

Abstract
The chemokine CX3CL1 and its receptor CX3CR1 are constitutively expressed in the nervous system. In this study, we used in vivo murine models of permanent middle cerebral artery occlusion (pMCAO) to investigate the protective potential of CX3CL1. We report that exogenous CX3CL1 reduced ischemia-induced cerebral infarct size, neurological deficits, and caspase-3 activation. CX3CL1-induced neuroprotective effects were long lasting, being observed up to 50 d after pMCAO in rats. The neuroprotective action of CX3CL1 in different models of brain injuries is mediated by its inhibitory activity on microglia and, in vitro, requires the activation of adenosine receptor 1 (A1R). We show that, in the presence of the A1R antagonist 1,3-dipropyl-8-cyclopentylxanthine and in A1R−/− mice, the neuroprotective effect of CX3CL1 on pMCAO was abolished, indicating the critical importance of the adenosine system in CX3CL1 protection also in vivo. In apparent contrast with the above reported data but in agreement with previous findings, cx3cl1−/− and cx3cr1GFP/GFP mice, respectively, deficient in CX3CL1 or CX3CR1, had less severe brain injury on pMCAO, and the administration of exogenous CX3CL1 increased brain damage in cx3cl1−/− ischemic mice. We also report that CX3CL1 induced a different phagocytic activity in wild type and cx3cl1−/− microglia in vitro during cotreatment with the medium conditioned by neurons damaged by oxygen-glucose deprivation. Together, these data suggest that acute administration of CX3CL1 reduces ischemic damage via an adenosine-dependent mechanism and that the absence of constitutive CX3CL1–CX3CR1 signaling changes the outcome of microglia-mediated effects during CX3CL1 administration to ischemic brain.
Introduction
After transient brain ischemia and during excitotoxic insult, neurons release soluble fractalkine/CX3CL1 (Chapman et al., 2000; Tarozzo et al., 2002; Zhu et al., 2009), a chemokine whose specific receptor CX3CR1 is expressed by microglia (Harrison et al., 1998). CX3CL1 reduces microglia toxicity and, consequently, neuronal damage both in vitro and in vivo models of neuropathologies and brain inflammation (Zujovic et al., 2000; Mizuno et al., 2003; Cardona et al., 2006; Bhaskar et al., 2010; Noda et al., 2011; Pabon et al., 2011). However, in murine models of transient cerebral ischemia and Alzheimer's disease (AD), CX3CL1 is reported to play opposite roles. In transient focal cerebral ischemia, genetic deletion of cx3cl1 (cx3cl1−/− mice) reduced post-ischemic brain injury (Soriano et al., 2002) and cx3cr1 deficiency (cx3cr1GFP/GFP mice, in which the cx3cr1 gene was replaced by GFP) significantly reduced ischemic damage, IL-1β, and TNF-α release (Dénes et al., 2008). In different genetic models of AD, cx3cr1 deficiency is associated with reduced β-amyloid deposition and neuron loss (Fuhrmann et al., 2010; Lee et al., 2010) but also with the enhanced microtubule-associated protein tau phosphorylation (Bhaskar et al., 2010), suggesting opposite effects at different phases of the disease. In both cases, the contribution of CX3CL1 to disease progression was only evaluated on transgenic mice. In vitro, CX3CL1 reduces neuronal death induced by glutamate (Limatola et al., 2005) and exerts its neurotrophic effect through microglia-derived protective factors, including adenosine, with the activation of adenosine A1 receptors (A1Rs) (Lauro et al., 2008, 2010).
Adenosine is an endogenous metabolite with several different roles in the nervous system, from neurotransmission to neuroprotection (Fredholm, 2007). Extracellular levels of adenosine rise in response to brain injury induced by hypoxia, stroke, trauma, and epileptic seizures, likely to counteract tissue damage, primarily through the involvement of A1R, A2AR, and A3R (Cunha, 2005).
In the present study, we investigated the neuroprotective role of CX3CL1 in permanent cerebral ischemia, focusing on brain infarct size, neurological deficits, and biochemical markers of cell death and analyzing microglia phagocytic activity during exposure to the medium conditioned by ischemic neurons in wild type (wt) and cx3cl1−/− mice. We confirmed that both cx3cr1GFP/GFP and cx3cl1−/− mice had less severe ischemic lesions also in the model of permanent ischemia and demonstrated that administration of exogenous CX3CL1 is neuroprotective in in vivo models of permanent ischemia in wt mice and rats with mechanisms similar to those described in vitro, requiring the activation of A1R. Furthermore, we report that CX3CL1 differently modulated in vitro the phagocytic activity and TNF-α release in wt and cx3cl1−/− microglia exposed to a conditioned medium (CM) of oxygen glucose-deprived (OGD) neuronal cultures. These data suggest that, in the ischemic microenvironment of the brain, microglia response is greatly influenced by CX3CR1/CX3CL1 signaling. This is the first demonstration of a neuroprotective activity of exogenous CX3CL1 in in vivo ischemia.
Materials and Methods
Materials
Recombinant rat CX3CL1 (chemokine domain) was from Merck, human and mouse CX3CL1 (chemokine domain) from PeproTech and R & D Systems, 1,3-dipropyl-8-cyclopentylxanthine (DPCPX) from Tocris Bioscience, BCA protein assay kit from Pierce, chloral hydrate from Carlo Erba, primary antibodies from Cell Signaling Technology, secondary antibodies from Dako, nitrocellulose paper and ECL from GE Healthcare, red fluorescent FluoSpheres (1 μm diameter) from Invitrogen, ELISA kit for TNF-α and IL-1β from R & D Systems, and all other reagents from Sigma-Aldrich.
Animals
Male Crl:CD (SD)BR rats (Charles River), A1R−/−, C57BL/6J (wt; Charles River), cx3cl1−/− (Cook et al., 2001; kindly provided by Dr. Richard M. Ransohoff, Cleveland, OH), and cx3cr1GFP/GFP knock-in (in which the cx3cr1 gene was replaced by GFP; Jung et al., 2000; The Jackson Laboratory) male mice were used. Procedures involving animals and their care were in accordance with institutional guidelines that are in compliance with national (decreto legislativo n.116, Gazzetta Ufficiale suppl. 40; February 18, 1992) and international (EEC Council Directive 86/609, OJ L 358,1; December 12, 1987; National Institutes of Health Guide for the Care and Use of Laboratory Animals, National Research Council, 1996) laws and policies.
Permanent middle cerebral artery occlusion
Rats (250–280 g, 7–8 weeks) were anesthetized with chloral hydrate (400 mg/kg, i.p.), and surgery was done as described previously (Villa et al., 2007). Briefly, the common carotid arteries (CCAs) were visualized, and the right one was occluded. A hole adjacent and rostral to the right orbit allowed visualization of the middle cerebral artery (MCA), which was cauterized distal to the rhinal artery. To produce a penumbra around this fixed MCA lesion, the contralateral CCA was occluded for 1 h using traction with fine forceps. Sham-operated rats were operated the same way, but the MCA and CCAs were not occluded. Mice (25–28 g, 11–12 weeks) were anesthetized with intraperitoneal Equitensine at 3.5 ml/kg (39 mm pentobarbital, 256 mm chloral hydrate, 86 mm MgSO4, 10% ethanol v/v, and 39.6% propyleneglycol v/v). The right MCA was permanently occluded by electrocoagulation as described previously (Storini et al., 2006). Both rats and mice were maintained at 37°C during surgery and killed at different times after permanent middle cerebral artery occlusion (pMCAO).
Drugs and administration protocols
CX3CL1 was dissolved in saline and intracerebroventricularly injected 20 min before pMCAO at 200 pmol/2 μl in rats (rat CX3CL1) and 70 pmol/2 μl in mice (human or mouse CX3CL1), similar to what was used previously (Lyons et al., 2009). For dose–response experiments, rats received 20–200 pmol and mice 15–150 pmol of CX3CL1, in the same volume of saline. Anesthetized animals were immobilized on a stereotaxic apparatus (David Kopf Instruments) and injected in the right cerebral ventricle (−0.9 mm anteroposterior, 1.5 mm lateral, 4 mm deep for rats; 1 mm lateral and 3 mm deep for mice, according to the atlas of Paxinos and Watson (1998). A constant rate of infusion (0.2 μl/min) was maintained with a pump (KD Scientific). DPCPX (0.1 mg/kg; Phillis, 1995) and DMSO (vehicle, 0.5 ml/kg) were given intraperitoneally 30 min before CX3CL1 or saline in rats. Sham-operated animals received only vehicles.
Neurological deficits
Rats were evaluated for neurological deficits using the foot-fault, Bederson's, and De Ryck's tests at different time points after induction of ischemia, from 24 h to 28 d (Villa et al., 2007). The foot-fault test measures the animal's ability to integrate motor responses. The rats were placed on a grid of 0.3-cm-diameter metal rods 2 cm apart and were observed for 2 min. With each weight-bearing step, the paw can fall or slip between the wires, and this was recorded as a foot fault. The number of foot faults for the paws contralateral and ipsilateral to the infarction was recorded with the number of successful steps, and the foot-fault index was calculated as the percentage of contralateral limb foot faults per limb step minus the percentage of ipsilateral limb foot faults per limb step. In the postural reflex test of Bederson, rats were scored as follows: grade 5, normal; grade 4, moderate (forelimb flexion and no other abnormalities); grade 3, severe (reduced resistance to lateral push toward the paretic side, and forelimb flexion); grade 2, severe (same behavior as grade 3, with circling toward the paretic side when pulling the tail on the table); grade 1, severe (same behavior as grade 2, with spontaneous circling); grade 0, no activity. De Ryck's limb-placing test examines sensorimotor integration in limb placing responses to visual, vibrissae, tactile, and proprioceptive stimuli. For each test, limb placing scores were as follows: 0, no placing; 1, incomplete and/or delayed (>2 s); or 2, immediate and complete placing. Each test was repeated for each paw up to 10 times, and, for each body side, the maximum limb placing score was 16. All the experiments were run by a trained investigator blinded to the experimental conditions.
Brain ischemic volume
Rats.
To evaluate the extent of cell injury in the brain, rats were killed 24 h after ischemia. The brains were removed and transferred to cold saline, and 12 serial 1-mm-thick sections were cut through the entire brain. Six alternate sections were stained with a solution of 1% (w/v) triphenyltetrazolium chloride (TTC) in 154 mm NaCl for 30 min at 37°C, as described previously (Villa et al., 2007). Injury was quantified blindly using a computerized image analysis system (AIS software; Imaging Research). The remaining six sections, divided into the right ischemic and left non-ischemic hemisphere, were rapidly frozen on dry ice and stored at −80°C for Western blotting analysis. Alternatively, 50 d after brain ischemia, rats were evaluated for ischemic volume by structural magnetic resonance imaging (MRI) analysis (see below).
Mice.
Mice were deeply anesthetized with Equitensine and transcardially perfused with ice-cold PBS (20 ml), pH 7.4, and paraformaldehyde (PFA; 4%, 50 ml) in PBS. The brains were carefully removed from the skull and transferred in 4% PFA at 4°C overnight, then to 30% sucrose in PBS at 4°C overnight for cryoprotection, frozen in isopentane at −45°C for 3 min, and then stored at −80°C until use. Twenty micrometer coronal brain cryosections were cut serially at 320 μm intervals and stained with cresyl violet. Infarct volumes were calculated by integration of the infarct areas on each brain slice, as described previously (Storini et al., 2006).
MRI measurements
Rats were anesthetized with isoflurane (5% induction, 1% maintenance) in a 2:1 mixture of N2O/O2. Stereotaxic ear bars were used to minimize movements during the imaging procedure. Temperature was maintained at 37°C by a feedback-controlled, water-circulated heating cradle. Imaging was performed on a 7 T small-bore animal scanner (Bruker Biospec). Two actively coupled radio frequency coils were used: a volume coil of 7.2 cm diameter used as transmitter and an anatomically shaped receive-only surface coil array (2 × 2 array) as receiver. ParaVision 5.1 software interface was used for data acquisition. A RARE T2-weighted sequence allowed the acquisition of 42 coronal slices (700 μm thick), which covered the whole rat brain volume. The morphological images were obtained with an in-plane resolution of 117 × 150 μm (matrix, 256 × 200; FOV, 3 × 3 cm), repetition time (TR) of 5000 ms, effective echo time (TE) of 36 ms, and eight averages. Diffusion tensor imaging (DTI) data were acquired using a DTI–EPI sequence. The brain coronal slices (1 mm thickness) were acquired with a TR of 3000 ms and a TE of 35,632 ms. Four averages were used to increase the signal-to-noise ratio. For diffusion, encoding b factors of 0 (b0 image) and 800 s/mm2 were used, and diffusion-sensitizing gradients were applied along 19 isotropic directions of three-dimensional space, resulting in an acquisition time of 15 min for each rat. The in-plane image resolution was 214 × 214 μm (FOV of 3 × 3 cm with an acquisition matrix of 140 × 140). Volume measurements of structural MRI images were obtained manually using custom-made software. Regions of interest (ROIs) were manually chosen and drawn on the images for quantitative comparison of the brain atrophy values. We studied the whole brain, the cortex, the hippocampus, the caudate–putamen, and computed lesion volume. An operator, blind to the drug treatment, performed the evaluation. Data from all the animals were averaged ROI area × slice thickness and presented as mean ± SE. The DTI was calculated using a freely available software (MedINRIA), and fractional anisotropy (FA) maps were generated. The orientation information was visualized through directional encoded images (color coded maps), which were generated by color coding the primary eigenvector (e1) direction. The eigenvector corresponding to the largest eigenvalue is used to define the color (x, y, and z components are mapped to the red–green–blue channel). ROIs were manually chosen for quantitative comparison of the DTI values. For each animal, the ROIs were chosen on FA maps, and the anatomy was checked further on the set of images with no diffusion gradient (b = 0) and on the color-coded images. The use of different image contrasts provided by different DTI indices represented a reproducible strategy in conducting ROI analysis consistently for all animals. We also computed the apparent diffusion coefficient (ADC) maps: the maps visually demonstrate changes in the area and corresponding MRI parameter values of ischemia damaged tissue and the continuity of ipsilateral ventricle expansion. The structure of the corpus callosum was analyzed in two separate region, the genu of the corpus callosum and the external capsula. Data from all the animals were averaged/ROI and presented as mean ± SE. All the ROIs chosen were divided in two separate ROIs: one for the ipsilateral hemisphere and one for the contralateral hemisphere.
Western blot analysis
For protein extraction, the same half-slice for each brain was homogenized in RIPA buffer in a glass Potter apparatus. The homogenate was centrifuged at 15,000 × g for 15 min, the supernatant sonicated for 10 s, and centrifuged again at 15,000 × g for 10 min. The same amount of proteins (50 μg/sample) was loaded onto 7.5 or 12% SDS polyacrylamide gel and electrophoretically transferred to nitrocellulose paper at 4°C for 2 h. Blots were then incubated for 1 h with 5% nonfat dry milk or 5% BSA in Tris-buffered saline containing 0.2% Tween 20 to block nonspecific binding sites and then incubated overnight at 4°C with specific primary antibodies. After washing, membranes were incubated with HRP-conjugated secondary antibodies, and the immunoreactivity was detected by ECL. Densitometric analysis of immune reactive bands was done with Chemi-Doc XRS, using the Quantity One software (Bio-Rad).
Cortical neuronal cultures and OGD experiments
Cortical neuronal cultures were prepared from the brain of newborn wt mice. Cerebral cortices were chopped and digested in 20 U/ml papain for 40 min at 37°C. Cells (2.5 × 105 cells per well) were seeded on dishes coated with poly-l-lysine (100 μg/ml) in basal medium Eagle supplemented with 1 mm sodium pyruvate, 30 mm glucose, 0.1% Mito serum extender, 10% FBS, 100 U/ml penicillin, 0.1 mg/ml streptomycin, and 10 mm HEPES-NaOH, pH 7.4. After 4 h, the medium was changed with Neurobasal medium supplemented with 1 mm glutamine, 0.1% Mito serum extender, 2.5% B27, 100 U/ml penicillin, and 0.1 mg/ml streptomycin. After 2 d, AraC (5 μm) was added to avoid the growth of glial cells. The percentage of neuronal cells obtained with this method is between 80 and 90%, as determined with β-tubulin III staining. After 10–11 d, cells were incubated in glucose-free Locke's buffer (Lauro et al., 2008) for 2 h at 37°C in a sealed humidified chamber (MIC-101; Billups-Rotenberg), saturated with an atmosphere of 95% N2 and 5% CO2 (OGD) or in standard Locke's buffer and normoxia. Cells were then reincubated for 24 h in the original medium with normal O2 concentration. This CM was then used for microglia treatment (see below), and cortical neurons were analyzed for viability by cell count and MTT assay (Lauro et al., 2010) to verify neuronal damage, ranging from 40 to 60%.
Phagocytosis and ELISAs
Pure (99%) cortical microglia, obtained from wt or cx3cl−/− mice (Lauro et al., 2010), were seeded on poly-l-lysine-treated 10 mm glass coverslips (8 × 104 cells) or 12 mm wells (2.5 × 105 cells), respectively, for phagocytosis and ELISAs. After 24 h, medium was replaced with CM from normoxia or OGD-treated cortical cultures in the presence or absence of CX3CL1 (100 nm). Medium was then analyzed for cytokine release (ELISA), and the cells were lysed for protein quantification. Alternatively, after additional 24 h, medium was removed, 0.05% red fluorescent FluoSpheres were added for 1 h in serum-free medium (0.1% BSA), and nuclei were stained by Hoechst (1:1000, 30 min). Cells were washed three times with PBS to remove nonphagocyted spheres, and cells were fixed in 4% PFA for 15 min. Phagocytosis was quantified counting both the number of phagocyting cells (scoring as positive only cells with at least three FluoSpheres; Labuzek et al., 2010) and the number of red FluoSpheres per cell in at least 10 random fields for coverslip (594 nm, Axioscope 2; Carl Zeiss).
Statistical analysis
Data are shown as the means ± SE. Vehicle- and CX3CL1-treated animals were analyzed using Student's t test and the nonparametric Mann–Whitney test for infarct volume and neurological scores, respectively, and/or using two-way ANOVA, followed by the Bonferroni's post hoc test to evaluate the interaction of intraperitoneal and intracerebroventricular treatments, for long-term neurological deficits and for MRI structural analysis. All other datasets were analyzed for normality, followed by Student's t test and one-way ANOVA. The differences between groups of data were considered highly significant with ***p < 0.001 and **p < 0.01 and significant with *p < 0.05.
Results
pMCAO in wt, cx3cr1GFP/GFP, and cx3cl1−/− mice
To analyze possible differences in the role of CX3CL1/CX3CR1 in transient and permanent ischemia models, we performed pMCAO on wt, cx3cr1GFP/GFP, and cx3cl1−/− mice. In accordance with previous studies (Soriano et al., 2002; Dènes et al., 2008), the absence of CX3CL1 or CX3CR1 significantly reduced infarct size compared with wt mice [Fig. 1, *p < 0.05 and **p < 0.01 for control (C) groups of different genotypes vs C wt], reinforcing the hypothesis that the constitutive absence of CX3CL1/CX3CR1 signaling results in phenotypes protected against the deleterious effects of both transient and permanent ischemia. Because we have demonstrated previously that exogenous CX3CL1 is neuroprotective against excitotoxicity death models in vitro (Limatola et al., 2005), we now investigated the effects of exogenous CX3CL1 in in vivo ischemia. When CX3CL1 (70 pmol) was injected intracerebroventricularly in wt mice shortly before pMCAO, there was a significant decrease in ischemic volume (Fig. 1, ##p < 0.01, CX3CL1 vs C). A significant reduction of ischemic volume is observed already at 15 pmol, whereas higher doses (150 pmol) are less effective (data not shown). In preliminary experiments, we also tested the efficacy of mouse versus human CX3CL1 in neuroprotection, and, because the two gave comparable results (data not shown) in all subsequent experiments, we used the human chemokine. We verified the specificity of chemokine activity treating pMCAO cx3cr1GFP/GFP mice with CX3CL1. Data obtained demonstrated that CX3CL1 was ineffective in these mice, thus confirming its specific activity on CX3CR1 (Fig. 1). We then investigated the effect of exogenous CX3CL1 treatment in cx3cl1−/− mice. The results, reported in Figure 1 (#p < 0.05, CX3CL1 vs C) prove that this treatment exacerbates ischemic damage, thus demonstrating that exogenous CX3CL1 induces opposite effects in wt vs cx3cl1−/− mice.
Figure 1.

Permanent MCAO in cx3cr1GFP/GFP and cx3cl1−/− mice. Brain infarct volume is reduced in both cx3cr1GFP/GFP and cx3cl1−/− mice during pMCAO (C) compared with wt mice. The effect of CX3CL1 treatment on infarct brain volume in pMCAO wt, cx3cr1GFP/GFP, and cx3cl1−/− mice is reported. Data are mean ± SE; n = 5–18, indicated in brackets on top of each column. Student's t test: *p < 0.05 and **p < 0.01 versus C wt; #p < 0.05 and ##p < 0.01 versus C for corresponding genotype.
Effects of CX3CL1 on wt and cx3cl1−/− microglia exposed to the CM of OGD-damaged neurons
To analyze whether the different effects induced by CX3CL1 treatment in vivo could be explained with altered responses of microglia to ischemic microenvironment in wt vs cx3cl1−/− mice, primary cultured microglia were treated with the CM of normoxia or OGD cortical cultures, stimulated or not with CX3CL1, and analyzed for phagocytosis of microspheres in vitro. It is reported that CX3CL1 reduces the phagocytic activity of microglia (Lee et al., 2010). In our experimental conditions, we confirmed these data on wt microglia exposed to OGD-CM, in which CX3CL1 reduced the percentage of phagocyting cells and the number of phagocyted FluoSpheres per cell (Fig. 2a,b, n = 6). However, both these effects were lost in cx3cl1−/− microglia, in which CX3CL1 treatment did not significantly change the phagocytic activity when considering either the percentage of phagocyting cells or the number of phagocyted FluoSpheres per cell (Fig. 2a,b, n = 7).
Figure 2.

Phagocytic activity and TNF-α release of wt and cx3cl1−/− microglia. Phagocytic activity of wt and cx3cl1−/− microglia exposed to OGD-CM. a, Results expressed as the percentage of phagocyting cells. b, Results expressed as the number of phagocyted microspheres per cell as detailed in Materials and Methods. Data are the mean ± SE, n = 6–7 (total number of cells counted for each condition: 1350 for wt; 900 for cx3cl1−/−). Student's t test: **p < 0.01. c, TNF-α released in culture medium of microglia, expressed as picograms per milliliter per milligram of total cell proteins. Data are the mean ± SE, n = 6–14. Student's t test: *p < 0.05.
No significant difference in the phagocytic activity was observed during CX3CL1 treatment of microglia under normoxia conditions (n = 7; data not shown). To gain insight into the possible mechanisms leading to the above described altered phagocytic activity, wt and cx3cl1−/− microglia were exposed to OGD-CM and analyzed for cytokine production with the same protocol used for phagocytosis assay (for details, see Materials and Methods). Data reported in Figure 2c demonstrated that CX3CL1 significantly reduced TNF-α release only in cx3cl1−/− microglia, with no significant effects being observed in wt microglia. No release of IL-1β was detected in these microglia cultures (data not shown).
CX3CL1 reduces neurological deficits and ischemic infarct size in rat brain after pMCAO
To better investigate the neuroprotective activity of exogenous CX3CL1, we investigated the effect of intracerebroventricular administration of CX3CL1 (200 pmol) in a rat model of permanent cerebral ischemia, on neurological deficits and ischemic infarct volume, with the experimental design illustrated in Figure 3a. CX3CL1-injected rats had better functional outcomes compared with vehicle-treated rats 23 h after ischemia in Bederson's, De Ryck's, and foot-fault tests (Fig. 3b, n = 7). Brains were then analyzed for ischemic volume at 24 h, a time point when the maximum infarction is reported for MCA occlusion (Barone et al., 1992). Figure 3c shows that the infarcted brain volumes in CX3CL1- and vehicle-treated rats were significantly different 24 h after pMCAO (n = 7, p < 0.01). Sham-operated rats had no signs of neurological deficits or cerebral injury (data not shown). When lower doses of CX3CL1 were injected (20 pmol), minor reductions of ischemic brain volume and neurological deficits were observed (data not shown).
Figure 3.

Effect of CX3CL1 on neurological deficits and ischemic brain volume after pMCAO in rats. a, Study paradigm. b, Results of Bederson's, De Ryck's, and foot-fault tests in vehicle-treated (black circles) and CX3CL1-treated (white circles) rats. Dotted lines indicate the maximal scores for sham rats. The foot-fault index for the six sham-operated rats was 4.1 ± 1.4 at 24 h. All data are mean ± SE from seven rats for each experimental condition. Mann–Whitney U test: **p < 0.01, ***p < 0.001. c, Infarct volume in vehicle- and CX3CL1-treated rats 24 h after pMCAO. Data are the mean ± SE from six to seven rats for each condition. Student's t test: **p < 0.01. Representative images of the ischemic area (white) in TTC-stained coronal brain sections are shown at the top.
To investigate whether the protective effects of CX3CL1 were long lasting, behavioral tests were repeated at different time points, up to 28 d (as summarized in Fig. 4a). Figure 4b demonstrates that improved performances in the three different tests were maintained at 7, 14, and 28 d after the insult. Because infarct volume measurements in rodents become inexact and influenced by many confounding factors (tissue shrinkage, glial scarring, cell infiltrates) at periods longer than 3 d after the ischemic insult, rats were analyzed for brain damage by MRI. Structural MRI data analysis revealed lesion volumes significantly reduced in CX3CL1-treated rats at 50 d after pMCAO, with specific reduction localized in the cortical region (Fig. 4c, n = 6). DTI data analysis also revealed a significant reduction of ADC, a marker of vasogenic edema directly correlated with tissue damage (Sood et al., 2009), in the cortex and the external capsula of the ipsilateral hemisphere during CX3CL1 treatment (Fig. 4d), with no significant changes in other brain regions (data not shown). A significant correlation was found between Bederson's, De Ryck's, and foot-fault tests evaluated 28 d after pMCAO and the lesion volume by MRI (p < 0.05, p < 0.01, and p < 0.05, respectively). All these data demonstrate that exogenous CX3CL1 has neuroprotective activities in cerebral ischemia and that its effects are durable.
Figure 4.
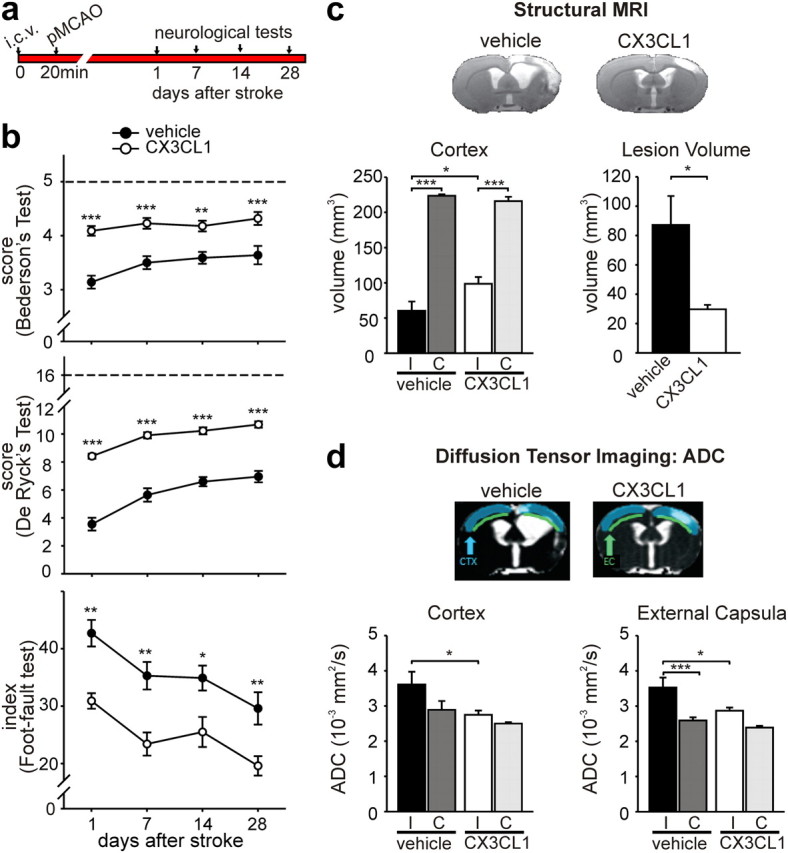
Long-term effects of CX3CL1 on ischemic brain. a, Study paradigm. b, Results of Bederson's (top), De Ryck's (middle), and foot-fault (bottom) tests in vehicle-treated (black circles) and CX3CL1-treated (white circles) rats at different days after pMCAO (n = 11). In the foot-fault test, values for sham-operated rats were similar to those obtained at 24 h. c, Results of structural MRI data analysis of brain lesions in ischemic rats treated with CX3CL1 and saline, 50 d after pMCAO. Volumes of cortex for ipsilateral (I) and contralateral (C) hemispheres and total lesion volume are shown for vehicle- and CX3CL1-treated rats. Representative images of ischemic brains are shown for vehicle- and CX3CL1-treated rats. d, DTI data analysis showing ADC of the ipsilateral and contralateral hemisphere of CX3CL1- and vehicle-treated rats in cortex and external capsula. Representative ADC images are shown. All data are mean ± SE from six rats for each experimental condition. Two-way ANOVA followed by the Bonferroni's post hoc test: *p < 0.05, **p < 0.01, ***p < 0.001.
CX3CL1-mediated neuroprotection requires the activation of A1R in vivo
To investigate whether the neuroprotective effect of CX3CL1 in cerebral ischemia was mediated by A1R, similarly to what we described recently in vitro (Lauro et al., 2008, 2010), the specific A1R antagonist DPCPX or vehicle (DMSO) were injected in rats before the ischemic insult. DPCPX has demonstrated efficacy in reaching the brain parenchyma and inhibiting A1R activity in the brain (Phillis, 1995). DPCPX abolished the protective effects of CX3CL1 (as shown Fig. 3) on both neurological deficits (Fig. 5a, n = 13) and ischemic volume (Fig. 5b, n = 12). DMSO alone did not alter CX3CL1-induced reduction of ischemia-induced neurological deficits (data not shown; n = 13, p < 0.001) and infarct size (data not shown; n = 12, p < 0.01).
Figure 5.

A1R inhibition or deficiency abolishes the neuroprotective effect of CX3CL1. a, Results of Bederson's, De Ryck's, and foot-fault tests 24 h after pMCAO in vehicle- and CX3CL1-treated rats, in the presence of DPCPX (n = 13). Dotted line indicates the maximal scores for sham-operated rats. Results for each experimental group are obtained from two independent sets of experiments. b, Infarct volume in vehicle- and CX3CL1-treated rats. Vehicle-treated (black bars) and CX3CL1-treated (white bars) rats received DPCPX (n = 12). Data are mean ± SE. c, A1R-deficient mice are not protected by CX3CL1 during pMCAO. Data are the mean ± SE from four to five mice. Mann–Whitney U and Student's t tests.
To further confirm the involvement of A1R in CX3CL1-mediated neuroprotection, pMCAO experiments were performed on A1R−/− mice. In contrast with what observed in wt (Fig. 1), in A1R−/− mice, CX3CL1 was ineffective in reducing brain ischemic volume during pMCAO (Fig. 5c, n = 4–5), confirming A1R involvement in CX3CL1-mediated neuroprotection also in vivo. Data in Figure 5c also demonstrated that pMCAO in A1R−/− mice induced a damage similar to wt mice, in accordance with previous findings (Olsson et al., 2004).
CX3CL1 inhibits pMCAO-induced caspase-3 activation
To see whether the neuroprotective effects of CX3CL1 involved the inhibition of ischemia-induced caspase-3 activation, we examined the levels of active caspase-3 in the ipsilateral hemisphere of ischemic rat brains by Western blotting. In the contralateral non-ischemic hemisphere (data not shown) and in the ipsilateral hemisphere of sham-operated rats (Fig. 6a), caspase-3 immunoreactivity was completely absent. During ischemia, caspase-3-positive signal appeared in the ipsilateral hemisphere and was significantly reduced in CX3CL1-treated rats (Fig. 6a, n = 7, p < 0.05). In contrast, in DPCPX-treated ischemic rats, the levels of active caspase-3 did not change during CX3CL1 treatment (Fig. 6a). We also investigated the cleavage of poly(ADP-ribose) polymerase (PARP), a specific caspase-3 substrate, quantified from the ratio of full-length PARP between the ipsilateral and contralateral hemisphere in rats. CX3CL1 treatment significantly reduced ischemia-induced PARP cleavage (Fig. 6b, n = 18, p < 0.01), and, again, it was not effective in the presence of DPCPX (Fig. 6b, n = 10). Representative blots are shown in Figure 6, a and b, with total actin or AKT as control for equal protein loading. Similar results for caspase-3 and PARP were obtained in control experiments performed on rats not treated with DMSO (n = 7; data not shown). These results show that CX3CL1 reduced ischemia-induced caspase activation and that DPCPX abolished this effect, reinforcing the hypothesis of a role for A1R in CX3CL1-mediated neuroprotective processes.
Figure 6.

CX3CL1-reduced activation of caspase 3 pathway during pMCAO is A1R dependent. Brain coronal sections from ischemic rats treated as in Figure 3 were analyzed by Western blot. The same amount of proteins (50 μg) was analyzed for each sample. a, Active form of caspase-3 (17–19 kDa) in the ipsilateral hemisphere of sham-operated (left) or pMCAO rats treated with vehicle or CX3CL1 in the presence of DMSO (control, C; middle) in the presence of DPCPX (right). Results are reported as arbitrary units (a.u.). The inset shows a representative blot: top, active form of caspase-3; bottom: actin, used to normalize signals. b, PARP (116 kDa) in sham-operated (left) or treated rats in the presence or in the absence of DPCPX. Data are expressed as the ratio of PARP in the ipsilateral (I) to the contralateral (C) hemisphere. The inset shows a representative blot. Top, Full-length form of PARP; bottom, AKT, used to normalize signals. Bars indicate the mean ± SE from 6 to 18 rats. Statistical differences are analyzed for each experimental condition for CX3CL1 versus vehicle; *p < 0.05, **p < 0.01 using Student's t test.
Discussion
In this study, we demonstrated that exogenous CX3CL1 has a long-lasting neuroprotective action against permanent cerebral ischemia in rodents. Previous studies on transient cerebral ischemia reported deleterious roles for the CX3CL1/CX3CR1 pair compared with wt animals, and cx3cl1−/− mice had a significant reduction of infarct lesion and a lower mortality (Soriano et al., 2002). Moreover, cx3cr1GFP/GFP mice had smaller infarct lesion, less blood–brain barrier damage, and fewer apoptotic cells and infiltrating leukocytes in the ischemic hemisphere, as well as lower expression of the proinflammatory cytokines IL-1β and TNF-α (Dènes et al., 2008). We confirmed that both cx3cl1−/− and cx3cr1GFP/GFP mice show less severe ischemic damage than wt mice. We further demonstrated that the CX3CL1/CX3CR1 pair has a detrimental role in cerebral ischemia in the absence of a constitutive CX3CL1/CX3CR1 signaling, because exogenous CX3CL1 increased brain damage in cx3cl1−/− ischemic mice. However, we also demonstrated that, when CX3CL1 is given exogenously to ischemic rats or wt mice, in which microglia exhibited, throughout development, a normal constitutive CX3CR1-mediated signaling, this chemokine exerts a protective role on the ischemic lesion. The genuine neuroprotective effect of CX3CL1 is corroborated by the long-lasting improvement in performances obtained in different sensorimotor tasks and by the reduction of ischemic volume, mainly in the cortical area, observed in CX3CL1-treated rats by brain slice staining and structural MRI analysis.
The basis for the different effect of CX3CL1 in wt versus cx3cl1−/− mice is likely that the inflammatory microenvironment induced by the ischemic insult is different in wt and cx3cl1−/− mice. CX3CL1 has been reported to reduce microglia activation, keeping these cells in an “off” state (Biber et al., 2007) and to inhibit the release of inflammatory cytokines (Zujovic et al., 2000; Mizuno et al., 2003; Cardona et al., 2006). In different experimental conditions, there is evidence of both harmful and protective roles of microglia in stroke. After stroke, acute and prolonged inflammatory response exacerbates neuronal injury, with microglia activation contributing through the production of reactive oxygen species, nitric oxide, and proinflammatory cytokines (Block et al., 2007). Conversely, minocycline, an antibiotic commonly used to “inhibit” microglia activation, strongly attenuates the damages induced by ischemia (Yrjänheikki et al., 1998), and both exogenous and resident proliferating microglia exert neuroprotection in brain ischemia, probably through the production of neurotrophic factors (Imai et al., 2007; Lalancette-Hébert et al., 2007; Narantuya et al., 2010). This dual activity of microglia might be explained by the different composition of the parenchymal milieu generated in injuries of different entity or at different stages of the pathology, which might have different effects on microglia activation (Lai and Todd, 2008). We provide evidence that, in OGD conditions in vitro, CX3CL1 deficiency changes the phagocytic response of microglia during CX3CL1 stimulation. We speculate that the CX3CL1-induced reduction of phagocytic activity in wt microglia could be beneficial for neurons, thus explaining the neuroprotective effects described in vivo and in vitro. A relation between the reduced phagocytic activity of microglia and neuron rescue was reported recently in cultured neurons exposed to the inflammatory stimuli lipoteichoic acid and lipopolysaccharide (LPS) (Neher et al., 2011). In different models of neuroinflammatory diseases, CX3CR1 deficiency correlated with enhanced microglia phagocytosis (Lee et al., 2010; Liu et al., 2010) and altered production of inflammatory cytokines (Cardona et al., 2006; Dénes et al., 2008; Bhaskar et al., 2010). In an in vitro models of excitotoxic damage, CX3CL1 was, however, reported to induce neuroprotection, increasing the clearance of neuronal debris, with an MFG-E8 (milk fat globule epidermal growth factor-8)-mediated mechanism (Noda et al., 2011). These differences might be explained by a different role played by microglia in different phases of the degenerative processes.
We also report that, in OGD conditions, CX3CL1 treatment of cx3cl1−/− microglia induces a significant reduction in TNF-α release, whereas no effect was observed in wt microglia. Previous studies reported that CX3CL1 reduces LPS-induced TNF-α release by microglia (Zujovic et al., 2000) and that the neuroprotective effect of CX3CL1 against inflammatory stimuli is correlated with a reduced synthesis of IL-1β (Cardona et al., 2006). A neuroprotective role has been described recently for TNF-α in cerebral ischemia (Lambertsen et al., 2009), leading us to hypothesize that the reduction in microglial TNF-α production during CX3CL1 treatment in OGD conditions might help to explain the increased cerebral damage observed in vivo in ischemic cx3cl1−/− mice. Even if we do not provide conclusive evidence of the direct involvement of TNF-α in the neuroprotective effect of CX3CL1 in our experimental systems, we speculate that the different response elicited in wt versus cx3cl1−/− microglia could explain the different effects on neuron viability. Together, these data confirm that CX3CL1/CX3CR1 pair is one of the key signaling involved in tuning microglia responses in different neuropathologies.
The localization of CX3CR1 on microglia (Harrison et al., 1998) and in vitro studies suggested that the neurotrophic action of CX3CL1 is indirect, requiring soluble factors released by microglial cells (Lauro et al., 2008). Among these, adenosine plays a role, being released from CX3CL1-stimulated microglia and acting on A1R (Lauro et al., 2008, 2010). In this study, we found that, in permanent ischemia, the neuroprotective effect of CX3CL1 is lost in rats treated with DPCPX and in A1R−/− mice, suggesting that the involvement of A1R in CX3CL1-induced neuroprotection is conserved from cellular to in vivo models. This agrees with data indicating the importance of A1R activation in neuroprotection, reducing neuronal injury in vitro and in vivo and being involved in mechanisms of ischemic preconditioning (Von Lubitz et al., 1996; Nakamura et al., 2002). Acute A1R activation reduces neuronal damage, whereas A1R antagonists potentiate damage in neuronal cultures, brain slices, and animal models of cerebral ischemia, epileptic models, and brain trauma (Olsson et al., 2004; Cunha, 2005). However, data on the effects of A1R antagonists on brain ischemia are not always unanimous: DPCPX is reported not to modify ischemia-induced damage (Yoshida et al., 2004; Wang et al., 2005; our data), whereas other antagonists, such as 8-cyclopentyltheophylline, do (Olsson et al., 2004). Furthermore, in both the present study and in a study of global ischemia (Olsson et al., 2004), there was no effect of the deletion of A1 receptors on cerebral damage. During brain ischemia, adenosine levels rise rapidly in the brain (Pearson et al., 2006), providing an endogenous break on excitatory neurotransmission that can provide neuroprotection in some experimental systems (Johansson et al., 2001). CX3CL1 may conceivably contribute to adenosine release in the ischemic brain, promoting activation of neuroprotective pathways in an A1R-dependent manner. The fact that the CX3CL1-induced reduction in damage was eliminated in the A1R−/− mice although there was no difference between A1R−/− and wt mice in the absence of CX3CL1 indicates that the exogenous chemokine activates the adenosine mechanism more powerfully or in different locations than does ischemia per se. However, from these data, we cannot exclude the possibility of peripheral effects of A1R inhibition, but given our previous in vitro data on the A1R-dependency of the protective effects of CX3CL1 on cultured neurons (Lauro et al., 2010), this is not the most likely possibility.
Cell death induced by cerebral ischemia results in necrosis within the ischemic core during the early phase of tissue degeneration and in apoptosis in the penumbra later (Dirnagl et al., 1999). Caspase-3 is a final effector of apoptotic signaling and appears to be activated in different experimental models of cerebral ischemia (Namura et al., 1998; Broughton et al., 2009). The inhibition of caspase-dependent pathways in LPS-induced brain inflammation has been correlated recently with microglia activation (Burguillos et al., 2011), thus suggesting that inhibiting caspase activation in these cells might drive protection from neuroinflammation rather than direct inhibition of cell death. We suggest that one possible mechanism of CX3CL1-mediated neuroprotection is inhibition of the caspase-3 activation and PARP cleavage induced by ischemia, both events again blocked by DPCPX. However, we cannot exclude that these effects are indirectly caused by cytokines or other factors released during CX3CL1 administration.
In conclusion, this study provides evidence that CX3CL1 has long-term neuroprotective properties in vivo, reducing neurological deficits and infarct size related with cerebral ischemia, with molecular mechanisms involving the inhibition of caspase-3 pathway and the activation of A1R. We also demonstrated that, in the absence of a constitutive CX3CL1/CX3CR1 signaling, microglial response to CX3CL1 during hypoxia is altered, likely explaining, at least in part, the opposite effects obtained with wt and CX3CL1/CX3CR1-deficient mice. Additional investigations, also involving studies of CX3CL1 administration subsequent to the ischemic insult, will be needed to define the therapeutic potential of CX3CL1 in human ischemic stroke.
Footnotes
This work was granted by Fondazione Cenci Bolognetti (C.Li.), Ministero Salute (Ricerca Finalizzata; Cinque per mille; Progetto Anti-doping) (C.Li.), and Fondazione Viva la Vita. We thank Drs. Myriam Catalano for initial in vivo experiments and Francesca Grassi for critical reading of this manuscript.
The authors declare no conflict of interest.
References
- Barone FC, Schmidt DB, Hillegass LM, Price WJ, White RF, Feuerstein GZ, Clark RK, Lee EV, Griswold DE, Sarau HM. Reperfusion increases neutrophils and leukotriene B4 receptor binding in rat focal ischemia. Stroke. 1992;23:1337–1348. doi: 10.1161/01.str.23.9.1337. [DOI] [PubMed] [Google Scholar]
- Bhaskar K, Konerth M, Kokiko-Cochran ON, Cardona A, Ransohoff RM, Lamb BT. Regulation of tau pathology by the microglial fractalkine receptor. Neuron. 2010;68:19–31. doi: 10.1016/j.neuron.2010.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biber K, Neumann H, Inoue K, Boddeke HW. WGM Neuronal “On” and “Off” signals control microglia. Trends Neurosci. 2007;30:596–602. doi: 10.1016/j.tins.2007.08.007. [DOI] [PubMed] [Google Scholar]
- Block ML, Zecca L, Hong JS. Microglia-mediated neurotoxicity: uncovering the molecular mechanisms. Nat Rev Neurosci. 2007;8:57–69. doi: 10.1038/nrn2038. [DOI] [PubMed] [Google Scholar]
- Broughton BR, Reutens DC, Sobey CG. Apoptotic mechanisms after cerebral ischemia. Stroke. 2009;40:e331–e339. doi: 10.1161/STROKEAHA.108.531632. [DOI] [PubMed] [Google Scholar]
- Burguillos MA, Deierborg T, Kavanagh E, Persson A, Hajji N, Garcia-Quintanilla A, Cano J, Brundin P, Englund E, Venero JL, Joseph B. Caspase signalling controls microglia activation and neurotoxicity. Nature. 2011;472:319–324. doi: 10.1038/nature09788. [DOI] [PubMed] [Google Scholar]
- Cardona AE, Pioro EP, Sasse ME, Kostenko V, Cardona SM, Dijkstra IM, Huang D, Kidd G, Dombrowski S, Dutta R, Lee JC, Cook DN, Jung S, Lira SA, Littman DR, Ransohoff RM. Control of microglia neurotoxicity by the fractalkine receptor. Nat Neurosci. 2006;9:917–924. doi: 10.1038/nn1715. [DOI] [PubMed] [Google Scholar]
- Chapman GA, Moores K, Harrison D, Campbell CA, Stewart BR, Strijbos PJ. Fractalkine cleavage from neuronal membranes represents an acute event in the inflammatory response to excitotoxic brain damage. J Neurosci. 2000;20(RC87):1–5. doi: 10.1523/JNEUROSCI.20-15-j0004.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook DN, Chen SC, Sullivan LM, Manfra DJ, Wiekowski MT, Prosser DM, Vassileva G, Lira SA. Generation and analysis of mice lacking the chemokine fractalkine. Mol Cell Biol. 2001;21:3159–3165. doi: 10.1128/MCB.21.9.3159-3165.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunha RA. Neuroprotection by adenosine in the brain: from A1 receptor activation to A2A receptor blockade. Purinergic Signal. 2005;1:111–134. doi: 10.1007/s11302-005-0649-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dénes A, Ferenczi S, Halász J, Környei Z, Kovács KJ. Role of CX3CR1 (fractalkine receptor) in brain damage and inflammation induced by focal cerebral ischemia in mouse. J Cereb Blood Flow Metab. 2008;28:1707–1721. doi: 10.1038/jcbfm.2008.64. [DOI] [PubMed] [Google Scholar]
- Dirnagl U, Iadecola C, Moskowitz MA. Pathobiology of ischemic stroke: an integrated view. Trends Neurosci. 1999;22:391–397. doi: 10.1016/s0166-2236(99)01401-0. [DOI] [PubMed] [Google Scholar]
- Fredholm BB. Adenosine, an endogenous distress signal, modulates tissue damage and repair. Cell Death Differ. 2007;14:1315–1323. doi: 10.1038/sj.cdd.4402132. [DOI] [PubMed] [Google Scholar]
- Fuhrmann M, Bittner T, Jung CK, Burgold S, Page RM, Mitteregger G, Haass C, LaFerla FM, Kretzschmar H, Herms J. Microglial CX3CR1 knockout prevents neuron loss in a mouse model of Alzheimer's disease. Nat Neurosci. 2010;13:411–413. doi: 10.1038/nn.2511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison JK, Jiang Y, Chen S, Xia Y, Maciejewski D, McNamara RK, Streit WJ, Salafranca MN, Adhikari S, Thompson DA, Botti P, Bacon KB, Feng L. Role for neuronally derived fractalkine in mediating interactions between neurons and CX3CR1-expressing microglia. Proc Natl Acad Sci U S A. 1998;95:10896–10901. doi: 10.1073/pnas.95.18.10896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imai F, Suzuki H, Oda J, Ninomiya T, Ono K, Sano H, Sawada M. Neuroprotective effect of exogenous microglia in global brain ischemia. J Cereb Blood Flow Metab. 2007;27:488–500. doi: 10.1038/sj.jcbfm.9600362. [DOI] [PubMed] [Google Scholar]
- Johansson B, Halldner L, Dunwiddie TV, Masino SA, Poelchen W, Giménez-Llort L, Escorihuela RM, Fernández-Teruel A, Wiesenfeld-Hallin Z, Xu XJ, Hårdemark A, Betsholtz C, Herlenius E, Fredholm BB. Hyperalgesia, anxiety, and decreased hypoxic neuroprotection in mice lacking the adenosine A1 receptor. Proc Natl Acad Sci U S A. 2001;98:9407–9412. doi: 10.1073/pnas.161292398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung S, Aliberti J, Graemmel P, Sunshine MJ, Kreutzberg GW, Sher A, Littman DR. Analysis of fractalkine receptor CX(3)CR1 function by targeted deletion and green fluorescent protein reporter gene insertion. Mol Cell Biol. 2000;20:4106–4114. doi: 10.1128/mcb.20.11.4106-4114.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labuzek K, Liber S, Gabryel B, Adamczyk J, Okopień B. Metformin increases phagocytosis and acidifies lysosomal/endosomal compartments in AMPK-dependent manner in rat primary microglia. Naunyn Schmied Arch Pharmacol. 2010;381:171–186. doi: 10.1007/s00210-009-0477-x. [DOI] [PubMed] [Google Scholar]
- Lai AY, Todd KG. Differential regulation of trophic and proinflammatory microglial effectors is dependent on severity of neuronal injury. Glia. 2008;56:259–270. doi: 10.1002/glia.20610. [DOI] [PubMed] [Google Scholar]
- Lalancette-Hébert M, Gowing G, Simard A, Weng YC, Kriz J. Selective Ablation of proliferating microglial cells exacerbates ischemic injury in the brain. J Neurosci. 2007;27:2596–2605. doi: 10.1523/JNEUROSCI.5360-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambertsen KL, Clausen BH, Babcock AA, Gregersen R, Fenger C, Nielsen HH, Haugaard LS, Wirenfeldt M, Nielsen M, Dagnaes-Hansen F, Bluethmann H, Faergeman NJ, Meldgaard M, Deierborg T, Finsen B. Microglia protect neurons against ischemia by synthesis of tumor necrosis factor. J Neurosci. 2009;29:1319–1330. doi: 10.1523/JNEUROSCI.5505-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauro C, Cipriani R, Catalano M, Trettel F, Chece G, Brusadin V, Antonilli L, van Rooijen N, Eusebi F, Fredholm BB, Limatola C. Adenosine A1 receptors and microglial cells mediate CX3CL1-induced protection of hippocampal neurons against Glu-induced death. Neuropsychopharmacology. 2010;35:1550–1559. doi: 10.1038/npp.2010.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauro C, Di Angelantonio S, Cipriani R, Sobrero F, Antonilli L, Brusadin V, Ragozzino D, Limatola C. Activity of adenosine receptor type 1 is required for CX3CL1-mediated neuroprotection and neuromodulation in hippocampal neurons. J Immunol. 2008;180:7590–7596. doi: 10.4049/jimmunol.180.11.7590. [DOI] [PubMed] [Google Scholar]
- Lee S, Varvel NH, Konerth ME, Xu G, Cardona AE, Ransohoff RM, Lamb BT. CX3CR1 deficiency alters microglia activation and reduces beta-amyloid deposition in two Alzheimer's disease mouse models. Am J Pathol. 2010;177:2549–2562. doi: 10.2353/ajpath.2010.100265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Limatola C, Lauro C, Catalano M, Ciotti MT, Bertollini C, Di Angelantonio S, Ragozzino D, Eusebi F. Chemokine CX3CL1 protects rat hippocampal neurons against glutamate-mediated excitotoxicity. J Neuroimmunol. 2005;166:19–28. doi: 10.1016/j.jneuroim.2005.03.023. [DOI] [PubMed] [Google Scholar]
- Liu Z, Condello C, Schain A, Harb R, Grutzendler J. CX3CR1 in microglia regulates brain amyloid deposition through selective protofibrillar amyloid-β phagocytosis. J Neurosci. 2010;30:17091–17101. doi: 10.1523/JNEUROSCI.4403-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyons A, Lynch AM, Downer EJ, Hanley R, O'Sullivan JB, Smith A, Lynch MA. Fractalkine-induced activation of the phosphatidylinositol-3 kinase pathway attenuates microglial activation in vivo and in vitro. J Neurochem. 2009;110:1547–1556. doi: 10.1111/j.1471-4159.2009.06253.x. [DOI] [PubMed] [Google Scholar]
- Mizuno T, Kawanokuchi J, Numata K, Suzumura A. Production and neuroprotective functions of Fractalkine in the central nervous system. Brain Res. 2003;979:65–70. doi: 10.1016/s0006-8993(03)02867-1. [DOI] [PubMed] [Google Scholar]
- Nakamura M, Nakakimura K, Matsumoto M, Sakabe T. Rapid tolerance to focal cerebral ischemia in rats is attenuated by Adenosine A1 receptor antagonist. J Cereb Blood Flow Metab. 2002;22:161–170. doi: 10.1097/00004647-200202000-00004. [DOI] [PubMed] [Google Scholar]
- Namura S, Zhu J, Fink K, Endres M, Srinivasan A, Tomaselli KJ, Yuan J, Moskowitz MA. Activation and cleavage of caspase-3 in apoptosis induced by experimental cerebral ischemia. J Neurosci. 1998;18:3659–3668. doi: 10.1523/JNEUROSCI.18-10-03659.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narantuya D, Nagai A, Sheikh AM, Masuda J, Kobayashi S, Yamaguchi S, Kim SU. Human microglia transplanted in rat focal ischemia brain induce neuroprotection and behavioral improvement. PLoS One. 2010;5:e11746. doi: 10.1371/journal.pone.0011746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neher JJ, Neniskyte U, Zhao JW, Bal-Price A, Tolkovsky AM, Brown GC. Inhibition of microglial phagocytosis is sufficient to prevent inflammatory neuronal death. J Immunol. 2011;186:4973–4983. doi: 10.4049/jimmunol.1003600. [DOI] [PubMed] [Google Scholar]
- Noda M, Doi Y, Liang J, Kawanokuchi J, Sonobe Y, Takeuchi H, Mizuno T, Suzumura A. Fractalkine attenuates excite-neurotoxicity via microglia clearance of damaged neurons and antioxidant enzyme Heme Oxigenase-1 expression. J Biol Chem. 2011;286:2308–2319. doi: 10.1074/jbc.M110.169839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsson T, Cronberg T, Rytter A, Asztély F, Fredholm BB, Smith ML, Wieloch T. Effects of adenosine A1 receptor gene deletion in neuronal damage following global cerebral ischemia in mouse in vivo and in vitro. Eur J Neurosci. 2004;20:1197–1204. doi: 10.1111/j.1460-9568.2004.03564.x. [DOI] [PubMed] [Google Scholar]
- Pabon MM, Bachstetter AD, Hudson CE, Gemma C, Bickford PC. CX3CL1 reduces neurotoxicity and microglial activation in a rat model of Parkinson's disease. J Neuroinflammation. 2011;8:9. doi: 10.1186/1742-2094-8-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paxinos G, Watson C. The rat brain in stereotaxic coordinates. San Diego: Academic; 1998. [DOI] [PubMed] [Google Scholar]
- Pearson T, Damian K, Lynas RE, Frenguelli BG. Sustained elevation of extracellular adenosine and activation of A1 receptors underline the post-ischemic inhibition of neuronal function in rat hippocampus in vitro. J Neurochem. 2006;97:1357–1368. doi: 10.1111/j.1471-4159.2006.03823.x. [DOI] [PubMed] [Google Scholar]
- Phillis JW. The effects of selective A 1 and A 2a adenosine receptor antagonists on cerebral ischemic injury in the gerbil. Brain Res. 1995;705:79–84. doi: 10.1016/0006-8993(95)01153-6. [DOI] [PubMed] [Google Scholar]
- Sood R, Yang Y, Taheri S, Candelario-Jalil E, Estrada EY, Walker EJ, Thompson J, Rosenberg GA. Increased apparent diffusion coefficients on MRI linked with matrix metalloproteinases and edema in white matter after bilateral carotid artery occlusion in rats. J Cereb Blood Flow Metab. 2009;29:308–316. doi: 10.1038/jcbfm.2008.121. [DOI] [PubMed] [Google Scholar]
- Soriano SG, Amaravadi LS, Wang YF, Zhou H, Yu GX, Tonra JR, Fairchild-Huntress V, Fang Q, Dunmore JH, Huszar D, Pan Y. Mice deficient in fractalkine are less susceptible to cerebral ischemia-reperfusion injury. J Neuroimmunol. 2002;125:59–65. doi: 10.1016/s0165-5728(02)00033-4. [DOI] [PubMed] [Google Scholar]
- Storini C, Bergamaschini L, Gesuete R, Rossi E, Maiocchi D, De Simoni MG. Selective inhibition of plasma kallikrein protects brain from reperfusion injury. J Pharmacol Exp Ther. 2006;318:849–854. doi: 10.1124/jpet.106.105064. [DOI] [PubMed] [Google Scholar]
- Tarozzo G, Campanella M, Ghiani M, Bulfone A, Beltramo M. Expression of fractalkine and its receptor, CX3CR1, in response to ischaemia-reperfusion brain injury in the rat. Eur J Neurosci. 2002;15:1663–1668. doi: 10.1046/j.1460-9568.2002.02007.x. [DOI] [PubMed] [Google Scholar]
- Villa P, Triulzi S, Cavalieri B, Di Bitondo R, Bertini R, Barbera S, Bigini P, Mennini T, Gelosa P, Tremoli E, Sironi L, Ghezzi P. The interleukin-8 (IL(/CXCL8) receptor inhibitor reparixin improves neurological deficits and reduces long-term inflammation in permanent and transient cerebral ischemia in rats. Mol Med. 2007;13:125–133. doi: 10.2119/2007-00008.Villa. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Von Lubitz DK, Beenhakker M, Lin RC, Carter MF, Paul IA, Bischofberger N, Jacobson KA. Reduction of postischemic brain damage and memory deficits following treatment with the selective adenosine A1 receptor agonist. Eur J Pharmacol. 1996;302:43–48. doi: 10.1016/0014-2999(96)00101-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Xiong L, Chen S, Liu Y, Zhu X. Rapid tolerance to focal cerebral ischemia in rats is induced by preconditioning with electroacupuncture: window of protection and the role of adenosine. Neurosci Lett. 2005;381:158–162. doi: 10.1016/j.neulet.2005.02.019. [DOI] [PubMed] [Google Scholar]
- Yoshida M, Nakakimura K, Cui YJ, Matsumoto M, Sakabe T. Adenosine A(1) receptor antagonist and mitochondrial ATP-sensitive potassium channel blocker attenuate the tolerance to focal cerebral ischemia in rats. J Cereb Blood Flow Metab. 2004;24:771–779. doi: 10.1097/01.WCB.0000122742.72175.1B. [DOI] [PubMed] [Google Scholar]
- Yrjänheikki J, Keinänen R, Pellikka M, Hökfelt T, Koistinaho J. Tetracyclines inhibit microglial activation and are neuroprotective in global brain ischemia. Proc Natl Acad Sci U S A. 1998;95:15769–15774. doi: 10.1073/pnas.95.26.15769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J, Zhou Z, Liu Y, Zheng J. Fractalkine and CX3CR1 are involved in the migration of intravenously grafted human bone marrow stromal cells toward ischemic brain lesion in rats. Brain Res. 2009;1287:173–183. doi: 10.1016/j.brainres.2009.06.068. [DOI] [PubMed] [Google Scholar]
- Zujovic V, Benavides J, Vigé X, Carter C, Taupin V. Fractalkine modulates TNF-α secretion and neurotoxicity induced by microglial activation. Glia. 2000;29:305–315. [PubMed] [Google Scholar]