

Abstract
The mechanotransduction of vestibular sensory cells depends on the high endolymphatic potassium concentration ([K+]) maintained by a fine balance between K+ secretion and absorption by epithelial cells. Despite the crucial role of endolymph as an electrochemical motor for mechanotransduction, little is known about the processes that govern endolymph formation. To address these, we took advantage of an organotypic rodent model, which regenerates a genuine neonatal vestibular endolymphatic compartment, facilitating the determination of endolymphatic [K+] and transepithelial potential (Vt) during endolymph formation. While mature Vt levels are almost immediately achieved, K+ accumulates to reach a steady [K+] by day 5 in culture. Inhibition of sensory cell K+ efflux enhances [K+] regardless of the blocker used (FM1.43, amikacin, gentamicin, or gadolinium). Targeting K+ secretion with bumetanide partially and transiently reduces [K+], while ouabain application and Kcne1 deletion almost abolishes it. Immunofluorescence studies demonstrate that dark cells do not express Na-K-2Cl cotransporter 1 (the target of bumetanide) in cultured and young mouse utricles, while Na/K-ATPase (the target of ouabain) is found in dark cells and transitional cells. This global analysis of the involvement of endolymphatic homeostasis actors in the immature organ (1) confirms that KCNE1 channels are necessary for K+ secretion, (2) highlights Na/K-ATPase as the key endolymphatic K+ provider and shows that Na-K-2Cl cotransporter 1 has a limited impact on K+ influx, and (3) demonstrates that transitional cells are involved in K+ secretion in the early endolymphatic compartment.
Introduction
Most neurons and sensory cells are depolarized by inward sodium or calcium currents. However, in the vestibule, an unusual potassium influx governs the excitability of sensory hair cells. This relies on the ionic composition of the endolymph, the luminal medium of vestibular cavities, which bathes the apical surface of the hair cells and contains >100 mm K+ and a few millimolars of Na+. Mechanosensitive hair cells bear stereocilia possessing stretch-activated channels, which open when stereocilia tilt in response to appropriate mechanical stimuli. The activation of these channels allows K+ influx, which is driven by the electrochemical gradient between the endolymph and the hair cell cytosol. K+ entrance depolarizes the hair cells, resulting in excitation of their innervating neurons. Ultimately, the resting membrane potential the hair cells recovers by extruding K+ into the perilymph (a low K+-containing interstitial medium surrounding basolateral membranes) through calcium- and voltage-gated K+ channels (Lewis and Hudspeth, 1983). Potassium ions also leave the endolymph through the transitional cells of the nonsensory epithelium, presumably in case of overstimulation (Lee et al., 2001). From the perilymph, K+ is recycled into the endolymph by the dark cells, located in the nonsensory epithelium. At their basolateral pole, these cells take up K+ by the Na/K-ATPase and Na-K-2Cl cotransporter 1 (NKCC1). K+ is then apically released into the endolymph through KCNE1/KCNQ1 channels and, presumably, other accessory K+ channels (Wangemann, 1995, 2002; Lang et al., 2007). The importance of such K+ cycling in endolymph maintenance is illustrated by the endolymph anomalies occurring in Menière's disease and Pendred syndromes, both of which comprise vestibular dysfunction among their symptoms. Interestingly, endolymph anomalies are also linked to the dysfunction of proteins involved in endolymphatic K+ secretion (Vetter et al., 1996; Delpire et al., 1999; Wangemann et al., 2004; Singh and Wangemann, 2008; Teggi et al., 2008, 2010; Yang et al., 2009).
The mechanisms of endolymph homeostasis are well described in the mature vestibule. However, the process of endolymph formation in the developing vestibule has received scarce attention (Masetto et al., 2005). This prompted us to study the cellular mechanisms underlying this process. We developed a rat utricular culture model in which cysts are formed by an organotypically organized vestibular epithelium made up of healthy hair cells and nonsensory cells with an isolated K+ rich luminal compartment (Gaboyard et al., 2005). This epithelium constitutes the boundary between endolymph and perilymph and permits the polar bathing of epithelial cells. Therefore, by gathering together all the cellular players, this model can be used to examine the contribution of each player toward the maintenance of endolymph homeostasis. Here we adapted this model to the mouse utricle in order to study the utricle of genetically modified animals. Using physiological, transgenic, and immunocytological approaches, the present data highlights the role of Na/K-ATPase and transitional cells in endolymph formation in the immature vestibule.
Materials and Methods
Animals.
Experiments were performed on newborn, young, and adult wild-type rodents of either sex (Wistar rats, Swiss and C57BL/6J mice) and on Kcne1 gene knock-out mice in which the expression of the K+ channel subunit KCNE1 was abolished (Vetter et al., 1996). Procedures involving animals were performed in accordance with the regulations of French Ministry of Agriculture and the European Community Council Directive 86/609/EEC. All efforts were made to minimize the number of animals used and their suffering.
Reagents.
Growth factor-reduced Matrigel matrix was purchased from Becton Dickinson. Agarose, Leibovitz medium, N2 supplement, DMEM, Ham's F-12 nutrient (F12), and N-(3-triethylammoniumpropyl)-4-(4-(dibutylamino)styryl)pyridinium dibromide (FM1-43) were purchased from Invitrogen. Amikacin was purchased from Bristol Myers Squibb. Dichlorodimethylsilane and I-Cocktail B ionophore were purchased from Fluka. Bumetanide, gadolinium, laminine, ouabain, and gentamicin sulfate were purchased from Sigma-Aldrich. Alexa-conjugated secondary antibodies were purchased from Invitrogen.
Statistical analysis of the data was performed by ANOVA. Data are given as mean ± SEM of repeated experiments.
Transgenic mice genotyping.
In newborn Kcne1 mutant mice, the phenotype of vestibular dysfunction cannot be easily discriminated by shaker/waltzer behavior or swimming disability (Deol, 1968; Lim et al., 1978). Disruption of the Kcne1 gene leads to inner ear defects that only lead to obvious behavioral differences when the maturation of the balance and motor functions reach the stage of steady locomotion (Vetter et al., 1996). Hence, genotyping of newborn littermates was performed using the REDExtract-N-Amp tissue PCR kit (Sigma) and the following primers: IsK1: 5′GAGTTCATAATGGCTGG 3′, IsK2: 5′ATGCCTGTAAACTGACC 3′, and Neo: 5′ TCCCGCTTCCATTGCTCA 3′. DNA was extracted from tissue samples collected at the time of death. The presence of the disrupted allele was characterized by PCR amplification of a 537 bp genomic DNA fragment using the primers Isk1 and Neo. The wild-type allele was characterized by the amplification of a 375 bp fragment with the primers IsK1 and IsK2.
Utricular cyst culture.
Vestibular end organs were aseptically removed from animals and placed in Leibovitz medium. Utricles were further dissected by removing the ampullae, thus breaking the endolymphatic continuum. Nerve fascicles were stripped from the stromal surface of the utricles. Care was taken to preserve the otolithic membranes. Explants composed of sensory and nonsensory epithelia were placed on 5 μl of growth factor-reduced Matrigel on laminin (10 μg/ml)-coated glass coverslips. These preparations were incubated for 30 min at 37°C in a 95%/5% O2/CO2 atmosphere at saturating moisture. Explants were then cultured in DMEM and F12 (50%/50% v/v) media supplemented with N2 mixture (2%). The medium was renewed every 4 d. After 2–3 d in vitro (DIV), the utricles developed vesicles that gave rise to utricular cysts.
Electrophysiological recordings.
Potassium concentration was determined by electrophysiological recordings. Glass microelectrodes were pulled from borosilicate capillaries with filaments (GC150F-10; World Precision Instruments) using a P-97 Sutter Instrument puller. The electrode tip diameters ranged from 2 to 6 μm. Electrodes were baked at 200°C for 2 h. Hydrophobicity of electrodes was achieved by evaporating dichlorodimethylsilane (5 min, 100°C) inside microelectrodes. Electrodes were baked again for 4 h at 200°C. The very tip of the silanized microelectrode was backfilled with I-Cocktail B K+-selective ion exchanger and the microelectrode barrel was filled with 150 mm KCl. The barrel was linked to an Ag/AgCl wire, which was connected to the inputs of a dual microprobe system (model Ks-700; WPI). The reference electrode was a hematocrite glass capillary filled with 2% agar in 1 m KCl. Before intracystic recordings, the signal of the electrode was calibrated with increasing concentrations of KCl: 0, 5, 10, 20, 50, 100, and 150 mm. All reference solutions contained 150 mm cation, supplemented with NaCl as appropriate. The transepithelial potential (Vt) of the cyst was recorded as the difference of potential between the intracystic compartment and extracystic bathing medium using a glass microelectrode pulled from a hematocrite capillary filled with 150 mm KCl, using the same setup as above.
Microelectrodes were advanced into the cyst using a micromanipulator under optical control on an upright microscope (Axioskop; Zeiss). The smallest cysts (∼30–50 μm long) were excluded from the study because their lumens were too small to allow visual control of electrode position. Values of cystic potential differences were consistently determined 15 s after the electrode was positioned in the cyst. Although the change in potential is immediate, the 15 s delay was required to visually confirm the correct position of the electrode in the lumen. Because piercing the cyst wall with the electrode initiates a leak of endocystic fluid into the medium, recordings did not exceed 1 min. Thus, these leaks prohibited long-term measurements required for assessing the reversibility of drug effects on an individual cyst.
Pharmacological manipulations.
To decipher the molecular processes involved in the maturation of the endolymph, [K+] and Vt were determined in cysts exposed to the Na/K-ATPase inhibitor, ouabain, and the Na-K-2Cl cotransporter 1 inhibitor, bumetanide, that perturb transmembrane K+ transport. Gadolinium, FM1-43, and two aminoglycosides (gentamicin and amikacin) known to block the hair cell stretch-activated channel (Kroese et al., 1989; Kimitsuki et al., 1996; Gale et al., 2001; Géléoc and Holt, 2003; Meyers et al., 2003) were also used to assay K+ efflux.
Immunocytochemistry.
Ten-day-old and adult inner ears were explanted, as were the kidneys for use as a positive control. These cysts and tissues were cultured for 7 DIV and then fixed by immersion in 4% paraformaldehyde in PBS (0.1 mm, pH 7.4) for 1 h. The inner ears were further dissected to isolate vestibular organs. Samples were embedded in 4% agarose/PBS, and 40-μm-thick sections were cut using a vibrating blade microtome (Vibratome series 1000; Technical Products International). Free-floating sections were permeabilized by incubation for 1 h in 4% Triton X-100/PBS for adult tissues and 1% Triton X-100/PBS for cysts and young vestibular tissues. Nonspecific binding was prevented by immersion of samples in blocking solutions [PBS containing 0.5% fish gelatin, 1% bovine serum albumin, and Triton X-100 (0.5% for mature tissues and 0.1% young tissues and cysts)] for 1 h at room temperature. Samples were then incubated in their corresponding blocking solutions, supplemented with primary antibodies: anti-Na/K-ATPase rabbit polyclonal serum (dilution 1/200; a gift from Dr. Geering, University of Lausanne, Lausanne, Switzerland), anti-Na-K-2Cl cotransporter 1 monoclonal mouse antibody (T4, dilution 1/175; Developmental Studies Hybridoma Bank) and anti-calretinin goat polyclonal serum (dilution 1/200; Millipore Bioscience Research Reagents) for 48 h at 4°C. For controls, primary antibodies were omitted. After rinsing with PBS, sections were incubated overnight at 4°C in blocking solutions containing the appropriate secondary antibodies, as follows: Alexa 488-conjugated donkey anti-rabbit serum (dilution 1/700), Alexa 594-conjugated donkey anti-mouse serum (dilution 1/700), and Alexa 647-conjugated donkey anti-goat serum (dilution 1/700). After rinsing with PBS, sections were mounted in Moviol (Calbiochem) and observed with a Zeiss 5 live duo laser scanning confocal microscope. Final image processing was performed with Adobe Photoshop software.
Results
Time course of cyst formation
The ability of the mouse utricle to generate cysts was compared with that of the rat organ in a developmental study. Utricles were dissected from newborn (P0) to 11-d-old (P11) animals and cultured for 4 d, after which the cysts that appeared were counted under a microscope (Fig. 1A). The capacity for cyst formation was then calculated by dividing the number of newly formed cysts by the number of utricles initially placed in culture. The resulting ratios were expressed as a percentage (100% being the same number of cysts as that of utricles put into culture; the number of cultured utricles ranged from 6 to 12 utricles per experiment). For each developmental stage, individual experiments were repeated several times; the number of experiments and average of the numbers of cysts per experiment are given in Figure 1B.
Figure 1.

Developing utricles generate endolymph-containing cysts. Utricles were dissected from newborn to 11-d-old mice and rats and were cultured for 4 d. A, After 4 DIV, cysts had emerged from the dissected epithelia. The cyst formation was assessed by observation under dissecting microscope (note the presence of shiny otoconia inside the vesicle). Scale bar, 75 μm. B, The number of formed vesicles out of the total number of utricles cultured is given as function of animal age. The higher yield of cyst formation is found in the early postnatal days; subsequently, the capacity for cyst formation progressively declines. Asterisks indicate significant differences between mouse and rat data (**p < 0.01; ***:p < 0.001). C, Cysts obtained from newborn rats and mice (1- to 3-d-old pups) were found to concentrate K+; this was detected as soon as the cysts appeared (by 2 or 3 DIV) for mouse and rat tissues. [K+]ec increased until 5 DIV, then remained relatively stable until 8 DIV. D, During the same period, Vt remained at negative values during the first 9 d of culture. From then on, Vt values oscillated, presumably indicating cyst degeneration, as may be the case for [K+]ec (see C). B–D, Data are expressed as the means ± SEM of repeated experiment, the numbers of which are indicated above or below each symbol. Error bars are not visible when they are smaller than symbols.
The pattern of cyst formation was similar for both rodents. High yields were obtained during the first 4 d for rat and mouse, but in the latter, high yields continued for another 1.5 d (Fig. 1B). During these early days, the yield of cyst formation declined minimally in both species: 84 ± 5% to 63 ± 6% for the mouse utricle and 65 ± 10% to 42 ± 6% for the rat. Cyst formation yield in the mouse was higher than that of rat, with significant differences occurring at P1 (p < 0.001), P3 (p < 0.01), and P5.5 (p < 0.001). Later, cyst formation sharply decreased and finally disappeared at P7.5 and P11 for the rat and mouse utricles, respectively.
Endocystic potassium accumulation
In our previous study, K+ accumulation was shown to occur in rat cysts cultured for 6, 8, and 10 d. Here, we extend our findings by assessing [K+] during the early stages of rat cyst formation (Fig. 1C). Utricular vesicles first appeared by 3 DIV and displayed a lumen large enough to insert the recording electrode. A substantial [K+] of 47.7 ± 12.5 mm was recorded. K+ accumulation plateaued at 5 DIV and maintained a constant level until 8 DIV (mean plateau level: 78.2 ± 4.7 mm). [K+] then varied and declined. At 20 DIV, ∼40 mm K+ was detected; however, the number of surviving cysts was very low (∼5% survival). The present data, obtained with single-barrel K+ selective microelectrodes, was statistically comparable to data previously recorded by means of double-barrel K+ selective microelectrodes at 6, 8, and 10 DIV (Gaboyard et al., 2005). Single- versus double-barrel data were as follows (in mm): 82.5 ± 11.8 versus 85.7 ± 17.4 at 6 DIV, 75.6 ± 6.6 versus 78.3 ± 15.0 at 8 DIV, and 73.3 ± 7.7 versus 93.3 ± 11.3 at 10 DIV, respectively. In addition, concerning [K+] recording in the mouse cysts, as Vt remains small and negative throughout the main duration of the study (see Transepithelial potential in mouse cysts), the impact of Vt on [K+] measurement would only result in a slight underestimation of [K+] that does not detract from the physiological meaning of the data. Moreover, as mouse cysts are half the size of rat cysts (the maximum length of mouse cyst is ∼300 μm; Fig. 1A), single-barrel electrodes had the advantage of being less invasive.
A time course analysis of the capacity of mouse cysts to concentrate K+ was performed (Fig. 1C) using utricles taken from P2–P4 animals (time of peak cyst yield; Fig. 1B). The size of a given cyst remained unchanged throughout the duration of the study, except during the first hours of cyst growth: the cyst lumen is first distinguishable after 24 h in vitro, when the lumen is too small to allow reliable recordings. By 48 h, the cyst walls completed their growth and from then on, both the size and the shape of the cyst remained constant. By 2 DIV, a substantial [K+] of 19.1 ± 4.0 mm was measured in the endocystic medium. By 5 DIV, K+ accumulation increases to reach values that remained fairly steady for ∼4 DIV. Such a time course of intraluminal K+ accumulation is consistent with pioneer studies showing that endolymphatic [K+] rise occurred between P4 and P8 in mouse (Anniko and Nordemar, 1980). During the plateau period, the average [K+] of 82.0 ± 2.8 mm was close to previously reported in vivo values ranging from 101 to 118 mm (Marcus et al., 2002; Royaux et al., 2003; Dravis et al., 2007). After 11 DIV, [K+] decreased as cultures degenerated and no reliable recordings were possible after the end of the second week of culture.
Transepithelial potential in mouse cysts
Differences in the potential between the endocystic compartment and the culture medium were recorded for 13 DIV. Cysts were grown from P2 to P4 utricles and the recordings began at 2 DIV as cysts first appeared. The Vt was predominantly negative during the course of the study. Furthermore, Vt was quite stable until 9 DIV; its amplitude ranged from −1.4 ± 1 to −3.7 ± 0.5 mV with a mean potential of −2.2 ± 0.2 mV over the 9 DIV period (Fig. 1D). This mean potential was similar to in vivo endo-utricular potentials recorded between 0.4 and −5 mV (Marcus et al., 2002; Royaux et al., 2003; Dravis et al., 2007; Nakaya et al., 2007). Past 9 DIV, Vt notably varied with time, displaying values that were significantly different (p < 0.001) from the mean. From 10 to 13 DIV, the potential oscillated from −6.0 ± 1.0 mV to 3.6 ± 1.0 mV. Such a variation may reflect deregulations in both the negative (dictated by hair cells) and positive (born by dark cells) components of Vt, foreshadowing cyst degeneration and precluding recordings past 13 DIV.
FM1-43, gadolinium, and aminoglycosides increase endocystic K+ content
To assess the impact of K+ efflux mechanisms in the developing K+ endolymphatic homeostasis, we tested the effects of hair cell transducer channel blockers on the endocystic [K+] ([K+]ec). Seven-DIV-old cysts were incubated in medium containing either FM1-43 (5 μm), gadolinium (50 and 100 μm), or aminoglycosides. Two aminoglycosides were used—gentamicin (1 mm) and amikacin (1 mm)—because the former is harmful in vivo for vestibular hair cells while the latter is not. In addition to gadolinium at 50 μm, which was ineffective regardless of the duration of exposure (data not shown), [K+]ec significantly increased following 45 min of incubation for FM1.43; it is considered that it reaches stereocilia within 15 min (Fig. 2A) and 60 min incubation with other components (Fig. 2B). The maximal increase in K+ content obtained were as follow: FM1.43 mediated a 35.9% increase at 45 min (98.8 ± 7.4 mm vs control: 72.7 ± 3.5 mm, p < 0.01), gentamicin produced a 38.6% increase at 90 min (99.4 ± 2.4 mm vs control: 71.7 ± 3.8 mm, p < 0.001), amikacin caused a 40.4% increase at 120 min (113.0 ± 4.3 mm vs control: 80.5 ± 4.5 mm, p < 0.001), and 100 μm gadolinium increased [K+] by 56.1% at 120 min (114.9 ± 7.4 mm vs control: 73.6 ± 3.5 mm, p < 0.001).
Figure 2.
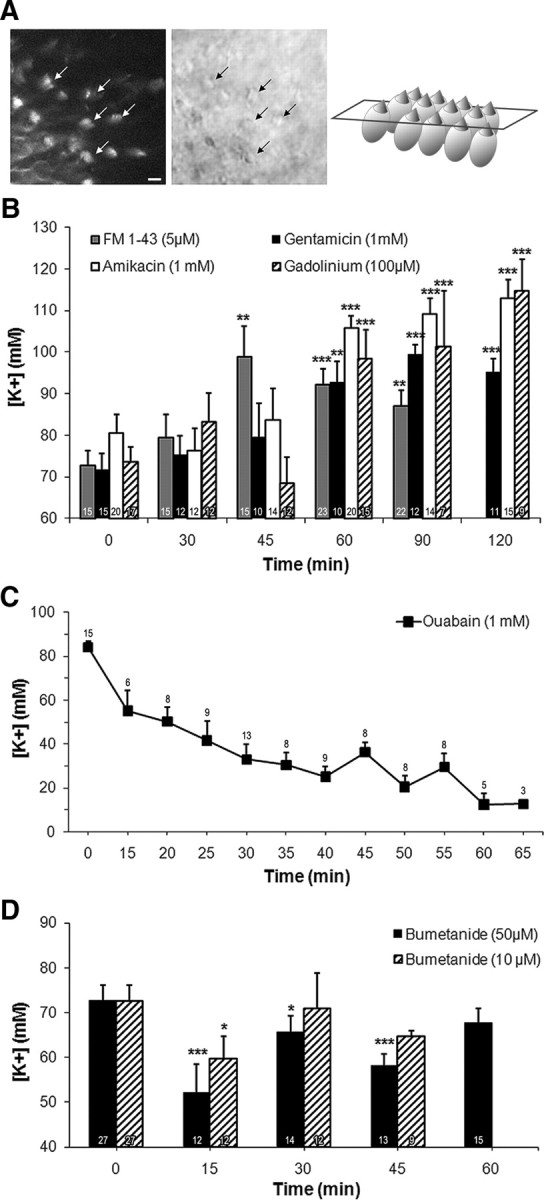
Inhibiting the transduction channels, Na/K-ATPase and NKCC1 alters [K+]ec. Cysts obtained from 2- to 3-d-old mice were cultured for 7 DIV. A, Left, Confocal imaging of FM1-43 fluorescence; middle, differential interference contrast imaging; right, schematic representation of the imaged sensory epithelium. When added to the media cated, FM1.43 (5 μm) reached the hair cell stereocilia within 15 min (arrows indicate hair bundles). Scale bar, 10 μm. B, FM1-43, gentamicin (1 mm), amikacin (1 mm), and gadolinium (100 μm) significantly increased [K+]ec when added to culture medium (45 min for FM1-43, 60 min for all other compounds) with respect to their corresponding untreated control levels (shown at t = 0). These control levels were not statistically significantly different. C, Incubation with ouabain (1 mm) caused a time-dependent decrease in [K+]ec. After as little as 15 min of treatment, ouabain significantly reduced [K+] to 55.1 ± 9.2 mm (p < 0.01). This corresponds to a significant 34% decrease with respect to control level (value plotted at t = 0 min). Maximum inhibition was recorded at 60 min when [K+]ec was reduced to 12.3 ± 5.2 mm (p < 0.001). D, Fifteen minutes incubation with bumetanide caused transient 17% (10 μm) and 28% (50 μm) decrease in the [K+]ec compared with the control levels (plotted at t = 0 min). Because the effects of both concentrations of bumetanide were assessed in parallel in the time course experiment, the control level was the same in both cases and was plotted twice at t = 0 min for graphic simplicity. [K+]s becomes similar to control levels when bumetanide was applied for 30 min at10 μm and for 60 min at 50 μm. Data are represented as mean ± SEM of several measures, the number of which are indicated at the bottom of each bar (i, D) or above each symbol (C). *p < 0.05; **p < 0.01; ***p < 0.001. Note that the scales of the y-axes are different in B–D.
Blocking the Na/K-ATPase pump and the Na-K-2Cl cotransporter 1 reduced K+ endocystic storage
Inhibition of two main ion transporters, the Na/K-ATPase pump and the Na-K-2Cl cotransporter 1 (NKCC1), by ouabain (1 mm) and bumetanide (10 and 50 μm), respectively, were performed in mouse cysts cultured for 7 d. Electrophysiological determination of [K+] in ouabain-treated cysts showed a dramatic time-dependent decrease of [K+]ec (Fig. 2C). After 15 min of treatment, ouabain induced a significant 34% decrease in [K+] (from control level 84.0 ± 2.9 mm to 55.1 ± 9.2 mm, p < 0.01). Maximum inhibition of 85% was recorded at 60 min when [K+]ec was reduced to 12.3 ± 5.2 mm (p < 0.001). Similarly, 15 min incubation with 10 and 50 μm bumetanide caused significant 17% and 28% reduction in K+ endocystic content, respectively (from control level 72.6 ± 3.5 mm to 59.8 ± 4.9 mm, p < 0.05, with 10 μm bumetanide and to 52.1 ± 6.4 mm, p < 0.001, with 50 μm bumetanide; Fig. 2D). However, unlike the permanent effect of ouabain, bumetanide-mediated inhibition was transient, as significant effects disappeared after 30 min exposure to 10 μm bumetanide and 60 min to 50 μm.
Cysts were simultaneously treated with ouabain (1 mm) and bumetanide (50 μm) for 15 min (time of exposure where bumetanide displayed the most prominent effect) to block both ion transporters. In the presence of both inhibitors, K+ accumulation reached 54.2 ± 6.2 mm, which was not different from levels obtained with either inhibitor alone (Table 1). This result was in agreement with the fact that Na/K-ATPase provides the driving force necessary for NKCC1 function when it pumps Na+ out of the cytosol. Hence, it was not surprising that blocking NKCC1 in addition to Na/K-ATPase did not reduce [K+]ec further.
Table 1.
Dual inhibition of K+ transfer systems lacks synergy in 7 DIV cysts
| Targets | Inhibitors | 15 min treatment |
Controls |
Significance (vs controls) | Inhibition (%) | ||
|---|---|---|---|---|---|---|---|
| [K+] (mm) | N | [K+] (mm) | n | ||||
| Na/K-ATPase | Ouabain (1 mM) | 55.1 ± 9.2 | 16 | 84.0 ± 2.9 | 29 | ** | 34 |
| NKCC1 | Bumetanide (50 μm) | 52.1 ± 6.4 | 12 | 72.6 ± 3.5 | 27 | *** | 28 |
| Na/K-ATPase and NKCC1 | Ouabain (1mM) and bumetanide (50 μm) | 54.2 ± 6.2 | 15 | 75.9 ± 4.9 | 12 | ** | 29 |
**p < 0.01;
***p < 0.001.
Expression of Na/K-ATPase α subunit and Na-K-2Cl cotransporter 1 in utricular epithelial cells
Immunofluorescence studies to detect the pump and the cotransporter were performed in 7-DIV-old cysts, dissected from P3 pups, as well as in age-matched (P10) and adult utricles. Staining in kidney was used as a positive control (Fig. 3A) because the Na/K-ATPase α subunit and NKCC1 are highly expressed in the thick ascending limb of the loop of Henle; negative controls were obtained by omitting the primary antibodies (data not shown). In adult utricle, the Na/K-ATPase α subunit and NKCC1 were expressed in both dark and transitional cells (at a higher level in dark cells) as well as in the hair cell-containing sensory epithelium (Fig. 3B). This epithelium was identified by means of antibodies directed against calretinin, which is synthesized by hair cells. In young vestibular epithelia, the Na/K-ATPase α subunit was detected in the sensory epithelium, in transitional cells, and dark cells. In contrast, NKCC1 was expressed in the sensory epithelium and in transitional cells, but not in dark cells (Fig. 3C). Consistently in cysts, the Na/K-ATPase α subunit was produced by hair cells, transitional cells, and dark cells, whereas NKCC1 was only found in transitional cells (Fig. 4).
Figure 3.
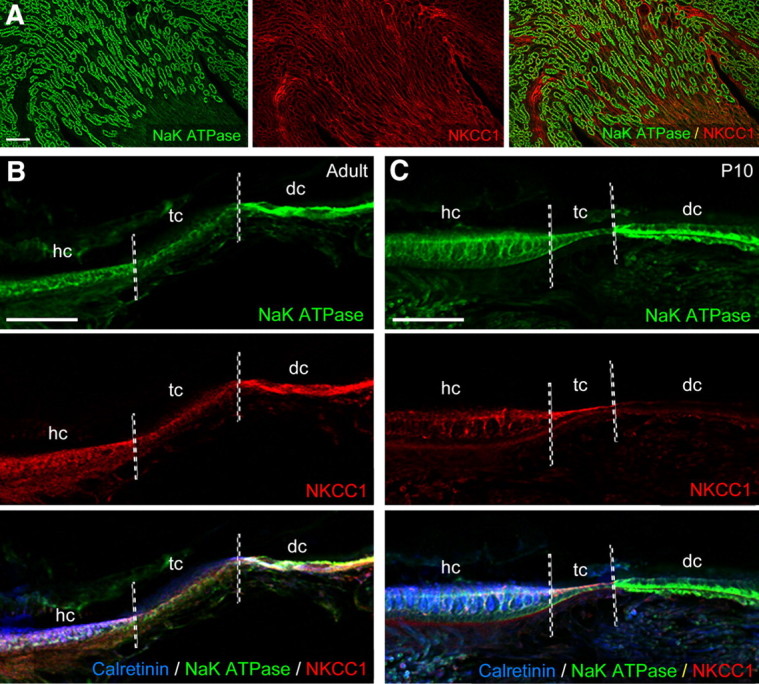
Expression of Na/K-ATPase and NKCC1 in mature and immature mouse utricles. A, Immunofluorescence detections of the pump and the cotransport were performed on kidney as positive controls (left, Na/K-ATPase; middle, NKCC1; right, merge). B, In adult utricles, Na/K-ATPase (top) was expressed in the sensory epithelium, localized by means of calretinin immunoreactivity (bottom, merge; blue) that identifies hair cells, in the adjacent transitional cells, and at a relative higher level in dark cells. Staining for NKCC1 was found in dark cells and, at a lower intensity, in the sensory epithelia and transitional cells (middle). C, Top, In P10 utricles, Na/K-ATPase was detected is in the sensory epithelium, transitional cells, and dark cells. Middle, NKCC1 was expressed in the sensory epithelium and in transitional cells, but not in dark cells. Bottom, Merged image summarizing the areas labeled in the immature utricle. Scale bars, 100 μm. hc, Hair cell; tc, transitional cell; dc, dark cell.
Figure 4.

Expression of Na/K-ATPase and NKCC1 in mouse cysts. A, Cross section of a cyst labeled with antibodies against Na/K-ATPase (left) and NKCC1 (middle). Right, Merged picture, supplemented with a staining of hair cells obtained by using antibodies against calretinin. B, High magnifications of the area delineated by a white square in A. Left, Na/K-ATPase expression was observed in dark cells and at lower level in transitional cells and hair cells. Middle, NKCC1 expression was restricted to transitional cells. Right, Merged picture. Scale bars: A, 100 μm; B, 10 μm. hc, Hair cell; tc, transitional cell; dc, dark cell; L, lumen.
Deletion of Kcne1 gene abolishes K+ accumulation in cysts
The involvement of the Kcne1 channel subunit in K+ secretion was assessed by measuring the of [K+] in cysts obtained from Kcne1−/− mice. As in the Kcne1−/− vestibule, where morphological degradation occurs as early as P3 (Vetter et al., 1996), degeneration of Kcne1−/− cysts begins at 5 DIV. At this time, the cysts become brownish under the microscope; the darkening of this tissue prevented accurate visualization of the lumen for optimal electrode placement. Therefore, measurements of the physiological features of the endocystic compartment were performed at 4 DIV, when Kcne1−/− cysts appeared as healthy as control cysts. Potassium concentration in Kcne1−/− cysts was drastically reduced to 9.3% of the [K+] of cysts grown from wild-type C57BL/6J littermates (Table 2). The level of endocystic K+ accumulation in this background did not statistically differ from that obtained in the Swiss background at 4 DIV. Kcne1−/− cysts displayed relatively higher Vt than C57BL/6J cysts (p < 0.05), whereas the transepithelial potentials of C57BL/6J and Swiss wild-type cysts were comparable (Table 2).
Table 2.
Deletion of the Kcne1 gene alters both endocystic [K+] and transepithelial potential in 4 DIV cysts
| N | Statistical significance | ||
|---|---|---|---|
| Endocystic [K+] (mm) | |||
| Kcne1−/− | 6.3 ± 1 | 3 | versus C57BL/6J: p < 0.01 |
| C57BL/6J | 67.8 ± 8.9 | 8 | |
| Swiss | 65.8 ± 5.5 | 9 | versus C57BL/6J: nonsignificant |
| Transepithelial potential (mV) | |||
| Kcne1−/− | −0.8 ± 0.8 | 8 | versus C57BL/6J: p < 0.05 |
| C57BL/6J | −2.5 ± 1.0 | 14 | |
| Swiss | −2.5 ± 1.4 | 18 | versus C57BL/6J: nonsignificant |
Pharmacological alterations of the mouse cyst transepithelial potential
We assessed whether the [K+]ec alterations caused by pharmacological agents (see above; Fig. 2) also provoked changes to the transepithelial potential. To achieve this goal, utricles were dissected from P2–P3 mice and cultured for 7 DIV. At this time, they were incubated with ouabain (1 mm, 40 min), bumetanide (10 and 50 μm, 15 min), FM1-43 (5 μm, 45 min), gadolinium (100 μm, 60 min), gentamicin (1 mm, 90 min), and amikacin (1 mm, 90 min). For each drug, the selected treatment time corresponded to those where the molecule saliently impacts the [K+] (Fig. 2B–D). Regardless of the compound tested, Vts remained negative (Fig. 5). FM1-43 and gentamicin actions were not significantly different from control levels (−1.3 ± 0.5 mV), whereas the other molecules significantly diminished Vt. The transepithelial potentials were as follows: ouabain, −7.9 ± 2.0 mV; bumetanide, −9.0 ± 1.6 mV (50 μm) and −4.6 ± 0.7 mV (10 μm); gadolinium, −4.7 ± 0.3 mV; and amikacin, −4.3 ± 0.6 mV. Concerning the influence of Vt on [K+] measurements obtained with single-barrel electrodes, the drug-induced changes in Vt would only slightly overestimate the ouabain- and bumetanide-induced [K+] decreases and underestimate the gadolinium- and amikacin-evoked [K+] increases without detracting from the physiological meaning of the data.
Figure 5.

Modulations of [K+]ec alter the endocystic potential. Utricles were dissected from P2–P3 mice and cultured for 7 DIV. At this time, they were incubated with ouabain (1 mm, 40 min), bumetanide (Bum; 10 and 50 μm, 15 min), FM1-43 (5 μm, 45 min), gadolinium (100 μm, 60 min), gentamicin (1 mm, 90 min), and amikacin (1 mm, 90 min). For each drug, the selected time of use corresponded to the times when the drug saliently impacts the [K+] (for details, see Fig. 2 and the Results). For simplicity, the changes in [K+]s are summarized in the upper graph. Note that the control K+ level used was the mean of all control data from the experiments depicted in Figure 2 and was equal to 72.4 ± 3.8. The lower graph shows the Vt recorded in cysts exposed to various pharmacological agents. Control Vt was −1.3 ± 0.5 mV in untreated cysts. Significant decreases in potential were mediated by ouabain (−7.9 ± 2.0 mV), bumetanide (−9.0 ± 1.6 mV at 10 μm and −4.6 ± 0.7 mV at 50 μm), gadolinium (−2.7 ± 0.3 mV), and amikacin (−4.3 ± 0.6 mV). Data are represented as mean ± SEM of several measures, the number of which are indicated at the top or bottom of each bar. *p < 0.05, **p < 0.01; ***p < 0.001.
Discussion
The vestibular cyst is a developmental model maintaining an endolymph-like medium with Vt and [K+] reaching levels close to in vivo values (Marcus et al., 2002; Royaux et al., 2003; Dravis et al., 2007; Nakaya et al., 2007). Moreover, [K+]ec is regulated by the same mechanisms controlling in vivo endolymph homeostasis (Wangemann, 1995, 2002; Lang et al., 2007): efflux through FM1.43-, gadolinium-, and aminoglycoside-sensitive channels (transduction channels), influx relying on ouabain- and bumetanide-sensitive transporters (Na/K-ATPase and NKCC1), and KCNE1. However, experiments performed with cysts and P10 utricles underline the key role of Na/K-ATPase in endolymph formation as well as particular developmental functions of dark and transitional cells during endolymph maturation.
In late embryonic development, hair cell mechanotransduction is acquired when transduction channel begin to be expressed (Géléoc and Holt, 2003). Later, when electrophysiological requirements are met, K+ flow through transduction channels becomes the main efflux challenging the K+ endolymph homeostasis. Here, the capacity for such efflux in the hair cells of the cyst model was assessed by means of various transduction channel blockers. All compounds raise [K+]ec, indicating that the loss of endolymphatic K+ through transducer channels was impeded. Significant increases in [K+] were recorded 45–60 min after drugs application. Such long latencies may reflect the time required for channel antagonists to accumulate to an efficient level in the cyst lumen. The pathways by which antagonists may enter the endolymph remain mostly unknown, apart for aminoglycoside entry; dark cells may be involved, since they take up transtympanically administered gentamicin (Roehm et al., 2007). Whereas aminoglycoside action as a transduction channel permeant blocker (Kroese et al., 1989; Gale et al., 2001; Géléoc and Holt, 2003) is consistent with the enhancement of [K+]ec, the contribution of aminoglycoside ototoxicity should also be considered. Neomycin provokes apical lesions of cochlear hair cells and 48 h gentamicin treatment damages stereocilia in cultured utricles (Richardson and Russell, 1991; Taura et al., 2006). Thus, stereocilia destruction may annihilate K+ efflux through the transducer channels. In our model however, this effect can be excluded: both vestibulotoxic gentamicin and nonvestibulotoxic amikacin (Aran et al., 1995) increased [K+]ec. Conversely, the secreting capability of dark cells may not be drastically altered because their Na/K-ATPase expression appeared unaffected following aminoglycoside administration (Yoshihara et al., 1994). Consequently, K+ efflux, through hair cell' transduction channels, is functional in the cyst. Moreover, our data are consistent with the small fraction of transduction channels that are open at rest (Roberts et al., 1988), as only the open state is compatible with blockage by aminoglycosides and FM1-43 (Gale et al., 2001; Meyers et al., 2003). Likewise, the observation that transduction channel blockers moderately raised [K+]ec are in accordance with the idea that a restricted portion of transduction channels are open at rest in the cyst.
At adulthood, the primary K+ influx through dark cells is drastically reduced when bumetanide blocks NKCC1 expressed at their basolateral membranes (Marcus and Shen, 1994; Marcus et al., 1994; Crouch et al., 1997; Delpire et al., 1999). Unlike these reported robust bumetanide-evoked inhibitions of K+ flux, bumetanide moderately and transiently decreased [K+]ec in our hands. While differences in species used (gerbil vs mouse) may account for this discrepancy, an explanation is also provided by NKCC1 immunolabeling in the nonsensory epithelium. In cysts and age-matched young animals, NKCC1 is found solely in transitional cells as opposed to adult mice where NKCC1 is produced by both dark and transitional cells (Delpire et al., 1999; Crouch et al., 1997; Choi et al., 2005; our data). Such immaturity in NKCC1 expression seems analogous to that reported in the cochlear stria vascularis, where NKCC1 expression reaches maturity by P12 (Sakaguchi et al., 1998). Thus, absence of NKCC1 in immature dark cells may explain the reduced impact of bumetanide. Importantly, it appears that transitional cells secrete K+ in the endolymph at immature stages, since bumetanide still decreases [K+]ec despite the lack of NKCC1 expressed in dark cells. In this secretion, NKCC1 loads transitional cells with K+ that may flow into the endolymph via apical KCNK channels (Nicolas et al., 2004; Popper et al., 2008). Such findings are in accord with the above-mentioned findings by Marcus and colleagues (Marcus and Shen, 1994; Marcus et al., 1994), which were obtained with mature animals, and with investigations of transitional cell ion fluxes that did not detect K+ secretion because they were performed in the presence of bumetanide (Lee et al., 2001).
A compensatory action of Na/K-ATPase may explain the transient nature of bumetanide inhibition. Indeed, the pump could be upregulated by [K+] increases in the basolateral microenvironment due to the inhibition of NKCC1. The Na/K-ATPase α subunits are sensitive to K+, with a half-maximal activation being ∼2 mm external K+ (Glitsch, 2001). At the dark cell level, basolateral K+ elevations increase transepithelial K+ transfer (Marcus et al., 1994; Vetter et al., 1996; Wangemann et al., 1996). Moreover, heterozygous loss-of-function demonstrates that the pump has priority over NKCC1 in regulating the cochlear endolymph (Diaz et al., 2007). Last, coadministration of ouabain and bumetanide failed to increase [K+]ec inhibition, indicating that the pump drives NKCC1 (Table 1).
Assigning the strong impact of ouabain, which caused the most dramatic drop in [K+]ec, solely to the inhibition of dark cell Na/K-ATPase would not take into account other vestibular targets. Na/K-ATPase is also expressed in transitional cells and hair cells (Spicer et al., 1990; Kawasaki et al., 1992; Pitovski and Kerr, 2002). We found similar expression in immature and mature utricles and in cysts. The ouabain block of the K+ secreting potential of transitional cells demonstrated in the present study may be the consequence of blocked intracellular K+ loading, as is the case for dark cell pumps. Alternatively, ouabain depolarizes the transitional cells (Wangemann and Marcus, 1989), thus reducing the driving force on K+ and the putative K+ absorption through P2X2 and nonselective cation channels (Lee et al., 2001), but the existence of such a process in developing utricles needs further investigation. Inhibition of hair cell Na/K-ATPase should result in failure of hair cell recovery from depolarization, hence reducing the K+ efflux through transduction channels. Dampening this efflux may moderate the decrease in endolymphatic [K+] due to ouabain-mediated dark cell inhibition. Consequently, the global impact of ouabain on the [K+]ec may comprise the combined disruption of cation absorptions and K+ secretions. With the latter overpowering the former, the overall ouabain effect on cyst endolymph is the observed fall in [K+]. Finally, NKCC1 is incapable of compensating for the ouabain-mediated inhibition since NKCC1 relies on the pump activity to drive the Na+ gradient necessary for its function (Gamba, 2005). The induction of the most pronounced alteration in [K+]ec by Na/K-ATPase inhibition highlights the executive role of this pump in endolymph homeostasis, as shown for the cochlear endolymph (Diaz et al., 2007). This conclusion is supported by the deregulation of Na/K-ATPase, which is mediated by anomalies affecting endogenous ouabain and may be involved in Menière's disease (Teggi et al., 2010).
The observed alterations in [K+]ec and Vt mediated by ouabain and bumetanide suggest that K+ drives Vt. Indeed, both molecules reduce [K+]ec and Vt, suggesting that the potential becomes more negative due to cation loss. However, this does not hold when K+ efflux is manipulated (FM1.43, gadolinium, and aminoglycosides treatments). These compounds increase [K+]ec but not Vt. Currently uncharacterized mechanisms triggered by a rise in [K+]ec may limit Vt changes. For instance, anion influxes may electrostatically compensate [K+] increases. Canal/wall cells and transitional cells may contribute to this phenomenon because canal cells secrete Cl− (Milhaud et al., 2002) and transitional cells express pendrin, a Cl−/HCO3− transporter, and carbonic anhydrase (Lim et al., 1983; Takumida et al., 1989; Hsu, 1991; Royaux et al., 2003; Wangemann et al., 2004). In addition, Vt regulation may overcompensate the triggering of [K+]ec changes, since both amikacin and gadolinium, which globally produce the most salient K+ elevation, move Vt toward the most negative values. Such overcompensation would prevent an increase in the driving force on endolymphatic K+ and the related hair cell hyperstimulation that would, otherwise, result in vestibular dysfunction.
In conclusion, during development, [K+] appears primarily regulated by Na/K-ATPase, as opposed to the adult stage where NKCCI also contributes to K+ homeostasis. At the beginning of the endolymph formation, transitional cells may assist the dark cells to secrete K+ in the endolymph. This partnership may occur solely when the secreting power of dark cells is not at its peak due to immature NKCC1 expression. Later, transitional cells might switch from K+ secretion to K+ absorption to shield hair cells from overstimulation; but this requires downregulation of their basolateral K+ uptake (Fig. 6). This downregulation could involve Na/K-ATPase regulators such as endogenous ouabain (Bagrov et al., 2009) or FXDY proteins, which modulate the stability and ionic affinity of the pump (Delprat et al., 2007; Geering, 2008).
Figure 6.
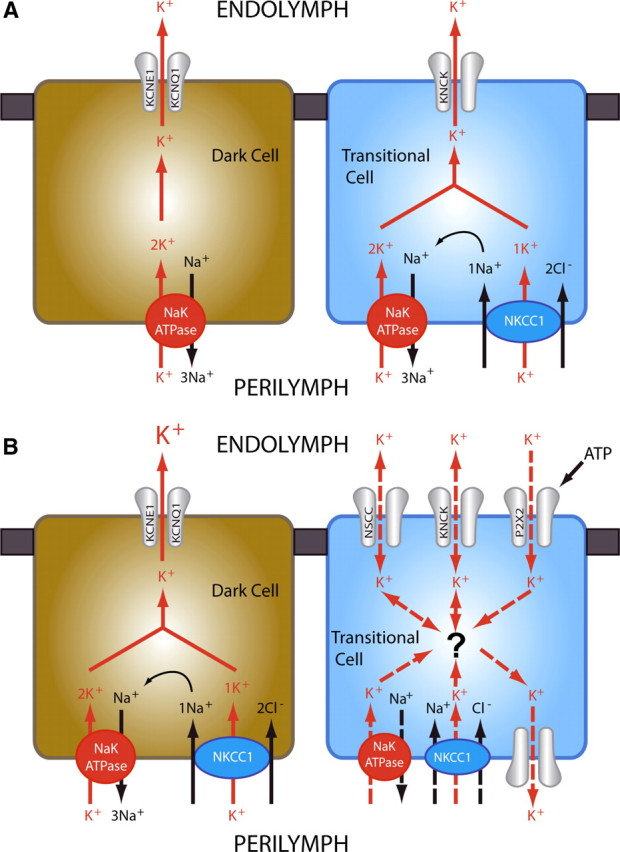
Models of K+ secretion in the vestibular endolymph. A, During the postnatal formation of the endolymphatic compartment, K+ secretion involves both dark cells and transitional cells. The former have not attained their maximal secreting power because they do not yet express NKCC1; the latter express both NKCC1 and Na/K-ATPase, which take K+ up into transitional cells. K+ then leaks from the transitional cells into the forming endolymph through KCNK channels (Nicolas et al., 2004). K+ flux from dark cells to the endolymph is already performed by the KCNE1/KCNQ1 channels, since these proteins are expressed during the late gestational development (Nicolas et al., 2001). In conclusion, in the early postnatal period, transitional cells could be considered as K+-secreting cells that assist dark cells in the process of endolymph maturation. B, At maturity, dark cells express both NKCC1 and Na/K-ATPase, which enable them to carry out the complete endolymph K+ secretion, whereas transitional cell are endowed with a constitutive K+ absorption capability through nonselective cation channels (NSCC), P2X2 receptors (Lee et al., 2001), and KCNK channels (Nicolas et al., 2004; Popper et al., 2008). Absorbed K+ may leak in the perilymph through background K+ channels that remain to be characterized. However, since transitional cells still express NKCC1 and Na/K-ATPase, they may mask their secreting potential by means of a currently unknown mechanism (indicated with a question mark). Active basolateral K+ uptake would create a [K+] gradient that would prevent K+ absorption. Hence, at adulthood, transitional cells could be considered as modulator cells of the endolymph [K+], absorbing K+ in case of overstimulation to protect hair cells and switching to K+ secretion if dark cells alone are incapable of handling this task.
Footnotes
This work was supported by a grant from the Centre National des Etudes Spatiales (to M.C.). We are grateful to Drs. Vasiliki Kalatzis, R. Fernando, B. Delprat and J. Dyhrfjeld-Johnsen for scientific discussions and critical reading of the manuscript. We thank Dr. K. Geering (University of Lausanne, Lausanne, Switzerland) for providing the antibodies against the Na/K-ATPase α subunit and JL Pasquier for his help with the figures.
The authors declare no financial conflicts of interest.
References
- Anniko M, Nordemar H. Embryogenesis of the inner ear. IV. Post-natal maturation of the secretory epithelia of the inner ear in correlation with the elemental composition in the endolymphatic space. Arch Otorhinolaryngol. 1980;229:281–288. doi: 10.1007/BF02565531. [DOI] [PubMed] [Google Scholar]
- Aran JM, Chappert C, Dulon D, Erre JP, Aurousseau C. Uptake of amikacin by hair cells of the guinea pig cochlea and vestibule and ototoxicity: comparison with gentamicin. Hear Res. 1995;82:179–183. doi: 10.1016/0378-5955(94)00175-p. [DOI] [PubMed] [Google Scholar]
- Bagrov AY, Shapiro JI, Federova OV. Endogenous cardiotonic steroids: physiology, pharmacology and novel therapeutic targets. Pharmacol Rev. 2009;61:9–38. doi: 10.1124/pr.108.000711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crouch JJ, Sakaguchi N, Lytle C, Schulte BA. Immunohistochemical localization of the Na-K-Cl co-transporter (NKCC1) in the gerbil inner ear. J Histochem Cytochem. 1997;45:773–778. doi: 10.1177/002215549704500601. [DOI] [PubMed] [Google Scholar]
- Delpire E, Lu J, England R, Dull C, Thorne T. Deafness and imbalance associated with inactivation of the secretory Na-K-2Cl cotransporter. Nat Genet. 1999;22:192–195. doi: 10.1038/9713. [DOI] [PubMed] [Google Scholar]
- Delprat B, Schaer D, Roy S, Wang J, Puel JL, Geering K. FXYD6 is a novel regulator of Na,K-ATPase expressed in the inner ear. J Biol Chem. 2007;282:7450–7456. doi: 10.1074/jbc.M609872200. [DOI] [PubMed] [Google Scholar]
- Deol MS. Inherited diseases of the inner ear in man in light of studies on the mouse. J Med Genet. 1968;5:137–158. doi: 10.1136/jmg.5.2.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaz RC, Vazquez AE, Dou H, Wei D, Cardell EL, Lingrel J, Shull GE, Doyle KJ, Yamoah EN. Conservation of hearing by simultaneous mutation of NaK-ATPase and NKCC1. J Assoc Res Otolaryngol. 2007;8:422–434. doi: 10.1007/s10162-007-0089-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dravis C, Wu T, Chumley MJ, Yokoyama N, Wei S, Wu DK, Marcus DC, Henkemeyer M. EphB2 and ephrin-B2 regulate the ionic homeostasis of vestibular endolymph. Hear Res. 2007;223:93–104. doi: 10.1016/j.heares.2006.10.007. [DOI] [PubMed] [Google Scholar]
- Gaboyard S, Chabbert C, Travo C, Bancel F, Lehouelleur J, Yamauchi D, Marcus DC, Sans A. Three-dimensional culture of newborn rat utricle using an extracellular matrix promotes formation of a cyst. Neuroscience. 2005;133:253–265. doi: 10.1016/j.neuroscience.2005.02.011. [DOI] [PubMed] [Google Scholar]
- Gale JE, Marcotti W, Kennedy HJ, Kros CJ, Richardson GP. FM1-43 dye behaves as a permeant blocker of the hair-cell mechanotransducer channel. J Neurosci. 2001;21:7013–7025. doi: 10.1523/JNEUROSCI.21-18-07013.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gamba G. Molecular physiology and physiopathology of electroneutral cation-chloride cotransporters. Physiol Rev. 2005;85:423–493. doi: 10.1152/physrev.00011.2004. [DOI] [PubMed] [Google Scholar]
- Geering K. Functional roles of Na,K-ATPase subunits. Curr Opin Nephrol Hypertens. 2008;17:526–532. doi: 10.1097/MNH.0b013e3283036cbf. [DOI] [PubMed] [Google Scholar]
- Géléoc GS, Holt JR. Developmental acquisition of sensory transduction in hair cells of the mouse inner ear. Nat Neurosci. 2003;6:1019–1020. doi: 10.1038/nn1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glitsch HG. Electrophysiology of the sodium-potassium-ATPase in cardiac cells. Physiol Rev. 2001;81:1791–1826. doi: 10.1152/physrev.2001.81.4.1791. [DOI] [PubMed] [Google Scholar]
- Hsu CJ. Ultrastructural study of cytochemical localization of carbonic anhydrase in the inner ear. Acta Otolaryngol. 1991;111:75–84. doi: 10.3109/00016489109137357. [DOI] [PubMed] [Google Scholar]
- Kawasaki K, Yamamoto A, Omori K, Iwano T, Kumazawa T, Tashiro Y. Quantitative immunoelectron microscopic localization of Na, K-ATPase alpha-subunit in the epithelial cells of rat vestibular apparatus. Hear Res. 1992;60:64–72. doi: 10.1016/0378-5955(92)90059-v. [DOI] [PubMed] [Google Scholar]
- Kimitsuki T, Nakagawa T, Hisashi K, Komune S, Komiyama S. Gadolinium blocks mechano-electric transducer current in chick cochlear hair cells. Hear Res. 1996;101:75–80. doi: 10.1016/s0378-5955(96)00134-7. [DOI] [PubMed] [Google Scholar]
- Kroese AB, Das A, Hudspeth AJ. Blockage of the transduction channels of hair cells in the bullfrog's sacculus by aminoglycoside antibiotics. Hear Res. 1989;37:203–217. doi: 10.1016/0378-5955(89)90023-3. [DOI] [PubMed] [Google Scholar]
- Lang F, Vallon V, Knipper M, Wangemann P. Functional significance of channels and transporters expressed in the inner ear and kidney. Am J Physiol Cell Physiol. 2007;293:C1187–C1208. doi: 10.1152/ajpcell.00024.2007. [DOI] [PubMed] [Google Scholar]
- Lee JH, Chiba T, Marcus DC. P2X2 receptor mediates stimulation of parasensory cation absorption by cochlear outer sulcus cells and vestibular transitional cells. J Neurosci. 2001;21:9168–9174. doi: 10.1523/JNEUROSCI.21-23-09168.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis RS, Hudspeth AJ. Voltage- and ion-dependent conductances in solitary vertebrate hair cells. Nature. 1983;304:538–541. doi: 10.1038/304538a0. [DOI] [PubMed] [Google Scholar]
- Lim DJ, Erway LC, Clark DL. Tilted head mice with genetic otoconial anomaly: behavioral and morphological correlates. In: Hood JD, editor. Vestibular mechanisms in health and disease. New York: Academic; 1978. pp. 195–206. [Google Scholar]
- Lim DJ, Karabinas C, Trune DR. Histochemical localization of carbonic anhydrase in the inner ear. Am J Otolaryngol. 1983;4:33–42. doi: 10.1016/s0196-0709(83)80005-2. [DOI] [PubMed] [Google Scholar]
- Marcus DC, Shen Z. Slowly activating voltage-dependent K+ conductance is apical pathway for K+ secretion in vestibular dark cells. Am J Physiol. 1994;267:C857–C864. doi: 10.1152/ajpcell.1994.267.3.C857. [DOI] [PubMed] [Google Scholar]
- Marcus DC, Liu J, Wangemann P. Transepithelial voltage and resistance of vestibular dark cell epithelium from the gerbil ampulla. Hear Res. 1994;73:101–108. doi: 10.1016/0378-5955(94)90287-9. [DOI] [PubMed] [Google Scholar]
- Marcus DC, Wu T, Wangemann P, Kofuji P. KCNJ10 (Kir4.1) potassium knockout abolishes endocochlear potential. Am J Physiol Cell Physiol. 2002;282:C403–C407. doi: 10.1152/ajpcell.00312.2001. [DOI] [PubMed] [Google Scholar]
- Masetto S, Zucca G, Bottà L, Valli P. Endolymphatic potassium of the chicken vestibule during embryonic development. Int J Dev Neurosci. 2005;23:439–448. doi: 10.1016/j.ijdevneu.2005.05.002. [DOI] [PubMed] [Google Scholar]
- Meyers JR, MacDonald RB, Duggan A, Lenzi D, Standaert DG, Corwin JT, Corey DP. Lighting up the senses: FM1-43 loading of sensory cells through nonselective ion channels. J Neurosci. 2003;23:4054–4065. doi: 10.1523/JNEUROSCI.23-10-04054.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milhaud PG, Pondugula SR, Lee JH, Herzog M, Lehouelleur J, Wangemann P, Sans A, Marcus DC. Chloride secretion by semicircular canal duct epithelium is stimulated via beta 2-adrenergic receptors. Am J Physiol Cell Physiol. 2002;283:C1752–C1760. doi: 10.1152/ajpcell.00283.2002. [DOI] [PubMed] [Google Scholar]
- Nakaya K, Harbidge DG, Wangemann P, Schultz BD, Green ED, Wall SM, Marcus DC. Lack of pendrin HCO3− transport elevates vestibular endolymphatic [Ca2+] by inhibition of acid-sensitive TRPV5 and TRPV6 channels. Am J Physiol Renal Physiol. 2007;292:F1314–F1321. doi: 10.1152/ajprenal.00432.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicolas M, Demêmes D, Martin A, Kupershmidt S, Barhanin J. KCNQ1/KCNE1 potassium channels in mammalian vestibular dark cells. Hear Res. 2001;153:132–145. doi: 10.1016/s0378-5955(00)00268-9. [DOI] [PubMed] [Google Scholar]
- Nicolas MT, Lesage F, Reyes R, Barhanin J, Demêmes D. Localization of Trek-1, a two pore domain K+ channel in the peripheral vestibular system of mouse and rat. Brain Res. 2004;1017:46–52. doi: 10.1016/j.brainres.2004.05.012. [DOI] [PubMed] [Google Scholar]
- Pitovski DZ, Kerr TP. Sodium and potassium activated ATPase in the mammalian vestibular system. Hear Res. 2002;171:51–65. doi: 10.1016/s0378-5955(02)00352-0. [DOI] [PubMed] [Google Scholar]
- Popper P, Winkler J, Erbe CB, Lerch-Gaggl A, Siebeneich W, Wackym PA. Distribution of two pore domain potassium channel in the adult rat vestibular periphery. Hear Res. 2008;246:1–8. doi: 10.1016/j.heares.2008.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson GP, Russell IJ. Cochlear cultures as a model system for studying aminoglycoside induced ototoxicity. Hear Res. 1991;53:293–311. doi: 10.1016/0378-5955(91)90062-e. [DOI] [PubMed] [Google Scholar]
- Roberts WM, Howard J, Hudspeth AJ. Hair cells: transduction, tuning, and transmission in the inner ear. Annu Rev Cell Biol. 1988;4:63–92. doi: 10.1146/annurev.cb.04.110188.000431. [DOI] [PubMed] [Google Scholar]
- Roehm P, Hoffer M, Balaban CD. Gentamicin uptake in the chinchilla inner ear. Hear Res. 2007;230:43–52. doi: 10.1016/j.heares.2007.04.005. [DOI] [PubMed] [Google Scholar]
- Royaux IE, Belyantseva IA, Wu T, Kachar B, Everett LA, Marcus DC, Green ED. Localization and functional studies of pendrin in the mouse inner ear provide insight about the etiology of deafness in pendred syndrome. J Assoc Res Otolaryngol. 2003;4:394–404. doi: 10.1007/s10162-002-3052-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakaguchi N, Crouch JJ, Lytle C, Schulte BA. Na-K-Cl cotransporter expression in the developing and senescent gerbil cochlea. Hear Res. 1998;118:114–122. doi: 10.1016/s0378-5955(98)00022-7. [DOI] [PubMed] [Google Scholar]
- Singh R, Wangemann P. Free radical stress-mediated loss of Kcnj10 protein expression in the stria vascularis contributes to deafness in Pendred syndrome mouse model. Am J Physiol Renal Physiol. 2008;294:F139–F148. doi: 10.1152/ajprenal.00433.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spicer SS, Schulte BA, Adams JC. Immunolocalization of Na+,K+-ATPase and carbonic anhydrase in the gerbil's vestibular system. Hear Res. 1990;43:205–217. doi: 10.1016/0378-5955(90)90229-i. [DOI] [PubMed] [Google Scholar]
- Takumida M, Bagger-Sjöbäck D, Wersäll J, Harada Y. Ultrastructural localization of carbonic anhydrase in the vestibular end organs of the guinea pig. Arch Otorhinolaryngol. 1989;246:56–60. doi: 10.1007/BF00454136. [DOI] [PubMed] [Google Scholar]
- Taura A, Kojima K, Ito J, Ohmori H. Recovery of hair cell function after damage induced by gentamicin in organ culture of rat vestibular maculae. Brain Res. 2006;1098:33–48. doi: 10.1016/j.brainres.2006.04.090. [DOI] [PubMed] [Google Scholar]
- Teggi R, Lanzani C, Zagato L, Delli Carpini S, Manunta P, Bianchi G, Bussi M. Gly460Trp a-adducin mutation as a possible mechanism leading to endolymphatic hydrops in Ménière's syndrome. Otol Neurotol. 2008;29:824–828. doi: 10.1097/MAO.0b013e318180a4b1. [DOI] [PubMed] [Google Scholar]
- Teggi R, Zagato L, Delli Carpini S, Messaggio E, Casamassima N, Lanzani C, Manunta P, Bussi M. Endogenous ouabain in Ménière's disease. Otol Neurotol. 2010;31:153–156. doi: 10.1097/MAO.0b013e3181c0eaba. [DOI] [PubMed] [Google Scholar]
- Vetter DE, Mann JR, Wangemann P, Liu J, McLaughlin KJ, Lesage F, Marcus DC, Lazdunski M, Heinemann SF, Barhanin J. Inner ear defects induced by null mutation of isk gene. Neuron. 1996;17:1251–1264. doi: 10.1016/s0896-6273(00)80255-x. [DOI] [PubMed] [Google Scholar]
- Wangemann P. Comparison of ion transport mechanisms between vestibular dark cells and strial marginal cells. Hear Res. 1995;90:149–157. doi: 10.1016/0378-5955(95)00157-2. [DOI] [PubMed] [Google Scholar]
- Wangemann P. K(+) cycling and its regulation in the cochlea and the vestibular labyrinth. Audiol Neurootol. 2002;7:199–205. doi: 10.1159/000063736. [DOI] [PubMed] [Google Scholar]
- Wangemann P, Marcus DC. Membrane potential measurements of transitional cells from the crista ampullaris of the gerbil: effects of barium, quinidine, quinine, tetraethylammonium, cesium, ammonium, thallium and ouabain. Pflugers Arch. 1989;414:656–662. doi: 10.1007/BF00582132. [DOI] [PubMed] [Google Scholar]
- Wangemann P, Shen Z, Liu J. K(+)-induced stimulation of K+ secretion involves activation of the IsK channel in vestibular dark cells. Hear Res. 1996;100:201–210. doi: 10.1016/0378-5955(96)00127-x. [DOI] [PubMed] [Google Scholar]
- Wangemann P, Itza EM, Albrecht B, Wu T, Jabba SV, Maganti RJ, Lee JH, Everett LA, Wall SM, Royaux IE, Green ED, Marcus DC. Loss of KCNJ10 protein expression abolishes endocochlear potential and causes deafness in Pendred syndrome mouse model. BMC Med. 2004;2:30–44. doi: 10.1186/1741-7015-2-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang T, Gurrola JG, 2nd, Wu H, Chiu SM, Wangemann P, Snyder PM, Smith RJ. Mutations of KCNJ10 together with mutations of SLC26A4 cause digenic nonsyndromic hearing loss associated with enlarged vestibular aqueduct syndrome. Am J Hum Genet. 2009;84:651–657. doi: 10.1016/j.ajhg.2009.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshihara T, Kaname H, Ishii T, Igarashi M. Effect of gentamicin on vestibular dark cells and melanocytes: an ultrastructural and cytochemical study. ORL J Otorhinolaryngol Relat Spec. 1994;56:24–30. doi: 10.1159/000276603. [DOI] [PubMed] [Google Scholar]
- Young Choi J, Ho Jung S, Namkung W, Lee JH, Jin Son E, Wook Shin J, Park HY, Sang Lee W, Kim HN. Vestibular malformation in mice lacking Na-K-2Cl cotransporter 1 and expression of Na-K-2Cl cotranspoter 1 in human vestibular end organs. Acta Otolaryngol. 2005;125:1252–1257. doi: 10.1080/00016480510012309. [DOI] [PubMed] [Google Scholar]