

Abstract
A hallmark of meiosis is the rearrangement of parental alleles to ensure genetic diversity in the gametes. These chromosome rearrangements are mediated by the repair of programmed DNA double-strand breaks (DSBs) as genetic crossovers between parental homologs. In mice, humans, and many other mammals, meiotic DSBs occur primarily at hotspots, determined by sequence-specific binding of the PRDM9 protein. Without PRDM9, meiotic DSBs occur near gene promoters and other functional sites. Studies in a limited number of mouse strains showed that functional PRDM9 is required to complete meiosis, but despite its apparent importance, Prdm9 has been repeatedly lost across many animal lineages. Both the reason for mouse sterility in the absence of PRDM9 and the mechanism by which Prdm9 can be lost remain unclear. Here, we explore whether mice can tolerate the loss of Prdm9. By generating Prdm9 functional knockouts in an array of genetic backgrounds, we observe a wide range of fertility phenotypes and ultimately demonstrate that PRDM9 is not required for completion of male meiosis. Although DSBs still form at a common subset of functional sites in all mice lacking PRDM9, meiotic outcomes differ substantially. We speculate that DSBs at functional sites are difficult to repair as a crossover and that by increasing the efficiency of crossover formation at these sites, genetic modifiers of recombination rates can allow for meiotic progression. This model implies that species with a sufficiently high recombination rate may lose Prdm9 yet remain fertile.
Meiotic recombination is essential for the production of euploid gametes. Recombination requires the repair of programmed double-stranded DNA breaks (DSBs), and this, in turn, results in an interchange of genetic information between homologous chromosomes (Bolcun-Filas and Schimenti 2012). In mice, humans, and several other mammals, the positions of recombination-initiating DSBs are primarily determined by DNA sequence-specific binding of the meiosis-specific histone methyltransferase PRDM9 (Baudat et al. 2010; Myers et al. 2010; Parvanov et al. 2010; Grey et al. 2018). All evidence to date has suggested that PRDM9 is essential for meiotic progression in mice because knocking out or disrupting the Prdm9 gene results in extensive asynapsis of homologous chromosomes and in complete meiotic arrest (Hayashi et al. 2005; Fairfield et al. 2011; Flachs et al. 2012; Weiss et al. 2012; Sun et al. 2015; Imai et al. 2017; Diagouraga et al. 2018; Thibault-Sennett et al. 2018). Nonetheless, dogs, swordtail fish, and even a human female (Muñoz-Fuentes et al. 2011; Narasimhan et al. 2016; Baker et al. 2017) have lost functional Prdm9, yet remain fertile. Thus, the question of why PRDM9 is important for meiotic recombination in mice is unresolved.
In species naturally lacking PRDM9, and in mice in which PRDM9 has been disrupted, meiotic recombination occurs at sites of non-PRDM9-mediated histone 3 lysine 4 trimethylation (H3K4me3). This includes gene promoters and other functional sites. It has been proposed that DSB repair as a crossover is inefficient at functional sites; thus, by sequestering the DSB-repair machinery, PRDM9 ensures fertility (Brick et al. 2012). However, this role is difficult to reconcile with the fertility of dogs and other species in which Prdm9 is absent. Phenotypic diversity across inbred mouse strains can result in different outcomes of a genetic knockout on different mouse genetic backgrounds (Guénet and Bonhomme 2003; Doetschman 2009). Thus, to gain insights into the role of Prdm9 in fertility, we examined whether Prdm9 is consistently essential for fertility in mice other than those previously studied.
Results
PRDM9 is dispensable for sperm production in PWD males
A loss-of-function allele of Prdm9, Prdm9tm1Ymat (henceforth, Prdm9tm), causes a complete arrest of spermatogenesis in C57BL/6 (B6) mice (B6tm/tm) (Table 1; Sun et al. 2015) and in mice with a mixed B6 and 129P2 genetic background (Hayashi et al. 2005). We crossed this allele into distantly related PWD/Ph (PWD) and found that male PWD mice homozygous for this loss-of-function Prdm9 allele (PWDtm/tm) produced sperm (Table 1, column SC). In contrast, no sperm were found in B6tm/tm mice, indicating a partial rescue of meiotic progression in the PWD genetic background. No PRDM9 protein was detected in PWDtm/tm (Supplemental Fig. S1); therefore, the phenotypic differences between B6tm/tm and PWDtm/tm were not the result of Prdm9 trans-activation or duplication of the Prdm9 locus in PWD. We validated the partial rescue with a second Prdm9-loss-of-function allele (Prdm9em) (see B6em/em, B6em/tm , and ((B6em/wt × PWD) × PWD)N3F1em/em in Table 1). Testicular tubule sections confirmed the presence of tubules with sperm and spermatids in PWDtm/tm but not in B6tm/tm mice (Fig. 1). Nonetheless, many tubules were devoid of spermatids in PWDtm/tm, and the sperm count in PWDtm/tm was far lower than that in wild-type mice. None of the four tested PWDtm/tm males produced offspring.
Table 1.
The effect of Prdm9 removal on male mouse fertility

Figure 1.

Partial meiotic arrest in Prdm9-deficient PWD male mice. (A) Scheme of spermatogenesis. Prophase I proceeds from prepachytene (leptotene and zygotene) spermatocytes via DSB formation and DSB repair (yellow stars) accompanied by partial synapsis (green rectangles) to pachytene with full synapsis and the repair of some DSB(s) to crossover(s) and of the remaining to noncrossovers. (B) Spermatogenesis can progress beyond the spermatocyte (SC) stages to the spermatid (ST) stages in PWDtm/tm, but not in B6tm/tm males. The evaluation of sections from adult testes (C–H) stained with hematoxylin and eosin (H/E). The tubule types were sorted according to their most advanced cell type excluding spermatozoa. (C–H) Representative tubule sections (higher magnification in F–H compared to C–E). The blue and red arrows point to round and elongated spermatids, respectively; these cell types are absent from B6tm/tm.
PRDM9 is important for meiotic synapsis in PWD males and females
Meiosis arrests at pachytene in B6tm/tm male mice. This arrest manifests as a failure to synapse all pairs of homologous autosomes and a failure to form the specialized chromatin compartment for the sex chromosomes (sex body) (Hayashi et al. 2005; Flachs et al. 2012). Both full synapsis and sex-body formation are required for normal meiotic progression in spermatocytes (for review, see Burgoyne et al. 2009). Although just 2% of pachytene nuclei appeared to be normal (fully synapsed autosomes and observable sex body) in B6tm/tm males (Table 1, column nPS; Supplemental Fig. S2), 41% were normal in PWDtm/tm. Homologous chromosome synapsis in pachynema depends on the repair of programmed DSBs (Mahadevaiah et al. 2001). PWDtm/tm males displayed 13% more DSB repair foci than equivalent nuclei from B6tm/tm (B6tm/tm = 193 ± 24 foci; PWDtm/tm = 219 ± 35 foci; P = 0.003, Wilcoxon test). A similar increase in DSBs was seen in PWDwt/wt compared to B6wt/wt (B6wt/wt = 201 ± 30 foci; PWDwt/wt = 225 ± 34 foci; P = 0.001, Wilcoxon test) (also see Balcova et al. 2016), implying that the loss of Prdm9 function was not responsible for the increase (Fig. 2A). One speculative hypothesis is that more DSBs would allow more chromosomes to synapse; for example, if two extra chromosomes could synapse in B6tm/tm pachytene cells (from the 13% increase in DSBs), close to 9% of pachytene nuclei would be fully synapsed. Finally, we found that the crossover rate (number of MLH1 foci) (see Methods) in normal PWDtm/tm pachytene spermatocytes was similar to that in PWDwt/wt males (Table 1; Fig. 2B). In addition, the density of MLH1 foci on synapsed chromosomes was lower in abnormal PWDtm/tm pachytene spermatocytes (1.33 foci/autosome, n = 245 autosomes), than in normal PWDtm/tm (1.56 foci/autosome, n = 1330) or PWDwt/wt pachynemas (1.54 foci/autosome, n = 1311) (Supplemental Table S1), suggesting that cells with a higher recombination rate may better tolerate Prdm9 loss. MLH1 foci were rarely seen (2/30 cells) on the synapsed chromosomes of abnormal B6tm/tm pachynemas. In conclusion, PWD males lacking functional PRDM9 exhibit a partial meiotic arrest. This contrasts mice with the same PRDM9 defect in a B6 background, where the meiotic arrest is close to absolute.
Figure 2.

Prdm9 dosage effect on crossover rate and DSB rate in PWD males. (A) PWD males have more DSBs than B6, independent of Prdm9 genotype (P = 0.006 for wild-type cells, P = 0.003 for Prdm9-deficient cells; Linear regression model). DNA meiotic recombinase 1/RAD51 recombinase (DMC1/RAD51) foci mark the sites of meiotic DSBs. The mean numbers of foci per genotype are shown as black dots ± SD. Values for individual cells are depicted as colored dots. These data were obtained from pooled prepuberal testes because these are enriched for zygotene spermatocytes. Loss of functional PRDM9 affects meiotic progression in juveniles similarly to adults, as there were more pachytene spermatocytes in 13-d-old PWDwt/wt (9%) versus PWDtm/tm testes (3%, P = 0.0001, χ2 test). The right panel depicts representative immunolabeling of a zygotene spermatocyte nucleus from PWDwt/tm. (B) The crossover rate (autosomal MLH1 foci per cell) is unchanged in PWDwt/wt and PWDtm/tm mice. The black dots with bars represent the mean ± SD; blue dots depict per-cell values (different shade for each individual male). See Supplemental Table S1 for values. The right panel depicts representative immunolabeling of a pachytene spermatocyte nucleus from PWDwt/tm for synaptonemal complex (SYCP1) and centromeres (CENT).
We next examined meiotic progression in B6tm/tm and PWDtm/tm females. At birth, the number of oocytes in both B6tm/tm and PWDtm/tm was comparable to wild type (Fig. 3D; Supplemental Fig. S3). Two days after birth, there was a notable reduction in both backgrounds (Fig. 3C; Supplemental Fig. S3), and all oocytes were completely eliminated in Prdm9-deficient adults (2–5 mo), unlike in wild-type controls (Fig. 3B; Hayashi et al. 2005). The post-birth oocyte loss is likely mediated by mechanisms that detect oocytes in which DSB repair and/or homolog synapsis were aberrant (Di Giacomo et al. 2005; Kogo et al. 2012; Wojtasz et al. 2012; Bolcun-Filas et al. 2014; Cloutier et al. 2015; Rinaldi et al. 2017; Qiao et al. 2018). Both B6tm/tm (Hayashi et al. 2005; Diagouraga et al. 2018) and PWDtm/tm adult females had small ovaries (Fig. 3A,B), and none of four mated PWDtm/tm females produced offspring.
Figure 3.

Similar meiotic arrest in B6 and PWD females lacking functional Prdm9. (A) Reduced adult ovary weight in both B6tm/tm and PWDtm/tm females indicative of arrested meiosis. There is no significant difference between B6tm/tm and PWDtm/tm. Black dots with bars symbolize mean ± SD; color dots depict individuals. Tukey's box-and-whisker plots show median values and quartiles. H/E-stained sections of paraffin-embedded ovaries of adult (B) and 2 d after birth (dpp) (C) mice. Arrows point to oocytes present in the controls but mostly absent in both PWDtm/tm and B6tm/tm. (D) Oocytes from newborn ovaries immunostained with anti-MSY2 (YBX2) antibody Abcam (ab33164) and DAPI. The number of oocytes was comparable in all tested females at birth (D), but decreased in both types of Prdm9-deficient mice compared to littermate controls 2 d after birth (C), suggesting pachytene arrest attributable to oocyte attrition. (E) Reduced homologous chromosome synapsis in PWDtm/tm oocytes (∼18 d post coitum). Colors distinguish oocytes derived from individual embryos (2 PWDwt/wt, 4 PWDwt/tm, and 6 PWDtm/tm). Black dots represent mean values, and bars represent SDs. (F) Representative spread nuclei from the PWDtm/tm and PWDwt/tm oocytes in E. Nuclei were immunostained for the synaptonemal complex lateral element (SYCP3), central element (SYCP1; yellow color indicates synapsis), and gamma H2AFX (a chromatin marker of DNA damage). (G) Decreased chromosomal synapsis in both PWD and B6 newborn oocytes lacking functional PRDM9. (H) Representative nuclei from the PWDtm/tm and PWDwt/tm oocytes in G immunostained for the synaptonemal complex central element (SYCP1; green indicates synapsis), recombination nodules (MLH1), and centromeres (CENT). See Supplemental Fig. S4 for nuclei from B6tm/tm and B6wt/wt.
In PWDwt/wt embryonic oocytes (∼18 d post coitum), pachytene synapsis was often incomplete. On average, 14/20 chromosomes were fully synapsed and just 14% of oocytes (9/64) exhibited a full complement of synapsed chromosomes. Like in males, the loss of functional Prdm9 exacerbated asynapsis at pachytene (Fig. 3E,F); PWDtm/tm oocytes had, on average, 3/20 synapsed chromosomes, and none of the 149 PWDtm/tm oocytes studied had more than 14 synapsed chromosomes (Fig. 3). In males, we only considered nuclei with more than nine synapsed chromosomes as “pachytene-like.” Only 10/149 PWDtm/tm oocytes met this strict criterion because of the severity of the defect in females. To compare with B6 mice, we examined ovaries from newborn mice. Both PWDtm/tm and B6tm/tm mice had fewer fully synapsed pachytene nuclei (PWDtm/tm = 0/28; B6tm/tm = 2/85 oocytes) than their wild-type littermates (PWDwt/wt = 6/30; B6wt/wt = 21/32 oocytes) (Fig. 3G,H; Supplemental Fig. S4). Thus, the lack of functional Prdm9 appears to result in synapsis defects and sterility both in B6 and PWD female mice.
Functional Prdm9 is not required for fertility in male mice
Having discovered that mice can make few sperm without functional Prdm9, we next hypothesized that hybrid vigor may help to restore full fertility to mice lacking Prdm9. By backcrossing (N) and intercrossing (F) B6 and PWD mice heterozygous for functional Prdm9 (for the breeding scheme, see Supplemental Fig. S5), we indeed obtained fertile Prdm9-deficient mice (((B6em/wt × PWD) × PWD)N3F1em/em) (Table 1). In an effort to increase fertility of these mice, we bred ((B6em/wt × PWD) × PWD)em/em males with their heterozygous sisters for several generations (for the breeding scheme, see Supplemental Fig. S6). These mice were designated (B6 × PWD)F7em/em (Table 1) and indeed had a higher sperm count (P = 0.014, Wilcoxon test) than ((B6em/wt × PWD) × PWD)N3F1em/em. Three of six (B6 × PWD)F7em/em males sired pups. To test the generality of this phenomenon, we performed a similar crossing strategy with two other pairs of mouse strains: B6 crossed with PWK/Ph (PWK), and C3H crossed with PWD. Each strategy yielded fertile mice that lacked functional Prdm9; 2/6 ((PWK × B6tm/wt) × PWK)N3F1tm/tm mice and 6/6 (C3H × PWD)F1tm/tm sired pups. In conclusion, mice can fully tolerate the loss of Prdm9 and remain fertile.
DSB hotspot distribution is similar in fertile and sterile Prdm9-deficient mice
The only known function of the PRDM9 protein is to designate the genomic locations at which meiotic DSBs occur. In the absence of functional PRDM9, meiotic DSBs occur at functional sites in the genome (Brick et al. 2012). It has been proposed that DSBs at functional sites may be difficult to repair, and that this may cause the sterility of B6tm/tm mice (Brick et al. 2012). We mapped meiotic DSB hotspots in the testes of sterile (B6tm/tm), sperm-producing but infertile (PWDtm/tm), and fully fertile ((B6 × PWD)F7em/em) Prdm9 knockout mice. Meiotic DSB hotspots occurred at similar locations in all three mice (83%–93% overlap) (Fig. 4A, B) and did not coincide with hotspot locations defined by PRDM9 (Fig. 4C). As expected, these hotspots mostly coincided with non-PRDM9-mediated H3K4me3-marked histones, often at transcription start sites (Fig. 4A,C). Thus, we conclude that meiotic DSBs at functional sites are not a complete impediment to meiotic progression.
Figure 4.

Hotspot locations are consistent in all mice lacking functional PRDM9. (A) DSB hotspots in wild-type mice (B6wt/wt and PWDwt/wt) are absent in mice lacking functional PRDM9 (B6tm/tm, PWDtm/tm, (B6 × PWD)F7em/em). Raw coverage is shown in 150-bp windows. Each panel is scaled to the maximum value. DSB hotspots are the peaks in DMC1-SSDS coverage. Red arrows represent Ensembl gene models. (B) Hotspot locations were conserved in all mice lacking functional PRDM9. The maximum reciprocal hotspot overlap (±500 bp) between pairs of samples is shown. (C) Hotspots in mice lacking functional PRDM9 occur at non-PRDM9-mediated H3K4me3 (non-hotspot) marks and transcription start sites (TSSs). All H3K4me3 peaks (Methods) that did not coincide with a wild-type DSB hotspot were considered non-hotspot H3K4me3. For TSSs, the overlap with Ensembl transcript's 5′ ends ± 500 bp is shown. These features rarely coincide with DSB hotspots in wild-type mice. Note that B6wt/wt H3K4me3 ChIP-seq data are from (Baker et al. 2014). B6wt/wt and PWDwt/wt DMC1-SSDS data are from Smagulova et al. (2016).
A locus on Chromosome X affects fertility of Prdm9-deficient male mice
Prdm9 governs hybrid sterility in male F1 hybrids of B6 and PWD mice (Mihola et al. 2009; Flachs et al. 2012). Genetic mapping identified variants of a locus on Chromosome X (Hstx2) that modulates fertility in these mice (Bhattacharyya et al. 2014). We therefore investigated whether this locus may also modulate fertility in mice lacking Prdm9.
We first examined meiotic progression in Prdm9-deficient male F1 mice from reciprocal crosses of PWDwt/tm and B6wt/tm. In F1 mice with a PWD mother (and therefore, a PWD Chromosome X and Hstx2PWD), just 2% of pachytene spermatocytes exhibited full synapsis and no sperm were found ((PWD × B6)F1tm/tm in Table 1). In the reciprocal cross, there was a substantial improvement in meiotic progression as 37% of pachytene spermatocytes were normal and sperm was produced ((B6 × PWD)F1tm/tm in Table 1). We next bred B6wt/tm to two subconsomic strains of B6 mice that carry either the Hstx2B6 or Hstx2PWD allele (Gregorová et al. 2008). These two strains carry the proximal 64.9 Mb (B6.PWD-ChrX.1wt/tm) and 69.6 Mb (B6.PWD-ChrX.1swt/tm) of PWD Chromosome X, respectively; the additional 4.7-Mb extension of the PWD region in the second strain is the Hstx2PWD locus. Each strain was used in place of female B6wt/tm in crosses with PWDwt/tm males to test whether the Hstx2 locus affects meiotic progression (for the breeding scheme, see Supplemental Fig. S7). As in (B6 × PWD)F1tm/tm males, we found that sperm was produced in the mice carrying Hstx2B6 ((B6.PWD-ChrX.1 × PWD)F1em/tm). In contrast, no sperm was found in mice carrying Hstx2PWD ((B6.PWD-ChrX.1s × PWD)F1tm/tm), thus implying that Hstx2B6 facilitates meiotic progression in mice lacking functional Prdm9. This mirrors a role of Hstx2B6 in facilitating fertility in otherwise sterile (PWD × B6)F1 hybrids (Bhattacharyya et al. 2014).
The Hstx2 locus controls the recombination rate in mouse; specifically, wild-type B6 mice or B6.PWD-ChrX.1 mice with the Hstx2B6 allele have more crossovers per meiosis than subconsomic B6 mice carrying the Hstx2PWD allele (B6.PWD-ChrX.1s) (Balcova et al. 2016). This could imply that an elevated crossover rate is sufficient to subvert the requirement for functional Prdm9 in meiosis. We therefore checked the crossover rate in our Prdm9-deficient mice by counting autosomal MLH1 foci in normal pachytene nuclei (Table 1). Note that all mice studied have the same number of chromosomes and chromosome arms (all chromosomes are acrocentric). In mice that did not produce normal pachytene nuclei, we assessed the crossover rate in genetically matched Prdm9wt/wt or Prdm9wt/- males. We felt that this was justified because in “normal” pachytene nuclei in mice that tolerate Prdm9 loss, the crossover rate is the same as in wild type (Fig. 2B). We found that the crossover rate was significantly higher in Prdm9-deficient mice that produced sperm than in Prdm9-deficient mice that did not (P = 0.019, Wilcoxon test) (Fig. 5; Table 1). These data are consistent with a model in which an elevated recombination rate helps to overcome the meiotic block imposed by the loss of functional Prdm9.
Figure 5.
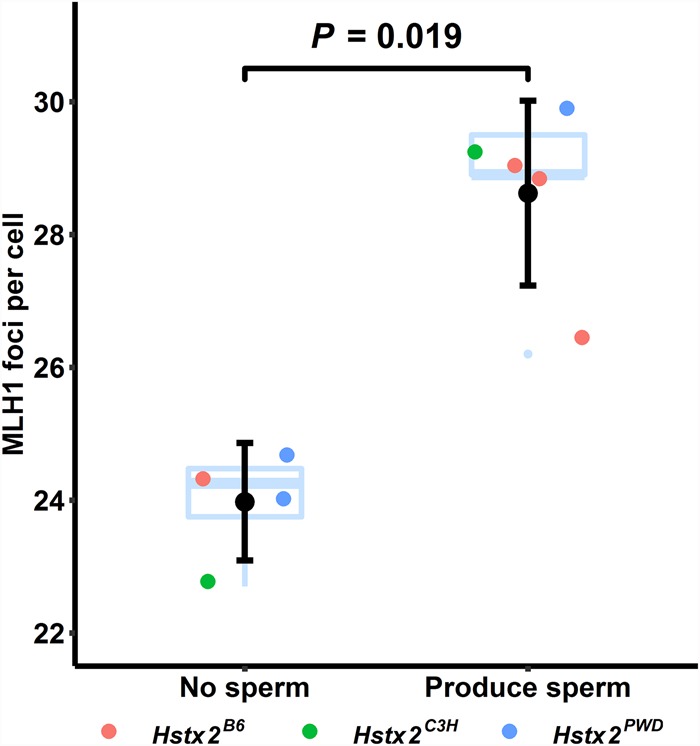
Sperm-producing Prdm9-deficient mice have a relatively high crossover rate. Black dots depict means, and error bars represent SDs. Each point represents a single mouse genetic background (data from Table 1). All mice carrying epididymal sperm (sperm count above the detection limit of a few thousands) were assigned to the “Produce Sperm” group. The MLH1 count values for the “No sperm” group were determined from genetically matched Prdm9wt males (Supplemental Table S1). Hstx2 is not the only locus affecting crossover rate in these mice, as judged from the distribution of its alleles in the groups. The mean crossover rate in animals that “Produce Sperm” was significantly higher than that in animals that did not (P, probability, Wilcoxon test).
Discussion
In this work, we identified male mice with multiple genetic backgrounds that are fertile despite lacking a functional PRDM9 protein. This unambiguously demonstrates that PRDM9 is not required for completion of meiosis in male mice. The only known function of PRDM9 is to target meiotic DSBs, and in its absence, meiotic DSBs occur at functional sites in the genome. We demonstrated that this redirection of DSBs away from functional sites is not necessary for the successful completion of meiosis, because DSB hotspots occurred at broadly similar sites in all mice lacking functional PRDM9. Indeed, this is consistent with the fertility of species that lack PRDM9 such as dogs, swordtail fish, and birds (Muñoz-Fuentes et al. 2011; Axelsson et al. 2012; Singhal et al. 2015; Baker et al. 2017), where recombination is favored at gene promoters. In humans, it has been proposed that an alternative mechanism may redirect DSBs from functional sites in the absence of PRDM9 to ensure fertility (Narasimhan et al. 2016). Our data showed that no such mechanism is required in mice.
A generalizable phenotype of Prdm9 loss in mice, consistent across all mice studied, is a quantitative defect in the number of chromosomes that can synapse correctly in pachynema; even in sterile B6tm/tm mice, most chromosomes synapse fully at a pachytene-like stage. Homologous chromosome synapsis at pachynema requires the formation and subsequent repair of programmed meiotic DSBs. Although there does not appear to be a DSB formation defect in mice that lack functional PRDM9 (Fig. 2), the pervasive unrepaired DSBs on asynapsed pachytene chromosomes in all Prdm9 knockout mice implies that DSB repair is defective. The nature of this defect is unclear; however, in Prdm9-deficient male mice with either more DSBs (Fig. 2A) or more crossovers (Fig. 5), the repair defect is partially alleviated and more pachytene nuclei successfully synapse all pairs of homologs (Table 1; Supplemental Fig. S8). These data imply that the loss of functional Prdm9 reduces the efficiency at which DSBs can be repaired from the homolog in such a way as to allow synapsis (likely as a crossover). If this efficiency dips below a critical threshold, one or more chromosomes fail to synapse and pachynema cannot be completed. In our model, the shortest chromosomes would be least likely to receive a repair-competent DSB, because the length of chromosomes is proportionate to the number of DSBs per meiosis (Supplemental Fig. S9). Thus, short chromosomes would be less likely to synapse with their homolog during pachytene. In mice with reduced DSB formation (Spo11-hypomorph) (Kauppi et al. 2013), short chromosomes asynapsis occurs disproportionately frequently. This is consistent with our model.
The extent of the pachytene defect resulting from Prdm9 deficiency does not linearly correlate with the observed sperm count and/or fertility of the animals; for example, 40% of pachytene spermatocytes synapse fully in PWDtm/tm mice, but the sperm count is just 1.2% of that in wild-type mice (sperm count SC = 0.4 million). In contrast, sperm production is far more robust in mice with just slightly more normal pachytene cells ((C3H × PWD)F1tm/tm, nPS = 49%, sperm count = 7.6 million; and (B6 × PWD)F7em/em, nPS = 46%, sperm count = 7.3 million). Previous studies have demonstrated that the failure to complete pachytene in a critical percentage of spermatocytes can effectively shut down spermatogenesis. This is thought to result from apoptotic signaling from the spindle assembly checkpoint (SAC; senses misaligned chromosomes), and this signal may affect “normal cells” because of communication among spermatocytes in the germinal syncytium (Faisal and Kauppi 2016). Indeed, Faisal and Kauppi (2016) found that the critical percentage of normal pachytene cells required for sperm production is somewhere between 30% (infertile) and 50% (fertile). Thus, we conclude that the removal of functional Prdm9 makes it more difficult for cells to complete full chromosome synapsis in pachytene. How this is translated into fertility is likely dependent on other factors, such as SAC potency, and can result in a broad range of fertility phenotypes ranging from sterility to fertility.
The Hstx2 locus, also known as meiotic recombination 1(Meir1), is a 4.7-Mb region (containing multiple genes) on Chromosome X that regulates the crossover rate in mice (Balcova et al. 2016). Consistent with published data, we found that in male mice differing at the Hstx2 locus, those carrying the Hstx2B6 allele had a 30% higher crossover rate than those carrying the Hstx2PWD allele. We argue that this difference resulted in sperm production in the Prdm9-deficient Hstx2B6 carrying mice, but not in mice with the Hstx2PWD allele. The Hstx2B6 allele was previously shown to alleviate hybrid incompatibilities governed by Prdm9 genotype that result in the sterility of male F1 hybrid mice from a cross between PWD females and B6 males (Bhattacharyya et al. 2014). We propose that this alludes to a unifying principle of fertility rescue in these two systems in which sterility arises in a Prdm9-dependent manner. In both systems, there is a clear DSB repair defect; in Prdm9 knockout mice, this may result from difficulties in repairing DSBs at functional sites; in sterile hybrids, DSB repair as a crossover is compromised at loci where Prdm9 binding sites have been destroyed in one parental lineage (Davies et al. 2016; Smagulova et al. 2016). In both systems, some but not all homologous chromosomes synapse at pachytene, and in both systems, fertility can apparently be rescued by increasing the recombination rate. We propose that in both systems, increasing the crossover rate (or perhaps the number of DSBs) elevates the likelihood of a crossover on every pair of homologous chromosomes, and thus increases the number of viable pachytene spermatocytes. Sufficient rescue can overcome the critical threshold of “normal” pachytene cells to assure robust sperm production and fertility.
Females lacking functional PRDM9 were sterile, with a similar pachytene asynapsis defect as in males. However, the number of pachytene nuclei with asynapsed chromosomes was far higher in females than in males. In our working model, this defect could theoretically be rescued by increasing the DSB/crossover rate or by subverting the SAC. The alternate explanation for the observed sex dimorphism is that the requirement for Prdm9 differs between males and females.
One implication from our model is that DSB repair as a crossover is more efficient in the presence of functional PRDM9. Thus, one of the roles of PRDM9 may be to facilitate a lower recombination rate than would otherwise be possible. This would help to explain why Prdm9 is widely conserved but repeatedly lost during evolutionary history, because species with a sufficiently high recombination rate could lose functional PRDM9 with little or no reproductive consequences. It remains to be seen whether this is the case.
Methods
Ethics statement
The European Community Council Directive 86/609/EEC, Appendix A of the Council of Europe Convention ETS123, and the Czech Republic Act for Experimental Work with Animals (Decree No. 207/2004 Sb, and the Acts Nos. 246/92 Sb and 77/2004 Sb) were obeyed during the laboratory animal care and experiments. Permissions Nos. 61/2013 and 10/2016 were issued by the ethics committee for the work with animals of the Institute of Molecular Genetics in Prague.
Mice
The PWD/Ph and PWK/Ph mouse strains were described in Gregorová and Forejt (2000). The C57BL/6J strain was from The Jackson Laboratory (Stock no. 000664), C3H/N was from Velaz, Czech Republic. The knockout line Prdm9tm1Ymat (MGI allele No. 3623909; here Prdm9tm) was generated in a 129P2/OlaHsd background by replacement of the first five coding exons with LacZ. This was maintained on the pure B6 background. The Prdm9tm line was transferred by 10-times-repeated backcrossing to PWD; the N7 generation was devoid of 129- and B6-specific sequences except for Chr 17 (proximal 31.4 Mb harboring Prdm9), and this differential segment was reduced in the N12 generation to about 11.7 Mb (position 15.1–26.8 Mb). The fertility parameters of the Prdm9-deficient mice resulting from the strains carrying the differential segments of 31.4 and 11.7 Mb were similar (Supplemental Table S2). The Prdm9em1Fore allele (abbreviated Prdm9em) strain was prepared by injecting programmed artificial zinc-finger endonuclease (Sigma-Aldrich) into C57BL/6NCrl zygotes, which resulted in a 4-bp deletion in the ninth exon of Prdm9 encoding the PR/SET domain (MGI allele No. 5501109). The (B6 × PWD)F7 line was prepared by repeated intercrossing of fertile Prdm9-deficient ((B6em × PWD) × PWD)em/em males to heterozygous littermates (for the breeding scheme, see Supplemental Fig. S6). The resulting (B6 × PWD)F7 mice carried the Hstx2B6 allele. C57BL/6J mice carrying different sections of the PWD Chromosome X were previously generated (Gregorová et al. 2008). We abbreviate these mice as B6.PWD-Chr# (where # indicates the introgressed chromosome fragment from PWD). F1 hybrids carrying regions of Chr X differing at Hstx2 and lacking PRDM9 were constructed by outcrossing the subconsomic strains B6.PWD-ChrX.1 (carries Hstx2B6) and B6.PWD-ChrX.1s (carries Hstx2PWD) to B6wt/tm, intercrossing the Prdm9wt/tm female offspring to PWDwt/tm males, and then genotyping their F1 offspring for Prdm9tm/tm and Chr X (for the breeding scheme, see Supplemental Fig. S7).
Genotyping and phenotyping
PCR genotyping conditions have been published (Flachs et al. 2012), except for genotyping primers for the Prdm9em allele: 5′-ACTCTAGGTATGTGAACTGTGC-3′ and 5′-CACAACCTGGTCTGATAACCC-3′; these primers amplify 111 bp and 107 bp from the wild type and mutant, respectively; annealing is carried out at 58°C, and products can be resolved in 5% agarose. Body weight (BW) and testis weight (TW) were taken for adult males (older than 11 wk). Sperm count (SC) was obtained from both epididymides. Embryos were dissected from mothers 18 d after the beginning of mating (mating lasted 1–3 d). Chromosome spreads (slides with surface-spread nuclei) were prepared from embryonic ovaries or whole adult testis using hypotonic treatment (Anderson et al. 1999). Spreads were stained with anti-DMC1 (Santa Cruz sc-22768), anti-RAD51 (Santa Cruz sc-8349), anti-SYCP3 (Santa Cruz sc-74569), and anti-centromere (Antibodies Incorporated 15-235) antibodies. DMC1/RAD51 foci (DSB repair intermediates) were counted in midzygotene nuclei with no fully synapsed autosome pairs and with fewer than five partially synapsed autosome pairs. Isotonic treatment (Turner et al. 2005) was used to prepare spermatocyte spreads for the analysis of MLH1 foci. Spreads were stained with anti-MLH1 (Abcam ab14206), anti-SYCP3 (Abcam ab15090), and anti-centromere (see above) antibodies. The MLH1 protein marks the sites of all genetic crossovers (Anderson et al. 1999; Wang et al. 2017). MLH1 foci were analyzed by confocal microscopy (Leica TCS SP5 AOBS Tandem) to ensure that only MLH1 foci that colocalized with the chromosomal axis (SYCP1) were counted. In cells with fully synapsed autosomes, we omitted cells in which any chromosome lacked an MLH1 focus. This allows for counting of properly stained, closely matched, and staged cells. In abnormal pachytene cells (carrying 1–4 asynapsed autosomal pairs), MLH1 foci were counted per synapsed autosomal pair. In the studies of male pachytene synapsis, cells carrying more than 10 asynapsed chromosomal pairs were considered as zygotene. Immunocytochemistry and histology were performed as described previously (Flachs et al. 2012, 2014; Bhattacharyya et al. 2013, 2014). The anti-MSY2 (YBX2) antibody was from Abcam (ab33164).
Chromatin immunoprecipitation (ChIP) and western blotting
DMC1 ChIP SSDS was done using snap-frozen adult testes as described previously (Khil et al. 2012; Brick et al. 2018). H3K4me3 was assayed by ChIP-seq in 13- to 15-d-old testes according to Baker et al. (2015b). Western blotting was performed as described (Baker et al. 2015a).
Sequencing data analyses
DMC1-SSDS samples were aligned to the mm10 reference genome, peak calling was performed, and hotspot strength was calculated as described (Brick et al. 2018; for sample quality metrics and hotspot counts, see Supplemental Table S3). H3K4me3 ChIP-seq data were aligned to the reference mm10 genome using BWA 0.7.12 (Li 2013). Peaks for H3K4me3 (used in Fig. 4) were called as follows. H3K4me3 ChIP-seq data from 12 dpp B6wt/wt mice (SRR1035576) (Baker et al. 2014) and corresponding input DNA data (SRR1035578) (Baker et al. 2014) were aligned to the reference mm10 genome using BWA 0.7.12 (Li 2013). Aligned BAM files were converted to BED files with BEDTools (Quinlan and Hall 2010) and duplicate reads were discarded. NCIS (Liang and Keleş 2012) was used to determine the background correction ratio (RBG). MACS2 version 2.1.0.20150731 (Zhang et al. 2008) was used with the following arguments to call peaks: --ratio {RBG} -g mm --bw 1000 --keep-dup all --slocal 5000 -t {SRR1035576 BED file} -c {SRR1035578 BED file}.
Data access
ChIP data generated in this study have been submitted to the NCBI Gene Expression Omnibus (GEO; https://www.ncbi.nlm.nih.gov/geo/) under accession number GSE109874.
Supplementary Material
Acknowledgments
We thank K. Tresnak, M. Fickerova, P. Valtrova, and L. Sebestova for technical assistance; employees of the animal facility of the Institute of Molecular Genetics of the Czech Academy of Sciences (IMG CAS) for mouse keeping; I. Beck for preparing the Prdm9em knockout; and J. Forejt and anonymous reviewers for comments. Some data were produced in the Microscopy Centre, IMG CAS. This work utilized the computational resources of the NIH HPC Biowulf cluster (http://hpc.nih.gov). O.M., P.F., E.L, R.S., and Z.T. were supported by CSF (14-20728S, 16-06548S), by CAS (RVO 68378050), by MEYS (LM2015040 CCP), and by ERDF plus MEYS (CZ.1.05/1.1.00/02.0109 BIOCEV, CZ.1.05/2.1.00/19.0395); C.L.B., K.P., and P.M.P. by NIH grants P01 GM099640 and R01 GM078452; F.P., K.B., and R.D.C.-O. by the NIDDK Intramural Research Program.
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.244426.118.
References
- Anderson LK, Reeves A, Webb LM, Ashley T. 1999. Distribution of crossing over on mouse synaptonemal complexes using immunofluorescent localization of MLH1 protein. Genetics 151: 1569–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Axelsson E, Webster MT, Ratnakumar A, LUPA Consortium, Ponting CP, Lindblad-Toh K. 2012. Death of PRDM9 coincides with stabilization of the recombination landscape in the dog genome. Genome Res 22: 51–63. 10.1101/gr.124123.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker CL, Walker M, Kajita S, Petkov PM, Paigen K. 2014. PRDM9 binding organizes hotspot nucleosomes and limits Holliday junction migration. Genome Res 24: 724–732. 10.1101/gr.170167.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker CL, Kajita S, Walker M, Saxl RL, Raghupathy N, Choi K, Petkov PM, Paigen K. 2015a. PRDM9 drives evolutionary erosion of hotspots in Mus musculus through haplotype-specific initiation of meiotic recombination. PLoS Genet 11: e1004916 10.1371/journal.pgen.1004916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker CL, Petkova P, Walker M, Flachs P, Mihola O, Trachtulec Z, Petkov PM, Paigen K. 2015b. Multimer formation explains allelic suppression of PRDM9 recombination hotspots. PLoS Genet 11: e1005512 10.1371/journal.pgen.1005512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker Z, Schumer M, Haba Y, Bashkirova L, Holland C, Rosenthal GG, Przeworski M. 2017. Repeated losses of PRDM9-directed recombination despite the conservation of PRDM9 across vertebrates. eLife 6: e24133 10.7554/eLife.24133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balcova M, Faltusova B, Gergelits V, Bhattacharyya T, Mihola O, Trachtulec Z, Knopf C, Fotopulosova V, Chvatalova I, Gregorova S, et al. 2016. Hybrid sterility locus on Chromosome X controls meiotic recombination rate in mouse. PLoS Genet 12: e1005906 10.1371/journal.pgen.1005906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baudat F, Buard J, Grey C, Fledel-Alon A, Ober C, Przeworski M, Coop G, de Massy B. 2010. PRDM9 is a major determinant of meiotic recombination hotspots in humans and mice. Science 327: 836–840. 10.1126/science.1183439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharyya T, Gregorova S, Mihola O, Anger M, Sebestova J, Denny P, Simecek P, Forejt J. 2013. Mechanistic basis of infertility of mouse intersubspecific hybrids. Proc Natl Acad Sci 110: E468–E477. 10.1073/pnas.1219126110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharyya T, Reifova R, Gregorova S, Simecek P, Gergelits V, Mistrik M, Martincova I, Pialek J, Forejt J. 2014. X chromosome control of meiotic chromosome synapsis in mouse inter-subspecific hybrids. PLoS Genet 10: e1004088 10.1371/journal.pgen.1004088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolcun-Filas E, Schimenti JC. 2012. Genetics of meiosis and recombination in mice. Int Rev Cell Mol Biol 298: 179–227. 10.1016/B978-0-12-394309-5.00005-5 [DOI] [PubMed] [Google Scholar]
- Bolcun-Filas E, Rinaldi VD, White ME, Schimenti JC. 2014. Reversal of female infertility by Chk2 ablation reveals the oocyte DNA damage checkpoint pathway. Science 343: 533–536. 10.1126/science.1247671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brick K, Smagulova F, Khil P, Camerini-Otero RD, Petukhova GV. 2012. Genetic recombination is directed away from functional genomic elements in mice. Nature 485: 642–645. 10.1038/nature11089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brick K, Pratto F, Sun CY, Camerini-Otero RD, Petukhova G. 2018. Analysis of meiotic double-strand break initiation in mammals. Methods Enzymol 601: 391–418. 10.1016/bs.mie.2017.11.037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgoyne PS, Mahadevaiah SK, Turner JM. 2009. The consequences of asynapsis for mammalian meiosis. Nat Rev Genet 10: 207–216. 10.1038/nrg2505 [DOI] [PubMed] [Google Scholar]
- Cloutier JM, Mahadevaiah SK, ElInati E, Nussenzweig A, Tóth A, Turner JM. 2015. Histone H2AFX links meiotic chromosome asynapsis to prophase I oocyte loss in mammals. PLoS Genet 11: e1005462 10.1371/journal.pgen.1005462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies B, Hatton E, Altemose N, Hussin JG, Pratto F, Zhang G, Hinch AG, Moralli D, Biggs D, Diaz R, et al. 2016. Re-engineering the zinc fingers of PRDM9 reverses hybrid sterility in mice. Nature 530: 171–176. 10.1038/nature16931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Giacomo M, Barchi M, Baudat F, Edelmann W, Keeney S, Jasin M. 2005. Distinct DNA-damage-dependent and -independent responses drive the loss of oocytes in recombination-defective mouse mutants. Proc Natl Acad Sci 102: 737–742. 10.1073/pnas.0406212102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diagouraga B, Clément JAJ, Duret L, Kadlec J, de Massy B, Baudat F. 2018. PRDM9 methyltransferase activity is essential for meiotic DNA double-strand break formation at its binding sites. Mol Cell 69: 853–865.e6. 10.1016/j.molcel.2018.01.033 [DOI] [PubMed] [Google Scholar]
- Doetschman T. 2009. Influence of genetic background on genetically engineered mouse phenotypes. Methods Mol Biol 530: 423–433. 10.1007/978-1-59745-471-1_23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fairfield H, Gilbert GJ, Barter M, Corrigan RR, Curtain M, Ding Y, D'Ascenzo M, Gerhardt DJ, He C, Huang W, et al. 2011. Mutation discovery in mice by whole exome sequencing. Genome Biol 12: R86 10.1186/gb-2011-12-9-r86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faisal I, Kauppi L. 2016. Sex chromosome recombination failure, apoptosis, and fertility in male mice. Chromosoma 125: 227–235. 10.1007/s00412-015-0542-9 [DOI] [PubMed] [Google Scholar]
- Flachs P, Mihola O, Šimeček P, Gregorová S, Schimenti JC, Matsui Y, Baudat F, de Massy B, Piálek J, Forejt J, et al. 2012. Interallelic and intergenic incompatibilities of the Prdm9 (Hst1) gene in mouse hybrid sterility. PLoS Genet 8: e1003044 10.1371/journal.pgen.1003044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flachs P, Bhattacharyya T, Mihola O, Piálek J, Forejt J, Trachtulec Z. 2014. Prdm9 incompatibility controls oligospermia and delayed fertility but no selfish transmission in mouse intersubspecific hybrids. PLoS One 9: e95806 10.1371/journal.pone.0095806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregorová S, Forejt J. 2000. PWD/Ph and PWK/Ph inbred mouse strains of Mus m. musculus subspecies—a valuable resource of phenotypic variations and genomic polymorphisms. Folia Biol (Praha) 46: 31–41. [DOI] [PubMed] [Google Scholar]
- Gregorová S, Divina P, Storchova R, Trachtulec Z, Fotopulosova V, Svenson KL, Donahue LR, Paigen B, Forejt J. 2008. Mouse consomic strains: exploiting genetic divergence between Mus m. musculus and Mus m. domesticus subspecies. Genome Res 18: 509–515. 10.1101/gr.7160508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grey C, Baudat F, de Massy B. 2018. PRDM9, a driver of the genetic map. PLoS Genet 14: e1007479 10.1371/journal.pgen.1007479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guénet JL, Bonhomme F. 2003. Wild mice: an ever-increasing contribution to a popular mammalian model. Trends Genet 19: 24–31. 10.1016/S0168-9525(02)00007-0 [DOI] [PubMed] [Google Scholar]
- Hayashi K, Yoshida K, Matsui Y. 2005. A histone H3 methyltransferase controls epigenetic events required for meiotic prophase. Nature 438: 374–378. 10.1038/nature04112 [DOI] [PubMed] [Google Scholar]
- Imai Y, Baudat F, Taillepierre M, Stanzione M, Toth A, de Massy B. 2017. The PRDM9 KRAB domain is required for meiosis and involved in protein interactions. Chromosoma 126: 681–695. 10.1007/s00412-017-0631-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kauppi L, Barchi M, Lange J, Baudat F, Jasin M, Keeney S. 2013. Numerical constraints and feedback control of double-strand breaks in mouse meiosis. Genes Dev 27: 873–886. 10.1101/gad.213652.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khil PP, Smagulova F, Brick KM, Camerini-Otero RD, Petukhova GV. 2012. Sensitive mapping of recombination hotspots using sequencing-based detection of ssDNA. Genome Res 22: 957–965. 10.1101/gr.130583.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kogo H, Tsutsumi M, Ohye T, Inagaki H, Abe T, Kurahashi H. 2012. HORMAD1-dependent checkpoint/surveillance mechanism eliminates asynaptic oocytes. Genes Cells 17: 439–454. 10.1111/j.1365-2443.2012.01600.x [DOI] [PubMed] [Google Scholar]
- Li H. 2013. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv:1303.3997v2 [q-bio.GN], pp. 1–3.
- Liang K, Keleş S. 2012. Normalization of ChIP-seq data with control. BMC Bioinformatics 13: 199 10.1186/1471-2105-13-199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahadevaiah SK, Turner JM, Baudat F, Rogakou EP, de Boer P, Blanco-Rodríguez J, Jasin M, Keeney S, Bonner WM, Burgoyne PS. 2001. Recombinational DNA double-strand breaks in mice precede synapsis. Nat Genet 27: 271–276. 10.1038/85830 [DOI] [PubMed] [Google Scholar]
- Mihola O, Trachtulec Z, Vlcek C, Schimenti JC, Forejt J. 2009. A mouse speciation gene encodes a meiotic histone H3 methyltransferase. Science 323: 373–375. 10.1126/science.1163601 [DOI] [PubMed] [Google Scholar]
- Muñoz-Fuentes V, Di Rienzo A, Vila C. 2011. Prdm9, a major determinant of meiotic recombination hotspots, is not functional in dogs and their wild relatives, wolves and coyotes. PLoS One 6: e25498 10.1371/journal.pone.0025498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers S, Bowden R, Tumian A, Bontrop RE, Freeman C, MacFie TS, McVean G, Donnelly P. 2010. Drive against hotspot motifs in primates implicates the PRDM9 gene in meiotic recombination. Science 327: 876–879. 10.1126/science.1182363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narasimhan VM, Hunt KA, Mason D, Baker CL, Karczewski KJ, Barnes MR, Barnett AH, Bates C, Bellary S, Bockett NA, et al. 2016. Health and population effects of rare gene knockouts in adult humans with related parents. Science 352: 474–477. 10.1126/science.aac8624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parvanov ED, Petkov PM, Paigen K. 2010. Prdm9 controls activation of mammalian recombination hotspots. Science 327: 835 10.1126/science.1181495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiao H, Rao H, Yun Y, Sandhu S, Fong JH, Sapre M, Nguyen M, Tham A, Van BW, Chng TYH, et al. 2018. Impeding DNA break repair enables oocyte quality control. Mol Cell 72: 211–221.e3. 10.1016/j.molcel.2018.08.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan AR, Hall IM. 2010. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26: 841–842. 10.1093/bioinformatics/btq033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rinaldi VD, Hsieh K, Munroe R, Bolcun-Filas E, Schimenti JC. 2017. Pharmacological inhibition of the DNA damage checkpoint prevents radiation-induced oocyte death. Genetics 206: 1823–1828. 10.1534/genetics.117.203455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singhal S, Leffler EM, Sannareddy K, Turner I, Venn O, Hooper DM, Strand AI, Li Q, Raney B, Balakrishnan CN, et al. 2015. Stable recombination hotspots in birds. Science 350: 928–932. 10.1126/science.aad0843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smagulova F, Brick K, Pu Y, Camerini-Otero RD, Petukhova GV. 2016. The evolutionary turnover of recombination hot spots contributes to speciation in mice. Genes Dev 30: 266–280. 10.1101/gad.270009.115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun F, Fujiwara Y, Reinholdt LG, Hu J, Saxl RL, Baker CL, Petkov PM, Paigen K, Handel MA. 2015. Nuclear localization of PRDM9 and its role in meiotic chromatin modifications and homologous synapsis. Chromosoma 124: 397–415. 10.1007/s00412-015-0511-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thibault-Sennett S, Yu Q, Smagulova F, Cloutier J, Brick K, Camerini-Otero RD, Petukhova GV. 2018. Interrogating the functions of PRDM9 domains in meiosis. Genetics 209: 475–487. 10.1534/genetics.118.300565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner JM, Mahadevaiah SK, Fernandez-Capetillo O, Nussenzweig A, Xu X, Deng CX, Burgoyne PS. 2005. Silencing of unsynapsed meiotic chromosomes in the mouse. Nat Genet 37: 41–47. 10.1038/ng1484 [DOI] [PubMed] [Google Scholar]
- Wang S, Hassold T, Hunt P, White MA, Zickler D, Kleckner N, Zhang L. 2017. Inefficient crossover maturation underlies elevated aneuploidy in human female meiosis. Cell 168: 977–989.e17. 10.1016/j.cell.2017.02.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss J, Hurley LA, Harris RM, Finlayson C, Tong M, Fisher LA, Moran JL, Beier DR, Mason C, Jameson JL. 2012. ENU mutagenesis in mice identifies candidate genes for hypogonadism. Mamm Genome 23: 346–355. 10.1007/s00335-011-9388-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wojtasz L, Cloutier JM, Baumann M, Daniel K, Varga J, Fu J, Anastassiadis K, Stewart AF, Remenyi A, Turner JM, et al. 2012. Meiotic DNA double-strand breaks and chromosome asynapsis in mice are monitored by distinct HORMAD2-independent and -dependent mechanisms. Genes Dev 26: 958–973. 10.1101/gad.187559.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, et al. 2008. Model-based Analysis of ChIP-Seq (MACS). Genome Biol 9: R137 10.1186/gb-2008-9-9-r137 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.