

Abstract
The endocannabinoid (eCB) system and the cannabinoid CB1 receptor (CB1R) play key roles in the modulation of brain functions. Although actions of eCBs and CB1Rs are well described at the synaptic level, little is known of their modulation of neural activity at the network level. Using microelectrode arrays, we have examined the role of CB1R activation in the modulation of the electrical activity of rat and mice cortical neural networks in vitro. We find that exogenous activation of CB1Rs expressed on glutamatergic neurons decreases the spontaneous activity of cortical neural networks. Moreover, we observe that the net effect of the CB1R antagonist AM251 inversely correlates with the initial level of activity in the network: blocking CB1Rs increases network activity when basal network activity is low, whereas it depresses spontaneous activity when its initial level is high. Our results reveal a complex role of CB1Rs in shaping spontaneous network activity, and suggest that the outcome of endogenous neuromodulation on network function might be state dependent.
Introduction
Neuromodulatory systems are thought to play a pivotal role in adjusting neural network activity to changing conditions (McCormick, 1992). The endocannabinoid (eCB) system is involved in the regulation of many physiological processes, including the modulation of neural circuits (Di Marzo et al., 1998; Katona and Freund, 2008). Endocannabinoids are produced and released on demand following sustained levels of electrical stimulation, whereupon they decrease the release of glutamate and GABA on a short- or long-term scale by acting in a retrograde fashion on presynaptic CB1Rs (Heifets and Castillo, 2009; Kano et al., 2009). How CB1R might influence ongoing spontaneous network activity, however, remains poorly understood. Moreover, since CB1Rs are expressed on GABAergic as well as on glutamatergic synaptic terminals and their activation decreases the release of both neurotransmitters, how the eCB system might affect the integrated activity of an ensemble of neurons remains unclear.
We have addressed these issues by combining extracellular recordings of the electrical activity of cortical neuron networks grown on microelectrode arrays (MEAs) with pharmacology and the use of conditional CB1R knock-out mice. We find that agonist activation of CB1Rs on glutamatergic neurons depresses spontaneous bursting in cultured cortical networks. Moreover, we find that endogenous activation of CB1Rs modulates bursting and that the net effect of this modulation is inversely correlated with the basal level of activity in the network. Our results indicate that eCBs and CB1Rs may shape spontaneous network activity in a bidirectional and state-dependent fashion.
Materials and Methods
Microelectrode arrays and cell culture.
Extracellular recordings of cortical neural networks in culture were performed on planar 60-channel microelectrode arrays (MEAs) (MultiChannel Systems; electrode distance 200 μm, diameter 30 μm). Cortices were dissected from Sprague Dawley rat brains (4–6 pups within 24 h of birth), and tissues were enzymatically digested for 20–40 min at 36.5°C in a medium containing 20 U/ml papain. After papain inactivation with trypsin inhibitor (125 mm) and bovine serum albumin (0.25%), cortices were mechanically triturated in the culture medium. Cells (105) were seeded in a 100 μl drop on the precoated (poly l-lysine + laminin) recording area of each MEA. Cells were left to sediment for up to 2 h before the MEA chambers were complemented to 1 ml of culture medium. Neurons were grown as previously described (Sinagra et al., 2005; Groc et al., 2007) in Minimum Essential Medium (Invitrogen Invitrogen) supplemented with 5% of Serum Supreme (Biowhittaker), 28.3 mm d-glucose, and 0.1% of Mito+ Serum Extender (Becton Dickinson). Media were fully renewed 24–48 h after plating and half-exchanged every 48 h. Cultures were kept in a humidified atmosphere of 5% CO2 at 36.5°C until recording. Chemicals were purchased from Sigma-Aldrich.
Transgenic mice.
Conditional mutant mice in which the CB1R gene is lacking in either cortical glutamatergic (CB1loxP/loxP; Nex-Cre; referred to as Glu-CB1R KO) or in forebrain GABAergic neurons (CB1loxP/loxP; Dlx-Cre; referred to as GABA-CB1R KO) (Monory et al., 2006) were used. Cortical tissues from individual transgenic mice were cultured separately using the protocols described above. Genotyping was carried out post hoc by PCR as previously described (Monory et al., 2006).
Recordings and data analysis.
Extracellular signals were recorded with an MEA-1060-Inv amplifier and digitized at 25–40 kHz with an MC_Card 64. Data were acquired with the MC_Rack program (MultiChannel Systems) and stored on a personal computer. Recordings were performed in the culture medium at 37°C.
Data were analyzed off-line. For each MEA, 3–10 channels were selected based on the quality of their signal-to-noise ratio. Traces were high-pass filtered in MC_Rack (300 Hz cutoff) and subsequently analyzed in Spike2 software (CED). Signals were considered as events when crossing a negative threshold defined in each recording as a multiple (typically 8–10) of the standard deviation of the electrical noise of the electrodes (Novellino et al., 2007). In agreement with previous work (Turnbull et al., 2005; Volmer et al., 2007), we defined “bursts” as a series of events, fulfilling the following criteria: burst onset when two events occurred within 25–100 ms of one another; and burst termination when events within a burst were separated by >50–200 ms, with at least 3–10 events per burst. Burst parameters were determined empirically in each MEA during the control period to optimize detection and kept constant throughout the recording. No spike sorting was attempted because of the density of our cultures (∼1500 cells/mm2) and the diameter of the microelectrodes (30 μm).
Experiments were repeated in cultures from 2–8 different litters of rats (14–26 d in vitro) and 3–5 animals from 2–3 different litters of transgenic mice (14–23 d in vitro). Statistical analyses were performed using GraphPad Prism 5 (GraphPad Software). Drug effects were assessed using Student's paired t test or repeated-measures ANOVA with Newman–Keuls post hoc test where appropriate. Group comparisons were performed using one-way ANOVA with Newman–Keuls post hoc test. In the figures, *, **, and *** represent p < 0.05, p < 0.01, and p < 0.001, respectively. Values given are mean ± SEM. Traces in figures are 30-s-long stretches of extracellular recordings from three channels separated by >200 μm.
Drugs.
4-Aminopyridine (4-AP), tetrodotoxin (TTX), 2,3-dioxo-6-nitro-1,2,3,4-tetrahydrobenzo[f]quinoxaline-7-sulfonamide disodium (NBQX), dl-2-amino-5-phosphonopentanoic acid (AP5; TOCRIS Bioscience), and muscimol (Sigma Aldrich) were dissolved to a stock concentration in water (4-AP, muscimol, NBQX, and TTX) and in NaOH (AP5), and diluted to the appropriate concentration in culture medium. WIN 55,212-2, AM251, CGP54626, cyclothiazide (TOCRIS Bioscience), and picrotoxin (Sigma Aldrich) were dissolved in DMSO and diluted to the appropriate concentration in culture medium. DMSO final concentration (typically 0.1–0.3‰) was kept constant throughout the recording. In the majority of the experiments, drugs were applied by slow perfusion (0.25 ml/min) and in a few cases by direct injection into the MEA chamber (100 μl volume). The results obtained with these two modes of drug application were indistinguishable and the data were pooled.
Results
We performed extracellular recordings of the network activity of rat cortical neurons grown on MEAs. After 2–3 weeks in vitro, cortical neurons displayed a stereotypical spontaneous activity, alternating periods of relative quiescence and periods of intense synchronous firing (network bursts, Fig. 1B). Basal spontaneous burst rates ranged between 0.03 and 0.81 Hz (mean 0.34 ± 0.02 Hz; n = 65) and were distributed as illustrated in Figure 1A. Network activity was completely blocked by 1 μm TTX, indicating that network bursts depended on action potential firing (Fig. 1D; n = 4). Bursting was also abolished by 10 μm NBQX and 50 μm AP5, antagonists of ionotropic AMPA and NMDA glutamate receptors, respectively, revealing that such electrical activity was dependent on glutamatergic synaptic transmission (Fig. 1B,D; n = 4).
Figure 1.

Spontaneous network activity of cortical neurons in culture. A, Distribution of basal burst rates (n = 65). Bars represent the number of recordings within individual 0.05 Hz bins. The distribution was well fitted by a single Gaussian function (black line; R2 = 0.77). B, Sample traces illustrating synchronous spontaneous bursting (upper panel). Application of NBQX and AP5 abolished network bursting (lower panel). C, Higher-resolution traces illustrating the network burst framed in B. D, Summary graph of the effect of NBQX + AP5 (n = 4) and of TTX (n = 4) on spontaneous network bursting.
GABAergic synaptic transmission obscures CB1R-mediated network inhibition
We examined the effects of activating CB1Rs on spontaneous network activity. Bath application of WIN 55,212-2 (WIN; 400 nm), a cannabinoid receptor agonist, had heterogeneous effects on bursting. Bursting was decreased in seven individual experiments, whereas it seemed unaffected or increased in four recordings (Fig. 2A). The direction of the effect was not age dependent (linear regression; p > 0.05; data not shown), and overall, the average effect of WIN was not significant (88.9 ± 14.0% of control; n = 11; p > 0.05; Fig. 2A,C). Because activation of CB1Rs affects both excitatory and inhibitory synaptic transmission, the variability we observed might result from the relative inhibition of the release of different neurotransmitters. We therefore tested the effect of WIN in the presence of picrotoxin (Ptx, 50 μm) and of CGP 54626 (CGP; 5 μm), GABAA and GABAB receptor antagonists, respectively. Under these conditions, WIN (400 nm) inhibited network activity in 7 of 8 MEAs (Fig. 2A,B) and had no apparent effect in the remaining recording. On average, WIN significantly decreased burst rates to 58.8 ± 13.7% of control (n = 8; p < 0.05; Fig. 2), an effect prevented in the presence of 2 μm AM251, a CB1R-selective antagonist (91.7 ± 5.5% of control; n = 4; p > 0.05; Fig. 2A,C). These results indicate that activation of CB1Rs can alter network activity by decreasing spontaneous burst rates and suggest that the relative contribution of CB1Rs located on GABAergic or glutamatergic terminals determined the global network effect.
Figure 2.

Activation of CB1R on glutamatergic neurons inhibits network bursting. A, Histogram of normalized spontaneous burst rates in different experimental conditions. Connected circles represent individual experiments. WIN had no effect on average in naive conditions (n = 11) but significantly decreased bursting in disinhibited cultures (in Ptx + CGP; n = 8). This effect was prevented by AM251 (n = 4). B, Sample traces illustrating the inhibitory effect of WIN in the presence of Ptx and CGP. C, Summary histogram of the average effect of WIN in different experimental conditions in cultures from rat cortices (left) and in cultures from cortices of GABA-CB1R KO, Glu-CB1R KO, and their respective wild-type (wt) littermates (right).
Activation of CB1R on glutamatergic neurons depresses network bursting
To further investigate the respective contribution of CB1Rs expressed on glutamatergic and on GABAergic neurons, we used conditional mutant mice that lack CB1R in either glutamatergic (Glu-CB1R KO) or in GABAergic neurons (GABA-CB1R KO) (Monory et al., 2006; Bellocchio et al., 2010). Networks of cortical neurons from GABA-CB1R KO, Glu-CB1R KO, and their respective wild-type littermates displayed a spontaneous electrical activity similar to that of rat cortical neurons, and no significant difference in the basal burst rates in the different genotypes was found (not shown).
In cultures from GABA-CB1R KO mice, WIN (400 nm) inhibited spontaneous bursting to the same extent as in cultures from their wild-type littermates (33.2 ± 9.2% of control vs 52.7 ± 8.1% of control, respectively; n = 5 each; Fig. 2C). In contrast, the effect of WIN was significantly different in cultures from Glu-CB1R KO mice than in those from their wild-type littermates (146.8 ± 32.3% of control vs 40.2 ± 12.3% of control, respectively; n = 3 and 5, respectively; p < 0.001). In fact, in cortical networks without CB1R on glutamatergic neurons, WIN did not inhibit bursting (p > 0.05; Fig. 2C).
These results indicate that CB1Rs on GABAergic neurons are not necessary for the inhibition of network bursting induced by WIN, whereas activation of those expressed on glutamatergic neurons is sufficient and necessary for inhibiting spontaneous network activity.
Endogenous modulation of network bursting by eCBs and CB1Rs
To examine whether CB1Rs were engaged in the endogenous modulation of spontaneous network activity, we used the CB1R antagonist AM251 (400 nm). Bath application of AM251 had variable effects on burst rates: blockade of CB1Rs increased burst rates in seven MEAs, decreased it in five, and had no clear effect in the remaining recording (Fig. 3A). Overall, AM251 had no significant effect on bursting (Fig. 3B; average effect of AM251: 115.3 ± 8.6% of control; n = 13; p > 0.05). The variability was not due to age differences (linear regression; p > 0.05; data not shown). We found, however, that the effect of AM251 was inversely correlated with the initial level of activity (p < 0.001; Pearson r = −0.84; Fig. 3C). Plotting these data as a function of the initial burst rate in control condition revealed that AM251 was excitatory in MEAs with low basal burst rates (<0.35 Hz; 133.3 ± 9.1% of control; n = 8; p < 0.05), whereas it was inhibitory in MEAs with higher basal burst rates (>0.35 Hz; 86.5 ± 3.9% of control; n = 5; p < 0.05; Fig. 3C). No such correlation was found for the effect of WIN (p > 0.05). These results suggest that the effect of blocking CB1R with AM251 on bursting was activity dependent.
Figure 3.

Endogenous modulation of network activity by CB1Rs. Aa, Sample traces illustrating the excitatory effect of AM251. Ab, Sample traces from a different culture where AM251 caused a decrease in burst rates. B, Summary graph of 13 different experiments (left; connected symbols represent individual recordings). The bar on the right illustrates the average effect of AM251 on burst rates. C, Effect of AM251 expressed as a function of basal burst rate. AM251 increased bursting in cultures with lower basal burst rates (<0.35 Hz; n = 8; white circles), whereas it was inhibitory in cultures with higher basal burst rates (>0.35 Hz; n = 5; white squares). Bold symbols with error bars illustrate the averages for the two groups. Solid and dashed lines represent the linear regression and 95% confidence interval, respectively.
Based on that observation, we pharmacologically decreased or increased basal network activity and tested the effect of AM251. Bath application of the GABAA receptor agonist muscimol (0.5 μm) dramatically decreased burst rates (from 0.46 ± 0.05 to 0.12 ± 0.03 Hz, p < 0.05, Fig. 4A) as reported previously (Rijal and Gross, 2008). Consistent with our hypothesis, subsequent application of AM251 (400 nm) in the presence of muscimol increased bursting to >200% of the burst rate in muscimol alone (0.25 ± 0.07 Hz in muscimol + AM251; n = 8; p < 0.05; Fig. 4A). Moreover, both cyclothiazide (CTZ) and 4-AP increased burst rates (from 0.42 ± 0.12 to 1.02 ± 0.12 Hz and from 0.48 ± 0.07 to 0.92 ± 0.06 Hz, p < 0.01 and 0.05, respectively, Fig. 4B,C). Application of AM251 in these conditions, however, reduced network activity (0.62 ± 0.06 Hz in CTZ + AM251; 0.68 ± 0.12 Hz in 4-AP + AM251; n = 5 each; p < 0.01 and 0.05, respectively; Fig. 4B,C). Tuning basal network activity to low or high levels was therefore sufficient to cause the effect of AM251 to be excitatory or inhibitory, respectively. By combining the data obtained in the presence of muscimol, CTZ, and 4-AP with the data obtained in naive conditions (Fig. 3), we found an inverse correlation between burst rates and the effect of AM251 (Fig. 4D, n = 31, p < 0.0001, Pearson r = −0.82), confirming that the effect of blocking CB1Rs switches from excitatory to inhibitory when the basal level of network activity increases.
Figure 4.
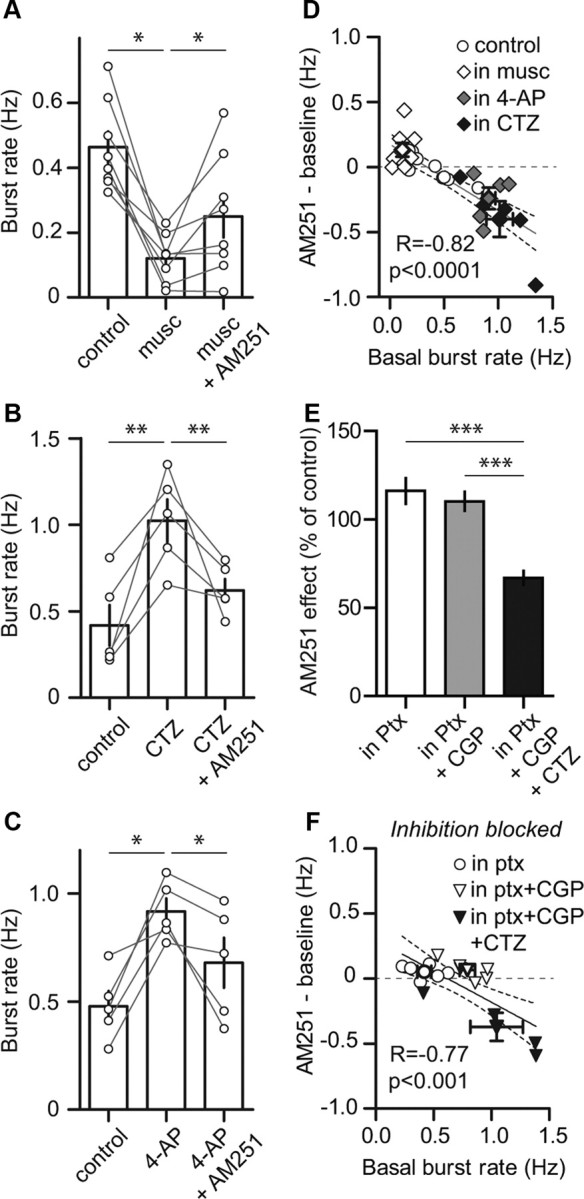
Basal level of network activity dictates direction of AM251 effect. A–C, Histograms of the effect of muscimol (A, n = 8), CTZ (B, n = 5), and 4-AP (C, n = 5) on spontaneous burst rates and of the effect of a subsequent application of AM251 (connected circles represent individual experiments). AM251 excited cultures with low basal burst rates (in muscimol), whereas it inhibited those with high basal burst rates (in CTZ and in 4-AP). D, Net effect of AM251 expressed as a function of basal burst rate. A significant inverse correlation was found when the data of all the AM251 experiments were pooled (n = 31). E, Histogram summarizing the effect of AM251 when inhibitory synaptic transmission was blocked. AM251 significantly increased bursting in Ptx (n = 6), had no effect in Ptx + CGP (n = 6), and significantly decreased bursting in Ptx + CGP + CTZ (n = 4). F, Net effect of AM251 expressed as a function of basal burst rate in disinhibited cultures. A significant inverse correlation was found when the data of all the AM251 experiments in disinhibited cultures were pooled (n = 16). Bold symbols with error bars represent the averages for their respective experimental conditions. Solid and dashed lines represent the linear regression and 95% confidence interval, respectively.
We next tested whether such inverse correlation could be seen when inhibitory synaptic transmission is blocked. Treating cortical networks with Ptx alone, Ptx + CGP, or Ptx + CGP + CTZ gave rise to graded levels of basal burst rates (0.42 ± 0.06 Hz n = 6, 0.80 ± 0.07 Hz n = 6, 1.05 ± 0.23 Hz n = 4, respectively; p < 0.01, not illustrated). Application of AM251 increased burst rates in Ptx (116.10 ± 7.31% of control, p < 0.05), had no effect in Ptx + CGP (110.20 ± 5.38% of control) and decreased burst rates in Ptx + CGP + CTZ (66.84 ± 7.69% of control, p < 0.05). The effect of AM251 was different in the three conditions (p < 0.001; Fig. 4E). Plotting these data as a function of the initial level of activity revealed that the effect of AM251 was inversely correlated to the basal burst rate (n = 16, p < 0.001, Pearson r = −0.77; Fig. 4F). These results suggest that CB1Rs on GABAergic neurons could not account for the bidirectional effect of AM251.
Discussion
We report here that exogenous activation of CB1Rs inhibits spontaneous bursting in cortical neuron networks. In naive conditions, this effect is observed in a subset of networks (∼65%), whereas it is seen in nearly all recordings (∼90%) when GABAergic synaptic transmission is inhibited, suggesting that CB1Rs on GABAergic neurons may be responsible for this variability. Although we cannot rule out their involvement in the modulation of subthreshold events, the role of CB1Rs on GABAergic neurons in the modulation of network activity remains unclear in our experiments. However, CB1Rs expressed by glutamatergic neurons are likely to be the main mediators of the inhibitory effect of WIN, since the inhibition is observed when GABAergic transmission is blocked and the effect of WIN is present in cultures from mice lacking CB1Rs in GABAergic neurons, but absent in cultures from mice lacking CB1Rs in glutamatergic neurons.
The effect of CB1R activation we see here is consistent with known effects on neurotransmitter release (Kano et al., 2009) and on neuronal firing (Kreitzer et al., 2002; Bacci et al., 2004; Marinelli et al., 2009). However, because we find that glutamatergic excitatory synaptic transmission is central to the generation of network bursts, we speculate that WIN-induced inhibition of bursting is mainly caused by depression of glutamate release.
In the second part of this study, we use AM251, a CB1R antagonist, to reveal endogenous activation of these receptors. To avoid potential confounding side effects of this drug (Pertwee, 2005), we used a low dose (400 nm) of the antagonist. Correlation analyses reveal that the net effect of antagonizing CB1Rs varies with the initial level of activity in the network. Indeed, blocking CB1Rs was excitatory in networks that had low basal burst rates, whereas it was inhibitory in networks with higher basal burst rates. Consistent with this, AM251 was excitatory when network activity was reduced with muscimol, whereas it was inhibitory when bursting was enhanced with CTZ or 4-AP. These results suggest that CB1Rs can modulate network activity in a bidirectional and state-dependent manner. Intriguingly, no correlation was found between the effect of WIN and the basal burst rates. We speculate that this results from indiscriminate decrease of neurotransmitter release at every CB1R-expressing synapse during WIN application. In contrast, AM251 would have a functional effect only at sites undergoing eCB-mediated modulation.
Although we cannot rule out the involvement of CB1Rs on GABAergic neurons, our data suggest a key role of CB1Rs on glutamatergic neurons in this phenomenon, since an inverse correlation between the effect of AM251 and basal burst rates is still observed in disinhibited networks. Burst rates in cultures lacking CB1Rs on glutamatergic neurons were no different from burst rates in cultures from their wild-type littermates. However, differences in cell density, wiring, and connectivity (Berghuis et al., 2007; Mulder et al., 2008), as well as compensation potentially occurring during 2 weeks in vitro in cultures lacking CB1Rs in either glutamatergic or in GABAergic neurons, obscure any interpretation of this finding.
Retrograde activation of presynaptic CB1Rs by eCBs provides a negative feedback on neurotransmitter release and is thought to act as a “circuit breaker” (Katona and Freund, 2008). Although this is consistent with the effect of AM251 we report in networks with low initial burst rates, the observation that AM251 inhibits bursting in networks with higher levels of activity is difficult to reconcile with negative feedback. Both experimental work (Staley et al., 1998) and modeling studies (Rubin et al., 2009) indicate that burst termination relies in part on activity-dependent depression of recurrent excitatory synaptic transmission. This implies that the burst rate in the network may be limited by recovery from synaptic depression. Because presynaptic inhibition (Brenowitz et al., 1998), including that mediated by CB1Rs (Oliet et al., 2007), may enhance the efficacy of synaptic transmission during bursts of presynaptic activity, it is possible that activation of CB1Rs in fast bursting networks sets the release of neurotransmitters, in particular glutamate, to limit synaptic depression and to optimally sustain high levels of ongoing activity. Previously described activity dependent plasticity of CB1R signaling may also be involved (Losonczy et al., 2004; Foldy et al., 2006). Alternatively, but not exclusively, intense network activity may affect the distribution and/or the function of CB1Rs or eCB release, thereby altering eCB signaling. Further studies are needed to determine the mechanisms involved in the state-dependent modulation of network activity by CB1Rs.
Collectively our results indicate that CB1R-mediated modulation of spontaneous network activity depends both on the cellular distribution of these receptors (following exogenous activation) and on the initial state of the network (during endogenous modulation). Our data, along with other recent studies (Stewart and Plenz, 2006; Antal et al., 2010) suggest that state-dependent multimodality might be a common feature of modulatory systems (Goldman-Rakic et al., 2000).
Footnotes
This work was supported by grants from Fondation pour la Recherche Medicale (R.P.), Agence Nationale de la Recherche Projet ANR-06-NEURO-043-01 (O.J.M., G.M.), and Conseil Régional d'Aquitaine (O.J.M., G.M.). The sequencing was performed at the Sequencing Facility of Bordeaux (Conseil Régional d'Aquitaine Grants 20030304002FA and 20040305003FA, and European Union, FEDER Grant 2003227). We thank N. Aubailly and her team for her assistance with mutant animals and D. Gonzales and her team for genotyping and sequencing. We also thank Danièle Verrier for her help with cell culture, Alexandre Brochard for his help with burst analysis, Dr. Karl Iremonger for discussion, and Aimée Ward for editing this manuscript.
References
- Antal M, Acuna-Goycolea C, Pressler RT, Blitz DM, Regehr WG. Cholinergic activation of M2 receptors leads to context-dependent modulation of feedforward inhibition in the visual thalamus. PLoS Biol. 2010;8:e1000348. doi: 10.1371/journal.pbio.1000348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bacci A, Huguenard JR, Prince DA. Long-lasting self-inhibition of neocortical interneurons mediated by endocannabinoids. Nature. 2004;431:312–316. doi: 10.1038/nature02913. [DOI] [PubMed] [Google Scholar]
- Bellocchio L, Lafenêtre P, Cannich A, Cota D, Puente N, Grandes P, Chaouloff F, Piazza PV, Marsicano G. Bimodal control of stimulated food intake by the endocannabinoid system. Nat Neurosci. 2010;13:281–283. doi: 10.1038/nn.2494. [DOI] [PubMed] [Google Scholar]
- Berghuis P, Rajnicek AM, Morozov YM, Ross RA, Mulder J, Urbán GM, Monory K, Marsicano G, Matteoli M, Canty A, Irving AJ, Katona I, Yanagawa Y, Rakic P, Lutz B, Mackie K, Harkany T. Hardwiring the brain: endocannabinoids shape neuronal connectivity. Science. 2007;316:1212–1216. doi: 10.1126/science.1137406. [DOI] [PubMed] [Google Scholar]
- Brenowitz S, David J, Trussell L. Enhancement of synaptic efficacy by presynaptic GABA(B) receptors. Neuron. 1998;20:135–141. doi: 10.1016/s0896-6273(00)80441-9. [DOI] [PubMed] [Google Scholar]
- Di Marzo V, Melck D, Bisogno T, De Petrocellis L. Endocannabinoids: endogenous cannabinoid receptor ligands with neuromodulatory action. Trends Neurosci. 1998;21:521–528. doi: 10.1016/s0166-2236(98)01283-1. [DOI] [PubMed] [Google Scholar]
- Foldy C, Neu A, Jones MV, Soltesz I. Presynaptic, activity-dependent modulation of cannabinoid type 1 receptor-mediated inhibition of GABA release. J Neurosci. 2006;26:1465–1469. doi: 10.1523/JNEUROSCI.4587-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldman-Rakic PS, Muly EC, 3rd, Williams GV. D(1) receptors in prefrontal cells and circuits. Brain Res Brain Res Rev. 2000;31:295–301. doi: 10.1016/s0165-0173(99)00045-4. [DOI] [PubMed] [Google Scholar]
- Groc L, Choquet D, Stephenson FA, Verrier D, Manzoni OJ, Chavis P. NMDA receptor surface trafficking and synaptic subunit composition are developmentally regulated by the extracellular matrix protein Reelin. J Neurosci. 2007;27:10165–10175. doi: 10.1523/JNEUROSCI.1772-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heifets BD, Castillo PE. Endocannabinoid signaling and long-term synaptic plasticity. Annu Rev Physiol. 2009;71:283–306. doi: 10.1146/annurev.physiol.010908.163149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kano M, Ohno-Shosaku T, Hashimotodani Y, Uchigashima M, Watanabe M. Endocannabinoid-mediated control of synaptic transmission. Physiol Rev. 2009;89:309–380. doi: 10.1152/physrev.00019.2008. [DOI] [PubMed] [Google Scholar]
- Katona I, Freund TF. Endocannabinoid signaling as a synaptic circuit breaker in neurological disease. Nat Med. 2008;14:923–930. doi: 10.1038/nm.f.1869. [DOI] [PubMed] [Google Scholar]
- Kreitzer AC, Carter AG, Regehr WG. Inhibition of interneuron firing extends the spread of endocannabinoid signaling in the cerebellum. Neuron. 2002;34:787–796. doi: 10.1016/s0896-6273(02)00695-5. [DOI] [PubMed] [Google Scholar]
- Losonczy A, Biro AA, Nusser Z. Persistently active cannabinoid receptors mute a subpopulation of hippocampal interneurons. Proc Natl Acad Sci U S A. 2004;101:1362–1367. doi: 10.1073/pnas.0304752101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marinelli S, Pacioni S, Cannich A, Marsicano G, Bacci A. Self-modulation of neocortical pyramidal neurons by endocannabinoids. Nat Neurosci. 2009;12:1488–1490. doi: 10.1038/nn.2430. [DOI] [PubMed] [Google Scholar]
- McCormick DA. Neurotransmitter actions in the thalamus and cerebral cortex and their role in neuromodulation of thalamocortical activity. Prog Neurobiol. 1992;39:337–388. doi: 10.1016/0301-0082(92)90012-4. [DOI] [PubMed] [Google Scholar]
- Monory K, Massa F, Egertová M, Eder M, Blaudzun H, Westenbroek R, Kelsch W, Jacob W, Marsch R, Ekker M, Long J, Rubenstein JL, Goebbels S, Nave KA, During M, Klugmann M, Wölfel B, Dodt HU, Zieglgänsberger W, Wotjak CT, et al. The endocannabinoid system controls key epileptogenic circuits in the hippocampus. Neuron. 2006;51:455–466. doi: 10.1016/j.neuron.2006.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulder J, Aguado T, Keimpema E, Barabas K, Ballester Rosado CJ, Nguyen L, Monory K, Marsicano G, Di sMarzo V, Hurd YL, Guillemot F, Mackie K, Lutz B, Guzmán M, Lu HC, Galve-Roperh I, Harkany T. Endocannabinoid signaling controls pyramidal cell specification and long-range axon patterning. Proc Natl Acad Sci U S A. 2008;105:8760–8765. doi: 10.1073/pnas.0803545105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novellino A, D'Angelo P, Cozzi L, Chiappalone M, Sanguineti V, Martinoia S. Connecting neurons to a mobile robot: an in vitro bidirectional neural interface. Comput Intell Neurosci. 2007;2007:12725. doi: 10.1155/2007/12725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliet SH, Baimoukhametova DV, Piet R, Bains JS. Retrograde regulation of GABA transmission by the tonic release of oxytocin and endocannabinoids governs postsynaptic firing. J Neurosci. 2007;27:1325–1333. doi: 10.1523/JNEUROSCI.2676-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pertwee RG. Inverse agonism and neutral antagonism at cannabinoid CB1 receptors. Life Sci. 2005;76:1307–1324. doi: 10.1016/j.lfs.2004.10.025. [DOI] [PubMed] [Google Scholar]
- Rijal SO, Gross GW. Dissociation constants for GABA(A) receptor antagonists determined with neuronal networks on microelectrode arrays. J Neurosci Methods. 2008;173:183–192. doi: 10.1016/j.jneumeth.2008.05.025. [DOI] [PubMed] [Google Scholar]
- Rubin JE, Hayes JA, Mendenhall JL, Del Negro CA. Calcium-activated nonspecific cation current and synaptic depression promote network-dependent burst oscillations. Proc Natl Acad Sci U S A. 2009;106:2939–2944. doi: 10.1073/pnas.0808776106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinagra M, Verrier D, Frankova D, Korwek KM, Blahos J, Weeber EJ, Manzoni OJ, Chavis P. Reelin, very-low-density lipoprotein receptor, and apolipoprotein E receptor 2 control somatic NMDA receptor composition during hippocampal maturation in vitro. J Neurosci. 2005;25:6127–6136. doi: 10.1523/JNEUROSCI.1757-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staley KJ, Longacher M, Bains JS, Yee A. Presynaptic modulation of CA3 network activity. Nat Neurosci. 1998;1:201–209. doi: 10.1038/651. [DOI] [PubMed] [Google Scholar]
- Stewart CV, Plenz D. Inverted-U profile of dopamine-NMDA-mediated spontaneous avalanche recurrence in superficial layers of rat prefrontal cortex. J Neurosci. 2006;26:8148–8159. doi: 10.1523/JNEUROSCI.0723-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turnbull L, Dian E, Gross G. The string method of burst identification in neuronal spike trains. J Neurosci Methods. 2005;145:23–35. doi: 10.1016/j.jneumeth.2004.11.020. [DOI] [PubMed] [Google Scholar]
- Volmer R, Prat CM, Le Masson G, Garenne A, Gonzalez-Dunia D. Borna disease virus infection impairs synaptic plasticity. J Virol. 2007;81:8833–8837. doi: 10.1128/JVI.00612-07. [DOI] [PMC free article] [PubMed] [Google Scholar]