

Abstract
Nucleotide excision repair (NER) is one of the main DNA repair pathways that protect cells against genomic damage. Disruption of this pathway can contribute to the development of cancer and accelerate aging. Mutational characteristics of NER-deficiency may reveal important diagnostic opportunities, as tumors deficient in NER are more sensitive to certain treatments. Here, we analyzed the genome-wide somatic mutational profiles of adult stem cells (ASCs) from NER-deficient Ercc1−/Δ mice. Our results indicate that NER-deficiency increases the base substitution load twofold in liver but not in small intestinal ASCs, which coincides with the tissue-specific aging pathology observed in these mice. Moreover, NER-deficient ASCs of both tissues show an increased contribution of Signature 8 mutations, which is a mutational pattern with unknown etiology that is recurrently observed in various cancer types. The scattered genomic distribution of the base substitutions indicates that deficiency of global-genome NER (GG-NER) underlies the observed mutational consequences. In line with this, we observe increased Signature 8 mutations in a GG-NER-deficient human organoid culture, in which XPC was deleted using CRISPR-Cas9 gene-editing. Furthermore, genomes of NER-deficient breast tumors show an increased contribution of Signature 8 mutations compared with NER-proficient tumors. Elevated levels of Signature 8 mutations could therefore contribute to a predictor of NER-deficiency based on a patient's mutational profile.
The genome is continuously exposed to mutagenic processes, which can damage the DNA and can ultimately result in mutation accumulation. To counteract these processes, cells exploit multiple DNA repair pathways that each repair specific lesions. Deficiency of these pathways can contribute to cancer initiation and progression. To increase insight into the cellular processes that underlie mutation accumulation, genome-wide mutational patterns of tumors can be characterized (Alexandrov et al. 2013; Nik-Zainal et al. 2016). To date, systematic analyses of tumor genomes have revealed 30 signatures of base substitutions and six rearrangement signatures of mutational processes in cancer genomes (Alexandrov et al. 2013; Nik-Zainal et al. 2016). Some links between mutational signatures and DNA repair pathways have been discovered with large-scale tumor genome analyses (Alexandrov et al. 2013; Kim et al. 2016; Davies et al. 2017). Mutational signatures associated with DNA repair deficiencies may have important diagnostic value. For example, several signatures have been associated with BRCA1/2 inactivity and can consequently be predictive for a response to PARP inhibition or cisplatin treatment (Waddell et al. 2015; Davies et al. 2017). However, linking DNA repair deficiencies to specific mutational signatures remains complicated, as tumors are genomically highly unstable and multiple processes have contributed to mutation accumulation, typically in a tissue-specific manner (Alexandrov et al. 2013; Nik-Zainal et al. 2016).
Nucleotide excision repair (NER) is one of the main DNA repair pathways (Iyama and Wilson 2013) and has been suggested to underlie multiple mutational signatures, based on large-scale tumor mutation analyses (Alexandrov et al. 2013). NER consists of two subpathways: global-genome NER (GG-NER), which repairs bulky helix-distorting lesions throughout the genome, and transcription-coupled NER (TC-NER), which resolves RNA polymerase blocking lesions during transcription (Hoeijmakers 2009; Iyama and Wilson 2013; Marteijn et al. 2014). Somatic mutations in ERCC2, a key factor of NER, were previously associated with Signature 5 in urothelial tumors (Kim et al. 2016). However, not all NER-deficient tumors are characterized by a high Signature 5 contribution (Kim et al. 2016), suggesting that NER-deficiency might be associated with other mutational signatures as well.
We have previously shown that clonal organoid cultures can be used to measure mutations that have accumulated during life or during culturing in adult stem cells (ASCs) (Blokzijl et al. 2016; Drost et al. 2017; Jager et al. 2018). Tissue-specific ASCs maintain a stable genome both in vivo and in vitro and therefore provide a versatile system to study mutational processes in detail (Huch et al. 2015; Blokzijl et al. 2016). Furthermore, ASCs constitute a relevant cell source to study mutational patterns, as these cells are believed to be the cell-of-origin for specific types of cancer (Barker et al. 2009; Adams et al. 2015; Zhu et al. 2016).
Here, we used whole-genome sequencing analysis of Ercc1−/Δ mouse organoids and XPC-knockout (XPCKO) human organoids to systematically characterize the mutational consequences of NER-deficiency. ERCC1 plays a crucial role in the core NER pathway involving both GG-NER and TC-NER (Aboussekhra et al. 1995; Sijbers et al. 1996a; Kirschner and Melton 2010; Iyama and Wilson 2013). ERCC1 is mutated in ∼4.5% of all human tumors, and single nucleotide polymorphisms in ERCC1 have been linked to an increased risk of developing colorectal cancer (Ni et al. 2014). Ercc1−/Δ mice are hemizygous for a single truncated Ercc1 allele, which largely corrupts protein function (Weeda et al. 1997; Dollé et al. 2011) and results in decreased NER-activity (Su et al. 2012). Ercc1−/Δ mice have five times shorter lives than wild-type (WT) littermates (Dollé et al. 2011; Vermeij et al. 2016). The livers of Ercc1−/Δ mice display various aging-like characteristics (Weeda et al. 1997; Niedernhofer et al. 2006; Dollé et al. 2011; Gregg et al. 2012), whereas other organs do not show an obvious pathological phenotype. Thus, the consequences of loss of ERCC1 differ considerably between tissues, yet the reason for this remains unclear. XPC is involved in the recognition of bulky DNA adducts in the GG-NER pathway specifically (Iyama and Wilson 2013; Puumalainen et al. 2016). Germline mutations in this gene cause xeroderma pigmentosum, a disorder characterized by development of various cancer types at an early age (Sands et al. 1995; Melis et al. 2008; Dupuy and Sarasin 2015).
In addition to the mutational analyses of mouse and human NER-deficiency models, we substantiated our findings by characterizing the genome-wide mutational differences between NER-deficient and NER-proficient tumors from a breast cancer cohort (Nik-Zainal et al. 2016).
Results
Loss of NER protein ERCC1 increases the number of base substitutions in liver but not in small intestinal mouse ASCs
To characterize the mutational consequences of NER-deficiency, we generated clonal organoid cultures from single liver and small intestinal ASCs of three Ercc1−/Δ mice and three WT littermates (Fig. 1A). Whole-genome sequencing (WGS) analysis of DNA isolated from the clonal organoid cultures allows for reliable determination of the somatic mutations that were accumulated during life in the original ASCs (Blokzijl et al. 2016; Jager et al. 2018). Subclonal mutations acquired after the single-cell step will only be present in a subpopulation of the cells and are filtered out based on low allele frequencies (Jager et al. 2018). We also sequenced the genomes of polyclonal biopsies from the tail of each mouse, which served as control samples to exclude germline variants.
Figure 1.

Experimental setup and tissue-specific expression of Ercc1 in mouse ASCs. (A) Schematic overview of the experimental setup used to determine the mutational patterns in single ASCs from the liver and small intestine of mice. Biopsies from the liver and small intestine of six 15-wk-old female mice (three Ercc1−/Δ mice and three WT littermates) were cultured in bulk for ∼1.5 wk to enrich for ASCs. Subsequently, clonal organoids were derived from these bulk organoid cultures and expanded for ∼1 mo, until there were enough cells to perform both WGS and RNA sequencing. As a control sample a biopsy of the tail of each mouse was also subjected to WGS. (B) Box plots depicting normalized Ercc1 expression in ASC organoid cultures derived from liver and small intestine of Ercc1−/Δ mice (n = 3 and n = 3, respectively) and WT littermates (n = 3 and n = 4, respectively). Asterisks represent significant differences (P < 0.05, negative binomial test). (C) Western blot analysis of ERCC1 in Ercc1−/Δ and WT small intestinal and liver mouse organoids.
We performed RNA sequencing on one clonal organoid culture from each tissue of each mouse. Ercc1 is significantly differentially expressed between WT and Ercc1−/Δ in both liver and small intestinal ASCs (P < 0.05, negative binomial test) (Fig. 1B), confirming the anticipated effects of the Ercc1 mutations at the mRNA level. While there is some Ercc1 expression in Ercc1−/Δ ASCs, the C-terminal domain of ERCC1 is essential in ERCC1-XPF complex formation, and disruption of this interaction reduces the stability of ERCC1 protein (Sijbers et al. 1996b; de Laat 1998; Tripsianes et al. 2005). Indeed, ERCC1 protein is not detectable by immunoblotting in Ercc1−/Δ organoid cultures of both tissues (Fig. 1C). No other DNA repair genes were differentially expressed between WT and Ercc1−/Δ ASCs (Supplemental Data S1). However, the expression of eight out of nine core NER genes, including Ercc1, is higher in WT liver ASCs than WT small intestinal ASCs (Supplemental Fig. S1; Supplemental Table S1).
WGS analysis on the clonally expanded organoid cultures revealed 4238 somatic base substitutions in the autosomal genome of 11 clonal ASC samples (Fig. 2A; Supplemental Table S2). With targeted deep-sequencing, we validated ∼97.5% of these base substitutions (Supplemental Data S2). Liver ASCs of WT mice acquired 19.5 ± 4.1 (mean ± standard deviation) base substitutions per week. This rate is similar in ASCs of the small intestine, at 16.1 ± 3.1 mutations per week, and is in line with the observation that human liver and intestinal ASCs have similar mutation accumulation rates in vivo (Blokzijl et al. 2016). Loss of ERCC1 induced a twofold increase (45.5 ± 3.0 base substitutions per week) in the number of base substitutions in ASCs of the liver (Fig. 2A; Supplemental Fig. S2A). We also observed a significant increase in the number of double base substitutions in liver ASCs lacking ERCC1 (q < 0.05, t-test, FDR correction) (Fig. 2B; Supplemental Fig. S2B; Supplemental Table S3). Ercc1−/Δ liver ASCs acquire 0.49 ± 0.06 double base substitutions per week, while WT liver ASCs acquire only 0.05 ± 0.04 double base substitutions per week. The increased number of double base substitutions in the liver ASCs remained significant after normalizing for the total number of base substitutions (q < 0.05, t-test, FDR correction) (Supplemental Fig. S2C), indicating a liver-specific enrichment of double base substitutions in Ercc1−/Δ ASCs compared with WT. In contrast, we did not observe a difference in mutation load between small intestinal ASCs of Ercc1−/Δ and WT mice (Fig. 2A; Supplemental Fig. S2A) or in the number of double base substitutions (Fig. 2B; Supplemental Fig. S2B).
Figure 2.

Somatic mutation rates in the genomes of ASCs from liver and small intestine of WT and Ercc1−/Δ mice. (A) Mean number of base substitutions, (B) double base substitutions, (C) indels, and (D) SVs acquired per autosomal genome per week in ASCs of WT liver (n = 3), Ercc1−/Δ liver (n = 3), WT small intestine (n = 2), and Ercc1−/Δ small intestine (n = 3). Error bars represent standard deviations. Asterisks represent significant differences (q < 0.05, two-sided t-test, FDR correction). (n.s.) Nonsignificant (q ≥ 0.05, two-sided t-test, FDR correction).
In addition to the 4238 base substitutions, we identified 2116 small insertions and deletions (indels) and 21 larger deletions (≥100 bp) in the autosomal genome of the 11 clonal ASC samples (Supplemental Table S2). We observed similar indel numbers in WT and Ercc1−/Δ ASCs of both tissues (Fig. 2C; Supplemental Fig. S2D). Of note, accurate identification of indels is more challenging than base substitutions, and, as a result, these calls may contain more false positives. ASCs in the small intestine and liver of the mice acquire approximately 13.3 ± 3.4 indels per week, independent of Ercc1 mutation status. Likewise, loss of ERCC1 did not influence the number or type of structural variations (SVs) in ASCs of the small intestine and the liver (Fig. 2D; Supplemental Fig. S2E; Supplemental Table S2). Mouse ASCs carried 0–6 deletions (median length of 539 bp) (Supplemental Table S4). Finally, a genome-wide copy-number profile was generated to identify chromosomal gains and losses. These profiles indicated that all WT and Ercc1−/Δ ASCs were karyotypically stable during life (Supplemental Fig. S3). Nevertheless, some subclonal aneuploidies were detected in both a WT and Ercc1−/Δ liver organoid sample, which most likely occurred in vitro after the clonal step, irrespective of Ercc1 mutation status.
Mouse ASCs lacking NER protein ERCC1 show increased Signature 8 mutations
To further dissect the mutational consequences of NER-deficiency, we characterized the mutation spectra in the mouse ASCs. Regardless of tissue-type, the mutation spectra of all assessed ASCs are predominantly characterized by C:G > A:T mutations and C:G > T:A mutations (Fig. 3A). However, the mutation spectra of NER-proficient and NER-deficient ASCs differed significantly for both tissues (q < 0.05, χ2 test, FDR correction). Indeed, there are some differences, such as an increased contribution of T:A > A:T mutations in Ercc1−/Δ ASCs compared with WT ASCs (Fig. 3A).
Figure 3.

Mutational patterns of base substitutions acquired in the genomes of ASCs from liver and small intestine of WT and Ercc1−/Δ mice. (A) Mean relative contribution of the indicated mutation types to the mutation spectrum for each mouse ASC group. Error bars represent standard deviations. The total number of mutations and total number of ASCs (n) per group is indicated. Asterisks indicate significant differences in mutation spectra (q < 0.05, χ2 test, FDR correction). (B) Relative contribution of the indicated COSMIC mutational signatures to the average 96-channel mutational profiles of each mouse ASC group. Asterisks indicate significantly different signature contributions; P-values were obtained using a bootstrap resampling approach (Methods; Supplemental Fig. S6E,F). (C) Absolute contribution of the indicated COSMIC mutational signatures to the average 96-channel mutational profiles of each mouse ASC group. (D) Absolute contribution of two mutational signatures that were identified by nonnegative matrix factorization (NMF) analysis of the average 96-channel mutational profiles of each mouse ASC group. (E) Relative contribution of each indicated context-dependent base substitution type to mutational Signature 8 and Signature 8*.
To gain insight into these differences, we generated 96-channel mutational profiles of all ASCs (Supplemental Figs. S4, S5) and assessed the contribution of each COSMIC mutational signature (https://cancer.sanger.ac.uk/cosmic/signatures_v2) to the average 96-channel mutational profile per group (Supplemental Fig. S6B). We could reconstruct the original profiles well with the 30 COSMIC signatures (average cosine similarity = 0.95) (Supplemental Fig. S6A). The contribution of the COSMIC signatures is significantly different between NER-proficient and NER-deficient ASC groups for both liver and small intestine (d > dWT_0.05 and d > dMUT_0.05, bootstrap resampling method) (Methods; Supplemental Fig. S6C,D). We could reconstruct the 96-channel mutational profiles with the top 10 most contributing COSMIC mutational signatures comparably well (average cosine similarity = 0.95) (Fig. 3B,C; Supplemental Fig. S6A).
The 96-channel mutational profiles of NER-deficient liver ASCs closely resemble Signature 8 (cosine similarity of 0.92) (Supplemental Fig. S7), and Signature 8 can almost fully explain the increase in base substitutions in NER-deficient liver ASCs (Fig. 3C). The number of Signature 8 mutations is also increased in all small intestinal ASCs of Ercc1−/Δ mice compared with WT small intestinal ASCs (Fig. 3C). This finding shows that NER-deficiency can result in elevated numbers of Signature 8 mutations in ASCs, regardless of tissue-type or overall increase in mutational load.
In addition, we performed an unbiased signature analysis by extracting two mutational signatures de novo from the mouse mutation catalogs using nonnegative matrix factorization (NMF) (Supplemental Data S3; Supplemental Fig. S8). One of the identified signatures, Signature X, contributes approximately 100 mutations to the mutational profiles of liver ASCs and 200 mutations to small intestinal ASCs, in both WT and Ercc1−/Δ mice (Fig. 3D), suggesting that this signature represents a mutational process that is generally active in mouse ASCs. In line with this, Signature X is highly similar to 96-channel mutational profiles of ASCs of the small intestine of old mice (cosine similarity = 0.95) (Supplemental Fig. S8B; Behjati et al. 2014). As expected, this mouse signature is not similar to any of the known COSMIC signatures identified in human tumor sequencing data (Supplemental Fig. S8B). The other signature, Signature 8*, is highly similar to COSMIC Signature 8 (cosine similarity = 0.91) (Fig. 3E; Supplemental Fig. S8B) and has an increased contribution in Ercc1−/Δ liver ASCs compared with WT (Fig. 3D; Supplemental Fig. S8C). Moreover, the contribution of Signature 8* mutations is also increased in Ercc1−/Δ small intestinal ASCs in comparison to WT small intestinal ASCs (Fig. 3D; Supplemental Fig. S8C). These findings confirmed that NER-deficiency can result in the accumulation of base substitutions that show a 96-channel profile similar to COSMIC Signature 8.
Mutations are distributed nonrandomly throughout the genome in cancer cells and in human ASCs (Schuster-Böckler and Lehner 2012; Blokzijl et al. 2016). NER is one of the pathways suggested to underlie this nonrandom distribution of mutations (Zheng et al. 2014; Perera et al. 2016). First, NER-activity has been linked to a local enrichment of mutations at gene promoters (Perera et al. 2016). However, we do not observe any significant differences in the depletion of mutations in promoters, promoter-flanking, or enhancer regions between NER-proficient and NER-deficient ASCs (Supplemental Fig. S9A). Second, activity of TC-NER typically results in a depletion of mutations in expressed genes, as this pathway repairs lesions on the transcribed strand during transcription (Pleasance et al. 2010). Mutations are indeed depleted in genic regions of NER-proficient WT mouse ASCs, but the depletion is not significantly different in NER-deficient ASCs (n.s., Poisson test, FDR correction) (Supplemental Fig. S9A). Moreover, the average expression levels of genes in which the somatic mutations are located do not differ between Ercc1−/Δ and WT ASCs (n.s., t-test, FDR correction) (Supplemental Fig. S9B), suggesting that Ercc1−/Δ ASCs do not accumulate more mutations in expressed genes. Finally, there are no obvious changes in transcriptional strand bias, although the mutation numbers are too low to be conclusive (n = 104–660 base substitutions per mouse ASC type) (Supplemental Fig. S9C). NER-deficiency thus influences both the mutation load and types but not the genomic distribution in mouse ASCs, suggesting that the contribution of TC-NER to the mutational consequences is minimal in these cells.
Human ASCs lacking GG-NER protein XPC show increased Signature 8 mutations
To further evaluate the link between NER-deficiency and Signature 8 in human ASCs, we generated a human GG-NER-deficient XPCKO ASC using CRISPR-Cas9 gene-editing in a human small intestinal organoid culture (Fig. 4A). After confirming absence of XPC protein (Fig. 4B), we passaged the XPCKO clone for ∼2 mo to allow the accumulation of sufficient mutations for downstream analyses. Similar to the Ercc1−/Δ mouse ASCs, the human ASC lacking XPC show an increased number of base substitutions acquired per week (Fig. 4C; Supplemental Table S5). In addition, the number of double base substitutions acquired per week was approximately 17 times higher (Fig. 4D; Supplemental Tables S5, S6). We did not observe a marked change in the genomic distribution of acquired mutations as a result of XPC deletion in human ASCs, nor a change in transcriptional strand bias (Supplemental Fig. S10C,D). In total, ∼39% of the increase in base substitutions in the XPCKO ASC can be explained by Signature 8 (Fig. 4E; Supplemental Fig. S10B).
Figure 4.
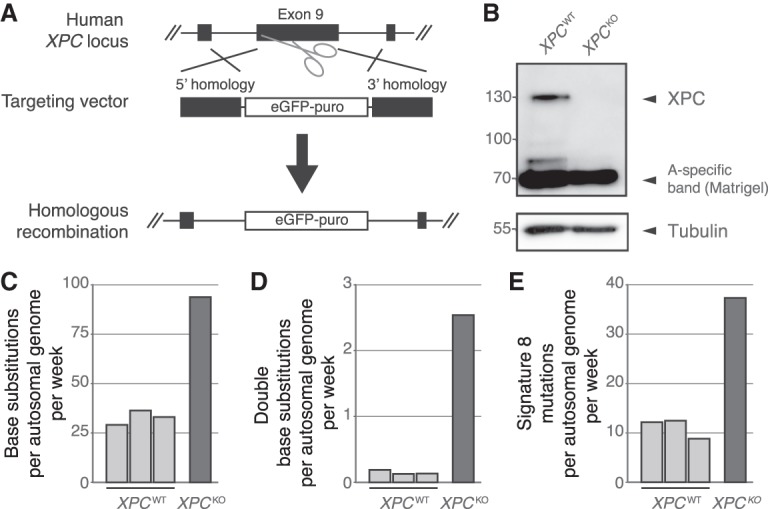
Mutational consequences of XPCKO in human intestinal organoid cultures in vitro. (A) Targeting strategy for the generation of XPCKO organoid cultures using CRISPR-Cas9 gene-editing. (B) Western blot analysis of XPC in human XPCWT and XPCKO organoids. (C) Number of base substitutions, (D) double base substitutions, and (E) Signature 8 mutations acquired per autosomal genome per week in human XPCWT ASCs (n = 3) and an XPCKO ASC (n = 1) in vitro.
NER-deficient human breast tumors show higher levels of Signature 8 mutations
To identify whether NER-deficiency can be linked to an increase in Signature 8 mutations in human cancer as well, we looked into publicly available whole-genome sequencing data of 344 breast tumors (Nik-Zainal et al. 2016). Approximately 70% of these tumors have accumulated Signature 8 mutations (Nik-Zainal et al. 2016). NER-status was predicted by assessing the presence of protein-coding mutations and the copy number status of 66 NER-genes (Pearl et al. 2015). We identified 27 NER-deficient samples, 43 NER-proficient samples, and 274 with obscure NER-status.
NER-proficient and NER-deficient breast cancers have accumulated a median of 3399 base substitutions (mean 3968, standard deviation 2708) and 4368 base substitutions (mean 6405, standard deviation 6666) per sample, respectively (Supplemental Fig. S11A). To characterize whether NER-status affects the accumulation of Signature 8 mutations (https://cancer.sanger.ac.uk/cosmic/signatures_v2), 96-channel mutational profiles of the somatic mutations were generated for all 344 breast tumors and the contribution of the top 18 contributing COSMIC mutational signature was assessed (Fig. 5A; Methods). In line with previous observations, NER-deficient tumors have acquired ∼208 additional Signature 8 mutations in comparison to NER-proficient tumors (P = 0.02, Wilcoxon rank-sum test) (Fig. 5B). Furthermore, Signature 8 has the largest effect size of all 18 COSMIC mutational signatures (Supplemental Fig. S11B).
Figure 5.

Mutation accumulation in predicted NER-deficient and NER-proficient breast cancer whole-genomes. (A) Relative contribution of each indicated context-dependent base substitution type to the average 96-channel mutational profiles of NER-deficient and NER-proficient breast cancer samples. (B) Number of Signature 8 mutations in NER-deficient and NER-proficient breast cancer whole-genomes (n = 27 and n = 43, respectively). Asterisk indicates significant difference (P < 0.05, Wilcoxon rank-sum test).
Discussion
We deleted specific NER components in an otherwise normal genetic background, providing us with the unique opportunity to directly characterize the mutational consequences of NER-deficiency. Our results show that loss of ERCC1 induces a significant increase in the accumulation of base substitutions, specifically in liver ASCs, which coincides with the pathological aging phenotype observed in the liver of Ercc1−/Δ mice (Dollé et al. 2011; Gregg et al. 2012). Liver ASCs might be more dependent on DNA repair facilitated by ERCC1 compared with small intestinal ASCs, e.g., as a result of tissue-specific mutagen exposure. In line with this, WT liver ASCs show a higher basal expression of Ercc1 and other NER genes compared with WT small intestinal ASCs. Alternatively, liver ASCs might cope differently with unrepaired DNA damage as a result of loss of ERCC1, such as the utilization of alternative DNA repair mechanisms to bypass polymerase-blocking lesions or differential induction of apoptosis or senescence.
ERCC1 is involved in multiple DNA repair pathways, including TC-NER, GG-NER, SSA, and crosslink repair (Al-Minawi et al. 2008; Rahn et al. 2010). Previously, it has been shown that SSA- and crosslink repair-deficiencies result in an increased number of indels and SVs in mice, whereas NER-deficiency introduces base substitutions (Dollé et al. 2006). Since we only observe an increase in base substitutions and Ercc1−/Δ and WT mice show a similar depletion of base substitutions in genes, the observed mutational consequences of impaired ERCC1 are most likely an effect of defective GG-NER. In line with this, we show that GG-NER-deficiency can also induce an increase in the number of base substitutions in a human small intestinal organoid culture deleted for GG-NER component XPC. More specifically, the increased base substitution load can be largely explained by an increased contribution of Signature 8 in both systems. In line with these observations, a mutational signature similar to Signature 8 has been shown to increase with age in neurons of NER-deficient patients (Lodato et al. 2018).
Until now, the etiology of Signature 8 was unknown (https://cancer.sanger.ac.uk/cosmic/signatures_v2). As Signature 8 mutations are also detected in healthy human and mouse ASCs (Figs. 3C, 4E), this signature most likely represents a mutagenic process that is generally active in normal cells and not completely error-free repaired. Signature 8 is characterized by C:G > A:T mutations and is associated with double base substitutions, particularly CC:GG > AA:TT double base substitutions (Alexandrov et al. 2013; Nik-Zainal et al. 2016). C:G > A:T mutations have been linked to several processes, including oxidative stress (Kamiya et al. 1995; Degtyareva et al. 2013). NER has been suggested to play a role in the repair of tandem DNA lesions that result from oxidative stress (Bergeron et al. 2010; Cadet et al. 2012). If left unrepaired, these lesions can block regular DNA polymerases but can be bypassed by error-prone TLS polymerases, resulting in increased incorporation of tandem mutations (Cadet et al. 2012). Moreover, it has been shown that oxidative stress results in increased induction of double base substitutions in NER-deficient human fibroblasts (Lee 2002). In line with this, we observe a significant increase in the double base substitution load in mouse liver ASCs and a similar trend in the human ASC culture as a result of NER-deficiency, although the number of double base substitutions is low (n = 0–23 per ASC). Thus, Signature 8 could reflect oxidative DNA damage bypassed by TLS in the absence of NER. Of note, we did not observe an enrichment for CC:GG > AA:TT double base substitutions such as described for Signature 8 (Nik-Zainal et al. 2016), which may reflect different activity of DNA damage processes and/or additional repair deficiencies in human tumors but could also be due to the low number of double base substitutions in our data set.
We did not observe a high contribution of signatures that have been previously observed in liver cancer in ASCs of Ercc1−/Δ livers (https://cancer.sanger.ac.uk/cosmic/signatures_v2) (Supplemental Fig. S6B). This finding suggests that the mutational processes that underlie these signatures are only active after oncogenic transformation or that mutagen exposure in liver cancer (progenitor) cells is different from in vivo mouse ASCs and in vitro human ASCs. Liver cancer-specific Signature 24, for example, is associated with aflatoxin intake (Huang et al. 2017), a substance to which our mice and organoids were not exposed. In addition, Signature 1 and Signature 5, which have been previously associated with age (Alexandrov et al. 2015; Blokzijl et al. 2016), did not have an increased contribution in the ASCs of progeroid Ercc1−/Δ mice. Finally, a high contribution of mutational Signature 5 has been linked to the presence of somatic mutations in ERCC2, a key factor in both TC-NER and GG-NER, in human urothelial cancer (Iyama and Wilson 2013; Kim et al. 2016). However, we did not observe an increase in Signature 5 contribution in the ASCs without ERCC1 or XPC. This discrepancy in mutational consequences could reflect various differences between these systems, such as different effects of the mutations on protein function, distinct roles of the proteins, or tumor- and/or tissue-specific activity of mutagenic damage and/or DNA repair processes.
The challenge of coupling mutational signatures to mutational processes based on genome sequencing data of tumors is illustrated by our analyses of the breast cancer genomes. As the number of mutations attributed to a signature typically increases at a higher mutational load and the mutational loads differ greatly between tumor types (Alexandrov et al. 2013), it is important to compare signature contributions between samples within a single tumor type. Our analyses show that genomes of NER-deficient breast cancer patients have an elevated number of Signature 8 mutations, which is in line with our observations in the ASCs.
Signature 8 is found in many tumor types, including medulloblastoma, bladder cancer, and bone cancer (Alexandrov et al. 2013, 2018). Furthermore, Signature 8 contributes to the mutational profile of the majority of breast cancer tumors (Alexandrov et al. 2013; Nik-Zainal et al. 2016). Our results show that, besides the mutational status of core NER genes, elevation of the number of Signature 8 mutations as compared to the average number of such mutations in a tumor type might contribute to future predictors of (GG-)NER-deficiency. It should be noted that the presence of Signature 8 mutations alone is not conclusive for NER status. Further optimization of mutational signature definitions may aid to fully discriminate NER-deficient from NER-proficient tumors. Furthermore, clinical studies will be required to demonstrate the added predictive value of Signature 8 for NER-deficiency detection and treatment response stratification.
Methods
Mouse and human tissue material
Ercc1−/Δ mice were generated and maintained as previously described (Vermeij et al. 2016). The tissues were harvested from three female Ercc1−/Δ mice and three female WT littermates at the age of 15 wk, which is the time at which Ercc1−/Δ mice generally start to die as a consequence of early aging pathologies (Vermeij et al. 2016). Experiments were performed in accordance with the Principles of Laboratory Animal Care and within the guidelines approved by the Dutch Ethical Committee in full accordance with European legislation.
Human endoscopic biopsies were performed at the University Medical Center Utrecht and the Wilhelmina Children's Hospital. The patients’ informed consent was obtained, and this study was approved by the ethical committee of University Medical Center Utrecht.
Generation of clonal Ercc1−/Δ and WT mouse organoid cultures
Single liver ASCs were isolated from livers as described previously (Kuijk et al. 2016). Liver organoid cultures were initiated by culturing the liver ASCs in BME overlaid with mouse liver culture initiation medium (Supplemental Table S7). One and a half weeks after culture initiation, clonal organoid liver cultures were generated and expanded according to protocol (Jager et al. 2018) in mouse liver expansion medium (Supplemental Table S7). Crypts were isolated from small intestines as described previously (Sato et al. 2009). Small intestinal organoid cultures were initiated by culturing the small intestinal ASCs in Matrigel overlaid with mouse small intestine medium (Supplemental Table S7). Clonal small intestinal organoid cultures were generated by picking single organoids manually and clonally expanding these organoid cultures according to protocol in mouse small intestine medium (Jager et al. 2018). Culture expansion failed for the small intestine of mouse WT1.
Generation of a clonal and subclonal XPCKO organoid culture
Clonal XPCKO organoid cultures were generated from a small intestinal bulk organoid culture derived previously (Blokzijl et al. 2016) using the CRISPR-Cas9 gene-editing technique as described in Drost et al. (2017). One clonal human XPCKO organoid culture was obtained and cultured for 72 d in human small intestinal organoid medium (Supplemental Table S7). Subsequently, we derived subclonal cultures of single ASCs and expanded these until sufficient DNA could be isolated for WGS. This approach allowed us to catalog the mutations that specifically accumulated between the two clonal expansion steps in the absence of XPC (Supplemental Fig. S10A; Blokzijl et al. 2016; Drost et al. 2017; Jager et al. 2018). As a control, WGS data of three previously established XPCWT organoid cultures of the same human donor (donor 6) (downloaded from https://wgs11.op.umcutrecht.nl/mutational_patterns_ASCs/) were used (Blokzijl et al. 2016).
RNA sequencing and differential expression analysis of Ercc1−/Δ and WT mouse organoid cultures
For each mouse (three Ercc1−/Δ mice and three WT littermates), we performed RNA sequencing on one clonal organoid culture from the liver and the small intestine. An additional small intestinal organoid clone was sequenced of mice WT2 and WT3 to increase the amount of replicates for differential expression analysis, as culture expansion failed for the small intestine of WT1. Details on the standard procedures of RNA isolation, library preparation, sequencing, and data (pre)processing can be found in the Supplemental Methods.
A DESeq nbinomTest was used to test for differential expression (1) of Ercc1 between Ercc1−/Δ and WT liver ASCs, (2) of Ercc1 between Ercc1−/Δ and WT small intestinal ASCs, (3) of 83 other DNA repair genes (Casorelli et al. 2006) between Ercc1−/Δ and WT liver ASCs and (4) between Ercc1−/Δ and WT small intestinal ASCs, and (5) of nine NER genes between the WT liver and WT small intestinal ASCs (Anders and Huber 2010). Differentially expressed genes with q < 0.05 (Benjamini-Hochberg FDR multiple-testing correction) were considered significant (Benjamini and Hochberg 1995).
WGS and read alignment
Details on the standard procedures of DNA isolation, library preparation, sequencing, and data (pre)processing can be found in the Supplemental Methods. The WGS data of the tails confirmed that the Ercc1−/Δ mice have compound heterozygous mutations in Ercc1 and the WT littermates do not (Supplemental Fig. S12).
Variant calling and base substitution filtering
For both human and mouse samples, base substitutions and indels were multi-sample-called with GATK HaplotypeCaller v3.4.46 (Van der Auwera et al. 2013; see Supplemental Methods for settings). The callable genome was determined for all sequenced samples as previously described (Jager et al. 2018; see Supplemental Material for details on this procedure). Approximately 90 ± 1% of the autosomal genome was surveyed in every mouse clone (Supplemental Table S2), and 73%–88% of the autosomal genome was surveyed in each human subclone (Supplemental Table S5).
To obtain high-quality catalogs of somatic base substitutions, we applied a comprehensive filtering procedure on the basis of several quality parameters using SNVFI (https://github.com/UMCUGenetics/SNVFI) (Supplemental Methods; Supplemental Code). For the mouse samples, we also excluded variants with any evidence in another organoid sample or control (tail) sample of the same mouse to remove germline variants. Finally, we excluded positions with a variant allele frequency (VAF) < 0.3 in the organoid sample to exclude mutations that were induced after the clonal step and remaining noise. In total, 4130 out of 4238 remaining base substitutions of the mouse samples (97.5%) were confirmed using an independent sequencing approach (Supplemental Data S2; Supplemental Methods).
To remove germline variants in the human samples, all variants with evidence in the control (blood) sample were excluded for both the clonal and subclonal organoid cultures. Subsequently, for both the clonal and subclonal cultures, all variants with a VAF < 0.3 were excluded. Finally, the resulting somatic base substitution catalogs of the clonal and subclonal cultures were compared and all events unique to the subclonal organoid were considered to be accumulated after the XPC deletion, that is, between the two sequential clonal expansion steps.
Clonality of organoid cultures
We validated whether the organoid samples were clonal based on the VAF of somatic base substitutions, before the final filter step (VAF < 0.3). Each cell acquires its own set of somatic mutations and the reads supporting a mutation will be diluted in the WGS data of nonclonal samples, resulting in a low VAF. After extensive filtering of somatic base substitutions, liver organoid samples from WT1, WT2, and Ercc1−/Δ2 showed a shift in the VAF-peak away from 0.5, and therefore these samples were excluded from further analyses (Supplemental Fig. S13). An additional liver organoid culture from these mice was sequenced, and these samples were confirmed to be clonal (Supplemental Fig. S13).
Double base substitutions
We selected base substitutions from the filtered variant call format (VCF) files that were called on consecutive bases in the mouse or human reference genome. The double base substitutions were subsequently manually checked in the Integrative Genomics Viewer (IGV) (Robinson et al. 2011) to exclude double base substitutions present in the control sample and/or with many base substitutions or indels in the region, as these are (likely) false-positives.
Indel filtering of Ercc1−/Δ and WT mouse organoid cultures
We only considered positions on the autosomal genome that were callable and had a sequencing depth of ≥20X in both the organoid sample and the control (tail) sample. We excluded positions that overlap with a base substitution. Furthermore, we only considered positions with a filter “PASS” from VariantFiltration, a GATK phred-scaled quality score >250 and a sample-level genotype quality of 99 in both the organoid sample and the control (tail) sample. We subsequently excluded indels that are located within 50 base pairs of an indel called in another organoid sample and indels with any evidence in another organoid sample or a control (tail) sample. Finally, we excluded positions with a VAF < 0.3 in the organoid sample.
SV calling and filtering of Ercc1−/Δ and WT mouse organoid cultures
SVs were called with DELLY v0.7.2 with settings “type DEL DUP INV TRA INS”, “map-qual 1”, “mad-cutoff 9”, “min-flank 13”, and “geno-qual 5” (Rausch et al. 2012). We only considered SVs of at least 100 bp on the autosomal chromosomes that were called with a filter “PASS” and a sample-specific genotype quality of at least 90 in the organoid culture and the control sample. We subsequently excluded positions with any evidence in the control (tail) sample. The filtered SVs were finally checked manually in IGV to reduce false-positives, and we excluded SVs present in the tail sample, with no visible change in the read-depth (for duplications and deletions), and/or with many base substitutions in the region.
Genome-wide copy number profiles of Ercc1−/Δ and WT mouse organoid cultures
To generate a virtual karyotype, genome-wide copy number states were determined using Control-FREEC v7.2 with settings “ploidy 2”, “window 1000”, and “telocentromeric 50,000” (Boeva et al. 2012). Subsequently, the average copy number across bins of 500,000 bp was calculated and plotted to assess genome stability.
Base substitution types
We retrieved the base substitution types from all the filtered VCF files, converted them to the six types of base substitutions that are distinguished by convention, and generated a mutation spectrum (the C > T changes at NpCpG sites are considered separately from C > T changes at other sites) for the four ASC groups (Ercc1−/Δ liver, Ercc1−/Δ small intestine, WT liver, and WT small intestine), as well as XPCKO, XPCWT1, XPCWT2, and XPCWT3 ASCs. χ2 tests were performed to determine whether the mutation spectra differ significantly between (1) mouse WT and Ercc1−/Δ liver ASCs, and (2) mouse WT and Ercc1−/Δ small intestinal ASCs. P-values were corrected for multiple testing using Benjamini-Hochberg FDR correction, and differences in mutation rates between Ercc1−/Δ and WT mouse ASCs with q < 0.05 were considered significant. We also retrieved the sequence context for all base substitutions to generate the 96-channel mutational profiles for each assessed ASC. In addition, we generated mutation spectra and 96-channel mutational profiles of base substitutions with a VAF < 0.3 that likely represent subclonal mutations or noise (Supplemental Fig. S14). Of note, the NER-deficiency will likely also affect in vitro mutation accumulation during culturing in the mutant organoids.
The centroid of the 96-channel mutational profiles of mutations with a VAF ≥ 0.3 was calculated per mouse ASC group. Pairwise cosine similarities of all 96-channel mutational profiles and of all centroids were computed. We also calculated the cosine similarities of the 96-channel mutational profiles and centroids with all 30 COSMIC mutational signatures (https://cancer.sanger.ac.uk/cosmic/signatures_v2) (Supplemental Fig. S7). These analyses were performed with the R package MutationalPatterns (Blokzijl et al. 2018).
De novo mutational signature extraction
We extracted two signatures using nonnegative matrix factorization (NMF) from the 96-channel mutational profiles of the mouse ASCs. Although the number of base substitutions is low for this dimension reduction approach, it does provide an unbiased method to characterize the mutational processes that have been active in the ASCs. Subsequently, we computed the absolute contribution of these de novo extracted signatures to the centroids of the mouse ASC groups. We also calculated the cosine similarity of these two mutational signatures to the 30 COSMIC mutational signatures (https://cancer.sanger.ac.uk/cosmic/signatures_v2) and to the 96-channel centroid of six small intestinal ASCs from two old mice that was published previously (Behjati et al. 2014). These analyses were performed with MutationalPatterns (Blokzijl et al. 2018).
Quantification of the contribution of COSMIC mutational signatures to the 96-channel mutational profiles
We estimated the contribution of the 30 COSMIC mutational signatures (https://cancer.sanger.ac.uk/cosmic/signatures_v2) to the centroids of each mouse ASC group and to the 96-channel mutational profiles of the human organoids using MutationalPatterns (Supplemental Figs. S6B, S10B; Blokzijl et al. 2018). We ranked the COSMIC signatures based on the total contribution of these signatures to the centroids of the mouse samples. Next, we iteratively reconstructed the centroids of the ASC groups, first using the top two COSMIC signatures, and in each iteration the next COSMIC signature was included until all 30 signatures were used. The cosine similarity was calculated between the original and the reconstructed centroid for each mouse ASC group (Supplemental Fig. S6A). As expected, the addition of more signatures increases the similarity of the reconstructed centroids with the original centroids, but after 10 COSMIC signatures, the cosine similarities plateau (Supplemental Fig. S6A). Therefore, we used the signature contribution with this subset of 10 COSMIC signatures to the centroids of the four ASC groups (Fig. 3B,C).
We have also performed the mutational signature analyses with the 60 SBS COSMIC mutational signatures (v3, https://cancer.sanger.ac.uk/cosmic/signatures/SBS/) (Alexandrov et al. 2018). We observe a similar increase in mutations of this latest version of Signature 8 (Supplemental Fig. S15). However, as the manuscript of COSMIC signatures v3 has not been published yet, these SBS signatures may be subject to change.
Determination of the statistical significance of differences in signature contributions
A bootstrap resampling—similar to that performed in Zou et al. (2018)—was applied to generate 7000 replicas of the 96-channel mutational profile of each WT liver ASC (n = 3), which yielded 21,000 WT liver replicas in total. Subsequently, three replicas were randomly selected and the relative contribution of 30 COSMIC signatures was determined for their centroid. Euclidean distance dWT was calculated between the relative signature contributions of the replicas centroid and that of the original centroid. This was repeated 10,000 times to construct a distribution of dWT (Supplemental Fig. S6C). Next, the threshold distance with P-value = 0.05, dWT_0.05, was identified. The same approach was taken to generate 7000 replicas of each Ercc1−/Δ (MUT) liver ASC (n = 3) and construct a distribution of dMUT (Supplemental Fig. S6C). The Euclidean distance d between the relative signature contributions of the original WT and Ercc1−/Δ liver centroids was considered to be significantly different when d > dMUT and d > dWT. Similarly, bootstrap distributions were generated for WT and Ercc1−/Δ (MUT) small intestine (Supplemental Fig. S6D), with the exception that, for the generation of the dMUT distribution, only two replicas were randomly selected in each permutation, as there are only two WT small intestinal ASC samples in the original set. Finally, we repeated the same analyses for the relative contributions of the subset of 10 COSMIC signatures for both liver (Supplemental Fig. S6E) and small intestine (Supplemental Fig. S6F).
Genomic distribution of base substitutions
To test whether the base substitutions appear more or less frequently than expected in genes, promoters, promoter-flanking, and enhancer regions, we loaded the UCSC Known Genes tables as TxDb objects for mm10 and hg19 and the regulatory features for mm10 and hg19 from Ensembl using biomaRt (Durinck et al. 2005, 2009). We tested for enrichment or depletion of base substitutions in the genomic regions per ASC group (Ercc1−/Δ liver, Ercc1−/Δ small intestine, WT liver, WT small intestine, XPCKO, and XPCWT) using a one-sided binomial test with MutationalPatterns (Blokzijl et al. 2018), which corrects for the surveyed genomic areas (Supplemental Figs. S9A, S10C). Two-sided Poisson tests were performed to test for significant differences in the ratio of base substitutions within a genomic region divided by the total number of base substitutions between (1) mouse WT and Ercc1−/Δ liver ASCs and (2) mouse WT and Ercc1−/Δ small intestinal ASCs (Supplemental Fig. S9A). Differences in mutation rates with q < 0.05 (Benjamini-Hochberg FDR multiple-testing correction) were considered significant.
To test whether base substitutions occur more frequently in more highly expressed genes in the NER-deficient mouse ASCs, we first selected base substitutions that occurred within genes in the mouse ASCs. Per ASC group, we next determined the average reads per kilobase per million (RPKM) mapped reads of these genes. Two-sided t-tests were performed to test for significant difference in the average expression of genes that carry a somatic mutation between (1) mouse WT and Ercc1−/Δ liver ASCs and (2) mouse WT and Ercc1−/Δ small intestinal ASCs (Supplemental Fig. S9B). Differences in gene expression distributions with q < 0.05 (Benjamini-Hochberg FDR multiple-testing correction) were considered significant.
Transcriptional strand bias of base substitutions
For the base substitutions within genes, we determined whether the mutations are located on the transcribed or the nontranscribed strand. To this end, we determined whether the mutated “C” or “T” base is on the same strand as the gene definition, which is untranscribed, or the opposite strand, which is transcribed. We generated a 192-channel mutational profile per ASC group with the relative contribution of each mutation type with separate bars for the mutations on the transcribed and untranscribed strand and calculated the significance of the strand bias using a two-sided Poisson test with MutationalPatterns (Supplemental Figs. S9C, S10D; Blokzijl et al. 2018). Furthermore, we performed two-sided Poisson tests to test whether there is a significant difference in strand bias per mutation type between (1) mouse WT and Ercc1−/Δ liver ASCs and (2) mouse WT and Ercc1−/Δ small intestinal ASCs (Supplemental Fig. S9C). Differences in strand bias with an adjusted P-value q < 0.05 (Benjamini–Hochberg FDR multiple-testing correction) were considered significant.
Comparison of mutation rates
Two-tailed t-tests were performed to determine whether the mutation rates (see Supplemental Methods) differ significantly between (1) mouse WT and Ercc1−/Δ liver ASCs and (2) mouse WT and Ercc1−/Δ small intestinal ASCs. Of note, these tests assume that the data is normally distributed. Differences in mutation rates between Ercc1−/Δ and WT mouse ASCs with q < 0.05 (Benjamini-Hochberg FDR multiple-testing correction) were considered significant.
Analysis of mutational patterns and signatures in breast cancer whole-genome sequences
In the analysis, we included 344 breast cancer samples with publicly available SNV, indels, and CNV calls obtained from tumor-normal sample pairs (Nik-Zainal et al. 2016). Samples with a biallelic inactivation (biallelic deletion, biallelic nonsense, splice site, nonsynonymous mutation, or frameshift indel, or two or more independent mutations of these types) of at least one NER-related gene (66 genes; GTF2H5 was excluded because of missing CNV calls) (Pearl et al. 2015) are considered as NER-deficient. Samples with no copy number depletions and no variants other than intronic SNVs and indels in any of the 66 NER-related genes are considered as NER-proficient. The remaining 274 samples (∼80%) carried a single mutation in a NER-gene, and since the other copy might be inactivated through, e.g., epigenetic silencing, the NER functionality is unknown and therefore these samples were excluded from the analysis.
The number of base substitutions was extracted from each VCF file and a Wilcoxon rank-sum test was performed to determine whether the number of base substitutions is different between NER-proficient and NER-deficient samples. The 96-channel mutational profile of each sample was generated as described in the subsection “Base substitution types.” Subsequently, the 96-channel mutational profile of each sample was reconstructed using the 30 mutational signatures from COSMIC, as described in the subsection “Quantification of the contribution of COSMIC mutational signatures to the 96-channel mutational profiles.” Signatures with a contribution of <10% in all 344 samples were excluded (signatures 4, 7, 10, 11, 14, 15, 22–25, 27, 28), and the 96-channel mutational profiles were finally reconstructed using the remaining 18 signatures. The cosine similarity between the observed 96-channel mutational profile and the reconstructed profile was >0.95 for all samples, which indicates a very good fit of the signatures.
Based on this, the number of mutations per signature was estimated for each sample. Then, for each signature, the number of mutations was compared between the NER-deficient and NER-proficient samples using the median difference (the median of all pairwise differences between NER-deficient and NER-proficient samples). A Wilcoxon rank-sum test was performed to determine whether the number of Signature 8 mutations differs significantly between NER-deficient and NER-proficient breast tumors. Signature 20 is excluded from the analysis because none of the NER-deficient or NER-proficient samples have a contribution of Signature 20.
Data access
The raw sequencing data of the mouse samples generated in this study have been submitted to the European Nucleotide Archive (ENA; https://www.ebi.ac.uk/ena) under accession number ERP021379. The raw sequencing data of the human samples generated in this study have been submitted to the European Genome-phenome Archive (EPA; https://www.ebi.ac.uk/ega/home) under accession number EGAS00001002681. The filtered VCF files of the samples generated in this study have been submitted to Zenodo under DOI 10.5281/zenodo.2628460. The VCF files of base substitutions with a low VAF in the mouse samples have been submitted to Zenodo under DOI 10.5281/zenodo.2632952. All analysis scripts are available in the Supplemental Code File and on https://github.com/UMCUGenetics/NER-deficiency.git, https://github.com/UMCUGenetics/SNVFI or https://github.com/johannabertl/BRCA_DNA_repair.
Supplementary Material
Acknowledgments
We thank the animal caretakers of the Erasmus MC for taking care of the mice and the Utrecht Sequencing Facility for providing the sequencing service and data. The Utrecht Sequencing Facility is subsidized by the University Medical Center Utrecht, Hubrecht Institute, and Utrecht University. This study was financially supported by the NWO Zwaartekracht program Cancer Genomics.nl and a VIDI grant of the Netherlands Organisation for Scientific Research (NWO) (no. 016.Vidi.171.023) to R.v.B.
Author contributions: M.J., E.K., M.V., N.B., and R.v.B. performed organoid culturing. N.B. and R.v.B. generated western blots and sequenced the organoid cultures. M.J., F.B., J.B., R.J., S.B., J.d.L., and R.v.B. performed bioinformatic analyses. M.J., F.B., E.K., J.S.P., J.H., J.P., R.v.B., and E.C. were involved in the conceptual design of this study. M.J., F.B., R.v.B., and E.C. wrote the manuscript.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.246223.118.
Freely available online through the Genome Research Open Access option.
References
- Aboussekhra A, Biggerstaff M, Shivji MK, Vilpo JA, Moncollin V, Podust VN, Protić M, Hübscher U, Egly JM, Wood RD. 1995. Mammalian DNA nucleotide excision repair reconstituted with purified protein components. Cell 80: 859–868. 10.1016/0092-8674(95)90289-9 [DOI] [PubMed] [Google Scholar]
- Adams PD, Jasper H, Lenhard Rudolph K. 2015. Aging-induced stem cell mutations as drivers for disease and cancer. Cell Stem Cell 16: 601–612. 10.1016/j.stem.2015.05.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SAJR, Behjati S, Biankin AV, Bignell GR, Bolli N, Borg A, Børresen-Dale A-L, et al. 2013. Signatures of mutational processes in human cancer. Nature 500: 415–421. 10.1038/nature12477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexandrov LB, Jones PH, Wedge DC, Sale JE, Campbell PJ, Nik-Zainal S, Stratton MR. 2015. Clock-like mutational processes in human somatic cells. Nat Genet 47: 1402–1407. 10.1038/ng.3441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexandrov L, Kim J, Haradhvala NJ, Huang MN, Ng AWT, Boot A, Covington KR, Gordenin DA, Bergstrom E, Lopez-Bigas N, et al. 2018. The repertoire of mutational signatures in human cancer. bioRxiv 10.1101/322859 [DOI]
- Al-Minawi AZ, Saleh-Gohari N, Helleday T. 2008. The ERCC1/XPF endonuclease is required for efficient single-strand annealing and gene conversion in mammalian cells. Nucleic Acids Res 36: 1–9. 10.1093/nar/gkm888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders S, Huber W. 2010. Differential expression analysis for sequence count data. Genome Biol 11: R106 10.1186/gb-2010-11-10-r106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barker N, Ridgway RA, van Es JH, van de Wetering M, Begthel H, van den Born M, Danenberg E, Clarke AR, Sansom OJ, Clevers H. 2009. Crypt stem cells as the cells-of-origin of intestinal cancer. Nature 457: 608–611. 10.1038/nature07602 [DOI] [PubMed] [Google Scholar]
- Behjati S, Huch M, van Boxtel R, Karthaus W, Wedge DC, Tamuri AU, Martincorena I, Petljak M, Alexandrov LB, Gundem G, et al. 2014. Genome sequencing of normal cells reveals developmental lineages and mutational processes. Nature 513: 422–425. 10.1038/nature13448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamini Y, Hochberg Y. 1995. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc Ser B (Methodol) 57: 289–300. 10.1111/j.2517-6161.1995.tb02031.x [DOI] [Google Scholar]
- Bergeron F, Auvré F, Radicella JP, Ravanat J-L. 2010. HO• radicals induce an unexpected high proportion of tandem base lesions refractory to repair by DNA glycosylases. Proc Natl Acad Sci 107: 5528–5533. 10.1073/pnas.1000193107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blokzijl F, de Ligt J, Jager M, Sasselli V, Roerink S, Sasaki N, Huch M, Boymans S, Kuijk E, Prins P, et al. 2016. Tissue-specific mutation accumulation in human adult stem cells during life. Nature 538: 260–264. 10.1038/nature19768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blokzijl F, Janssen R, van Boxtel R, Cuppen E. 2018. MutationalPatterns: comprehensive genome-wide analysis of mutational processes. Genome Med 10: 33 10.1186/s13073-018-0539-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boeva V, Popova T, Bleakley K, Chiche P, Cappo J, Schleiermacher G, Janoueix-Lerosey I, Delattre O, Barillot E. 2012. Control-FREEC: a tool for assessing copy number and allelic content using next-generation sequencing data. Bioinformatics 28: 423–425. 10.1093/bioinformatics/btr670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cadet J, Ravanat J-L, TavernaPorro M, Menoni H, Angelov D. 2012. Oxidatively generated complex DNA damage: tandem and clustered lesions. Cancer Lett 327: 5–15. 10.1016/j.canlet.2012.04.005 [DOI] [PubMed] [Google Scholar]
- Casorelli I, Tenedini E, Tagliafico E, Blasi MF, Giuliani A, Crescenzi M, Pelosi E, Testa U, Peschle C, Mele L, et al. 2006. Identification of a molecular signature for leukemic promyelocytes and their normal counterparts: focus on DNA repair genes. Leukemia 20: 1978–1988. 10.1038/sj.leu.2404376 [DOI] [PubMed] [Google Scholar]
- Davies H, Glodzik D, Morganella S, Yates LR, Staaf J, Zou X, Ramakrishna M, Martin S, Boyault S, Sieuwerts AM, et al. 2017. HRDetect is a predictor of BRCA1 and BRCA2 deficiency based on mutational signatures. Nat Med 23: 517–525. 10.1038/nm.4292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Degtyareva NP, Heyburn L, Sterling J, Resnick MA, Gordenin DA, Doetsch PW. 2013. Oxidative stress-induced mutagenesis in single-strand DNA occurs primarily at cytosines and is DNA polymerase ζ-dependent only for adenines and guanines. Nucleic Acids Res 41: 8995–9005. 10.1093/nar/gkt671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Laat W. 1998. Mapping of interaction domains between human repair proteins ERCC1 and XPF. Nucleic Acids Res 26: 4146–4152. 10.1093/nar/26.18.4146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dollé MET, Busuttil RA, Garcia AM, Wijnhoven S, van Drunen E, Niedernhofer LJ, van der Horst G, Hoeijmakers JHJ, van Steeg H, Vijg J. 2006. Increased genomic instability is not a prerequisite for shortened lifespan in DNA repair deficient mice. Mutat Res 596: 22–35. 10.1016/j.mrfmmm.2005.11.008 [DOI] [PubMed] [Google Scholar]
- Dollé MT, Kuiper R, Roodbergen M, Robinson J, de Vlugt S, Wijnhoven SP, Beems RB, de la Fonteyne L, de With P, van der Pluijm I, et al. 2011. Broad segmental progeroid changes in short-lived Ercc1−/Δ7 mice. Pathobiol Aging Age Relat Dis 1: 7219 10.3402/pba.v1i0.7219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drost J, van Boxtel R, Blokzijl F, Mizutani T, Sasaki N, Sasselli V, de Ligt J, Behjati S, Grolleman JE, van Wezel T, et al. 2017. Use of CRISPR-modified human stem cell organoids to study the origin of mutational signatures in cancer. Science 358: 234–238. 10.1126/science.aao3130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dupuy A, Sarasin A. 2015. DNA damage and gene therapy of xeroderma pigmentosum, a human DNA repair-deficient disease. Mutat Res 776: 2–8. 10.1016/j.mrfmmm.2014.08.007 [DOI] [PubMed] [Google Scholar]
- Durinck S, Moreau Y, Kasprzyk A, Davis S, De Moor B, Brazma A, Huber W. 2005. BioMart and Bioconductor: a powerful link between biological databases and microarray data analysis. Bioinformatics 21: 3439–3440. 10.1093/bioinformatics/bti525 [DOI] [PubMed] [Google Scholar]
- Durinck S, Spellman PT, Birney E, Huber W. 2009. Mapping identifiers for the integration of genomic datasets with the R/Bioconductor package biomaRt. Nat Protoc 4: 1184–1191. 10.1038/nprot.2009.97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregg SQ, Gutiérrez V, Robinson AR, Woodell T, Nakao A, Ross MA, Michalopoulos GK, Rigatti L, Rothermel CE, Kamileri I, et al. 2012. A mouse model of accelerated liver aging caused by a defect in DNA repair. Hepatology 55: 609–621. 10.1002/hep.24713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoeijmakers JHJ. 2009. DNA damage, aging, and cancer. N Engl J Med 361: 1475–1485. 10.1056/NEJMra0804615 [DOI] [PubMed] [Google Scholar]
- Huang MN, Yu W, Teoh WW, Ardin M, Jusakul A, Ng AWT, Boot A, Abedi-Ardekani B, Villar S, Myint SS, et al. 2017. Genome-scale mutational signatures of aflatoxin in cells, mice, and human tumors. Genome Res 27: 1475–1486. 10.1101/gr.220038.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huch M, Gehart H, van Boxtel R, Hamer K, Blokzijl F, Verstegen MMA, Ellis E, van Wenum M, Fuchs SA, de Ligt J, et al. 2015. Long-term culture of genome-stable bipotent stem cells from adult human liver. Cell 160: 299–312. 10.1016/j.cell.2014.11.050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iyama T, Wilson DM III. 2013. DNA repair mechanisms in dividing and non-dividing cells. DNA Repair 12: 620–636. 10.1016/j.dnarep.2013.04.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jager M, Blokzijl F, Sasselli V, Boymans S, Besselink N, Janssen R, Clevers H, van Boxtel R, Cuppen E. 2018. Measuring mutation accumulation in single human adult stem cells by whole-genome sequencing of organoid cultures. Nat Protoc 13: 59–78. 10.1038/nprot.2017.111 [DOI] [PubMed] [Google Scholar]
- Kamiya H, Murata-Kamiya N, Koizume S, Inoue H, Nishimura S, Ohtsuka E. 1995. 8-Hydroxyguanine (7,8-dihydro-8-oxoguanine) in hot spots of the c-Ha-ras gene: effects of sequence contexts on mutation spectra. Carcinogenesis 16: 883–889. 10.1093/carcin/16.4.883 [DOI] [PubMed] [Google Scholar]
- Kim J, Mouw KW, Polak P, Braunstein LZ, Kamburov A, Kwiatkowski DJ, Rosenberg JE, Van Allen EM, D'Andrea A, Getz G. 2016. Somatic ERCC2 mutations are associated with a distinct genomic signature in urothelial tumors. Nat Genet 48: 600–606. 10.1038/ng.3557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirschner K, Melton DW. 2010. Multiple roles of the ERCC1-XPF endonuclease in DNA repair and resistance to anticancer drugs. Anticancer Res 30: 3223–3232. [PubMed] [Google Scholar]
- Kuijk EW, Rasmussen S, Blokzijl F, Huch M, Gehart H, Toonen P, Begthel H, Clevers H, Geurts AM, Cuppen E. 2016. Generation and characterization of rat liver stem cell lines and their engraftment in a rat model of liver failure. Sci Rep 6: 22154 10.1038/srep22154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee D-H. 2002. Oxidative DNA damage induced by copper and hydrogen peroxide promotes CG → TT tandem mutations at methylated CpG dinucleotides in nucleotide excision repair-deficient cells. Nucleic Acids Res 30: 3566–3573. 10.1093/nar/gkf478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lodato MA, Rodin RE, Bohrson CL, Coulter ME, Barton AR, Kwon M, Sherman MA, Vitzthum CM, Luquette LJ, Yandava C, et al. 2018. Aging and neurodegeneration are associated with increased mutations in single human neurons. Science 359: 555–559. 10.1126/science.aao4426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marteijn JA, Lans H, Vermeulen W, Hoeijmakers JHJ. 2014. Understanding nucleotide excision repair and its roles in cancer and ageing. Nat Rev Mol Cell Biol 15: 465–481. 10.1038/nrm3822 [DOI] [PubMed] [Google Scholar]
- Melis JPM, Wijnhoven SWP, Beems RB, Roodbergen M, van den Berg J, Moon H, Friedberg E, van der Horst GTJ, Hoeijmakers JHJ, Vijg J, et al. 2008. Mouse models for xeroderma pigmentosum group A and group C show divergent cancer phenotypes. Cancer Res 68: 1347–1353. 10.1158/0008-5472.CAN-07-6067 [DOI] [PubMed] [Google Scholar]
- Ni M, Zhang W-Z, Qiu J-R, Liu F, Li M, Zhang Y-J, Liu Q, Bai J. 2014. Association of ERCC1 and ERCC2 polymorphisms with colorectal cancer risk in a Chinese population. Sci Rep 4: 4112 10.1038/srep04112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niedernhofer LJ, Garinis GA, Raams A, Lalai AS, Robinson AR, Appeldoorn E, Odijk H, Oostendorp R, Ahmad A, van Leeuwen W, et al. 2006. A new progeroid syndrome reveals that genotoxic stress suppresses the somatotroph axis. Nature 444: 1038–1043. 10.1038/nature05456 [DOI] [PubMed] [Google Scholar]
- Nik-Zainal S, Davies H, Staaf J, Ramakrishna M, Glodzik D, Zou X, Martincorena I, Alexandrov LB, Martin S, Wedge DC, et al. 2016. Landscape of somatic mutations in 560 breast cancer whole-genome sequences. Nature 534: 47–54. 10.1038/nature17676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearl LH, Schierz AC, Ward SE, Al-Lazikani B, Pearl FMG. 2015. Therapeutic opportunities within the DNA damage response. Nat Rev Cancer 15: 166–180. 10.1038/nrc3891 [DOI] [PubMed] [Google Scholar]
- Perera D, Poulos RC, Shah A, Beck D, Pimanda JE, Wong JWH. 2016. Differential DNA repair underlies mutation hotspots at active promoters in cancer genomes. Nature 532: 259–263. 10.1038/nature17437 [DOI] [PubMed] [Google Scholar]
- Pleasance ED, Cheetham RK, Stephens PJ, McBride DJ, Humphray SJ, Greenman CD, Varela I, Lin M-L, Ordóñez GR, Bignell GR, et al. 2010. A comprehensive catalogue of somatic mutations from a human cancer genome. Nature 463: 191–196. 10.1038/nature08658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puumalainen M-R, Rüthemann P, Min J-H, Naegeli H. 2016. Xeroderma pigmentosum group C sensor: unprecedented recognition strategy and tight spatiotemporal regulation. Cell Mol Life Sci 73: 547–566. 10.1007/s00018-015-2075-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahn JJ, Adair GM, Nairn RS. 2010. Multiple roles of ERCC1-XPF in mammalian interstrand crosslink repair. Environ Mol Mutagen 51: 567–581. 10.1002/em.20583 [DOI] [PubMed] [Google Scholar]
- Rausch T, Zichner T, Schlattl A, Stütz AM, Benes V, Korbel JO. 2012. DELLY: structural variant discovery by integrated paired-end and split-read analysis. Bioinformatics 28: i333–i339. 10.1093/bioinformatics/bts378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson JT, Thorvaldsdóttir H, Winckler W, Guttman M, Lander ES, Getz G, Mesirov JP. 2011. Integrative Genomics Viewer. Nat Biotechnol 29: 24–26. 10.1038/nbt.1754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sands AT, Abuin A, Sanchez A, Conti CJ, Bradley A. 1995. High susceptibility to ultraviolet-induced carcinogenesis in mice lacking XPC. Nature 377: 162–165. 10.1038/377162a0 [DOI] [PubMed] [Google Scholar]
- Sato T, Vries RG, Snippert HJ, van de Wetering M, Barker N, Stange DE, van Es JH, Abo A, Kujala P, Peters PJ, et al. 2009. Single Lgr5 stem cells build cryptvillus structures in vitro without a mesenchymal niche. Nature 459: 262–265. 10.1038/nature07935 [DOI] [PubMed] [Google Scholar]
- Schuster-Böckler B, Lehner B. 2012. Chromatin organization is a major influence on regional mutation rates in human cancer cells. Nature 488: 504–507. 10.1038/nature11273 [DOI] [PubMed] [Google Scholar]
- Sijbers AM, de Laat WL, Ariza RR, Biggerstaff M, Wei Y-F, Moggs JG, Carter KC, Shell BK, Evans E, de Jong MC, et al. 1996a. Xeroderma pigmentosum group F caused by a defect in a structure-specific DNA repair endonuclease. Cell 86: 811–822. 10.1016/S0092-8674(00)80155-5 [DOI] [PubMed] [Google Scholar]
- Sijbers AM, van der Spek PJ, Odijk H, van den Berg J, van Duin M, Westerveld A, Jaspers NG, Bootsma D, Hoeijmakers JH. 1996b. Mutational analysis of the human nucleotide excision repair gene ERCC1. Nucleic Acids Res 24: 3370–3380. 10.1093/nar/24.17.3370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su Y, Orelli B, Madireddy A, Niedernhofer LJ, Schärer OD. 2012. Multiple DNA binding domains mediate the function of the ERCC1-XPF protein in nucleotide excision repair. J Biol Chem 287: 21846–21855. 10.1074/jbc.M111.337899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tripsianes K, Folkers G, Ab E, Das D, Odijk H, Jaspers NGJ, Hoeijmakers JHJ, Kaptein R, Boelens R. 2005. The structure of the human ERCC1/XPF interaction domains reveals a complementary role for the two proteins in nucleotide excision repair. Structure 13: 1849–1858. 10.1016/j.str.2005.08.014 [DOI] [PubMed] [Google Scholar]
- Van der Auwera GA, Carneiro MO, Hartl C, Poplin R, Del Angel G, Levy-Moonshine A, Jordan T, Shakir K, Roazen D, Thibault J, et al. 2013. From FastQ data to high confidence variant calls: the Genome Analysis Toolkit best practices pipeline. Curr Protoc Bioinformatics 43: 11.10.1–11.10.33. 10.1002/0471250953.bi1110s43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vermeij WP, Dollé MET, Reiling E, Jaarsma D, Payan-Gomez C, Bombardieri CR, Wu H, Roks AJM, Botter SM, van der Eerden BC, et al. 2016. Restricted diet delays accelerated ageing and genomic stress in DNA-repair-deficient mice. Nature 537: 427–431. 10.1038/nature19329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waddell N, Pajic M, Patch A-M, Chang DK, Kassahn KS, Bailey P, Johns AL, Miller D, Nones K, Quek K, et al. 2015. Whole genomes redefine the mutational landscape of pancreatic cancer. Nature 518: 495–501. 10.1038/nature14169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weeda G, Donker I, de Wit J, Morreau H, Janssens R, Vissers CJ, Nigg A, van Steeg H, Bootsma D, Hoeijmakers JHJ. 1997. Disruption of mouse ERCC1 results in a novel repair syndrome with growth failure, nuclear abnormalities and senescence. Curr Biol 7: 427–439. 10.1016/S0960-9822(06)00190-4 [DOI] [PubMed] [Google Scholar]
- Zheng CL, Wang NJ, Chung J, Moslehi H, Sanborn JZ, Hur JS, Collisson EA, Vemula SS, Naujokas A, Chiotti KE, et al. 2014. Transcription restores DNA repair to heterochromatin, determining regional mutation rates in cancer genomes. Cell Rep 9: 1228–1234. 10.1016/j.celrep.2014.10.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu L, Finkelstein D, Gao C, Shi L, Wang Y, López-Terrada D, Wang K, Utley S, Pounds S, Neale G, et al. 2016. Multi-organ mapping of cancer risk. Cell 166: 1132–1146.e7. 10.1016/j.cell.2016.07.045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou X, Owusu M, Harris R, Jackson SP, Loizou JI, Nik-Zainal S. 2018. Validating the concept of mutational signatures with isogenic cell models. Nat Commun 9: 1744 10.1038/s41467-018-04052-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.