

Abstract
We describe the development and application of a novel series of vectors that facilitate CRISPR-Cas9-mediated genome editing in mammalian cells, which we call CRISPR-Bac. CRISPR-Bac leverages the piggyBac transposon to randomly insert CRISPR-Cas9 components into mammalian genomes. In CRISPR-Bac, a single piggyBac cargo vector containing a doxycycline-inducible Cas9 or catalytically dead Cas9 (dCas9) variant and a gene conferring resistance to Hygromycin B is cotransfected with a plasmid expressing the piggyBac transposase. A second cargo vector, expressing a single-guide RNA (sgRNA) of interest, the reverse-tetracycline TransActivator (rtTA), and a gene conferring resistance to G418, is also cotransfected. Subsequent selection on Hygromycin B and G418 generates polyclonal cell populations that stably express Cas9, rtTA, and the sgRNA(s) of interest. We show that CRISPR-Bac can be used to knock down proteins of interest, to create targeted genetic deletions with high efficiency, and to activate or repress transcription of protein-coding genes and an imprinted long noncoding RNA. The ratio of sgRNA-to-Cas9-to-transposase can be adjusted in transfections to alter the average number of cargo insertions into the genome. sgRNAs targeting multiple genes can be inserted in a single transfection. CRISPR-Bac is a versatile platform for genome editing that simplifies the generation of mammalian cells that stably express the CRISPR-Cas9 machinery.
Keywords: CRISPR, piggyBac, genome editing, stem cells, regulatory element, lncRNA
INTRODUCTION
Within the last decade, the CRISPR (clustered regularly interspaced short palindromic repeat) bacterial immune system has provided researchers with multiple new methods to control gene expression in mammalian genomes. Coexpression of the Cas9 (CRISPR-associated protein 9) nuclease from Streptococcus pyogenes along with an engineered single guide RNA (sgRNA) that targets a protein-coding exon is an effective way to introduce frameshift mutations in proteins of interest, owing to the fact that repair of the DNA break introduced by Cas9 often results in small deletions surrounding the cut site. Coexpression of Cas9 and multiple sgRNAs can also be used to excise larger regions from genes of interest, or to excise DNA regulatory elements (Ran et al. 2013; Canver et al. 2014; Aparicio-Prat et al. 2015; Zhu et al. 2016; Gasperini et al. 2017). Expression of a catalytically dead Cas9 (dCas9) fused to a transcriptional activation or repression domain can be used to up- or down-regulate gene expression when sgRNAs are targeted to promoters or regulatory elements of interest (Hsu et al. 2014; Wright et al. 2016).
Owing to the broad utility of CRISPR, multiple methods have been developed to deliver the CRISPR-Cas9 machinery to mammalian cells. Transient transfection of Cas9- and sgRNA-expressing plasmids, or of Cas9 protein and in vitro synthesized sgRNAs, are useful when the efficiency of transfection for the cell type of interest is high and when the desired endpoint can be reached via transient expression of Cas9 and the sgRNA. Lentiviral delivery of Cas9/sgRNA vectors is also possible, and provides distinct advantages when transfection efficiency is low, or when the desired endpoint requires stable expression and or integration of Cas9/sgRNAs into the genome, such as for studies performed in vivo or for genome-wide phenotypic screens (Hartenian and Doench 2015; Joung et al. 2017). However, delivery of the CRISPR machinery via lentivirus requires additional hands-on time, expertise, safety precautions, and cost relative to delivery via transient transfection.
The piggyBac transposon is a broadly used tool that allows DNA cargos up to 100 kilobases in length to be inserted into “AATT” sequences that are preferentially located in euchromatic regions of mammalian genomes (Ding et al. 2005; Cadinaños and Bradley 2007; Wilson et al. 2007; Wang et al. 2008; Li et al. 2011). Owing to its high efficiency of transposition, piggyBac has been used in a wide range of applications, including in the stable expression of multisubunit protein complexes, in the generation of transgenic mice and induced pluripotent stem cells, and in the large-scale production of recombinant proteins (Ding et al. 2005; Kaji et al. 2009; Yusa et al. 2009; Kahlig et al. 2010; Li et al. 2013). Most recently, piggyBac has begun to be used for CRISPR-based applications; piggyBac vectors have been used to study CRISPR off-target effects (Wu et al. 2014), to engineer mutations in human induced pluripotent stem cells (Wang et al. 2017), and to perform multiplexed activation of protein-coding and noncoding genes (Li et al. 2017).
Herein, we describe the creation and validation of a piggyBac-based system for inducible editing of mammalian genomes by CRISPR-Cas9. In the system, which we call “CRISPR-Bac,” two separate piggyBac cargo vectors, one that expresses an inducible Cas9 or dCas9 variant, and another that expresses an sgRNA and the reverse-tetracycline transactivator (rtTA; Gossen et al. 1995), are transfected into cells along with a plasmid that expresses the piggyBac transposase. A short period of selection is used to obtain cells that stably express both the Cas9 and sgRNA cargo vectors. Our CRISPR-Bac vectors provide a simple way to rapidly insert the CRISPR-Cas9 machinery into mammalian genomes to knock down proteins, delete kilobase-sized genomic regions, and activate or repress transcription of protein-coding genes and long noncoding RNAs (lncRNAs), without the additional cost and labor involved in the packaging and delivery of lentiviral particles to cells.
RESULTS
Cloning of CRISPR-Bac
We modeled CRISPR-Bac (Fig. 1) after the pX330 plasmid system in which a humanized version of the Cas9 enzyme from Streptococcus pyogenes is coexpressed with a chimeric sgRNA driven by a U6 promoter (Cong et al. 2013). We cloned the Cas9 from pX330 into a doxycycline-inducible expression cassette in a piggyBac cargo vector that also expresses a gene conferring resistance to Hygromycin B. We then converted the dual BbsI sites in pX330, which are used to clone the sgRNA targeting sequence into that vector, into dual BsmbI sites. Like BbsI, BsmbI is a Type IIS restriction enzyme, and it generates overhanging ends that are identical to those generated by BbsI. We cloned the BsmbI-modified sgRNA expression cassette into a piggyBac cargo from Kirk et al. (2018) that expresses a bicistronic message which encodes the rtTA3 gene and a gene conferring resistance to G418 (originally cloned from Addgene plasmid #25735; Shin et al. 2006). The conversion of the pX330 BbsI sites, which are not unique in the rtTA-expressing vector, to BsmbI sites, allows the exact sgRNA design and cloning protocol for pX330 (Cong et al. 2013) to be used to clone sgRNAs into CRISPR-Bac.
FIGURE 1.

Experimental pipeline used for CRISPR-Bac. In a CRISPR-Bac experiment, a Cas9-expressing piggyBac cargo vector (or dCas9 variant) is cotransfected with a sgRNA- and rtTA-expressing piggyBac cargo vector and with a piggyBac transposase plasmid. Growth in Hygromycin B and G418 for 7 to 12 d selects for a population of cells that stably express an sgRNA of interest, and inducibly express a Cas9 or dCas9 variant. TR, piggyBac inverted terminal repeat. HS4, chicken βglobin insulator element. TRE, tetracycline responsive element (i.e., doxycycline inducible promoter). hCas9, dCas-VP160, dCas-KRAB from Cheng et al. (2013), Cong et al. (2013), and Kearns et al. (2014). EF1, EF1α promoter. HygroR, Hygromycin B resistance gene. SV40pA, SV40 polyadenylation signal. hU6-chimeric sgRNA from Cong et al. (2013) with BsmbI sites replacing BbsI sites. hUbiC-rtTA3-IRES-G418 cassette was from pSLIK-Neo (Shin et al. 2006).
Knockdown of a protein-coding gene using CRISPR-Bac
We tested whether CRISPR-Bac could be used to knock down a protein of interest in mouse embryonic stem cells (ESCs). We designed three sgRNAs targeting different exons of the Ezh2 gene (Supplemental Fig. S1; Supplemental Table S1) and cloned them into our sgRNA-rtTA-expressing vector using the protocol outlined in Cong et al. (2013). We then cotransfected our inducible Cas9-expressing piggyBac vector, a plasmid expressing the piggyBac transposase, and either each sgRNA-expressing vector separately, or a pool of all three sgRNAs into ESCs. As a control, we transfected an sgRNA-rtTA-expressing vector into which we did not clone a specific sgRNA-targeting sequence (our “no sgRNA” control). After selecting ESCs on Hygromycin B and G418 for 10 d, we removed the selection drugs and added 1 µg/mL of doxycycline to the media for 4 d to induce the expression of Cas9. To assess the extent of EZH2 knockdown, we performed western blot and immunofluorescence (IF). Relative to the control ESCs, we observed >60% reduction in EZH2 protein levels in the three lines expressing individual sgRNAs and more than 90% loss in the line expressing the pool of sgRNAs (Fig. 2A). In repeat transfections of the sgRNA pool, we consistently observed >90% loss of EZH2 protein levels (Fig. 2B). IF to EZH2 confirmed our western blot analysis (Fig. 2C). We compared the levels of EZH2 knockdown obtained via CRISPR-Bac to those obtained via transient transfection of the same sgRNA pool cloned into the widely used pX330 vector (Cong et al. 2013). Four days after transfection of pX330, we harvested cells and performed western blot and IF. Relative to pX330 lacking a gene-targeting sgRNA (“No sgRNA” control), we measured 25%–35% reduction in EZH2 protein (Fig. 2D,E). These results demonstrate that CRISPR-Bac can be used to inducibly knock down a protein-coding gene of interest.
FIGURE 2.

Inducible protein-coding gene knockdown with CRISPR-Bac. See Supplemental Table S2 for details on replicates and experimental design for each figure panel. (A) Western blot of CRISPR-Bac assay in which three separate sgRNAs targeting Ezh2, a pool of all three sgRNAs, or a nontargeting sgRNA control (“No sgRNA”) were cotransfected along with the inducible Cas9-expressing piggyBac cargo into E14 mouse ESCs. Western blots to EZH2 and TBP were performed on protein extracted from stably selected cells, after 4 d of Cas9 induction with doxycycline. Values underneath blots represent knockdown of EZH2 relative to no sgRNA controls and normalized for loading with TBP protein levels. (B) Biological replicates of experiment in A for sgRNA pool. “Pool rep #1” is the same sample as in A. (C) Representative IF image showing EZH2 knockdown in nontargeting sgRNA control (“no sgRNA”) or pooled sgRNA cells from replicate #2 in B. Scale bar, 10 µm. (D,E) Western blot and IF to EZH2 in transient transfection experiments with pX330.
Targeted deletion of genetic elements using CRISPR-Bac
An important use of CRISPR-Cas9 is to create targeted deletions of regulatory elements (Ran et al. 2013; Canver et al. 2014; Hsu et al. 2014; Aparicio-Prat et al. 2015; Wright et al. 2016; Zhu et al. 2016; Gasperini et al. 2017). To test the utility of CRISPR-Bac in this application, we cloned into CRISPR-Bac pairs of sgRNAs that flank multiple different regulatory elements (RE1, RE2, RE3, RE4), to delete 2331 bp, 2480 bp, 1222 bp, and 2609 bp regions, respectively (Supplemental Fig. S1; Supplemental Table S1). We created ESCs that stably express the different sgRNA pairs along with doxycycline-inducible Cas9. We induced expression of Cas9 for 4 d, collected genomic DNA, and performed quantitative PCR (qPCR) using amplicons within the deleted regions. By comparing qPCR results between the sgRNA-expressing ESCs and nontargeting sgRNA control ESCs, we approximated the extent that each targeted region was deleted in a polyclonal cell population. For four of four deletions, we observed more than 40% reduction in signal, indicating that close to half of the alleles in the cell population were deleted (Fig. 3A; two-sided t-test).
FIGURE 3.

Targeted deletion of DNA regulatory elements using CRISPR-Bac. See Supplemental Table S2 for details on replicates and experimental design for each figure panel. (A) qPCR results from polyclonal populations of ESCs expressing Cas9 and pairs of sgRNAs flanking four separate regulatory elements (RE). Primers for qPCR sit entirely inside of the expected deletion. Individual qPCR data points are shown in box-and-whisker format representing the mean and the interquartile range. Data from the nontargeting sgRNA control (NG) and sgRNA-expressing cells (Del) are plotted relative to the average of the signal in the NG control cells. (***) P < 0.001 from a two-sided t-test between NG and Del. (B,C) Agarose gels showing genotyping PCR products for the NG control and RE3 Del polyclonal populations from A and 36 clones isolated from RE3 deletion ESCs. The UCSC browser tracks above each gel show the location of RE3, the location of the expected deletion, and the location of primers used in the corresponding genotyping PCR. Scale bar, 500 bps. Gel in B identifies clones that have a deleted allele (four possible band sizes based on the combination of sgRNAs that cut). Gel in C identifies clones that have a wild-type allele (primer pairs are the same as used for RE3 in A). Many clones showed weak wild-type bands, which we presume is due to MEF genomic DNA and not due to the presence of a wild-type allele. (D) qPCR results from RE3 clones in panels B and C classified as wild-type (#1 and #2), heterozygous (#7 and #8), and homozygous (#3, #5, and #13). Primers pairs are the same as in A and C to detect wild-type alleles. Data are plotted as in A.
To assess deletion efficiency in single cells, we isolated 36 individual colonies from cells transfected with sgRNAs to the RE3 element and extracted their genomic DNA. To assess whether a deletion occurred on at least one allele, we performed PCR using primers that flanked the expected deletion. Twenty-one of 36 clones (58%) showed a band within the expected size range for a deletion (310–426 bp; Fig. 3B), signifying that these clones were at least heterozygous for the deletion. To distinguish between clones that were heterozygous versus homozygous for the deletion, we used a pair of primers that amplify inside the deletion. Twelve out of the 21 clones (33% of the 36 clones) did not show a band, indicating that no wild-type allele was present, and the cells were homozygous for the deletion (Fig. 3C). Many homozygous clones showed weak wild-type bands, which we presume were due to genomic DNA from MEF feeder cells and not due to the presence of a wild-type allele in the clones. In support of this notion, we performed qPCR to detect the wild-type allele on seven total clones: two clones classified as wild-type, two as heterozygous, and three as homozygous. Relative to heterozygous and wild-type clones, all three homozygous clones showed a 10 and 11 cycle difference, respectively, signifying that the clones that we genotyped as homozygous indeed lacked wild-type alleles (Fig. 3D). These data demonstrate that CRISPR-Bac can be used to generate targeted genomic deletions with high efficiency.
Activation and repression of protein-coding gene transcription using CRISPR-Bac
In addition to creating targeted genomic deletions, the CRISPR-Cas9 system can be used to up- or down-regulate genes from their endogenous promoters, by targeting dCas9 fused to effector domains that recruit transcriptional coactivators or corepressors. We cloned one such transcriptional activator fusion, dCas9-VP160 from Cheng et al. (2013), and one such transcriptional repressor fusion, dCas9-KRAB from Kearns et al. (2014), into the same piggyBac-based inducible expression vector that we used to express catalytically active Cas9 (Fig. 1). We then tested our ability to up-regulate Ascl1, a silent gene in ESCs, with dCas9-VP160, and we tested our ability to down-regulate Oct4, an active gene in ESCs, with dCas9-KRAB (Fig. 4A). Using CRISPR-Bac, we routinely observed 350-fold up-regulation of Ascl1 relative to nontargeting sgRNA control cells (Fig. 4B). This level of activation was similar to that obtained using transient transfection of dCas9-VP160 and SPgRNA vectors from Cheng et al. (2013) and Perez-Pinera et al. (2013) (Fig. 4B). Using multiple sets of published and in-house-designed sgRNAs, the maximum level of Oct4 down-regulation we achieved was two- to threefold (Fig. 4C and data not shown). Relative to the 4 d used for protein knockdown and genomic deletions, we observed that for transcriptional modulation experiments, 2 d of doxycycline treatment was sufficient to detect maximal effects induced by dCas9-VP160 and -KRAB. These data show that CRISPR-Bac can be used to up- and down-regulate transcription of protein-coding genes of interest.
FIGURE 4.

Activation and repression of protein-coding gene transcription using CRISPR-Bac. See Supplemental Table S2 for details on replicates and experimental design for each figure panel. Individual qPCR data points are shown in box-and-whisker format representing the mean and the interquartile range. (***) P < 0.001 from a two-sided t-test between no sgRNA and sgRNA-expressing cells. (A) qPCR results showing endogenous expression of Ascl1 and Oct4 in ESCs relative to the average Ascl1 signal. (B) qPCR showing transcriptional activation of Ascl1 using stable transfection of CRISPR-Bac dCas9-VP160 and a pool of four Ascl1-targeting sgRNAs (Perez-Pinera et al. 2013) versus transient transfection of dCas9-VP160 and pooled SPgRNA-Ascl1. Data from the nontargeting sgRNA control (No sgRNA) and sgRNA-expressing cells are plotted relative to the average of the signal in the No sgRNA control cells. (C) qPCR showing transcriptional repression of Oct4 using dCas9-KRAB. Data are plotted as in B.
Activation and repression of lncRNA transcription using CRISPR-Bac
We next examined whether we could use CRISPR-Bac to activate and repress transcription of a lncRNA using dCas9-VP160 and dCas9-KRAB, respectively. We chose to target a lncRNA called Airn in two cell types: mouse ESCs, in which Airn is expressed at low levels, and mouse trophoblast stem cells (TSCs), in which Airn is more highly expressed and active (Fig. 5A; Latos et al. 2009; Calabrese et al. 2015; Andergassen et al. 2017). In ESCs, we were able to activate Airn ∼15-fold above its levels in nontargeting sgRNA control cells (Fig. 5B), but we were not able to repress Airn, likely due to its low endogenous expression (data not shown; Latos et al. 2009). Compared to transient transfection of dCas9-VP160 and SPgRNA vectors from (Cheng et al. 2013; Perez-Pinera et al. 2013), we achieved a greater level of activation with CRISPR-Bac (Fig. 5B). In TSCs, we were able to repress Airn to 10% of its normal expression and activate Airn 2.5-fold relative to nontargeting sgRNA control cells (Fig. 5C). Therefore, CRISPR-Bac can be used to activate and repress transcription of lncRNAs.
FIGURE 5.

Activation and repression of lncRNA transcription using CRISPR-Bac. See Supplemental Table S2 for details on replicates and experimental design for each figure panel. All individual qPCR data points are shown in box-and-whisker format representing the mean and the interquartile range. (***) P < 0.001; (**) P < 0.01 from a two-sided t-test between no sgRNA and sgRNA-expressing cells. (A) qPCR showing relative levels of Airn expression in wild-type ESCs and TSCs. (B) qPCR measuring Airn transcriptional activation in ESCs with stably transfected CRISPR-Bac constructs, dCas9-VP160 and Airn-targeting sgRNA, versus transient transfection of dCas9-VP160 and SPgRNA-Airn. Data from the nontargeting sgRNA control (No sgRNA) and sgRNA-expressing cells are plotted relative to the average of the signal in the No sgRNA control cells. (C) qPCR results for TSCs with stably transfected dCas9-KRAB or dCas9-VP160 and Airn-targeting sgRNA. Data are plotted as in B. (D) Representative RNA FISH image showing Airn and Kcnq1ot1 RNA in ESCs harvested alongside ESCs from B. Numbers in parentheses correspond to spots counted by Imaris software for Airn and Kcnq1ot1 in each cell line. Scale bar, 10 µm.
Under normal physiological conditions, the Airn lncRNA is monoallelically expressed due to a process called genomic imprinting that leads to methylation of its promoter and gene silencing specifically on the maternally inherited allele (Stoger et al. 1993; Lee and Bartolomei 2013). To assess whether activation of Airn via CRISPR-Bac led to mono- or biallelic activation of the lncRNA, we performed RNA fluorescence in situ hybridization (FISH) in ESCs stably expressing dCas9-VP160 and either a nontargeting sgRNA or an Airn-targeting sgRNA. We performed a two-color RNA FISH experiment where one probe was complementary to the Airn lncRNA, and the other probe was complementary to the Kcnq1ot1 lncRNA. Kcnq1ot1, like Airn, is also imprinted and monoallelically expressed (Lee and Bartolomei 2013). Unlike Airn, Kcnq1ot1 is robustly expressed in ESCs under normal conditions (Umlauf et al. 2004). Kcnq1ot1 therefore served as a control to gauge the extent of Airn monoallelism upon activation by CRISPR-Bac. After taking z-stacks on a widefield microscope and deconvolving the resultant images, we used an automated pipeline to identify puncta whose RNA FISH signal surpassed a specified threshold. In two images taken of cells expressing the nontargeting sgRNA control, we counted zero puncta of Airn relative to 100 puncta of Kcnq1ot1, confirming prior data that show Kcnq1ot1 is robustly expressed in ESCs while Airn is not (Umlauf et al. 2004; Latos et al. 2009). In contrast, in two images taken of cells expressing the Airn-targeting sgRNA, we counted 97 puncta of Airn relative to 130 puncta of Kcnq1ot1 (Fig. 5D). These data support the notion that CRISPR-Bac activates expression of Airn on the unmethylated paternal allele, and that the methylated maternal allele of Airn remains resistant to activation (Stoger et al. 1993).
sgRNA titration to achieve variable levels of lncRNA induction
The number of piggyBac cargos inserted into the genome can be controlled by altering the ratio of cargo vector to transposase plasmid (Cadinaños and Bradley 2007; Wilson et al. 2007; Wang et al. 2008). The CRISPR-Bac platform relies on simultaneous delivery of two cargo vectors: one vector expressing the sgRNA and rtTA/G418 resistance genes, and the other vector expressing the Cas9/dCas9 variant and hygromycin resistance genes (Fig. 1). We sought to determine whether the extent of activation of a target gene of interest could be altered by altering the ratios of sgRNA, Cas9, and piggyBac transposase vectors in transfections. We tested a range of sgRNA-to-dCas9VP160-to-transposase ratios, using the Airn lncRNA as our target gene for activation (Supplemental Table S3). We found modest but significant differences in the level of Airn activation when we transfected higher amounts of sgRNA and dCas9-VP160 plasmids relative to the piggyBac transposase plasmid (Fig. 6A; see table of adjusted P-values from Tukey's HSD test), and these differences were accompanied by increased numbers of sgRNA and dCas9-VP160 cargo insertions per cell (Fig. 6B). Thus, the extent of target gene activation using CRISPR-Bac can be partly controlled by changing the ratios of sgRNA/rtTA, Cas9, and transposase plasmids in transfections.
FIGURE 6.

Cargo to transposase ratio controls the extent of activation and multiplex gene activation by CRISPR-Bac. See Supplemental Table S2 for details on replicates and experimental design for each figure panel. Each replicate in the no sgRNA control and sgRNA-expressing cells is shown relative to the average of the signal in “No sgRNA.” (***) P < 0.001; (**) P < 0.01 from a two-sided t-test between no sgRNA and sgRNA-expressing cells. (A) qPCR measuring Airn activation. X-axis gives the transfection ratio of rtTA-sgRNA to dCas9-VP160 to transposase for each experiment. Ratios are plotted in ascending order based on the summed cargo (rtTA-sgRNA plus dCas9-VP160) to transposase ratio. “1:1:2 no sgRNA” and “8:2:1” data are the same as shown in Figure 5B. Corresponding table gives adjusted P-values from Tukey's HSD post-hoc test for all comparisons, where P ≤ 0.05 are highlighted in green. (B) Bar plot showing DNA copy-number per cell for the rtTA-sgRNA and dCas9-VP160 cargos under each transfection condition. Numbers over each bar give the average copy-number calculated from three technical qPCR replicates. The tables below correspond to the bar plot showing the transfection ratio and total number of DNA cargos inserted (copy-number of rtTA-sgRNA plus dCas9-VP160). (C) Simultaneous activation of Ascl1 and Airn transcription upon cotransfection of a pool of four Ascl1 sgRNAs from Perez-Pinera et al. (2013) and one Airn. Data from the nontargeting sgRNA control (No sgRNA) and sgRNA-expressing cells are plotted relative to the average of the signal in the No sgRNA control cells. Individual qPCR data points are shown.
Simultaneous up-regulation of two genes via CRISPR-Bac
By coexpression of multiple sgRNAs, CRISPR can be used to activate or repress multiple genes simultaneously (Cheng et al. 2013). To test if CRISPR-Bac is capable of multiplexed gene activation, we created ESCs expressing dCas9-VP160 and sgRNAs targeting the Ascl1 and Airn promoters (same sgRNAs as in Figs. 4B, 5B,C, 6A). Relative to nontargeting sgRNA controls, qPCR demonstrated simultaneous 411-fold activation of Ascl1 and 9.5-fold activation of Airn when sgRNAs for both targets were cotransfected (Fig. 6C). This confirms that CRISPR-Bac can be used to target multiple genes in a single experiment.
CRISPR-Bac can be used in human cells
To determine if the CRISPR-Bac system could be used in human cells, we tested Cas9-mediated knockdown and dCas9-VP160-mediated up-regulation in SUM-159 cells, a commonly used cell line in breast cancer research (Grigoriadis et al. 2012). Similar to our experiments in ESCs, we cotransfected the inducible Cas9-expressing piggyBac vectors into SUM-159 cells, a plasmid expressing the piggyBac transposase, and a pool of four sgRNAs targeting human EZH2 exons, then selected the cells with Hygromycin B and G418 for at least 10 d, and induced Cas9 expression with 1 µg/mL of doxycycline for 4 d. Via western blot, we detected >70% reduction in EZH2 protein levels in both replicates, which was confirmed via IF (Fig. 7A,B). In parallel, we cotransfected the dCas9-VP160, transposase, and a pool of two sgRNAs targeting the promoter of Il1RN, drug selected for at least 10 d, and induced dCas9-VP160 expression for 2 d. Il1RN was activated ∼54-fold relative to the no dCas9 control (Fig. 7C). In these experiments, the Cas9/EZH2 sgRNA cells served as a negative control in the dCas9-VP160/Il1RN sgRNA experiment, and vice versa. These data show that the CRISPR-Bac system can be used in human cells.
FIGURE 7.
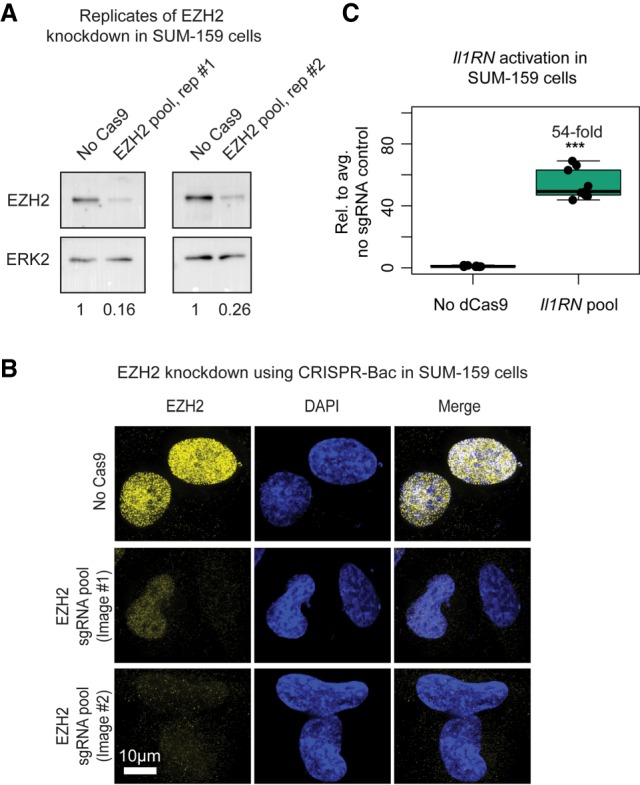
The CRISPR-Bac system functions in human cell lines. See Supplemental Table S2 for details on replicates and experimental design for each figure panel. (A) Western blots to EZH2 and ERK2 loading control from two replicate CRISPR-Bac experiments in SUM-159 cells. “No Cas9” refers to measurements taken from dCas9-VP160/rtta-Il1RN sgRNA expressing SUM-159 cells that were cultured in parallel to those expressing Cas9 and the EZH2 sgRNA pool. Values underneath blots represent knockdown of EZH2 relative to No Cas9 control and normalized for loading with ERK2 protein levels. (B) Representative IF images showing EZH2 knockdown from “No Cas9” and “EZH2 pool, rep #1” SUM-159 cells in A. Image #1 shows a cell with partial knockdown next to a cell with full knockdown, and image #2 shows two cells with full knockdown. Scale bar, 10 µm. (C) qPCR results showing transcriptional activation of Il1RN in SUM-159 cells. In panel C, “No dCas9” refers to measurements taken from Cas9/rtta-EZH2 sgRNA expressing SUM-159 cells that were cultured in parallel to those expressing dCas9 and the Il1RN sgRNAs. Data from the nontargeting sgRNA control (No sgRNA) and sgRNA-expressing cells are plotted relative to the average of the signal in the No sgRNA control cells. Individual qPCR data points are shown in box-and-whisker format. (***) P < 0.001 from a two-sided t-test between no sgRNA and sgRNA-expressing cells.
Conclusions
In Mus musculus-derived ESCs and TSCs, we have shown that CRISPR-Bac can be used to knock down proteins through frameshift/deletion, to delete kilobase-sized regulatory elements with high efficiency, and to up- and down-regulate the transcription of protein-coding genes and an imprinted lncRNA. Levels of CRISPR-induced activation could partly be controlled through delivery of different ratios of CRISPR-Bac vectors. It seems likely that the use of different promoter elements within CRISPR-Bac (for example, a constitutive CMV promoter driving dCas9-VP160 instead of a TRE) might afford additional levels of control. It may also be possible to engineer CRISPR-Bac vectors that express multiple sgRNAs, as has been done elsewhere (Kabadi et al. 2014; Sakuma et al. 2014; Albers et al. 2015). Although in this work we only tested CRISPR-Bac in a limited number of cell types, it seems reasonable to presume that the CRISPR-Bac vectors or their modified derivatives would be functional in other mammalian cell types, given the broad activity of the piggyBac transposase (Ding et al. 2005; Cadinaños and Bradley 2007; Wilson et al. 2007; Wang et al. 2008; Kaji et al. 2009; Yusa et al. 2009; Kahlig et al. 2010; Li et al. 2011, 2013). Indeed, CRISPR-Bac facilitated efficient knockdown and transcriptional up-regulation in at least one human cell line, SUM159 (Grigoriadis et al. 2012). In our view, the main utility of CRISPR-Bac over other genome editing platforms is that CRISPR-Bac allows the generation of stable cell lines without the need to package CRISPR-Cas9 components into lentiviral delivery systems. It also preserves the sgRNA cloning strategy from the widely used pX330/335 systems, facilitating horizontal transfer of sgRNAs between the two platforms (Cong et al. 2013). Relative to prior studies that have used piggyBac to carry out CRISPR in mammalian cells (Wu et al. 2014; Li et al. 2017; Wang et al. 2017), our study describes a single platform with interchangeable functionalities that has been optimized for protein knockdown, regulatory element deletion, and the up- and down-regulation of protein-coding and noncoding gene transcription.
MATERIALS AND METHODS
Construction of CRISPR-Bac vectors
To create the doxycycline-inducible Cas9, dCas9-VP160, and dCas9-KRAB piggyBac vectors, a parent piggyBac vector was created in which a bGH-polyA signal and an EF1α promoter driving expression of a hygromycin resistance gene were ligated into the cumate-inducible piggyBac transposon vector from System Biosciences after its digestion with HpaI and SpeI, which cut just downstream from each chicken β-globin insulator sequence and removed all other internal components of the original vector. The TRE from pTRE-Tight (Clontech) was cloned upstream of the bGH-poly(A) site, and Cas9 from pX330 (Addgene plasmid # 42230; Cong et al. 2013; gift from Feng Zhang), dCas9-VP160 from Addgene plasmid # 48225 (Cheng et al. 2013; gift from Rudolf Jaenisch), and dCas9-KRAB from Addgene plasmid # 50917 (Kearns et al. 2014; gift from Rene Maehr and Scot Wolfe) were each cloned behind the TRE by digestion with AgeI and SalI (NEB) followed by Gibson Assembly (NEB), to generate piggyBac cargo vectors capable of inducibly expressing Cas9, dCas9-VP160, and dCas9-KRAB, respectively, upon addition of doxycycline.
To create the rtTA-sgRNA expressing piggyBac vector, the dual BbsI sites in pX330 were converted to BsmbI sites using oligonucleotides, and the entire U6 expression cassette was cloned via Gibson assembly into the PacI site upstream of the rtTA3-IRES-Neo cassette in the rtTA-piggyBac-Cargo vector described in Kirk et al. (2018). The rtTA3-IRES-Neo cassette was originally cloned from pSLIK-Neo and was a gift from Iain Fraser (Addgene plasmid # 25735). Oligonucleotides used for cloning are in Supplemental Table S1.
We have submitted four plasmids to Addgene: (1) PB_rtTA_BsmBI, #126028, (2) PB_tre_Cas9, #126029, (3) PB_ tre_dCas9_KRAB, #126030, and (4) PB_ tre_dCas9_VP160, #126031.
sgRNA design
Oligonucleotides used for sgRNA cloning are listed in Supplemental Table S1, and their location relative to gene features are shown in Supplemental Figure S1. Protein knockdown sgRNAs were designed using Desktop Genetics, and all other sgRNAs were designed using the CRISPOR program or taken from published sources (Supplemental Table S1; Haeussler et al. 2016).
Embryonic stem cell (ESC) culture
ESCs were grown on gelatin coated plates at 37°C in a humidified incubator at 5% CO2. Media were changed daily and consisted of DMEM high glucose plus sodium pyruvate, 0.1 mM nonessential AA, 100 µ/mL penicillin-streptomycin, 2 mM l-glutamine, 0.1 mM 2-mercaptoethanol, 15% ES-qualified FBS, and 1:500 LIF conditioned media produced from Lif-1Cα (COS) cells. ESCs were split at an approximate ratio of 1:6 every 48 h.
Trophoblast stem cell (TSC) culture
TSCs were cultured as in Quinn et al. (2006). Briefly, TSCs were cultured at 37°C on preplated irradiated MEF feeder cells in TSC media (RPMI [Invitrogen], 20% Qualified FBS [Invitrogen], 100 µ/mL penicillin-streptomycin, 1 mM sodium pyruvate [Invitrogen], 100 µM β-mercaptoethanol [Sigma], and 2 mM l-glutamine) supplemented with Fgf4 (25 ng/mL; Invitrogen) and Heparin (1 µg/mL; Sigma) just before use. At passage, TSCs were trypsinized with 0.125% Trypsin (Invitrogen) for 3 min at room temperature and gently dislodged from their plate with a sterile, cotton-plugged Pasteur pipette (Thermo Fisher). To deplete MEF feeder cells from TSCs prior to RNA isolation, TSCs were preplated for 40 min and cultured for 3 d in 70% MEF-conditioned TSC media supplemented with Fgf4 (25 ng/mL; Invitrogen) and Heparin (1 µg/mL; Sigma).
SUM-159 cell culture
SUM-159 cells were maintained in DMEM/F12 medium (Gibco, Thermo Fisher Scientific) supplemented with 5% FBS, 5 µg/mL insulin, 1 µg/mL hydrocortisone, and antibiotic:antimycotic cocktail (Gemini Bio Products) as in Zawistowski et al. (2017).
Stable transfections of CRISPR-Bac components
To generate stable CRISPR-Bac E14 ESC lines, 5 × 105 cells were seeded in a single well of a six-well plate, and the next day transfected with piggyBac cargo vectors and pUC19-piggyBac transposase from Kirk et al. (2018), totaling 2.5 µg of plasmid DNA (see exact amounts in Supplemental Table S3), using Lipofectamine 3000 (Invitrogen) according to manufacturer instructions. Cells were subsequently selected on Hygromycin (150 µg/mL; Gibco) and G418 (200 µg/mL; Gibco) for 7 to 12 d. Due to the efficiency of piggyBac cargo integration and the rapidity of Hygromycin selection, most observable death from drug selection occurred within ∼3 d after addition of Hygromycin and G418 (i.e., cells with Hygromycin resistance were invariably resistant to G418).
To generate stable CRISPR-Bac TSC lines, 7.5 × 105 cells were coelectroporated using the Neon instrument (electroporation program: 1300 V, 40 msec, 1 pulse; Invitrogen) with 5 µg of plasmid DNA at a 1:1:2 ratio of rtTA-sgRNA to dCas9 to transposase. Cells were selected on Hygromycin (150 µg/mL; Gibco) and G418 (200 µg/mL; Gibco) for 9 d.
To generate stable CRISPR-Bac SUM-159 cells, 5 × 105 cells were seeded per well of a six-well plate and the next day transfected with 2.5 µg of plasmid DNA at a 1:1:2 ratio of rtTA-sgRNA to Cas9 to transposase using Lipofectamine 3000 (Invitrogen). Cells were subsequently selected on Hygromycin (250 µg/mL; Gibco) and G418 (600 µg/mL; Gibco) for at least 10 d.
Transient transfections
For transient transfections using pX330, 5 × 105 ESCs were seeded in a single well of a six-well plate and transfected the next day using Lipofectamine 3000 with 2.5 µg of the pX330 empty vector (“no sgRNA”) or a pool of pX330 vectors expressing the 3 sgRNAs to mouse Ezh2. Cells were harvested 4 d after transfection. For transient transfections using VP160, 5 × 105 ESCs were seeded in a single well of a six-well plate and transfected the next day using Lipofectamine 3000 with 1.25 µg of dCas9-VP160 (Cheng et al. 2013) and 1.25 µg of the SPgRNA empty vector (“no sgRNA”; Perez-Pinera et al. 2013) or SPgRNA containing sgRNAs targeting either Airn or Ascl1. Cells were harvested 2 d after transfection.
Protein isolation and western blotting
To isolate protein for western blotting, cells were washed with PBS, and then lysed with RIPA buffer (10 mM Tris-Cl (pH 7.5), 1 mM EDTA, 0.5 mM EGTA, 1% NP40, 0.1% sodium deoxycholate, 0.1% SDS, 140 mM NaCl) supplemented with 1 mM PMSF (Fisher Scientific) and 1× protease inhibitor cocktail (Sigma) for 15 min at 4°C, 4 d after induction with 1 µg/mL doxycycline. Prior to western blotting, protein levels were quantified using the DC assay from Bio-Rad. For western blotting, primary and secondary antibody incubations were done for 1 h at room temperature. Antibodies used were EZH2 (Cell Signaling #5246, 1:1000 dilution), TBP (Abcam ab818, 1:2000 dilution), ERK2 (Santa Cruz; sc-1647; 1:500), donkey anti-mouse IgG-HRP secondary (Santa Cruz; sc-2314; 1:2500), and donkey anti-rabbit IgG-HRP secondary (Santa Cruz; sc-2313; 1:2500).
Genomic DNA isolation and qPCR
To isolate genomic DNA, 400 µL of ESC lysis buffer (100 mM Tris-HCl, pH 8.1, 5 mM EDTA, pH 8.0, 200 mM NaCl, 0.2% SDS) supplemented with 80 µL proteinase K (Denville) was used per 24-well of ESCs, four days after induction with 1 µg/mL doxycycline. Lysed ESCs were incubated at 55°C overnight, cells were boiled at 100°C for 1 h to degrade RNA, and DNA was precipitated by addition of 2 volumes of 100% ethanol. DNA was pelleted and resuspended in 1× TE (10 mM Tris-HCl 1 mM EDTA pH 8.0) overnight at room temperature prior to qPCR. qPCR was performed using 100 ng of DNA per reaction and iTaq Universal SYBR Green Supermix (Bio-Rad), with primers specified in Supplemental Table S1. All related plots were generated using R version 3.4.1 (The R Development Core Team 2017).
qPCR for DNA copy-number analysis
Genomic DNA was prepared as in Genomic DNA isolation and qPCR section above. qPCR signal (SsoFast, Bio-Rad) from the genomic DNA was compared to signal from a molar standard amplified from increasing amounts of the corresponding dCas9-VP160 and rtTA plasmids. Primers used are listed in Supplemental Table S1. All related plots were generated using R version 3.4.1 (The R Development Core Team 2017).
Generation of clonal ESCs with targeted genomic deletions and genotyping
After 4 d of dox induction, RE3 deletion E14 cells were cultured 2 d in the absence of dox to ensure that Cas9 was fully depleted. Then, 2000 cells were plated on a 10 cm plate with preplated irradiated MEF feeder cells. After 4 d, individual colonies were picked and plated on irMEFs. Clonal lines were passaged twice off of MEFs before genomic DNA was prepared as in Genomic DNA isolation and qPCR section above.
Genotyping PCR reactions were performed with gDNA using Apex Taq DNA Polymerase (Genesee Scientific). The first set of primers flanked the deletion and identified clonal lines with at least one allele deleted. The second set only amplified a wild-type allele, with both primers sitting inside the deletion. Many clones showed weak wild-type bands, likely due to MEF gDNA and not due to the presence of a wild-type allele in the ESC clone. Primers used are listed in Supplemental Table S1.
RNA isolation and qPCR
RNA was isolated using TRIzol (Invitrogen). For RT-qPCR assays, 1–2 µg of RNA was reverse transcribed using MultiScribe RT (Applied Biosystems), and qPCR was performed using iTaq Universal SYBR Green (Bio-Rad) and primers specified in Supplemental Table S1. All related plots were generated using R version 3.4.1 (The R Development Core Team 2017).
RNA FISH
Fosmid Wl1-2156F18 (Airn) and BAC RP23-101N20 (Kcnq1ot1) were ordered from the BACPAC resource center and fingerprinted with restriction digestion prior to use to verify inserted DNA. Fluorescent labeling was performed using BioPrime (Invitrogen). ESCs were fixed on coverslips for 10 min in 4% paraformaldehyde/PBS, followed by a 10-min permeabilization on ice in 0.5% TritonX-100 in PBS and 1:200 Ribonucleoside Vanadyl Complex (NEB). Coverslips were stored at −20°C in 70% ethanol until use.
To initiate the RNA FISH protocol, coverslips were dehydrated by serial 3-min incubations with 75%, 85%, 95%, and 100% ethanol, and air-dried for 5 min. RNA FISH probes were added, and coverslips were placed cell-side down in a chamber humidified with 50% formamide/2×SSC overnight at 37°C. After overnight incubation, coverslips were washed 3× with 50% formamide/2×SSC at 42°C and 3× with 1×SSC at 50°C. Each wash was 5 min long. Coverslips were then rinsed 1× with PBS before a 2 min incubation in DAPI stock diluted 1:1000 in water. Coverslips were rinsed twice more and affixed to glass slides using Vectashield (VectorLabs), then sealed with nail polish.
Four-dimensional data sets were acquired by taking multichannel Z-stacks on an Olympus BX61 widefield fluorescence microscope using a Plan-Aprochromat 60X/1.4 oil objective and a Hamamatsu ORCA R2 camera, controlled by Volocity 6.3 software. Excitation was provided by a mercury lamp, and the following filters were used for the three fluorescent channels that were imaged: 377/25 ex, 447/30 em for DAPI (DAPI-5060B Semrock filter); 482/17 ex, 536/20 em for AlexaFluor488 (Semrock FITC-3540B filter); 562/20 ex, 642/20 em for Cy3 (Semrock TXRED-4040B filter). Pixel size was 0.108 µm, Z spacing was 0.2 μm, and images had 1344 × 1024 pixels. Between 46–49 Z-stacks were acquired for each image. Z-stacks were deconvolved using the iterative-constrained algorithm (Mediacy AutoQuantX3) with default algorithm settings. Sample settings for the deconvolution were: peak emissions for dyes (570, 519, 461 nm for Cy3, AlexaFluor 488 and DAPI, respectively), widefield microscopy mode, NA = 1.4, RI of oil = 1.518, and RI of sample = 1.45. After deconvolution, RNA FISH signals were located using the “Spots” function in Imaris software (version 8.3.1) and marked with equal sized spheres. To initially call spots on all images, spot detection values were set at 0.5 µm for xy and 1.5 µm for z, and background subtraction and auto quality settings were used. We manually optimized the quality/sensitivity setting to call Kcnq1ot1 spots, and then used the same quality threshold to call Airn spots for the same image. Images are shown as maximum intensity projections made using ImageJ (Schindelin et al. 2012).
Immunofluorescence (IF)
Cells were fixed on coverslips the same as for RNA FISH (see above). To initiate the IF protocol, coverslips were washed twice in PBS and blocked for 30 min at room temperature in blocking solution (1× PBS with 0.2% Triton X-100, 1% goat serum, and 6 mg/mL IgG-free BSA). Then, coverslips were washed in 0.2% triton/1× PBS and incubated with EZH2 antibody (Cell Signaling #5246; 1:200 in block solution) for 1 h at RT. Coverslips were washed 3× in 0.2% triton/1× PBS for 4 min each and incubated with secondary antibody (AlexaFluor 647 goat anti-rabbit, A-21245, 1:1000 in block solution for ESCs and AlexaFluor 488 goat anti-rabbit, A-11034, 1:1000 in block solution) for 30 min at RT. After incubation, coverslips were washed 3× in 0.2% triton/1× PBS for 4 min each and rinsed 1× with PBS before a 2 min incubation in DAPI stock diluted to 5 ng/mL in water. Coverslips were rinsed twice more and mounted to glass slides using Prolong Gold (Thermo Fisher Scientific P10144). Imaging and deconvolution were performed the same as described in the RNA FISH section with the below exceptions. The filters used for the two fluorescent channels that were imaged are 377/25 ex, 447/30 em for DAPI (DAPI-5060B Semrock filter), 482/17 ex, 536/20 em for AlexaFluor488, (Semrock FITC-3540B filter), and 628/20 ex, 692/20 em for AlexaFluor 647 (Semrock Cy5 4040A filter). Approximately 40 Z-stacks were acquired for each image. Sample settings for the deconvolution included the following peak emissions for dyes: 670, 519, and 461 nm for AlexaFluor 647, AlexaFluor 488, and DAPI, respectively. Images are shown as maximum intensity projections made using ImageJ (Schindelin et al. 2012).
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
Supplementary Material
ACKNOWLEDGMENTS
We thank UNC colleagues for discussions and the Gary Johnson laboratory for the SUM-159 cell line. This work was supported by National Institutes of Health (NIH) grant GM121806, Basil O'Connor award #5100683 from the March of Dimes Foundation, and funds from the Lineberger Comprehensive Cancer Center and UNC Department of Pharmacology (to J.M.C.). E.T., D.M.L., and E.R.H. were supported in part by the National Institute of General Medical Sciences (NIGMS) training award T32 GM007092, K.C.A.B. and R.M.M. by the NIGMS training award T32 GM119999, and E.C.M.V. by the National Institute of Environmental Health Sciences (NIEHS) Toxicology Training grant T32 ES007126 and the NIGMS training award R25 GM055336. The Microscopy Services Laboratory was supported in part by National Cancer Institute (NCI) grant P30 CA016086.
Author contributions: M.D.S. and J.M.C. conceived the study; M.D.S., E.T., K.C.A.B., D.M.L., E.R.H., R.M.M., S.O.K., and E.C.M.V. performed the experiments; M.D.S. and J.M.C. performed the analysis; and M.D.S. and J.M.C. wrote the paper.
Footnotes
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.068932.118.
REFERENCES
- Albers J, Danzer C, Rechsteiner M, Lehmann H, Brandt LP, Hejhal T, Catalano A, Busenhart P, Gonçalves AF, Brandt S, et al. 2015. A versatile modular vector system for rapid combinatorial mammalian genetics. J Clin Invest 125: 1603–1619. 10.1172/JCI79743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andergassen D, Dotter CP, Wenzel D, Sigl V, Bammer PC, Muckenhuber M, Mayer D, Kulinski TM, Theussl HC, Penninger JM, et al. 2017. Mapping the mouse Allelome reveals tissue-specific regulation of allelic expression. eLife 6: e25125 10.7554/eLife.25125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aparicio-Prat E, Arnan C, Sala I, Bosch N, Guigó R, Johnson R. 2015. DECKO: single-oligo, dual-CRISPR deletion of genomic elements including long non-coding RNAs. BMC Genomics 16: 846 10.1186/s12864-015-2086-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cadinaños J, Bradley A. 2007. Generation of an inducible and optimized piggyBac transposon system. Nucleic Acids Res 35: e87 10.1093/nar/gkm446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calabrese JM, Starmer J, Schertzer MD, Yee D, Magnuson T. 2015. A survey of imprinted gene expression in mouse trophoblast stem cells. G3 (Bethesda) 5: 751–759. 10.1534/g3.114.016238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canver MC, Bauer DE, Dass A, Yien YY, Chung J, Masuda T, Maeda T, Paw BH, Orkin SH. 2014. Characterization of genomic deletion efficiency mediated by clustered regularly interspaced short palindromic repeats (CRISPR)/Cas9 nuclease system in mammalian cells. J Biol Chem 289: 21312–21324. 10.1074/jbc.M114.564625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng AW, Wang H, Yang H, Shi L, Katz Y, Theunissen TW, Rangarajan S, Shivalila CS, Dadon DB, Jaenisch R. 2013. Multiplexed activation of endogenous genes by CRISPR-on, an RNA-guided transcriptional activator system. Cell Res 23: 1163–1171. 10.1038/cr.2013.122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, Hsu PD, Wu X, Jiang W, Marraffini LA, et al. 2013. Multiplex genome engineering using CRISPR/Cas systems. Science 339: 819–823. 10.1126/science.1231143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding S, Wu X, Li G, Han M, Zhuang Y, Xu T. 2005. Efficient transposition of the piggyBac (PB) transposon in mammalian cells and mice. Cell 122: 473–483. 10.1016/j.cell.2005.07.013 [DOI] [PubMed] [Google Scholar]
- Gasperini M, Findlay GM, McKenna A, Milbank JH, Lee C, Zhang MD, Cusanovich DA, Shendure J. 2017. CRISPR/Cas9-mediated scanning for regulatory elements required for HPRT1 expression via thousands of large, programmed genomic deletions. Am J Hum Genet 101: 192–205. 10.1016/j.ajhg.2017.06.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gossen M, Freundlieb S, Bender G, Muller G, Hillen W, Bujard H. 1995. Transcriptional activation by tetracyclines in mammalian cells. Science 268: 1766–1769. 10.1126/science.7792603 [DOI] [PubMed] [Google Scholar]
- Grigoriadis A, Mackay A, Noel E, Wu PJ, Natrajan R, Frankum J, Reis-Filho JS, Tutt A. 2012. Molecular characterisation of cell line models for triple-negative breast cancers. BMC Genomics 13: 619 10.1186/1471-2164-13-619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haeussler M, Schönig K, Eckert H, Eschstruth A, Mianné J, Renaud JB, Schneider-Maunoury S, Shkumatava A, Teboul L, Kent J, et al. 2016. Evaluation of off-target and on-target scoring algorithms and integration into the guide RNA selection tool CRISPOR. Genome Biol 17: 148 10.1186/s13059-016-1012-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartenian E, Doench JG. 2015. Genetic screens and functional genomics using CRISPR/Cas9 technology. FEBS J 282: 1383–1393. 10.1111/febs.13248 [DOI] [PubMed] [Google Scholar]
- Hsu PD, Lander ES, Zhang F. 2014. Development and applications of CRISPR-Cas9 for genome engineering. Cell 157: 1262–1278. 10.1016/j.cell.2014.05.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joung J, Konermann S, Gootenberg JS, Abudayyeh OO, Platt RJ, Brigham MD, Sanjana NE, Zhang F. 2017. Genome-scale CRISPR-Cas9 knockout and transcriptional activation screening. Nat Protoc 12: 828–863. 10.1038/nprot.2017.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabadi AM, Ousterout DG, Hilton IB, Gersbach CA. 2014. Multiplex CRISPR/Cas9-based genome engineering from a single lentiviral vector. Nucleic Acids Res 42: e147 10.1093/nar/gku749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahlig KM, Saridey SK, Kaja A, Daniels MA, George AL Jr, Wilson MH. 2010. Multiplexed transposon-mediated stable gene transfer in human cells. Proc Natl Acad Sci 107: 1343–1348. 10.1073/pnas.0910383107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaji K, Norrby K, Paca A, Mileikovsky M, Mohseni P, Woltjen K. 2009. Virus-free induction of pluripotency and subsequent excision of reprogramming factors. Nature 458: 771–775. 10.1038/nature07864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kearns NA, Genga RM, Enuameh MS, Garber M, Wolfe SA, Maehr R. 2014. Cas9 effector-mediated regulation of transcription and differentiation in human pluripotent stem cells. Development 141: 219–223. 10.1242/dev.103341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirk JM, Kim SO, Inoue K, Smola MJ, Lee DM, Schertzer MD, Wooten JS, Baker AR, Sprague D, Collins DW, et al. 2018. Functional classification of long non-coding RNAs by k-mer content. Nat Genet 50: 1474–1482. 10.1038/s41588-018-0207-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Latos PA, Stricker SH, Steenpass L, Pauler FM, Huang R, Senergin BH, Regha K, Koerner MV, Warczok KE, Unger C, et al. 2009. An in vitro ES cell imprinting model shows that imprinted expression of the Igf2r gene arises from an allele-specific expression bias. Development 136: 437–448. 10.1242/dev.032060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JT, Bartolomei MS. 2013. X-inactivation, imprinting, and long noncoding RNAs in health and disease. Cell 152: 1308–1323. 10.1016/j.cell.2013.02.016 [DOI] [PubMed] [Google Scholar]
- Li MA, Turner DJ, Ning Z, Yusa K, Liang Q, Eckert S, Rad L, Fitzgerald TW, Craig NL, Bradley A. 2011. Mobilization of giant piggyBac transposons in the mouse genome. Nucleic Acids Res 39: e148 10.1093/nar/gkr764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Michael IP, Zhou D, Nagy A, Rini JM. 2013. Simple piggyBac transposon-based mammalian cell expression system for inducible protein production. Proc Natl Acad Sci 110: 5004–5009. 10.1073/pnas.1218620110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Zhang A, Xue H, Li D, Liu Y. 2017. One-step piggyBac transposon-based CRISPR/Cas9 activation of multiple genes. Mol Ther Nucleic Acids 8: 64–76. 10.1016/j.omtn.2017.06.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Pinera P, Kocak DD, Vockley CM, Adler AF, Kabadi AM, Polstein LR, Thakore PI, Glass KA, Ousterout DG, Leong KW, et al. 2013. RNA-guided gene activation by CRISPR-Cas9-based transcription factors. Nat Methods 10: 973–976. 10.1038/nmeth.2600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinn J, Kunath T, Rossant J. 2006. Mouse trophoblast stem cells. Methods Mol Med 121: 125–148. 10.1385/1-59259-983-4:123 [DOI] [PubMed] [Google Scholar]
- The R Development Core Team. 2017. R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria: https://www.r-project.org. [Google Scholar]
- Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F. 2013. Genome engineering using the CRISPR-Cas9 system. Nat Protoc 8: 2281–2308. 10.1038/nprot.2013.143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakuma T, Nishikawa A, Kume S, Chayama K, Yamamoto T. 2014. Multiplex genome engineering in human cells using all-in-one CRISPR/Cas9 vector system. Sci Rep 4: 5400 10.1038/srep05400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, et al. 2012. Fiji: an open-source platform for biological-image analysis. Nat Methods 9: 676–682. 10.1038/nmeth.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin KJ, Wall EA, Zavzavadjian JR, Santat LA, Liu J, Hwang JI, Rebres R, Roach T, Seaman W, Simon MI, et al. 2006. A single lentiviral vector platform for microRNA-based conditional RNA interference and coordinated transgene expression. Proc Natl Acad Sci 103: 13759–13764. 10.1073/pnas.0606179103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoger R, Kubicka P, Liu CG, Kafri T, Razin A, Cedar H, Barlow DP. 1993. Maternal-specific methylation of the imprinted mouse Igf2r locus identifies the expressed locus as carrying the imprinting signal. Cell 73: 61–71. 10.1016/0092-8674(93)90160-R [DOI] [PubMed] [Google Scholar]
- Umlauf D, Goto Y, Cao R, Cerqueira F, Wagschal A, Zhang Y, Feil R. 2004. Imprinting along the Kcnq1 domain on mouse chromosome 7 involves repressive histone methylation and recruitment of Polycomb group complexes. Nat Genet 36: 1296–1300. 10.1038/ng1467 [DOI] [PubMed] [Google Scholar]
- Wang W, Lin C, Lu D, Ning Z, Cox T, Melvin D, Wang X, Bradley A, Liu P. 2008. Chromosomal transposition of PiggyBac in mouse embryonic stem cells. Proc Natl Acad Sci 105: 9290–9295. 10.1073/pnas.0801017105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang G, Yang L, Grishin D, Rios X, Ye LY, Hu Y, Li K, Zhang D, Church GM, Pu WT. 2017. Efficient, footprint-free human iPSC genome editing by consolidation of Cas9/CRISPR and piggyBac technologies. Nat Protoc 12: 88–103. 10.1038/nprot.2016.152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson MH, Coates CJ, George AL Jr. 2007. PiggyBac transposon-mediated gene transfer in human cells. Mol Ther 15: 139–145. 10.1038/sj.mt.6300028 [DOI] [PubMed] [Google Scholar]
- Wright AV, Nuñez JK, Doudna JA. 2016. Biology and applications of CRISPR systems: harnessing nature's toolbox for genome engineering. Cell 164: 29–44. 10.1016/j.cell.2015.12.035 [DOI] [PubMed] [Google Scholar]
- Wu X, Scott DA, Kriz AJ, Chiu AC, Hsu PD, Dadon DB, Cheng AW, Trevino AE, Konermann S, Chen S, et al. 2014. Genome-wide binding of the CRISPR endonuclease Cas9 in mammalian cells. Nat Biotechnol 32: 670–676. 10.1038/nbt.2889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yusa K, Rad R, Takeda J, Bradley A. 2009. Generation of transgene-free induced pluripotent mouse stem cells by the piggyBac transposon. Nat Methods 6: 363–369. 10.1038/nmeth.1323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zawistowski JS, Bevill SM, Goulet DR, Stuhlmiller TJ, Beltran AS, Olivares-Quintero JF, Singh D, Sciaky N, Parker JS, Rashid NU, et al. 2017. Enhancer remodeling during adaptive bypass to MEK inhibition is attenuated by pharmacologic targeting of the P-TEFb complex. Cancer Discov 7: 302–321. 10.1158/2159-8290.CD-16-0653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu S, Li W, Liu J, Chen CH, Liao Q, Xu P, Xu H, Xiao T, Cao Z, Peng J, et al. 2016. Genome-scale deletion screening of human long non-coding RNAs using a paired-guide RNA CRISPR-Cas9 library. Nat Biotechnol 34: 1279–1286. 10.1038/nbt.3715 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.