg/kg. Antinociception was again assessed after 30 min, and mice were again injected with fentanyl (0.2 mg/kg, s.c.) to yield a final cumulative dose of 0.3 mg/kg. After 30 min, antinociception was assessed once more. Acute antinociceptive tolerance was induced by treating MORS375A/S375A mice and their wild-type littermates (MOR+/+) with saline or morphine (100 mg/kg, s.c.). After 12 h, all mice were treated with a challenge dose of morphine (10 mg/kg, s.c.), and hot plate latencies (Ugo Basile) were recorded. Twenty-four hours after morphine treatment, both genotypes had returned to their basal nociceptive latencies.
Analgesia—electrical tail root stimulation test.
A 0.1-mm-thick stainless steel wire was subcutaneously drawn through the root of the tail under pentobarbital anesthesia (40 mg/kg, i.p.) 3 d before the start of the experiments. The second electrode was the plate of the restraining tube (diameter = 3 cm, length = 8 cm). The plate of the tube and the subcutaneously implanted electrode were connected with a stimulating current apparatus (TuR RS12). The current intensity (rectangular pulses, 50 Hz, 50 ms impulse width) was continuously increased until the animal vocalized. The maximum stimulation was set at 300 mA (impulse peak). After vocalization or after reaching the impulse peak, the current was immediately switched off. On test days, pain thresholds were determined as mean of three electrical stimulations performed in 1 min intervals before and 30 min after drug application. Data are reported as the maximum possible effect (% MPE), which was determined by accounting for each individual's basal response as well as the imposed maximum stimulation using the following calculation: 100 × [(drug response current − basal response current)/(300 mA − basal response current)] = % MPE. Chronic antinociceptive tolerance was induced by daily subcutaneous treatment with 30 mg/kg morphine or 30 μg/kg etonitazene for 12 consecutive days. Antinociceptive activity was evaluated on days 1, 3, 5, 8, 10, and 12, 30 min after application of challenge doses of 15 mg/kg morphine or 15 μg/kg etonitazene. The rest of the daily dose was injected after the behavioral test. Chronic antinociceptive tolerance was induced by daily intracerebroventricular application of 20 nmol of morphine, 10 nmol of etonitazene, or 10 nmol of DAMGO in a volume of 2.5 μl. One week before the experiments, microcannulas for intracerebroventricular application of the substances were implanted under pentobarbital anesthesia into the right lateral ventricle [coordinates: AP = −0.2 mm, lateral = 0.2 mm (relative to bregma), vertical = 2.5 mm]. Microcannulas were fixed with a socket of acrylic dental cement. Antinociceptive activity was evaluated on days 1, 4, 8, 11, and 16, 30 min after drug treatment.
Results
μ-Opioid receptors are upregulated during chronic morphine treatment
To facilitate detection of endogenous μ-opioid receptors in mouse brain, we have extensively characterized the rabbit monoclonal anti-MOR antibody {UMB-3} (Doll et al., 2011b; Lupp et al., 2011). When crude brain homogenates from MOR+/+ mice were immunoprecipitated with UMB-3, the guinea pig anti-MOR antibody {GP6} detected a broad band migrating at Mr 70,000–80,000 in the subsequent immunoblot (Fig. 2A). In contrast, no such band was detectable in brain homogenates prepared from MOR−/− mice under otherwise identical conditions (Fig. 2A). When MOR+/+ mice were treated for 9 d with escalating doses of morphine, μ-opioid receptor expression was strongly upregulated as compared to saline-treated animals (Fig. 2B,C). In contrast, such an upregulation was not seen in animals that had been treated for 9 d with escalating doses of etonitazene (Fig. 2B,C).
Figure 2.

Upregulation of μ-opioid receptor expression during chronic morphine treatment. A, Immunoprecipitation of μ-opioid receptors from cell-lysate free samples (no lysate) and brain homogenates prepared from MOR+/+ or MOR−/− mice. Homogenates were prepared from entire brain after removal of the cerebellum. μ-Opioid receptors were immunoprecipitated with UMB-3 bound to protein A agarose beads. Samples were separated on 8% SDS-polyacrylamide gels and blotted. Blots were incubated with guinea pig anti-MOR antiserum {GP6}. Incubations were performed in the absence (−) or presence (+) of the peptide antigen. Note that UMB-3 selectively detects μ-opioid receptors and does not cross-react with other proteins present in tissue extracts prepared from MOR−/− mice. B, MOR+/+ mice were treated for 9 d with saline (lanes 1–4), escalating doses of morphine (10–90 mg/kg, s.c.) (lanes 5–9), or escalating doses of etonitazene (10–90 μg/kg, s.c.) (lanes 10–13). One hour after the last injection, brains were dissected. Homogenates were prepared from entire brain after removal of the cerebellum. μ-Opioid receptors were immunoprecipitated with UMB-3 and immunoblotted with guinea pig anti-MOR antiserum {GP6} (top). After UMB-3 immunoprecipitation, lysates were subsequently incubated with wheat germ lectin agarose beads to enrich glycoproteins. These samples were then probed with an anti-TFR antibody to confirm that equal amounts of lysate were used for UMB-3 immunoprecipitation (bottom). Each lane represents one animal. C, Blots were quantified by densitometry and expressed as percentage of μ-opioid receptor expression in saline-treated animals. Data correspond to the mean ± SEM. Results were analyzed by one-way ANOVA followed by the Bonferroni post hoc test (*p < 0.05). Note that μ-opioid receptor expression is upregulated during chronic morphine but not during chronic etonitazene. The positions of the molecular mass markers are indicated on the left (in kilodaltons).
Dose-dependent S375 phosphorylation in vivo
To facilitate detection of S375-phosphorylated μ-opioid receptors in UMB-3 immunoprecipitates from mouse brain, we generated a guinea pig polyclonal anti-pS375 antibody {GP2}. When brain homogenates from MOR+/+ mice were immunoprecipitated with UMB-3, the guinea pig anti-pS375 antibody {GP2} detected a broad band migrating at Mr 70,000–80,000 only in morphine- or etonitazene-treated but not in saline-treated animals (Fig. 3A). In contrast, no such band was detectable in brain homogenates prepared from MORS375A/S375A mice after identical drug treatment (Fig. 3A). These findings clearly show that S375 phosphorylation of the μ-opioid receptor occurs also in mouse brain after administration of morphine. MORS375A/S375A mice exhibited similar densities of μ-opioid receptors with a similar cellular and subcellular distribution in the spinal cord, and all other investigated brain regions, including dorsal root ganglia, thalamus, striatum, and cortex, were devoid of any detectable anatomical abnormality compared to MOR+/+ mice (Fig. 3A,B). When MOR+/+ mice were treated with increasing doses of morphine (2–100 mg/kg, s.c.) or fentanyl (0.02–1 mg/kg, s.c.), a dose-dependent increase in S375 phosphorylation was observed (Fig. 4A). S375 phosphorylation of the μ-opioid receptor was completely blocked by coadministration of naloxone (Fig. 4A). The level of S375 phosphorylation correlated well to the dose-dependent antinociceptive responses evoked by a single injection of morphine or fentanyl in MOR+/+ mice, suggesting that S375 phosphorylation occurs at pharmacologically relevant doses (Fig. 4A,B).
Figure 3.

Detection of agonist-induced S375 phosphorylation in vivo. A, MOR+/+ and MORS375A/S375A mice were treated with saline, morphine (30 mg/kg, s.c.), or etonitazene (30 μg/kg, s.c.). After 30 min, brains were dissected. Homogenates were prepared from entire brain after removal of the cerebellum. μ-Opioid receptors were immunoprecipitated with UMB-3 and immunoblotted with guinea pig anti-pS375 antibody {GP2} (top). Blots were stripped and reprobed with the phosphorylation-independent guinea pig anti-MOR antibody {GP6} to confirm equal loading of the gel (bottom). Note that both morphine and etonitazene stimulated S375 phosphorylation of the μ-opioid receptor. No such band was detectable in saline-treated MOR+/+ or in MORS375A/S375A mice. The positions of the molecular mass markers are indicated on the left (in kilodaltons). B, Spinal cords from MOR−/−, MOR+/+, and MORS375A/S375A mice were dissected, fixed in formalin, and embedded in paraffin. Sections were dewaxed, microwaved in citric acid, and incubated with anti-MOR antibody {UMB-3). Specimens were then sequentially treated with biotinylated anti-rabbit IgG and peroxidase-conjugated avidin and developed in AEC. Note that μ-opioid receptors have similar density and distribution in MOR+/+ and in MORS375A/S375A mice. Scale bar: B, 200 μm.
Figure 4.

Greater dose-dependent antinociceptive responses to morphine and fentanyl in MORS375A/S375A mice. A, MOR+/+ mice were treated with the indicated doses of morphine or fentanyl. When indicated the μ-opioid receptor antagonist naloxone was administered immediately before drug application. After 30 min, brains were dissected. Homogenates were prepared from entire brain after removal of the cerebellum. μ-Opioid receptors were immunoprecipitated with UMB-3 and immunoblotted with guinea pig anti-pS375 antibody {GP2} (top). Blots were stripped and reprobed with the phosphorylation-independent guinea pig anti-MOR antibody {GP6} to confirm equal loading of the gel (bottom). Note that S375 phosphorylation was dose dependent after both morphine and fentanyl and completely blocked by naloxone. The positions of molecular mass markers are indicated on the left (in kilodaltons). B, The degree of antinociception was determined by measuring the latency of hot-plate responses at 56°C. In morphine dose–response studies, withdrawal latencies were measured 30 min after a first dose of morphine (1 mg/kg, s.c.); at this time point, animals were injected with morphine (4 mg/kg, s.c.) for a cumulative dose of 5 mg/kg. Antinociception was again assessed after 30 min, and mice were again injected with morphine (5 mg/kg, s.c.) to yield a final cumulative dose of 10 mg/kg. After 30 min, antinociception was assessed once more. In fentanyl dose–response studies, withdrawal latencies were measured 30 min after a first dose of fentanyl (0.05 mg/kg, s.c.); at this time point, animals were injected with fentanyl (0.05 mg/kg, s.c.) for a cumulative dose of 0.1 mg/kg. Antinociception was again assessed after 30 min, and mice were again injected with fentanyl (0.2 mg/kg, s.c.) to yield a final cumulative dose of 0.3 mg/kg. After 30 min, antinociception was assessed once more. Response was defined by the animal either licking or flicking his hindpaws. To avoid tissue damage, a maximum time for exposure was imposed, which prevented the animal from contact with the plate for longer than 30 s. Data are reported as % MPE. Data are presented as the means ± SEM from MOR+/+mice (morphine: n = 7, fentanyl: n = 7) and MORS375A/S375A mice (morphine: n = 8, fentanyl: n = 8). Differences between genotypes were analyzed by two-way ANOVA followed by the Bonferroni post hoc test (*p < 0.05).
Enhanced acute antinociceptive responses in MORS375A/S375A mice
We then compared acute antinociceptive responses in MORS375A/S375A and MOR+/+ mice. Initial experiments revealed that MORS375A/S375A and MOR+/+ exhibited similar basal pain responses as determined using the hot plate test (data not shown). Next, opioid-induced antinociception was evaluated after administration of increasing doses of morphine (1–10 mg/kg, s.c.) or fentanyl (0.05–0.3 mg/kg, s.c.). Under these conditions, it became apparent that knock-in mice expressing the phosphorylation-deficient S375A mutant of the μ-opioid receptor exhibited greater dose-dependent antinociceptive responses to morphine and fentanyl than their wild-type littermates (Fig. 4B). These results suggest that S375 phosphorylation is required for acute desensitization of the μ-opioid receptor after exposure to both morphine and fentanyl.
Acute tolerance to morphine is retained in MORS375A/S375A mice
We then examined whether MORS375A/S375A mice would develop acute tolerance to morphine. To examine this, we injected a “high” dose of morphine (100 mg/kg, s.c.) and evaluated the antinociceptive effect of a “normal” dose of morphine (10 mg/kg, s.c.) 12 h later. When a “high” dose of morphine was injected, we observed a rapid and transient S375 phosphorylation (Fig. 5A). As depicted in Figure 5B, morphine pretreatment resulted in a similar robust acute tolerance in both MORS375A/S375A and MOR+/+ mice. When comparing the results from Figure 5, A and B, it becomes apparent that S375 phosphorylation declines within 8 h, whereas acute tolerance is still detectable 12 h after morphine application.
Figure 5.

Acute tolerance to morphine is retained in MORS375A/S375A mice. A, MOR+/+ mice received an injection of morphine (100 mg/kg, s.c.). Mice were killed after the indicated time intervals, and brains were dissected. Homogenates were prepared from entire brain after removal of the cerebellum. μ-Opioid receptors were immunoprecipitated with UMB-3 and immunoblotted with guinea pig anti-pS375 antibody {GP2} (top). Blots were stripped and reprobed with the phosphorylation-independent guinea pig anti-MOR antibody {GP6} to confirm equal loading of the gel (bottom). Note that S375 phosphorylation occurs shortly after morphine administration and declines after 8 h. The positions of the molecular mass markers are indicated on the left (in kilodaltons). B, Acute tolerance in MOR+/+ and MORS375A/S375A mice was determined after injection of saline or morphine (100 mg/kg, s.c.). After 12 h, all animals were challenged with morphine (10 mg/kg, s.c.), and hot plate response latencies were recorded at 56°C 30 min after morphine injection. Data are presented as the mean ± SEM from MOR+/+mice (saline: n = 7, morphine: n = 7) and MORS375A/S375A mice (saline: n = 7, morphine: n = 7). Differences between saline- and morphine-pretreated groups were analyzed by Student's t test (*p < 0.05).
Chronic tolerance to high-efficacy agonists but not to morphine is diminished in MORS375A/S375A mice
Analgesic tolerance typically develops over the course of repeated administration of opioids during the treatment of chronic pain. Thus, we evaluated the development of tolerance after daily subcutaneous administration of equieffective doses of morphine (30 mg/kg) or etonitazene (30 μg/kg) over 12 d. Unlike fentanyl, etonitazene has a similar duration of action as morphine when applied subcutaneously at equieffective doses. Consequently, etonitazene was used instead of fentanyl for chronic experiments. Given that a decrease in hot plate latency is known to occur with repeated testing, opioid-induced antinociception was evaluated by the electrical tail root stimulation test in addition to the hot plate test (Grecksch et al., 2006). Under these conditions, analgesic tolerance to morphine developed rapidly in both MORS375A/S375A and MOR+/+ mice (Fig. 6A). Similar to that previously observed in rats (Grecksch et al., 2006), tolerance to etonitazene developed at a slower time course in MOR+/+ mice than tolerance to morphine. Nevertheless, the development of antinociceptive tolerance to etonitazene in MORS375A/S375A mice was significantly reduced (Fig. 6B). DAMGO is a prototypical highly selective agonist often used to study μ-opioid receptor regulation. We therefore evaluated the development of tolerance after daily intracerebroventricular administration of morphine (20 nmol), etonitazene (10 nmol), or DAMGO (10 nmol) over 16 d. Again, analgesic tolerance to morphine developed rapidly in both MORS375A/S375A and MOR+/+ mice (Fig. 7A), whereas the development of antinociceptive tolerance to intracerebroventricular administration of etonitazene in MORS375A/S375A mice was significantly reduced (Fig. 7B). Robust analgesic tolerance also developed in MOR+/+ mice after repeated intracerebroventricular administration of DAMGO (Fig. 7C). In contrast, MORS375A/S375A mice did not develop tolerance to the antinociceptive effects of DAMGO during the 16 d treatment period. Similar results were obtained with both hot plate and electrical tail root stimulation test. These findings suggest that tolerance induced by DAMGO or etonitazene requires agonist-driven phosphorylation of S375, whereas the development of antinociceptive tolerance to morphine occurs independent of S375 phosphorylation.
Figure 6.

Chronic tolerance to subcutaneous etonitazene but not to morphine is diminished in MORS375A/S375A mice. A, B, chronic tolerance in MOR+/+ and MORS375A/S375A mice was determined after daily subcutaneous injection of morphine (30 mg/kg) or etonitazene (30 μg/kg) for 12 d. On days 1, 3, 5, 8, 10, and 12, antinociceptive response was determined 30 min after application challenge doses of 15 mg/kg morphine or 15 μg/kg etonitazene using electrical tail root stimulation. The rest of the daily dose was injected after the behavioral test. The current intensity was continuously increased until the animal vocalized. To avoid tissue damage, a maximum stimulation of 300 mA was imposed. After vocalization or after reaching the impulse peak, the current was immediately switched off. On test days, pain thresholds were determined as mean of three electrical stimulations performed in intervals of exactly 1 min before and 30 min after drug application. Data are reported as % MPE. Data are presented as the means ± SEM from MOR+/+ mice (morphine: n = 12, etonitazene: n = 10) and MORS375A/S375A mice (morphine: n = 16, etonitazene: n = 16). Results were analyzed by two-way ANOVA with repeated measures (time × treatment: p < 0.01) followed by the Bonferroni post hoc test (A, status effect by morphine treatment: not significant; B, status effect by etonitazene treatment: *p < 0.011).
Figure 7.
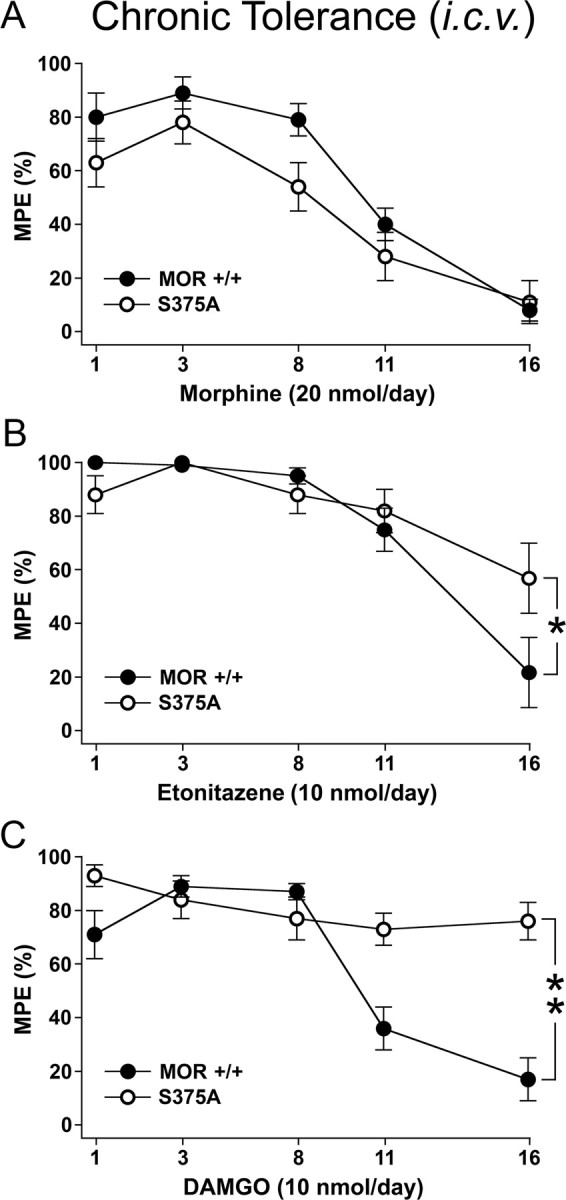
Chronic tolerance to high-efficacy agonists but not to morphine is diminished in MORS375A/S375A mice. A–C, Chronic tolerance in MOR+/+ and MORS375A/S375A mice was determined after daily intracerebroventricular application of 20 nmol of morphine, 10 nmol of etonitazene, or 10 nmol of DAMGO in a volume of 2.5 μl for 16 d. On days 1, 3, 8, 11, and 16, antinociceptive response was determined 30 min after drug application using electrical tail root stimulation. The current intensity was continuously increased until the animal vocalized. To avoid tissue damage, a maximum stimulation of 300 mA was imposed. After vocalization or after reaching the impulse peak, the current was immediately switched off. On test days, pain thresholds were determined as mean of three electrical stimulations performed in intervals of exactly 1 min before and 30 min after drug application. Data are reported as % MPE. Data are presented as the means ± SEM from MOR+/+mice (morphine: n = 12, entonitazene: n = 12, DAMGO: n = 12) and MORS375A/S375A mice (morphine: n = 13, entonitazene: n = 13, DAMGO: n = 14). Results were analyzed by two-way ANOVA with repeated measures (time × treatment: p < 0.001; treatment × time: p < 0.02; genotype × treatment: p < 0.003) followed by the Bonferroni post hoc test (morphine × DAMGO and etonitazene, p < 0.001; A, status effect by morphine treatment: not significant; B, status effect by etonitazene treatment: *p < 0.04; C, status effect by DAMGO treatment, **p < 0.001).
Discussion
For many clinically used drugs such as β2 agonists, GnRH analogs, or oxytocin, tolerance has been associated with phosphorylation, internalization, and ultimately downregulation of their targeted GPCRs. Morphine is unique in that it is a poor inducer of μ-opioid receptor internalization, but a potent inducer of cellular tolerance in vivo. In the present study, we clearly show that chronic morphine produces a significant upregulation rather than downregulation of μ-opioid receptors in vivo under conditions that induce profound cellular tolerance. This is in contrast to the majority of previous binding studies, which detected no change in functional μ-opioid receptor binding sites in morphine-tolerant mice (Law et al., 1983; Werling et al., 1989). Given that morphine blocks receptor internalization, it is possible that in the continuous presence of morphine nonfunctional μ-opioid receptors accumulate at the cell surface. Thus, changes in total μ-opioid receptor protein may be detectable only by immunoprecipitation and not by receptor binding.
Recently, we have used novel phosphosite-specific antibodies in combination with siRNA knock down screening to identify the G-protein-coupled receptor kinases (GRKs) involved in agonist-dependent phosphorylation of the μ-opioid receptor (C. Doll, F. Poll, and S. Schulz, unpublished work). We found that the morphine-activated μ-opioid receptor acquires a conformation that is an efficient substrate for phosphorylation by GRK5 but a poor substrate for phosphorylation by GRK2/3 (C. Doll, F. Poll, and S. Schulz, unpublished work). GRK5 phosphorylates μ-opioid receptors selectively on S375, which is not sufficient to facilitate receptor sequestration. Conversely, μ-opioid receptors activated by high-efficacy agonists acquire a conformation that is an efficient substrate for phosphorylation by GRK2/3 but a poor substrate for phosphorylation by GRK5 (C. Doll, F. Poll, and S. Schulz, unpublished work). GRK2/3 phosphorylate μ-opioid receptors at a number of C-terminal phosphate acceptor sites, including T370 and S375, which in turn facilitates a robust receptor endocytosis (C. Doll, F. Poll, and S. Schulz, unpublished work). We have also shown that μ-opioid receptor phosphorylation is a hierarchical process (Doll et al., 2011). Upon exposure to DAMGO, S375 is phosphorylated more rapidly than T370. T370 phosphorylation and internalization is strongly reduced in the S375A mutant, suggesting that S375 is the primary site of phosphorylation and that phosphorylation of T370 is in part dependent on the prior phosphorylation of S375 (Schulz et al., 2004; Doll et al., 2011).
Here, we clearly show that S375 phosphorylation of the μ-opioid receptor occurs in vivo in mouse brain shortly after opioid administration. S375 phosphorylation was dose dependent and occurred at pharmacologically relevant doses. We hypothesized that S375 phosphorylation would be an initial event in opioid-mediated μ-opioid receptor desensitization. Thus, to directly assess the contribution of S375 phosphorylation to the development of morphine antinociceptive tolerance, we generated a novel knock-in mouse expressing the S375A mutant of the μ-opioid receptor. In fact, S375A mutant mice exhibit greater dose-dependent antinociceptive responses to morphine and fentanyl than their wild-type littermates, supporting the idea that S375 phosphorylation is involved in acute μ-opioid receptor desensitization. In contrast to our initial hypothesis, however, we found that acute and chronic tolerance to morphine was retained in S375A mutant mice. Nevertheless, we also found that antinociceptive tolerance after repeated subcutaneous application of etonitazene or repeated intracerebroventricular application of DAMGO or etonitazene was diminished. The phenotype we observed in S375A knock-in mice resembles closely that reported for GRK3 knock-out mice, in which the development of tolerance to the high-efficacy agonist fentanyl is strongly reduced, whereas acute and chronic tolerance to morphine is retained (Terman et al., 2004). Similar, intracerebroventricular injection of a small-molecule GRK2 inhibitor reversed tolerance to DAMGO but not to morphine (Hull et al., 2010). Conversely, mice lacking β-arrestin-2 show diminished antinociceptive tolerance to morphine only but not to high-efficacy agonists such as fentanyl or methadone (Bohn et al., 1999, 2000; Dang et al., 2011; Raehal and Bohn, 2011). Moreover, inhibition of PKC reversed tolerance to morphine but not to DAMGO (Bailey et al., 2006, 2009; Hull et al., 2010).
Together, tolerance to μ agonists with different efficacy develops through distinct pathways. Tolerance induced by high-efficacy agonists such as DAMGO, fentanyl, or etonitazene requires prototypical GRK2/3-mediated phosphorylation of S375. In contrast, the development and maintenance of antinociceptive tolerance to morphine occurs independent of S375 phosphorylation.
Footnotes
This work was supported by the Deutsche Forschungsgemeinschaft (SCHU924/10-1), European Regional Development Fund, and the Doktor Robert Pfleger-Stiftung. We thank Heidrun Guder, Heike Stadler, and Gabriele Schulze for excellent technical assistance.
The authors declare no competing financial interests.
References
- Bailey CP, Smith FL, Kelly E, Dewey WL, Henderson G. How important is protein kinase C in mu-opioid receptor desensitization and morphine tolerance? Trends Pharmacol Sci. 2006;27:558–565. doi: 10.1016/j.tips.2006.09.006. [DOI] [PubMed] [Google Scholar]
- Bailey CP, Oldfield S, Llorente J, Caunt CJ, Teschemacher AG, Roberts L, McArdle CA, Smith FL, Dewey WL, Kelly E, Henderson G. Involvement of PKC alpha and G-protein-coupled receptor kinase 2 in agonist-selective desensitization of mu-opioid receptors in mature brain neurons. Br J Pharmacol. 2009;158:157–164. doi: 10.1111/j.1476-5381.2009.00140.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohn LM, Lefkowitz RJ, Gainetdinov RR, Peppel K, Caron MG, Lin FT. Enhanced morphine analgesia in mice lacking beta-arrestin 2. Science. 1999;286:2495–2498. doi: 10.1126/science.286.5449.2495. [DOI] [PubMed] [Google Scholar]
- Bohn LM, Gainetdinov RR, Lin FT, Lefkowitz RJ, Caron MG. Mu-opioid receptor desensitization by beta-arrestin-2 determines morphine tolerance but not dependence. Nature. 2000;408:720–723. doi: 10.1038/35047086. [DOI] [PubMed] [Google Scholar]
- Dang VC, Chieng B, Azriel Y, Christie MJ. Cellular morphine tolerance produced by βarrestin-2-dependent impairment of μ-opioid receptor resensitization. J Neurosci. 2011;31:7122–7130. doi: 10.1523/JNEUROSCI.5999-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doll C, Konietzko J, Pöll F, Koch T, Höllt V, Schulz S. Agonist-selective patterns of micro-opioid receptor phosphorylation revealed by phosphosite-specific antibodies. Br J Pharmacol. 2011b doi: 10.1111/j.1476-5381.2011.01382.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Kouhen R, Burd AL, Erickson-Herbrandson LJ, Chang CY, Law PY, Loh HH. Phosphorylation of Ser363, Thr370, and Ser375 residues within the carboxyl tail differentially regulates mu-opioid receptor internalization. J Biol Chem. 2001;276:12774–12780. doi: 10.1074/jbc.M009571200. [DOI] [PubMed] [Google Scholar]
- Grecksch G, Bartzsch K, Widera A, Becker A, Höllt V, Koch T. Development of tolerance and sensitization to different opioid agonists in rats. Psychopharmacology (Berl) 2006;186:177–184. doi: 10.1007/s00213-006-0365-8. [DOI] [PubMed] [Google Scholar]
- Hull LC, Llorente J, Gabra BH, Smith FL, Kelly E, Bailey C, Henderson G, Dewey WL. The effect of protein kinase C and G protein-coupled receptor kinase inhibition on tolerance induced by mu-opioid agonists of different efficacy. J Pharmacol Exp Ther. 2010;332:1127–1135. doi: 10.1124/jpet.109.161455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keith DE, Murray SR, Zaki PA, Chu PC, Lissin DV, Kang L, Evans CJ, von Zastrow M. Morphine activates opioid receptors without causing their rapid internalization. J Biol Chem. 1996;271:19021–19024. doi: 10.1074/jbc.271.32.19021. [DOI] [PubMed] [Google Scholar]
- Kelly E. The subtleties of micro-opioid receptor phosphorylation. Br J Pharmacol. 2011 doi: 10.1111/j.1476-5381.2011.01387.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch T, Widera A, Bartzsch K, Schulz S, Brandenburg LO, Wundrack N, Beyer A, Grecksch G, Höllt V. Receptor endocytosis counteracts the development of opioid tolerance. Mol Pharmacol. 2005;67:280–287. doi: 10.1124/mol.104.004994. [DOI] [PubMed] [Google Scholar]
- Law PY, Hom DS, Loh HH. Opiate receptor down-regulation and desensitization in neuroblastoma X glioma NG108–15 hybrid cells are two separate cellular adaptation processes. Mol Pharmacol. 1983;24:413–424. [PubMed] [Google Scholar]
- Lupp A, Richter N, Doll C, Nagel F, Schulz S. UMB-3, a novel rabbit monoclonal antibody, for assessing mu-opioid receptor expression in mouse, rat and human formalin-fixed and paraffin-embedded tissues. Regul Pept. 2011;167:9–13. doi: 10.1016/j.regpep.2010.09.004. [DOI] [PubMed] [Google Scholar]
- Martini L, Whistler JL. The role of mu opioid receptor desensitization and endocytosis in morphine tolerance and dependence. Curr Opin Neurobiol. 2007;17:556–564. doi: 10.1016/j.conb.2007.10.004. [DOI] [PubMed] [Google Scholar]
- McPherson J, Rivero G, Baptist M, Llorente J, Al-Sabah S, Krasel C, Dewey WL, Bailey CP, Rosethorne EM, Charlton SJ, Henderson G, Kelly E. mu-opioid receptors: correlation of agonist efficacy for signalling with ability to activate internalization. Mol Pharmacol. 2010;78:756–766. doi: 10.1124/mol.110.066613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nestler EJ. Under siege: the brain on opiates. Neuron. 1996;16:897–900. doi: 10.1016/s0896-6273(00)80110-5. [DOI] [PubMed] [Google Scholar]
- Raehal KM, Bohn LM. The role of beta-arrestin2 in the severity of antinociceptive tolerance and physical dependence induced by different opioid pain therapeutics. Neuropharmacology. 2011;60:58–65. doi: 10.1016/j.neuropharm.2010.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulz S, Mayer D, Pfeiffer M, Stumm R, Koch T, Höllt V. Morphine induces terminal micro-opioid receptor desensitization by sustained phosphorylation of serine-375. EMBO J. 2004;23:3282–3289. doi: 10.1038/sj.emboj.7600334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terman GW, Jin W, Cheong YP, Lowe J, Caron MG, Lefkowitz RJ, Chavkin C. G-protein receptor kinase 3 (GRK3) influences opioid analgesic tolerance but not opioid withdrawal. Br J Pharmacol. 2004;141:55–64. doi: 10.1038/sj.bjp.0705595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werling LL, McMahon PN, Cox BM. Selective changes in mu opioid receptor properties induced by chronic morphine exposure. Proc Natl Acad Sci U S A. 1989;86:6393–6397. doi: 10.1073/pnas.86.16.6393. [DOI] [PMC free article] [PubMed] [Google Scholar]