

Abstract
Altered splicing contributes to the pathogenesis of human blood disorders including myelodysplastic syndromes (MDS) and leukemias. Here we characterize the transcriptomic regulation of PRPF40B, which is a splicing factor mutated in a small fraction of MDS patients. We generated a full PRPF40B knockout (KO) in the K562 cell line by CRISPR/Cas9 technology and rescued its levels by transient overexpression of wild-type (WT), P383L or P540S MDS alleles. Using RNA sequencing, we identified hundreds of differentially expressed genes and alternative splicing (AS) events in the KO that are rescued by WT PRPF40B, with a majority also rescued by MDS alleles, pointing to mild effects of these mutations. Among the PRPF40B-regulated AS events, we found a net increase in exon inclusion in the KO, suggesting that this splicing factor primarily acts as a repressor. PRPF40B-regulated splicing events are likely cotranscriptional, affecting exons with A-rich downstream intronic motifs and weak splice sites especially for 5′ splice sites, consistent with its PRP40 yeast ortholog being part of the U1 small nuclear ribonucleoprotein. Loss of PRPF40B in K562 induces a KLF1 transcriptional signature, with genes involved in iron metabolism and mainly hypoxia, including related pathways like cholesterol biosynthesis and Akt/MAPK signaling. A cancer database analysis revealed that PRPF40B is lowly expressed in acute myeloid leukemia, whereas its paralog PRPF40A expression is high as opposed to solid tumors. Furthermore, these factors negatively or positively correlated with hypoxia regulator HIF1A, respectively. Our data suggest a PRPF40B role in repressing hypoxia in myeloid cells, and that its low expression might contribute to leukemogenesis.
Keywords: alternative splicing, PRPF40B, hypoxia, myelodysplastic syndromes, acute myeloid leukemia
INTRODUCTION
Alternative splicing (AS) generates more than one mRNA and often more than one protein per gene, so it largely contributes to the complexity of the human transcriptome and proteome (Nilsen and Graveley 2010; Lee and Rio 2015). AS can be sorted into distinct types, such as cassette-exon inclusion, intron retention (IR), or use of alternative 5′ or 3′ splice sites (5′ss, 3′ss), which can generate protein isoforms with altered, antagonistic, or unrelated functions. Despite advances in computational predictions (Xiong et al. 2015), AS profiles for each cellular condition still rely on high-throughput methods (Licatalosi and Darnell 2010; Barbosa-Morais et al. 2012; Merkin et al. 2012).
AS patterns are established by the combinatorial effects of (i) cis-acting splicing elements, including 5′ss or 3′ss at either side of intron, branchpoint sequence (BPS) and exonic or intronic splicing enhancers or silencers; (ii) cognate trans-acting splicing factors (SFs) that bind to these sequences to either activate or repress splice-site usage (Busch and Hertel 2012); (iii) structure of the pre-mRNA substrate; and (iv) transcription and chromatin modifications, because splicing largely occurs during nascent transcript synthesis with the carboxy-terminal domain (CTD) of RNA polymerase II (Pol II) acting as platform for loading SFs (Naftelberg et al. 2015). Cotranscriptional AS was evidenced by promoter-dependent AS events, and exons in genomic regions are enriched in nucleosomes and certain chromatin modifications. The speed of Pol II elongation affects AS, as found by Pol II mutants with slow polymerization, and by small molecules that affect its kinetics such as 5,6-dichloro-1-β-d-ribofuranosylbenzimidazole (DRB) and camptothecin (CPT) (Dujardin et al. 2014; Tang et al. 2016). The main kinetic model of AS proposes that slow and fast Pol II elongation respectively determine high or low inclusion of cassette exons, by positive AS regulators only assembling onto the substrates with weak splice sites or other signals under slow Pol II elongation (de la Mata et al. 2003). Other cassette exons are more included in fast Pol II elongation because a splicing repressor can only bind to slowly elongating pre-mRNAs (Dujardin et al. 2014). The physiological significance and pathological implications of cotranscriptional AS remain to be elucidated. Nevertheless, it is now clear that mutations affecting both cis-acting elements and trans-acting SFs are common in genetic diseases and cancer (Anczuków and Krainer 2016), with many mutations in blood disorders.
Myelodysplastic syndromes (MDS) are a heterogeneous set of clonal hematopoietic stem cell (HSC) disorders with myeloid cell dysplasia, cytopenia, and risk of acute myeloid leukemia (AML) (Inoue et al. 2016). MDS genomic studies identified frequent and recurrent mutations in several SFs, which spurred widespread efforts to link the most common mutations to clinical features and to characterize their tumorigenesis mechanisms (Inoue et al. 2016). Over half of MDS patients suffer heterozygous, mutually exclusive mutations in four SF genes, which disrupt the transcriptome as follows: (i) the frequent SF3B1 mutations in mild MDS activate cryptic 3′ss (Darman et al. 2015; DeBoever et al. 2015; Alsafadi et al. 2016); mutations in (ii) SRSF2 and (iii) U2AF1 in aggressive MDS change the RNA-binding affinities and specificities (Ilagan et al. 2015; Kim et al. 2015; Shirai et al. 2015; Zhang et al. 2015); and (iv) less common inactivating mutations in ZRSR2 affect only minor spliceosome introns (Madan et al. 2015). Each mutant SF results in a distinct pathogenesis and prognosis with almost no common themes, implying that several aberrant splicing pathways lead to MDS. Among the less common mutant SFs in MDS stands DDX41 RNA helicase (Cheah et al. 2017), PRPF40B (PRP40 pre-mRNA processing factor 40 homolog B) as found in the landmark study (Yoshida et al. 2011), and LUC7L2 (Singh et al. 2013). While most MDS-mutated SFs act via the 3′ss or BPS, the two latter SFs are involved in recognition of 5′ss on the other intron side, suggesting even more diverse pathogenic mechanisms.
Most information of PRPF40A/B is inferred from their budding yeast homolog PRP40 (Becerra et al. 2016), which is part of the U1 small nuclear ribonucleoprotein (snRNP) (Kao and Siliciano 1996) that recognizes the 5′ss early in splicing (Roca et al. 2013; Tan et al. 2016). PRP40 bridges between U1 on the 5′ss side, and branchpoint-binding protein (BBP) and Mud2p on the BPS and 3′ss (Abovich and Rosbash 1997). Prp40 contains two WW and four FF domains in its primary sequence, and it binds BBP directly through the WW domain whereas the binding to Mud2p occurs indirectly (Becerra et al. 2016).
The Prp40 protein structure is shared with a relatively small number of proteins in humans, including PRPF40A, PRPF40B, and hTCERG1, all involved in the coupling of splicing and transcription elongation (Becerra et al. 2016). The closest human ortholog PRPF40A (PRP40 pre-mRNA processing factor 40 homolog A) has a similar function as yeast Prp40. The WW domain of PRPF40A is essential for its localization to nuclear speckles (Mizutani et al. 2004). PRPF40A interacts with SF1 and U2AF and it is present in the spliceosome complexes A and B but not C, which is consistent with a role of Prp40 in early spliceosome assembly (Lin et al. 2004; Sánchez-Álvarez et al. 2010). PRPF40A immunoprecipitates together with U2 snRNP and proteins involved in transcription elongation (Makarov et al. 2012).
Despite sharing 65% identity, PRPF40B is far less characterized than PRPF40A (Becerra et al. 2016). Both PRPF40A and PRPF40B regulate cotranscriptional splicing by interacting with elongation complexes via Pol II CTD (Morris and Greenleaf 2000; Allen et al. 2002), and both bind to the expanded huntingtin polyglutamine tracts in Huntington's disease brains and are involved in other neurodegenerative syndromes, with unknown significance (Faber et al. 1998; Becerra et al. 2016). Even though PRPF40B does not possess any RNA recognition motif (RRM), it regulates AS likely by bridging between U1 at the 5′ss and SF1 bound to the BPS (Padgett 2012). PRPF40B can bind to SF1 through its WW domain while a weak binding to U2AF is modulated by the FF domain (Becerra et al. 2015). In addition, nuclear speckle localization of PRPF40B is mediated by the second FF domain, as opposed to PRPF40A for which the WW domain is required. Importantly, PRPF40B controls the selection of alternative splice sites of important apoptotic genes such as Fas (Becerra et al. 2015), as this SF was proposed to regulate AS in exons with weak splice sites.
Here we characterize the transcriptomic regulation of PRPF40B in K562 cells upon knockout (KO) and rescue with MDS mutations, to reveal a set of targets with weak splice sites and A-rich motifs. Importantly, PRPF40B KO up-regulated a hypoxia signature which, together with its low expression in AML, suggests a role of this SF in myeloid leukemias.
RESULTS
Generation of PRPF40B knockout and rescue in K562 cells for transcriptomic analysis
To investigate the role of PRPF40B in regulating AS, we devised a pipeline to identify the affected AS events upon KO plus rescue with MDS mutants in K562 cells. We used CRISPR/Cas9 technology (Hsu et al. 2014) to knock PRPF40B out, by introducing two double-stranded DNA cuts flanking a fragment encompassing exons 5–8 (Fig. 1A). For three different clones, we confirmed the deletion of this fragment at DNA, RNA, and protein level (Fig. 1B–D). PCR genotyping along the PRPF40B gene confirmed that the engineered deletion was confined to the intron 4–intron 8 fragment (Fig. 1B), with no traces of this internal fragment detected in any of the three KO clones. mRNA expression measured by real-time PCR revealed that expression of PRPF40B exons 7–8 (within deletion, qRT II) was undetectable in the three KO clones (Fig. 1C). In turn, the three KO clones showed a >60% reduction in RNA levels for PRPF40B exons 3–4 (upstream of deletion, qRT I) and exons 16–17 (downstream from deletion, qRT III) compared to parental K562. This reduction is likely caused by nonsense-mediated mRNA decay (NMD) because the deletion introduces an in-frame stop codon at the beginning of exon 9, which is followed by many other exons. The deletion also generates an mRNA potentially encoding a peptide of 77 amino acids and 8 KDa, which cannot be detected by western blotting because of (i) its small size and (ii) the monoclonal antibody epitope at the C terminus falling outside this 8 KDa fragment (Fig. 1A,D).
FIGURE 1.

Design and validation of PRPF40B-KO and rescue in K562 cells. (A) Schematic of PRPF40B gene, mRNA, and protein. We depict the PRPF40B gene with exons on scale, with the translation start sites of isoforms A and C. We indicate the position of the sgRNAs for the CRISPR/Cas9 KO that flank the deletion including exons 5–8 in red, and the pairs of primers for genomic PCR validations. The mRNA shows the natural stop codon in last exon 26, the termination codon in exon 9 introduced by the deletion (KO), and the positions of the three amplimers for the real-time RT-PCR. mRNA exons are aligned to their WW and FF protein domains, with the MDS mutations and the monoclonal antibody epitope for western blot. (B) PCR genotyping of PRPF40B-KO clones 1–3. The PCR number matches the primer pairs in Figure 1A. (–) Negative control, water template; (+) positive control, parental K562 genomic DNA. Ladders are shown on each side of gel, with the band sizes in base pairs in the middle. PCRs 1, 2, 5, and 6 fall outside the deletion fragment and are detected in all KO clones. PCR 3, with primers flanking the deletion, shows the full-length band in parental cells, and a much smaller band in the KO clones because of the deletion. PCR 4, with one primer inside the deletion, only generates a band in parental but not KO cells. The identity of specific bands was confirmed by sequencing. Asterisk, unspecific amplimers. (C) Real-time RT-PCR measurement of PRPF40B RNA levels for the qRT I–III regions in Figure 1A. We plotted PRPF40B expression ratios relative to β-actin and with parental K562 values normalized to 1. (D) Representative western blots of the parental K562 and three PRPF40B-KO clones (left), and of overexpression samples with WT (isoforms A or C) and mutant protein (right), both with GAPDH as loading control. (E) Diagram of the samples with altered PRPF40B expression, including KO by CRISPR/Cas9 in K562 cell line, and rescue by overexpression in the KO cells of PRPF40B either as WT (Rescue WT) or the indicated MDS mutations alone or mixed with WT at 1:1 ratio. We performed RNA-sequencing with all these seven conditions.
Then we used PRPF40B isoform C to restore expression of wild-type (WT) PRPF40B and to express the PRPF40B alleles with the P383L and P540S MDS mutations (Yoshida et al. 2011). After optimization experiments (available upon request), we decided to use 100 ng of PRPF40B plasmids and 72 h after transient transfection of K562-PRPF40B-KO cells, to obtain a relatively similar amount of PRPF40B RNA and protein compared to parental K562 cells (Fig. 1D).
We then generated samples from the seven conditions as follows (Fig. 1E): (i) parental K562 cells; (ii) K562 cells with PRPF40B-KO; (iii) KO cells transfected with PRPF40B-WT plasmid; (iv) KO cells transfected with PRPF40B-P383L; (v) KO cells transfected with PRPF40B-P540S; (vi) KO cells transfected with 1:1 ratio of PRPF40B-WT and P383L plasmids; (vii) KO cells transfected with 1:1 ratio of PRPF40B-WT and P540S plasmids. The last two samples with WT and MDS-mutant plasmid mixed at 1:1 ratio aim to recapitulate heterozygous expression of mutant PRPF40B found in MDS patients.
We performed RNA sequencing from total mRNA extracted from three samples (independent replicates) of each condition, all with RNA integrity number (RIN) higher than eight. We applied a similar bioinformatics pipeline as previously published by our group (Liu et al. 2018), to obtain a majority of reads aligned to the hg19 genome, and a high correlation between replicates (details available upon request).
Differentially expressed genes upon knockout and rescue with WT or MDS-mutant PRPF40B
PRPF40B-KO only led to subtle changes in gene expression, as the differentially expressed genes (DEGs) are affected by a low magnitude of change. We found a significant set of DEGs upon KO of PRPF40B (≤−0.3- or ≥0.3-fold change and P-value ≤ 0.05), with a fraction that are rescued when PRPF40B was reintroduced in the KO cells (Fig. 2A). The use of such a low 0.3-fold-change cutoff rather than more standard one- or twofold leads to a higher fraction of genes rescued by WT protein among genes altered in KO and allows for a more comprehensive pathway analysis. Almost all DEGs rescued by overexpressing the PRPF40B-WT gene were also rescued by the MDS mutants, and surprisingly, the mixed WT-MDS mutant rescued the DEGs to a slightly lesser extent compared to the MDS mutants alone (Fig. 2B). These data suggest that PRPF40B regulates directly or indirectly the expression of a certain subset of genes.
FIGURE 2.

Experimental procedure to investigate the role of human PRPF40B in the regulation of AS and of two MDS mutations. (A) Number of DEGs upon KO (K562 vs. KO), in rescue WT cells (KO vs. WT) and those in common between the two comparisons which are under control of PRPF40B (DEGs Rescued) (fc ≤ 0.3, fc ≤ −0.3, P-value ≤ 0.05). (B) Number of DEGs regulated by PRPF40B (DEGs Rescued) which are rescued (blue) or not (red) by the MDS mutations. (C) Gene ontology of the DEGs regulated by PRPF40B shows the genes are mainly involved in cholesterol biogenesis, glycolysis, and hypoxia.
One of the DEGs is Formin-like 3 (FMNL3), whose genomic location overlaps with the 3′ portion of PRPF40B in the antisense direction, and its end is <3 Kbp downstream from our CRISPR/Cas9-deleted fragment. FMNL3 expression is low in parental K562 cells (FPKM 2.16), and it is reduced in the KO (FPKM 1.42) but rescued by WT and mutant PRPF40B (FPKM ranging from 1.88 to 2.37, with 2.23 in WT PRPF40B rescue). This indicates that altered expression of FMNL3 in the KO is not due to the introduced genomic alteration, but instead FMNL3 is one of the DEGs under the regulation of PRPF40B.
Gene set over-representation analysis for the 405 rescued DEGs, performed by the online tool Enrichr (Fig. 2C; Kuleshov et al. 2016) and subsequently confirmed by ConsensusPathDB (Kamburov et al. 2011) (data not shown), revealed that the top two enriched pathways are cholesterol biogenesis and HIF 1-α (HIF1-α/A) signaling, with the latter as a master hypoxia regulator. HIF1A signaling was also highly enriched in DEGs with onefold change cutoff (not shown). As elevated cholesterol biogenesis may be a consequence of hypoxia (Hwang et al. 2017), we focused on the latter pathway. Of these 405 genes, 106 were down-regulated while 299 up-regulated in the KO cells, suggesting that PRPF40B is mainly repressing gene expression.
The hypoxia gene signature under PRPF40B regulation is illustrated by the heatmap of important selected genes involved in hypoxia according to the literature, including DEGs and other genes that did not pass the statistical cutoffs (Fig. 3A). This heatmap reveals a large cluster of genes up-regulated in the KO which are rescued by WT and mutant PRPF40B, while other genes are instead down-regulated and others barely change expression. We validated DEGs that regulate different pathways in hypoxia by real-time RT-PCR (Fig. 3B), which included five genes in the rescued DEGs, and one (CA9) that clearly changed in the KO, but the expression changes in the PRPF40B-WT rescue did not pass the statistical cutoff. Among the proteins encoded by these selected DEGs, BNIP3 regulates autophagy (Zhang and Ney 2009), ENO2 and PGK1 are involved in glycolysis (Yeh et al. 2008), CA9 regulates pH (Swietach et al. 2007), TGF-β3 is involved in extracellular matrix (ECM) remodeling (Karamichos et al. 2011), and COL1A2 in collagen production (Bauman et al. 2010). Excluding TGF-β3, which shows an opposite trend in the first 3 samples compared to RNA-seq, and in part ENO2 which does not show rescue by real-time RT-PCR, we validated the DEGs found by RNA-seq. Real-time RT-PCR for CA9 showed clear expression changes that were rescued by PRPF40B-WT, indicating that our set of rescued DEGs are just the reliable fraction of a larger set of genes under control of PRPF40B.
FIGURE 3.

PRPF40B regulates hypoxia and other relevant pathways. (A) Heatmap of DEGs with a role in hypoxia. PRPF40B-KO induces the up-regulation of hypoxic genes that are down-regulated when the expression of PRPF40B-WT gene is reintroduced in the KO cells. In many cases, overexpression of the MDS mutants also reversed the expression changes in the KO. The six DEGs selected for validation are highlighted in red. (B) Validation by real-time RT-PCR of six selected hypoxic genes with different functions as indicated. The y-axis indicates expression of the gene for each condition, relative to that on parental cells normalized to 1. (C) DEGs that are targets of KLF1, showing strong up- or down-regulation in the KO that are rescued by WT and MDS-mutant PRPF40B. (D) Homeobox (HOX) genes as DEGs regulated by PRPF40B. (E) DEGs involved in iron metabolism are mostly up-regulated upon PRPF40B-KO. (F) Cytokines and chemokines under expression control of PRPF40B.
By an in-depth analysis using the gene set enrichment analysis (GSEA) tool (Subramanian et al. 2005), we found that the PRPF40B-regulated DEGs are enriched in targets of the transcription factor KLF1 (Fig. 3C; Pilon et al. 2008) which acts as master regulator of erythroid differentiation (Siatecka and Bieker 2011) and also in genes involved in development (HOX genes) (Fig. 3D) and iron metabolism (Fig. 3E). In addition, we observed up-regulation of specific cytokines and chemokines (Fig. 3F), such as CCL4/5 and CXCL1/8, which can induce angiogenesis (Benelli et al. 2006). All these are likely secondary pathways activated downstream from hypoxia upon PRPF40B-KO (see Discussion).
PRPF40B regulates hundreds of alternative splicing events mostly as repressor
Next, we applied the common splicing tool rMATS (Shen et al. 2014) on our RNA-seq data to calculate the differential AS events (DASEs) with >10% ΔPSI (percent spliced in) and false discovery rate (FDR) < 1 cutoff. We found a total of 5236 DASEs between parental and PRPF40B-KO K562 cells, among which 1367 (26%) were rescued by PRPF40B-WT (Table 1). The largest number of rescued DASEs belong to cassette exons (also known as skipped exons, CA or SE), followed by mutually exclusive exons (MXE), alternative 5′ or 3′ splice sites (A5SS and A3SS), and last retained introns (IR). For cassette exons, we found 2544 DASEs in the KO with 764 DASEs (30%) rescued by WT PRPF40B (Fig. 4A), which we named PRPF40B-regulated DASEs. Similarly to DEGs, the majority of DASEs rescued by WT were also rescued by MDS-mutant PRPF40B, with a lower rescue with the mixed WT-MDS-mutant mix (Fig. 4B). The 764 DASEs belong to 463 unique genes. This data set includes three genes mutated in MDS, namely, AXSL1, SF1, and LUC7L2, but we did not further study these DASEs because the splicing changes in the KO were rescued by MDS-mutant PRPF40B. Among the total of 764 DASEs, 455 DASEs show more exon inclusion in the KO while 309 display more exon skipping, implying that PRPF40B mostly acts as a splicing repressor.
TABLE 1.
Summary of alternative splicing events under regulation by PRPF40B
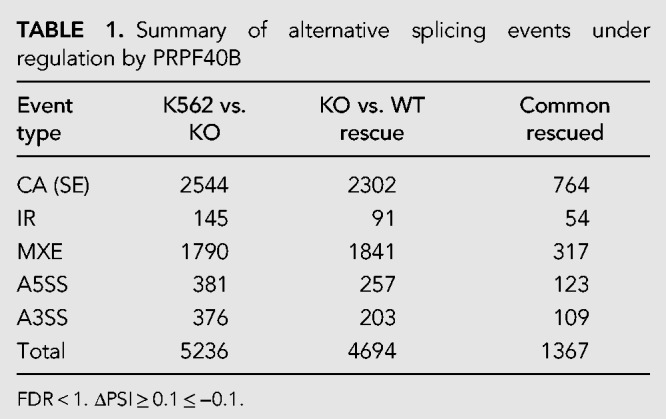
FIGURE 4.

PRPF40B regulates differential AS events (DASEs). (A) Number of cassette-exon DASEs upon KO (K562 vs. KO), in rescue WT cells (KO vs. WT rescue) and those in common between the two comparisons which are regulated by PRPF40B (AS events Rescued) (FDR < 1, ΔPSI ≥ 0.1 ≤ −0.1). (B) Number of cassette exons regulated by PRPF40B (AS events Rescued) which are (blue) or are not (red) rescued by the MDS mutations. (C) Gene Ontology of the cassette-exon DASEs regulated by PRPF40B. We noted an enrichment in genes with binding sites for the Pol II subunit POLR2A based on Chip-seq data, with histone marker H2AFZ, and which are involved in MAPK signaling cascade. (D) Network of PPIs of the cassette exons regulated by PRPF40B. Genes with same function are grouped in clusters (dotted circles) with labels. Right shows a zoomedin view of two selected clusters, ubiquitination, and Akt/MAPK1.
In addition, in ∼2/3 of the 1780 DASEs affected by PRPF40B-KO but not rescued (Fig. 4A), the change in splicing (or ΔPSI) in the KO is partially reversed by WT PRPF40B, yet below the statistical thresholds. Similarly, ∼2/3 of the 1538 “unrelated splicing events” found in the WT rescue but not in the KO show a reversal trend in the splicing change in the KO. This emphasizes that our number of PRPF40B-dependent DASEs is a “conservative” yet reliable data set. And finally, ∼2/3 of DASEs found by either of the four MDS-mutant PRPF40B rescue (including unrelated events not found in KO) also show a similar splicing change by WT PRPF40B (not shown). These results further suggest that PRPF40B MDS mutations do not confer a substantial gain of function or change in splicing specificity over WT.
Pathway analysis by Enrichr (Kuleshov et al. 2016) and confirmed by ConsensusPathDB (Kamburov et al. 2011) showed that PRPF40B-regulated DASEs are enriched in genes with binding sites for the POLR2A subunit of RNA Pol II based on a Chip-seq data in K562 cells (Fig. 4C). Furthermore, the genes with DASEs are enriched in the H2AFZ histone marker and are involved in the MAPK signaling pathway and RNA binding. We also built a protein–protein interaction (PPI) network of the cassette exons regulated by PRPF40B using the STRING database (Szklarczyk et al. 2017), as AS events tend to map to regions where a protein interaction domain or a short linear motif is present (Resch et al. 2004). Among the clusters in the PRPF40B network, we found that this SF mainly regulates splicing targets in the Akt/MAPK1 cascade and ubiquitination (Fig. 4D).
We attempted validation by RT-PCR for a total of 42 DASEs, including DASEs rescued by PRPF40B-WT (Fig. 5A for 16 cases), and other DASEs with a significant change in splicing upon KO but not rescued (Fig. 5B for eight validated cases). Overall we obtained a validation rate of 67% based on correlation between RT-PCR and RNA-seq ΔPSIs for the seven measured conditions (Fig. 5C,D). The nonvalidated DASEs had a poor correlation (<0.4) between RNA-seq and RT-PCR, or only showed a single band by RT-PCR. Importantly, some validated DASEs from the nonrescued group, most clearly for SPTAN1, showed RT-PCR evidence of reversing the AS change in the KO by PRPF40B-WT. This suggests that our rescued data set is also a smaller yet more reliable subset of the total AS events regulated by PRPF40B.
FIGURE 5.

Validation of DASEs regulated by PRPF40B. (A) Validation by RT-PCR of 16 selected DASEs altered by PRPF40B-KO and rescued by WT protein. All DASEs in all panels are cassette exons except the indicated IR. Upper bars depict the RNA-seq–derived PSIs (scale 0–1), while bottom numbers indicate PSIs (scale 0–100) by RT-PCR quantification (PSIs are not identical because PCR biases against larger isoforms). (B) RT-PCR validation of eight selected DASEs not rescued by PRPF40B-WT. (C, right table) For each tested DASE gene, we indicate the correlation coefficient between the PSIs by RNA-seq and RT-PCR for the seven conditions. Smaller left table shows the gene names of nonvalidated DASEs that only showed a single band by RT-PCR. Genes in red show poor correlation between RNA-seq and RT-PCR PSIs, so are not validated. (D) Breakdown of correlation coefficients for the 42 tested DASEs as parameter for validation, showing a total of 67% (24 + 43) validated DASEs with correlation coefficient >0.4.
PRPF40B regulates splicing of cassette exons flanked by weak splice sites and A-rich downstream introns
We carried out detailed analyses with the subset of cassette exons that are affected by PRPF40B-KO and rescued by WT PRPF40B. First, by examining the splice-site strength scores using the common MAXENT algorithm (Yeo and Burge 2004), we found that AS events are associated with weak splice sites, but that PRPF40B-regulated DASEs are even weaker than the bulk of AS events (Fig. 6A,B). The MAXENT score differences between all AS events and PRPF40B-regulated DASEs were stronger for 5′ss (Fig. 6A) than for 3′ss (Fig. 6B). However, we did not find any correlation between splice-site strength and rMATS ΔPSI in the 764 PRPF40B-regulated DASEs (Fig. 6C). The introns upstream the PRPF40B-regulated cassette exons were shorter than those for other AS exons (Fig. 6D), yet the introns downstream or the exon length showed no difference (Fig. 6E).
FIGURE 6.

Features of the cassette exons regulated by PRPF40B. (A) Measurement of the 5′ss scores by maximum entropy model (MAXENT) for constitutive and alternative exons, regulated or not by PRPF40B, shows that PRPF40B-regulated exons have even weaker 5′ss than other alternative exons (Wilcoxon test). (B) MAXENT scores for 3′ss show that PRPF40B-regulated exons also have weaker 3′ss than background constitutive exons and other AS events (Wilcoxon test). (C) Lack of correlation between the strengths of the 5′ss of the PRPF40B-regulated cassette exons and their ΔPSI values in absolute numbers (t-test). (D) Cassette exons regulated by PRPF40B have shorter upstream but not downstream introns (Wilcoxon test). (E) The length of the PRPF40B-regulated cassette exons is similar to exons not regulated by PRPF40B (Wilcoxon test). (F) Top MEME motif found to be enriched in the PRPF40B-regulated cassette exons and flanking 200-nt-long intronic segments. The width of arrows indicates that the A-rich motifs are especially enriched in downstream introns. (G) Pol II inhibition assay in K562 WT, KO, and rescued cells. hnRNP DL exon 8 was used as a positive control because its inclusion increases upon slow transcription.
In order to identify the most enriched motifs in the sequences of the PRPF40B-regulated cassette exons, we performed motif analysis by DREME (Bailey et al. 2009), using exon sequences of the 764 DASEs with 200 bp of the flanking upstream and downstream introns. The most enriched motif (Fig. 6F) was AAAWRAAA (where W is A or T/U, and R is A or G) by a very large difference in significance to the second motif (e-26 for AAAWRAAA vs. e-12 for RCAGARA), which in turn was similar to many other motifs. Feeding DREME with the upstream/downstream intronic and exonic sequences separately unveiled that the high frequency of A-rich motifs mostly applies to the downstream introns, while the upstream introns and exon bodies also tend to have GA-rich motifs yet with a much lower significance (data not shown). Hence, our data strongly suggest that PRPF40B regulates cassette exons via downstream A-rich introns.
To further study the PRFP40B mechanisms of AS regulation, we constructed few minigenes with three exons and two introns, with the middle exon from the list of PRPF40B-regulated cassette exons. Using two different minigene designs, their derived transcripts did not recapitulate the AS patterns or did not show the AS difference between parental and PRPF40B-KO K562 cells (data not shown). Hence, we could not use these minigenes to study PRPF40B AS regulation.
To further gain insight into PRPF40B mechanisms, we examined whether the PRPF40B-regulated DASEs are regulated by transcription elongation rates. Hence, we checked whether the 463 unique genes whose splicing is regulated by PRPF40B are enriched in a publically available bromouridine-sequencing (Bru-seq) data set in K562 cells (Veloso et al. 2014). Bru-seq methodology maps sequences of nascent RNA transcripts to determine the relative transcription rate of each gene. We observed that our 463 genes have a higher transcription rate than the average rate of the genes in the data set. However, we found the same trend with all AS events, including those not regulated by PRPF40B (data not shown). We thus concluded that a faster elongation rate is associated to AS events, but this phenomenon is not specific to PRPF40B.
We also tested the connection between PRPF40B and transcription elongation by slowing down Pol II elongation with DRB and CPT. We used exon 8 of hnRNP DL as a positive control, whose inclusion increases upon inhibition of Pol II (Ip et al. 2011). Inclusion of this exon increased with DRB in parental K562, KO and rescue cells, and with CPT in parental and rescue cells (Fig. 6G). Similarly, DRB and CPT increased the PSI of AFMID exon 5 in parental K562, KO and rescue, despite that the KO shows a higher inclusion already. PRPF40B-KO decreased inclusion of SPTAN1 exon 8, which was exacerbated by DRB and CPT, and these drugs showed similar effects in the rescue cells. Based on these limited target tests, we conclude that DRB and CPT alter AS in parental K562, KO, and rescue cells to a similar extent, hence the potential cotranscriptional AS regulation of PRPF40B might not act via speed of Pol II elongation.
PRPF40B expression is low in acute myeloid leukemia and might induce hypoxia
As shown above, the DEGs in PRPF40B null cells point to a role of this protein in preventing hypoxia. Next, we searched for RNA expression patterns of PRPF40B in cancer samples by mining the data in The Cancer Genome Atlas (TCGA) in cBioportal (Cerami et al. 2012). We found that PRPF40B is expressed at low levels in AML samples but not solid tumors, and that is anti-correlated with PRPF40A as this SF is expressed highly in AML relative to the other cancers (Fig. 7A). We did not detect any clear change in PRPF40A expression at the protein level after KO of PRPF40B (data not shown), suggesting that PRPF40B does not regulate PRPF40A, and its loss is not compensated by increase in PRPF40A. Finally, PRPF40B expression in AML samples negatively correlates with HIF1A (Fig. 7B), while PRPF40A positively correlates with this hypoxia gene driver (Fig. 7C), suggesting a role of these SFs in hypoxia in the context of AML.
FIGURE 7.

mRNA expression of PRPF40B and its paralog PRPF40A in various human cancers by RNA-seq (TCGA). (A) In AML, PRPF40A is highly expressed at higher levels than solid tumors, while PRPF40B expression is lower. (B) Inverse correlation between PRPF40B and HIF1A mRNA levels in AML (t-test). (C) Positive correlation between PRPF40A and HIF1A mRNA levels in AML (t-test).
DISCUSSION
We characterized the transcriptome regulation by PRPF40B, by identifying a subset of PRPF40B-regulated DEGs involved in hypoxia, and DASEs with weak splice sites. We carried out such analyses in the K562 chronic myeloid leukemia cell line, as this cell line has been used as MDS model (Dolatshad et al. 2015), and it can be differentiated to both erythroid and myeloid cells (Sutherland et al. 1986). We successfully knocked out all PRPF40B alleles (Fig. 1B–D) despite K562 being hypotriploid (Naumann et al. 2001) and derived a highly reliable set of PRPF40B-regulated DEGs and DASEs.
We found 764 cassette-exon DASEs that were altered in the PRPF40B-KO and rescued by WT protein. The use of a transient transfection system for the rescue likely limited the number of rescued DEGs and DASEs, yet the reliability of these data is high as validated independently. Most of the DASEs rescued by WT PRPF40B were also rescued by the two MDS mutations, like for DEGs (Figs. 2B, 4B), so the effects of the MDS mutations onto PRPF40B function are very mild at best. We could not draw any pattern for the DASEs regulated by WT versus mutant PRPF40B, so further studies are required to shed light onto PRPF40B-dependent MDS pathogenesis. This lack of insight could also be due to the use of K562 cell line versus the actual MDS cells in patients. Based on their mild effects on splicing and their very low frequency in MDS patients, we cannot rule out that the MDS PRPF40B mutations are “passengers” rather than “drivers,” or that they may have a weak contribution to MDS. Unexpectedly, the rescue by either MDS mutant together with WT PRPF40B was lower than the rescue by either of the two MDS mutants alone, both in DEGs and DASEs. Even though further analyses are needed to confirm and extend the implications of this observation, it is possible that the effects of MDS-mutant PRFP40B are exacerbated by mixing with WT PRPF40B, perhaps by forming dimers or oligomers of WT and mutant protein with more impaired function.
This PRPF40B-regulated transcriptome allowed us to decipher some properties of the regulated AS events. PRPF40B-regulated cassette exons show more inclusion in the KO, unveiling that this SF mostly acts as a repressor. These cassette exons are flanked by both weak 5′ss and 3′ss (Fig. 6A,B), as reported in few AS events (Becerra et al. 2015), and consistent with its role in bridging splice sites across introns (Abovich and Rosbash 1997). The P-value for 5′ss scores is much lower than that for 3′ss, suggesting a closer functional link between PRPF40B and 5′ss as reported for yeast and humans (Kao and Siliciano 1996; Becerra et al. 2016). Our data are partly consistent with a role of PRPF40B in cotranscriptional splicing (Becerra et al. 2016): the lack of AS regulation in minigenes versus endogenous genes suggests that the endogenous promoter, genomic location and chromatin configuration of the regulated DASE are essential for PRPF40B regulation. However, our DRB and CPT treatments altered AS in the PRPF40B-KO to a similar degree than in parental K562 cells, suggesting that cotranscriptional effects of PRPF40B are not mediated by the speed of Pol II elongation. Also, we found that PRPF40B-regulated cassette exons are preceded by shorter introns, while the cotranscriptional model would support long introns flanking a cassette exon to give time to Pol II to modulate AS via its speed. We did not find a significant connection between our PRPF40B-dependent transcriptome and previously published AS data set of fast transcription. PRPF40B-regulated cassette exons are enriched in Pol II and H2AFZ (Fig. 4C), suggesting a functional link to this histone variant. Finally, PRPF40B-regulated exons are associated to A-rich motifs, especially in the downstream intron, hinting at a functional link between this SF and RBPs that bind these sequences, such as TRA2 (Grellscheid et al. 2011), SRSF10 (Shin and Manley 2002) or even poly(A)-binding proteins, which were identified in complex with RANBP9 SF to regulate AS (Bao et al. 2014). However, we did not detect a change in Fas AS like it was reported (Becerra et al. 2015), probably due to the use of different cell lines (HEK293T in the previous study vs. K562 in ours). Future studies relying on our transcriptomic data should further elucidate the mechanisms of AS regulation by PRPF40B, including regulatory motifs and cotranscriptional effects.
Our DEG analyses in the KO and WT rescue revealed that PRPF40B loss in K562 induces subtle changes in DEGs, and specifically induces hypoxia-related pathways such as glycolysis, autophagy, pH regulation, etc. (Fig. 3A,B). Furthermore, the PRPF40B-dependent DEGs suggest that this SF regulates KLF1-driven erythroid differentiation and iron metabolism. These effects are likely a consequence of hypoxia gene activation in the KO cells, as low oxygen in the body sends signals to the bone marrow to increase red blood cell (RBC) production through the regulation of homeostasis of iron (Haase 2013), whose increase enables high rates of hemoglobin synthesis in erythroid progenitors (Rybinska and Cairo 2017). Among the highly up-regulated DEGs in the PRPF40B-KO are H19, a long noncoding RNA induced by hypoxia with a role in cancer progression (Matouk et al. 2015), as well as genes encoding various metallothionines (MT1X, MT2A, MT1G, MT1E), a family of proteins activated by hypoxia which protect against oxidants and act in cell proliferation and apoptosis (Murphy et al. 1999). Furthermore, the PRPF40B-regulated cytokines and chemokines support an indirect role of this SF in driving angiogenesis via hypoxia (Liao and Johnson 2007).
We did not find substantial changes in any of the known hypoxia master regulators, but we found a PPI clustering of PRPF40B-regulated DASEs in the Akt/MAPK pathway, which is known to regulate hypoxia reciprocally (Alvarez-Tejado et al. 2001; Dai et al. 2008). Hence, the hypoxia signature may be up-regulated in PRPF40B-KO cells by splicing changes in the Akt/MAPK pathway, to be further tested in the future. In turn, the closest paralog PRPF40A induces hypoxia in lung cancer and its expression correlates with hypoxia markers (Oleksiewicz et al. 2017). However, we did not find any overlap between this PRPF40A-dependent hypoxia data set and our PRPF40B-regulated DEGs (data not shown), likely because in the lung model only a few targets were analyzed by real-time RT-PCR. PRPF40A was also linked to hypoxia in whole blood in connection to obstructive sleep apnea (Perry et al. 2013). In addition, we found that PRPF40A and PRPF40B are, respectively, high and low in AML compared to solid tumors (Fig. 7A). These SFs, respectively, correlate positively and negatively with the major hypoxia regulator HIF1A, which is quite remarkable considering the high heterogeneity in gene expression from AML samples of different subtypes. Bone marrow is low in oxygen due to limited blood supply and HSCs actively using oxygen, and in turn, hypoxia triggers the secretion of factors for HSC maintenance and protects cells from mutations by reactive oxygen species (Zhang and Sadek 2014). AML tumorigenicity also relies on hypoxia via regulation of metabolism, cell cycle and immune functions like chemotaxis (Deynoux et al. 2016), and HIF1A predicts poor prognosis for MDS and induces transition to AML (Tong et al. 2012). Some of our PRPF40B-regulated cytokine/chemokine DEGs are HIF1A targets in primary human AML cells (Hatfield et al. 2010) and can induce angiogenesis (Benelli et al. 2006). Finally, our data do not support a direct activation of a full-fledged hypoxia response only by KO of PRPF40B, as HIF1A stabilization could not be detected in these cells (data not shown), but they suggest that PRPF40B would predispose or enhance the hypoxia response during AML tumorigenesis via other triggers and in combination with high PRPF40A, which remains to be tested in follow-up projects.
In brief, we present the first transcriptome regulated by a Prp40 ortholog in mammalian cells. We derived few properties of the PRPF40B-regulated AS events, while many mechanistic questions would be addressed by further mining these data sets. Finally, the role of PRPF40B in preventing hypoxia genes and its expression levels relative to those in PRPF40A in human cancers suggest that these SFs play important roles in leukemia tumorigenesis via hypoxia, to be characterized in future studies.
MATERIALS AND METHODS
PRPF40B single-guide RNA (sgRNA) expression vector construction
We designed two 20-bp guide sequences (5′-GAGCCGTCTGCCGAAGAAAG-3′) and (5′-AGGGTGAGATGTCCTACGGG-3′), respectively, targeting genomic DNA within intron 4 and intron 8 of the human PRPF40B gene, using ChopChop (Montague et al. 2014). We ordered from Integrated DNA Technologies two pairs of complementary oligos (5′-CACCGGAGCCGTCTGCCGAAGAAAG-3′ and 5′-AAACCTTTCTTCGGCAGACGGCTCC-3′) and (5′-CACCGAGGGTGAGATGTCCTACGGG-3′ and 5′-AAACCCCGTAGGACATCTCACCCTC-3′), containing the PRPF40B guide sequences and BbsI ligation adapters. We annealed 100 µmol of each oligo by T4 PNK polynucleotide kinase (New England Biolabs) and 10× T4 Ligation Buffer in 10 µL, at 37°C for 30 min, then 95°C for 5 min followed by cooling at room temperature. We diluted the solution 250 times and used 1 µL for ligation into the BbsI-digested pX458 vector with T4 Ligase (New England Biolabs). We transformed chemically competent DH5α cells (Life Technologies) with 5 µL of the ligation mixture. After plasmid DNA extraction (Qiagen), we verified the construct sequences by Sanger sequencing (1st Base Asia), with sequencing primer in the U6 promoter (5′-GACGTAATACGACTCACTATAGGGC).
Nucleofection and cell sorting
We maintained K562 cells in Dulbecco's modified Eagle's medium (DMEM), 1% penicillin/streptomycin (Pen/Strep) (Gibco, Life Technologies) and 10% fetal bovine serum (FBS), within a concentration of 1 × 106 and 2 × 106 cells/mL. We nucleofected the cells when they reached a maximum of 0.7 × 106 cells/mL. Briefly, we resuspended 2 × 106 K562 cells in 100 µL SF reagent (Lonza) with 2.5 µg of both Cas9/sgRNA CRISPR plasmids. We performed nucleofection according to manufacturer's instructions by using the FF-120 program with Amaxa 4D-Nucleofector (Lonza). After 24 h, we sorted cells for green fluorescent protein (GFP) expression and seeded them into a 12-well plate to recover for 24 h. Subsequently, we sorted again the top 2% of the GFP positive population from the previous day to enrich for cells highly expressing Cas9. We then grew sorted cells as a bulk in six-well plates or as single cells in 96-well plates. We expanded single cells to obtain individual clones cultured in DMEM, 1% Pen/Strep and 20% FBS.
Clone validation by genotyping PCR
From edited clones and nonedited K562 control cells, we extracted genomic DNA by incubating cells with 200 µL of lysis buffer (100 mM NaCl, 100 mM Tris-HCl [pH 7.5], and 1 mM EDTA [pH 7.5)] at 55°C for 1 h. We then subjected DNA to ethanol precipitation with 2× volume of 100% ethanol and 0.1× volume of 3 M sodium acetate (pH 5.2) and incubated samples at −20°C overnight. We centrifuged DNA at 13,500 rpm for 15 min and then washed the pellets twice with 400 µL of 70% ethanol before resuspending them in 150 µL of water. We performed two different genotyping PCRs (Fig. 1A). In the first PCR, we used a forward primer upstream (5′-TCTCAACAAAATACTCTCACTTGAG-3′) and a reverse primer downstream (5′-CCACAGTGTAGAGATCATGGCGACAG-3′) of the sgRNA cleavage site. In the second PCR, we paired one forward primer (5′-CTTAGTCTCTAACCATATTCATGAT-3′) targeting a sequence inside the deleted region, with a downstream reverse primer targeting the portion downstream from the deletion. We carried out four more genotyping PCRs to confirm that other parts of the PRPF40B gene were unaffected by the deletion. We designed two sets of primers targeting two regions upstream of the deletion, and the other two sets targeting downstream regions: For intron 1 forward and reverse primers were 5′-GCTCCTTACTGGTGGCTATTT-3′ and 5′-GACAAGAATGGAGAGGCAGTAG-3′; For intron 2–4, 5′-TAGAGGTAGGAGAAGTGGAAGG-3′ and 5′-GAGGGAAAGAATGGAGGTCATAG-3′; For intron 15–16, 5′-GGACAGCATCGTTGAAACATTAG-3′ and 5′-GAAGAGGCAGGGACAATCAA-3′; and for intron 23–26, 5′-CCTTTGAGGGTAGACTGGATTG-3′ and 5′-CACCAAGAGCAACCTTGAGA-3′. PCR program was as follows: initial denaturation at 95°C for 2 min, followed by 40 cycles of denaturation at 95°C for 30 sec, annealing at 58/50°C for 50 sec and extension at 72°C for 50 sec, plus final extension at 72°C for 5 min. We resolved WT genomic fragment for K562 and truncated genomic fragments for KO clones by 1.5% (w/v) agarose gels and visualized them in a gel docking station.
PRPF40B rescue plasmid
We purchased the cDNA open reading frame (ORF) clone for the longest isoform A of human PRPF40B from GenScript (Clone ID OHu20176, RefSeq accession number NM_001031698.2), within the pcDNA3.1+/C-(K) plasmid backbone with DYK flag. We derived the canonical shorter isoform C (RefSeq Accession XM_011538141.1) with the KAPA HiFi DNA polymerase (KAPA Biosystems) and the following forward and reverse primers: 5′-CTCGGATCCGCCACCATGATGCCACCACC-3′ and 5′-GGGTGGTGGCATCATGGTGGCGGATCCGAG-3′. We introduced a CAG sequence missing in the long isoform by use of the downstream from a pair of tandem alternative 3′ss with NAGNAG sequence (Hiller et al. 2004), by PCR mutagenesis (5′-CCTACAGGTGCTGGAGCAGACTGAATGCCAGCACCTCC-3′) and (5′-GGAGGTGCTGGCATTCAGTCTGCTCCAGCACCTGTAGG-3′). Next, we separately introduced the P383L and P540S MDS mutations (Yoshida et al. 2011) into the WT PRPF40B gene, using (5′-AGGTCTGGGCTGTGGTCCTTGAGAGGGATCGAAAAGAGGTTTATGATG-3′) and (5′-CATCATAAACCTCTTTTCGATCCCTCTCAAGGACCACAGCCCAGACCT-3′) primers for P383L, and (5′-GCCAGCCGGGCTCCACCTCTCTGGACTTATTCAAGTTCTATGTGGAGGAG-3′) and (5′-CTCCTCCACATAGAACTTGAATAAGTCCAGAGAGGTGGAGCCCGGCTGGC-3′) for P540S. We used the following PCR mutagenesis conditions: one cycle at 95°C for 2 min, 18 cycles at 98°C for 30 sec followed by 50°C or 55°C annealing for 1 min and extension at 72°C for 5 min, plus final extension for 5 min. We treated PCR products with 20 U DpnI (New England Biolabs) to remove methylated PCR templates followed by DH5α transformation. After overnight growth with Lysogeny broth (LB) and 70 µg/mL ampicillin, we purified plasmid DNA by miniprep (Omega Bio-Tek) and confirmed all sequences by Sanger sequencing with specific primers (1st Base Asia).
RNA extraction and RT-PCR
We collected 2 × 106 cells for each condition, washed them with 1× PBS, and extracted total RNA using PureLink RNA Mini Kit (Life Technologies) following the kit's manual. Briefly, we lysed cells by incubating them for 2 min with 600 µL lysis buffer with 2-mercaptoethanol (Sigma). We then homogenized the samples, followed by addition of equal volume of 70% ethanol. We passed the lysate through the RNeasy mini-column to allow RNA binding to the membrane. We discarded the flow through and applied wash buffers to wash the column. We then carefully transferred the RNeasy column to a new 1.5 mL collection tube and eluted them with RNase-free water. In order to remove genomic DNA contaminations, we treated samples with TurboDNase (Life Technologies) according to the manufacturer's protocol. Briefly, we added TurboDNase (2 U/µL) to each sample and incubated them at 37°C for 30 min. Subsequently, we inactivated reactions at 72°C for 10 min and resuspended RNA in RNase-free water. For RNA-sequencing samples, we assessed RNA quality after DNase treatment by gel electrophoresis and by absorbance using a Nanodrop Spectrometer (Thermo Scientific). Finally, we purified and concentrated samples by RNA Clean & Concentrator-5 (Zymo Research) according to the manufacturer's protocol. After RNA concentration, we assessed RNA quality by Picogreen assay and determined RIN for each sample by Bioanalyzer 2100 system (Agilent). We reverse transcribed 1 µg of RNA to cDNA by heating it up with deoxynucleotide (dNTPs, 10 nM) and oligodT (40 µM) at 65°C for 5 min in order to disassociate RNA secondary structures. Subsequently, we performed reverse transcription by adding 1 µL of M-MuLV reverse transcriptase (New England Biolabs) and 10× buffer containing 1 mM dithiothreitol (DTT) for 2 h at 42°C. We performed quantitative RT-PCR using the SYBR GreenMaster Mix (Thermo Fisher Scientific), according to the kit's manual, on CFX96 Touch Real-Time PCR Detection System (Bio-Rad). All amplicons were shorter than 300 bp, with primer sequences available upon request. We used hACTB (β-actin) as real-time RT-PCR reference gene. We calculated the relative expression of three technical replicates of each target gene by the following equation: 2(Cq(β-actin) – Cq(target gene)). We plotted means and standard deviations. We performed semi-quantitative RT-PCR to validate AS events using primer pairs mapping to the flanking exons of each target cassette exon, with sequences available upon request.
Transfection with overexpression plasmids
Initially, we performed titration and time-course experiments by mixing PRPF40B-WT and mock pUC19 plasmids in different ratios in order to find the optimal amount of plasmid and time to extract total RNA and proteins, so that exogenous PRPF40B expression by transient transfection matches that in parental K562 cells. Transfection efficiency was acceptable at ∼50%, yet each cell would uptake a different amount of plasmid and/or express it at different levels. Despite these limitations, a number of DEGs and DASEs in the KO could be rescued by this system. We performed rescue experiments in triplicates by overexpressing PRPF40B both as WT and with two MDS mutations. We overexpressed mutants both alone as homozygous-like and mixed in a 1:1 ratio with WT plasmid as heterozygous-like. We seeded K562-PRPF40B-KO cells at a density of 0.2 × 106 cells/mL in a six-well plate before transfection in DMEM, 1% Pen/Strep and 10% FBS. We mixed plasmid DNA with 100 µL Opti-MEM (Life Technologies) and XtremeGENE HP transfection reagent (Roche) in a 1:2 DNA:reagent ratio. We then incubated the transfection mixture at room temperature for 40 min before adding it to cells in a drop-wise manner.
Western blotting
We lysed cells in RIPA buffer [10 mM Tris-Cl (pH 8.0), 1 mM EDTA, 1% Triton X-100, 0.1% sodium deoxycholate, 0.1% SDS, 140 mM NaCl, 1 mM PMSF] for 60 min on ice, and collected supernatants after centrifugation at maximum speed for 30 min. We determined protein concentrations by Bradford Assay (Bio-Rad). Before gel loading, we boiled 40 µg of protein per sample for 10 min with sodium dodecyl sulfate (SDS)-loading buffer. We separated cell lysates by SDS-PAGE on an 8% gel (Acrylamide/Bis 37.5:1; Bio-Rad) in 1× SDS running buffer (25 mM Tris, 192 mM glycine, 0.1% SDS), and transferred proteins to a polyvinylidene difluoride (PVDF) membrane (Bio-Rad) in transfer buffer (25 mM Tris, 192 mM glycine, 20% methanol) at 100 V for 1.5 h using a wet-tank electrotransfer system (Bio-Rad). We blocked membranes in 5% milk in 1× TBST (144 mM NaCl, 20 mM Tris, 0.05% Tween 20, pH 7.6) for 1 h and washed them with 1× TBST thrice, each for 5 min. We incubated membranes with the primary antibody anti-rabbit PRPF40B (Abcam) or anti-mouse GAPDH (Sigma) in 1× TBST at 4°C overnight followed by three washes with 1× TBST. We incubated membranes with secondary anti-mouse/rabbit IgG-HRP antibodies (BioLegend, Sigma, Santa Cruz Biotechnology) diluted in 1× TBST with or without 5% milk. After three washes, we detected proteins by Western Lightning Plus ECL (PerkinElmer).
Inhibition of Pol II mRNA transcription
We seeded nonedited K562, K562-PRPF40B-KO and rescued cells at a concentration of 0.5 × 106 cells/mL and incubated with either dimethyl sulfoxide (DMSO), 50 µM DRB or 1 µM CPT for 5 h to inhibit transcription (Tang et al. 2016). After RNA extraction and cDNA synthesis, we used primers designed in exons flanking the alternative exon 8 of hnRNPDL to detect the efficiency of the drugs by RT-PCR (FW-hnRNPDL.Ex7 5′-GCCAACAGAGCCACTTATGG-3′, RW-hnRNPDL.Ex9 5′-ACAGTTGGACACAATGGTGTC-3′). Primers for AFMID and SPTAN1 were the same as in Figure 5.
RNA-sequencing library preparation
We prepared RNA libraries using the TruSeq Stranded mRNA Library Prep Kit (Illumina). In brief, we purified poly(A) containing mRNAs by poly(T) oligonucleotides attached to magnetic beads. We cleaved and fragmented mRNAs into small pieces, followed by reverse transcription to generate the first cDNA strand using random primers. To track the strand information in the following steps, we replaced dTTP with dUTP in the second strand synthesis by using DNA polymerase I and RNase H. We ligated the cDNA fragments with the adapter and purified them. We enriched the molecules by PCR to generate the final cDNA library. We used HiSeq 2500 System Rapid mode (Illumina) for RNA-sequencing, giving rise to paired-end reads of 101 bp.
RNA-seq data analysis
As mentioned before (Liu et al. 2018), we first confirmed the original read quality of our RNA-seq data by FastQC (Simon Andrews, Babraham Bioinformatics). For gene expression analysis, we aligned the paired-end reads to human genome (UCSC-hg19) using Tophat2 (Trapnell et al. 2009) with Bowtie2 (Langmead 2010). In RNA-seq data analysis, we used the default parameters for each tool unless otherwise stated. For Tophat2, we allowed a maximum of two mismatches and set two parameters as –library-type=fr-firststrand and –read-realign-edit-dist=0. We used Cufflinks, Cuffmerge, and Cuffdiff (Trapnell et al. 2012) for transcripts assembly and quantification of the Tophat2 output. We obtained more than 92% aligned reads to the genome with Tophat2 (details available upon request). We used the common FPKM (fragments per kilobase of transcript per million fragments mapped) as the expression levels measured for each gene or transcript. We obtained the information about the correlation between samples with CummeRbund. The log10(FPKM) values for each individual condition across replicates highlighted the strong reproducibility of our data. To detect DASEs, we used the software rMATS (Shen et al. 2014), using the alignment outputs of Tophat2. Our read type is paired-end (-t paired) and with a length of 101 bp (-len 101), while the analysis type was the default unpaired. We obtained information about changes in AS for each type of AS event (AA, AD, SE, IR, MXE), with total numbers available upon request. For the rMATS AS list, we applied the cutoffs of ΔPSI ≥ 0.10 or ΔPSI ≤ −0.10 and FDR < 1. We sorted the data using the R package dplyr to obtain a list of PRPF40B-regulated DASEs having common coordinates between comparisons of the WT versus KO and KO versus Rescue. Then we compared overlapping SE events with the lists of cassette exons before FDR and ΔPSI cutoff for each rescue with PRPF40B mutants.
We performed GSEA of the DEGs and DASEs regulated by PRPF40B both by the online tool ConsensusPathDB (Max Plank Institute for Molecular Genetics) (Kamburov et al. 2011) and Enrichr (Kuleshov et al. 2016). We generated the heatmaps of the most important selected genes involved in hypoxia according to the literature using the R package heatmap2. We built PPI network for the DASEs regulated by PRPF40B with the online database STRING v10.5 (https://string-db.org/) with a high confidence score of 0.7 and a K-means clustering score of 10 (Szklarczyk et al. 2017). From the human genome hg19, we retrieved sequences of the 5′ss and 3′ss for all the PRPF40B-regulated cassette exons with BEDTools package (Quinlan and Hall 2010). As background, we used a list of constitutive exons present on the same mRNA transcript where the DASEs were detected. As a list of DASEs that are not regulated by PRPF40B, we used events from the RNA-seq which did not pass the cutoff of FDR < 1. We used the splice sites as input for the online tool MaxEntScan (Yeo and Burge 2004) to calculate the entropy score for each single sequence, in order to obtain the 5′ss and 3′ss scores. We performed statistical analysis to obtain P-values between the different comparisons by Wilcoxon test, and generated boxplots with the R package ggplot2 (Ito and Murphy 2013). We retrieved from the genome the sequences of the PRPF40B-regulated cassette exons flanked by 200 bp in the upstream and downstream introns with the BEDTools package. We performed motifs analysis by DREME, a subprogram of the online web portal MEME Suite v4.12.0 (Bailey et al. 2009) in order to elucidate the most enriched motifs in these sequences. DREME automatically uses shuffled sequences as a background. We mined the TCGA data with the online tool cBioPortal (Cerami et al. 2012) and other databases such as Oncogeo, Gent, and Gepia (Shin et al. 2011; Tang et al. 2017).
DATA DEPOSITION
We deposited the raw fastq files in the Sequence Read Archives (SRA) of the National Center for Biotechnology Information (NCBI) under accession number SRP197235 of Bioproject PRJNA540831. All DEGs and DASEs in Excel files are available at Data Repository-NTU (DR-NTU): https://doi.org/10.21979/N9/MUULD6 and https://doi.org/10.21979/N9/3RYIVP.
ACKNOWLEDGMENTS
We thank all members of the Roca laboratory for advice. We are grateful to Daniela Moses for RNA library preparation and for running the Illumina HiSeq machine. We also thank Fursham Hamid for advice. This work was supported by Academic Research Fund Tier 2 [MOE2013-T2-1-101 ARC 45/13 and MOE2016-T2-2-104(S)], both from Singapore's Ministry of Education (https://www.olga.moe.gov.sg/T2/default.aspx). The funders played no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Author contributions: P.A.L. performed most of the wet-lab and in silico experiments. R.S.E.C. performed most of the RT-PCR validation experiments and analysis. J.Y.Y. also contributed some RT-PCRs, and C.W.T. performed the genomic and expression validations of the knockout. F.Z. helped in the AS analyses under supervision of J.Z. X.R. designed the study, supervised all work, analyzed data, and wrote the manuscript with the help of P.A.L. All other authors read and approved the manuscript.
Footnotes
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.069534.118.
Freely available online through the RNA Open Access option.
REFERENCES
- Abovich N, Rosbash M. 1997. Cross-intron bridging interactions in the yeast commitment complex are conserved in mammals. Cell 89: 403–412. 10.1016/S0092-8674(00)80221-4 [DOI] [PubMed] [Google Scholar]
- Allen M, Friedler A, Schon O, Bycroft M. 2002. The structure of an FF domain from human HYPA/FBP11. J Mol Biol 323: 411–416. 10.1016/S0022-2836(02)00968-3 [DOI] [PubMed] [Google Scholar]
- Alsafadi S, Houy A, Battistella A, Popova T, Wassef M, Henry E, Tirode F, Constantinou A, Piperno-Neumann S, Roman-Roman S, et al. 2016. Cancer-associated SF3B1 mutations affect alternative splicing by promoting alternative branchpoint usage. Nat Commun 7: 10615 10.1038/ncomms10615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez-Tejado M, Naranjo-Suárez S, Jiménez C, Carrera AC, Landázuri MO, del Peso L. 2001. Hypoxia induces the activation of the phosphatidylinositol 3-kinase/Akt cell survival pathway in PC12 cells: protective role in apoptosis. J Biol Chem 276: 22368–22374. 10.1074/jbc.M011688200 [DOI] [PubMed] [Google Scholar]
- Anczuków O, Krainer AR. 2016. Splicing-factor alterations in cancers. RNA 22: 1285–1301. 10.1261/rna.057919.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey TL, Boden M, Buske FA, Frith M, Grant CE, Clementi L, Ren J, Li WW, Noble WS. 2009. MEME SUITE: tools for motif discovery and searching. Nucleic Acids Res 37: W202–W208. 10.1093/nar/gkp335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao J, Tang C, Li J, Zhang Y, Bhetwal BP, Zheng H, Yan W. 2014. RAN-binding protein 9 is involved in alternative splicing and is critical for male germ cell development and male fertility. PLoS Genet 10: e1004825 10.1371/journal.pgen.1004825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbosa-Morais NL, Irimia M, Pan Q, Xiong HY, Gueroussov S, Lee LJ, Slobodeniuc V, Kutter C, Watt S, Colak R, et al. 2012. The evolutionary landscape of alternative splicing in vertebrate species. Science 338: 1587–1593. 10.1126/science.1230612 [DOI] [PubMed] [Google Scholar]
- Bauman JA, Li SD, Yang A, Huang L, Kole R. 2010. Anti-tumor activity of splice-switching oligonucleotides. Nucleic Acids Res 38: 8348–8356. 10.1093/nar/gkq731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becerra S, Montes M, Hernández-Munain C, Suñé C. 2015. Prp40 pre-mRNA processing factor 40 homolog B (PRPF40B) associates with SF1 and U2AF65 and modulates alternative pre-mRNA splicing in vivo. RNA 21: 438–457. 10.1261/rna.047258.114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becerra S, Andrés-Leon E, Prieto-Sánchez S, Hernández-Munain C, Suñé C. 2016. Prp40 and early events in splice site definition. Wiley Interdiscip Rev RNA 7: 17–32. 10.1002/wrna.1312 [DOI] [PubMed] [Google Scholar]
- Benelli R, Lorusso G, Albini A, Noonan DM. 2006. Cytokines and chemokines as regulators of angiogenesis in health and disease. Curr Pharm Des 12: 3101–3115. 10.2174/138161206777947461 [DOI] [PubMed] [Google Scholar]
- Busch A, Hertel KJ. 2012. Evolution of SR protein and hnRNP splicing regulatory factors. Wiley Interdiscip Rev RNA 3: 1–12. 10.1002/wrna.100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, et al. 2012. The cBio Cancer Genomics Portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov 2: 401–404. 10.1158/2159-8290.CD-12-0095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheah JJC, Hahn CN, Hiwase DK, Scott HS, Brown AL. 2017. Myeloid neoplasms with germline DDX41 mutation. Int J Hematol 106: 163–174. 10.1007/s12185-017-2260-y [DOI] [PubMed] [Google Scholar]
- Dai T, Zheng H, Fu GS. 2008. Hypoxia confers protection against apoptosis via the PI3K/Akt pathway in endothelial progenitor cells. Acta Pharmacol Sin 29: 1425–1431. 10.1111/j.1745-7254.2008.00904.x [DOI] [PubMed] [Google Scholar]
- Darman RB, Seiler M, Agrawal AA, Lim KH, Peng S, Aird D, Bailey SL, Bhavsar EB, Chan B, Colla S, et al. 2015. Cancer-associated SF3B1 hotspot mutations induce cryptic 3′ splice site selection through use of a different branch point. Cell Rep 13: 1033–1045. 10.1016/j.celrep.2015.09.053 [DOI] [PubMed] [Google Scholar]
- DeBoever C, Ghia EM, Shepard PJ, Rassenti L, Barrett CL, Jepsen K, Jamieson CH, Carson D, Kipps TJ, Frazer KA. 2015. Transcriptome sequencing reveals potential mechanism of cryptic 3′ splice site selection in SF3B1-mutated cancers. PLoS Comput Biol 11: e1004105 10.1371/journal.pcbi.1004105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de la Mata M, Alonso CR, Kadener S, Fededa JP, Blaustein M, Pelisch F, Cramer P, Bentley D, Kornblihtt AR. 2003. A slow RNA polymerase II affects alternative splicing in vivo. Mol Cell 12: 525–532. 10.1016/j.molcel.2003.08.001 [DOI] [PubMed] [Google Scholar]
- Deynoux M, Sunter N, Hérault O, Mazurier F. 2016. Hypoxia and hypoxia-inducible factors in leukemias. Front Oncol 6: 41 10.3389/fonc.2016.00041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolatshad H, Pellagatti A, Fernandez-Mercado M, Yip BH, Malcovati L, Attwood M, Przychodzen B, Sahgal N, Kanapin AA, Lockstone H, et al. 2015. Disruption of SF3B1 results in deregulated expression and splicing of key genes and pathways in myelodysplastic syndrome hematopoietic stem and progenitor cells. Leukemia 29: 1092–1103. 10.1038/leu.2014.331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dujardin G, Lafaille C, de la Mata M, Marasco LE, Munoz MJ, Le Jossic-Corcos C, Corcos L, Kornblihtt AR. 2014. How slow RNA polymerase II elongation favors alternative exon skipping. Mol Cell 54: 683–690. 10.1016/j.molcel.2014.03.044 [DOI] [PubMed] [Google Scholar]
- Faber PW, Barnes GT, Srinidhi J, Chen J, Gusella JF, MacDonald ME. 1998. Huntingtin interacts with a family of WW domain proteins. Hum Mol Genet 7: 1463–1474. 10.1093/hmg/7.9.1463 [DOI] [PubMed] [Google Scholar]
- Grellscheid SN, Dalgliesh C, Rozanska A, Grellscheid D, Bourgeois CF, Stevenin J, Elliott DJ. 2011. Molecular design of a splicing switch responsive to the RNA binding protein Tra2β. Nucleic Acids Res 39: 8092–8104. 10.1093/nar/gkr495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haase VH. 2013. Regulation of erythropoiesis by hypoxia-inducible factors. Blood Rev 27: 41–53. 10.1016/j.blre.2012.12.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatfield KJ, Bedringsaas SL, Ryningen A, Gjertsen BT, Bruserud O. 2010. Hypoxia increases HIF-1α expression and constitutive cytokine release by primary human acute myeloid leukaemia cells. Eur Cytokine Netw 21: 154–164. 10.1684/ecn.2010.0204 [DOI] [PubMed] [Google Scholar]
- Hiller M, Huse K, Szafranski K, Jahn N, Hampe J, Schreiber S, Backofen R, Platzer M. 2004. Widespread occurrence of alternative splicing at NAGNAG acceptors contributes to proteome plasticity. Nat Genet 36: 1255–1257. 10.1038/ng1469 [DOI] [PubMed] [Google Scholar]
- Hsu PD, Lander ES, Zhang F. 2014. Development and applications of CRISPR-Cas9 for genome engineering. Cell 157: 1262–1278. 10.1016/j.cell.2014.05.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang S, Nguyen AD, Jo Y, Engelking LJ, Brugarolas J, DeBose-Boyd RA. 2017. Hypoxia-inducible factor 1α activates insulin-induced gene 2 (Insig-2) transcription for degradation of 3-hydroxy-3-methylglutaryl (HMG)-CoA reductase in the liver. J Biol Chem 292: 9382–9393. 10.1074/jbc.M117.788562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ilagan JO, Ramakrishnan A, Hayes B, Murphy ME, Zebari AS, Bradley P, Bradley RK. 2015. U2AF1 mutations alter splice site recognition in hematological malignancies. Genome Res 25: 14–26. 10.1101/gr.181016.114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue D, Bradley RK, Abdel-Wahab O. 2016. Spliceosomal gene mutations in myelodysplasia: molecular links to clonal abnormalities of hematopoiesis. Genes Dev 30: 989–1001. 10.1101/gad.278424.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ip JY, Schmidt D, Pan Q, Ramani AK, Fraser AG, Odom DT, Blencowe BJ. 2011. Global impact of RNA polymerase II elongation inhibition on alternative splicing regulation. Genome Res 21: 390–401. 10.1101/gr.111070.110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito K, Murphy D. 2013. Application of ggplot2 to pharmacometric graphics. CPT Pharmacometrics Syst Pharmacol 2: e79 10.1038/psp.2013.56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamburov A, Pentchev K, Galicka H, Wierling C, Lehrach H, Herwig R. 2011. ConsensusPathDB: toward a more complete picture of cell biology. Nucleic Acids Res 39: D712–D717. 10.1093/nar/gkq1156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kao HY, Siliciano PG. 1996. Identification of Prp40, a novel essential yeast splicing factor associated with the U1 small nuclear ribonucleoprotein particle. Mol Cell Biol 16: 960–967. 10.1128/MCB.16.3.960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karamichos D, Hutcheon AE, Zieske JD. 2011. Transforming growth factor-β3 regulates assembly of a non-fibrotic matrix in a 3D corneal model. J Tissue Eng Regen Med 5: e228–e238. 10.1002/term.429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim E, Ilagan JO, Liang Y, Daubner GM, Lee SC, Ramakrishnan A, Li Y, Chung YR, Micol JB, Murphy ME, et al. 2015. SRSF2 mutations contribute to myelodysplasia by mutant-specific effects on exon recognition. Cancer Cell 27: 617–630. 10.1016/j.ccell.2015.04.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuleshov MV, Jones MR, Rouillard AD, Fernandez NF, Duan Q, Wang Z, Koplev S, Jenkins SL, Jagodnik KM, Lachmann A, et al. 2016. Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res 44: W90–W97. 10.1093/nar/gkw377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B. 2010. Aligning short sequencing reads with Bowtie. Curr Protoc Bioinformatics 32: 11.7.1–11.7.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee Y, Rio DC. 2015. Mechanisms and regulation of alternative pre-mRNA splicing. Annu Rev Biochem 84: 291–323. 10.1146/annurev-biochem-060614-034316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao D, Johnson RS. 2007. Hypoxia: a key regulator of angiogenesis in cancer. Cancer Metastasis Rev 26: 281–290. 10.1007/s10555-007-9066-y [DOI] [PubMed] [Google Scholar]
- Licatalosi DD, Darnell RB. 2010. RNA processing and its regulation: global insights into biological networks. Nat Rev Genet 11: 75–87. 10.1038/nrg2673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin KT, Lu RM, Tarn WY. 2004. The WW domain-containing proteins interact with the early spliceosome and participate in pre-mRNA splicing in vivo. Mol Cell Biol 24: 9176–9185. 10.1128/MCB.24.20.9176-9185.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H, Lorenzini PA, Zhang F, Xu S, Wong MSM, Zheng J, Roca X. 2018. Alternative splicing analysis in human monocytes and macrophages reveals MBNL1 as major regulator. Nucleic Acids Res 46: 6069–6086. 10.1093/nar/gky401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madan V, Kanojia D, Li J, Okamoto R, Sato-Otsubo A, Kohlmann A, Sanada M, Grossmann V, Sundaresan J, Shiraishi Y, et al. 2015. Aberrant splicing of U12-type introns is the hallmark of ZRSR2 mutant myelodysplastic syndrome. Nat Commun 6: 6042 10.1038/ncomms7042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makarov EM, Owen N, Bottrill A, Makarova OV. 2012. Functional mammalian spliceosomal complex E contains SMN complex proteins in addition to U1 and U2 snRNPs. Nucleic Acids Res 40: 2639–2652. 10.1093/nar/gkr1056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matouk IJ, Halle D, Gilon M, Hochberg A. 2015. The non-coding RNAs of the H19-IGF2 imprinted loci: a focus on biological roles and therapeutic potential in lung cancer. J Transl Med 13: 113 10.1186/s12967-015-0467-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merkin J, Russell C, Chen P, Burge CB. 2012. Evolutionary dynamics of gene and isoform regulation in mammalian tissues. Science 338: 1593–1599. 10.1126/science.1228186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizutani K, Suetsugu S, Takenawa T. 2004. FBP11 regulates nuclear localization of N-WASP and inhibits N-WASP-dependent microspike formation. Biochem Biophys Res Commun 313: 468–474. 10.1016/j.bbrc.2003.11.139 [DOI] [PubMed] [Google Scholar]
- Montague TG, Cruz JM, Gagnon JA, Church GM, Valen E. 2014. CHOPCHOP: a CRISPR/Cas9 and TALEN web tool for genome editing. Nucleic Acids Res 42: W401–W407. 10.1093/nar/gku410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris DP, Greenleaf AL. 2000. The splicing factor, Prp40, binds the phosphorylated carboxyl-terminal domain of RNA polymerase II. J Biol Chem 275: 39935–39943. 10.1074/jbc.M004118200 [DOI] [PubMed] [Google Scholar]
- Murphy BJ, Andrews GK, Bittel D, Discher DJ, McCue J, Green CJ, Yanovsky M, Giaccia A, Sutherland RM, Laderoute KR, et al. 1999. Activation of metallothionein gene expression by hypoxia involves metal response elements and metal transcription factor-1. Cancer Res 59: 1315–1322. [PubMed] [Google Scholar]
- Naftelberg S, Schor IE, Ast G, Kornblihtt AR. 2015. Regulation of alternative splicing through coupling with transcription and chromatin structure. Annu Rev Biochem 84: 165–198. 10.1146/annurev-biochem-060614-034242 [DOI] [PubMed] [Google Scholar]
- Naumann S, Reutzel D, Speicher M, Decker HJ. 2001. Complete karyotype characterization of the K562 cell line by combined application of G-banding, multiplex-fluorescence in situ hybridization, fluorescence in situ hybridization, and comparative genomic hybridization. Leuk Res 25: 313–322. 10.1016/S0145-2126(00)00125-9 [DOI] [PubMed] [Google Scholar]
- Nilsen TW, Graveley BR. 2010. Expansion of the eukaryotic proteome by alternative splicing. Nature 463: 457–463. 10.1038/nature08909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oleksiewicz U, Liloglou T, Tasopoulou KM, Daskoulidou N, Gosney JR, Field JK, Xinarianos G. 2017. COL1A1, PRPF40A, and UCP2 correlate with hypoxia markers in non-small cell lung cancer. J Cancer Res Clin Oncol 143: 1133–1141. 10.1007/s00432-017-2381-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padgett RA. 2012. New connections between splicing and human disease. Trends Genet 28: 147–154. 10.1016/j.tig.2012.01.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry JC, Guindalini C, Bittencourt L, Garbuio S, Mazzotti DR, Tufik S. 2013. Whole blood hypoxia-related gene expression reveals novel pathways to obstructive sleep apnea in humans. Respir Physiol Neurobiol 189: 649–654. 10.1016/j.resp.2013.08.012 [DOI] [PubMed] [Google Scholar]
- Pilon AM, Arcasoy MO, Dressman HK, Vayda SE, Maksimova YD, Sangerman JI, Gallagher PG, Bodine DM. 2008. Failure of terminal erythroid differentiation in EKLF-deficient mice is associated with cell cycle perturbation and reduced expression of E2F2. Mol Cell Biol 28: 7394–7401. 10.1128/MCB.01087-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan AR, Hall IM. 2010. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26: 841–842. 10.1093/bioinformatics/btq033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Resch A, Xing Y, Modrek B, Gorlick M, Riley R, Lee C. 2004. Assessing the impact of alternative splicing on domain interactions in the human proteome. J Proteome Res 3: 76–83. 10.1021/pr034064v [DOI] [PubMed] [Google Scholar]
- Roca X, Krainer AR, Eperon IC. 2013. Pick one, but be quick: 5′ splice sites and the problems of too many choices. Genes Dev 27: 129–144. 10.1101/gad.209759.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rybinska I, Cairo G. 2017. Mutual cross talk between iron homeostasis and erythropoiesis. Vitam Horm 105: 143–160. 10.1016/bs.vh.2017.01.001 [DOI] [PubMed] [Google Scholar]
- Sánchez-Álvarez M, Montes M, Sánchez-Hernández N, Hernández-Munain C, Suñé C. 2010. Differential effects of sumoylation on transcription and alternative splicing by transcription elongation regulator 1 (TCERG1). J Biol Chem 285: 15220–15233. 10.1074/jbc.M109.063750 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen S, Park JW, Lu ZX, Lin L, Henry MD, Wu YN, Zhou Q, Xing Y. 2014. rMATS: robust and flexible detection of differential alternative splicing from replicate RNA-Seq data. Proc Natl Acad Sci 111: E5593–E5601. 10.1073/pnas.1419161111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin C, Manley JL. 2002. The SR protein SRp38 represses splicing in M phase cells. Cell 111: 407–417. 10.1016/S0092-8674(02)01038-3 [DOI] [PubMed] [Google Scholar]
- Shin G, Kang TW, Yang S, Baek SJ, Jeong YS, Kim SY. 2011. GENT: gene expression database of normal and tumor tissues. Cancer Inform 10: 149–157. 10.4137/CIN.S7226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirai CL, Ley JN, White BS, Kim S, Tibbitts J, Shao J, Ndonwi M, Wadugu B, Duncavage EJ, Okeyo-Owuor T, et al. 2015. Mutant U2AF1 expression alters hematopoiesis and pre-mRNA splicing in vivo. Cancer Cell 27: 631–643. 10.1016/j.ccell.2015.04.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siatecka M, Bieker JJ. 2011. The multifunctional role of EKLF/KLF1 during erythropoiesis. Blood 118: 2044–2054. 10.1182/blood-2011-03-331371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh H, Lane AA, Correll M, Przychodzen B, Sykes DB, Stone RM, Ballen KK, Amrein PC, Maciejewski J, Attar EC. 2013. Putative RNA-splicing gene LUC7L2 on 7q34 represents a candidate gene in pathogenesis of myeloid malignancies. Blood Cancer J 3: e117 10.1038/bcj.2013.16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, et al. 2005. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci 102: 15545–15550. 10.1073/pnas.0506580102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutherland JA, Turner AR, Mannoni P, McGann LE, Turc JM. 1986. Differentiation of K562 leukemia cells along erythroid, macrophage, and megakaryocyte lineages. J Biol Response Mod 5: 250–262. [PubMed] [Google Scholar]
- Swietach P, Vaughan-Jones RD, Harris AL. 2007. Regulation of tumor pH and the role of carbonic anhydrase 9. Cancer Metastasis Rev 26: 299–310. 10.1007/s10555-007-9064-0 [DOI] [PubMed] [Google Scholar]
- Szklarczyk D, Morris JH, Cook H, Kuhn M, Wyder S, Simonovic M, Santos A, Doncheva NT, Roth A, Bork P, et al. 2017. The STRING database in 2017: quality-controlled protein-protein association networks, made broadly accessible. Nucleic Acids Res 45: D362–D368. 10.1093/nar/gkw937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan J, Ho JX, Zhong Z, Luo S, Chen G, Roca X. 2016. Noncanonical registers and base pairs in human 5′ splice-site selection. Nucleic Acids Res 44: 3908–3921. 10.1093/nar/gkw163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang SJ, Luo S, Ho JX, Ly PT, Goh E, Roca X. 2016. Characterization of the regulation of CD46 RNA alternative splicing. J Biol Chem 291: 14311–14323. 10.1074/jbc.M115.710350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang Z, Li C, Kang B, Gao G, Li C, Zhang Z. 2017. GEPIA: a web server for cancer and normal gene expression profiling and interactive analyses. Nucleic Acids Res 45: W98–W102. 10.1093/nar/gkx247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong H, Hu C, Zhuang Z, Wang L, Jin J. 2012. Hypoxia-inducible factor-1α expression indicates poor prognosis in myelodysplastic syndromes. Leuk Lymphoma 53: 2412–2418. 10.3109/10428194.2012.696637 [DOI] [PubMed] [Google Scholar]
- Trapnell C, Pachter L, Salzberg SL. 2009. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics 25: 1105–1111. 10.1093/bioinformatics/btp120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, Pimentel H, Salzberg SL, Rinn JL, Pachter L. 2012. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protoc 7: 562–578. 10.1038/nprot.2012.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veloso A, Kirkconnell KS, Magnuson B, Biewen B, Paulsen MT, Wilson TE, Ljungman M. 2014. Rate of elongation by RNA polymerase II is associated with specific gene features and epigenetic modifications. Genome Res 24: 896–905. 10.1101/gr.171405.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong HY, Alipanahi B, Lee LJ, Bretschneider H, Merico D, Yuen RK, Hua Y, Gueroussov S, Najafabadi HS, Hughes TR, et al. 2015. RNA splicing. The human splicing code reveals new insights into the genetic determinants of disease. Science 347: 1254806 10.1126/science.1254806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeh CS, Wang JY, Chung FY, Lee SC, Huang MY, Kuo CW, Yang MJ, Lin SR. 2008. Significance of the glycolytic pathway and glycolysis related-genes in tumorigenesis of human colorectal cancers. Oncol Rep 19: 81–91. [PubMed] [Google Scholar]
- Yeo G, Burge CB. 2004. Maximum entropy modeling of short sequence motifs with applications to RNA splicing signals. J Comput Biol 11: 377–394. 10.1089/1066527041410418 [DOI] [PubMed] [Google Scholar]
- Yoshida K, Sanada M, Shiraishi Y, Nowak D, Nagata Y, Yamamoto R, Sato Y, Sato-Otsubo A, Kon A, Nagasaki M, et al. 2011. Frequent pathway mutations of splicing machinery in myelodysplasia. Nature 478: 64–69. 10.1038/nature10496 [DOI] [PubMed] [Google Scholar]
- Zhang J, Ney PA. 2009. Role of BNIP3 and NIX in cell death, autophagy, and mitophagy. Cell Death Differ 16: 939–946. 10.1038/cdd.2009.16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang CC, Sadek HA. 2014. Hypoxia and metabolic properties of hematopoietic stem cells. Antioxid Redox Signal 20: 1891–1901. 10.1089/ars.2012.5019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Lieu YK, Ali AM, Penson A, Reggio KS, Rabadan R, Raza A, Mukherjee S, Manley JL. 2015. Disease-associated mutation in SRSF2 misregulates splicing by altering RNA-binding affinities. Proc Natl Acad Sci 112: E4726–E4734. 10.1073/pnas.1514105112 [DOI] [PMC free article] [PubMed] [Google Scholar]