

Abstract
In addition to analgesia, opioids may also produce paradoxical pain amplification [opioid-induced hyperalgesia (OIH)] either on abrupt withdrawal or during continuous long-term application. Here, we assessed antinociceptive and pronociceptive effects of three clinically used opioids at C-fiber synapses in the rat spinal dorsal horn in vivo. During 60 min of intravenous infusions of remifentanil (450 μg·kg−1·h−1), fentanyl (48 μg·kg−1·h−1), or morphine (14 mg·kg−1·h−1), C-fiber-evoked field potentials were depressed and paired-pulse ratios (PPR) were increased, indicating a presynaptic inhibition by all three opioids. After withdrawal, postsynaptic responses were enhanced substantially for the remaining of the recording periods of at least 3 h. Withdrawal from remifentanil led to long-term potentiation (LTP) of synaptic strength in C-fibers via activation of spinal μ-opioid receptors (MORs) and spinal NMDA receptors (NMDARs). Fentanyl and morphine caused an enhancement of synaptic transmission at C-fibers, which involved two distinct mechanisms: (1) an opioid withdrawal LTP that also required activation of spinal MORs and NMDARs and that was associated with a decrease in PPR suggestive of a presynaptic mechanism of its expression, and (2) an immediate-onset, descending facilitation of C-fiber-evoked field potentials during and after intravenous infusion of fentanyl and morphine. Immediate-onset, descending facilitation was mediated by the activation of extraspinal MORs, descending serotonergic pathways, and spinal 5-hydroxytryptamine-3 receptors (5-HT3Rs). Our study identified fundamentally different pronociceptive effects of clinically used opioids and suggests that OIH can be prevented by the combined use of NMDAR and 5-HT3R antagonists.
Introduction
Opioids represent the gold standard for the treatment of acute and chronic pain. Opioids may, however, also cause paradoxical pain amplification (hyperalgesia) that develops either on their abrupt withdrawal or during their continuous long-term use. Opioid-induced hyperalgesia (OIH) has been reported in animal models and was confirmed in human subjects (Angst and Clark, 2006). We have recently discovered a spinal mechanism that likely contributes to OIH: synaptic long-term potentiation (LTP) after opioid withdrawal. When the ultra-short-acting μ-opioid receptor (MOR) agonist remifentanil is withdrawn abruptly in vivo, synaptic transmission between nociceptive C-fibers and neurons in superficial spinal dorsal horn is persistently potentiated (Drdla et al., 2009). Opioid withdrawal LTP requires activation of spinal MORs and spinal NMDA receptors (NMDARs). LTP at C-fiber synapses can also be induced by various types of noxious stimulation and constitutes a cellular model of hyperalgesia (Sandkühler, 2009).
It is presently unknown whether opioid withdrawal LTP also applies to other clinically used MOR agonists that differ considerably with respect to their pharmacological profile. For example, remifentanil has an ultra-short half-life of several minutes (Egan et al., 1993) because of its rapid metabolism by blood and tissue esterases (Feldman et al., 1991). Most clinically used opioids, including fentanyl and morphine, have half-lives in the range of hours (Trescot et al., 2008). Remifentanil and fentanyl but not morphine cause considerable internalization of MORs (Trafton et al., 2000; Zaki et al., 2000). Furthermore, morphine has a broader opioid receptor binding profile (Matthes et al., 1998) compared with fentanyl and remifentanil.
Clinically used opioids all bind to MORs that are present presynaptically on spinal terminals of nociceptive fibers and postsynaptically on spinal dorsal horn neurons. Acute opioid application depresses transmitter release from central terminals of nociceptive fibers (Kohno et al., 1999; Heinke et al., 2011). This mechanism correlates to their analgesic effect. In contrast, postsynaptic G-protein coupling, activation of postsynaptic NMDARs and postsynaptic Ca2+ rise are required for the induction of opioid withdrawal LTP at C-fiber synapses (Drdla et al., 2009). It is presently not clear whether the expression of withdrawal LTP is also postsynaptic or whether it involves any presynaptic mechanisms.
MORs are also expressed on neurons in various brain areas, including the rostral ventromedial medulla, that send descending projections to the spinal cord (Marinelli et al., 2002). MOR agonists may activate descending pathways that modulate spinal nociception. Several lines of evidence suggest that descending facilitation contributes to OIH (Ossipov et al., 2004). Accordingly, surgical lesion (Vanderah et al., 2001) or pharmacological blockade of descending facilitation by intrathecal injection of a 5-hydroxytryptamine-3 receptor (5-HT3R) antagonist (Vera-Portocarrero et al., 2007; Liang et al., 2011) prevents development of OIH. The spinal mechanisms of opioid-induced descending facilitation of nociception are, however, primarily unidentified.
The present study revealed that remifentanil, fentanyl, and morphine all enhance synaptic transmission at spinal C-fibers but via fundamentally different mechanisms. The expression of withdrawal LTP by fentanyl and morphine but not remifentanil may involve a presynaptic mechanism and was additionally boosted by descending facilitation via activation of spinal 5-HT3Rs.
Materials and Methods
Animals.
All procedures were in accordance with European Communities Council directives (86/609/EEC) and were approved by the Austrian Federal Ministry of Science and Research.
Male Sprague Dawley rats (Medical University of Vienna breeding facility) weighing between 150 and 250 g were used for all experiments. Animals were kept on a 12 h light/dark cycle, housed three to six per cage, and were provided food and water ad libitum.
Animal surgery in vivo.
Isoflurane (4 vol/%) in two-thirds N2O and one-third O2 was initially administered via a mask to induce anesthesia. Animals were intubated using a 16 gauge cannula and then mechanically ventilated at a rate of 75 strokes/min using a tidal volume of 4–6 ml. Anesthesia was maintained by 1.5 vol/% isoflurane. Body core temperature was kept at 37.5°C with a feedback-controlled heating blanket. Deep surgical level of anesthesia was verified by stable mean arterial blood pressure during noxious stimulation. Surgical procedures were performed as described previously (Ikeda et al., 2006). Briefly, a jugular vein and a carotid artery were cannulated to allow intravenous infusions and arterial blood pressure monitoring, respectively. Muscle relaxation was achieved by 2 μg·kg−1·h−1 intravenous pancuronium bromide. After cannulation, the left sciatic nerve was dissected free for bipolar electrical stimulation with a silver hook electrode. The lumbar segments L4 and L5 were exposed by laminectomy. The dura mater was carefully incised and retracted. Two metal clamps were used for fixation of the vertebral column in a stereotactic frame. An agarose pool was formed around the exposed spinal segments. The spinal cord was continuously superfused with 5 ml of artificial CSF in which additional drugs could be dissolved as indicated. At the end of each electrophysiological experiment, animals were decapitated under deep anesthesia. The spinal cord was removed and cryofixed for detection of a rhodamine B spot at the recording site under a fluorescence microscope. Only those experiments in which the recording site was located in laminae I or II were analyzed.
Drugs and drug administration.
For in vivo recordings, pancuronium bromide (Pancuronium-ratiopharm; Ratiopharm) was administered as an intravenous infusion (2 μg·kg−1·h−1). Remifentanil (Ultiva; kindly provided by GlaxoSmithKline) was dissolved in sterile NaCl and applied as a 30 μg/kg bolus injection, followed by a 1 h infusion at a rate of 450 μg·kg−1·h−1. Fentanyl dihydrogene citrate (Fentanyl-Janssen; Jannsen-Cilag Pharma) was applied as a 40 μg/kg bolus injection, followed by a 1 h infusion at a rate of 48 μg·kg−1·h−1. Morphine hydrochloride (Vendal; Lannacher) was applied as an 8 mg/kg bolus injection, followed by an infusion at a rate of 14 mg·kg−1·h−1 for 1 h. Naloxone (Tocris Bioscience) was dissolved in sterile NaCl before given intravenously (100 mg·kg−1·h−1).
All other drugs were purchased from Tocris Bioscience. They were dissolved in water and added directly to 5 ml of artificial CSF superfusate to obtain the desired concentration as indicated: the MOR antagonist d-Phe-Cys-Tyr-d-Trp-Orn-Thr-Pen-Thr-NH2 (CTOP; 10 μm), the competitive NMDAR antagonist d(−)-2-amino-5-phosphonopentanoic acid (d-AP-5; 100 μm), the opioid receptor antagonist naloxone hydrochloride (100 μm), and the 5-HT3R antagonists granisetron hydrochloride (1 mm) and ondansetron hydrochloride (1 mm).
Electrophysiological recording.
Electrophysiological recordings were performed as described previously (Ikeda et al., 2006). Briefly, C-fiber-evoked field potentials were recorded with glass electrodes (impedance of 2–3 MΩ) from laminae I and II of the spinal cord dorsal horn in response to stimulation of sciatic nerve fibers. The pipette solution consisted of 135 mm NaCl, 5.4 mm KCl, 1.8 mm CaCl2, 10 mm HEPES, 1 mm MgCl, and 0.2% rhodamine B. At the end of each electrophysiological experiment, the recording site was labeled by pressure application (300 mbar for 1 min) with 0.2% rhodamine B via the electrode. Electrodes were driven by a microstepping motor. Recordings were made with an ISO-DAM-amplifier (World Precision Instruments) using a bandwidth filter of 0.1–1000 Hz. Signals were monitored on a digital oscilloscope and digitized by an analog-to-digital converter. Afferent input from the hindpaw was identified by mechanical stimulation of the foot while acoustically evaluating the evoked responses with an audio monitor. Test stimuli were delivered to the sciatic nerve and consisted of pulses of 0.5 ms duration at 25 V applied every 5 min using an electrical stimulator (ISO-01D-100; NPI Electronic). For paired-pulse recordings, two consecutive test stimuli were applied at a 500 ms interval.
Behavioral tests.
Behavioral experiments were performed between 9:00 A.M. and 6:00 P.M. Animals were habituated to the facility for at least 3 d and handled by the experimenters during this time. For 2 d before the assessment of baseline thresholds, rats were habituated to the behavioral testing apparatus for 30 min. Mechanical thresholds were measured with calibrated von Frey monofilaments with incremental stiffness between 0.25 and 15 g (Stoelting) based on the up and down method of Dixon (1965) at regular intervals. Rats were placed in individual Plexiglas boxes on a wire mesh metal floor. The plantar surface of the hindpaw between the footpads was stimulated in a consistent manner for 10 s. A foot withdrawal not attributable to normal locomotion was counted as a positive response. A lower force hair was presented during a positive response and a higher force during a negative response. A 50% threshold in grams was calculated as described previously (Chaplan et al., 1994). Experiments were performed by an experimenter unaware of treatment groups. The response thresholds for each hindpaw were averaged.
Baseline threshold testing was initiated 3 h before the treatment. Anesthesia was induced and maintained as described above for the in vivo electrophysiology. Animals were intubated and mechanically ventilated. The same drugs and doses were administered for 1 h into the jugular vein as described above. For remifentanil, a saline infusion with the equivalent concentration of glycine (3.4 mg·kg−1·h−1 infusion) contained in Ultiva (Hahnenkamp et al., 2004), whereas for fentanyl a physiological saline infusion served as control. Cannulation was removed and the skin was sutured using sterile precautions. Animals with remifentanil or respective control were removed from artificial ventilation and anesthesia 15 min after withdrawal. Anesthesia and ventilation of fentanyl-treated animals or respective controls was terminated 1 h after the end of the infusion, when spontaneous breathing restarted. Behavioral testing was performed 4 h, 24 h, 72 h, and 7 d after withdrawal from opioids.
Statistical analyses.
Data were analyzed using SigmaStat 3.1 (Systat Software). For electrophysiological recordings, the area under the curve of C-fiber-evoked field potentials was determined offline using Clampfit 10 (Molecular Devices). The mean area under the curve of six consecutive stable field potentials before opioid application served as a baseline control. Responses were normalized to the baseline in every animal. Data were tested for normality using the Kruskal–Wallis test. Unless otherwise indicated, a one-way repeated-measures (RM) ANOVA was performed to compare the different experimental protocols and treatments. Nonparametric one-way RM-ANOVA on ranks was performed in the case of non-normality. Effects of treatments were assessed using a t test comparing final size of potentiation from treated and nontreated animals. In case normality failed, a Mann–Whitney U test was performed.
For analysis of the paired-pulse measurements, a PPR was calculated by dividing the area of the C-fiber-evoked field potential of the second response by the area of the first response. One-way RM-ANOVA was performed on the mean of six PPRs from the baseline, 15 min after the start of the opioid infusion, and 15 min after withdrawal.
Behavioral data were analyzed by using a two-way RM-ANOVA comparing treatments and time points.
One-way RM-ANOVAs were corrected using a Bonferroni's adjustment, and one-way RM-ANOVA on ranks was corrected by Dunnett's test. A p value of <0.001 was considered as statistically “highly significant” and a p value of <0.05 as “significant.” Values are expressed as mean ± SEM.
Results
Fentanyl and morphine enhance synaptic transmission during precipitated and unprecipitated withdrawal
Withdrawal from an intravenous infusion of remifentanil was followed by a persistent enhancement of C-fiber-evoked field potentials (to 214 ± 26% of control at 220–240 min, p < 0.001, n = 8; Fig. 1A). Our previous study revealed that this enhancement is attributable to synaptic LTP, which is triggered by withdrawal from the opioid and which we thus termed “opioid withdrawal LTP” (Drdla et al., 2009). We showed that abrupt termination of a remifentanil infusion, which leads to a fast recovery from opioid-induced depression, is crucial for remifentanil-induced withdrawal LTP. A tapered withdrawal regimen, however, fully prevents LTP induction. Recovery from depression is slower for fentanyl and morphine because of their longer half-life. For quick recovery from depression, we precipitated the withdrawal by spinal application of the specific MOR antagonist CTOP at the end of the opioid infusion. During precipitated withdrawal, the recovery from depression was as fast as that observed after remifentanil. Under these conditions, fentanyl and morphine also elicited a robust enhancement of C-fiber-evoked field potentials (to 381 ± 57% of control at 220–240 min, p < 0.001, n = 8 and to 356 ± 66% of control at 220–240 min, p < 0.001, n = 7, respectively; Fig. 1B,C). This enhancement of C-fiber-evoked field potentials induced by fentanyl and morphine was attributable to two distinct mechanisms: (1) an opioid withdrawal LTP that required activation of spinal MORs and spinal NMDARs and (2) an immediate-onset, descending facilitation that was induced by activation of extraspinal MORs (see below). We collectively named both effects “opioid-induced enhancement of synaptic transmission.”
Figure 1.

Acute withdrawal from intravenous remifentanil, fentanyl, or morphine induced an enhancement of synaptic transmission in superficial spinal dorsal horn. In all graphs, areas of C-fiber-evoked field potentials were normalized to pre-drug values (dotted line) and plotted against time (minutes). A, Left, Time course of withdrawal LTP of C-fiber-evoked field potentials after termination of intravenous remifentanil infusion (black bar; 30 μg/kg bolus injection, followed by 450 μg·kg−1·h−1 infusion). Right, Representative traces of field potentials from one animal at indicated time points. The late negative deflection represents C-fiber potential (arrows). B, MOR antagonist CTOP was applied topically to the spinal cord dorsum at the recording site (open bar; 10 μm) after termination of intravenous infusion of fentanyl (black bar; 40 μg/kg bolus injection, followed by 48 μg·kg−1·h−1 infusion). C, Same experiment as in B with intravenous morphine (black bar; 8 mg/kg bolus injection, followed by 14 mg·kg−1·h−1 infusion). D, Nonprecipitated withdrawal from intravenous fentanyl (black bar; dosing as in B) elicited an enhancement of synaptic transmission. E, After withdrawal from intravenous morphine (black bar; dosing as in C), C-fiber-evoked field potentials recovered from depression within 2 h and continued to rise in size subsequently.
We next tested whether precipitation of the withdrawal was necessary for the induction of opioid-induced enhancement of synaptic transmission. Fentanyl and morphine were thus withdrawn without application of the MOR antagonist. The magnitude of C-fiber-evoked field potentials reached predrug level within 30 min after stopping the fentanyl infusion. It was followed by an enhancement of synaptic transmission that lasted until the end of the recording period (to 227 ± 44% of control at 280–300 min, p = 0.006, n = 8; Fig. 1D). After termination of the morphine infusion, the baseline was reached within 2 h and was followed by an enhancement of synaptic transmission (to 215 ± 50% of control at 440–460 min, p = 0.049, n = 6; Fig. 1E).
These results demonstrate that precipitated withdrawal or fast recovery from depression was not essential for the enhancement of synaptic transmission by fentanyl or morphine.
Presynaptic expression of the enhancement of synaptic transmission by fentanyl or morphine but not by remifentanil
The induction of withdrawal LTP by the MOR-specific agonist [d-Ala2, N-Me-Phe4, Gly-ol]-enkephalin (DAMGO) in vitro requires postsynaptic signaling (Drdla et al., 2009). Here, we tested whether the expression of opioid-induced enhancement of synaptic transmission is presynaptic and/or postsynaptic in nature. We evaluated the PPR of field potentials evoked by C-fiber stimulation before, during, and after administration of remifentanil, fentanyl, and morphine. PPR increased during remifentanil infusion (from 0.82 ± 0.03 during baseline to 1.15 ± 0.4 during infusion, p = 0.011, n = 11), consistent with a presynaptic inhibition. After withdrawal from remifentanil, PPR fell back to predrug levels (0.78 ± 0.02, p = 1.00, n = 11; Fig. 2A). During fentanyl infusion, the PPR also significantly increased (from 0.83 ± 0.06 during baseline to 1.06 ± 0.06 during infusion, p = 0.001, n = 11). However, in contrast to remifentanil, CTOP precipitated withdrawal of fentanyl induced a drop in the PPR below control values to 0.66 ± 0.04 15 min after termination of the infusion (p = 0.01, n = 11; Fig. 2B). This reduction in the PPR suggests an enhanced neurotransmitter release from presynaptic terminals as a mechanism underlying the enhancement of synaptic transmission by fentanyl. Likewise, the PPR increased during morphine infusion (from 0.92 ± 0.06 during baseline to 1.18 ± 0.12 during infusion, p = 0.025, n = 7). After precipitated withdrawal from morphine, the PPR was depressed below baseline values to 0.65 ± 0.03 (p = 0.029, n = 7; Fig. 2C). CTOP application alone had no effect on the PPR [0.79 ± 0.04 during baseline and 0.74 ± 0.03 15 min after CTOP application, p = 0.123 (paired t test), n = 12; data not shown].
Figure 2.
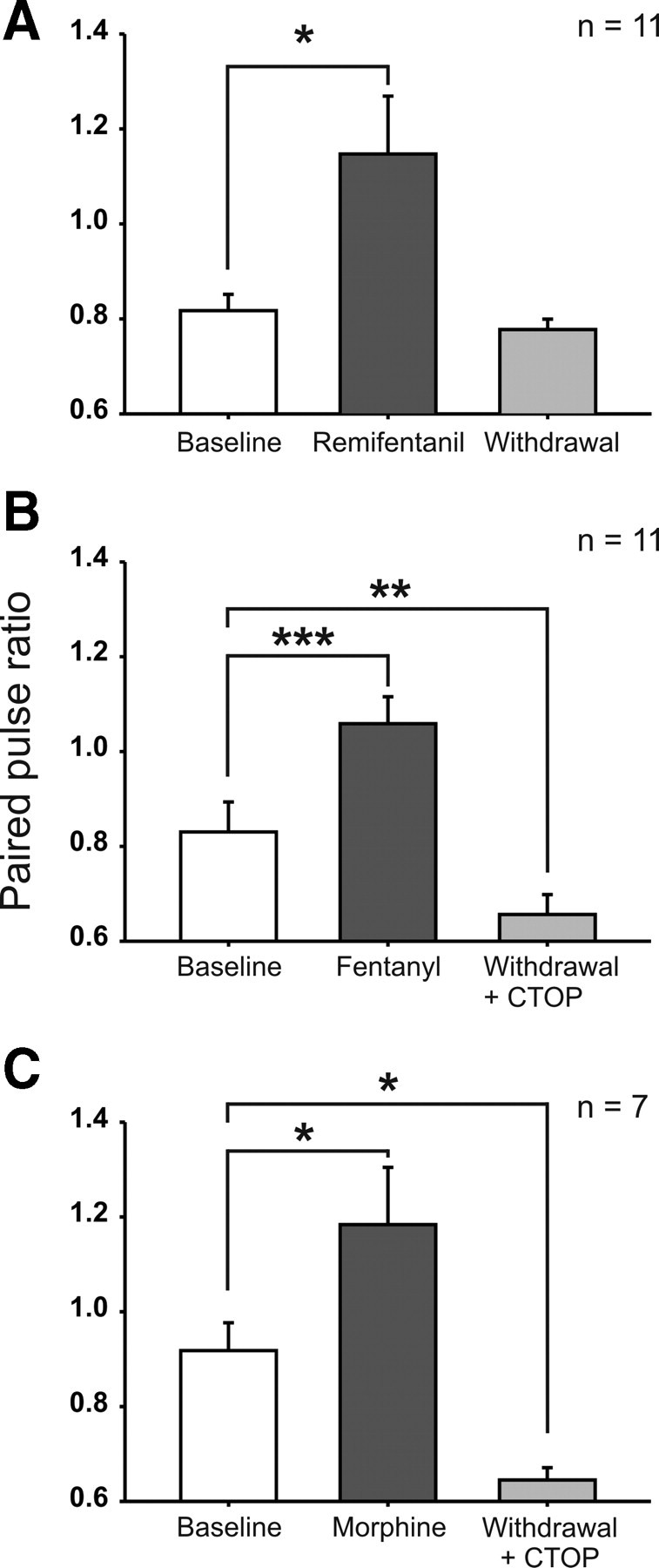
Intravenous opioids bidirectionally modulated the PPR of spinal C-fiber-evoked field potentials. Bar graphs represent the mean PPR during baseline (30 min before opioid infusion), during opioid infusion (at 15–45 min after onset of the infusion), and after withdrawal (at 15–45 min after termination of the infusion). A, During intravenous remifentanil infusion (dosing as in Fig. 1A), PPR was significantly increased and returned to baseline level after withdrawal. B, Fentanyl infusion (dosing as in Fig. 1B) was associated with an increased PPR. After withdrawal, precipitated by topic application of CTOP (10 μm), the PPR was depressed below baseline level. C, During morphine infusion (dosing as in Fig. 1C), the PPR increased significantly. After CTOP precipitated withdrawal, the PPR was decreased below baseline. *p < 0.05, **p < 0.01, ***p < 0.001, significant differences from baseline.
These results suggest that all three opioids depress C-fiber-evoked transmitter release presynaptically during their application. After withdrawal, fentanyl and morphine but not remifentanil may potentiate the transmitter release from C-fibers as a mechanism that enhances synaptic transmission.
Enhancement of synaptic transmission induced by fentanyl and morphine is partially independent of spinal NMDAR activation
Withdrawal LTP induced by remifentanil requires activation of spinal NMDARs (Drdla et al., 2009). In the present study, blockade of spinal NMDARs by topical application of d-AP-5 significantly reduced (p = 0.048) but failed to abolish enhancement of synaptic transmission after precipitated withdrawal from fentanyl (to 171 ± 29% of control at 220–240 min, p = 0.022, n = 6; Fig. 3A). Similarly, spinal d-AP-5 reduced (p = 0.004) but did not fully prevent enhancement of synaptic transmission after precipitated withdrawal from morphine (to 185 ± 23% of control at 220–240 min, p = 0.008, n = 6; Fig. 3B).
Figure 3.

Enhancement of synaptic transmission induced by withdrawal from fentanyl or morphine was reduced but not abolished by blockade of spinal NMDARs. In both graphs, areas of C-fiber-evoked field potentials were normalized to pre-drug values (dotted line) and plotted against time (minutes). A, Spinal superfusion with the NMDAR antagonist d-AP-5 (bottom white bar; 100 μm) failed to prevent enhancement of synaptic transmission induced by withdrawal, precipitated by topic application of the MOR antagonist CTOP (top white bar; 10 μm), after intravenous fentanyl (black bar; dosing as in Fig. 1B). B, As in A but with morphine infusion (black bar; dosing as in Fig. 1C).
These results suggest that, in contrast to remifentanil, fentanyl and morphine may not only trigger opioid withdrawal LTP but also an additional mechanism that is independent of spinal NMDAR activation.
Immediate-onset facilitation of C-fiber-evoked potentials during systemic fentanyl and morphine but not remifentanil infusion
We next studied the role of spinal MORs and applied the specific MOR antagonist CTOP directly onto the spinal cord at the recording site throughout the recording period. This abolished both the depression of C-fiber-evoked field potentials by systemically applied remifentanil and the induction of withdrawal LTP (96 ± 5% of control at 220–240 min, p = 0.360, n = 5; Fig. 4A). When spinal MORs were blocked by CTOP, the intravenous infusion of fentanyl induced, in contrast, an immediate-onset, slowly rising facilitation rather than a depression of C-fiber-evoked field potentials. This facilitation continued to increase after ending the infusion and thus contributed to the enhancement of synaptic transmission after opioid withdrawal (to 203 ± 26% of control at 220–240 min, p < 0.05, n = 6; Fig. 4C). Similarly, spinal CTOP blocked the depression by systemic morphine and unmasked an immediate-onset facilitation (to 149 ± 14% of control at 220–240 min, p = 0.002, n = 6; Fig. 4E). In the presence of CTOP, the PPR during remifentanil, fentanyl, and morphine infusion as well as after withdrawal remained unchanged (0.96 ± 0.07 during baseline, 0.98 ± 0.07 during remifentanil infusion, and 0.97 ± 0.08 15 min after withdrawal, p = 0.974, n = 5; 0.75 ± 0.04 during baseline, 0.71 ± 0.03 during fentanyl infusion, and 0.73 ± 0.03 15 min after withdrawal, p = 0.607, n = 6; 0.73 ± 0.04 during baseline, 0.69 ± 0.02 during morphine infusion, and 0.73 ± 0.03 15 min after withdrawal, p = 0.431 n = 6, respectively; Fig. 4B,D,F). These results suggest that the increase in PPR during intravenous infusion of remifentanil, fentanyl, and morphine depends on spinal MOR activation. They further imply that the depression of the PPR after withdrawal from intravenous fentanyl and morphine was mediated by spinal MORs.
Figure 4.

Blockade of spinal MORs prevented LTP induction after withdrawal from intravenous remifentanil and unmasked an immediate-onset facilitation by intravenous fentanyl and morphine. In graphs A, C, and E, areas of C-fiber-evoked field potentials were normalized to predrug values (dotted line) and plotted against time (minutes). A, Spinal superfusion with the MOR antagonist CTOP (white bar; 10 μm) abolished the depression during intravenous remifentanil (black bar; dosing as in Fig. 1A) and the subsequent withdrawal LTP. C, E, Intravenous infusion of fentanyl (black bar; intravenous dosing as in Fig. 1B) or morphine (black bar; dosing as in Fig. 1C) under spinal blockade of MORs by topically applied CTOP (white bar; 10 μm) induced an immediate-onset, descending facilitation that persisted until the end of the recording period. B, D, F, Bar graphs represent the corresponding mean PPR during baseline (30 min before opioid infusion), during opioid infusion (at 15–45 min after onset of the infusion), and after withdrawal (at 15–45 min after termination of the infusion). PPR remained unchanged during and after intravenous remifentanil, fentanyl, and morphine infusion when spinal MORs were blocked with CTOP.
Fentanyl-induced immediate-onset facilitation involves extraspinal opioid receptors
Spinal blockade of MORs unmasked the immediate-onset facilitation induced by systemic fentanyl. This facilitation could be caused by an opioid receptor-independent process, e.g., by active metabolites, or could involve activation of extraspinal opioid receptors. For a global blockade of opioid receptors, we applied naloxone intravenously, which in contrast to CTOP penetrates the blood–brain barrier. Systemic naloxone fully blocked both the depression and the immediate-onset facilitation induced by intravenous fentanyl (101 ± 14% of control at 220–240 min, p = 0.262, n = 6; Fig. 5A). Naloxone is also an antagonist at δ- and κ-opioid receptors albeit with lower affinity. To test whether blockade of spinal δ- and κ-opioid receptors plays any role, naloxone was next applied directly onto the spinal cord by superfusion. Systemic fentanyl still induced an immediate-onset facilitation (to 178 ± 14% of control at 220–240 min, n = 5, p = 0.001; data not shown). This demonstrates that the inhibition of the immediate-onset facilitation by systemic naloxone was not attributable to blockade of spinal δ- or κ-opioid receptors. The results rather suggest that fentanyl-induced immediate-onset facilitation requires activation of opioid receptors that are located outside of the spinal cord.
Figure 5.
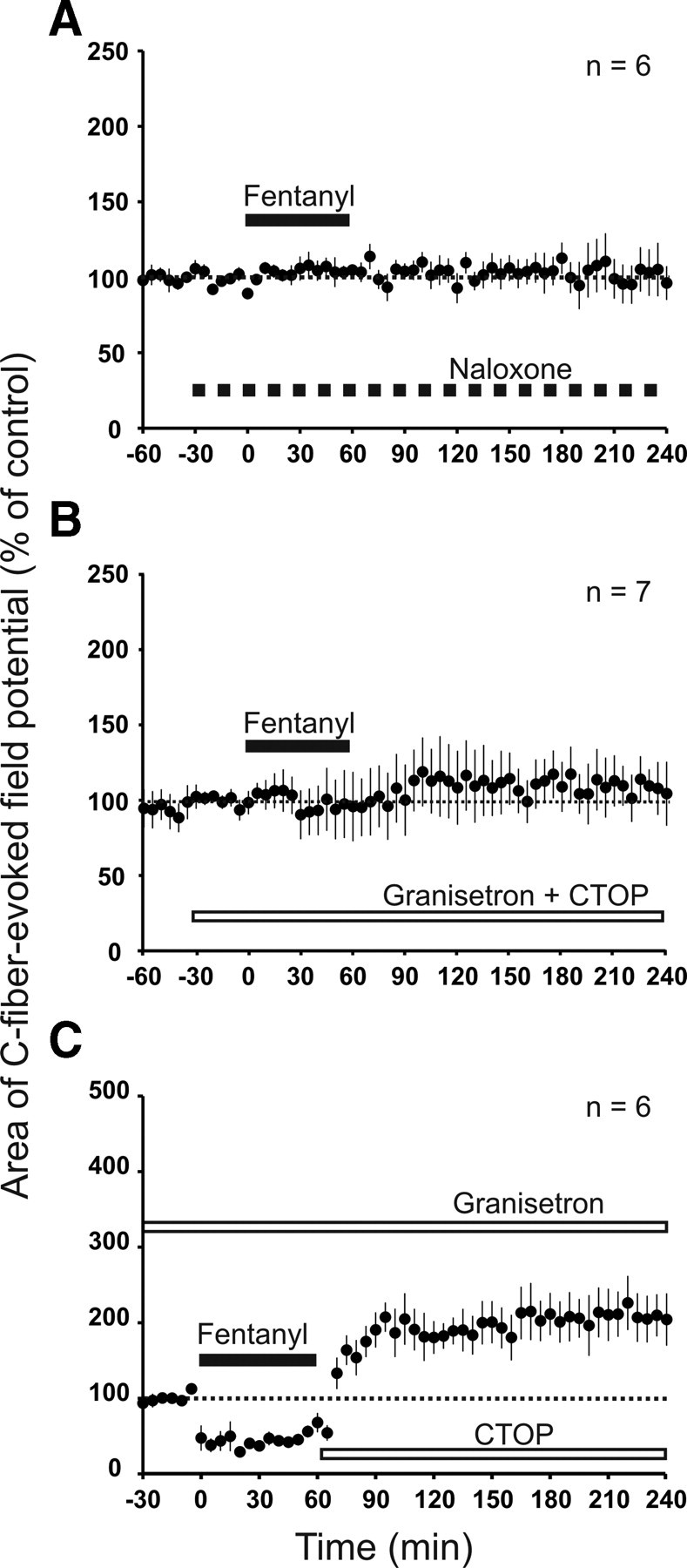
Immediate-onset, descending facilitation requires activation of extraspinal MORs and spinal 5-HT3Rs. Areas of C-fiber-evoked field potentials were normalized to predrug values (dotted line) and plotted against time (minutes). A, Intravenous infusion of the opioid receptor antagonist naloxone (hatched bar; 100 mg·kg−1·h−1) blocked depression induced by intravenous fentanyl (black bar; dosing as in Fig. 1B) as well as opioid-induced enhancement of synaptic transmission. B, Spinal superfusion with the 5-HT3R antagonist granisetron and MOR antagonist CTOP (white bar; 1 mm and 10 μm, respectively) abolished immediate-onset, descending facilitation induced by intravenous fentanyl (black bar). C, Granisetron was applied to the spinal cord dorsum (top white bar; 1 mm). Precipitated withdrawal from intravenous fentanyl with spinal CTOP (bottom white bar; 10 μm) induced withdrawal LTP.
Fentanyl but not remifentanil activates immediate-onset, descending facilitation via spinal 5-HT3Rs
It has been shown that descending pathways (King et al., 2005) and activation of spinal 5-HT3Rs (Vera-Portocarrero et al., 2007; Liang et al., 2011) are involved in OIH. To evaluate the contribution of descending serotonergic pathways to the immediate-onset facilitation induced by fentanyl, we superfused the spinal cord with the 5-HT3R antagonists ondansetron or granisetron in the presence of spinal CTOP. Fentanyl-induced immediate-onset facilitation was fully blocked by ondansetron (113 ± 19% of control at 220–240 min, p = 0.128, n = 7; data not shown) and by granisetron (107 ± 16% of control at 220–240 min, p = 0.563, n = 7; Fig. 5B). Thus, activation of descending serotonergic pathways and spinal 5-HT3Rs is essential for fentanyl-induced immediate-onset facilitation. We next asked whether the immediate-onset, descending facilitation interacts with the withdrawal LTP. We thus induced a precipitated withdrawal from fentanyl in the presence of spinal granisetron to block descending facilitation. At least two scenarios are conceivable. First, the spinal mechanisms of withdrawal LTP and immediate-onset facilitation could overlap and thus occlude each other. In this case, both effects should be sub-additive when leading to opioid-induced enhancement of synaptic transmission. Second, activation of spinal 5-HT3Rs could facilitate the expression of withdrawal LTP, which would lead to supra-additive effects. Under blockade of spinal 5-HT3Rs, we observed a robust withdrawal LTP to 208 ± 31% of control at 220–240 min (p = 0.005, n = 6; Fig. 5C). The immediate-onset, descending facilitation induced by fentanyl was to 203 ± 26% of control at 220–240 min (p < 0.05, n = 6) as shown in Figure 4C. Thus, both mechanisms have approximately additive effects when leading to opioid-induced enhancement of synaptic transmission. Under spinal 5-HT3R blockade, PPR was still depressed after withdrawal from intravenous fentanyl (from 0.75 ± 0.19 during baseline to 0.43 ± 0.06 after withdrawal, p = 0.023, n = 6; data not shown). Thus, 5-HT3R activation is not required for PPR depression induced by withdrawal of fentanyl. Together, these findings indicate that immediate-onset, descending facilitation and opioid withdrawal LTP are independent pronociceptive mechanisms of opioids.
Spinal granisetron did not affect (p = 0.513) remifentanil-induced withdrawal LTP (to 192 ± 19% of control at 220–240 min, p < 0.001, n = 8; data not shown), supporting that immediate-onset, descending facilitation is not activated by remifentanil.
Complete block of spinal pronociceptive effects of fentanyl and morphine
We next tested whether withdrawal LTP and immediate-onset, descending facilitation by fentanyl or morphine can be blocked fully without compromising inhibition. We simultaneously superfused the spinal cord with the NMDAR antagonist d-AP-5 to block withdrawal LTP and the 5-HT3R antagonist granisetron to block immediate-onset, descending facilitation. This fully prevented the rise of C-fiber-evoked field potentials after fentanyl (105 ± 15% of control at 220–240 min, p = 1.00, n = 6; Fig. 6A). The fentanyl-induced depression was, in contrast, fully preserved. Similar results were obtained for morphine (112 ± 19% of control at 220–240 min, p = 0.749, n = 7; Fig. 6B).
Figure 6.

Blockade of spinal 5-HT3Rs and NMDARs abolished immediate-onset, descending facilitation and withdrawal LTP after intravenous fentanyl and morphine. Areas of C-fiber-evoked field potentials were normalized to predrug values (dotted line) and plotted against time (minutes). A, The 5-HT3R antagonist granisetron and the NMDAR antagonist d-AP-5 were applied simultaneously to the spinal cord dorsum (top white bar; 1 mm and 100 μm, respectively). Intravenous infusion of fentanyl (black bar) still induced a depression, but withdrawal precipitated with spinal CTOP (bottom white bar; 10 μm) failed to elicit opioid-induced enhancement of synaptic transmission. B, Same experiment as in A but with intravenous morphine (black bar; dosing as in Fig. 1C).
Thus, the two pronociceptive mechanisms that are activated by fentanyl and morphine, i.e., the NMDAR-dependent withdrawal LTP, and the 5-HT3R-mediated immediate-onset, descending facilitation can be blocked pharmacologically without affecting opioid-induced synaptic depression.
Acute remifentanil or fentanyl applications induce persistent mechanical hyperalgesia
We next investigated whether the above used dosing regimen of remifentanil and fentanyl, respectively, induce hyperalgesia in behaving animal. As shown above, both treatments enhanced synaptic strength but through activation of distinct mechanisms. Withdrawal from short-term infusion of remifentanil (1 h) caused a decrease in response thresholds to von Frey hairs compared with baseline (from 13.7 ± 0.4 to 5.6 ± 0.5 g 7 d after withdrawal, p < 0.001, n = 11). There was no significant decrease in the response threshold after infusion of vehicle (14.4 ± 0.6 and 11.9 ± 1.5 g 7 d after withdrawal, p = 0.166, n = 8; Fig. 7A). Withdrawal from short-term infusion of fentanyl caused a significant lowering of response thresholds to mechanical stimuli (from 14.1 ± 0.3 to 6.8 ± 1.9 g 7 d after withdrawal, p = 0.006, n = 8). Response thresholds of saline-treated control group remained unchanged throughout the testing period compared with baseline (13.1 ± 0.6 and 11.8 ± 1.1 g 7 d after withdrawal, p = 1.00, n = 8; Fig. 7B). Our behavioral tests demonstrate that the presently used dosing regimen for remifentanil and fentanyl induce long-lasting increase in nociception of behaving animals.
Figure 7.
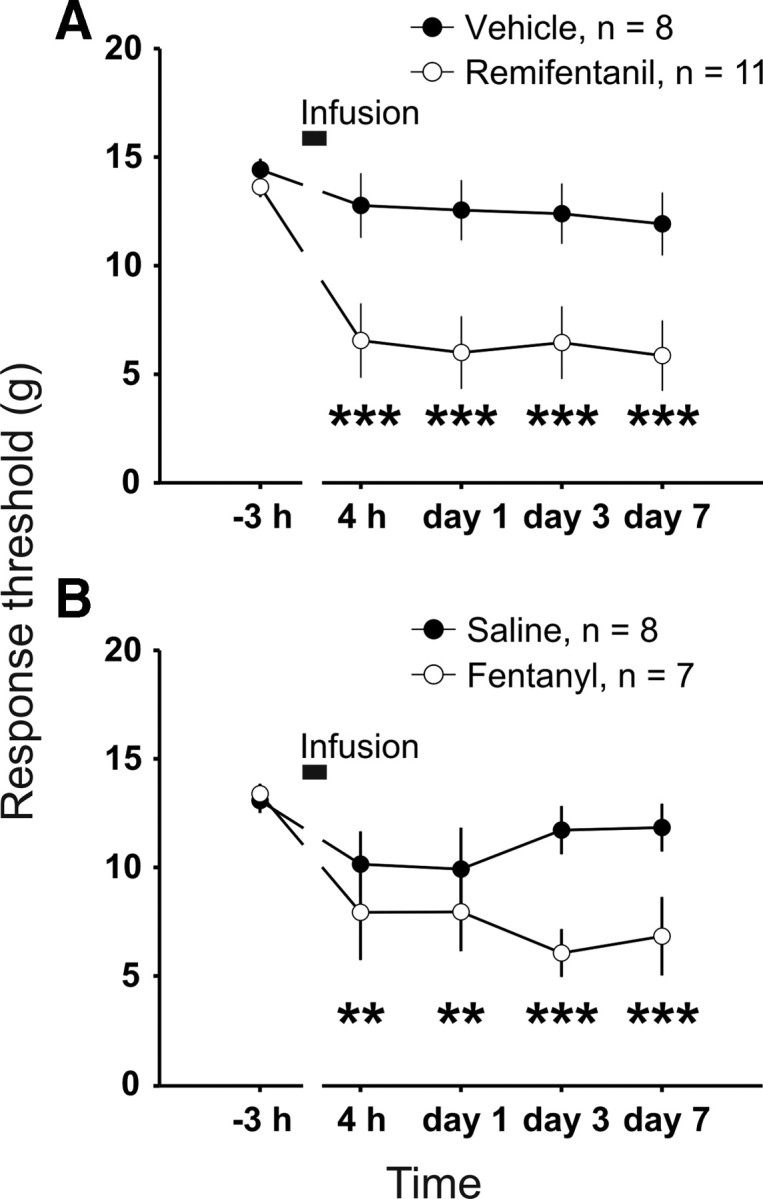
Remifentanil and fentanyl induced a long-lasting reduction in mechanical hindpaw response thresholds. The mean response thresholds to probing with von Frey filaments are plotted at different time points. Animals received a 1 h infusion into the jugular vein (black bars) under deep anesthesia. A, Response thresholds from remifentanil-treated animals (dosing as in Fig. 1A) were significantly reduced compared with baseline at all time points after the infusion. Response threshold for vehicle-treated group (3.4 mg·kg−1·h−1 glycine infusion) remained stable compared with pre-drug level. B, Fentanyl-treated animals (dosing as in Fig. 1B) showed significantly lower thresholds compared with predrug responses at all time points after the infusion. The saline-treated group showed no significant changes after infusion compared with baseline. **p < 0.01, *** p < 0.001.
Discussion
In the present study, we demonstrate that remifentanil, fentanyl, and morphine exert spinal pronociceptive effects that likely contribute to OIH. The three opioids involve, however, distinct mechanisms as judged by NMDAR dependence, presynaptic versus postsynaptic expression of opioid withdrawal LTP, and the role of descending facilitatory pathways.
Opioid-induced depression at spinal C-fiber synapses
In the superficial spinal dorsal horn, MORs are present on the terminals of afferent nerve fibers, including C-fibers, and postsynaptically on dorsal horn neurons (Besse et al., 1990; Scherrer et al., 2009). We and others have shown previously that, in vitro, the MOR agonist DAMGO induces a powerful presynaptic depression at C-fiber terminals (Ikoma et al., 2007; Heinke et al., 2011). Similarly, in the present study, depression of C-fiber-evoked field potentials by systemic remifentanil, fentanyl, and morphine required activation of spinal MORs and was associated with an increase in the PPR. The PPR is an indication to assess changes in neurotransmitter release probability in patch clamp as well as in in vivo field potential recordings (Zucker, 1973; Zucker and Regehr, 2002). Collectively, these data suggest that, after systemic application, the three opioids act on spinal MORs to depress neurotransmitter release from nociceptive C-fibers as a mechanism of antinociception.
Enhancement of synaptic transmission by remifentanil, fentanyl, and morphine
After withdrawal, all three opioids induced a robust enhancement of C-fiber-evoked field potentials. Compared with remifentanil, recovery from depression takes considerably longer for fentanyl and morphine because of their longer half-life. Our previous study revealed that LTP during abrupt withdrawal from remifentanil is fully prevented when a tapered withdrawal regimen is used, which leads to a slower recovery from depression (Drdla et al., 2009). The slower recovery from depression by morphine or fentanyl without precipitation was comparable with the recovery rate after tapered withdrawal from remifentanil. This did, however, not prevent opioid-induced enhancement of synaptic transmission and suggests that the rate of recovery from depression is not a determinant parameter for enhancement of synaptic transmission by fentanyl and morphine.
Enhancement of synaptic transmission by fentanyl was significantly reduced by the blockade of spinal NMDARs as well as by the blockade of spinal 5-HT3Rs. However, only simultaneous blockade of both receptors could fully prevent opioid-induced enhancement of synaptic transmission, indicating that two distinct mechanisms mediate this effect: opioid withdrawal LTP and immediate-onset, descending facilitation.
Withdrawal LTP by remifentanil, fentanyl, and morphine
The withdrawal LTP induced by the three opioids differed with respect to their impact on the PPR. In contrast to remifentanil, the expression of morphine- and fentanyl-induced withdrawal LTP was associated with a decreased PPR, suggesting an enhanced transmitter release probability (Oleskevich et al., 2000; Thomson, 2000). When spinal MORs were blocked by CTOP, fentanyl and morphine induced neither a paired-pulse facilitation nor a paired-pulse depression. This indicates that spinal MORs mediate both phenomena. Our results are in contrast to those of Zhou et al. (2010) who reported that the MOR agonist DAMGO enhanced PPR, during both acute synaptic depression and withdrawal LTP in a spinal cord slice preparation. Surprisingly, these authors interpreted the increased PPR as a sign for presynaptic expression of LTP (see also our eLetter in response to their report at http://www.jneurosci.org/content/30/12/4460.long/reply#jneuro_el_71584). Nevertheless, the data suggest that different opioids can have diverging effects on the PPR at C-fiber synapses. We have shown previously that postsynaptic signaling is essential for the induction of withdrawal LTP (Drdla et al., 2009). In the present work, we extended these findings by showing that enhanced transmitter release likely contributes to the expression of withdrawal LTP induced by fentanyl and morphine but not by remifentanil at C-fiber synapses.
We suggest that opioid withdrawal LTP underlies OIH because both can be induced by identical dosing regimen and involve overlapping signaling pathways, including activation of NMDARs (Trujillo and Akil, 1991; Célèrier et al., 1999) and protein kinase C (Mao et al., 1994; Sweitzer et al., 2004; Drdla et al., 2009). Fentanyl and morphine but not remifentanil activate in addition serotonergic descending facilitatory pathways, which may further boost OIH.
Immediate-onset, descending facilitation induced by morphine and fentanyl but not by remifentanil
Several days after continuous application of opioids, the initial analgesia may not only vanish but may actually turn into OIH (Ossipov et al., 2004; King et al., 2005). OIH may involve activation of descending serotonergic facilitatory systems because it can be blocked by surgical disruption of descending pathways (Vanderah et al., 2001) or by blocking spinal 5-HT3Rs (Vera-Portocarrero et al., 2007; Liang et al., 2011). The present study identified a novel, immediate-onset, descending facilitation of synaptic strength in C-fibers activated by opioids. This process could also well underlie the slowly developing loss of analgesic efficacy (apparent tolerance) and OIH. Within minutes of systemic application, both fentanyl and morphine triggered a facilitation that progressively increased during the application and continued to increase throughout the recording period after termination of the opioid infusion. This immediate-onset, descending facilitation by morphine and fentanyl was normally masked by the concomitant depression of spinal nociception but became apparent when spinal MORs were blocked.
Systemic but not spinal opioid-receptor blockade abolished immediate-onset, descending facilitation induced by intravenous infusion of fentanyl, demonstrating the involvement of extraspinal opioid receptors. Good candidates are MORs in the rostral ventromedial medulla, a brainstem region that sends serotonergic pathways to the spinal dorsal horn (Fields and Basbaum, 2000) and that has been implicated in the expression of OIH (Porreca et al., 2002). Descending serotonergic pathways can be activated by ascending pathways involving neurokinin 1 receptor-positive projection neurons (Suzuki et al., 2002). Previous studies using combined electrophysiological and immunohistological approaches indicate that serotonergic descending pathways can also be disinhibited by MOR agonists (Marinelli et al., 2002). Disinhibition has a potentially rapid onset that could correspond well to the time course of the presently described immediate-onset facilitation.
Immediate-onset, descending facilitation identified in the present study required activation of spinal 5-HT3Rs, and it is safe to say that it involves descending serotonergic pathways because they are the only relevant source of serotonin in the spinal dorsal horn (Millan, 2002). Some studies propose that 5-HT3R activation enhances transmitter release from presynaptic terminals of afferent fibers (Nayak et al., 1999; Suzuki et al., 2004). The immediate-onset, descending facilitation shown in the present study was, however, not associated with any change in the PPR. This suggests that 5-HT3R activation during descending facilitation did not enhance transmitter release from presynaptic terminals but could have rather affected superficial spinal dorsal neurons expressing the 5-HT3R (Kia et al., 1995; Miquel et al., 2002; Conte et al., 2005). 5-HT3Rs are not coupled to G-proteins but directly to nonselective cationic channels (Derkach et al., 1989). Activation of 5-HT3Rs located on somadendritic regions of spinal dorsal horn neurons induce an inward current that may exert both pronociceptive and antinociceptive effects because 5-HT3Rs are expressed on spinal GABAergic and on putatively excitatory interneurons (Fukushima et al., 2009).
Differential pronociceptive mechanisms activated by remifentanil, fentanyl, and morphine
We have found major differences in the spinal pronociceptive mechanisms of remifentanil on the one hand and fentanyl and morphine on the other. The question arises which properties might cause this grouping. Characteristics that are not shared by fentanyl and morphine can be excluded, such as the low potency to induce MOR internalization (Trafton et al., 2000; Zaki et al., 2000), the broad opioid receptor subtype binding (Matthes et al., 1998), and the production of active metabolites (Yaksh et al., 1986), which are all typical for morphine but not for fentanyl. Lipophilicity is also an unlikely grouping variable because fentanyl but not morphine and remifentanil is characterized by a very high lipophilicity. One distinguishing characteristic could be that the three opioids do not act on the same sites in the CNS because of an unequal ability to activate different splice variants of the MOR gene (Pasternak, 2004). The relevance of this is, however, presently unknown.
The present study revealed substantial differences in the mechanisms that underlie the enhancement of spinal nociception by three systemically applied opioids. Although remifentanil selectively induced an NMDAR-dependent withdrawal LTP via activation of spinal MORs, fentanyl and morphine in addition activated descending, facilitatory, serotonergic pathways via extraspinal MORs. Our findings add to the list of distinguishing features between opioids and provide an additional rationale for opioid rotation in pain patients. Our data further suggest that a combination of NMDAR antagonists with opioids, such as fentanyl or morphine, may reduce but may not fully prevent OIH. An additional antagonist at the 5-HT3R may prove to be useful (Liang et al., 2011; Vera-Portocarrero et al., 2011). Clinically used 5-HT3R antagonists are available for the treatment of emesis and pruritus, e.g., caused by opioids, and include granisetron, dolasetron, ondansetron, palonosetron, and tropisetron. The present findings provide another motivation for combining these drugs with opioids from early on.
Footnotes
This project was supported by Austrian Science Fund Grant P22306-B19 (J.S.).
The authors declare no conflict of interests.
References
- Angst MS, Clark JD. Opioid-induced hyperalgesia: a qualitative systematic review. Anesthesiology. 2006;104:570–587. doi: 10.1097/00000542-200603000-00025. [DOI] [PubMed] [Google Scholar]
- Besse D, Lombard MC, Zajac JM, Roques BP, Besson JM. Pre- and postsynaptic distribution of μ, δ and κ opioid receptors in the superficial layers of the cervical dorsal horn of the rat spinal cord. Brain Res. 1990;521:15–22. doi: 10.1016/0006-8993(90)91519-m. [DOI] [PubMed] [Google Scholar]
- Célèrier E, Laulin J, Larcher A, Le Moal M, Simonnet G. Evidence for opiate-activated NMDA processes masking opiate analgesia in rats. Brain Res. 1999;847:18–25. doi: 10.1016/s0006-8993(99)01998-8. [DOI] [PubMed] [Google Scholar]
- Chaplan SR, Bach FW, Pogrel JW, Chung JM, Yaksh TL. Quantitative assessment of tactile allodynia in the rat paw. J Neurosci Methods. 1994;53:55–63. doi: 10.1016/0165-0270(94)90144-9. [DOI] [PubMed] [Google Scholar]
- Conte D, Legg ED, McCourt AC, Silajdzic E, Nagy GG, Maxwell DJ. Transmitter content, origins and connections of axons in the spinal cord that possess the serotonin (5-hydroxytryptamine) 3 receptor. Neuroscience. 2005;134:165–173. doi: 10.1016/j.neuroscience.2005.02.013. [DOI] [PubMed] [Google Scholar]
- Derkach V, Surprenant A, North RA. 5-HT3 receptors are membrane ion channels. Nature. 1989;339:706–709. doi: 10.1038/339706a0. [DOI] [PubMed] [Google Scholar]
- Dixon WJ. The up-and-down method for small samples. J Am Statist Assoc. 1965;60:967–978. [Google Scholar]
- Drdla R, Gassner M, Gingl E, Sandkühler J. Induction of synaptic long-term potentiation after opioid withdrawal. Science. 2009;325:207–210. doi: 10.1126/science.1171759. [DOI] [PubMed] [Google Scholar]
- Egan TD, Lemmens HJ, Fiset P, Hermann DJ, Muir KT, Stanski DR, Shafer SL. The pharmacokinetics of the new short-acting opioid remifentanil (GI87084B) in healthy adult male volunteers. Anesthesiology. 1993;79:881–892. doi: 10.1097/00000542-199311000-00004. [DOI] [PubMed] [Google Scholar]
- Feldman PL, James MK, Brackeen MF, Bilotta JM, Schuster SV, Lahey AP, Lutz MW, Johnson MR, Leighton HJ. Design, synthesis, and pharmacological evaluation of ultrashort- to long-acting opioid analgetics. J Med Chem. 1991;34:2202–2208. doi: 10.1021/jm00111a041. [DOI] [PubMed] [Google Scholar]
- Fields HL, Basbaum AI. Central nervous system mechanisms of pain modulation. In: Wall PD, Melzack R, editors. Textbook of pain. London: Churchill Livingstone; 2000. pp. 243–257. [Google Scholar]
- Fukushima T, Ohtsubo T, Tsuda M, Yanagawa Y, Hori Y. Facilitatory actions of serotonin type 3 receptors on GABAergic inhibitory synaptic transmission in the spinal superficial dorsal horn. J Neurophysiol. 2009;102:1459–1471. doi: 10.1152/jn.91160.2008. [DOI] [PubMed] [Google Scholar]
- Hahnenkamp K, Nollet J, Van Aken HK, Buerkle H, Halene T, Schauerte S, Hahnenkamp A, Hollmann MW, Strümper D, Durieux ME, Hoenemann CW. Remifentanil directly activates human N-methyl-d-aspartate receptors expressed in Xenopus laevis oocytes. Anesthesiology. 2004;100:1531–1537. doi: 10.1097/00000542-200406000-00028. [DOI] [PubMed] [Google Scholar]
- Heinke B, Gingl E, Sandkühler J. Multiple targets of μ-opioid receptor mediated presynaptic inhibition at primary afferent Aδ- and C-fibers. J Neurosci. 2011;31:1313–1322. doi: 10.1523/JNEUROSCI.4060-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeda H, Stark J, Fischer H, Wagner M, Drdla R, Jäger T, Sandkühler J. Synaptic amplifier of inflammatory pain in the spinal dorsal horn. Science. 2006;312:1659–1662. doi: 10.1126/science.1127233. [DOI] [PubMed] [Google Scholar]
- Ikoma M, Kohno T, Baba H. Differential presynaptic effects of opioid agonists on Aδ- and C-afferent glutamatergic transmission to the spinal dorsal horn. Anesthesiology. 2007;107:807–812. doi: 10.1097/01.anes.0000286985.80301.5e. [DOI] [PubMed] [Google Scholar]
- Kia HK, Miquel MC, McKernan RM, Laporte AM, Lombard MC, Bourgoin S, Hamon M, Vergé D. Localization of 5-HT3 receptors in the rat spinal cord: immunohistochemistry and in situ hybridization. Neuroreport. 1995;6:257–261. doi: 10.1097/00001756-199501000-00008. [DOI] [PubMed] [Google Scholar]
- King T, Ossipov MH, Vanderah TW, Porreca F, Lai J. Is paradoxical pain induced by sustained opioid exposure an underlying mechanism of opioid antinociceptive tolerance? Neurosignals. 2005;14:194–205. doi: 10.1159/000087658. [DOI] [PubMed] [Google Scholar]
- Kohno T, Kumamoto E, Higashi H, Shimoji K, Yoshimura M. Actions of opioids on excitatory and inhibitory transmission in substantia gelatinosa of adult rat spinal cord. J Physiol. 1999;518:803–813. doi: 10.1111/j.1469-7793.1999.0803p.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang DY, Li X, Clark JD. 5-Hydroxytryptamine type 3 receptor modulates opioid-induced hyperalgesia and tolerance in mice. Anesthesiology. 2011;114:1180–1189. doi: 10.1097/ALN.0b013e31820efb19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao J, Price DD, Mayer DJ. Thermal hyperalgesia in association with the development of morphine tolerance in rats: roles of excitatory amino acid receptors and protein kinase C. J Neurosci. 1994;14:2301–2312. doi: 10.1523/JNEUROSCI.14-04-02301.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marinelli S, Vaughan CW, Schnell SA, Wessendorf MW, Christie MJ. Rostral ventromedial medulla neurons that project to the spinal cord express multiple opioid receptor phenotypes. J Neurosci. 2002;22:10847–10855. doi: 10.1523/JNEUROSCI.22-24-10847.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthes HW, Smadja C, Valverde O, Vonesch JL, Foutz AS, Boudinot E, Denavit-Saubié M, Severini C, Negri L, Roques BP, Maldonado R, Kieffer BL. Activity of the δ-opioid receptor is partially reduced, whereas activity of the κ-receptor is maintained in mice lacking the μ-receptor. J Neurosci. 1998;18:7285–7295. doi: 10.1523/JNEUROSCI.18-18-07285.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millan MJ. Descending control of pain. Prog Neurobiol. 2002;66:355–474. doi: 10.1016/s0301-0082(02)00009-6. [DOI] [PubMed] [Google Scholar]
- Miquel MC, Emerit MB, Nosjean A, Simon A, Rumajogee P, Brisorgueil MJ, Doucet E, Hamon M, Vergé D. Differential subcellular localization of the 5-HT3-As receptor subunit in the rat central nervous system. Eur J Neurosci. 2002;15:449–457. doi: 10.1046/j.0953-816x.2001.01872.x. [DOI] [PubMed] [Google Scholar]
- Nayak SV, Rondé P, Spier AD, Lummis SC, Nichols RA. Calcium changes induced by presynaptic 5-hydroxytryptamine-3 serotonin receptors on isolated terminals from various regions of the rat brain. Neuroscience. 1999;91:107–117. doi: 10.1016/s0306-4522(98)00520-x. [DOI] [PubMed] [Google Scholar]
- Oleskevich S, Clements J, Walmsley B. Release probability modulates short-term plasticity at a rat giant terminal. J Physiol. 2000;524:513–523. doi: 10.1111/j.1469-7793.2000.00513.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ossipov MH, Lai J, King T, Vanderah TW, Malan TP, Jr, Hruby VJ, Porreca F. Antinociceptive and nociceptive actions of opioids. J Neurobiol. 2004;61:126–148. doi: 10.1002/neu.20091. [DOI] [PubMed] [Google Scholar]
- Pasternak GW. Multiple opiate receptors: déjà vu all over again. Neuropharmacology. 2004;47(Suppl 1):312–323. doi: 10.1016/j.neuropharm.2004.07.004. [DOI] [PubMed] [Google Scholar]
- Porreca F, Ossipov MH, Gebhart GF. Chronic pain and medullary descending facilitation. Trends Neurosci. 2002;25:319–325. doi: 10.1016/s0166-2236(02)02157-4. [DOI] [PubMed] [Google Scholar]
- Sandkühler J. Models and mechanisms of hyperalgesia and allodynia. Physiol Rev. 2009;89:707–758. doi: 10.1152/physrev.00025.2008. [DOI] [PubMed] [Google Scholar]
- Scherrer G, Imamachi N, Cao YQ, Contet C, Mennicken F, O'Donnell D, Kieffer BL, Basbaum AI. Dissociation of the opioid receptor mechanisms that control mechanical and heat pain. Cell. 2009;137:1148–1159. doi: 10.1016/j.cell.2009.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki R, Morcuende S, Webber M, Hunt SP, Dickenson AH. Superficial NK1-expressing neurons control spinal excitability through activation of descending pathways. Nat Neurosci. 2002;5:1319–1326. doi: 10.1038/nn966. [DOI] [PubMed] [Google Scholar]
- Suzuki R, Rygh LJ, Dickenson AH. Bad news from the brain: descending 5-HT pathways that control spinal pain processing. Trends Pharmacol Sci. 2004;25:613–617. doi: 10.1016/j.tips.2004.10.002. [DOI] [PubMed] [Google Scholar]
- Sweitzer SM, Wong SM, Tjolsen A, Allen CP, Mochly-Rosen D, Kendig JJ. Exaggerated nociceptive responses on morphine withdrawal: roles of protein kinase C ε and γ. Pain. 2004;110:281–289. doi: 10.1016/j.pain.2004.04.004. [DOI] [PubMed] [Google Scholar]
- Thomson AM. Facilitation, augmentation and potentiation at central synapses. Trends Neurosci. 2000;23:305–312. doi: 10.1016/s0166-2236(00)01580-0. [DOI] [PubMed] [Google Scholar]
- Trafton JA, Abbadie C, Marek K, Basbaum AI. Postsynaptic signaling via the μ-opioid receptor: responses of dorsal horn neurons to exogenous opioids and noxious stimulation. J Neurosci. 2000;20:8578–8584. doi: 10.1523/JNEUROSCI.20-23-08578.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trescot AM, Datta S, Lee M, Hansen H. Opioid pharmacology. Pain Physician. 2008;11(Suppl 2):S133–S153. [PubMed] [Google Scholar]
- Trujillo KA, Akil H. Inhibition of morphine tolerance and dependence by the NMDA receptor antagonist MK-801. Science. 1991;251:85–87. doi: 10.1126/science.1824728. [DOI] [PubMed] [Google Scholar]
- Vanderah TW, Suenaga NM, Ossipov MH, Malan TP, Jr, Lai J, Porreca F. Tonic descending facilitation from the rostral ventromedial medulla mediates opioid-induced abnormal pain and antinociceptive tolerance. J Neurosci. 2001;21:279–286. doi: 10.1523/JNEUROSCI.21-01-00279.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vera-Portocarrero LP, Zhang ET, King T, Ossipov MH, Vanderah TW, Lai J, Porreca F. Spinal NK-1 receptor expressing neurons mediate opioid-induced hyperalgesia and antinociceptive tolerance via activation of descending pathways. Pain. 2007;129:35–45. doi: 10.1016/j.pain.2006.09.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vera-Portocarrero LP, Ossipov MH, Lai J, King T, Porreca F. Descending facilitatory pathways from the rostroventromedial medulla mediate naloxone-precipitated withdrawal in morphine-dependent rats. J Pain. 2011;12:667–676. doi: 10.1016/j.jpain.2010.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yaksh TL, Harty GJ, Onofrio BM. High dose of spinal morphine produce a nonopiate receptor-mediated hyperesthesia: clinical and theoretic implications. Anesthesiology. 1986;64:590–597. doi: 10.1097/00000542-198605000-00008. [DOI] [PubMed] [Google Scholar]
- Zaki PA, Keith DE, Jr, Brine GA, Carroll FI, Evans CJ. Ligand-induced changes in surface μ-opioid receptor number: relationship to G protein activation? J Pharmacol Exp Ther. 2000;292:1127–1134. [PubMed] [Google Scholar]
- Zhou HY, Chen SR, Chen H, Pan HL. Opioid-induced long-term potentiation in the spinal cord is a presynaptic event. J Neurosci. 2010;30:4460–4466. doi: 10.1523/JNEUROSCI.5857-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zucker RS. Changes in the statistics of transmitter release during facilitation. J Physiol. 1973;229:787–810. doi: 10.1113/jphysiol.1973.sp010167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zucker RS, Regehr WG. Short-term synaptic plasticity. Annu Rev Physiol. 2002;64:355–405. doi: 10.1146/annurev.physiol.64.092501.114547. [DOI] [PubMed] [Google Scholar]