

Abstract
Previous studies have shown that neuron-specific deletion of serum response factor (SRF) results in deficits in tangential cell migration, guidance-dependent circuit assembly, activity-dependent gene expression, and synaptic plasticity in the hippocampus. Furthermore, SRF deletion in mouse embryonic stem cells causes cell death in vitro. However, the requirement of SRF for early neuronal development including neural stem cell homeostasis, neurogenesis, and axonal innervations remains unknown. Here, we report that SRF is critical for development of major axonal tracts in the forebrain. Conditional mutant mice lacking SRF in neural progenitor cells (Srf-Nestin-cKO) exhibit striking deficits in cortical axonal projections including corticostriatal, corticospinal, and corticothalamic tracts, and they show a variable loss of the corpus callosum. Neurogenesis and interneuron specification occur normally in the absence of SRF and the deficits in axonal projections were not due to a decrease or loss in cell numbers. Radial migration of neurons and neocortical lamination were also not affected. No aberrant cell death was observed during development, whereas there was an increase in the number of proliferative cells in the ventricular zone from embryonic day 14 to day 18. Similar axonal tract deficits were also observed in mutant mice lacking SRF in the developing excitatory neurons of neocortex and hippocampus (Srf-NEX-cKO). Together, these findings suggest distinct roles for SRF during neuronal development; SRF is specifically required in a cell-autonomous manner for axonal tract development but is dispensable for cell survival, neurogenesis, neocortical lamination, and neuronal differentiation.
Introduction
Neuronal development in the CNS is an intricately coordinated process that involves proliferation and maintenance of neural precursor cells (NPCs), neurogenesis, growth and extension of axons and dendrites, and structural organization within specific brain regions. At the molecular level, these processes are regulated by several extracellular cues through activation of specific transcriptional programs (Goldberg et al., 2002; Zhou and Snider, 2006). Serum response factor (SRF) is a stimulus-dependent transcription factor belonging to the Mcm1-Agamous-Deficiens-SRF-domain family of transcriptional regulators. Thus far, the roles of SRF in CNS development remain poorly understood because of early embryonic lethality of SRF-null mice (Arsenian et al., 1998). Recent studies using conditional SRF mutant mice have begun to elucidate the importance of SRF in CNS development and adult function. Perinatal neuron-specific deletion of SRF results in several developmental abnormalities, including defects in tangential neuronal migration along the rostral migratory stream, deficits in axon guidance within the hippocampal mossy fiber circuitry, hippocampal lamination and dendritic complexity of hippocampal pyramidal neurons, and ultimately resulting in lethality by 3 weeks of age (Alberti et al., 2005; Knöll et al., 2006; Stritt and Knöll, 2010).
In contrast, mice carrying postnatal forebrain-specific deletion of SRF are viable and fertile, and do not exhibit any of the above developmental abnormalities (Ramanan et al., 2005; Etkin et al., 2006). Instead, these mice exhibit specific deficits in activity-dependent expression of several immediate early genes (IEG), including c-Fos, Egr-1, and Arc, in the hippocampus and neocortex (Ramanan et al., 2005). SRF ablation does not affect basal synaptic transmission but disrupts both early and late phases of LTP and LTD in hippocampus and in cultured cerebellar Purkinje neurons (Ramanan et al., 2005; Etkin et al., 2006; Smith-Hicks et al., 2010). Interestingly, SRF loss does not affect neuronal cell survival and maintenance (Ramanan et al., 2005). Defects in activity-dependent transcription and synaptic plasticity are the likely underlying causes of learning and memory deficits observed in these mice (Etkin et al., 2006; Johnson et al., 2011).
Despite these advances, the role of SRF in neural progenitor cell homeostasis, neurogenesis, and neuronal maturation during early brain development remains unknown. In this study, we conditionally deleted SRF in NPCs using a nestin-cre transgenic line to investigate earlier developmental roles of SRF. Srf-Nestin-cKO mutants exhibited neonatal lethality along with several abnormalities in brain architecture. Closer analysis revealed that loss of SRF affected the development of major CNS axonal fiber tracts. However, neurogenesis, neuronal subtype specification, and neuronal survival were unaffected. Similarly, Srf-NEX-cKO mutant mice, lacking SRF only in postmitotic glutamatergic neurons in the neocortex and hippocampus, also exhibited defects in axonal projections suggesting a cell-autonomous role of SRF in axon growth in vivo. Contrary to previous findings, neocortical lamination occurs normally in both these lines of mutant mice. Last, examination of NPCs revealed an accumulation of precursors in Srf-Nestin-cKO mutants suggesting that SRF plays an important role in NPC homeostasis. Thus, our study reveals a critical role for SRF in NPC maintenance and axon outgrowth during CNS development.
Materials and Methods
Animals.
Srff/f mice (control) were maintained as a homozygous colony as previously described (Ramanan et al., 2005). The Srf f/f were crossed to the nestin-Cre transgenic mouse strain (Tronche et al., 1999) (The Jackson Laboratory, Stock # 003771) to generate Srf f/+; Nestin-Cre double heterozygous mice. The double heterozygous mice were bred to Srf f/f mice to obtain Srf f/f; Nestin-Cre (Srf-Nestin-cKO) mutant mice in the expected Mendelian ratio. Similarly, Srf f/f mice were bred to the NEX-cre transgenic mice to generate Srf f/f; NEX-Cre (Srf-NEX-cKO) mice (Goebbels et al., 2006). The Srf-NEX-cKO mice were viable and were bred to Srf f/f mice to propagate the colony. For experiments that required embryos of various developmental stages, we set up timed pregnancies with the day following detection of a vaginal plug being identified as embryonic day 0.5 (E0.5). All experiments were approved by the Animals Studies Committee, Division of Comparative Medicine, Washington University School of Medicine, St. Louis, MO.
Immunohistochemistry.
Immunohistochemistry was performed as previously described (Ramanan et al., 2005). Briefly, postnatal day 0.5 (P0.5) and older animals were fixed by transcardial perfusion. The brains were cryopreserved in 30% sucrose, frozen, and stored at −80°C until use. For staining, 12–16 μm cryosections were made and incubated in blocking/permeabilization solution containing 3% normal goat serum and 0.3% Triton-X in PBS. Embryos until E18.5 were drop-fixed in 4% PFA followed by cryopreservation in 30% sucrose. The following primary antibodies were used: NeuN (1:1000, Millipore Bioscience Research Reagents), SRF (1:1500, Santa Cruz Biotechnology), 2H3/Neurofilament (1:1000, DSHB), activated-Caspase 3 (1:1500, Millipore), Tbr2/EOMES (1:50, Santa Cruz Biotechnology), Tbr1 (1:250, Santa Cruz Biotechnology), Cux1 (1:5000, Santa Cruz Biotechnology), 40E-C/Vimentin (1:50, DSHB), somatostatin (SST; 1:600, Santa Cruz Biotechnology), calbindin (1:2000, Sigma-Aldrich), parvalbumin (1:1000, Sigma-Aldrich), Gad-6 (1:500, DHSB), p-histone H3 (1:500, Sigma-Aldrich), Ki67 (1:500, BD Biosciences), and Sox2 (1:100, Santa Cruz Biotechnology). The following secondary antibodies were used: anti-goat Cy3 (1:300, Jackson ImmunoResearch), anti-mouse Alexa Fluor 594 and Alexa Fluor 488 (1:500, Invitrogen), and anti-rabbit Alexa Fluor 488 and Alexa Fluor 594 (1:500, Invitrogen).
TUNEL.
Embryonic and neonatal brains were perfused with 4% PFA and cryoprotected in 30% sucrose. Samples were sectioned at 12–16 μm. Before staining, sections were permeabilized with 0.1 m citrate buffer, pH 6.0, at 80°C for 30 min. Slides were rinsed with PBS and immersed in 0.1 m Tris-HCl containing 3% BSA and 20% bovine serum for 30 min at room temperature. Finally, 50–100 μl of terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) reaction mixture (Roche) was added per slide and incubated at 37°C in a humidified chamber in dark to complete the staining.
In situ hybridization.
In situ hybridization was performed as previously described (Ramanan et al., 2005). The cDNA clones of Cux2, Klf6, Lhx5, Lmo4, Nfix, Nr4a2, and Sox5 for riboprobes were generously provided by Paul Gray, Washington University School of Medicine, St. Louis, MO. Both sense and antisense riboprobes were synthesized and hybridized and sense strand probes did not produce any signal above that of the background.
Cell counts.
High-magnification (10× or 20×) images of 10 nonconsecutive bregma axis-matched sections were taken using a Nikon 80i epifluorescence microscope. A universal threshold determined by signal to background ratio was applied to all images from control and knock-out samples. Positive cells based on their nuclear staining were counted using Analyze Particle function with constraints on the particle size in pixels (300–2000 pixels) and circularity of the particle (0.4–1.0) in ImageJ software. Total number of counts per area in pixel square was computed and converted to counts per square micrometers based on the magnification of the image.
DiI labeling.
Tiny crystals of 1,1-dioctadecyl-3,3,3′,3′-tetramethyl-indocarbocyanine perchlorate (DiI) similar in size were placed on the surface of the motor and the visual cortices (ipsilateral hemisphere) of neonatal Srf-Nestin-cKO, Srf-NEX-cKO, and control littermate brains, using an insect needle pin. Control and knock-out littermate brain pairs were positioned next to each other to ensure crystal placements were as comparable as possible. Samples were incubated in 37°C for 2–4 weeks and then sectioned coronally, sagittally, or horizontally at 100 μm thickness using a vibratome. Sections were collected as floating sections and mounted serially on glass microscope slides using Vectashield mounting medium with DAPI (Vector Laboratories). For DiI staining of thalamocortical axons in sections, glass beads (250 μm, acid-washed, Supelco) were coated with DiI (2 mg of DiI in 100 μl of methylene chloride to coat 300 mg of glass beads). A single DiI-coated bead was placed in the ventral thalamus of 100 μm paraformaldehyde-fixed coronal section and incubated for 3 weeks at 37°C.
Quantification of axonal projections.
DiI-labeled corticostriatal projections in 2–3 slices were measured for projection length using ImageJ to track and record the absolute length in pixels and then converted to micrometers. For measuring the target innervation of corticothalamic projection, DiI-labeled thalamic area was measured using ImageJ in square pixels and then converted to square micrometers. Mean of projection length or target innervation area of comparable sections from three pairs of control and mutant animals was calculated to quantify for the difference between control and mutants.
Nissl staining.
Fresh frozen brains were sectioned at 20 μm and mounted on gelatin-coated glass slides. After overnight drying, slides were immersed in 0.5% cresyl violet in water for 10 min; rinsed in H2O; dehydrated serially in 50%, 75%, 95%, and 100% ethanol (2 min each), followed by two rinses in xylenes (3 min each); and then coverglass mounted with permount histology mounting medium.
Statistical analyses.
The mean, SD and SEM for cell counts were calculated from images, which were sampled serially to encompass a structural region. Pups of either sex (n = 3–5) from at least two different litters were used in all experiments. Statistical significance between control and mutant pair was determined by Student's t test.
Results
Ablation of SRF in Srf-Nestin-cKO mutant brain
To determine the role of SRF in neurogenesis and CNS development, we deleted SRF using a nestin-Cre transgenic mouse line, in which Cre-mediated recombination has been shown to occur ∼E9.5 to E11.5 (Tronche et al., 1999). Srf-Nestin-cKO animals were born in the expected Mendelian ratio, but the mutant mice did not survive beyond P1 due to unknown reasons. The Srf-Nestin-cKO mice were physically indistinguishable from control littermates (Srf f/f) but exhibited neonatal hemorrhage starting ∼12–16 h after birth (Fig. 1A). In the Srf-Nestin-cKO mutant mice, SRF deletion begins ∼E12.5 and is complete by E14.5 in the brain and spinal cord as determined by immunohistochemistry (data not shown). SRF loss was also confirmed by immunoblotting using whole brain lysates from neonatal control and Srf-Nestin-cKO mice (Fig. 1B). A closer examination of spatial deletion of SRF at P0.5 in Srf-Nestin-cKO brains by immunostaining using anti-SRF antibody demonstrates loss of SRF in all regions examined, including the neocortex, hippocampus, striatum, thalamus, and hypothalamus (Fig. 1C, data not shown). Coimmunostaining for NeuN, a marker of mature neurons, and SRF showed absence of SRF expression in neurons throughout the brain (Fig. 1D, data not shown).
Figure 1.
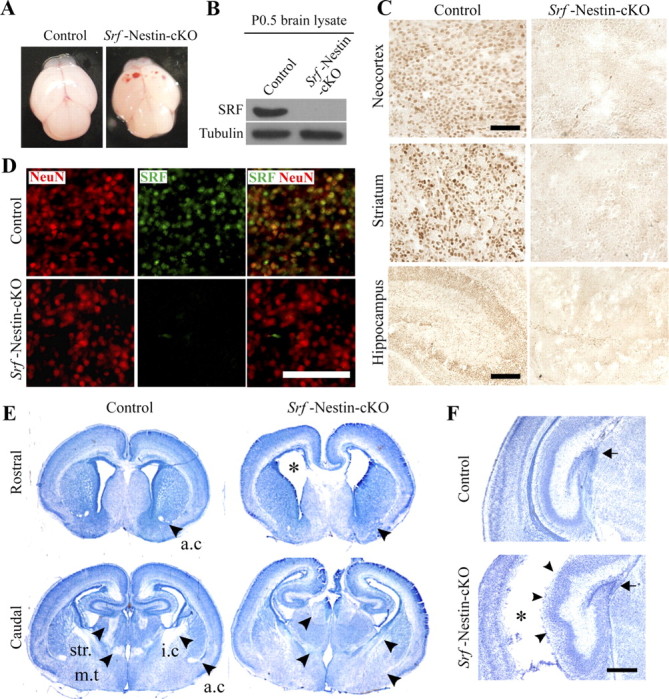
SRF deletion in Srf-Nestin-cKO mutants. A, Srf-Nestin-cKO mice exhibited neonatal hemorrhage starting 12–16 h after birth while the brains of control littermates were normal. The mutant mice died by 18–24 h. The weight of the brains was comparable. B, Immunoblotting using whole brain lysates shows complete loss of SRF in P0.5 Srf-Nestin-cKO mice. C, Immunostaining using a SRF-specific antibody shows loss of SRF expression in forebrain regions including the neocortex, striatum, and hippocampus. Scale bars: Neocortex, 50 μm; others, 150 μm. D, Coimmunofluorescence staining for SRF and the neuron-specific marker, NeuN, shows deletion of SRF in neurons of Srf-Nestin-cKO mice compared with control littermates. Scale bar, 30 μm. E, Forebrain nissl staining reveals several abnormalities in the mutant brains. Compared with control littermates, Srf-Nestin-cKO mice exhibit enlarged lateral ventricles (asterisk) and greatly diminished anterior commissure (a.c), stria medullaris (str.), mammilothalamic tract (m.t), and internal capsule (i.c). F, Closer examination of the mutant hippocampus shows that the dentate gyrus is more compact (arrow) and cellular lamination of CA3 and CA1 fields are more aberrant (arrowheads) than those of control littermates. Asterisk indicates enlarged lateral ventricle in the caudal forebrain. Scale bar, 150 μm.
To analyze the consequence of SRF ablation on brain morphology, we performed nissl staining of P0.5 Srf-Nestin-cKO and control brains. The mutant mice exhibited enlarged lateral ventricles and the corpus callosum was present only in the most rostral part of the brain (Fig. 1E). In the Srf--Nestin-cKO mutant brain, several of the white matter tracts were notably absent or greatly diminished compared with control mice. In the rostral forebrain, the anterior commissure was absent and the lateral ventricular zones were also enlarged in Srf-Nestin-cKO mice (Fig. 1E). More caudally, the stria medullaris, mammilothalamic tract, internal capsule, and anterior commissure were also markedly diminished in Srf-Nestin-cKO mice compared with those in control littermates (Fig. 1E). The hippocampus was smaller and the dentate gyrus was more compact while the CA1 pyramidal cell layer was disorganized (Fig. 1F).
SRF is required for proper establishment of cortical axonal innervations
Given these deficits in white matter tracts, we further analyzed axon growth in Srf-Nestin-cKO mice. Immunostaining using anti-neurofilament antibody (anti-2H3) revealed striking loss of entorhinal-hippocampal or perforant path innervations in the hippocampus of Srf-Nestin-cKO mutants (Fig. 2A, n = 3 animals). The entorhinal-hippocampal pathway, which is important for hippocampal plasticity (Bliss and Gardner-Medwin, 1973), is comprised of the axonal projections primarily from the entorhinal cortex layer II/III neurons that innervate all fields of CA1 pyramidal neurons, granular neurons of the dentate gyrus, and subicular neurons in the hippocampus (Witter et al., 2000). Furthermore, we found a substantial reduction in corticostriatal innervations, which arise mainly from layer V cortical pyramidal neurons in the Srf-Nestin-cKO mutants compared with control littermates (Fig. 2B, n = 3 animals). As observed with nissl staining, anti-2H3 staining also revealed a lack of or deficits in anterior commissure, fasciculus retroflexis, and internal capsule in Srf-Nestin-cKO mice compared with control mice (Fig. 2C, n = 3 mice). We also performed anti-β-III tubulin (anti-Tuj1) immunostaining of sagittal brain sections to visualize the corticospinal tract projections of pyramidal neurons in layer V of the motor cortex. We observed significantly less abundant corticospinal projections through the internal capsule and cerebral peduncle in the Srf-Nestin-cKO brains as compared with control littermates (data not shown).
Figure 2.
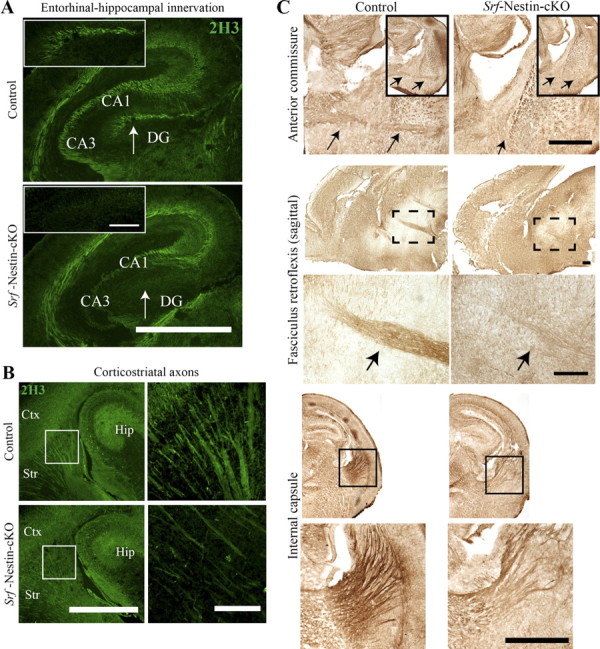
Srf-Nestin-cKO mutant mice exhibit axonal growth deficits. A, Neurofilament immunostaining using anti-2H3 antibody reveals absence of entorhinal-hippocampal innervation in the hippocampus of Srf-Nestin-cKO mice. Inset shows the magnified view of the region indicated by the arrow. B, Examination of neurofilament expression in striatum shows less abundant corticostriatal projections in Srf-Nestin-cKO brains than in control littermates, n = 3 mice. Right, Magnified views of the boxed regions in B, showing sparse axonal projections in the Srf-Nestin-cKO mice compared with control littermates. C, Anti-2H3 neurofilament staining shows absence or highly reduced anterior commissure (arrows), fasciculus retroflexis (arrows), and internal capsule. Enlarged view of the boxed region is shown for each fiber tract. Scale bars: A and C, large panels, and B, left, 500 μm; A, insets, and B,right, 100 μm. DG, Dentate gyrus; Ctx, neocortex; Str, striatum; Hip, hippocampus.
In addition to immunostaining, we used DiI labeling to visualize axonal projections in vivo. DiI crystals were placed on the surface of the brain corresponding to the motor and visual cortices of one hemisphere (Fig. 3A). To ensure the comparisons between control and mutant brain sections are made accurately, we cross-examined all coronal serial sections from the anterior to the posterior forebrain. As observed for anti-neurofilament immunostaining (anti-2H3), DiI labeling showed significant deficits in the corticostriatal projections in Srf-Nestin-cKO brains. In the anterior forebrain region of control mice, the projections from the cortical neurons clearly innervated the striatum. However, in Srf-Nestin-cKO brains these projections mostly terminate in the lateral corpus callosum and fail to reach their targets in the striatum (Fig. 3B,D; projection length: control, 460 ± 13 μm; Srf-Nestin-cKO, 248 ± 55 μm; n = 2 mice; p < 0.05). In the posterior forebrain of control mice, we observed robust corticothalamic projections innervating the thalamus. Strikingly, these corticothalamic innervations were absent in Srf-Nestin-cKO brain (Fig. 3C,D; projection area: control, 20,875 ± 1127 μm2; Srf-Nestin-cKO, 1619 ± 2894 μm2; p < 0.01). Furthermore, we also observed lack of innervations to the dorsal hippocampal commissure in the Srf-Nestin-cKO mice (Fig. 3C). DiI labeling further confirmed the deficits in anterior commissure and corpus callosum observed using anti-2H3 immunostaining (Fig.3 E,F). We then asked whether projections to the cortex were also affected in the mutant mice. DiI-coated beads placed in the ventral thalamus revealed striking deficits in thalamocortical projections in Srf-Nestin-cKO mutant mice compared with control (Fig. 3G, n = 2 mice). The axonal projection deficits in the Srf-Nestin-cKO mutant mice were consistent in all animals analyzed. Together, these observations suggest that SRF plays a critical role in the formation of axonal tracts during neural development.
Figure 3.
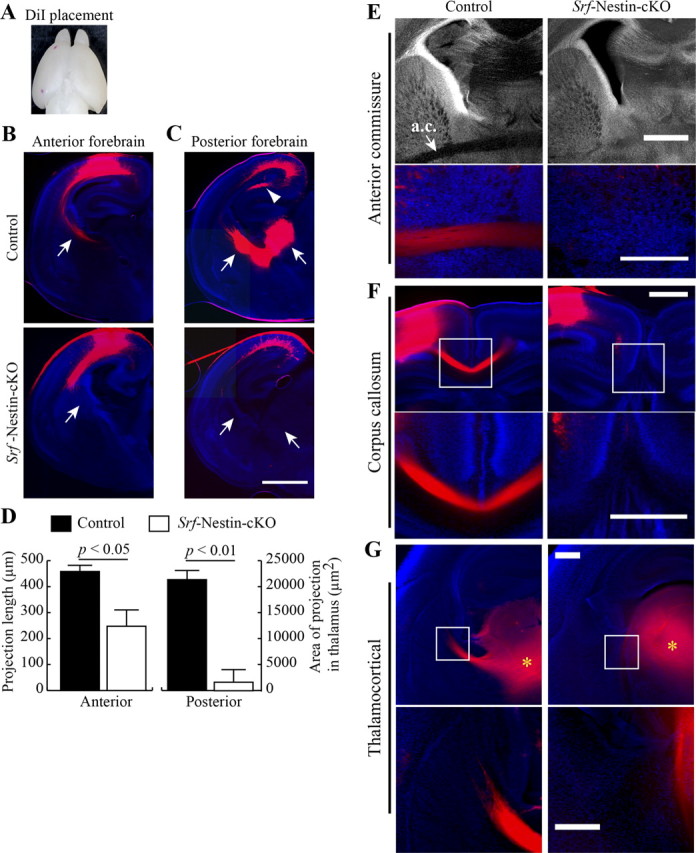
Corticostriatal, corticothalamic, and thalamocortical projections are impaired in Srf-Nestin-cKO mice. A, DiI-labeled P0.5 brain showing the position of the DiI crystals. DiI crystals were placed on the surface of the brain in motor and visual cortices of control and Srf-Nestin-cKO mice. B, Coronal section of the anterior forebrain shows robust corticostriatal projection (arrows) into the striatum in the control, but not in Srf-Nestin-cKO brain. Instead, innervations mostly terminate in the corpus callosum in Srf-Nestin-cKO mice. C, Examination of the posterior forebrain reveals a striking absence of corticothalamic projection (arrows) and lack of innervation into the dorsal hippocampal commissure (arrowhead) in the mutant brain. D, Quantification of axon length from the position of the DiI crystal in the anterior region (B) and the area of target innervation in the posterior region (C) of the brain are shown (n = 3 mice). E, F, DiI labeling shows absence of anterior commissure (E) and corpus callosum (F) in Srf-Nestin-cKO mice compared with control littermates. Scale bar, 200 μm. G, DiI labeling from the thalamus reveals deficits in thalamocortical projections in Srf-Nestin-cKO mutant mice while robust projections are seen in control littermates. Asterisk indicates position of the DiI-coated bead. Scale bar, 100 μm.
Defects in neuronal projections are not a result of cell death or defects in neurogenesis
The deficits in axonal projections could result from a requirement of SRF for axon growth or from cell loss as a result of apoptosis. A previous study has shown that SRF is required for the survival of mouse embryonic stem cells and that SRF promotes cell survival by regulating the expression of the anti-apoptotic gene, Bcl-2 (Schratt et al., 2004). Also, SRF has been shown to promote survival of neocortical neurons under conditions of trophic deprivation in vitro (Chang et al., 2004). To determine whether deficits in axonal projections are due to cell loss during development, we first assessed cell death using TUNEL assay and immunostaining for activated-caspase 3 expression at P0.5. We did not find any increase in the number of TUNEL-positive and activated caspase-3-positive cells in the brains of Srf-Nestin-cKO brains as compared with control littermates (Fig. 4A). To ascertain whether increased cell death could have occurred earlier during brain development, we analyzed the brains of control and Srf-Nestin-cKO mice at E14.5, E16.5, and E18.5. We did not observe any difference in cell death at any of these stages between Srf-Nestin-cKO and control littermates, suggesting that SRF-deletion does not cause apoptotic cell death during brain development (Fig. 4B–D).
Figure 4.

Loss of SRF does not cause apoptotic cell death during brain development. A, TUNEL cell death assay and immunostaining for cleaved activated-caspase3 (act-Casp3) show no detectable elevation of apoptotic cell death in vivo in the absence of SRF at birth. Scale bar, 100 μm. B–D, Both TUNEL assay and immunostaining against activated-Caspase3 at E18.5, E16.5, and E14.5 indicate no significant elevation in the number of apoptotic cells in Srf-Nestin-cKO neocortex. CP, Cortical plate; IZ, intermediate zone; SVZ, subventricular zone; VZ, ventricular zone. Scale bar, 100 μm.
Although we did not see any increased cell death in the brains of Srf-Nestin-cKO mice during development, it is possible that the deficits in axonal tract formation observed in these mutant mice could be due to deficits in the total number of neurons generated. To investigate the effect of SRF loss on neurogenesis, we determined the number of neurons in control and Srf-Nestin-cKO brains by immunostaining for NeuN, a marker for mature neuronal cell nuclei. We found that the total number of NeuN-positive cells was similar in both Srf-Nestin-cKO and control littermates (Fig. 5A–D; neocortex: control 100 ± 4.5% and knock-out 108.95 ± 6.2%; striatum: control 100 ± 3.5% and knock-out 105.2 ± 4.4%; thalamus: control 100 ± 6.0% and knock-out 106.1 ± 2.7%; hippocampus: control 100 ± 5.9% and knock-out 103.5 ± 8.0%; and dentate gyrus: control 100 ± 7.6% and knock-out 92.8 ± 3.1%. Data shown are mean ± SEM as a percentage of mean; n = 3 mice). Although the dentate gyrus in Srf-Nestin-cKO mice appeared smaller, it had a higher cell density than that of control animals and there were no appreciable differences in total neuronal numbers (Fig. 5C, data not shown). We also analyzed the number of intermediate neuronal precursors (INPs), as identified by Tbr2 (or Eomes) expression, and found no statistically significant difference in the total number of INPs within the neocortex of Srf-Nestin-cKO mice and control littermates (Fig. 5E; control 100 ± 6.5% and knock-out 96 ± 5.8%; p < 0.001; n = 3 mice). Together, these results demonstrate that SRF is not required for cell survival and neurogenesis and that the deficits in axonal projections observed in Srf-Nestin-cKO brains reflect a specific requirement for SRF for axon growth.
Figure 5.
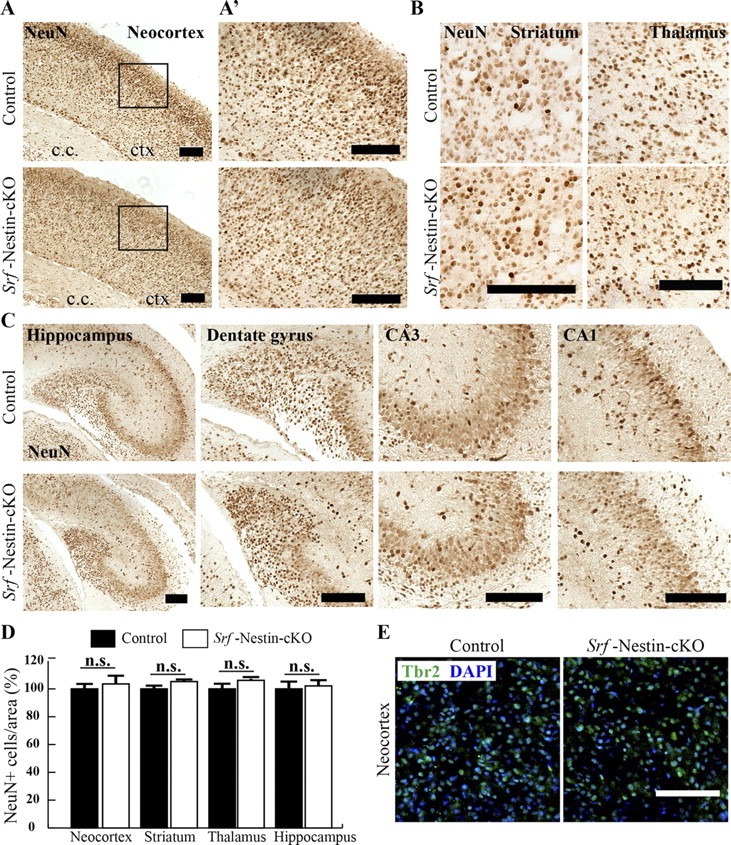
Loss of SRF does not affect neurogenesis. A, Srf-Nestin-cKO mutants exhibit no significant changes in the number of neurons generated, as indicated by NeuN immunostaining, in the neocortex. ctx, Neocortex; c.c, corpus callosum. A', Magnified view of boxed regions shown in A. B, C, Immunostaining for NeuN in the striatum and thalamus (B) and hippocampus (C) of control and Srf-Nestin-cKO mice. Although the dentate gyrus is more compact in Srf-Nestin-cKO mice, there is no significant difference in the number of cells compared with control mice. Scale bars: A–C, 100 μm. D, Quantification of total number of NeuN-positive cells in neocortex, striatum, thalamus, and hippocampus. Error bars represent SEM. Student's t test analysis between control and Srf-Nestin-cKO animals shows no statistically significant differences. E, Immunostaining for Tbr2 (or Eomes), a marker for intermediate neuronal progenitors, showed no difference in total number of committed neuronal precursors in Srf-Nestin-cKO mice and control littermates. Scale bar, 50 μm.
SRF is not required for projection neuron subtype specification and cortical lamination
We next asked whether SRF was required for neuronal subtype specification. The excitatory projection neurons reside in defined layers of the neocortex and make intracortical, subcortical, or subcerebral connections (Molyneaux et al., 2007). A number of transcription factors, including Fezf2 and Ctip2, are critical for specification of cortical projection neuron subtypes and their loss results in absence of subcerebral and subcortical projections (Arlotta et al., 2005; Chen et al., 2005; Molyneaux et al., 2005; Chen et al., 2008). Therefore, it is possible that the lack of cortical projections observed in Srf-Nestin-cKO mutants could be attributed to a switch in projection neuron identity, a result that could affect neocortical lamination. To study this, we probed for expression of Tbr1, a deep-layer neuronal marker, and Cux1, which is specifically expressed in neocortical layers 2–4 (Hevner et al., 2001; Ferrere et al., 2006). Immunostaining of P0.5 brains revealed no differences in the layer-specific expression patterns of both Tbr1 and Cux1 in Srf-Nestin-cKO mice and control littermates (Fig. 6A,B). Additionally, we also probed for expression of other neocortical layer-specific transcription factors by in situ hybridization (Gray et al., 2004). Expression patterns of several transcription factors, including Cux2, Klf6, Lhx5, Lmo4, Nfix, Nr4a2, and Sox5, which specify the identity and position of projection neuron subtypes, were similar between Srf-Nestin-cKO mice and control littermates (Fig. 6C, data not shown). These results indicate that there are no changes in the establishment of layer-specific neuronal subtypes and neocortical lamination at P0.5 in the brains of Srf-Nestin-cKO mice.
Figure 6.
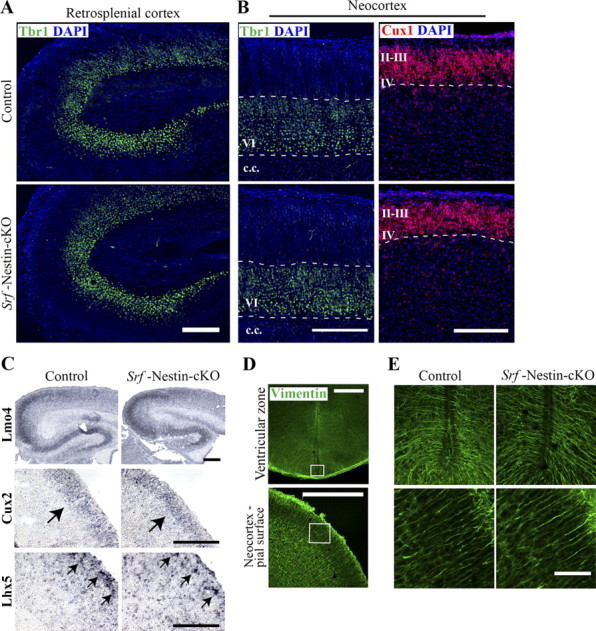
SRF is dispensable for neocortical lamination and projection neuron subtype specification. A, Immunostaining for Tbr1 expression, a marker of deep layer neurons in the neocortex, shows normal layer VI lamination in Srf-Nestin-cKO and control brains. Shown here is the retrosplenial cortex. B, Immunofluorescence staining for expression of Tbr1 and Cux 1 (a marker for superficial layer neurons) in the neocortex shows that both layer 2/3 and layer VI neurons are specified and positioned normally in knock-out mice. c.c., Corpus callosum. C, In situ hybridization of layer-specific transcription factors, including Lmo4, Cux2, and Lhx5, shows that neocortical lamination and the specification of those subtypes are normal in the absence of SRF. Arrows point to Cux2- or Lhx5-expressing upper layer neurons. Scale bars: A, C, 200 μm; B, 100 μm. D, Immunostaining using anti-vimentin antibody, expressed in radial glial processes, show that the structural integrity of radial glia is normal in Srf-Nestin-cKO brain. E, Magnified images of boxed regions in D show normal appearance of radial glial processes in Srf-Nestin-cKO mice compared with control mice. Scale bar, 20 μm.
The proper lamination of the neocortex in Srf-Nestin-cKO mice suggested that radial migration of neurons is not affected in the absence of SRF. We found that SRF is also deleted in radial glial cells in Srf-Nestin-cKO mice (data not shown). We immunostained P0.5 brains using anti-vimentin antibody, which labels radial glial processes, and found no gross alteration in the abundance of radial glial processes or the integrity of projections within multiple regions examined including the ventricular zone, neocortex, hippocampus, and corpus callosum (Fig. 6D,E, data not shown). Together, these findings suggest that loss of SRF does not affect radial glial projections and thereby, the radial migration of neurons and lamination of neocortex.
SRF is not required for interneuron subtype specification
Given the role of SRF in mediating differentiation and development of several cell types in other tissues, we next sought to determine whether SRF is required for specification of interneurons. Interneurons, which show astonishing differences in their electrophysiological, morphological, and molecular properties, are primarily derived from the medial and caudal ganglionic eminences during brain development and then migrate to populate the neocortex as the brain matures (Marín and Rubenstein, 2003; Wonders and Anderson, 2006). Since Srf-Nestin-cKO mutants do not survive beyond P1, we restricted our analysis to interneuron populations in the striatum. We used immunostaining for anti-Gad-6 to identify the expression of GAD, an enzyme that synthesizes GABA neurotransmitters in all interneurons (Fig. 7A). We found no significant difference in the overall numbers of interneurons between control and Srf-Nestin-cKO mice (Fig. 7A,C; control 100 ± 6.8% and knock-out 98.8 ± 10.8%). Next, we analyzed for different interneuron subtypes, including SST-positive, parvalbumin-positive, and calbindin-positive cells. We observed no difference in their numbers between Srf-Nestin-cKO mutants and control littermates (Fig. 7B,C; SST+ cells: control 100 ± 7.3% and knock-out 92.6 ± 7.5%; parvalbumin+ cells: control 100 ± 6.4% and knock-out 117.8 ± 11.2%; calbindin: control 100 ± 7.8% and knock-out 116.0 ± 10.1%). Together, these findings demonstrate that, in addition to projection neuron subtype specification, the establishment of interneuron subtypes occurs normally in the absence of SRF during neuronal lineage commitment.
Figure 7.

SRF is not required for interneuron subtype specification. A, Immunostaining for Gad-6, which labels all GABAergic interneurons, showed normal expression of Gad-6 in Srf-Nestin-cKO and control mice. Inset shows enlarged image of a single stained neuron. B, Immunohistochemistry staining showing expression of SST, parvalbumin, and calbindin, which label unique subtypes of interneurons, suggests no apparent change in the population of interneurons in brains of Srf-Nestin-cKO mice. The striatal region from control and mutant is shown as magnified images. Inset shows enlarged image of a single stained neuron. C, Quantification of cell numbers for different interneurons subtypes for B. Student's t test showed no statistically significant difference in the number of each interneuron subtype between paired control and knock-out animals. Error bars represent SEM.
Neural precursor cell population increases in Srf-Nestin-cKO mutant mice
Quiescent cells that rest in the G0 state can be induced by extracellular stimuli to express immediate early gene (IEG) programs (Herschman, 1991). A number of transcription factor-encoding IEGs, such as cFos, c-Myc, Egr-1, and JunB, and are then responsible for activating gene cascades that enable cell progression to the G1 state (Greenberg and Ziff, 1984; Lau and Nathans, 1985). SRF-mediated transcription was demonstrated to be necessary for inducing IEG expression in embryonic stem cells and in neurons (Norman et al., 1988; Schratt et al., 2001; Ramanan et al., 2005). Sequestration of functional SRF was also shown to impede rat embryonic fibroblast and myoblast proliferation, but not self-renewal of embryonic stem cells (Gauthier-Rouvière et al., 1991; Soulez et al., 1996). We therefore assessed whether SRF deletion affects NPC growth and maintenance in vivo. Proliferating progenitor cells are identified by the expression of phospho-histone H3, which is a modification event that occurs specifically during cell division events of both mitosis and meiosis (Hans and Dimitrov, 2001). Surprisingly, we observed an increase in phospho-histone H3-positive cells in the proliferative subventricular zone (SVZ) of Srf-Nestin-cKO brains at both E14.5 and E18.5 (Fig. 8A–D). Quantitative analyses of the number of phospho-histone H3-positive cells per area of the parameter of the SVZ revealed a 20% and 80% increase in mutants at E14.5 and E18.5, respectively (Fig. 8E; E14.5 p-histone H3 normalized: control 100 ± 7.3% and knock-out 120.7 ± 7.8%; and E18.5 p-histone H3 normalized: control 100 ± 3.0% and knock-out 180.6 ± 4.2%). We confirmed the increase in proliferative progenitor cell numbers using two additional markers: Ki-67, which is expressed by cells in the cell-cycle phases G1, S, and G2 and in mitosis; and Sox2, a transcription factor expressed in NPC. At E14.5, immunohistochemistry staining of Ki-67 in Srf-Nestin-cKO brains showed an increase in NPC numbers per square micrometer in the SVZ and the neocortex compared with control (Fig. 8E,F; Ki-67 normalized: control 100 ± 18.0% and knock-out 140 ± 6.3%). Similarly, neocortex and SVZ in Srf-Nestin-cKO showed markedly increased numbers of Sox2-expressing cells compared with those in control littermates (Fig. 8F). These observations suggest that loss of SRF affects NPC homeostasis during development without affecting NPC survival.
Figure 8.
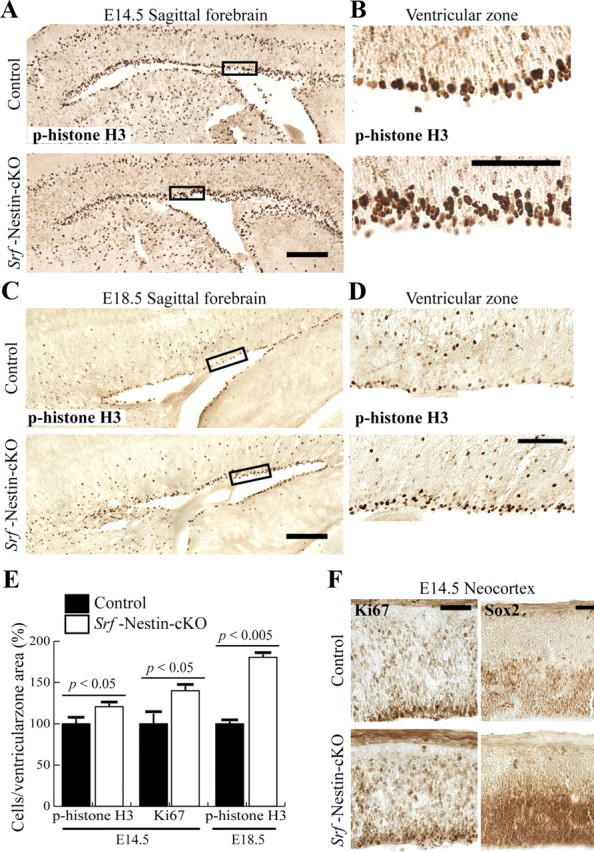
Loss of SRF results in an increase in the number of neural precursor cells. A, Proliferating NPCs are identified in the control and Srf-Nestin-cKO forebrains using anti-p-histone H3 antibody at E14.5. B, Magnification of the boxed regions in A. C, Comparison of NPC populations at E18.5. D, Magnified view of the boxed regions in C. E, Cell count analyses of the number of p-histone-H3- and Ki67-positive cells at E14.5 indicate a statistically significant increase in the number of proliferating cells in the ventricular zone of Srf-Nestin-cKO mice. The difference in relative numbers of NPCs between the control and mutant brain is more pronounced at E18.5. F, Immunostaining for two additional proteins, Ki67, a marker of cells in the active phase of cell cycle, and Sox2, a marker of neural precursor cells, was used to visualize proliferating cells in the neocortex at E14.5. Mutant brains not only display more Ki67-positive cells but also show a broadened layer of Sox2-positive cells. Scale bars, 50 μm.
Conditional deletion of SRF in developing forebrain neurons
Our analyses of the Srf-Nestin-cKO mice suggested that SRF plays a specific role in the development of axonal tracts without affecting neurogenesis, neuronal survival, and neuronal subtype specification. However, since SRF deletion occurs in all neural precursor cells before cellular differentiation occurs, it is possible that axon growth defects in Srf-Nestin-cKO mice could be due to a non-cell autonomous requirement of SRF for axon growth. To ascertain whether SRF is required cell autonomously for axon growth, we generated a neuron-specific deletion of SRF using the NEX-Cre transgenic mouse. In the NEX-Cre mouse, cre expression is controlled by the onset of expression of the NEX gene (also known as Math2 or NeuroD6), an early neuronal basic helix-loop-helix gene expressed specifically in differentiating neurons (Schwab et al., 1998; Goebbels et al., 2006). Cre recombinase-mediated excision in the NEX-Cre mouse has been shown to take place starting at ∼E11.5 and is restricted only to the glutamatergic neurons in the neocortex and hippocampus, whereas cre expression is not observed in the interneurons and glial cells (Brockschnieder et al., 2004; Goebbels et al., 2006; Kashani et al., 2006). The Srf-NEX-cKO mice were born in the expected Mendelian ratio but unlike the Srf-Nestin-cKO mice, these mice did not exhibit neonatal lethality and grew to adulthood. We first confirmed deletion at P0.5 by immunostaining and found that SRF deletion was restricted to the neocortex and hippocampus but not in the striatum and other regions of the brain, consistent with previous findings (Fig. 9A—D, data not shown).
Figure 9.
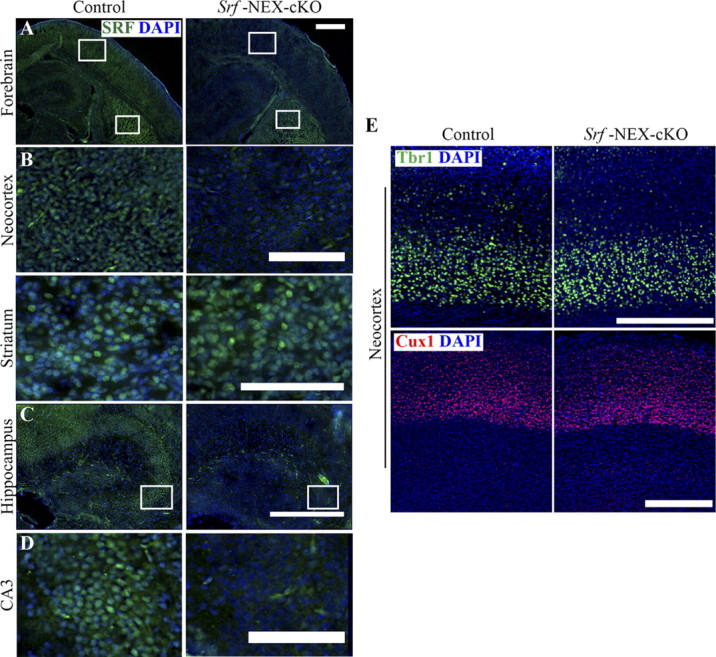
Ablation of SRF in Srf-NEX-cKO. A, Immunofluorescence staining at P0.5 using anti-SRF antibody shows that SRF is deleted in the neocortex but not in the striatum of Srf-NEX-cKO mutants. B, Magnified views of boxed regions in A. C, SRF expression is also abolished in the hippocampus in mutants. D, A magnified view of CA3 neurons of control and Srf-NEX-cKO mice. E, Immunostaining of P21 brains using anti-Tbr1 and anti-Cux1 shows normal lamination of neocortex in Srf-NEX-cKO mice and control littermates. Scale bars, 50 μm.
We next asked whether lamination occurs normally in Srf-NEX-cKO mice. We performed immunostaining for laminar-specific markers, Cux1 and Tbr1, on P21 brains. We did not find any deficits in neocortical lamination in Srf-NEX-cKO mice as compared with control littermates (Fig. 9E). At birth, the lack of defects in lamination in Srf-NEX-cKO mice was similar to that observed for Srf-Nestin-cKO mutant mice (data not shown). Together, these findings suggest neocortical lamination is properly established in the absence of SRF.
SRF mediates cortical neuron target innervation cell autonomously
We next analyzed axonal projections in Srf-NEX-cKO mice at P0.5 using DiI labeling. Two weeks following DiI labeling, sagittal sections of Srf-NEX-cKO mouse brains revealed greatly diminished corticospinal projections, and cortical motor neuron axons passing through the internal capsule were less abundant with very few projections reaching the cerebral peduncle (Fig. 10A,A',A”; n = 3 mice). Comparison of parallel serial sections from the lateral side to the medial region of the forebrain between control and Srf-NEX-cKO mice showed a severe reduction in corticospinal projections in the mutant neocortex (Fig. 10B; n = 3 mice).
Figure 10.
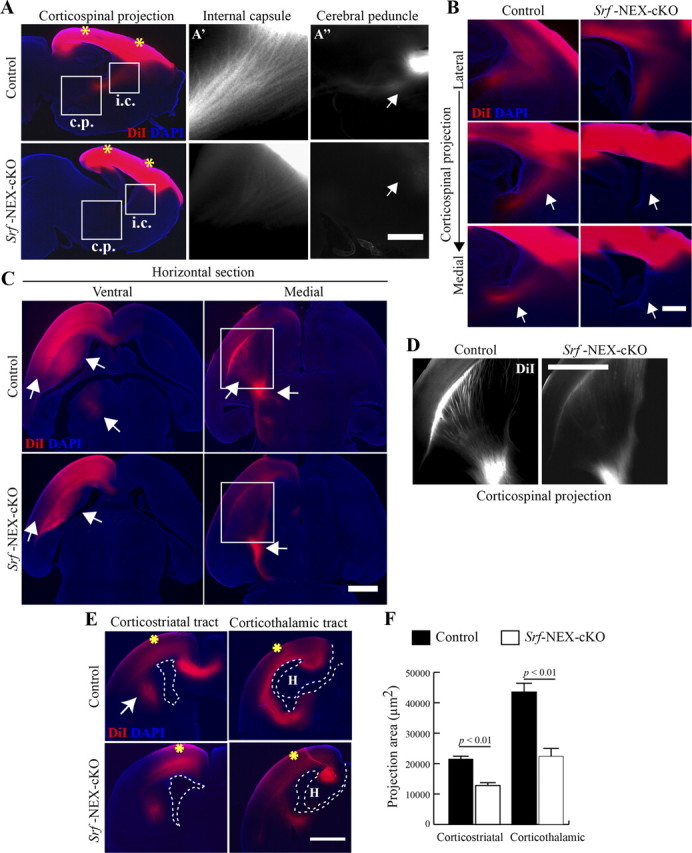
Dil labeling shows impairment in axonal projections in Srf-NEX-cKO mutants. A, DiI crystals were placed on the brain surface in the regions of the motor and the visual cortices (indicated by asterisks) in P0.5 Srf-NEX-cKO knock-out and control littermates. Two weeks after labeling, brains were sectioned sagittally. Impaired corticospinal innervation was observed in the knock-out brain. Magnifications of the internal capsule (i.c) and cerebral peduncle (c.p) regions are shown in A' and A”. Projections through the cerebral peduncle are seen in the brains of control but Srf-NEX-cKO mice. B, Serial sagittal sections from lateral to medial regions of the brain show lack of corticostriatal projections (arrows) in Srf-NEX-cKO mice. No misguided axons were observed in the mutant mice. C, After 6 weeks of labeling, control and Srf-NEX-cKO brains were sectioned horizontally. Arrows show diminished projections within the neocortex, corticostriatal projections, and innervations to the thalamus in the mutant. Medial horizontal section shows impaired projections through the internal capsule and the cerebral peduncle. D, Magnified views of the boxed regions in C showing the corticospinal projections. E, Coronal sections from caudal regions of the brain reveal diminished corticostriatal as well as corticothalamic tracts (arrows). Asterisks indicate sites of crystal placement; dotted lines outline the ventricular zone and the hippocampus (H). F, Quantification of area of innervation by corticostriatal and corticothalamic axons in E (n = 3 mice).
We then examined horizontal sections of control and Srf-NEX-cKO brains after 6 weeks of DiI labeling. On the ventral side of the brain, we observed that the intracortical and corticostriatal connections in Srf-NEX-cKO mice were less abundant and shorter than those observed in control littermates. In particular, a region of the thalamus is clearly innervated in the control brain; however, this innervation is less prominent in the Srf-NEX-cKO brain (Fig. 10C; n = 3 mice). In the medial region of the brains, similar to that observed in 2-week-old sagittal sections, it was evident that corticospinal innervations through the striatum to the cerebral peduncle were greatly reduced overall in Srf-NEX-cKO mutants compared with control littermates (Fig. 10D).
We further examined serial coronal sections of control and Srf-NEX-cKO brains at 4 weeks after DiI labeling. In rostral sections, DiI tracing showed that callosal innervations that form the corpus callosum are diminished and shorter in the Srf-NEX-cKO brains (Fig. 10E). We found less abundant corticostriatal projections in the Srf-NEX-cKO brain (Fig. 10E,F; projection area: control, 205,737 ± 496 μm2; Srf-NEX-cKO, 12,148 ± 427 μm2, n = 3 mice), an observation that is consistent with that made in brains of Srf-Nestin-cKO mice using anti-2H3 immunostaining. Toward the caudal end of the forebrain, retrograde DiI labeling revealed that the corticothalamic connections, which are important relays of sensory information between the visual cortex and the lateral geniculate nucleus of the thalamus, were also less robustly established in the Srf-NEX-cKO mice compared with control mice (Fig. 10E,F; projection area: control, 41,690 ± 1643 μm2; Srf-NEX-cKO, 21,229 ± 1207 μm2, n = 2 mice). These observations were consistent in all the mutant mice analyzed. These neuroanatomical tracing experiments demonstrate that SRF plays a critical cell-autonomous role in regulating axonal growth and establishment of axonal projections in vivo.
Discussion
Neuronal development in the CNS involves several critical stages including neurogenesis and maturation of neurons, growth and extension of axons, and structural organization within the brain. Our current understanding of the role of SRF in neuronal development comes mainly from studies using mice carrying conditional neuron-specific deletion of SRF in late gestation or in postnatal brain. However, the requirement of SRF for early stages of neuronal development remains unknown. In the present study, we show that conditional deletion of SRF in neural precursor cells (Srf-Nestin-cKO) results in severe deficits in the development of major axonal projections in the forebrain, including corticospinal, corticothalamic, corticostriatal, and thalamocortical tracts along with a variable loss of the corpus callosum. Axonal deficits were seen as early as E14.5 in the Srf-Nestin-cKO mice and there was little or no cell death during development. Interestingly, there was a significant increase in the number of proliferating cells in the ventricular zone in Srf-Nestin-cKO mice. Conditional deletion of SRF in embryonic forebrain neurons (Srf-NEX-cKO) also resulted in severe deficits in major axonal projections. Neurogenesis, radial neuronal migration in the neocortex, neocortical lamination, and neuronal subtype specification were unaffected by SRF loss. Together, these findings suggest that SRF is required in a cell-autonomous manner for axon growth and extension. Similar to that observed in mice with prenatal and postnatal deletion of SRF, SRF is dispensable for neuronal survival. Our study identifies a specific role for SRF in promoting axon growth during neuronal development without affecting neurogenesis and neuronal differentiation.
Previous studies have shown that neuron-specific SRF deletion during late gestation in the brain causes deficits in terminal targeting of mossy fiber axons in the hippocampus, while SRF loss in developing sensory neurons in the peripheral nervous system affects NGF-dependent terminal arborization and target innervation (Knöll et al., 2006; Wickramasinghe et al., 2008). However, proximal axon growth in the peripheral nervous system is not affected while the role of SRF in axon growth in the CNS remains unknown. We found that deleting SRF in neural precursor cells results in severe deficits in axon growth and targeting of cortical axon projections. Observations made in cultured hippocampal neurons have shown that SRF is required for contact-mediated axon repulsion (Knöll et al., 2006). We did not observe any mistargeted axonal tracts in the brains of Srf-Nestin-cKO mice, suggesting that the lack of target innervation seen in the SRF mutant mice is primarily due to defects in axon growth and not due to defects in axon guidance. We found similar axonal growth defects when SRF was deleted in developing postmitotic neurons in neocortex and hippocampus in Srf-NEX-cKO mutant mice. Unlike Srf-Nestin-cKO mice, the Srf-NEX-cKO mice survived to adulthood, and in preliminary observations, we found that adult Srf-NEX-cKO mice exhibited clasping of limbs in a dystonic manner when subjected to the tail suspension test, which is suggestive of motor dysfunction (Carter et al., 1999; Yamamoto et al., 2000). Furthermore, consistent with previous observations, we also found that SRF-deficient neurons exhibit highly attenuated axon growth in culture (Knöll et al., 2006) (C. Li and N. Ramanan, unpublished observations). The similarities in deficits in axon growth in the brains of Srf-Nestin-cKO and Srf-NEX-cKO mice suggested that SRF-dependent transcription plays a cell-intrinsic role in axon growth.
The molecular mechanisms underlying SRF-dependent axon growth remain poorly understood. One mechanism by which SRF potentially regulates axon growth is through association with specific cofactors. We found that blocking the functions of the Ternary Complex Factor-family cofactors of SRF, including Elk-1, does not affect axon growth in cultured neurons (C. Li and N. Ramanan, unpublished observations). However, we and others have found that blocking the functions of myocardin-family cofactors, MKL1 (also known as MAL/MRTF-A) and MKL2 (MRTF-B), by dominant-negative or knockdown approaches or by gene deletion attenuates axon growth in vitro (Knöll et al., 2006; Shiota et al., 2006; Wickramasinghe et al., 2008; Mokalled et al., 2010) (C. Li and N. Ramanan, unpublished observations). Mutant mice that lack both MKL1 and MKL2 in the brain exhibit deficits in dendritic growth in the neocortex and hippocampus as assessed by MAP2 and Golgi staining (Mokalled et al., 2010). However, the effect of MKL1/MKL2 loss on axon growth in vivo has not been reported in these mice. In the peripheral nervous system, SRF has been shown to function downstream of NGF-signaling to regulate terminal arborization of axons and target innervation (Wickramasinghe et al., 2008). Furthermore, NGF signaling to SRF is dependent on both ERK/MEK and MAL/MKL1 signaling pathways. The findings from the peripheral nervous system raise an interesting question as to which extracellular signals might stimulate SRF-dependent transcription during axon growth in the brain. Currently we lack sufficient knowledge on the nature of the extracellular signals and the identities of SRF target genes critical for axon growth in the CNS. It is likely that SRF functions downstream of growth factors such as BDNF to regulate axon growth. SRF could also regulate axon growth by regulating the expression of components of the actin cytoskeleton, including β-actin, γ-actin, paxillin, vinculin, and talin (Schratt et al., 2002). In fact, previous studies including our own have shown that β-actin expression is reduced in SRF knock-out neurons (Alberti et al., 2005; Ramanan et al., 2005; Knöll et al., 2006), and it was hypothesized that reduction in actin levels was one of the underlying causes for neurite outgrowth deficits observed in SRF-deficient neurons. However, overexpression of actin was found to be insufficient to rescue the growth deficits of SRF-null neurons (Knöll et al., 2006; Stern et al., 2009). Since SRF regulates the expression of several cytoskeletal proteins (Schratt et al., 2002), it is possible that the neuronal growth deficits exhibited by SRF-deficient neurons could be due to a breakdown in cytoskeletal apparatus critical for growth and extension.
We did not observe any increased cell death in the brains of Srf-Nestin-cKO and Srf-NEX-cKO mutant mice during development. There was also no noticeable difference in neuronal cell numbers in older Srf-NEX-cKO mice, and this is consistent with our previous findings that SRF deletion does not result in cell death or neurodegeneration in the CNS (Ramanan et al., 2005). Interestingly, SRF deletion in neural precursor cells did not cause apoptotic cell death, a phenotype that contrasts with observations made in SRF-deficient embryonic stem cells (Schratt et al., 2004). SRF-deficient mouse ES cells exhibited apoptotic cell death both in vitro and in vivo (Schratt et al., 2004). Our findings suggest that SRF is dispensable for survival of NPCs both in vitro and in vivo (our unpublished observations). In contrast, we observed an increase in the total number of p-histone-H3 and Sox2-positive cells in Srf-Nestin-cKO mice. A recent elegant study showed that SRF deletion in neurons affects oligodendrocyte differentiation in a paracrine manner (Stritt et al., 2009). Consistent with this observation, we also observed a decrease in Olig2+ cells at birth in Srf-Nestin-cKO mice (our unpublished observations). Therefore, a likely explanation for the increase in NPC numbers in Srf-Nestin-cKO mice is that SRF loss in NPCs affects oligodendrocyte differentiation, thereby resulting in an increase in undifferentiated neural precursor cells. Together, these observations suggest that there are distinct requirements for SRF in ES cells and in NPCs for cell survival.
Previous studies have shown that SRF has a profound role in regulating cell-type specific gene expression that underlies the development of many cell types. A number of tissue-specific inactivation studies later elucidated essential functions of SRF for the development of cardiac muscle cells (Niu et al., 2005, 2008; Zhao et al., 2005), the differentiation of smooth muscles (Miano et al., 2004; Parlakian et al., 2004), and the normal proliferation and differentiation of keratinocytes (Koegel et al., 2009). We observed no differences in total number of NeuN-positive cells in Srf-Nestin-cKO mice, suggesting that SRF is dispensable for neurogenesis in the brain. We also found that neuronal subtype specification and both interneuron and neocortical lamina-specific neuron identities were properly established in the absence of SRF. The findings that SRF-deficient neurons negatively influence oligodendrocyte differentiation suggest that SRF-dependent transcription can promote cell-type specification in the brain (Stritt et al., 2009).
SRF deletion in developing neurons has been shown to affect tangential cell migration along the rostral migratory stream (Alberti et al., 2005). We also observed similar tangential migration deficits in the Srf-Nestin-cKO mice (our unpublished observations). However, we found that radial migration of neurons in the neocortex was not affected and neocortical lamination was established normally in both Srf-Nestin-cKO and Srf-NEX-cKO mutant mice. Our observations differ from those of a recent study in which cortical lamination was shown to be affected in mice carrying neuron-specific deletion of SRF (Stritt and Knöll, 2010). In this study, calbindin-positive cells were reduced in SRF-mutant neocortex while immunostaining for the neurofilament protein SMI-32, which also labels a subpopulation of cortical neurons (Campbell and Morrison, 1989) in layers III and V, showed mislocalization of Smi-32-positive cells between layers III and V. Calbindin-positive interneurons are mainly generated in the medial ganglionic eminences before they tangentially migrate to populate the neocortex (Marín and Rubenstein, 2003; Wonders and Anderson, 2006), and we did not find any change in the total numbers of striatal calbindin-positive cells in Srf-Nestin-cKO mice at birth. In this study, we used both in situ hybridization and immunohistochemistry for several transcription factors that are expressed in specific cortical layers during mouse development (Gray et al., 2004). We did not find any lamination defects in the neocortex of either Srf-Nestin-cKO or Srf-NEX-cKO mice at P0.5 or in 3-week-old Srf-NEX-cKO mice. If SRF is critical for neocortical lamination, this phenotype should be more severe in the Srf-Nestin-cKO mice, since SRF is deleted in all major cell types in the brain starting at E12.5 (our unpublished observations). Based on our observations, we conclude that SRF loss in neural precursor cells and in developing neurons does not affect layering of the neocortex. We also found that SRF deletion in radial glial cells in Srf-Nestin-cKO mice did not affect their morphology, suggesting that SRF-dependent transcription is not required for extension of radial glial processes.
Our study identifies specific roles for SRF during neuronal development. SRF plays a critical role in neural precursor cell homeostasis and in the formation of major axonal tracts in the brain. SRF is dispensable for neurogenesis and cell survival but contrary to recent findings, SRF is not required for neocortical lamination.
Footnotes
This work was supported by Andrew B. and Virginia C. Craig Faculty Fellowship (N.R.) and a Whitehall Foundation Award (N.R.). We thank Anna Oldenborg and Deanna Young for expert technical assistance, Dr. A. Burkhalter with help with DiI labeling, and B. Hall (Tulane University) for the NEX-Cre mice. We thank members of the Ramanan laboratory for comments on this manuscript and useful discussions.
References
- Alberti S, Krause SM, Kretz O, Philippar U, Lemberger T, Casanova E, Wiebel FF, Schwarz H, Frotscher M, Schütz G, Nordheim A. Neuronal migration in the murine rostral migratory stream requires serum response factor. Proc Natl Acad Sci U S A. 2005;102:6148–6153. doi: 10.1073/pnas.0501191102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arlotta P, Molyneaux BJ, Chen J, Inoue J, Kominami R, Macklis JD. Neuronal subtype-specific genes that control corticospinal motor neuron development in vivo. Neuron. 2005;45:207–221. doi: 10.1016/j.neuron.2004.12.036. [DOI] [PubMed] [Google Scholar]
- Arsenian S, Weinhold B, Oelgeschläger M, Rüther U, Nordheim A. Serum response factor is essential for mesoderm formation during mouse embryogenesis. EMBO J. 1998;17:6289–6299. doi: 10.1093/emboj/17.21.6289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bliss TV, Gardner-Medwin AR. Long-lasting potentiation of synaptic transmission in the dentate area of the unanaestetized rabbit following stimulation of the perforant path. J Physiol. 1973;232:357–374. doi: 10.1113/jphysiol.1973.sp010274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brockschnieder D, Lappe-Siefke C, Goebbels S, Boesl MR, Nave KA, Riethmacher D. Cell depletion due to diphtheria toxin fragment A after Cre-mediated recombination. Mol Cell Biol. 2004;24:7636–7642. doi: 10.1128/MCB.24.17.7636-7642.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell MJ, Morrison JH. Monoclonal antibody to neurofilament protein (SMI-32) labels a subpopulation of pyramidal neurons in the human and monkey neocortex. J Comp Neurol. 1989;282:191–205. doi: 10.1002/cne.902820204. [DOI] [PubMed] [Google Scholar]
- Carter RJ, Lione LA, Humby T, Mangiarini L, Mahal A, Bates GP, Dunnett SB, Morton AJ. Characterization of progressive motor deficits in mice transgenic for the human Huntington's disease mutation. J Neurosci. 1999;19:3248–3257. doi: 10.1523/JNEUROSCI.19-08-03248.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang SH, Poser S, Xia Z. A novel role for serum response factor in neuronal survival. J Neurosci. 2004;24:2277–2285. doi: 10.1523/JNEUROSCI.4868-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen B, Schaevitz LR, McConnell SK. Fezl regulates the differentiation and axon targeting of layer 5 subcortical projection neurons in cerebral cortex. Proc Natl Acad Sci U S A. 2005;102:17184–17189. doi: 10.1073/pnas.0508732102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen B, Wang SS, Hattox AM, Rayburn H, Nelson SB, McConnell SK. The Fezf2-Ctip2 genetic pathway regulates the fate choice of subcortical projection neurons in the developing cerebral cortex. Proc Natl Acad Sci U S A. 2008;105:11382–11387. doi: 10.1073/pnas.0804918105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Etkin A, Alarcón JM, Weisberg SP, Touzani K, Huang YY, Nordheim A, Kandel ER. A role in learning for SRF: deletion in the adult forebrain disrupts LTD and the formation of an immediate memory of a novel context. Neuron. 2006;50:127–143. doi: 10.1016/j.neuron.2006.03.013. [DOI] [PubMed] [Google Scholar]
- Ferrere A, Vitalis T, Gingras H, Gaspar P, Cases O. Expression of Cux-1 and Cux-2 in the developing somatosensory cortex of normal and barrel-defective mice. Anat Rec A Discov Mol Cell Evol Biol. 2006;288:158–165. doi: 10.1002/ar.a.20284. [DOI] [PubMed] [Google Scholar]
- Gauthier-Rouvière C, Cavadore JC, Blanchard JM, Lamb NJ, Fernandez A. p67SRF is a constitutive nuclear protein implicated in the modulation of genes required throughout the G1 period. Cell Regul. 1991;2:575–588. doi: 10.1091/mbc.2.7.575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goebbels S, Bormuth I, Bode U, Hermanson O, Schwab MH, Nave KA. Genetic targeting of principal neurons in neocortex and hippocampus of NEX-Cre mice. Genesis. 2006;44:611–621. doi: 10.1002/dvg.20256. [DOI] [PubMed] [Google Scholar]
- Goldberg JL, Espinosa JS, Xu Y, Davidson N, Kovacs GT, Barres BA. Retinal ganglion cells do not extend axons by default: promotion by neurotrophic signaling and electrical activity. Neuron. 2002;33:689–702. doi: 10.1016/s0896-6273(02)00602-5. [DOI] [PubMed] [Google Scholar]
- Gray PA, Fu H, Luo P, Zhao Q, Yu J, Ferrari A, Tenzen T, Yuk DI, Tsung EF, Cai Z, Alberta JA, Cheng LP, Liu Y, Stenman JM, Valerius MT, Billings N, Kim HA, Greenberg ME, McMahon AP, Rowitch DH, et al. Mouse brain organization revealed through direct genome-scale TF expression analysis. Science. 2004;306:2255–2257. doi: 10.1126/science.1104935. [DOI] [PubMed] [Google Scholar]
- Greenberg ME, Ziff EB. Stimulation of 3T3 cells induces transcription of the c-fos proto-oncogene. Nature. 1984;311:433–438. doi: 10.1038/311433a0. [DOI] [PubMed] [Google Scholar]
- Hans F, Dimitrov S. Histone H3 phosphorylation and cell division. Oncogene. 2001;20:3021–3027. doi: 10.1038/sj.onc.1204326. [DOI] [PubMed] [Google Scholar]
- Herschman HR. Primary response genes induced by growth factors and tumor promoters. Annu Rev Biochem. 1991;60:281–319. doi: 10.1146/annurev.bi.60.070191.001433. [DOI] [PubMed] [Google Scholar]
- Hevner RF, Shi L, Justice N, Hsueh Y, Sheng M, Smiga S, Bulfone A, Goffinet AM, Campagnoni AT, Rubenstein JL. Tbr1 regulates differentiation of the preplate and layer 6. Neuron. 2001;29:353–366. doi: 10.1016/s0896-6273(01)00211-2. [DOI] [PubMed] [Google Scholar]
- Johnson AW, Crombag HS, Smith DR, Ramanan N. Effects of serum response factor (SRF) deletion on conditioned reinforcement. Behav Brain Res. 2011;220:312–318. doi: 10.1016/j.bbr.2011.02.015. [DOI] [PubMed] [Google Scholar]
- Kashani AH, Qiu Z, Jurata L, Lee SK, Pfaff S, Goebbels S, Nave KA, Ghosh A. Calcium activation of the LMO4 transcription complex and its role in the patterning of thalamocortical connections. J Neurosci. 2006;26:8398–8408. doi: 10.1523/JNEUROSCI.0618-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knöll B, Kretz O, Fiedler C, Alberti S, Schütz G, Frotscher M, Nordheim A. Serum response factor controls neuronal circuit assembly in the hippocampus. Nat Neurosci. 2006;9:195–204. doi: 10.1038/nn1627. [DOI] [PubMed] [Google Scholar]
- Koegel H, von Tobel L, Schäfer M, Alberti S, Kremmer E, Mauch C, Hohl D, Wang XJ, Beer HD, Bloch W, Nordheim A, Werner S. Loss of serum response factor in keratinocytes results in hyperproliferative skin disease in mice. J Clin Invest. 2009;119:899–910. doi: 10.1172/JCI37771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau LF, Nathans D. Identification of a set of genes expressed during the G0/G1 transition of cultured mouse cells. EMBO J. 1985;4:3145–3151. doi: 10.1002/j.1460-2075.1985.tb04057.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marín O, Rubenstein JL. Cell migration in the forebrain. Annu Rev Neurosci. 2003;26:441–483. doi: 10.1146/annurev.neuro.26.041002.131058. [DOI] [PubMed] [Google Scholar]
- Miano JM, Ramanan N, Georger MA, de Mesy Bentley KL, Emerson RL, Balza RO, Jr, Xiao Q, Weiler H, Ginty DD, Misra RP. Restricted inactivation of serum response factor to the cardiovascular system. Proc Natl Acad Sci U S A. 2004;101:17132–17137. doi: 10.1073/pnas.0406041101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mokalled MH, Johnson A, Kim Y, Oh J, Olson EN. Myocardin-related transcription factors regulate the Cdk5/Pctaire1 kinase cascade to control neurite outgrowth, neuronal migration and brain development. Development. 2010;137:2365–2374. doi: 10.1242/dev.047605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molyneaux BJ, Arlotta P, Hirata T, Hibi M, Macklis JD. Fezl is required for the birth and specification of corticospinal motor neurons. Neuron. 2005;47:817–831. doi: 10.1016/j.neuron.2005.08.030. [DOI] [PubMed] [Google Scholar]
- Molyneaux BJ, Arlotta P, Menezes JR, Macklis JD. Neuronal subtype specification in the cerebral cortex. Nat Rev Neurosci. 2007;8:427–437. doi: 10.1038/nrn2151. [DOI] [PubMed] [Google Scholar]
- Niu Z, Yu W, Zhang SX, Barron M, Belaguli NS, Schneider MD, Parmacek M, Nordheim A, Schwartz RJ. Conditional mutagenesis of the murine serum response factor gene blocks cardiogenesis and the transcription of downstream gene targets. J Biol Chem. 2005;280:32531–32538. doi: 10.1074/jbc.M501372200. [DOI] [PubMed] [Google Scholar]
- Niu Z, Iyer D, Conway SJ, Martin JF, Ivey K, Srivastava D, Nordheim A, Schwartz RJ. Serum response factor orchestrates nascent sarcomerogenesis and silences the biomineralization gene program in the heart. Proc Natl Acad Sci U S A. 2008;105:17824–17829. doi: 10.1073/pnas.0805491105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norman C, Runswick M, Pollock R, Treisman R. Isolation and properties of cDNA clones encoding SRF, a transcription factor that binds to the c-fos serum response element. Cell. 1988;55:989–1003. doi: 10.1016/0092-8674(88)90244-9. [DOI] [PubMed] [Google Scholar]
- Parlakian A, Tuil D, Hamard G, Tavernier G, Hentzen D, Concordet JP, Paulin D, Li Z, Daegelen D. Targeted inactivation of serum response factor in the developing heart results in myocardial defects and embryonic lethality. Mol Cell Biol. 2004;24:5281–5289. doi: 10.1128/MCB.24.12.5281-5289.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramanan N, Shen Y, Sarsfield S, Lemberger T, Schütz G, Linden DJ, Ginty DD. SRF mediates activity-induced gene expression and synaptic plasticity but not neuronal viability. Nat Neurosci. 2005;8:759–767. doi: 10.1038/nn1462. [DOI] [PubMed] [Google Scholar]
- Schratt G, Weinhold B, Lundberg AS, Schuck S, Berger J, Schwarz H, Weinberg RA, Rüther U, Nordheim A. Serum response factor is required for immediate-early gene activation yet is dispensable for proliferation of embryonic stem cells. Mol Cell Biol. 2001;21:2933–2943. doi: 10.1128/MCB.21.8.2933-2943.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schratt G, Philippar U, Berger J, Schwarz H, Heidenreich O, Nordheim A. Serum response factor is crucial for actin cytoskeletal organization and focal adhesion assembly in embryonic stem cells. J Cell Biol. 2002;156:737–750. doi: 10.1083/jcb.200106008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schratt G, Philippar U, Hockemeyer D, Schwarz H, Alberti S, Nordheim A. SRF regulates Bcl-2 expression and promotes cell survival during murine embryonic development. EMBO J. 2004;23:1834–1844. doi: 10.1038/sj.emboj.7600188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwab MH, Druffel-Augustin S, Gass P, Jung M, Klugmann M, Bartholomae A, Rossner MJ, Nave KA. Neuronal basic helix-loop-helix proteins (NEX, neuroD, NDRF): spatiotemporal expression and targeted disruption of the NEX gene in transgenic mice. J Neurosci. 1998;18:1408–1418. doi: 10.1523/JNEUROSCI.18-04-01408.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiota J, Ishikawa M, Sakagami H, Tsuda M, Baraban JM, Tabuchi A. Developmental expression of the SRF co-activator MAL in brain: role in regulating dendritic morphology. J Neurochem. 2006;98:1778–1788. doi: 10.1111/j.1471-4159.2006.03992.x. [DOI] [PubMed] [Google Scholar]
- Smith-Hicks C, Xiao B, Deng R, Ji Y, Zhao X, Shepherd JD, Posern G, Kuhl D, Huganir RL, Ginty DD, Worley PF, Linden DJ. SRF binding to SRE 6.9 in the Arc promoter is essential for LTD in cultured Purkinje cells. Nat Neurosci. 2010;13:1082–1089. doi: 10.1038/nn.2611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soulez M, Rouviere CG, Chafey P, Hentzen D, Vandromme M, Lautredou N, Lamb N, Kahn A, Tuil D. Growth and differentiation of C2 myogenic cells are dependent on serum response factor. Mol Cell Biol. 1996;16:6065–6074. doi: 10.1128/mcb.16.11.6065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stern S, Debre E, Stritt C, Berger J, Posern G, Knöll B. A nuclear actin function regulates neuronal motility by serum response factor-dependent gene transcription. J Neurosci. 2009;29:4512–4518. doi: 10.1523/JNEUROSCI.0333-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stritt C, Knöll B. Serum response factor regulates hippocampal lamination and dendrite development and is connected with reelin signaling. Mol Cell Biol. 2010;30:1828–1837. doi: 10.1128/MCB.01434-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stritt C, Stern S, Harting K, Manke T, Sinske D, Schwarz H, Vingron M, Nordheim A, Knöll B. Paracrine control of oligodendrocyte differentiation by SRF-directed neuronal gene expression. Nat Neurosci. 2009;12:418–427. doi: 10.1038/nn.2280. [DOI] [PubMed] [Google Scholar]
- Tronche F, Kellendonk C, Kretz O, Gass P, Anlag K, Orban PC, Bock R, Klein R, Schütz G. Disruption of the glucocorticoid receptor gene in the nervous system results in reduced anxiety. Nat Genet. 1999;23:99–103. doi: 10.1038/12703. [DOI] [PubMed] [Google Scholar]
- Wickramasinghe SR, Alvania RS, Ramanan N, Wood JN, Mandai K, Ginty DD. Serum response factor mediates NGF-dependent target innervation by embryonic DRG sensory neurons. Neuron. 2008;58:532–545. doi: 10.1016/j.neuron.2008.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witter MP, Naber PA, van Haeften T, Machielsen WC, Rombouts SA, Barkhof F, Scheltens P, Lopes da Silva FH. Cortico-hippocampal communication by way of parallel parahippocampal-subicular pathways. Hippocampus. 2000;10:398–410. doi: 10.1002/1098-1063(2000)10:4<398::AID-HIPO6>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- Wonders CP, Anderson SA. The origin and specification of cortical interneurons. Nat Rev Neurosci. 2006;7:687–696. doi: 10.1038/nrn1954. [DOI] [PubMed] [Google Scholar]
- Yamamoto A, Lucas JJ, Hen R. Reversal of neuropathology and motor dysfunction in a conditional model of Huntington's disease. Cell. 2000;101:57–66. doi: 10.1016/S0092-8674(00)80623-6. [DOI] [PubMed] [Google Scholar]
- Zhao Y, Samal E, Srivastava D. Serum response factor regulates a muscle-specific microRNA that targets Hand2 during cardiogenesis. Nature. 2005;436:214–220. doi: 10.1038/nature03817. [DOI] [PubMed] [Google Scholar]
- Zhou FQ, Snider WD. Intracellular control of developmental and regenerative axon growth. Philos Trans R Soc Lond B Biol Sci. 2006;361:1575–1592. doi: 10.1098/rstb.2006.1882. [DOI] [PMC free article] [PubMed] [Google Scholar]