

Abstract
Spinal cord injury (SCI) triggers inflammatory responses that involve neutrophils, macrophages/microglia and astrocytes and molecules that potentially cause secondary tissue damage and functional impairment. Here, we assessed the contribution of the calcium-dependent K+ channel KCNN4 (KCa3.1, IK1, SK4) to secondary damage after moderate contusion lesions in the lower thoracic spinal cord of adult mice. Changes in KCNN4 mRNA levels (RT-PCR), KCa3.1 protein expression (Western blots), and cellular expression (immunofluorescence) in the mouse spinal cord were monitored between 1 and 28 d after SCI. KCNN4 mRNA and KCa3.1 protein rapidly increased after SCI; double labeling identified astrocytes as the main cellular source accounting for this upregulation. Locomotor function after SCI, evaluated for 28 d in an open-field test using the Basso Mouse Scale, was improved in a dose-dependent manner by treating mice with a selective inhibitor of KCa3.1 channels, TRAM-34 (triarylmethane-34). Improved locomotor function was accompanied by reduced tissue loss at 28 d and increased neuron and axon sparing. The rescue of tissue by TRAM-34 treatment was preceded by reduced expression of the proinflammatory mediators, tumor necrosis factor-α and interleukin-1β in spinal cord tissue at 12 h after injury, and reduced expression of inducible nitric oxide synthase at 7 d after SCI. In astrocytes in vitro, TRAM-34 inhibited Ca2+ signaling in response to metabotropic purinergic receptor stimulation. These results suggest that blocking the KCa3.1 channel could be a potential therapeutic approach for treating secondary damage after spinal cord injury.
Introduction
The intermediate conductance Ca2+-activated potassium channel, known as KCa3.1, IK1, or SK4 (KCNN4 for the gene), was first described from functional studies of human red blood cells (Gardos, 1958). Its gating is voltage independent and depends on Ca2+ binding to calmodulin molecules constitutively associated with the channel protein (Joiner et al., 1997; Xia et al., 1998; Khanna et al., 1999). Small increases in intracellular Ca2+ induce channel opening (Kd ∼300 nm), resulting in K+ efflux that maintains a driving force for subsequent Ca2+ influx. Therefore, the KCNN4/KCa3.1 channel could play a key role in regulating a plethora of cell functions mediated by Ca2+ influx. There is considerable evidence that KCa3.1 regulates activation and proliferation of T-lymphocyte subsets (Khanna et al., 1999; Ghanshani et al., 2000; Cahalan et al., 2001).
In contrast, there is very little information about KCa3.1 expression and roles in the CNS. We reported that KCNN4/KCa3.1 is expressed in microglia in vitro; and that a selective blocker [TRAM-34 (triarylmethane-34)] reduced microglial activation, inducible nitric oxide synthase (iNOS) expression, and the ability of microglia to kill neurons in vitro, and decreased retinal ganglion cell degeneration following optic nerve transection in vivo (Kaushal et al., 2007). In experimental autoimmune encephalomyelitis (EAE), a widely used animal model of CNS autoimmune disease multiple sclerosis, TRAM-34 reduced proinflammatory chemokine/cytokine expression in the spinal cord and reduced clinical disability (Reich et al., 2005). KCa3.1 blockers also reduced brain edema, intracranial pressure, and infarct volume in a model of acute subdural hematoma (Mauler et al., 2004). None of the studies examined the cell types that express KCa3.1 in the CNS in vivo or the changes in protein expression over time, or demonstrated its actions within the CNS or the mechanisms underlying the in vivo improvements. However, these studies suggest that KCNN4/KCa3.1 is a potential therapeutic target for reducing inflammation-mediated neurotoxicity in certain CNS conditions. It is important therefore to further investigate the expression and roles of KCNN4/KCa3.1 in other CNS conditions.
After spinal cord injury (SCI), the initial trauma induces an inflammatory response that can contribute to secondary tissue damage and worsen the functional outcome (Kwon et al., 2004; Popovich and Longbrake, 2008). Two resident cells (astrocytes and microglia) and infiltrating blood macrophages can contribute to CNS inflammation and secondary damage by producing interleukin 1β (IL-1β), tumor necrosis factor α (TNF-α), superoxide, nitric oxide (NO) (Klusman and Schwab, 1997; Yang et al., 2004a; Fu et al., 2007; Pineau and Lacroix, 2007), and other cytotoxic mediators (Lee et al., 2000; Liu et al., 2005; Donnelly and Popovich, 2008; Bao et al., 2009). For instance, production of peroxynitrite from superoxide and nitric oxide can cause lipid peroxidation, DNA damage, and protein nitration, actions that can damage both neurons and oligodendrocytes after spinal cord injury (Satake et al., 2000; Bao et al., 2004; Xu et al., 2006; Xiong et al., 2007; Xiong and Hall, 2009). KCa3.1 channels contribute to nitric oxide and peroxynitrite production, and to neurodegeneration caused by activated microglia in vitro (Kaushal et al., 2007). Therefore, we tested the hypothesis that KCNN4/KCa3.1 is expressed in the injured spinal cord, and that blocking the channel will reduce secondary damage and improve function after contusion injury in adult mice.
Materials and Methods
Spinal cord contusion injury and treatment with TRAM-34.
All surgical procedures were performed in accordance with guidelines from the Canadian Council on Animal Care and were approved by the McGill University Animal Care Committee. Young adult (6–8-week-old) female C57BL/6 mice (Charles River) were deeply anesthetized by intraperitoneal injection of ketamine (50 mg/kg), xylazine (5 mg/kg), and acepromazine (1 mg/kg), and a moderate contusion injury was made at the T11 thoracic vertebral level using the Infinite Horizons Impactor device (Precision Scientific Instrumentation) with a contusion force of 50 kilodynes and tissue displacement ranging between 400 and 600 μm, as previously described (Ghasemlou et al., 2005). Mice with injuries within these parameters were randomly assigned to either the control or experimental groups.
TRAM-34, a selective blocker of the KCa3.1 channel (Wulff et al., 2000; Chandy et al., 2004), was synthesized by Toronto Research Chemicals and prepared as previously described (Köhler et al., 2003; Toyama et al., 2008). After the surgery, one cohort of mice received intraperitoneal injections of TRAM-34, while the control cohort received the same volume of vehicle (peanut oil). We started with eight mice in each of the TRAM-34 (120 mg/kg/d dose) and vehicle-treated groups used for locomotor analyses (see Fig. 5). However, two TRAM-treated mice died, leaving six mice in this group. Additional mice were used for other types of analyses as indicated. TRAM was injected intraperitoneally at 60 mg/kg in 100 μl of peanut oil every 12 h (total of 120 mg/kg/d) for 4 weeks, starting 1 h after the injury. We selected this TRAM-34 dose based on the daily subcutaneous dose of 120 mg/kg used in a recent study on mice, which showed rapid turnover (circulating half-life ∼1 h) and low bioavailability (Toyama et al., 2008). For analysis of locomotor recovery using the Basso Mouse Scale (BMS) (see below), additional groups of mice were treated with lower TRAM-34 doses (3 mg/kg/d [TRAM-34 n = 11; vehicle n = 9]; 6 mg/kg/d [TRAM-34 n = 6; vehicle n = 7]; 30 mg/kg/d [TRAM-34 n = 8; vehicle n = 8]; and 120 mg/kg/d [TRAM-34 n = 6; vehicle n = 8]) to monitor dose-dependent effects. All the locomotor and histological analyses were performed in a blinded fashion.
Figure 5.
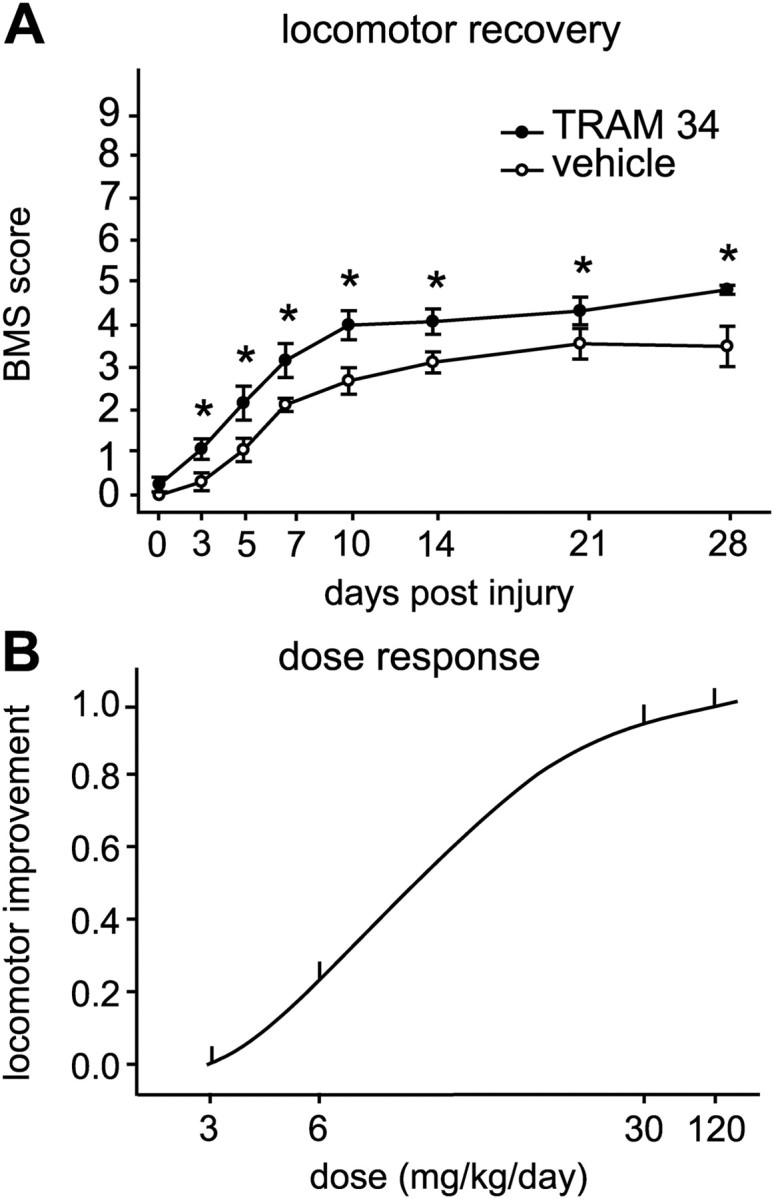
TRAM-34 treatment improves locomotor recovery after SCI. A, The time course of locomotor recovery in mice treated after SCI with TRAM-34 (120 mg/kg/d) or the vehicle (peanut oil) was assessed using the 9-point BMS. Animals treated with TRAM-34 had significantly greater locomotor recovery starting 3 d after SCI (*p < 0.01; TRAM-34 group, n = 6; vehicle group, n = 8). B, Dose dependence of the improvement in locomotor function with TRAM-34 treatment, assessed at 14 dpi. The mean BMS score of each TRAM-treated group was subtracted from the mean BMS score of the vehicle group. The number of mice used in each of the groups is indicated in Materials and Methods. The difference was only statistically significant at the 120 mg/kg/d dose.
Analysis of mRNA expression.
Animals were killed under deep anesthesia, and a 5 mm length of the spinal cord centered on the lesion site was dissected out at 1, 3, 5, 7, 14, 21, and 28 d postinjury (dpi), and from TRAM-34- (120 mg/kg/d) or vehicle-treated animals at 28 dpi. Spinal cord tissue from the same region from uninjured mice was also obtained for analysis. Total RNA was extracted from the frozen tissue (RNeasy, Qiagen) after degrading any contaminating DNA with DNaseI (0.1 U/ml, 15 min, 37°C; GE Healthcare). Gene-specific primers and Taqman probes were generated using PrimerQuest (Integrated DNA Technologies). Reverse transcription was immediately followed by PCR amplification with primers specifically designed for mouse KCNN4, IL-1β, and TNF-α. RT-PCR products were separated by electrophoresis on a 2% agarose ethidium bromide-stained gel and quantified with the gel analyzer AlphaImager 2200 (Cell Biosciences) and ImageQuant. Data were first normalized to levels of the housekeeping gene peptidylprolyl isomerase A (PPIA) and then expressed as the fold change (mean ± SD; three experiments were done with spinal cords of three different mice) compared with the uninjured spinal cord.
iNOS expression was measured by qRT-PCR in TRAM-34- and vehicle-treated spinal cords. Amplification was performed using Brilliant Probe-based Reagents and an MX4000 detection system (Stratagene). The threshold cycle for was determined and normalized to that of the housekeeping gene, glyceraldehyde 3-phosphate dehydrogenase (GAPDH), and then expressed as the fold change (mean ± SD; six experiments were done with spinal cords of six different mice) compared with the uninjured spinal cord.
Western blot analysis.
As above, a 5 mm length of the spinal cord tissue centered on the lesion site was dissected out from uninjured and injured animals at 1, 3, 7, 14, 21, and 28 dpi. Total protein was extracted using buffer containing protease inhibitors (Cocktail Tablet; Roche), phosphatase inhibitors (500 mm NaF and 100 mm Na3VO4), and 1% Triton in PBS. Equal concentrations of proteins, as determined by the Bio-Rad Bradford protein assay, were mixed with NuPAGE LDS sample buffer, sonicated, heated to 95°C for 10 min, and then separated by NuPAGE Bis-Tris gel (12% acrylamide) and electrotransferred (XCell SureLock, Invitrogen) onto Immobilon PVDF membrane. Membranes were first incubated in blocking solution (5% nonfat dry milk, 0.05% Tween 20 in 1× PBS) for 1 h at room temperature and then incubated (overnight, 4°C) with mouse anti-KCa3.1 (1:400; Alomone Labs) in the blocking solution. After rinsing with PBS-T (1× PBS, 0.05% Tween 20), blots were incubated (1 h, room temperature) with HRP-linked anti-rabbit Ig (1:500; Jackson Immunoresearch) in the blocking solution. After washing, the membranes were treated with Enhanced Chemiluminescence ECL Plus kit reagents (PerkinElmer) and exposed to ECL hyper film (VWR). Membranes were later reprobed with mouse anti-β actin (1:400; Sigma-Aldrich). Relative intensities of the KCa3.1 immunoreactivity were compared with β-actin controls, and densitometry was performed using ImageQuant Software (GE Healthcare). Analyses were run in triplicate with different animals (n = 3).
Isolation of macrophages from the lesion spinal cord and microglial purification.
Macrophages/microglia were isolated from the injured spinal cord at 7 dpi. Briefly, 5 mm of the spinal cord, centered on the lesion epicenter, were dissected and minced in cold Minimal Essential Medium (MEM; Invitrogen), centrifuged, and resuspended in a Percoll (Sigma-Aldrich) gradient (3.5 ml of 80% Percoll overlaid by 4 ml of 40% Percoll). After centrifugation (500 × g, 35 min), cells at the interface of the Percoll gradient were collected, resuspended in HBSS (Invitrogen), centrifuged again, and resuspended in RPMI-1640 medium containing 10% fetal bovine serum (FBS; Wisent), 1% vitamins, and 1% penicillin/streptomycin. Cells were then plated on coverslips and placed in an incubator for 2 h before double labeling with macrophage antigen-1 (Mac-1) and anti-KCa3.1 antibodies.
Cultures of bone marrow-derived macrophages (BMDMs) were prepared as described previously (Longbrake et al., 2007). Briefly, adult female C57BL/6 mice (6–8 weeks) were killed, and their femur and tibias were removed. The bone marrow plugs were flushed with RPMI-1640 medium containing 10% FBS and 10% L-cell-conditioned medium (a source for granulocyte macrophage-colony stimulating factor). After centrifugation (600 × g, 7 min), cells were cultured in 10 cm tissue Petri dishes. The next day, adherent cells were discarded and nonadherent cells were cultured with fresh medium supplemented with L-cell-conditioned medium for 7–10 d until cells matured into adherent macrophages. Cell purity was confirmed with Mac-1 labeling.
Purified cultures of microglia were prepared from 2-day-old mouse pups, using standard protocols. After removing the meninges, the brain was dissected, minced in cold MEM, centrifuged (300 × g, 10 min), and resuspended in MEM supplemented with 5% horse serum, 5% FBS, and 0.05 mg/ml gentamycin (Invitrogen). Two days later, cellular debris, nonadherent cells, and supernatant were removed, fresh medium was added to the flask, and the mixed cultures were allowed to grow for another 8–10 d. Microglia suspensions were harvested by shaking the flasks on an orbital shaker (65 rpm, 4–6 h, 37°C), and then seeded in MEM with 2% FBS.
Ca2+ signaling in astrocytes.
Primary astrocyte cultures were prepared as in our earlier work (Kaushal et al., 2007). Brain tissue from 1- to 2-d-old rat pups was mashed through a stainless steel sieve (100 mesh; Tissue Grinder Kit CD-1, Sigma-Aldrich), centrifuged (1000 × g, 12 min), resuspended, and seeded into flasks with culture medium containing DMEM (Invitrogen), 10% FBS, and 100 μm gentamycin (Invitrogen). The medium was replaced after 2 d, and the mixed cultures were incubated for a further 7–8 d. The flasks were shaken for 5–6 h to remove less adherent cells; and astrocytes were harvested by a 6 min trypsin digestion, centrifugation (1000 × g) for 10 min, washing, and resuspension in culture medium. Cells were seeded at a density of 2 × 104 cells/15 mm diameter coverslip, and cultured for a further 2–3 d. This yielded >80% astrocyte purity, as determined by GFAP labeling, and ∼20% microglia.
Fura-2 signaling was used to monitor intracellular Ca2+ (Ca2+i), according to our standard procedures (Ohana et al., 2009). Astrocytes were loaded for ∼45 min at room temperature with 3.5 μg/ml Fura-2-AM (Invitrogen) diluted in standard bath solution containing the following (in mm): 135 NaCl, 5 KCl, 1 MgCl2, 1 CaCl2, 5 glucose, 10 HEPES, adjusted to pH 7.4 with NaOH, and to ∼300 mOsm with sucrose. Coverslips bearing astrocytes were mounted in a perfusion chamber (model RC-25, Warner Instruments) on the stage of a Nikon Diaphot inverted microscope and superfused with fresh bath solution. Images were acquired at room temperature using a Nikon Diaphot inverted microscope with a 40× quartz objective lens, Retiga-EX camera (Q-Imaging), DG-4 arc lamp with excitation wavelength changer (Sutter Instruments), and Northern Eclipse image acquisition software (Empix Imaging). Images were captured every 1–2 s at 340 and 380 nm excitation wavelengths, and 340:380 nm ratios calculated. Data are presented as the mean ± SEM, and the statistical significance of differences was determined with Student's t test. After recording the baseline Fura-2 signal for 1 min, the perfusion solution was switched to 300 μm uridine triphosphate (UTP, Sigma-Aldrich) in standard bath solution for 2 min, followed by a 10 min wash, and then 1 μm thapsigargin (Sigma-Aldrich) for 2 min, followed by a 10 min wash. For TRAM-34-treated astrocytes, the same procedure was used, except that 1 μm TRAM-34 was present throughout the recordings. For each field of cells, astrocytes (morphologically distinct from the microglia) were selected for off-line Ca2+ analysis.
Analysis of locomotor recovery.
Locomotor function of the hindlimbs of TRAM-34-treated and control mice was evaluated in an open-field test using the 9-point BMS (Basso et al., 2006), which was specifically developed for locomotor testing after contusion injuries in mice. Before testing, bladders were expressed to avoid spontaneous bladder contraction that often accompanies hindlimb activity. The evaluations were performed by two individuals who were trained in the Basso laboratory at Ohio State University and who were blinded to the experimental groups. Locomotor function in both hindlimbs was scored, and the consensus score was taken. Hindlimb movements immediately after contact with the assessor were disregarded. The mean of right and left hindlimb scores was taken. The test was performed at 1, 3, 5, 7, 14, 21, and 28 dpi. The final score is presented as the mean ± SEM.
Histological and immunohistochemical analyses.
To assess cell type-specific expression of KCa3.1 after SCI, mice were deeply anesthetized with ketamine/xylazine/acepromazine at varying times after SCI, as indicated below, and perfused through the heart with 4% paraformaldehyde in 0.1 m PBS, pH 7.4. A 6 mm length of the spinal cord containing the lesion site was dissected out and cryoprotected with 30% sucrose in 0.1 m PBS. Serial cross sections (14 μm thick) were cut on a cryostat and picked up on to gelatin-coated glass slides. Double-immunofluorescence staining was performed as follows. Nonspecific binding was blocked by a 4 h incubation in 1% bovine serum albumin and 1% goat normal serum. Sections were then incubated overnight at 4°C with a rabbit polyclonal antibody against KCa3.1 (1:400; Santa Cruz Biotechnology), and one of the following antibodies: rat monoclonal anti-GFAP (1:400; Zymed Laboratories); rat monoclonal Mac-1 (against CD11b; 1:200; Serotec); mouse monoclonal Mac-2 against galectin (hybridoma supernatant); rabbit polyclonal anti-ionized Ca2+ binding adapter-1′ (Iba-1; 1:1000, Wako); mouse monoclonal anti-200 kDa neurofilament (1:500; NF200, Sigma-Aldrich); and mouse monoclonal anti-NeuN antibody (to label neurons; 1:500; Millipore). The sections were then rinsed three times with PBS-T and incubated with appropriate secondary antibodies (Alexa Fluor 568 goat anti-rat [1:400; Invitrogen]; Alexa Fluor 488 goat anti-rabbit [1:400; Invitrogen]; Alexa Fluor 568 goat anti-mouse [1:400; Invitrogen] for 2 h at room temperature, washed and mounted with DAPI (4′,6′-diamidino-2-phenylindole dihydrochloride)-Vectashield medium (Vector Laboratories). In some experiments, microglia and macrophages were labeled with tomato lectin (FITC conjugated; 1:500; Sigma-Aldrich).
To assess histological changes after TRAM-34 treatment (120 mg/kg/d), mice were perfused with fixative (4% paraformaldehyde in 0.1 m phosphate buffer) at 28 dpi. Cryostat cross sections of the spinal cord from TRAM-34- and vehicle-treated mice were immunostained with goat anti-rabbit GFAP to delineate the size of the lesion (1:400; Zymed Laboratories) or for 200 kDa neurofilaments (NF200, clone NE-14; 1:500; Sigma-Aldrich) to visualize spared axons. Tissue sections were also stained in 0.1% cresyl violet (Sigma-Aldrich) to quantify neuron survival in the ventral horns.
Image analysis and quantification.
Tissue sections were viewed with an Axioskop 2 Plus microscope (Zeiss), and images were captured using a QImaging Retiga 1300 camera with the same exposure time among images within a series, including tissue from treated and control animals. Quantification was done using BioQuant Nova Prime image analysis system (BioQuant Image Analysis). All analyses were done blinded to the treatment groups. The epicenter of injury in all spinal cords was identified using immunoreactivity for GFAP to label astrocytes, and CD11b (Mac-1 antibody) to label microglia and macrophages. Quantification was done at regular intervals over a maximum distance of 6 mm. Each type of analysis was performed in six to eight mice per group, as indicated in Results. The percentage of KCa3.1+/GFAP+ astrocytes was counted in cross sections. For quantification of astrocytes, individual cells were identified by their GFAP-labeled processes surrounding a DAPI+ nucleus. Spared CNS tissue was assessed by calculating the GFAP-labeled area as a percentage of the total area of the cross section, and the results were expressed as the percentage of spared tissue. The large neurons in the ventral horn were quantified in tissue sections stained with cresyl violet. Axonal sparing in the dorsal column in experimental and control mice was assessed by calculating the area occupied by NF200 labeling in a fixed area of the dorsal column, which is the site of direct impact from the contusion. The result is expressed as a percentage of the area occupied by spared axons based on the NF200 staining.
Statistical analyses.
Comparisons of two datasets were analyzed by Student's t test, while data with more than two variables (i.e., over time or distance) were analyzed by two-way repeated-measures ANOVA with post hoc Tukey's analysis (SigmaStat). Values are expressed as the mean ± SEM, except for RT-PCR, where they are mean ± SD. Statistical significance was set at p < 0.05. All analyses were performed blind to the treatment the mice had received.
Results
KCNN4/KCa3.1 is upregulated in reactive astrocytes after SCI
We first assessed changes in mRNA expression of KCNN4 in the injured spinal cord of adult C57BL/6 mice at 1, 3, 7, 14, 21, and 28 d after a moderate spinal cord contusion. Comparisons were made with the normal uninjured spinal cord. RT-PCR analysis showed a significant increase in KCNN4 expression following SCI, starting at 3 dpi and reaching a peak sixfold increase at 7 dpi (Fig. 1A,B). KCNN4 expression remained significantly elevated up to 28 dpi, the last time point examined (p < 0.05; n = 3 mice per time point). Western blot analysis also revealed a significant elevation in KCa3.1 protein from 5 to 21 dpi (p < 0.05; n = 3 mice per time point) (Fig. 1C,D).
Figure 1.
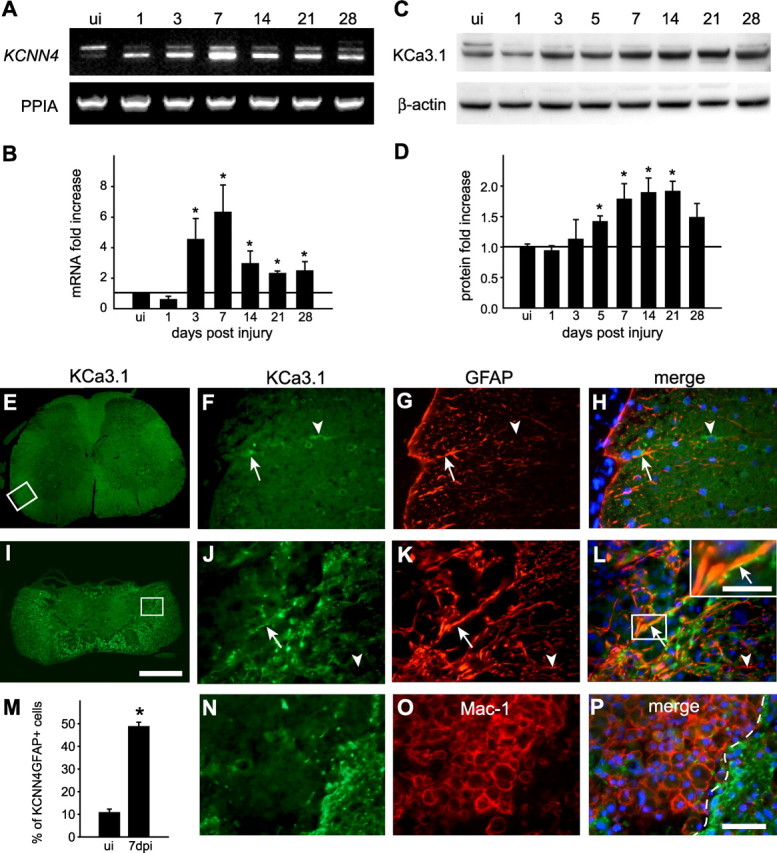
KCNN4/KCa3.1 expression increases after SCI. A, B, RT-PCR analysis of KCNN4 mRNA levels in spinal cord from uninjured (ui) mice and 1, 3, 5, 7, 14, and 21 dpi mice. PPIA was used as a loading control. A representative example is shown in A. For the quantification in B, data were first normalized to PPIA and then expressed as fold change (mean ± SD; 3 experiments, n = 3) compared with the uninjured spinal cord (horizontal line). Significant increases in KCNN4 expression are indicated as follows: *p < 0.05. C, Western blot analysis of the 47-kDa-molecular-weight band labeled with anti-KCa3.1 antibody in spinal cord homogenates. β-actin was used as the loading control. D, Quantification of KCa3.1 protein levels (*p < 0.05; n = 3). The horizontal line indicates the level in uninjured control spinal cords. E, Low-magnification image of the normal, uninjured spinal cord immunolabeled for KCa3.1. The area in the box is shown at higher magnification in F–H. Higher magnification showing double labeling for KCa3.1 (F) and the astrocyte marker GFAP (G) (arrow), and the merged image with DAPI-stained nuclei (H). Note also the single labeled cell (arrowhead). I, Low-magnification image of the injured spinal cord at 7 dpi immunolabeled for KCa3.1. The area in the box is shown at higher magnification in J–L. Higher-magnification images showing double labeling for KCa3.1 (J) and GFAP (K) (arrows), and the merged images with DAPI-stained nuclei (L). Note the increase in KCa3.1 labeling of GFAP+-reactive astrocytes (arrows); the inset in L shows double-labeled profiles at higher magnification (arrow). In the inset, note that the strongest labeling for KCa3.1 is on the membrane, while the GFAP labeling is intracellular. Arrowheads in J–L show a single-labeled GFAP+/KCa3.1− profile. M, Quantification of the percentage of GFAP+ cells colabeled for KCa3.1 at 7 dpi compared with the uninjured cord (*p < 0.001, n = 4). N–P, The lesion core at 7 d after SCI is double labeled for KCa3.1 (N) and the microglia/macrophage marker Mac-1 (O). The lesion core is to the left of the dashed line in the merged image (P), which also shows DAPI-stained nuclei. Scale bars: E, I, 500 μm; H, L, P, 50 μm; L inset, 30 μm.
To identify the cell types in which KCNN4/KCa3.1 was upregulated, double labeling was performed with cell-specific markers in spinal cords of uninjured mice, and at 7 d after the contusion injury, which is at the peak of the increase in KCNN4 expression. In the uninjured spinal cord, KCa3.1 colocalized with some GFAP+ astrocytes (Fig. 1E–H). In the injured cord (Fig. 1I–L), KCa3.1 was expressed around the lesion core, in a region that contains reactive astrocytes that form the glial scar. The number of KCa3.1-labeled astrocytes was approximately fivefold higher than in the uninjured spinal cord (p < 0.001; n = 4) (Fig. 1M). In the uninjured spinal cord, KCa3.1 was also detected in neurons, as previously reported for neurons isolated from rat brain (Kaushal et al., 2007), and in oligodendrocytes (Fig. 2A–F), but neither cell type showed altered staining in the injured spinal cord.
Figure 2.

KCa3.1 is expressed in neurons and some oligodendrocytes in the uninjured mouse spinal cord. A–C, Double labeling with antibodies against the neuronal marker NeuN (A) and the KCa3.1 channel (B) shows that the cell bodies of large neurons in the ventral horn express KCa3.1 (C). D–F, Double labeling with antibodies against the oligodendrocyte marker CC1 (D) and the KCa3.1 channel (E) shows KCa3.1 expression in the cell body and on the processes of some oligodendrocytes in the ventral white matter (F). This staining pattern was unchanged after SCI (data not shown). Scale bars, 50 μm.
Because previous studies have reported KCNN4/KCa3.1 expression in cultured microglia, we labeled sections from the injured spinal cord for KCa3.1 and several markers of macrophages/microglia. Mac-1, an antibody that recognizes CD11b on macrophages/microglia, did not appear to colocalize with KCa3.1 at 7 dpi (Fig. 1N–P), or with three other macrophage/microglial markers: Mac-2, Iba-1, or tomato lectin (Fig. 3A–I). While KCa3.1 labeling surrounded the lesion, it was not detected in the macrophages that filled the lesion. To rule out fixative-induced antigen masking, we next labeled unfixed spinal cord tissue at 7 dpi; KCa3.1 staining was not detected in macrophages/microglia (Fig. 3J–L). Finally, to address whether staining differences might occur under in vitro conditions, we examined KCNN4/KCa3.1 expression in macrophages/microglia harvested from the contused spinal cord at 7 dpi, and in cultures of murine BMDMs and microglia. Strong KCa3.1 staining was seen in Mac-1+ macrophages/microglia that were acutely isolated from the spinal cord and placed in culture for 2 h to allow time to attach to the coverslip (Fig. 4A–C). Since macrophages in the injured spinal cord could arise from activated microglia or from circulating cells derived from BMDMs, we cultured murine microglia from the neonatal brain, and BMDMs from the adult mouse. Cultured microglia also showed punctate immunoreactivity for KCa3.1 (Fig. 4D–F). Consistent with these observations, RT-PCR analysis showed KCNN4 expression in cultures of BMDMs and microglia, which appear to be somewhat upregulated by LPS (Fig. 4G,H).
Figure 3.

KCa3.1 immunoreactivity was not detected in macrophages and microglia in the injured mouse spinal cord. A–I, Double labeling with anti-KCa3.1 antibody (A, D, G) and different macrophage/microglial markers (B, E, H) in perfusion fixed tissue sections of the spinal cord at 7 d after SCI. The anti-Mac-2 antibody (B) recognizes phagocytic macrophages; tomato lectin (E) binds to macrophages and microglial cells; and Iba-1 (H) is upregulated in activated macrophages/microglia. The lesion core is to the right of the dashed lines in the merged images in C and F. Note that the macrophages/microglia in the lesion lack KCa3.1 staining. J–L, Unfixed tissue section of the spinal cord taken at 7 dpi was double labeled for KCa3.1 (J) and CD11b (Mac-1 antibody; K). Note the absence of double labeling of the Mac-1+ macrophages (L). Scale bars, 50 μm.
Figure 4.

KCNN4 mRNA and KCa3.1 immunoreactivity are expressed in ex vivo macrophages and in primary cultures of macrophages/microglia. A–C, Representative images of double labeling for CD11b (Mac-1 antibody) (A) and KCa3.1 (B) of macrophages isolated from spinal cord 7 d after SCI; the merged image (C) includes nuclear DAPI staining. Note that the large, round Mac-1+ macrophage shows cytoplasmic labeling for KCa3.1. Scale bar: (in C) A–C, 20 μm. D–F, Representative images of unstimulated cultured mouse microglia double labeled with tomato lectin (D) and anti-KCa3.1 antibody (E); the merged image (F) includes DAPI-stained nuclei. Scale bar: (in F) D–F, 10 μm. G, RT-PCR analysis shows mRNA expression of KCNN4 in primary cultures of BMDMs and cultured microglia, with and without activation by LPS treatment. H, Note the much higher KCNN4 expression in BMDMs than in microglia. Values are mean ± SEM, n = 3.
Blocking KCa3.1 with TRAM-34 improves locomotor function after contusion injury
To assess the role of KCNN4/KCa3.1 channels after SCI, mice with a moderate contusion injury were treated daily for 28 d with TRAM-34, a selective inhibitor of KCa3.1 (Wulff et al., 2000; Chandy et al., 2004). TRAM-34 treatment significantly improved locomotor recovery by as early as 3 dpi, and the improvement lasted up to the end of the assessment period at 28 dpi (Fig. 5A) (p < 0.01). TRAM-34-treated mice reached an average final BMS score of 4.8, which reflects the ability to step frequently and to exhibit some coordination. Vehicle-treated control mice reached an average final score of 3.4 (i.e., plantar placing of the paw, with or without weight support). There was a dose-dependent improvement in locomotor recovery measured over the 28 d period with different TRAM-34 amounts injected (Fig. 5B). The small but significant improvement in the BMS score seen at 3 dpi might be due to blocking KCa3.1 channels already present before the injury-induced increase seen beginning at 5 d. The increase in KCa3.1 protein is maintained from 5 to 21 d, and blocking these channels with TRAM-34 might contribute to the sustained improvements.
Blocking KCa3.1 reduces tissue damage after contusion injury
To assess whether TRAM-34 treatment reduces secondary tissue damage in the spinal cord, the extent of CNS tissue loss after SCI was examined by staining cross sections of the spinal cord with anti-GFAP. The GFAP-negative area outlines the cystic cavities or areas infiltrated with fibroblasts, which is seen after injury in mice (Fig. 6B,C). Quantification of the GFAP+ area showed that TRAM-34 treatment resulted in a statistically significant reduction in tissue loss over a 1200 μm distance spanning the lesion epicenter (Fig. 6A). Reduction of CNS tissue loss mediated by TRAM-34 extended between 600 μm rostral to 600 μm caudal to the epicenter (Fig. 6A) (p < 0.001).
Figure 6.
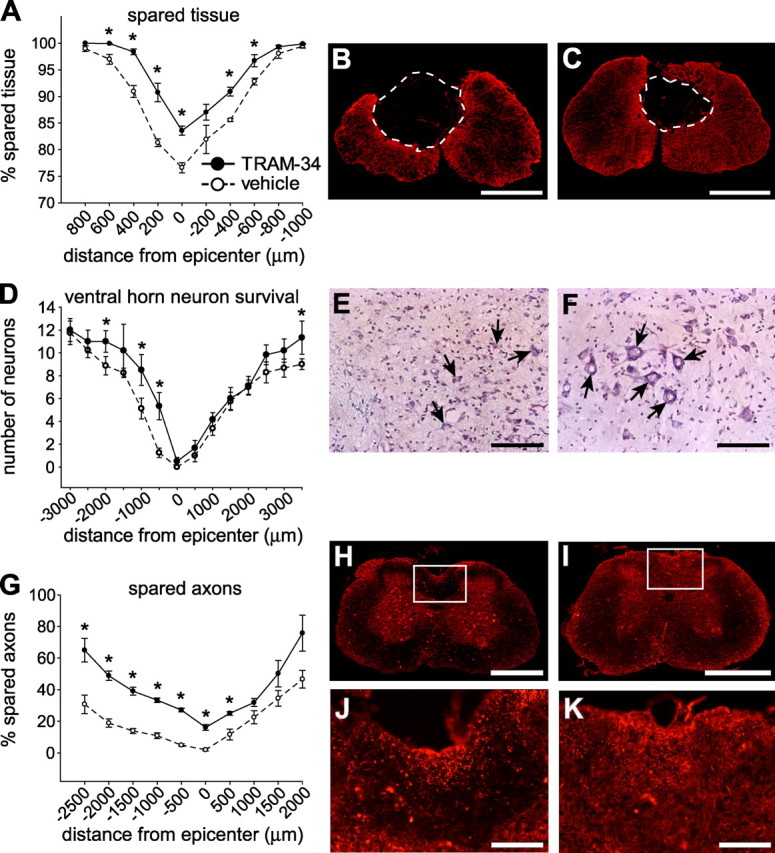
TRAM-34 treatment reduces secondary damage after SCI. All spinal cord histological sections were from mice at 28 dpi. A–C, Spinal cord sections taken at different distances from the lesion epicenter (at position 0) were labeled for GFAP, and the stained area was quantified (A). Data show significant sparing of tissue in TRAM-34-treated mice compared with vehicle-treated controls (p < 0.001). Micrographs show GFAP immunostaining at the epicenter of the injury in representative vehicle-treated (B) and TRAM-34-treated (C) mice. Note the smaller cystic cavity after treatment with TRAM-34. D–F, Tissue sections were stained with cresyl violet to quantify neurons in the ventral horn. Mice treated with TRAM-34 had greater neuron survival (D) in a region spanning 500-2000 μm rostral and 3000 μm caudal to the lesion epicenter (*p < 0.05). Micrographs showing cresyl violet staining of neurons in the ventral horn (arrows) at 500 μm rostral to the lesion epicenter in mice treated with vehicle (E) or TRAM-34 (F). G–K, Tissue sections were labeled for neurofilament 200 (NF200) to label axons. Mice treated with TRAM-34 had more spared axons (G) in the dorsal column white matter (*p < 0.001). Micrographs of NF-200 staining taken 500 μm rostral to the epicenter of the lesion. The areas outlined in the boxes for control (H) and TRAM-34-treated (I) mice are shown at higher magnification in J and K. More NF-200-stained profiles are present in the TRAM-34-treated mouse (K) than the vehicle-treated mouse (J). For all graphs, values are expressed as mean ± SEM (n = 6, TRAM-34 group; n = 8, vehicle group). Scale bars: B, C, H, I, 500 μm; E, F, J, K, 100 μm.
We next examined whether TRAM-34 treatment improved motor neuron survival in the ventral horn. Neuronal survival after SCI was assessed in cresyl violet-stained cross sections of the spinal cord. Quantification of the large motor neurons showed that TRAM-34-treated mice had significantly more neurons from 500 μm to 2 mm rostral and at a distance of 3 mm caudal to the lesion epicenter compared with vehicle-treated mice (Fig. 6D–F) (p < 0.05). Immunofluorescence labeling with an anti-neurofilament antibody was also performed to assess whether the TRAM-34 treatment improved axonal sparing in the dorsal column white matter after injury, which is the site of maximum damage from the contusion force. Quantification showed that TRAM-34 treatment resulted in up to 40% greater axonal sparing rostrally, as well as at the lesion epicenter and 500 μm caudally (Fig. 6G–K) (p < 0.001).
Blocking KCa3.1 reduces expression of proinflammatory mediators after SCI
The inflammatory response mediated by microglia/macrophages and astrocytes after SCI contributes to secondary damage. We therefore assessed whether TRAM-34 treatment reduces expression of IL-1β and TNF-α, two proinflammatory cytokines thought to play important roles in SCI. We measured mRNA expression at 12 h after SCI because cytokine levels in the spinal cord increase early after injury (Bartholdi and Schwab, 1997; Klusman and Schwab, 1997; Yang et al., 2004b; Pineau and Lacroix, 2007). As shown in Figure 7A,B, TRAM-34 treatment significantly reduced expression of both cytokines (p < 0.05).
Figure 7.
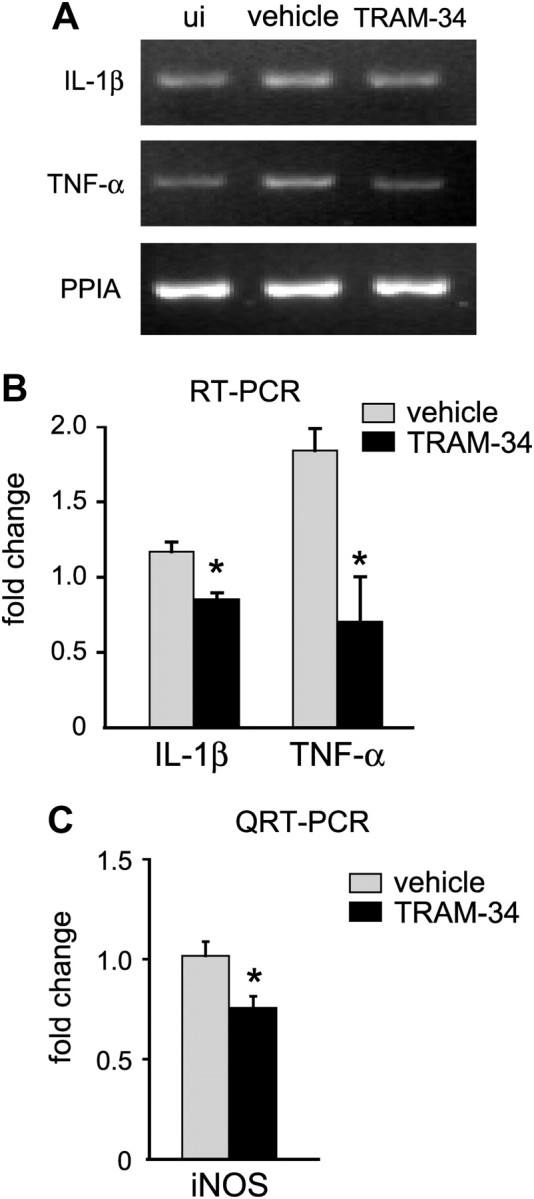
TRAM-34 treatment (120 mg/kg/d) reduces proinflammatory mediators after SCI. A, A representative RT-PCR analysis of mRNA expression of IL-1β and TNF-α, with PPIA used as a loading control. Spinal cord samples taken from an uninjured mouse (ui) or at 12 h after contusion injury from mice treated with the vehicle (peanut oil) or TRAM-34. B, Quantification of the RT-PCR data. Data were first normalized to levels of PPIA, and then expressed as the fold change (mean ± SD; 3 experiments, n = 3 mice per group) compared with the uninjured spinal cord. Both cytokines were reduced in TRAM-34-treated mice (*p < 0.05, Student's t test). C, Quantitative real-time PCR analysis of expression of iNOS at 7 d after SCI from mice treated with vehicle (peanut oil) or TRAM-34. Data were first normalized to the housekeeping gene (GAPDH) and then expressed as the fold change (mean ± SD; 3 experiments, n = 6 mice per group, compared with the uninjured spinal cord). TRAM-34 treatment reduced iNOS expression (*p < 0.05, Student's t test).
Reactive nitrogen species can contribute to secondary damage after SCI (Xu et al., 2001; Xu et al., 2005), and TRAM-34 reduces iNOS expression and nitric oxide production in microglia in vitro (Kaushal et al., 2007). We therefore assessed whether the observed neuroprotective effect of TRAM-34 after SCI corresponds with a reduction in iNOS. Quantitative real-time PCR analysis showed that TRAM-34 treatment significantly reduced iNOS expression in the spinal cord at 7 dpi compared with vehicle-treated control mice (Fig. 7C) (p < 0.05). This time was chosen because iNOS has been shown previously to be expressed up to 7–14 d after SCI (López-Vales et al., 2005; Yang et al., 2007; Rathore et al., 2008), and this is a time point when one sees robust astrocyte and macrophage responses. iNOS can be generated by several pathways, only one of which is likely to be blocked by TRAM-34, and this might account for the partial reduction seen. Furthermore, at 12 h after SCI (the time point used in our work) microglia and astrocytes express IL-1β (Pineau and Lacroix, 2007). Because KCa3.1 was upregulated only in astrocytes after SCI, TRAM-34 treatment might be expected to affect only the IL-1β produced by astrocytes, thus accounting for the modest reduction with treatment.
Blocking KCa3.1 alters Ca2+ signaling in astrocytes
KCa3.1 channels are thought to promote Ca2+ entry by maintaining a driving force and negative membrane potential. To test this hypothesis, we stimulated astrocytes with an agonist of metabotropic purinergic receptors (UTP) (Jiménez et al., 2000; Fam et al., 2003) that evokes IP3 production, release of Ca2+ from internal stores, and a Ca2+i rise mediated by store-operated Ca2+ entry (SOCE), which then refills the stores (Peuchen et al., 1996; Wu et al., 2007). The baseline Ca2+ signal (340:380 nm ratio) (Fig. 8A,B) did not differ between control (0.35 ± 0.05; mean ± SEM) and TRAM-34-treated astrocytes (0.33 ± 0.05; p > 0.4); and UTP evoked a biphasic Ca2+i rise in both control and TRAM-34-treated cells. The Ca2+i signal outlasted the application of UTP (as expected for SOCE), and then decayed and reached a plateau within 2 min under both conditions. TRAM-34 accelerated the Ca2+i decrease, as reflected by the half-time of decay (Fig. 8C). We predicted that blocking KCa3.1 would inhibit the SOCE, thereby reducing the ability of the Ca2+ stores to refill. To test this mechanism, in the second part of the experiment, the cells were washed for 10 min to allow store refilling, and then the sarcoplasmic/endoplasmic reticulum Ca2+-ATPase pump inhibitor, thapsigargin, was applied to release Ca2+ from the stores, and to evoke a second period of SOCE. Control astrocytes exhibited a robust and prolonged rise in Ca2+i (Fig. 8A,D), but, as predicted, TRAM-34-treated cells did not respond (Fig. 8B,D). ATP released from damaged cells can activate multiple purinergic receptors; therefore, our results show that blocking KCa3.1 in astrocytes reduces SOCE and refilling of Ca2+ stores after a stimulus that is relevant to spinal cord injury.
Figure 8.
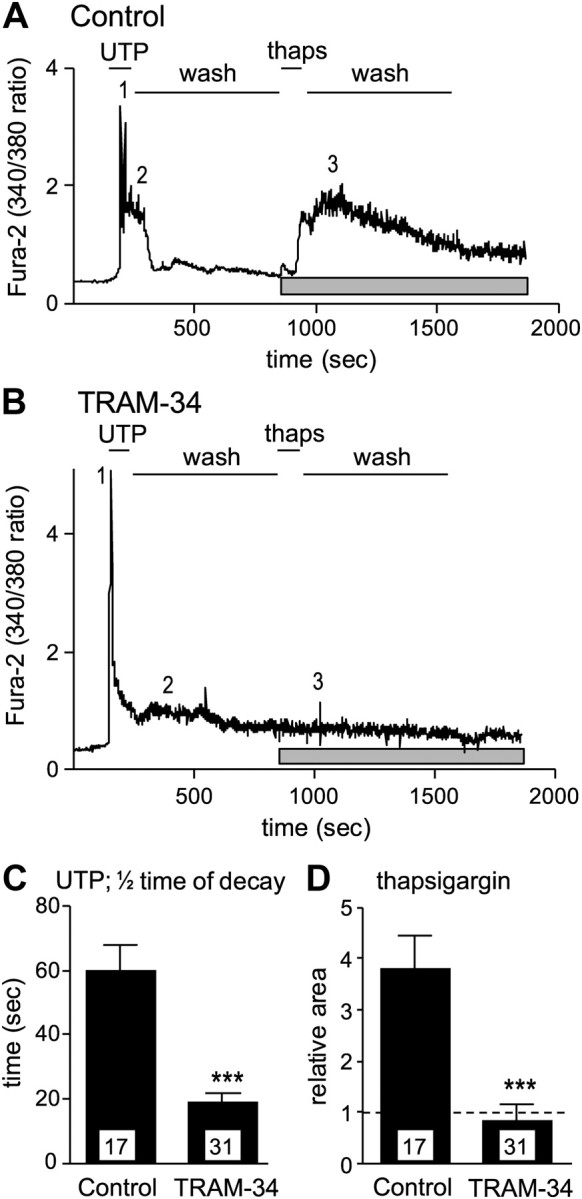
TRAM-34 inhibits Ca2+ signaling in cultured astrocytes. A, B, Ca2+ responses to stimulation with 300 μm UTP, a metabotropic purinergic receptor agonist, followed by the Ca2+ pump inhibitor thapsigargin (thaps). Representative astrocytes in normal bath solution (control; A) or in the continued presence of 1 μm TRAM-34 (B). In the control cell, note the biphasic response to UTP: 1 shows the peak, and 2 indicates the plateau phase; 3 indicates the peak response to thapsigargin; and the gray bar below each trace indicates the time over which the Ca2+ response was integrated to compare responses (in D). In the TRAM-34-treated cell (B), note the reduced plateau after UTP (2) and the lack of response to thapsigargin (3). C, The time taken for the Ca2+ signal to decrease to 50% of the peak value (mean ± SEM; n values indicated on each bar). Note the faster decay in the presence of TRAM-34 (***p < 0.001). D, The Ca2+ response to thapsigargin was integrated over the period indicated by the gray bars in A and B. Note the lack of response to thapsigargin in the presence of TRAM-34 (***p < 0.001). All statistical differences were analyzed with Student's t test.
Our salient results are as follows: (1) KCa3.1 is expressed in vivo in some astrocytes, motor neurons, and some oligodendrocytes in the uninjured adult murine spinal cord; (2) KCa3.1 expression increased in the spinal cord after contusion injury, especially in reactive astrocytes; (3) KCNN4/KCa3.1 expression was also detected in activated macrophages isolated ex vivo from the injured spinal cord, and in vitro in BMDMs and microglia; (4) blocking KCa3.1 after SCI with TRAM-34 improved locomotor recovery and reduced secondary damage, as judged by increased tissue and axon sparing and neuron survival; (5) the improved outcomes of TRAM-34 treatment were accompanied by reduced expression of the proinflammatory mediators IL-1β, TNF-α, and iNOS in the injured spinal cord; and (6) blocking KCa3.1 with TRAM-34 reduced Ca2+ signaling in isolated astrocytes in vitro.
Discussion
KCa3.1 is mainly expressed in nonexcitable cells, including some peripheral immune cells (Köhler et al., 1996; Joiner et al., 1997; Xia et al., 1998; Fanger et al., 1999; Khanna et al., 1999), and its role in homeostatic functions and activation of T cells is especially well characterized (Khanna et al., 1999; Ghanshani et al., 2000; Chandy et al., 2004). Two properties of KCa3.1 are especially important for nonexcitable cells: it is exquisitely sensitive to Ca2+ and, unlike many K+ channels, it's opening does not require depolarization. Among cells of monocytic lineage, KCa3.1 has been detected in cultured rat microglia (Khanna et al., 2001; Kaushal et al., 2007) and in murine macrophages in atherosclerotic lesions (Toyama et al., 2008). In cultured rat microglia, pharmacological blockade of KCa3.1 inhibits p38 MAPK activation, upregulation of iNOS, production of nitric oxide and superoxide, and the capacity of microglia to kill neurons through peroxynitrite formation and caspase 3 activation (Khanna et al., 2001; Kaushal et al., 2007).
The few published studies using TRAM-34 in vivo support the view that KCa3.1 is a promising therapeutic target for acute and chronic CNS disorders. In EAE, in which the spinal cord was examined after TRAM-34 treatment, expression of KCa3.1 was not assessed, and TRAM-34 was thought to act on T-cell and microglial responses (Reich et al., 2005). In the retina study, TRAM-34 rescued retinal ganglion cells from the retrograde effects of optic nerve transection, likely by affecting retinal microglial activation, but KCa3.1 expression was not assessed (Kaushal et al., 2007). The acute subdural hematoma study only examined KCNN4 mRNA changes at 1 and 7 d; cellular expression was not examined, and they assessed effects of blocking KCa3.1 on brain swelling and infarct volume but did not directly assess neuronal survival (Mauler et al., 2004). Here, we show that KCNN4/KCa3.1 is upregulated at the mRNA and protein levels within the first few days after SCI, and is maintained for up to 3 weeks. Unlike the other CNS studies, we found that after SCI, reactive astrocytes were the main cell type upregulating KCa3.1. Furthermore, treatment with TRAM-34 reduced tissue and axonal loss, and improved neuronal survival and locomotor recovery after SCI. This study is therefore the first to show the cell-type expression of KCa3.1 in vivo in the CNS. It is also the first to show in vivo changes in expression of KCa3.1 at the protein level, and the effects of blocking this ion channel on secondary damage and functional recovery after SCI. Further studies will be needed to expand on these findings, in particular to test alternative dosing (e.g., twice-daily treatments), and timing (i.e., assess various delay times before commencement of treatments) regimens to establish the therapeutic window.
When considering potential mechanisms by which TRAM-34 reduced inflammation and injury in the contused spinal cord, it is necessary to first consider the cellular expression of KCa3.1. We show that although KCa3.1 channels are expressed in some neurons and glia in the normal spinal cord, its expression is increased markedly in reactive astrocytes after spinal cord contusion injury. In nonexcitable cells, the main postulated mechanism of action of KCa3.1 channels is to maintain a negative membrane potential (and thus, driving force) to facilitate Ca2+ entry. Our results are entirely consistent with this hypothesis. Using isolated astrocytes, we showed that TRAM-34 inhibited Ca2+ entry after activation of metabotropic purinergic receptors, and prevented the refilling of Ca2+ stores. In regulating these processes, KCa3.1 channels are expected to be involved in multiple cell functions downstream of Ca2+ entry.
KCNN4/KCa3.1 was detected in activated microglia/macrophages isolated ex vivo from injured spinal cords but not in tissue sections at 7 and 14 d after SCI. We do not yet know the reasons for these differences in expression, but several possibilities for future analysis are as follows. (1) We might have missed earlier, transient, low-level or cell activation-state-dependent expression. (2) Microglia might express lower levels than macrophages. For instance, KCa3.1 was detected in tissue macrophages in atherosclerotic lesions, but not in circulating blood monocytes (Toyama et al., 2008). (3) In different cell types, the relative contribution of KCa3.1 channels likely differs and is not necessarily proportional to its expression level. Very few channels are needed to control the microglial membrane potential because the membrane resistance is extremely high (Newell and Schlichter, 2005) compared with astrocytes, neurons, and oligodendrocytes. Such low channel numbers in vivo might not be detectable by immunohistochemistry. (4) Specific roles of KCa3.1 channels likely differ in each cell type in which it is expressed. From in vitro studies, we identified several roles for KCa3.1 in microglial activation (Khanna et al., 2001; Kaushal et al., 2007), but its roles in astrocytes, neurons, and oligodendrocytes have not been addressed. Other SK channels (KCa2.2, KCa2.3) have been implicated in the slow afterhyperpolarization that regulates neuron firing frequency (Hammond et al., 2006; Faber, 2009).
There is very little information on the effects of TRAM-34 on expression of immune mediators in vivo. In one EAE study, chemokine/cytokine expression was assessed in the context of the autoimmune response in which these immune mediators play an important role (Reich et al., 2005). Interferon-γ, TNF-α, IL-1α and -1β, lymphotoxin-α and -β, and others were reduced and thought to be due to the effect of TRAM-34 on T cells (Reich et al., 2005). Changes in cytokines were not examined in the acute subdural hematoma study (Mauler et al., 2004) or the optic nerve study (Kaushal et al., 2007), two models in which an innate immune response occurs. In the latter study, iNOS expression was reduced by TRAM-34, but this work was done in vitro in LPS-stimulated microglia, and iNOS expression was not assessed in subdural hematoma (Mauler et al., 2004). We therefore show for the first time that that blocking KCa3.1 with TRAM-34 reduces in vivo levels of proinflammatory cytokines (TNF-α and IL-1β) and iNOS in spinal cord injury, in which secondary tissue damage is attributed to innate immunity. While we have not proven a link between these proinflammatory molecules and the TRAM-34-mediated reduction of neuron loss and functional impairment, the literature correlates these inflammatory molecules to outcome in spinal cord injury. TNF-α is involved in excitotoxicity and motoneuron cell death after spinal cord injury (Lee et al., 2000; Ferguson et al., 2008; Genovese et al., 2008). IL-1β contributes to glutamate-mediated damage following SCI, and blocking IL-1β improves locomotor recovery (Liu et al., 2008). Finally, reducing iNOS expression has been associated with neuroprotection and better functional outcomes after SCI (Isaksson et al., 2005; López-Vales et al., 2006). iNOS is not expressed in the healthy adult CNS, but after spinal cord injury, proinflammatory cytokines can induce it in microglia and astrocytes (Murphy, 2000; Xu et al., 2001; Rathore et al., 2008), and its expression is seen as late as 14 d after injury. The subsequent production of NO and NO-derived reactive nitrogen species (e.g., peroxynitrite) can kill neurons by inhibiting mitochondrial cytochrome oxidase (Brown and Cooper, 1994; Bal-Price and Brown, 2001), and by causing lipid peroxidation and DNA oxidative damage (Bolaños et al., 1995; Scott et al., 1999; Liu et al., 2000; Szabó, 2003). Therefore, there is evidence that reducing these immune modulators (TNF-α, IL-1β, iNOS), as we observed with TRAM-34 treatment after SCI, can contribute to neuroprotection and functional recovery.
There is considerable potential for cellular cross talk downstream of cytokine production. The close proximity of astrocytes and microglia means that inhibiting KCa3.1 on one cell type could alter the molecules they secrete and affect production of proinflammatory mediators (IL-1β, TNF-α, iNOS) on adjacent cells. In principle, by regulating the membrane potential, KCa3.1 channels can affect Ca2+ entry and influence many downstream signaling pathways. Ca2+ increases contribute to astrocyte–astrocyte and astrocyte–neuron communication by evoking release of transmitters such as glutamate and triggering receptor-mediated currents (Nedergaard et al., 2003; Halassa et al., 2007; Shigetomi et al., 2008), and to cytokine secretion from astrocytes (El-Hage et al., 2005; Paco et al., 2009). It is tempting to speculate that TRAM-34 might reduce TNF-α and IL-1β expression by reducing Ca2+ in astrocytes, microglia, and macrophages, thereby decreasing iNOS expression. However, it should be noted that the cellular and molecular changes after spinal cord contusion injury are very complex, involve many cell types, and change with time after injury.
The data presented in this study show that inhibition of KCa3.1 reduces proinflammatory mediators, reduces secondary tissue damage, and improves locomotor recovery after spinal cord contusion injury. The neuroprotective effects are modest, as is the case for many other targets reported in the SCI literature. This points to a more widely acknowledged view that multiple molecular targets or pathways will have to be targeted to achieve more substantial neuroprotection and functional improvements after SCI. For this to occur, we will need to identify potential targets and pathways that can contribute to secondary damage. The improvement in secondary damage and locomotor recovery reported here supports the hypothesis that KCa3.1 could be a potential therapeutic target in SCI. Future work will be necessary to elucidate the specific roles of KCa3.1 channels in the different CNS cells in which they are expressed.
Footnotes
This work was supported by a grant from the Canadian Institute of Health Research (CIHR) to S.D. D.B. was funded by a Post-Doctoral Fellowship from the Multiple Sclerosis Society of Canada; N.G. was funded by a Studentship from the CIHR; S.L. was funded by a CIHR Post-Doctoral Fellowship; A.R. and K.R. were funded by Studentships from the CIHR Neuroinflammation Training Program. We thank Catherine Vincent for the images in Figure 4G, and Margaret Attiwell for help with the illustrations.
References
- Bal-Price A, Brown GC. Inflammatory neurodegeneration mediated by nitric oxide from activated glia-inhibiting neuronal respiration, causing glutamate release and excitotoxicity. J Neurosci. 2001;21:6480–6491. doi: 10.1523/JNEUROSCI.21-17-06480.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao F, Chen Y, Dekaban GA, Weaver LC. Early anti-inflammatory treatment reduces lipid peroxidation and protein nitration after spinal cord injury in rats. J Neurochem. 2004;88:1335–1344. doi: 10.1046/j.1471-4159.2003.02240.x. [DOI] [PubMed] [Google Scholar]
- Bao F, Bailey CS, Gurr KR, Bailey SI, Rosas-Arellano MP, Dekaban GA, Weaver LC. Increased oxidative activity in human blood neutrophils and monocytes after spinal cord injury. Exp Neurol. 2009;215:308–316. doi: 10.1016/j.expneurol.2008.10.022. [DOI] [PubMed] [Google Scholar]
- Bartholdi D, Schwab ME. Expression of pro-inflammatory cytokine and chemokine mRNA upon experimental spinal cord injury in mouse: an in situ hybridization study. Eur J Neurosci. 1997;9:1422–1438. doi: 10.1111/j.1460-9568.1997.tb01497.x. [DOI] [PubMed] [Google Scholar]
- Basso DM, Fisher LC, Anderson AJ, Jakeman LB, McTigue DM, Popovich PG. Basso Mouse Scale for locomotion detects differences in recovery after spinal cord injury in five common mouse strains. J Neurotrauma. 2006;23:635–659. doi: 10.1089/neu.2006.23.635. [DOI] [PubMed] [Google Scholar]
- Bolaños JP, Heales SJ, Land JM, Clark JB. Effect of peroxynitrite on the mitochondrial respiratory chain: differential susceptibility of neurones and astrocytes in primary culture. J Neurochem. 1995;64:1965–1972. doi: 10.1046/j.1471-4159.1995.64051965.x. [DOI] [PubMed] [Google Scholar]
- Brown GC, Cooper CE. Nanomolar concentrations of nitric oxide reversibly inhibit synaptosomal respiration by competing with oxygen at cytochrome oxidase. FEBS Lett. 1994;356:295–298. doi: 10.1016/0014-5793(94)01290-3. [DOI] [PubMed] [Google Scholar]
- Cahalan MD, Wulff H, Chandy KG. Molecular properties and physiological roles of ion channels in the immune system. J Clin Immunol. 2001;21:235–252. doi: 10.1023/a:1010958907271. [DOI] [PubMed] [Google Scholar]
- Chandy KG, Wulff H, Beeton C, Pennington M, Gutman GA, Cahalan MD. K+ channels as targets for specific immunomodulation. Trends Pharmacol Sci. 2004;25:280–289. doi: 10.1016/j.tips.2004.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donnelly DJ, Popovich PG. Inflammation and its role in neuroprotection, axonal regeneration and functional recovery after spinal cord injury. Exp Neurol. 2008;209:378–388. doi: 10.1016/j.expneurol.2007.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Hage N, Gurwell JA, Singh IN, Knapp PE, Nath A, Hauser KF. Synergistic increases in intracellular Ca2+, and the release of MCP-1, RANTES, and IL-6 by astrocytes treated with opiates and HIV-1 Tat. Glia. 2005;50:91–106. doi: 10.1002/glia.20148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faber ES. Functions and modulation of neuronal SK channels. Cell Biochem Biophys. 2009;55:127–139. doi: 10.1007/s12013-009-9062-7. [DOI] [PubMed] [Google Scholar]
- Fam SR, Gallagher CJ, Kalia LV, Salter MW. Differential frequency dependence of P2Y1- and P2Y2-mediated Ca2+ signaling in astrocytes. J Neurosci. 2003;23:4437–4444. doi: 10.1523/JNEUROSCI.23-11-04437.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fanger CM, Ghanshani S, Logsdon NJ, Rauer H, Kalman K, Zhou J, Beckingham K, Chandy KG, Cahalan MD, Aiyar J. Calmodulin mediates calcium-dependent activation of the intermediate conductance KCa channel, IKCa1. J Biol Chem. 1999;274:5746–5754. doi: 10.1074/jbc.274.9.5746. [DOI] [PubMed] [Google Scholar]
- Ferguson AR, Christensen RN, Gensel JC, Miller BA, Sun F, Beattie EC, Bresnahan JC, Beattie MS. Cell death after spinal cord injury is exacerbated by rapid TNF α-induced trafficking of GluR2-lacking AMPARs to the plasma membrane. J Neurosci. 2008;28:11391–11400. doi: 10.1523/JNEUROSCI.3708-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu X, Zhu ZH, Wang YQ, Wu GC. Regulation of proinflammatory cytokines gene expression by nociceptin/orphanin FQ in the spinal cord and the cultured astrocytes. Neuroscience. 2007;144:275–285. doi: 10.1016/j.neuroscience.2006.09.016. [DOI] [PubMed] [Google Scholar]
- Gardos G. The function of calcium in the potassium permeability of human erythrocytes. Biochim Biophys Acta. 1958;30:653–654. doi: 10.1016/0006-3002(58)90124-0. [DOI] [PubMed] [Google Scholar]
- Genovese T, Mazzon E, Crisafulli C, Di Paola R, Muià C, Esposito E, Bramanti P, Cuzzocrea S. TNF-alpha blockage in a mouse model of SCI: evidence for improved outcome. Shock. 2008;29:32–41. doi: 10.1097/shk.0b013e318059053a. [DOI] [PubMed] [Google Scholar]
- Ghanshani S, Wulff H, Miller MJ, Rohm H, Neben A, Gutman GA, Cahalan MD, Chandy KG. Up-regulation of the IKCa1 potassium channel during T-cell activation. Molecular mechanism and functional consequences. J Biol Chem. 2000;275:37137–37149. doi: 10.1074/jbc.M003941200. [DOI] [PubMed] [Google Scholar]
- Ghasemlou N, Kerr BJ, David S. Tissue displacement and impact force are important contributors to outcome after spinal cord contusion injury. Exp Neurol. 2005;196:9–17. doi: 10.1016/j.expneurol.2005.05.017. [DOI] [PubMed] [Google Scholar]
- Halassa MM, Fellin T, Haydon PG. The tripartite synapse: roles for gliotransmission in health and disease. Trends Mol Med. 2007;13:54–63. doi: 10.1016/j.molmed.2006.12.005. [DOI] [PubMed] [Google Scholar]
- Hammond RS, Bond CT, Strassmaier T, Ngo-Anh TJ, Adelman JP, Maylie J, Stackman RW. Small-conductance Ca2+-activated K+ channel type 2 (SK2) modulates hippocampal learning, memory, and synaptic plasticity. J Neurosci. 2006;26:1844–1853. doi: 10.1523/JNEUROSCI.4106-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isaksson J, Farooque M, Olsson Y. Improved functional outcome after spinal cord injury in iNOS-deficient mice. Spinal Cord. 2005;43:167–170. doi: 10.1038/sj.sc.3101672. [DOI] [PubMed] [Google Scholar]
- Jiménez AI, Castro E, Communi D, Boeynaems JM, Delicado EG, Miras-Portugal MT. Coexpression of several types of metabotropic nucleotide receptors in single cerebellar astrocytes. J Neurochem. 2000;75:2071–2079. doi: 10.1046/j.1471-4159.2000.0752071.x. [DOI] [PubMed] [Google Scholar]
- Joiner WJ, Wang LY, Tang MD, Kaczmarek LK. hSK4, a member of a novel subfamily of calcium-activated potassium channels. Proc Natl Acad Sci U S A. 1997;94:11013–11018. doi: 10.1073/pnas.94.20.11013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaushal V, Koeberle PD, Wang Y, Schlichter LC. The Ca2+-activated K+ channel KCNN4/KCa3.1 contributes to microglia activation and nitric oxide-dependent neurodegeneration. J Neurosci. 2007;27:234–244. doi: 10.1523/JNEUROSCI.3593-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khanna R, Chang MC, Joiner WJ, Kaczmarek LK, Schlichter LC. hSK4/hIK1, a calmodulin-binding KCa channel in human T lymphocytes. Roles in proliferation and volume regulation. J Biol Chem. 1999;274:14838–14849. doi: 10.1074/jbc.274.21.14838. [DOI] [PubMed] [Google Scholar]
- Khanna R, Roy L, Zhu X, Schlichter LC. K+ channels and the microglial respiratory burst. Am J Physiol Cell Physiol. 2001;280:C796–C806. doi: 10.1152/ajpcell.2001.280.4.C796. [DOI] [PubMed] [Google Scholar]
- Klusman I, Schwab ME. Effects of pro-inflammatory cytokines in experimental spinal cord injury. Brain Res. 1997;762:173–184. doi: 10.1016/s0006-8993(97)00381-8. [DOI] [PubMed] [Google Scholar]
- Köhler M, Hirschberg B, Bond CT, Kinzie JM, Marrion NV, Maylie J, Adelman JP. Small-conductance, calcium-activated potassium channels from mammalian brain. Science. 1996;273:1709–1714. doi: 10.1126/science.273.5282.1709. [DOI] [PubMed] [Google Scholar]
- Köhler R, Wulff H, Eichler I, Kneifel M, Neumann D, Knorr A, Grgic I, Kämpfe D, Si H, Wibawa J, Real R, Borner K, Brakemeier S, Orzechowski HD, Reusch HP, Paul M, Chandy KG, Hoyer J. Blockade of the intermediate-conductance calcium-activated potassium channel as a new therapeutic strategy for restenosis. Circulation. 2003;108:1119–1125. doi: 10.1161/01.CIR.0000086464.04719.DD. [DOI] [PubMed] [Google Scholar]
- Kwon BK, Tetzlaff W, Grauer JN, Beiner J, Vaccaro AR. Pathophysiology and pharmacologic treatment of acute spinal cord injury. Spine J. 2004;4:451–464. doi: 10.1016/j.spinee.2003.07.007. [DOI] [PubMed] [Google Scholar]
- Lee YB, Yune TY, Baik SY, Shin YH, Du S, Rhim H, Lee EB, Kim YC, Shin ML, Markelonis GJ, Oh TH. Role of tumor necrosis factor-alpha in neuronal and glial apoptosis after spinal cord injury. Exp Neurol. 2000;166:190–195. doi: 10.1006/exnr.2000.7494. [DOI] [PubMed] [Google Scholar]
- Liu D, Ling X, Wen J, Liu J. The role of reactive nitrogen species in secondary spinal cord injury: formation of nitric oxide, peroxynitrite, and nitrated protein. J Neurochem. 2000;75:2144–2154. doi: 10.1046/j.1471-4159.2000.0752144.x. [DOI] [PubMed] [Google Scholar]
- Liu D, Bao F, Prough DS, Dewitt DS. Peroxynitrite generated at the level produced by spinal cord injury induces peroxidation of membrane phospholipids in normal rat cord: reduction by a metalloporphyrin. J Neurotrauma. 2005;22:1123–1133. doi: 10.1089/neu.2005.22.1123. [DOI] [PubMed] [Google Scholar]
- Liu S, Xu GY, Johnson KM, Echetebu C, Ye ZS, Hulsebosch CE, McAdoo DJ. Regulation of interleukin-1beta by the interleukin-1 receptor antagonist in the glutamate-injured spinal cord: endogenous neuroprotection. Brain Res. 2008;1231:63–74. doi: 10.1016/j.brainres.2008.07.035. [DOI] [PubMed] [Google Scholar]
- Longbrake EE, Lai W, Ankeny DP, Popovich PG. Characterization and modeling of monocyte-derived macrophages after spinal cord injury. J Neurochem. 2007;102:1083–1094. doi: 10.1111/j.1471-4159.2007.04617.x. [DOI] [PubMed] [Google Scholar]
- López-Vales R, García-Alías G, Forés J, Udina E, Gold BG, Navarro X, Verdú E. FK 506 reduces tissue damage and prevents functional deficit after spinal cord injury in the rat. J Neurosci Res. 2005;81:827–836. doi: 10.1002/jnr.20605. [DOI] [PubMed] [Google Scholar]
- López-Vales R, Garcia-Alias G, Guzman-Lenis MS, Fores J, Casas C, Navarro X, Verdu E. Effects of COX-2 and iNOS inhibitors alone or in combination with olfactory ensheathing cell grafts after spinal cord injury. Spine (Phila Pa 1976) 2006;31:1100–1106. doi: 10.1097/01.brs.0000216448.61860.1d. [DOI] [PubMed] [Google Scholar]
- Mauler F, Hinz V, Horváth E, Schuhmacher J, Hofmann HA, Wirtz S, Hahn MG, Urbahns K. Selective intermediate-/small-conductance calcium-activated potassium channel (KCNN4) blockers are potent and effective therapeutics in experimental brain oedema and traumatic brain injury caused by acute subdural haematoma. Eur J Neurosci. 2004;20:1761–1768. doi: 10.1111/j.1460-9568.2004.03615.x. [DOI] [PubMed] [Google Scholar]
- Murphy S. Production of nitric oxide by glial cells: regulation and potential roles in the CNS. Glia. 2000;29:1–13. doi: 10.1002/(sici)1098-1136(20000101)29:1<1::aid-glia1>3.0.co;2-n. [DOI] [PubMed] [Google Scholar]
- Nedergaard M, Ransom B, Goldman SA. New roles for astrocytes: redefining the functional architecture of the brain. Trends Neurosci. 2003;26:523–530. doi: 10.1016/j.tins.2003.08.008. [DOI] [PubMed] [Google Scholar]
- Newell EW, Schlichter LC. Integration of K+ and Cl- currents regulate steady-state and dynamic membrane potentials in cultured rat microglia. J Physiol. 2005;567:869–890. doi: 10.1113/jphysiol.2005.092056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohana L, Newell EW, Stanley EF, Schlichter LC. The Ca2+ release-activated Ca2+ current (I(CRAC)) mediates store-operated Ca2+ entry in rat microglia. Channels (Austin) 2009;3:129–139. doi: 10.4161/chan.3.2.8609. [DOI] [PubMed] [Google Scholar]
- Paco S, Margelí MA, Olkkonen VM, Imai A, Blasi J, Fischer-Colbrie R, Aguado F. Regulation of exocytotic protein expression and Ca2+-dependent peptide secretion in astrocytes. J Neurochem. 2009;110:143–156. doi: 10.1111/j.1471-4159.2009.06116.x. [DOI] [PubMed] [Google Scholar]
- Peuchen S, Clark JB, Duchen MR. Mechanisms of intracellular calcium regulation in adult astrocytes. Neuroscience. 1996;71:871–883. doi: 10.1016/0306-4522(95)00515-3. [DOI] [PubMed] [Google Scholar]
- Pineau I, Lacroix S. Proinflammatory cytokine synthesis in the injured mouse spinal cord: multiphasic expression pattern and identification of the cell types involved. J Comp Neurol. 2007;500:267–285. doi: 10.1002/cne.21149. [DOI] [PubMed] [Google Scholar]
- Popovich PG, Longbrake EE. Can the immune system be harnessed to repair the CNS? Nat Rev Neurosci. 2008;9:481–493. doi: 10.1038/nrn2398. [DOI] [PubMed] [Google Scholar]
- Rathore KI, Kerr BJ, Redensek A, López-Vales R, Jeong SY, Ponka P, David S. Ceruloplasmin protects injured spinal cord from iron-mediated oxidative damage. J Neurosci. 2008;28:12736–12747. doi: 10.1523/JNEUROSCI.3649-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reich EP, Cui L, Yang L, Pugliese-Sivo C, Golovko A, Petro M, Vassileva G, Chu I, Nomeir AA, Zhang LK, Liang X, Kozlowski JA, Narula SK, Zavodny PJ, Chou CC. Blocking ion channel KCNN4 alleviates the symptoms of experimental autoimmune encephalomyelitis in mice. Eur J Immunol. 2005;35:1027–1036. doi: 10.1002/eji.200425954. [DOI] [PubMed] [Google Scholar]
- Satake K, Matsuyama Y, Kamiya M, Kawakami H, Iwata H, Adachi K, Kiuchi K. Nitric oxide via macrophage iNOS induces apoptosis following traumatic spinal cord injury. Brain Res Mol Brain Res. 2000;85:114–122. doi: 10.1016/s0169-328x(00)00253-9. [DOI] [PubMed] [Google Scholar]
- Scott GS, Jakeman LB, Stokes BT, Szabó C. Peroxynitrite production and activation of poly (adenosine diphosphate-ribose) synthetase in spinal cord injury. Ann Neurol. 1999;45:120–124. [PubMed] [Google Scholar]
- Shigetomi E, Bowser DN, Sofroniew MV, Khakh BS. Two forms of astrocyte calcium excitability have distinct effects on NMDA receptor-mediated slow inward currents in pyramidal neurons. J Neurosci. 2008;28:6659–6663. doi: 10.1523/JNEUROSCI.1717-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szabó C. Multiple pathways of peroxynitrite cytotoxicity. Toxicol Lett. 2003;140–141:105–112. doi: 10.1016/s0378-4274(02)00507-6. [DOI] [PubMed] [Google Scholar]
- Toyama K, Wulff H, Chandy KG, Azam P, Raman G, Saito T, Fujiwara Y, Mattson DL, Das S, Melvin JE, Pratt PF, Hatoum OA, Gutterman DD, Harder DR, Miura H. The intermediate-conductance calcium-activated potassium channel KCa3.1 contributes to atherogenesis in mice and humans. J Clin Invest. 2008;118:3025–3037. doi: 10.1172/JCI30836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, Holstein JD, Upadhyay G, Lin DT, Conway S, Muller E, Lechleiter JD. Purinergic receptor-stimulated IP3-mediated Ca2+ release enhances neuroprotection by increasing astrocyte mitochondrial metabolism during aging. J Neurosci. 2007;27:6510–6520. doi: 10.1523/JNEUROSCI.1256-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wulff H, Miller MJ, Hansel W, Grissmer S, Cahalan MD, Chandy KG. Design of a potent and selective inhibitor of the intermediate-conductance Ca2+-activated K+ channel, IKCa1: a potential immunosuppressant. Proc Natl Acad Sci U S A. 2000;97:8151–8156. doi: 10.1073/pnas.97.14.8151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia XM, Fakler B, Rivard A, Wayman G, Johnson-Pais T, Keen JE, Ishii T, Hirschberg B, Bond CT, Lutsenko S, Maylie J, Adelman JP. Mechanism of calcium gating in small-conductance calcium-activated potassium channels. Nature. 1998;395:503–507. doi: 10.1038/26758. [DOI] [PubMed] [Google Scholar]
- Xiong Y, Hall ED. Pharmacological evidence for a role of peroxynitrite in the pathophysiology of spinal cord injury. Exp Neurol. 2009;216:105–114. doi: 10.1016/j.expneurol.2008.11.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong Y, Rabchevsky AG, Hall ED. Role of peroxynitrite in secondary oxidative damage after spinal cord injury. J Neurochem. 2007;100:639–649. doi: 10.1111/j.1471-4159.2006.04312.x. [DOI] [PubMed] [Google Scholar]
- Xu J, Kim GM, Chen S, Yan P, Ahmed SH, Ku G, Beckman JS, Xu XM, Hsu CY. iNOS and nitrotyrosine expression after spinal cord injury. J Neurotrauma. 2001;18:523–532. doi: 10.1089/089771501300227323. [DOI] [PubMed] [Google Scholar]
- Xu W, Chi L, Xu R, Ke Y, Luo C, Cai J, Qiu M, Gozal D, Liu R. Increased production of reactive oxygen species contributes to motor neuron death in a compression mouse model of spinal cord injury. Spinal Cord. 2005;43:204–213. doi: 10.1038/sj.sc.3101674. [DOI] [PubMed] [Google Scholar]
- Xu Z, Wang BR, Wang X, Kuang F, Duan XL, Jiao XY, Ju G. ERK1/2 and p38 mitogen-activated protein kinase mediate iNOS-induced spinal neuron degeneration after acute traumatic spinal cord injury. Life Sci. 2006;79:1895–1905. doi: 10.1016/j.lfs.2006.06.023. [DOI] [PubMed] [Google Scholar]
- Yang JY, Kim HS, Lee JK. Changes in nitric oxide synthase expression in young and adult rats after spinal cord injury. Spinal Cord. 2007;45:731–738. doi: 10.1038/sj.sc.3102036. [DOI] [PubMed] [Google Scholar]
- Yang L, Blumbergs PC, Jones NR, Manavis J, Sarvestani GT, Ghabriel MN. Early expression and cellular localization of proinflammatory cytokines interleukin-1beta, interleukin-6, and tumor necrosis factor-alpha in human traumatic spinal cord injury. Spine. 2004;29:966–971. doi: 10.1097/00007632-200405010-00004. [DOI] [PubMed] [Google Scholar]