

Abstract
The c-Jun NH2-terminal protein kinase (JNK), which belongs to the mitogen-activated protein kinase family, plays important roles in a broad range of physiological processes. JNK is controlled by two upstream regulators, mitogen-activated protein kinase kinase (MKK) 7 and MKK4. To elucidate the physiological functions of MKK7, we used Nestin-Cre to generate a novel mouse model in which the mkk7 gene was specifically deleted in the nervous system (Mkk7flox/flox Nestin-Cre mice). These mice were indistinguishable from their control littermates in gross appearance during embryogenesis but died immediately after birth without breathing. Histological examination showed that the mutants had severe defects in brain development, including enlarged ventricles, reduced striatum, and minimal axon tracts. Electron microscopy revealed abnormal accumulations of filamentous structures and autophagic vacuoles in Mkk7flox/flox Nestin-Cre brain. Further analysis showed that MKK7 deletion decreased numbers of TAG-1-expressing axons and delayed neuronal migration in the cerebrum. Neuronal differentiation was not altered. In utero electroporation studies showed that contralateral projection of axons by layer 2/3 neurons was impaired in the absence of MKK7. Moreover, MKK7 regulated axon elongation in a cell-autonomous manner in vivo, a finding confirmed in vitro. Finally, phosphorylation levels of JNK substrates, including c-Jun, neurofilament heavy chain, microtubule-associated protein 1B, and doublecortin, were reduced in Mkk7flox/flox Nestin-Cre brain. Our findings demonstrate that the phenotype of Mkk7flox/flox Nestin-Cre mice differs substantially from that of Mkk4flox/flox Nestin-Cre mice, and establish that MKK7-mediated regulation of JNK is uniquely critical for both axon elongation and radial migration in the developing brain.
Introduction
The activation of c-Jun NH2-terminal protein kinase (JNK) results in the phosphorylation of numerous important substrates, including the AP-1 transcription factor c-Jun (Hibi et al., 1993) and various microtubule-associated proteins (MAPs) (Kawauchi et al., 2003; Gdalyahu et al., 2004), that control cellular processes such as cell growth, apoptosis, differentiation, migration, and transformation. JNK activation in response to environmental stresses, growth factors, hormones, and proinflammatory cytokines is triggered by mitogen-activated protein kinase (MAPK) kinase 4 (MKK4) and MKK7 (Davis, 2000; Chang and Karin, 2001; Asaoka and Nishina, 2010). While MKK4 activates both JNK and another MAPK, p38 (Enslen et al., 1998), MKK7 regulates only the JNK signaling cascade.
MKK7 modulates JNK signaling by interacting with scaffold proteins such as the JNK-interacting protein (JIP) 1, 2, or 3 or filamin A (Whitmarsh et al., 1998; Ito et al., 1999; Yasuda et al., 1999; Nakagawa et al., 2010). Previously, we reported that MKK7 has an essential role in liver development, in that Mkk7 total knock-out mice die at E12.5–E13.5 with severely disorganized livers and reduced hepatoblast numbers (Wada et al., 2004). This study also revealed that hepatoblast proliferation depends on the MKK7-JNK-c-Jun pathway. However, the functions of MKK7 in other tissues remain unclear due to the early embryonic lethality of Mkk7 total knock-out mice.
Several lines of evidence have established the importance of JNK signaling in the mammalian brain. First, Jnk1−/− mice display an abnormality in the maintenance of telencephalic commissures (Chang et al., 2003) as well as altered dendritic architecture (Björkblom et al., 2005). Second, the dopaminergic neurons of both Jnk2−/− and Jnk3−/− mice resist MPTP-induced cell death (Hunot et al., 2004). Thirdly, Jnk1/Jnk2 double mutant mice die at E11.5 with defective neural tube morphogenesis and reduced apoptosis in the hindbrain but increased apoptosis and caspase activation in the forebrain (Kuan et al., 1999; Sabapathy et al., 1999), showing that JNK signaling has both pro- and anti-apoptotic effects on the developing brain. Last, JNK signaling is involved in neuronal migration during brain development. Cortical neuronal migration was retarded by overexpression of a dominant-negative form of JNK (Kawauchi et al., 2003), and specific deletion of MKK4 in the CNS caused a delay in neuronal radial migration in the cerebral cortex as well as misalignment of Purkinje cells in the cerebellum (Wang et al., 2007). Together, these reports demonstrate that the proper control of JNK signaling is required for normal brain development; however, the specific role of MKK7 in this control has yet to be defined.
Here we report the generation of a novel mouse model, Mkk7flox/flox Nestin-Cre mice, in which the murine mkk7 gene is specifically deleted in neural stem cells and postmitotic neurons. We found that Mkk7flox/flox Nestin-Cre mice display phenotypes different from those previously reported for Mkk4flox/flox Nestin-Cre mice, indicating that MKK7 has unique and crucial functions in the developing brain.
Materials and Methods
Animals.
Mice carrying the mkk7 flox allele were described previously (Schramek et al., 2011). Mkk7flox/flox mice were crossed to Nestin-Cre transgenic mice expressing Cre recombinase under the control of the mouse nestin promoter and the second intronic neural enhancer (Imai et al., 2006; Okada et al., 2006). The resulting control (Mkk7flox/+, Mkk7flox/flox and Mkk7flox/+ Nestin-Cre) and mutant (Mkk7flox/flox Nestin-Cre) mice were reared on a normal 12 h light/dark schedule. The day when a plug was observed in a female mouse was designated as embryonic day 0.5 (E0.5), and the day of birth was termed postnatal day 0 (P0). Mouse genotypes were determined by PCR and Southern blotting. For all experiments, only littermate mice from the same breeding were used. All procedures were performed in accordance with a protocol approved by the Tokyo Medical and Dental University Animal Care Committee.
In utero electroporation.
In utero electroporation was performed as described previously (Saito, 2006; Tabata and Nakajima, 2008) with slight modifications. Briefly, pregnant mice were anesthetized with sodium pentobarbital and the uterine horns were exposed. Approximately 1–2 μl of DNA solution (1–2.5 mg/ml) was injected into the lateral ventricle of each embryonic brain using a pulled glass micropipette. Each embryo within its uterus was then placed between tweezer-type electrodes with a diameter of 5 mm (CUY650-P5; NEPA Gene). Square electric pulses (45 V, 50 ms) were passed five times at 1 s intervals using an electroporator (ECM830, BTX). Care was taken to quickly place embryos back into the abdominal cavity to avoid excessive temperature loss. The wall and skin of the abdominal cavity were sutured, and embryos were allowed to develop normally.
In vivo neuronal migration.
Pregnant mice at E15.5 were injected intraperitoneally with bromodeoxyuridine (BrdU) (50 mg/kg of body weight). Embryos were dissected and fixed at E18.5 and brains were processed for paraffin sectioning. Deparaffinized and rehydrated 6 μm coronal sections were treated with 2N HCl at 37°C for 30 min and neutralized with 0.1% boric acid buffer, pH 8.5. After blocking in 2% skim milk in PBS containing 0.1% Triton X-100, sections were subjected to immunohistochemical analysis as described below.
Dissociation and culture of cortical neurons.
At E16.5, cortices were dissected from Mkk7flox/flox Nestin-Cre, Mkk7flox/flox, Mkk7flox/+, or wild-type embryos that had been electroporated at E15.5. The dissected cortices were placed in culture and incubated for 20 min with papain solution plus DNase I at 37°C, followed by mechanical dissociation in dissociation medium (EBSS; Sigma-Aldrich). Cortical neurons were plated onto poly-l-lysine-coated coverslips, and maintained in Basal Medium Eagle (Invitrogen) containing 1 mm l-glutamine (Sigma), HBSS (Invitrogen), 25% (v/v) horse serum (Invitrogen), 6.6 mg/ml dextrose (Sigma), penicillin/streptomycin (Invitrogen), and 1 mm HEPES, pH 7.4 (Invitrogen). Cortical neuron cultures were maintained in 5% CO2 at 37°C.
Immunoblotting.
Immunoblotting was performed as previously described (Ura et al., 2007) with slight modifications. Proteins were extracted from tissues in TNE buffer [20 mm Tris-HCl, pH 7.4, 150 mm NaCl, 1 mm EDTA, 1 mm EGTA, 0.5% Nonidet P40, 5% (w/v) glycerol, 1 mm PMSF, 200 mm NaF, 200 μm Na3VO4, 10 μg/ml aprotinin]. Total extracts were clarified by centrifugation (20,000 × g for 10 min at 4°C) and soluble proteins in the supernatants were quantified using the BCA Protein Assay Kit (PIERCE). Protein extracts were fractionated by SDS/PAGE and transferred to a PVDF membrane, which was incubated in blocking solution (2% skim milk in TBS) for 1 h. The blocked membrane was incubated overnight in TBS containing 5% BSA plus antibodies recognizing phospho-JNK (Cell Signaling Technology), JNK1 (46 kDa and 55 kDa splicing variants; Santa Cruz Biotechnology), MKK7 (Cell Signaling Technology), GAPDH (Millipore Bioscience Research Reagents), phospho-MKK4 (Cell Signaling Technology), MKK4 (Santa Cruz Biotechnology), phospho-DCX (Gdalyahu et al., 2004), DCX (Gdalyahu et al., 2004), phospho-neurofilament heavy chain (Covance), phospho-MAP1B (Covance), MAP1B (Sigma), or tubulin (Invitrogen). The membrane was then washed in TBS/Tween 20 (0.05%), incubated for 1 h with anti-mouse/rabbit horseradish peroxidase-conjugated antibodies (Jackson ImmunoResearch Laboratory), and washed three times in TBS/Tween 20. Proteins were visualized using Immobilon-HRP (Millipore) or the SuperSignal West Femto Kit (Pierce) and ChemiDoc XRS (Bio-Rad).
In vitro kinase assay.
Assay of MKK7 activity was as described previously (Nakagawa et al., 2010). Endogenous MKK7 proteins were immunoprecipitated with anti-MKK7 (KN-004) monoclonal antibody (Wada et al., 2001). Immunocomplexes were washed three times with lysis buffer and three times with kinase reaction buffer, which consisted of 10 mm MgCl2, 50 mm Tris-HCl, pH 7.5, and 1 mm EGTA. MKK7 activity on beads was measured using an in vitro MAPK kinase assay using 100 μm ATP and GST-JNK1 as the substrate. Reactions were terminated after 30 min at 30°C by the addition of 4× SDS gel sample buffer. The reaction product was detected using anti-phospho-JNK antibody.
Nissl staining.
Nissl staining was performed as described previously (Toda et al., 2008) with slight modifications. Brains were fixed at E18.5 and processed for paraffin sectioning. Deparaffinized and rehydrated 6 μm horizontal sections were subjected to Nissl staining with 0.2% cresyl violet.
Immunostaining.
Immunohistochemistry was performed as described previously (Kawasaki et al., 2000). For embryos, brains were isolated and fixed overnight in 4% paraformaldehyde. For postnatal mice, the animals were deeply anesthetized with pentobarbital and transcardially perfused with 4% paraformaldehyde. Fixed tissues were cryoprotected by overnight immersion in 30% sucrose, followed by embedding in OCT compound. Sections of 14 μm thickness were attached to glass slides, whereas sections of 50 μm thickness were floated on PBS. For cultures of dissociated neurons, cells were fixed by incubation in 4% paraformaldehyde for 5 min at 37°C.
For immunostaining, tissue sections and isolated cells were permeabilized with 0.1–0.5% Triton X-100 in PBS and incubated overnight with primary antibodies, including those recognizing phospho-JNK (Cell Signaling Technology), TAG-1 (DSHB), L1 (Millipore Bioscience Research Reagents), cleaved caspase-3 (PharMingen), β-tubulin III (Covance), Brn2 (Santa Cruz Biotechnology), Satb2 (Abcam), Ctip2 (Abcam), Foxp2 (Abcam), Tbr2 (Abcam), or GFP or RFP (both from MBL). Immunostained tissues and cells were then incubated with Alexa488- and/or Cy3-conjugated secondary antibodies and 1 μg/ml Hoechst 33342, followed by washing and mounting. Epifluorescent microscopy was performed using an Axioimager A1 microscope (Carl Zeiss) and BZ9000 (Keyence). Confocal microscopy was performed using an LSM510 microscope (Carl Zeiss).
Electron microscopy.
Tissues were fixed by immersion first in 1.5% paraformaldehyde plus 3% glutaraldehyde in 0.1 m phosphate buffer, pH 7.3, and then in an aqueous solution of 1% OsO4. Fixed samples were embedded in Epon812, and thin sections (70–80 nm) were cut and stained with uranyl acetate and lead citrate for observation under a Jeol-1010 electron microscope (Jeol) at 80 kV.
Plasmids.
All genes were expressed under the control of the CAG promoter. pCAG-NLS-Cre and pCAG-floxed-polyA-EGFP were kindly provided by Dr. Fumio Matsuzaki (RIKEN-CDB, Japan). pCAG-EGFP-MKK7, pCAG-EGFP-MKK4, pCAG-MKK7-JNK, pCAG-c-Jun-IRES-EGFP, and pCAG-EGFP-JBD were constructed in the Nishina laboratory using standard protocols. Plasmids were purified using the Endofree Plasmid Maxi kit (Qiagen).
Statistical analysis.
Data were analyzed using the paired Student's t test for two-tailed distributions. Significance levels were as follows: *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
Results
Mkk7flox/flox Nestin-Cre mice die at P0 without breathing
Because MKK7 total knock-out mice are embryonic lethal, it has not been possible to examine MKK7 functions in the brain. To confirm that MKK7-JNK signaling occurs during normal brain development, we established a time course of JNK phosphorylation and MKK7 activity in wild-type (WT) mouse brain. Extracts of brains of embryos from E12.5 to adult were prepared and immunoblotted to detect total and activated JNK. MKK7 activity was measured by in vitro kinase assay. We found that JNK and MKK7 were highly activated from the E16.5 to the adult stages (Fig. 1A,B, and data not shown). These data establish that MKK7-JNK signaling occurs during normal brain development.
Figure 1.

The MKK7-JNK signaling pathway is activated in the developing brain and essential for postnatal viability. A, Analysis of JNK activity during mouse development. Extracts of brains from WT mice at the indicated stages of development were prepared and immunoblotted to detect JNK activity. B, Analysis of MKK7 activity in WT adult mouse brain. MKK7 kinase activity was assayed as described in Materials and Methods. Testis, Negative control. C, CNS-specific targeting of the murine mkk7 gene. The genomic mkk7 locus, the mkk7 targeting vector, the predicted structure of the mutated mkk7 locus, and the floxed and deleted mkk7 alleles are depicted. Black boxes, mkk7 exons; black arrowheads, loxP sites; white arrowheads, FRT sites; X, XbaI; H, HindIII; B, BamHI; P, PstI. The Neo and DTA cassettes and the probe used for Southern blotting are indicated. D, Confirmation of MKK7 deletion. HindIII-restricted genomic DNA was prepared from mice of the indicated genotypes at E16.5 and subjected to Southern blotting with the probe in C. E, Suppression of JNK activity by MKK7 disruption in the CNS. Left, Extracts of brains of E18.5 embryos of the indicated genotypes were immunoblotted to detect the indicated proteins. Right, Normalized protein expression levels for MKK7/Gapdh, phospho-JNK/JNK, and phospho-MKK4/MKK4 in the extracts in the left panel were determined by densitometry and are shown in the histograms. F, Mkk7 disruption results in death immediately after caesarean section. Control and Mkk7flox/flox Nestin-Cre embryos were obtained by caesarean section at E18.5 and subjected to gross examination. All control mice started breathing (13/13), but most Mkk7flox/flox Nestin-Cre mice (9/10) did not and died immediately. Scale bar, 5 mm.
To investigate the role of this MKK7-JNK signaling in vivo, we made a mutant mouse carrying a mkk7 flox allele (Fig. 1C), and crossed it with a transgenic mouse line that expresses Cre under the control of the nestin promoter (Imai et al., 2006). We confirmed the deletion of mkk7 in Mkk7flox/flox Nestin-Cre brain at E16.5 by Southern blotting (Fig. 1D). As expected, levels of MKK7 and phosphorylated JNK proteins were markedly reduced in the brains of these mice, but, unexpectedly, phospho-MKK4 was upregulated (Fig. 1E). These results suggest that, in the absence of one JNK regulator (MKK7), the activation of another JNK regulator (MKK4) may increase in compensation.
To investigate whether MKK7 was important for postnatal viability, we intercrossed Mkk7flox/+ Nestin-Cre and Mkk7flox/flox mice and determined the genotypes of embryos and postnatal pups. Surprisingly, although Mkk7floxflox Nestin-Cre mice appeared normal through all embryonic stages, no mutants were detected on P1 (Table 1). To determine whether Mkk7flox/flox Nestin-Cre mice died at birth, we performed caesarean sections and found that Mkk7flox/flox Nestin-Cre mice were indistinguishable from controls at E18.5. However, all but one Mkk7flox/flox Nestin-Cre mouse died immediately without breathing after caesarean section (Fig. 1F). MKK7 is therefore vital for postnatal survival.
Table 1.
MKK7 is critical for postnatal viability
| Stage | No. of litters | Number of embryos or newborns with genotype |
||||
|---|---|---|---|---|---|---|
| Total | Mkk7flox/+ | Mkk7flox/flox | Mkk7flox/+ Nestin-Cre | Mkk7flox/flox Nestin-Cre | ||
| E12.5 | 2 | 15 | 6 | 3 | 4 | 2 |
| E14.5 | 6 | 46 | 10 | 11 | 17 | 8 |
| E16.5 | 6 | 51 | 12 | 17 | 9 | 13 |
| E18.5 | 7 | 54 | 14 | 9 | 14 | 17 |
| P1 | 5 | 29 | 9 | 7 | 13 | 0 |
Mkk7flox/+ Nestin-Cre mice were crossed with Mkk7flox/flox mice and the genotypes of embryos or neonates were determined by PCR at the indicated stages.
Abnormal brain development in Mkk7flox/flox Nestin-Cre mice
The death of Mkk7flox/flox Nestin-Cre mutants at birth suggested that there might be defects in the brains of these animals. To assess brain histology, sections of various brain structures were prepared from control (Mkk7flox/+, Mkk7flox/flox, and Mkk7flox/+ Nestin-Cre) and Mkk7flox/flox Nestin-Cre mice at E18.5 and stained with cresyl violet. Although Mkk7flox/flox Nestin-Cre whole brain was grossly indistinguishable from control littermate whole brain (Fig. 2A), histological analysis showed that Mkk7flox/flox Nestin-Cre brain had larger ventricles and a reduced striatum compared to control brain (Fig. 2B). In addition, whereas the corpus callosum (CC) and the anterior commissure (AC) were clearly formed in control E18.5 brain, these commissural axon tracts were barely detectable in E18.5 Mkk7flox/flox Nestin-Cre brain (Fig. 2B,C). Moreover, although there was no defect in the mutant hippocampal commissure (data not shown), the internal capsule (IC), which consists of axonal fibers connecting the cerebral cortex and subcortical brain regions, was reduced in size in Mkk7flox/flox Nestin-Cre brain (Fig. 2D). These data indicate that MKK7 regulates the formation of axon tracts during brain development.
Figure 2.

Lack of Mkk7 causes abnormal brain development and reduced axon tracts in the cerebrum. A, Gross appearance of whole brains of control and Mkk7flox/flox Nestin-Cre embryos at E18.5. Results are representative of embryos examined per genotype. Scale bar, 1 mm. B–D, Histological analysis of brain defects in Mkk7flox/flox Nestin-Cre mice. Horizontal sections of the brains of control and Mkk7flox/flox Nestin-Cre embryos at E18.5 were stained with cresyl violet. Left, Forebrains with inset boxes indicating CC (B), AC (C), and IC (D) are shown. Scale bar, 500 μm. Right, Inset boxes in left panels are magnified. Scale bar, 500 μm.
To further examine the effect of MKK7 deletion on axon tracts, we used electron microscopy to look for ultrastructural alterations in Mkk7flox/flox Nestin-Cre brain. We found that abnormally electron-dense axons were present in Mkk7flox/flox Nestin-Cre cortex and striatum (Fig. 3A,B), and that these structures contained an accumulation of intermediate filaments (Fig. 3A, inset). In addition, autophagic vacuoles and swollen mitochondria were detected in the striatum, cortex, and brainstem of Mkk7flox/flox Nestin-Cre brain (Fig. 3B and data not shown). Thus, MKK7-mediated regulation of JNK may be important for suppressing autophagy and controlling the distribution of intermediate filaments in the developing brain.
Figure 3.
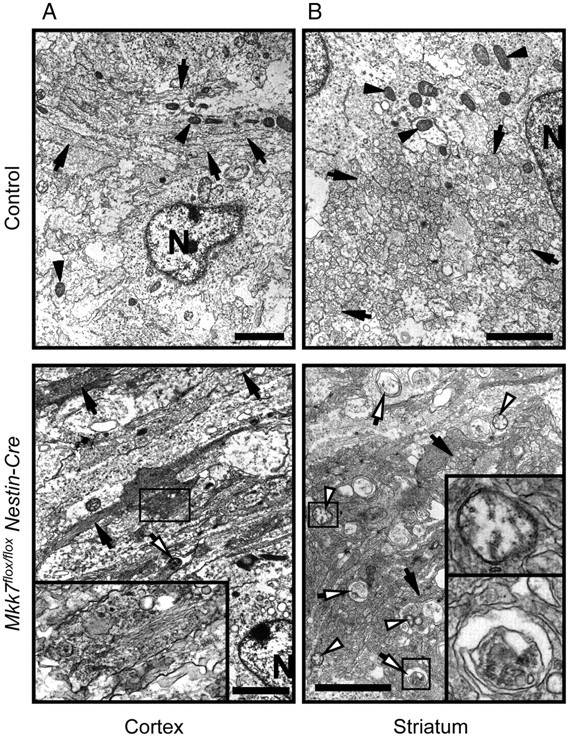
Loss of MKK7 causes accumulations of filamentous structures and autophagic vacuoles. Brains of control and Mkk7flox/flox Nestin-Cre embryos at E18.5 were analyzed by electron microscopy. The cortex (A) and striatum (B) are shown. Black arrows, Axons; black arrowheads, mitochondria; white arrows, autophagic vacuoles; white arrowheads, swollen mitochondria; N, nuclei. Inset boxes, A, Accumulation of filamentous structures; B, top, swollen mitochondrion; B, bottom, autophagic vacuole. Scale bar, 2 μm.
Impaired axon formation
Our histological analyses led us to hypothesize that MKK7-JNK signaling might normally be activated in the axons of WT developing brain. As expected, phospho-JNK coimmunostained with L1, which is a cell adhesion molecule expressed on axons, in control brain but was reduced in Mkk7flox/flox Nestin-Cre brain (Fig. 4A). We then performed a more detailed analysis by immunostaining to detect both L1 and transient axonal glycoprotein-1 (TAG-1), which is expressed in corticofugal axons (Wolfer et al., 1994; Denaxa et al., 2001). In Mkk7flox/flox Nestin-Cre brain at E18.5, we found that numbers of L1-positive as well as TAG-1-positive axons were decreased in the cortex and CC compared to controls (Fig. 4B,C). These results indicate that MKK7 has a particularly important role in the formation of TAG-1-positive axons.
Figure 4.
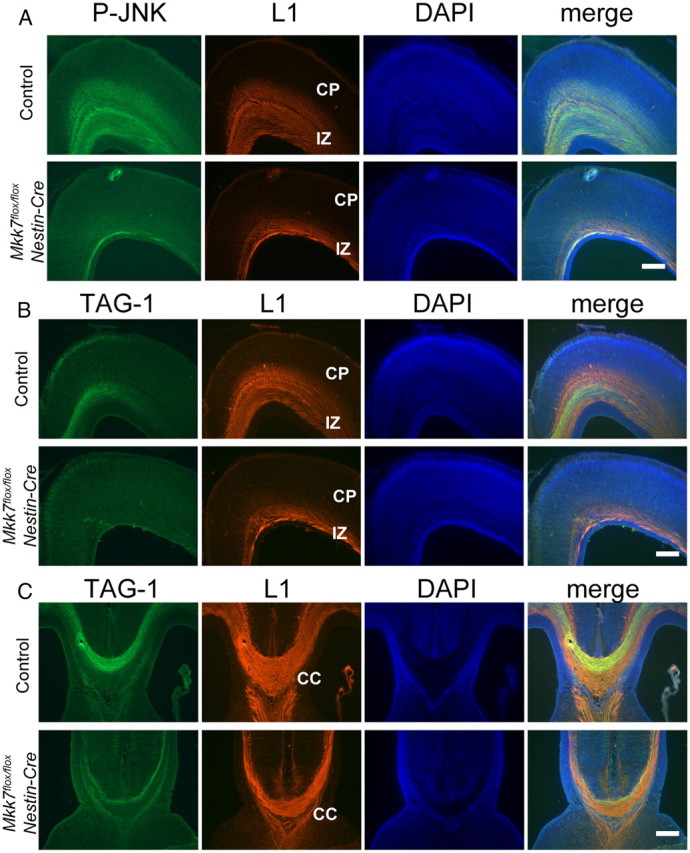
Axonal defects in the cortex and corpus callosum of Mkk7flox/flox Nestin-Cre mice. A, Suppression of JNK phosphorylation in axons. Coronal sections of the cortex in control and Mkk7flox/flox Nestin-Cre embryos at E18.5 were immunostained with anti-phospho-JNK and anti-L1 antibodies. DAPI staining was used to visualize nuclei. CP, Cortical plate; IZ, intermediate zone. B, C, Coronal sections of the cortex (B) and the CC (C) in control and Mkk7flox/flox Nestin-Cre embryos at E18.5 were immunostained with anti-TAG-1 and anti-L1 antibodies as for A. Scale bar: A–C, 200 μm.
Because previous reports implicated JNK in regulating apoptosis during brain development (Kuan et al., 1999; Sabapathy et al., 1999), we examined whether apoptosis was altered in Mkk7flox/flox Nestin-Cre brain. Immunostaining with antibody specific for cleaved caspase-3 revealed that, like control brain, there were few apoptotic cells in the Mkk7flox/flox Nestin-Cre cortex (Fig. 5A). Apoptotic cells were also rare in the striatum of both Mkk7flox/flox Nestin-Cre and control brains (data not shown). A few apoptotic cells were observed surrounding the mutant CC but were no more frequent than in control brains (Fig. 5B). Thus, a lack of MKK7 does not impair the regulation of apoptosis in the developing brain.
Figure 5.
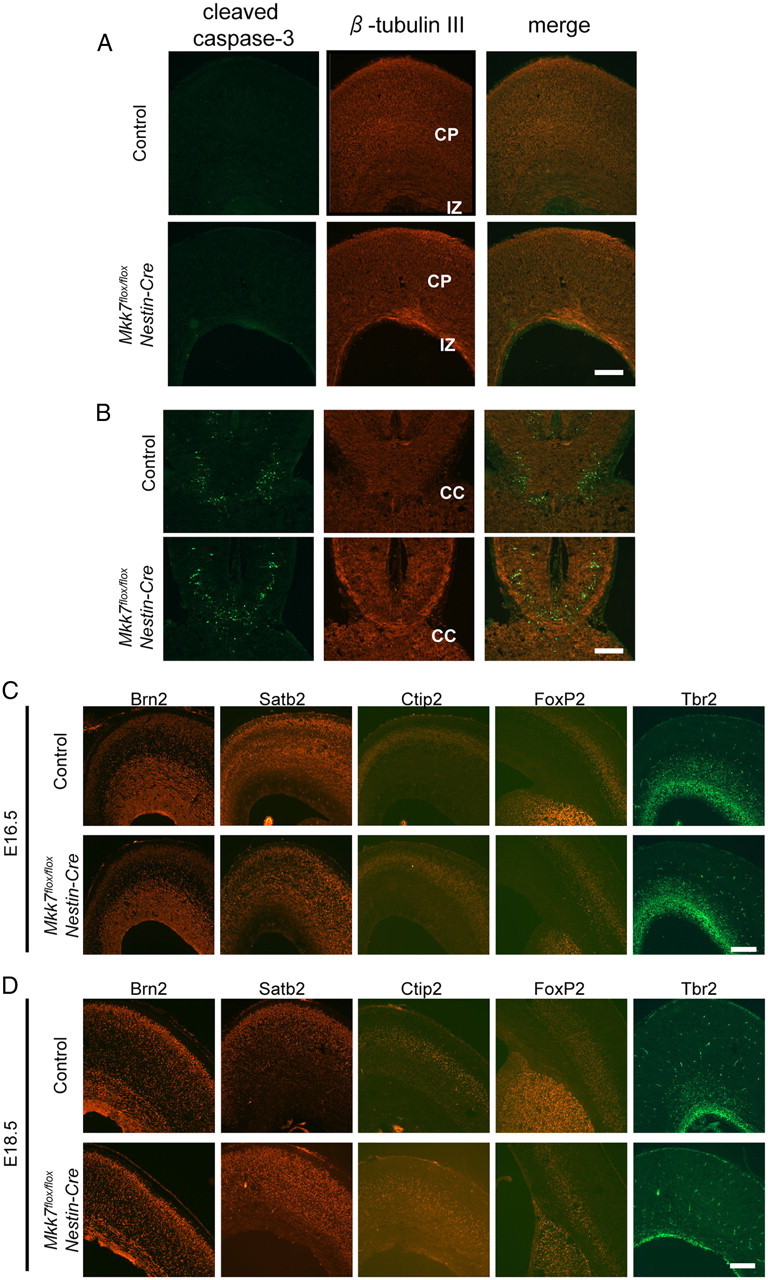
Loss of MKK7 does not influence apoptosis or neuronal differentiation in the brain. A, B, Coronal sections of cortex (A) and CC (B) in control and Mkk7flox/flox Nestin-Cre embryos at E18.5 were immunostained with anti-cleaved caspase-3 and anti-β-tubulin III antibodies. C, D, Coronal sections of the cortex in control and Mkk7flox/flox Nestin-Cre embryos at E16.5 (C) or E18.5 (D) were immunostained with antibodies recognizing the neuronal markers Brn2, Satb2, Ctip2, Foxp2, or Tbr2. Scale bar, 200 μm.
We next investigated whether loss of MKK7 caused any alterations to neuronal differentiation. Cortical neurons can be identified by immunostaining with antibodies recognizing neuronal markers such as Brn2, Satb2, Ctip2, Foxp2, and Tbr2 (Bulfone et al., 1999; McEvilly et al., 2002; Ferland et al., 2003; Arlotta et al., 2005; Britanova et al., 2005). We detected expression of all of these markers in Mkk7flox/floxNestin-Cre cortex (Fig. 5C,D), indicating that cortical neurons differentiate normally without MKK7. Next, because it was reported that the major JNK substrate c-Jun is involved in the specification of GABAergic/glutamatergic neurons in the spinal cord of Xenopus tropicalis (Marek et al., 2010), we performed an in situ hybridization analysis of Mkk7flox/flox Nestin-Cre spinal cord using probes for gad1 and vglut2, which are expressed in GABAergic or glutamatergic neurons, respectively. However, no significant alterations to expression levels of gad1 or vglut2 were detected between control and Mkk7flox/flox Nestin-Cre mice (data not shown). These data show that, as was true for cortical neurons, the differentiation of GABAergic and glutamatergic neurons is not affected by MKK7 deletion.
Delayed neuronal radial migration
Previous reports showed that overexpression of dominant-negative JNK or MKK4 disruption can delay neuronal radial migration in the cerebral cortex (Kawauchi et al., 2003; Wang et al., 2007). To determine whether MKK7 also affects this process, we performed a timed pregnancy study of BrdU incorporation in the developing cerebral cortex. Embryos were labeled in utero with BrdU at E15.5 and brain sections prepared at E18.5 were immunostained with anti-BrdU antibody. We found that, in Mkk7flox/flox Nestin-Cre brain, BrdU-positive cells were dispersed throughout the ventricular zone (VZ), intermediate zone, and cortical plate, with a small proportion of BrdU-positive cells located in Bin1. In control brain, a significantly larger proportion of BrdU-positive cells was located in Bin1 (Fig. 6A,B). Thus, like MKK4, MKK7 influences the radial migration of neurons in the cortex.
Figure 6.

MKK7 deletion delays neuronal radial migration in the cortex. A, Control and Mkk7flox/flox Nestin-Cre embryos were labeled in utero with BrdU at E15.5 and coronal sections were prepared at E18.5 and analyzed by immunostaining with anti-BrdU antibody. White, BrdU. Scale bar, 100 μm. CP, cortical plate; IZ, intermediate zone. B, Distribution of BrdU-positive nuclei. The immunostained cortices in A were divided into four equal bins from the pial side to the ventricular side. The percentage of the total number of immunostained nuclei in a particular bin was determined and the results plotted in a histogram. Data shown are the mean ± SD (n = 6). *p < 0.05; **p < 0.01.
MKK7 deletion in layer 2/3 neurons results in the disappearance of contralateral projecting axons in vivo
Our immunostaining experiments indicated that axon tracts were abnormal in the absence of MKK7. However, axon elongation is regulated not only by the neuron itself but also by environmental factors such as nerve growth factors. To determine whether the axon defect, we observed was due to the effects of MKK7 deletion on neuronal axon elongation or on environmental factors, we generated mice in which MKK7 disruption was induced specifically in layer 2/3 neurons. We used in utero electroporation to introduce pCAG-NLS-Cre and its reporter plasmid pCAG-floxed-polyA-EGFP into the VZ of control and Mkk7flox/flox embryos at E15.5. We then killed the pups at P4, P10, or P31 and examined them for GFP fluorescence (representing successful Cre recombination). GFP-positive neurons were detected in layer 2/3 neurons in both Mkk7flox/+ and Mkk7flox/flox brains through all postnatal stages examined. However, unlike controls, GFP-positive neurons in Mkk7flox/flox brain were not able to elongate axons to reach the contralateral cortex by P4 (Fig. 7A), nor was contralateral elongation detected at P10 or P31 in the mutant brains (Fig. 7B,C). Interestingly, when we investigated the morphology of these faulty axons in detail at P31, we found that both control and Mkk7flox/flox layer 2/3 neurons were able to elongate axons radially in the cerebral cortex such that their branches projected to layer 5 (Fig. 7D). However, in the mutant brain, axons running tangentially in the white matter had disappeared (Fig. 7E). When we attempted to determine whether the disappearance of these contralaterally projecting axons was due to a failure in axon guidance, we found no evidence of an axon guidance defect (data not shown). These results show in vivo that MKK7 regulates axon elongation in a cell-autonomous manner, and that MKK7 is required for the contralateral projection of axons of layer 2/3 neurons in the white matter.
Figure 7.
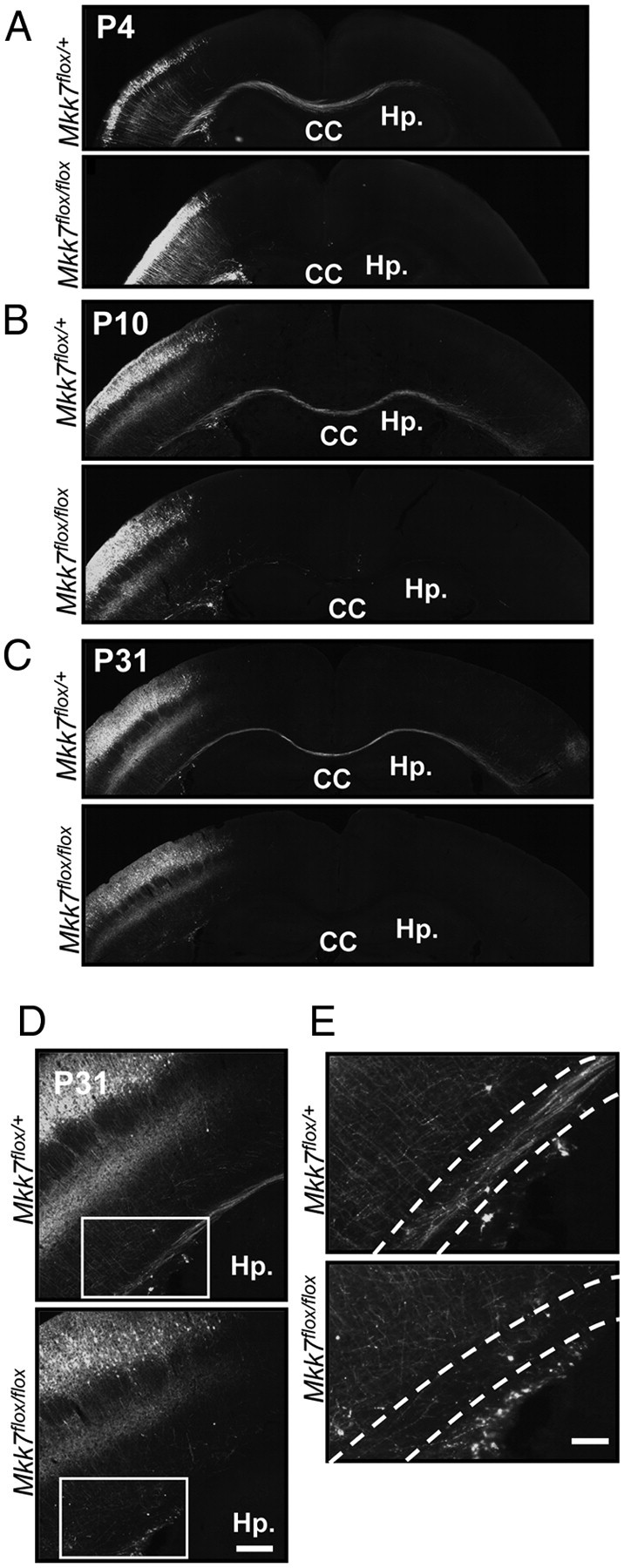
Deletion of mkk7 in layer 2/3 neurons prevents axon elongation in vivo. A–C, Layer-specific MKK7 deletion. pCAG-NLS-Cre and pCAG-floxed-polyA-EGFP were introduced by in utero electroporation into the VZ of control Mkk7flox/+ and Mkk7flox/flox embryos at E15.5. Brains were fixed on the indicated postnatal days and coronal sections of the cortex were immunostained with anti-GFP antibody. Hp., Hippocampus. Scale bar, 1 mm. D, Higher-magnification view of axons in the cortices of the Mkk7flox/+ and Mkk7flox/flox mice in C. Scale bar, 200 μm. E, Higher-magnification view of the inset boxes in D, focusing on the white matter (broken lines). Scale bar, 100 μm.
We also examined axon elongation of layer 2/3 neurons in vitro. We introduced GFP into control and Mkk7flox/flox Nestin-Cre brains by in utero electroporation at E15.5 and prepared cultures of dissociated neurons at E16.5. We then measured the lengths of the longest neurites appearing in the cultures and found that there were more neurons with shorter neurites in Mkk7flox/flox Nestin-Cre cultures after 2, 4, or 6 d in vitro culture (DIV) compared with control cultures (Fig. 8A–C). To determine whether this defect in axon growth was induced a cell-autonomous manner, we disrupted MKK7 in Mkk7flox/flox layer 2/3 neurons using in utero electroporation of NLS-Cre and floxed-polyA-EGFP, and cultured control and mutant GFP-positive neurons for 4 d. Upon measurement of the longest neurites appearing in these cultures, we found that there were more neurons with shorter neurites in Mkk7flox/flox cultures at 4 DIV compared with Mkk7flox/+ cultures (Fig. 8D). These results confirm in vitro that MKK7 regulates axon elongation in a cell-autonomous manner.
Figure 8.

Deletion of mkk7 in layer 2/3 neurons reduces neurite length in vitro. A–C, Reduced axon elongation. The VZ of control (n = 6) and Mkk7flox/flox Nestin-Cre (n = 3) embryos at E15.5 was electroporated with pCAG-EGFP. Brains were dissected at E16.5 and primary neuron cultures were established. A, After 2, 4, or 6 DIV, neurons were fixed and stained with anti-GFP antibody (upper panels; scale bar, 100 μm). B, The lengths of the longest neurites of individual neurons in the cultures in A were measured at 2, 4, and 6 DIV (>100 neurons examined in each 2 DIV and 4 DIV culture, and >40 neurons in each 6 DIV culture). The results are plotted as a histogram and are the mean ± SE for each time point (Mkk7flox/flox Nestin-Cre, n = 3; control, n = 6). C, The distribution of the longest neurite lengths was plotted for each set of cultures in A. Results are the mean ± SD (Mkk7flox/flox Nestin-Cre, n = 3; control, n = 6). D, Cell-autonomous defect. The VZ of control and Mkk7flox/flox embryos at E15.5 was electroporated with pCAG-NLS-Cre plus pCAG-floxed-polyA-EGFP, and primary neuron cultures were established at E16.5. After 4 DIV, neurons were fixed and stained with anti-GFP-antibody (upper; scale bar, 50 μm). The lengths of the longest neurites of individual neurons were measured, and the distribution of these lengths was plotted (lower). Results are the mean ± SD (n = 3). E, Effect of JNK inhibition. The VZ of WT embryos at E15.5 was electroporated with pCAG-EGFP or pCAG-EGFP-JBD, and primary neuron cultures were established at E16.5. After 2 DIV, neurons were fixed and stained (left; scale bar, 50 μm). The distribution of neurite lengths was analyzed as for E (right). Results are the mean ± SD (n = 3). For A–D, *p < 0.05, ***p < 0.001, ****p < 0.0001.
To determine whether our observations were due to effects on JNK signaling, we investigated whether inhibition of JNK caused the same neuronal phenotype as MKK7 disruption. WT layer 2/3 neurons were prepared after in utero electroporation of a plasmid expressing the JNK binding domain of JIP1 (JBD), which acts as a JNK inhibitory peptide (Westerlund et al., 2011). We found that, like MKK7 deletion, JNK inhibition resulted in more neurons with shorter axons (Fig. 8E). These results support our hypothesis that MKK7's effects on axon elongation are likely due to MKK7's regulation of its downstream target JNK.
Reduced phosphorylation of cytoskeletal components
We next determined whether the loss of MKK7 in neurons altered the phosphorylation of JNK substrates. Because phosphorylation of c-Jun was reduced in Mkk7flox/flox Nestin-Cre neurons (Fig. 9A), we performed rescue experiments in which plasmids expressing MKK7, MKK4, MKK7-JNK fusion protein (constitutively active JNK), or c-Jun were coelectroporated along with NLS-Cre and floxed-polyA-mCherry or -GFP into the VZ of E15.5 control and mutant embryos. Coelectroporation of MKK7 (Fig. 9B) and constitutively active JNK (Fig. 9D) allowed mutant neurons to recover their ability to contralaterally elongate axons, but the same was not true for coelectroporation of either MKK4 (Fig. 9C) or c-Jun (Fig. 9E). These data suggest that MKK7 regulates axon elongation through JNK, but that this signaling pathway does not involve transcription mediated by c-Jun.
Figure 9.
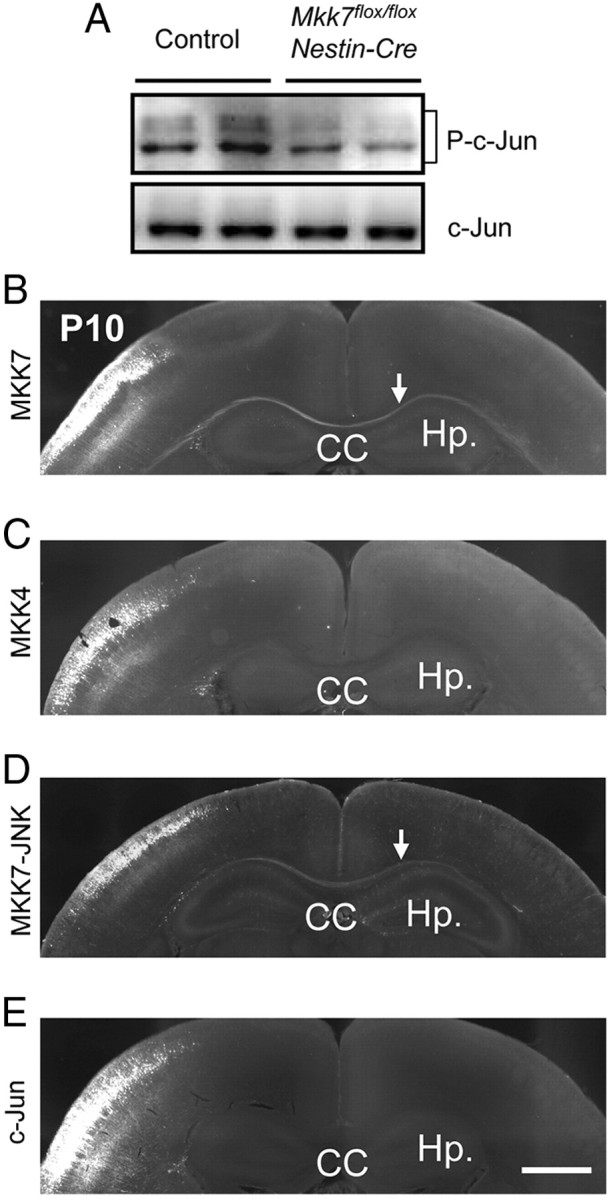
Contralateral axon projection is rescued by electroporation of MKK7 and active-JNK but not by MKK4 or c-Jun. A, Decreased c-Jun phosphorylation. Brain extracts prepared from control and Mkk7flox/flox Nestin-Cre embryos at E18.5 were immunoblotted to detect total c-Jun and phospho-c-Jun. B–E, Reversal of effects of MKK7 disruption. pCAG-NLS-Cre and pCAG-floxed-polyA-mCherry or -GFP were coelectroporated along with MKK7 (0.25 mg/ml) (B), MKK4 (0.25 mg/ml) (C), MKK7-JNK (0.05 mg/ml) (D), or c-Jun (1.0 mg/ml) (E) into the VZ of control and Mkk7flox/flox embryos at E15.5. Brains were fixed at P10 and coronal sections of cortices were immunostained with anti-RFP or GFP antibody. Arrows indicate axons rescued by coelectroporation. Hp, Hippocampus. Scale bar, 1 mm.
In Mkk4flox/flox Nestin-Cre mice, the phosphorylation of certain neurofilament and MAPs is reportedly altered (Wang et al., 2007). Neurofilament heavy chain (NF-H) is a neuron-specific intermediate filament whose C-terminal tail is constitutively phosphorylated in axons by several proline-directed kinases, including JNK. When we investigated phosphorylation levels of NF-H in Mkk7flox/flox Nestin-Cre brain, we found that NF-H phosphorylation was reduced (Fig. 10A). The axon elongation and radial migration required for normal brain development also rely on MAP-dependent regulation of microtubule dynamics, and MAP activity is controlled by phosphorylation mediated by a number of protein kinases, including JNK. When we tested whether MAP phosphorylation was altered in Mkk7flox/flox Nestin-Cre brain, we found that phosphorylation levels of MAP1B were reduced (Fig. 10B). In addition, the phosphorylation of doublecortin (DCX) was suppressed by loss of MKK7 (Fig. 10C), an alteration that does not occur after disruption of MKK4 (Wang et al., 2007). These data show that, within the developing brain, MKK7 regulates the phosphorylation of not only NF-H and MAP1B, which are also regulated by MKK4, but also DCX.
Figure 10.
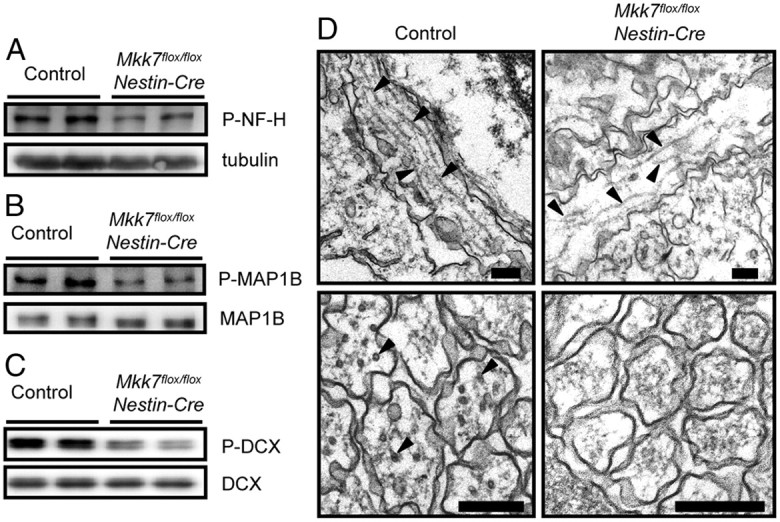
MKK7 regulates MAP phosphorylation. A–C, Brain extracts from control and Mkk7flox/flox Nestin-Cre embryos at E18.5 were immunoblotted to detect phospho-NF-H and tubulin (loading control) (A); phospho-MAP1B and total MAP1B (B); and phospho-DCX and total DCX (C). D, Defective microtubule structures in axons of Mkk7flox/flox Nestin-Cre brain. Electron micrographs of horizontal sections (upper) and cross-sections (lower) of axons in control and Mkk7flox/floxNestin-Cre brains are shown. Black arrowheads, Microtubules. Scale bars, 200 nm.
We next examined whether microtubule stabilization could ameliorate the axonal defects caused by MKK7 disruption. Cultures of Mkk7flox/flox Nestin-Cre primary neurons were treated with Taxol, a drug that stabilizes microtubules and induces axon formation (Hirai et al., 2011). We found that Taxol treatment did indeed increase the lengths of axons in Mkk7flox/flox Nestin-Cre brain, but that these structures were still shorter than axons in Taxol-treated control cultures (data not shown). Finally, we examined microtubule structures in sections of axons directly by electron microscopy and found that Mkk7flox/flox Nestin-Cre brain contained axons with fewer and shorter microtubules than control brain (Fig. 10D). Thus, the regulatory effects of MKK7 on molecules critical for microtubule structures involve more than mere microtubule stabilization and do not overlap the effects of MKK4.
Discussion
Role of MKK7 in axon elongation
In this study, we investigated the physiological functions of MKK7 in the developing brain. Our most striking finding is that MKK7 regulates the elongation of axons by layer 2/3 neurons. Mkk7flox/flox Nestin-Cre brains displayed defective formation of axon tracts and decreased numbers of TAG-1-positive axons (Figs. 2, 4), a phenotype never observed in JNK-deficient mice. Jnk1/2 double mutant mice die during early embryogenesis, precluding analysis of the developing brain. No brain defects have been reported for Jnk2 and/or Jnk3 knock-out mice (Hunot et al., 2004), and although Jnk1−/− mice display abnormalities of axon maintenance and radial migration, axon formation is normal in these mutants (Chang et al., 2003; Westerlund et al., 2011). These results indicate that there is redundancy among JNK1, 2, and 3 functions that obscures the effects of JNK signaling on axon elongation because all of these kinases are expressed in the brain. Our report on mice lacking the JNK activator MKK7 specifically in neural stem cells and postmitotic neurons is the first to demonstrate that JNK signaling regulates axon elongation in vivo.
By using in utero electroporation, we showed that MKK7 controls axon elongation in a cell-autonomous manner (Figs. 7, 8). Our work has also uncovered a very interesting phenotype affecting neuronal contralateral axon projection. It is well known that axons of layer 2/3 neurons run radially in the ipsilateral cortex to the white matter, and then run tangentially and project to the contralateral cortex. In the ipsilateral side, layer 2/3 axons normally extend branches projecting to layer 5 and form barrel nets, which are axonal trajectories made after the initial appearance of barrels (Sehara et al., 2010). In our study, layer 2/3 neurons lacking MKK7 did not extend axons running tangentially in the white matter or form contralateral projections, although the axons of these neurons did run radially in the ipsilateral cortex and formed branches and barrel nets (Fig. 7D,E). Our results suggest that axon elongation is regulated by different mechanisms in the cerebral cortex and in the white matter, and that MKK7 plays an important role only in the latter case.
Role of MKK7 in MAP regulation
Proper regulation of microtubule dynamics is critical for axon elongation and radial migration, and this control is mediated mainly by MAPs such as MAP1B and DCX that bind to microtubules and stabilize them. Double mutant mice such as the Map1b/Tau or Dcx/doublecortin-like kinase (Dclk) strains display axonal malformation during brain development (Takei et al., 2000; Koizumi et al., 2006). Importantly, the ability of MAPs to regulate microtubule dynamics depends on their phosphorylation by several protein kinases, including JNK (Kawauchi et al., 2003; Gdalyahu et al., 2004). Phosphorylation of a MAP protein induces its disassociation from microtubules, which causes the microtubules to enter a dynamic state. In our study, we found that the phosphorylation levels of both MAP1B and DCX were reduced (Fig. 10B,C), and that microtubule structures appeared abnormal in Mkk7flox/flox Nestin-Cre axons (Fig. 10D). Based on our results and the literature, we propose that MKK7-JNK signaling controls the phosphorylation of MAP1B and DCX that regulates microtubule dynamics, and that this phosphorylation may be one of several mechanisms promoting axon elongation.
Role of MKK7 in apoptosis
It has been previously reported that Jnk1/Jnk2 double mutant mice are embryonic lethal at E11.5 due to severely dysregulated apoptosis in the brain (Kuan et al., 1999; Sabapathy et al., 1999). The results of these studies indicated that JNK regulates apoptosis positively in the forebrain and negatively in the hindbrain during early brain development. However, when we examined the regulation of apoptosis in our Mkk7flox/flox Nestin-Cre mice, we found no abnormalities (Fig. 5), in line with previous analyses of Mkk4flox/flox Nestin-Cre mice (Wang et al., 2007). One explanation for this discrepancy is that JNK signaling may regulate apoptosis during early brain development but is not involved in such apoptosis at later stages. Another possibility is that, because MKK4 remains intact in MKK7flox/flox Nestin-Cre brain, weak JNK activation could have occurred that was sufficient for normal regulation of apoptosis in the developing brain. We are currently generating double conditional knock-out Mkk7flox/flox/Mkk4flox/flox Nestin-Cre mice to resolve this issue.
Role of MKK7 in neuronal differentiation
When we investigated whether MKK7 was involved in neuronal differentiation, all markers examined were expressed with essentially normal distributions (Fig. 5). These results indicate that neuronal differentiation is not affected by MKK7 disruption in the cortex. We also determined whether MKK7 was involved in regulating the activity of c-Jun needed for neurotransmitter specification in the dorsal spinal cord. A recent study of Xenopus tropicalis demonstrated that c-Jun regulates neurotransmitter specification through transcriptional regulation of Tlx3 in the dorsal spinal cord of this animal (Marek et al., 2010). This group showed that c-Jun activation induces the formation of GABAergic neurons and suppresses glutamatergic differentiation. Because c-Jun is a major substrate of JNK, we hypothesized that we might see a reduction of GABAergic neurons and an increase in glutamatergic neurons in the spinal cords of Mkk7flox/flox Nestin-Cre mice due to suppression of c-Jun activity caused by the lack of MKK7. However, our in situ hybridization analysis of gad1 and vglut2 expression showed no alterations in the dorsal spinal cords of Mkk7flox/flox Nestin-Cre mice (data not shown). Thus, neurotransmitter specification is not influenced by MKK7 deletion. It remains possible that low levels of c-Jun phosphorylation still occurred in Mkk7flox/flox Nestin-Cre mice that were sufficient for normal neurotransmitter specification. Alternatively, GABAergic/glutamatergic differentiation may be regulated by different mechanisms in Xenopus tropicalis and Mus musculus.
Ultrastructural alterations in Mkk7flox/flox Nestin-Cre mice
Electron microscopy revealed abnormal accumulations of intermediate filaments in the axons of Mkk7flox/flox Nestin-Cre brain (Fig. 3A), as well as reduced phosphorylation of NF-H (Fig. 10A). NF-H is a neuron-specific intermediate filament, and it has been reported that the C-terminal tail of NF-H regulates interactions among NFs and influences their axonal transport (Sasaki et al., 2009; Lee et al., 2011). It is therefore possible that JNK-mediated phosphorylation of NF-H is required for normal NF-H distribution, and that failure of this regulation due to loss of MKK7 induces the accumulation of filamentous structures within axons. Our electron microscopy analyses also showed that Mkk7flox/flox Nestin-Cre brain contained numerous autophagic vacuoles (Fig. 3B). Although it is possible that autophagy is a secondary effect induced by the abnormal accumulation of intermediate filaments in our mutants, it has been reported that JNK signaling is directly involved in the formation of autophagic vacuoles and autophagic cell death (Byun et al., 2009; Shimizu et al., 2010; Kim et al., 2011; Xu et al., 2011). We are currently studying our Mkk7flox/flox Nestin-Cre mice to determine whether MKK7-JNK signaling acts to suppress autophagy in the developing brain.
Comparison of MKK4 and MKK7 in the developing brain
MKK7 and MKK4 are both JNK activators, and complete activation of JNK requires the activities of both of these kinases (Kishimoto et al., 2003). In our Mkk7flox/flox Nestin-Cre mice, phosphorylated JNK was markedly reduced despite an upregulation of MKK4 (Fig. 1E). This result is consistent with that of Wang and colleagues, who showed that MKK4 deletion decreases JNK activation to 1/5 of control values (Wang et al., 2007). Together, these findings suggest that both MKK7 and MKK4 are required for optimal JNK activation in the developing brain. Furthermore, this optimal JNK activation is essential for normal neuronal radial migration in the cortex, since both Mkk7flox/flox Nestin-Cre mice (Fig. 6) and Mkk4flox/flox Nestin-Cre mice (Wang et al., 2007) show defects in this process. However, other phenotypes do not overlap between MKK7flox/flox Nestin-Cre and Mkk4flox/flox Nestin-Cre mice. Mkk7flox/flox Nestin-Cre mice die at birth (Table 1, Fig. 1F), whereas Mkk4flox/flox Nestin-Cre mice survive until age 3 weeks. Mkk7flox/flox Nestin-Cre mice (E18.5) display enlarged brain ventricles, diminished striatum, decreased forebrain axon tracts, and reduced corticofugal axons (Figs. 2, 4), but none of these defects has been observed (or at least has not been reported) for Mkk4flox/flox Nestin-Cre mice. Thus, MKK7 has unique functions in the developing brain that differ from those of MKK4.
Differences between MKK7 and MKK4 functions also appear at the molecular level. In Mkk4flox/flox Nestin-Cre brain, phosphorylation levels of MAP1B are reduced, but DCX phosphorylation is not altered (Wang et al., 2007). In contrast, we found that the phosphorylation levels of both MAP1B and DCX were reduced in Mkk7flox/flox Nestin-Cre brain (Fig. 10B,C), suggesting a difference in substrates affected by MKK7-JNK versus MKK4-JNK signaling. In line with this hypothesis, the scaffold protein JIP1 binds to JNK, MKK7, and DCX but not to MKK4 (Whitmarsh et al., 1998; Wang et al., 2007). We therefore propose that differences in scaffold proteins and/or substrates involved in MKK7-JNK versus MKK4-JNK signaling pathways could cause the phenotypic divergence observed between Mkk7flox/flox Nestin-Cre and Mkk4flox/flox Nestin-Cre mice.
In conclusion, our results reveal an important and unique role for MKK7 during axon elongation in the developing brain, and reinforce the complex nature of JNK signaling and regulation in vivo.
Footnotes
This work was supported in part by research grants from the Ministry of Education, Culture, Sports, Science, and Technology of Japan, the Ministry of Health, Labour, and Welfare of Japan, and the Japan Society for the Promotion of Science. We are grateful to numerous members of the Nishina, Kawasaki, and Katada laboratories for critical reading and helpful discussions. We also thank Dr. Syu-ichi Hirai (Yokohama City University, Kanagawa, Japan) for his generous and expert technical advice.
References
- Arlotta P, Molyneaux BJ, Chen J, Inoue J, Kominami R, Macklis JD. Neuronal subtype-specific genes that control corticospinal motor neuron development in vivo. Neuron. 2005;45:207–221. doi: 10.1016/j.neuron.2004.12.036. [DOI] [PubMed] [Google Scholar]
- Asaoka Y, Nishina H. Diverse physiological functions of MKK4 and MKK7 during early embryogenesis. J Biochem. 2010;148:393–401. doi: 10.1093/jb/mvq098. [DOI] [PubMed] [Google Scholar]
- Björkblom B, Ostman N, Hongisto V, Komarovski V, Filén JJ, Nyman TA, Kallunki T, Courtney MJ, Coffey ET. Constitutively active cytoplasmic c-Jun N-terminal kinase 1 is a dominant regulator of dendritic architecture: role of microtubule-associated protein 2 as an effector. J Neurosci. 2005;25:6350–6361. doi: 10.1523/JNEUROSCI.1517-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Britanova O, Akopov S, Lukyanov S, Gruss P, Tarabykin V. Novel transcription factor Satb2 interacts with matrix attachment region DNA elements in a tissue-specific manner and demonstrates cell-type-dependent expression in the developing mouse CNS. Eur J Neurosci. 2005;21:658–668. doi: 10.1111/j.1460-9568.2005.03897.x. [DOI] [PubMed] [Google Scholar]
- Bulfone A, Martinez S, Marigo V, Campanella M, Basile A, Quaderi N, Gattuso C, Rubenstein JL, Ballabio A. Expression pattern of the Tbr2 (Eomesodermin) gene during mouse and chick brain development. Mech Dev. 1999;84:133–138. doi: 10.1016/s0925-4773(99)00053-2. [DOI] [PubMed] [Google Scholar]
- Byun JY, Yoon CH, An S, Park IC, Kang CM, Kim MJ, Lee SJ. The Rac1/MKK7/JNK pathway signals upregulation of Atg5 and subsequent autophagic cell death in response to oncogenic Ras. Carcinogenesis. 2009;30:1880–1888. doi: 10.1093/carcin/bgp235. [DOI] [PubMed] [Google Scholar]
- Chang L, Karin M. Mammalian MAP kinase signalling cascades. Nature. 2001;410:37–40. doi: 10.1038/35065000. [DOI] [PubMed] [Google Scholar]
- Chang L, Jones Y, Ellisman MH, Goldstein LSB, Karin M. JNK1 is required for maintenance of neuronal microtubules and controls phosphorylation of microtubule-associated proteins. Dev Cell. 2003;4:521–533. doi: 10.1016/s1534-5807(03)00094-7. [DOI] [PubMed] [Google Scholar]
- Davis RJ. Signal transduction by the JNK group of MAP kinases. Cell. 2000;103:239–252. doi: 10.1016/s0092-8674(00)00116-1. [DOI] [PubMed] [Google Scholar]
- Denaxa M, Chan CH, Schachner M, Parnavelas JG, Karagogeos D. The adhesion molecule TAG-1 mediates the migration of cortical interneurons from the ganglionic eminence along the corticofugal fiber system. Development. 2001;128:4635–4644. doi: 10.1242/dev.128.22.4635. [DOI] [PubMed] [Google Scholar]
- Enslen H, Raingeaud J, Davis RJ. Selective activation of p38 mitogen-activated protein (MAP) kinase isoforms by the MAP kinase kinases MKK3 and MKK6. J Biol Chem. 1998;273:1741–1748. doi: 10.1074/jbc.273.3.1741. [DOI] [PubMed] [Google Scholar]
- Ferland RJ, Cherry TJ, Preware PO, Morrisey EE, Walsh CA. Characterization of Foxp2 and Foxp1 mRNA and protein in the developing and mature brain. J Comp Neurol. 2003;460:266–279. doi: 10.1002/cne.10654. [DOI] [PubMed] [Google Scholar]
- Gdalyahu A, Ghosh I, Levy T, Sapir T, Sapoznik S, Fishler Y, Azoulai D, Reiner O. DCX, a new mediator of the JNK pathway. EMBO J. 2004;23:823–832. doi: 10.1038/sj.emboj.7600079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hibi M, Lin A, Smeal T, Minden A, Karin M. Identification of an oncoprotein- and UV-responsive protein kinase that binds and potentiates the c-Jun activation domain. Genes Dev. 1993;7:2135–2148. doi: 10.1101/gad.7.11.2135. [DOI] [PubMed] [Google Scholar]
- Hirai S, Banba Y, Satake T, Ohno S. Axon formation in neocortical neurons depends on stage-specific regulation of microtubule stability by the dual leucine zipper kinase-c-Jun N-terminal kinase pathway. J Neurosci. 2011;31:6468–6480. doi: 10.1523/JNEUROSCI.5038-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunot S, Vila M, Teismann P, Davis RJ, Hirsch EC, Przedborski S, Rakic P, Flavell RA. JNK-mediated induction of cyclooxygenase 2 is required for neurodegeneration in a mouse model of Parkinson's disease. Proc Natl Acad Sci U S A. 2004;101:665–670. doi: 10.1073/pnas.0307453101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imai F, Hirai S, Akimoto K, Koyama H, Miyata T, Ogawa M, Noguchi S, Sasaoka T, Noda T, Ohno S. Inactivation of aPKClambda results in the loss of adherens junctions in neuroepithelial cells without affecting neurogenesis in mouse neocortex. Development. 2006;133:1735–1744. doi: 10.1242/dev.02330. [DOI] [PubMed] [Google Scholar]
- Ito M, Yoshioka K, Akechi M, Yamashita S, Takamatsu N, Sugiyama K, Hibi M, Nakabeppu Y, Shiba T, Yamamoto KI. JSAP1, a novel jun N-terminal protein kinase (JNK)-binding protein that functions as a scaffold factor in the JNK signaling pathway. Mol Cell Biol. 1999;19:7539–7548. doi: 10.1128/mcb.19.11.7539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawasaki H, Mizuseki K, Nishikawa S, Kaneko S, Kuwana Y, Nakanishi S, Nishikawa SI, Sasai Y. Induction of midbrain dopaminergic neurons from ES cells by stromal cell-derived inducing activity. Neuron. 2000;28:31–40. doi: 10.1016/s0896-6273(00)00083-0. [DOI] [PubMed] [Google Scholar]
- Kawauchi T, Chihama K, Nabeshima Y, Hoshino M. The in vivo roles of STEF/Tiam1, Rac1 and JNK in cortical neuronal migration. EMBO J. 2003;22:4190–4201. doi: 10.1093/emboj/cdg413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim MJ, Woo SJ, Yoon CH, Lee JS, An S, Choi YH, Hwang SG, Yoon G, Lee SJ. Involvement of autophagy in oncogenic K-Ras-induced malignant cell transformation. J Biol Chem. 2011;286:12924–12932. doi: 10.1074/jbc.M110.138958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kishimoto H, Nakagawa K, Watanabe T, Kitagawa D, Momose H, Seo J, Nishitai G, Shimizu N, Ohata S, Tanemura S, Asaka S, Goto T, Fukushi H, Yoshida H, Suzuki A, Sasaki T, Wada T, Penninger JM, Nishina H, Katada T. Different properties of SEK1 and MKK7 in dual phosphorylation of stress-induced activated protein kinase SAPK/JNK in embryonic stem cells. J Biol Chem. 2003;278:16595–16601. doi: 10.1074/jbc.M213182200. [DOI] [PubMed] [Google Scholar]
- Koizumi H, Tanaka T, Gleeson JG. Doublecortin-like kinase functions with doublecortin to mediate fiber tract decussation and neuronal migration. Neuron. 2006;49:55–66. doi: 10.1016/j.neuron.2005.10.040. [DOI] [PubMed] [Google Scholar]
- Kuan CY, Yang DD, Samanta Roy DR, Davis RJ, Rakic P, Flavell RA. The Jnk1 and Jnk2 protein kinases are required for regional specific apoptosis during early brain development. Neuron. 1999;22:667–676. doi: 10.1016/s0896-6273(00)80727-8. [DOI] [PubMed] [Google Scholar]
- Lee S, Sunil N, Shea TB. C-terminal neurofilament phosphorylation fosters neurofilament-neurofilament associations that compete with axonal transport. Cytoskeleton (Hoboken) 2011;68:8–17. doi: 10.1002/cm.20488. [DOI] [PubMed] [Google Scholar]
- Marek KW, Kurtz LM, Spitzer NC. cJun integrates calcium activity and tlx3 expression to regulate neurotransmitter specification. Nat Neurosci. 2010;13:944–950. doi: 10.1038/nn.2582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McEvilly RJ, de Diaz MO, Schonemann MD, Hooshmand F, Rosenfeld MG. Transcriptional regulation of cortical neuron migration by POU domain factors. Science. 2002;295:1528–1532. doi: 10.1126/science.1067132. [DOI] [PubMed] [Google Scholar]
- Nakagawa K, Sugahara M, Yamasaki T, Kajiho H, Takahashi S, Hirayama J, Minami Y, Ohta Y, Watanabe T, Hata Y, Katada T, Nishina H. Filamin associates with stress signalling kinases MKK7 and MKK4 and regulates JNK activation. Biochem J. 2010;427:237–245. doi: 10.1042/BJ20091011. [DOI] [PubMed] [Google Scholar]
- Okada S, Nakamura M, Katoh H, Miyao T, Shimazaki T, Ishii K, Yamane J, Yoshimura A, Iwamoto Y, Toyama Y, Okano H. Conditional ablation of Stat3 or Socs3 discloses a dual role for reactive astrocytes after spinal cord injury. Nat Med. 2006;12:829–834. doi: 10.1038/nm1425. [DOI] [PubMed] [Google Scholar]
- Sabapathy K, Jochum W, Hochedlinger K, Chang L, Karin M, Wagner EF. Defective neural tube morphogenesis and altered apoptosis in the absence of both JNK1 and JNK2. Mech Dev. 1999;89:115–124. doi: 10.1016/s0925-4773(99)00213-0. [DOI] [PubMed] [Google Scholar]
- Saito T. In vivo electroporation in the embryonic mouse central nervous system. Nat Protoc. 2006;1:1552–1558. doi: 10.1038/nprot.2006.276. [DOI] [PubMed] [Google Scholar]
- Sasaki T, Ishiguro K, Hisanaga S. Novel axonal distribution of neurofilament-H phosphorylated at the glycogen synthase kinase 3beta-phosphorylation site in its E-segment. J Neurosci Res. 2009;87:3088–3097. doi: 10.1002/jnr.22148. [DOI] [PubMed] [Google Scholar]
- Schramek D, Kotsinas A, Meixner A, Wada T, Elling U, Pospisilik JA, Neely GG, Zwick R-H, Sigl V, Forni G, Serrano M, Gorgoulis VG, Penninger JM. The stress kinase MKK7 couples oncogenic stress to p53 stability and tumor suppression. Nat Genet. 2011;43:212–219. doi: 10.1038/ng.767. [DOI] [PubMed] [Google Scholar]
- Sehara K, Toda T, Iwai L, Wakimoto M, Tanno K, Matsubayashi Y, Kawasaki H. Whisker-related axonal patterns and plasticity of layer 2/3 neurons in the mouse barrel cortex. J Neurosci. 2010;30:3082–3092. doi: 10.1523/JNEUROSCI.6096-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimizu S, Konishi A, Nishida Y, Mizuta T, Nishina H, Yamamoto A, Tsujimoto Y. Involvement of JNK in the regulation of autophagic cell death. Oncogene. 2010;29:2070–2082. doi: 10.1038/onc.2009.487. [DOI] [PubMed] [Google Scholar]
- Tabata H, Nakajima K. Labeling embryonic mouse central nervous system cells by in utero electroporation. Dev Growth Differ. 2008;50:507–511. doi: 10.1111/j.1440-169X.2008.01043.x. [DOI] [PubMed] [Google Scholar]
- Takei Y, Teng J, Harada A, Hirokawa N. Defects in axonal elongation and neuronal migration in mice with disrupted tau and map1b genes. J Cell Biol. 2000;150:989–1000. doi: 10.1083/jcb.150.5.989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toda T, Hayakawa I, Matsubayashi Y, Tanaka K, Ikenaka K, Lu QR, Kawasaki H. Termination of lesion-induced plasticity in the mouse barrel cortex in the absence of oligodendrocytes. Mol Cell Neurosci. 2008;39:40–49. doi: 10.1016/j.mcn.2008.05.014. [DOI] [PubMed] [Google Scholar]
- Ura S, Nishina H, Gotoh Y, Katada T. Activation of the c-Jun N-terminal kinase pathway by MST1 is essential and sufficient for the induction of chromatin condensation during apoptosis. Mol Cell Biol. 2007;27:5514–5522. doi: 10.1128/MCB.00199-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wada T, Nakagawa K, Watanabe T, Nishitai G, Seo J, Kishimoto H, Kitagawa D, Sasaki T, Penninger JM, Nishina H, Katada T. Impaired synergistic activation of stress-activated protein kinase SAPK/JNK in mouse embryonic stem cells lacking SEK1/MKK4: different contribution of SEK2/MKK7 isoforms to the synergistic activation. J Biol Chem. 2001;276:30892–30897. doi: 10.1074/jbc.M011780200. [DOI] [PubMed] [Google Scholar]
- Wada T, Joza N, Cheng HY, Sasaki T, Kozieradzki I, Bachmaier K, Katada T, Schreiber M, Wagner EF, Nishina H, Penninger JM. MKK7 couples stress signalling to G2/M cell-cycle progression and cellular senescence. Nat Cell Biol. 2004;6:215–226. doi: 10.1038/ncb1098. [DOI] [PubMed] [Google Scholar]
- Wang X, Nadarajah B, Robinson AC, McColl BW, Jin JW, Dajas-Bailador F, Boot-Handford RP, Tournier C. Targeted deletion of the mitogen-activated protein kinase kinase 4 gene in the nervous system causes severe brain developmental defects and premature death. Mol Cell Biol. 2007;27:7935–7946. doi: 10.1128/MCB.00226-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westerlund N, Zdrojewska J, Padzik A, Komulainen E, Björkblom B, Rannikko E, Tararuk T, Garcia-Frigola C, Sandholm J, Nguyen L, Kallunki T, Courtney MJ, Coffey ET. Phosphorylation of SCG10/stathmin-2 determines multipolar stage exit and neuronal migration rate. Nat Neurosci. 2011;14:305–313. doi: 10.1038/nn.2755. [DOI] [PubMed] [Google Scholar]
- Whitmarsh AJ, Cavanagh J, Tournier C, Yasuda J, Davis RJ. A mammalian scaffold complex that selectively mediates MAP kinase activation. Science. 1998;281:1671–1674. doi: 10.1126/science.281.5383.1671. [DOI] [PubMed] [Google Scholar]
- Wolfer DP, Henehan-Beatty A, Stoeckli ET, Sonderegger P, Lipp HP. Distribution of TAG-1/axonin-1 in fibre tracts and migratory streams of the developing mouse nervous system. J Comp Neurol. 1994;345:1–32. doi: 10.1002/cne.903450102. [DOI] [PubMed] [Google Scholar]
- Xu P, Das M, Reilly J, Davis RJ. JNK regulates FoxO-dependent autophagy in neurons. Genes Dev. 2011;25:310–322. doi: 10.1101/gad.1984311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasuda J, Whitmarsh AJ, Cavanagh J, Sharma M, Davis RJ. The JIP group of mitogen-activated protein kinase scaffold proteins. Mol Cell Biol. 1999;19:7245–7254. doi: 10.1128/mcb.19.10.7245. [DOI] [PMC free article] [PubMed] [Google Scholar]