

Abstract
Subanesthetic doses of NMDA receptor antagonist ketamine induce schizophrenia-like symptoms in humans and behavioral changes in rodents. Subchronic administration of ketamine leads to loss of parvalbumin-positive interneurons through reactive oxygen species (ROS), generated by the NADPH oxidase NOX2. However, ketamine induces very rapid alterations, in both mice and humans. Thus, we have investigated the role of NOX2 in acute responses to subanesthetic doses of ketamine. In wild-type mice, ketamine caused rapid (30 min) behavioral alterations, release of neurotransmitters, and brain oxidative stress, whereas NOX2-deficient mice did not display such alterations. Decreased expression of the subunit 2A of the NMDA receptor after repetitive ketamine exposure was also precluded by NOX2 deficiency. However, neurotransmitter release and behavioral changes in response to amphetamine were not altered in NOX2-deficient mice. Our results suggest that NOX2 is a major source of ROS production in the prefrontal cortex controlling glutamate release and associated behavioral alterations after acute ketamine exposure. Prolonged NOX2-dependent glutamate release may lead to neuroadaptative downregulation of NMDA receptor subunits.
Introduction
Alteration of glutamatergic neurotransmission is thought to be involved in the development of psychosis (Harte et al., 2007). This appears to be attributable to NMDA receptor (NMDAR) hypofunction, which decreases the activity of GABAergic interneurons, leading to an excessive glutamate release (Homayoun and Moghaddam, 2007) and subsequent cortical excitation, as well as schizophrenia-like behavioral and cognitive anomalies (Farber, 2003).
Studies on the effects of the NMDA receptor antagonists, such as ketamine and phencyclidine, support this theory (Olney et al., 1999). Controlled administration of ketamine in healthy volunteers leads to positive, negative, and cognitive symptoms similar to those observed in schizophrenic patients (Krystal et al., 2003). Subanesthetic ketamine administration also induces behavioral alterations in animals, and it is therefore used as a pharmacological animal model of schizophrenia (Bubeníková-Valesová et al., 2008; Gunduz-Bruce, 2009). Although psychotic symptoms are impossible to evaluate and replicate in rodents, alteration of motor behavior can be used as a read-out for the response to NMDA receptor antagonists (Krystal et al., 2003; Imre et al., 2006; Gunduz-Bruce, 2009). The effect of ketamine is mainly attributable to the activation of the prefrontal cortex and the limbic structures (Jentsch and Roth, 1999; Imre et al., 2006), in which increased levels of glutamate and dopamine have been detected after acute ketamine injections (Gunduz-Bruce, 2009). In addition, chronic exposure to low doses of ketamine leads to phenotypic changes in neurons, such as the loss of parvalbumin in GABAergic interneurons (Cochran et al., 2003; Kinney et al., 2006), also observed in schizophrenic patients (Lewis et al., 2005). Corroborating these results, genetic ablation of the NMDA receptor also confers schizophrenia-like phenotypes in mice (Gainetdinov et al., 2001; Belforte et al., 2010).
Emerging evidence indicates a contribution of reactive oxygen species (ROS) to the development of schizophrenia. Altered antioxidant defense and signs of oxidative insults are observed in patient samples (Yao et al., 2001). In this context, it has been shown that the activation of the ROS-producing NADPH oxidase NOX2 enzyme contributes to dysfunction of GABAergic interneurons after subchronic ketamine exposure in mice (Behrens et al., 2007, 2008). We have also recently demonstrated the involvement of NOX2 in behavioral and biochemical alterations caused by social isolation in rats (Schiavone et al., 2009). These two different animal models induce chronic neural stress and, in both cases, NOX2 upregulation appears to be responsible for the prolonged oxidative insult on GABAergic neurons. However, it has been reported that acute ketamine administration causes immediate formation of ROS (Zuo et al., 2007) and oxidative damage (de Oliveira et al., 2009). The mechanism of this rapid ROS generation is not known. Possible sources of ROS include NOX enzymes, which are constitutively expressed in the brain and are major generators of ROS in pathological conditions (Sorce and Krause, 2009).
The aim of this study was to understand how NOX2-dependent ROS production participates in the rapid modifications induced by acute ketamine administration. Our data show that the immediate behavioral and neurochemical abnormalities induced by ketamine are prevented in NOX2-deficient knock-out (KO) mice and that NOX2 is involved in neurotransmitter release.
Materials and Methods
Mice.
NOX2-deficient mice in the C57BL/6 background were purchased from The Jackson Laboratory. Mice were given access ad libitum to food and water in a quiet room at 25°C with a 12 h light/dark cycle. Mice were housed in groups of three to four. All experiments were performed using wild-type (WT) and KO NOX2 mice male littermates of 8–10 weeks of age. All experiments were performed according to institutional guidelines (Italian Legislative Decree 116, Official Journal of the Italian Republic, supplement 40, 18 February 1992; Ministerial Memorandum No. 8, Official Journal of the Italian Republic, 14 July 1994; EEC Council Directive 86/609, Official Journal Law 358, 1, December 12, 1987; Guide for the Care and Use of Laboratory Animals, U.S. National Research Council, 1996), and approved by the ethical committee and the veterinary office of the University of Foggia.
Behavioral analysis: open-field test.
The apparatus consisted of a circular arena, made of dark plastic located in a silent experimental room illuminated. Habituation trial was performed over 5 d before the test in the same experimental conditions. Animals were acclimated to the test room for 1 h before each test. The day of the test, mice were injected intraperitoneally with ketamine (30 mg/kg; Sigma-Aldrich), amphetamine (1 mg/kg; Sigma-Aldrich), or saline solution, as control. Drug doses were chosen according to previous reports (for ketamine, Irifune et al., 1991, 1998; Behrens et al., 2007; and for amphetamine, Sansone, 1980; Spielewoy et al., 2001; Coitinho et al., 2002). Behavioral test was performed 30 min and 4 h after injection. Motor activity was measured by placing the mouse into the center of the arena for a 20 min session. The scoring was performed using a specific software (Observer 3.0; Noldus Information Technology). To assess general locomotor activity, the following behavioral parameters (expressed as frequency in 20 min) were scored: number of square limit crossings with both forepaws, rearing (standing with the body inclined vertically, forequarters raised), total grooming activity consisting of face grooming (strokes along the snout), head washing (semicircular movements over the top of the head and behind the ears), and body grooming (body fur licking), and total sniffing (deep smelling activity both on the floor and on the walls of the arena). Behavioral test was performed by an investigator who was blind to the animal experimental group.
Immunohistochemistry.
Mice were perfused with a solution of 4% paraformaldehyde in PBS, pH 7.35, and brains were removed, paraffin embedded, and cut in 10-μm-thick sections.
Immunohistochemistry was performed as described previously (Schiavone et al., 2009). Briefly, after deparaffinization and heat-induced epitope retrieval, brain sections were incubated overnight at 4°C with one or two (for double staining) primary antibodies at the following concentrations: 8-hydroxy-2′-deoxyguanosine (8-OHdG), 1:10 (JaICA); NMDAR subunit 2A (NMDAR-2A), 1:400 (Abcam); and NMDAR-2B, 1:200 (Abcam). For 3,3′-diaminobenzidine (DAB) immunohistochemistry, sections were then incubated first for 1 h at room temperature with a biotinylated secondary antibody (Vector Laboratories) and then with the ABC complex solution (Vector Laboratories). Tissue-bound peroxidase was visualized using DAB (Sigma-Aldrich) and H2O2 (Sigma-Aldrich). For immunofluorescence detection, sections were incubated with specific fluorescent-labeled secondary antibody (1:1000, Alexa Fluor 488 or 555; Invitrogen). 4′,6′-Diamidino-2-phenylindole (DAPI) counterstaining was performed to visualize cell nuclei. Imaging was performed on an LSM 510 Meta confocal laser scanner mounted on an Axio Imager Z1 microscope (Carl Zeiss). Negative controls consisting of tissue incubated without primary antibodies were performed for each experiment (data not shown).
Real-time quantitative PCR.
Total RNA was isolated using RNeasy mini kit (Qiagen) according to the instructions of the manufacturer. Residual genomic DNA was removed using RNase-Free DNase set (Qiagen). Total RNA (1 μg) was reverse transcribed using the superscript II kit according to the instructions of the manufacturer (Invitrogen). Real-time quantitative PCR reactions were performed using Power SYBR Green PCR master mix (Applied Biosystems) and a Chromo 4TM Real-Time system (Bio-Rad). Quantification was performed at a threshold detection line (Ct value). The Ct value of each target genes (c-fos, parvalbumin) was normalized with relative levels of Rps9 (ribosomal protein S9) and Tbp (TATA-box binding protein) mRNAs used as housekeeping genes. Triplicates were performed for each condition. The list of the primers used is given in supplemental Table 1 (available at www.jneurosci.org as supplemental material).
Semiquantitative end-point PCR.
Extraction of RNA and reverse transcription were performed as described above. Semiquantitative end-point PCR was performed by determining the suitable number of PCR cycles giving linear cDNA amplification of each gene of interest using TaqDNA polymerase (Qiagen). Amplification of the housekeeping gene encoding the L32 ribosomal protein was used as control. The list of the primers used is given in supplemental Table 1 (available at www.jneurosci.org as supplemental material).
Microdialysis.
Mice were anesthetized intraperitoneally with 3.6 ml/kg Equithesin (composition: 1.2 g of sodium pentobarbital, 5.3 g of chloral hydrate, 2.7 g of MgSO4, 49.5 ml of propylene glycol, 12.5 ml of ethanol, and 58 ml of distilled water) and secured in a stereotaxic frame (David Kopf Instruments). The skin was shaved, disinfected, and cut with a sterile scalpel to expose the skull. A vertical dialysis probe was positioned in the prefrontal cortex of mice (Castane et al., 2008): from bregma, anteroposterior, +2.2 mm and mediolateral, ±0.2 mm; and from the skull surface, the extremity of the probe was pushed in dorsoventral, −3.4 mm. On the day of the experiment (20–24 h after implantation of the probes), microdialysis was performed in freely moving mice as described previously (Castane et al., 2008) by perfusing the fibers with an artificial CSF containing the following (in mm): 145 NaCl, 3 KCl, 1.26 CaCl2, 1 MgCl2, and 1.4 Na2HPO4 in distilled water; the solution was buffered at pH 7.3 with 0.6 mm NaH2PO4 and filtered (0.45 μm). The fibers were perfused at a constant flow rate of 1 μl/min using a CMA/100 microinjection pump (CMA Microdialysis). In all experiments, the microdialysis membrane was allowed to stabilize for 2 h at the flow rate of 1 μl/min without collecting samples. At the end of the stabilization period, samples were collected at 30 min intervals at a flow rate of 1 μl/min. Three baseline samples were collected to evaluate baseline release of neurotransmitters. After this time, ketamine (30 mg/kg, i.p.), amphetamine (1 mg/kg, i.p.), or vehicle (saline) injections were performed, and additional samples were collected. WT and KO NOX2 mice were treated and analyzed at the same time, but data in figures are presented as separate panels for better clarity. The position of the microdialysis probe was verified by histological procedures at the end of each experiment. Only mice in which probe tracks were exactly located in the target area were considered in Results.
Quantification of glutamate in the dialysate.
Glutamate concentrations were determined by HPLC using ODS-3 column (150 × 4.6 mm, 3 μm; INERTSIL) with fluorescence detection after derivatization with o-phthalaldehyde/mercaptopropionic acid (emission length, 4.60 nm; excitation length, 3.40 nm). The mobile phase gradient consisted of 50 mm sodium acetate buffer, pH 6.95, with methanol increasing linearly from 2 to 30% (v/v) over 40 min (Morrone et al., 2007). The flow rate was 0.5 ml/min. Results were analyzed by Borwin software (version 1.50; Jasco). Because no difference in extracellular basal levels of glutamate between experimental groups was detected (for original values and statistics, see supplemental Table 2, available at www.jneurosci.org as supplemental material), results were expressed as percentage of baseline.
Quantification of dopamine in the dialysate.
Dopamine concentrations were determined by HPLC using LC-18 DB column (150 × 4.6 mm, 5 μm; SUPELCOSIL) with ESA Coulometric detection (ESA) in oxidation/reduction mode (E1, +300 mV; E2, −300 mV). The mobile phase used consisted of 85 mm NaH2PO4, 1 mm SDS, 0.02 mm EDTA, 0.7 mm triethylamine, 15% acetonitrile, 13% methanol, solved in distilled water and buffered at pH 5.8 with 1N NaOH. The flow rate was 1 ml/min, and detection limit was ∼0.5 fmol of dopamine on column (signal-to-noise ratio of 2) (Bassareo and Di Chiara, 1997; Trabace et al., 2007). Results were analyzed by Chromeleon software (version 6.60; Dionex). Because no difference in extracellular basal levels of dopamine between experimental groups was detected (for original values and statistics, see supplemental Table 2, available at www.jneurosci.org as supplemental material), results were expressed as percentage of baseline.
Statistical analysis.
Data were analyzed using SigmaStat 3.1 software. The statistical tests (with F values and degrees of freedom when appropriate) are indicated in the figure legends. For all tests, p < 0.05 was taken as statistically significant. Results are expressed as means ± SEM.
Results
NOX2 deficiency prevents ketamine-induced behavioral abnormalities
An open-field test was performed 30 min after ketamine injection. Locomotor activity was evaluated as the frequency of crossings, rearings, grooming, and sniffings. In WT mice, all these parameters were markedly increased compared with saline control mice. In contrast, ketamine did not elicit similar responses in KO NOX2 mice because their locomotor behavior remained unaltered (Fig. 1). This absence of response by KO NOX2 mice was similarly observed at a later time point (4 h after ketamine injection) (supplemental Fig. 1, available at www.jneurosci.org as supplemental material), whereas behavioral abnormalities in WT mice could still be observed 4 h after ketamine injection.
Figure 1.

Behavioral alterations induced by ketamine exposure are prevented in NOX2-deficient mice. Thirty minutes after injection with ketamine (30 mg/kg, i.p.) or saline, mice were placed in the arena for open-field test. A–D, Bar graphs represent the frequency of grooming (A), rearing (B), crossing (C), and sniffing (D) recorded during the 20 min of the test in WT and KO NOX2 mice. For grooming: Ftreatment (tr) × Fgenotype (gen)(1,22) = 15.98, p < 0.001; Ftr(1,22) = 14.92, p < 0.001; Fgen(1,22) = 11.96, p < 0.01; not significant (NS): WT saline versus KO NOX2 saline, p = 0.742; KO NOX2 saline versus KO NOX2 ketamine, p = 0.928. For rearing: Ftr×gen(1,22) = 109.48, p < 0.001; Ftr(1,22) = 114.93, p < 0.001; Fgen(1,22) = 107.34, p < 0.001; NS: WT saline versus KO NOX2 saline, p = 0.950; KO NOX2 saline versus KO NOX2 ketamine, p = 0.864. For crossing: Ftr×gen(1,22) = 218.926, p < 0.001; Ftr(1,22) = 217.231, p < 0.001; Fgen(1,22) = 198.637, p < 0.001; NS: WT saline versus KO NOX2 saline, p = 0.669; KO NOX2 saline versus KO NOX2 ketamine, p = 0.970. For sniffing: Ftr×gen(1,22) = 23.76, p < 0.001; Ftr(1,22) = 38.26, p < 0.001; Fgen(1,22) = 27.63, p < 0.001. NS: WT saline versus KO NOX2 saline, p = 0.816; KO NOX2 saline versus KO NOX2 ketamine, p = 0.385. ***p < 0.001 WT ketamine versus WT saline and WT ketamine versus KO NOX2 ketamine using two-way ANOVA followed by Tukey's post hoc test (n = 5 WT saline; n = 9 WT ketamine; n = 4 KO NOX2 saline; n = 8 KO NOX2 ketamine).
Together, these results indicate that NOX2 is a key mediator of immediate ketamine-induced behavioral changes.
Stress-related markers after ketamine injection
Ketamine administration causes an increase in ROS production (Zuo et al., 2007) and oxidative damage (de Oliveira et al., 2009) in the brain. However, the source of the ROS generation is not known. Indirect markers of oxidative stress were measured 30 min after ketamine injection in WT and KO NOX2 mice. The staining for the DNA oxidation marker 8-OHdG was increased in the prefrontal cortex of WT mice and especially localized in cell nuclei. In contrast, no increase of 8-OHdG was detected in KO NOX2 mice (Fig. 2A–D). Consistent with the role of NOX2 as source of ROS in this condition, NOX2 and all the subunits required for its activity were detected in the prefrontal cortex (supplemental Fig. 2A,B, available at www.jneurosci.org as supplemental material). It is also known that ketamine injections leads to the upregulation of the transcription factor subunit c-fos (Farber, 2003), which is an early response gene known to be redox-sensitive (Nanduri et al., 2008). We have therefore quantified c-fos mRNA expression by real-time PCR. In WT mice, c-fos expression levels were increased in the prefrontal cortex after ketamine injection. In contrast, no change in c-fos expression was seen in mice lacking NOX2 (Fig. 2E). Under the same conditions, we have also measured the level of parvalbumin mRNA. Whereas subchronic exposure to ketamine induces a decrease in parvalbumin (Behrens et al., 2007), we did not detect a reduction in parvalbumin mRNA after acute ketamine exposure in neither control nor KO NOX2 mice (supplemental Fig. 3, available at www.jneurosci.org as supplemental material). Thus, c-fos expression is an early NOX2-dependent event in response to ketamine, whereas the decrease in parvalbumin appears to occur only at later time points (Behrens et al., 2007).
Figure 2.
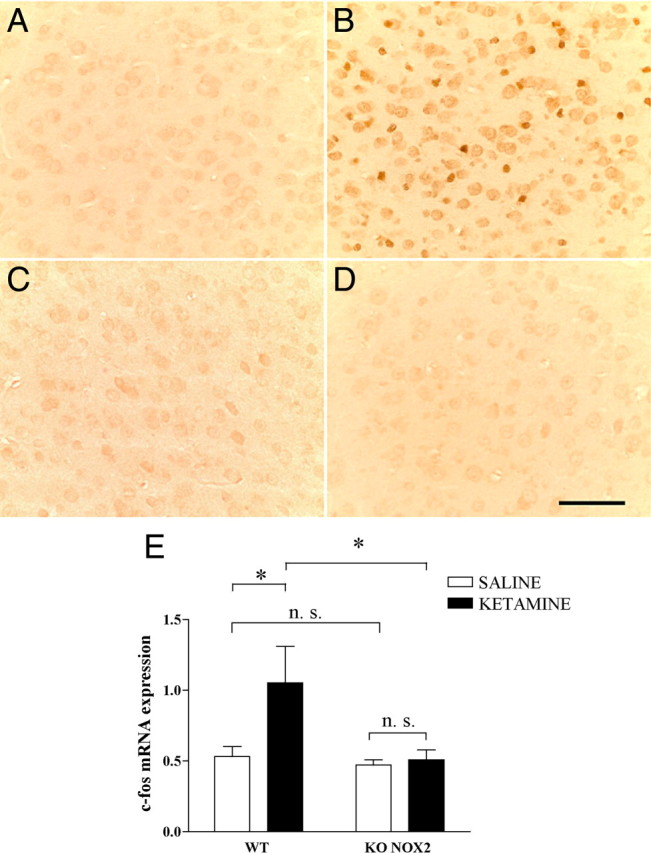
Stress markers are increased after ketamine injections in wild-type mice, and not in NOX2-deficient mice. A–D, Representative images of immunohistochemistry staining for 8-OHdG in the prefrontal cortex of WT (A, B) or KO NOX2 (C, D) mice treated with saline (A, C) or ketamine (B, D). n = 5 per condition. Scale bar, 65 μm. E, Real-time PCR quantification of the immediate early gene c-fos mRNA in prefrontal cortex after ketamine or saline injection in WT and KO NOX2 mice. Ftr×gen(1,14) = 1.976, p = 0.182; Ftr(1,14) = 5.653, p < 0.05; Fgen(1,14) = 3.463, p < 0.05. NS: WT saline versus KO NOX2 saline, p = 0.503; KO NOX2 saline versus KO NOX2 ketamine, p = 0.385. *p < 0.05 WT ketamine versus WT saline and WT ketamine versus KO NOX2 ketamine using two-way ANOVA followed by Tukey's post hoc test (n = 5 WT saline; n = 4 WT ketamine; n = 4 KO NOX2 saline; n = 5 KO NOX2 ketamine).
Ketamine-induced release of extracellular glutamate and dopamine depends on NOX2
Alterations of neurotransmitter levels in the cortex induced by ketamine are thought to determine psychotic symptoms (Gunduz-Bruce, 2009). To understand the cause of the different behavioral response between WT and KO NOX2 mice, we have performed microdialysis in freely moving animals to measure the extracellular concentration of neurotransmitters in mouse brain. Basal levels of glutamate were comparable in WT and KO NOX2 mice. After ketamine injection, an immediate increase in glutamate level was detected in the prefrontal cortex of WT mice. The elevated concentration was observed after 30 min and persisted during the subsequent 4 h from the injection (Fig. 3A). In contrast, ketamine did not elicit the same response in KO NOX2 mice because glutamate concentration remained at basal levels (Fig. 3B). Glutamatergic and dopaminergic systems are closely interconnected (Sesack et al., 2003), and it has been suggested that glutamate, once released, stimulates other non-NMDA glutamate receptors and thereby induces changes in dopamine levels (Olney et al., 1999). Therefore, we have also evaluated dopamine concentration. Similarly to glutamate, the levels of dopamine instantly increased after ketamine injection in WT brain (Fig. 3C) but remained at basal level in KO NOX2 mice (Fig. 3D). These results indicate that NOX2 influences the response to ketamine, by modulating glutamate and dopamine release.
Figure 3.

Glutamate and dopamine level elevation is prevented in NOX2-deficient mice. A–D, Time-dependent effect of ketamine or saline injection on extracellular glutamate (GLU; A, B) and dopamine (DA; C, D) levels was determined by microdialysis in the prefrontal cortex of WT (A, C) and KO NOX2 (B, D) mice. Data are expressed as the percentage of baseline (as described in Materials and Methods). E, F, Concentration of glutamate (E) and dopamine (F) in WT and KO NOX2 mice at basal level or 30 min after saline or ketamine injection. ***p < 0.001 using two-way ANOVA for repeated measures followed by Tukey's post hoc test (n = 6 WT saline and ketamine; n = 4 KO NOX2 saline and ketamine). For glutamate analysis: two-way ANOVA for repeated measures in KO NOX2 saline versus KO NOX2 ketamine: Ftr(1,66) = 2.259, p = 0.184; Ftime (t)(11,66) = 1.882, p = 0.058; Ftr×t(11,66) = 0.568, p = 0.847; in WT ketamine versus KO NOX2 ketamine: Fgen(1,88) = 10268.736, p < 0.001; Ft(11,88) = 345.017, p < 0.001; Ft×gen(11,88) = 327.118, p < 0.001; in WT ketamine versus KO NOX2 saline: Ftr(1,88) = 7557.223, p < 0.001; Ft(11,88) = 358.748, p < 0.001; Ft×tr(11,88) = 329.535, p < 0.001; in WT saline versus KO NOX2 ketamine: Ftr(1,88) = 15.944, p = 0.004; Ft(11,88) = 2.118, p = 0.027; Ftr×t(11,88) = 0.551, p = 0.863; however, Tukey's post hoc test did not reveal any significant difference for treatment and time; in WT saline versus KO NOX2 saline: Fgen(1,88) = 0.00160, p = 0.969; Ft(11,88) = 2.011, p = 0.036; Fgen×t(11,88) = 0.443, p = 0.932; however, Tukey's post hoc test did not reveal any significant difference for genotype and time; in WT ketamine versus WT saline: Ftr(1,110) = 16,702.806, p < 0.001; Ft(11,110) = 529.138, p < 0.001; Ftr×t(11,110) = 505.459, p < 0.001. For dopamine analysis: two-way ANOVA for repeated measures in KO NOX2 saline versus KO NOX2 ketamine: Ftr(1,66) = 0.00525, p = 0.945; Ft(11,66) = 0.633, p = 0.749; Ftr×t(11,66) = 0.440, p = 0.932; in WT ketamine versus KO NOX2 saline: Ftr(1,88) = 360.147, p < 0.001; Ft(11,88) = 16.156, p < 0.001; Ft×tr(11,88) = 14.890, p < 0.001; in WT saline versus KO NOX2 ketamine: Ftr(1,88) = 0.00714, p = 0.935; Ft(11,88) = 0.480, p = 0.911; Ftr×t(11,88) = 0.37, p = 0.965; in WT saline versus KO NOX2 saline: Fgen(1,88) = 0.00000392, p = 0.998; Ft(11,88) = 0.31, p = 0.982; Fgen×t(11,88) = 0.234, p = 0.994; in WT ketamine versus WT saline: Ftr(1,110) = 530.505, p < 0.001; Ft(11,110) = 20.573, p < 0.001; Ftr×t(11,110) = 18.919, p < 0.001; WT ketamine versus KO NOX2 ketamine: Fgen(1,88) = 331.637, p < 0.001; Ft(11,88) = 16.311, p < 0.001; Ft×gen(11,88) = 15.508, p < 0.001.
Amphetamine induces similar response in WT and KO NOX2 mice
To verify whether the lack of neurotransmitter increase after ketamine injection was attributable to a general defect in KO NOX2 mice, the response to amphetamine, another psychostimulant drug, was evaluated. In fact, amphetamine is known to induce a rapid efflux of dopamine from the ventral tegmental area toward the prefrontal cortex, in which subsequent glutamate is similarly released (Sesack et al., 2003; Sulzer et al., 2005). As illustrated in Figure 4, A and B, amphetamine injection caused a large increase in dopamine levels in both WT and KO NOX2 mice. Consequently, glutamate concentration was also elevated (Fig. 4C,D). Thus, NOX2-dependent alteration of glutamatergic and dopaminergic release is a specific response to ketamine. In addition, we have evaluated the behavioral alterations induced by amphetamine. In agreement with the microdialysis data, WT and KO NOX2 mice displayed a similar increase of locomotor activity after amphetamine injection (Fig. 5). Hence, NOX2 deficiency does not prevent behavioral changes in response to amphetamine.
Figure 4.
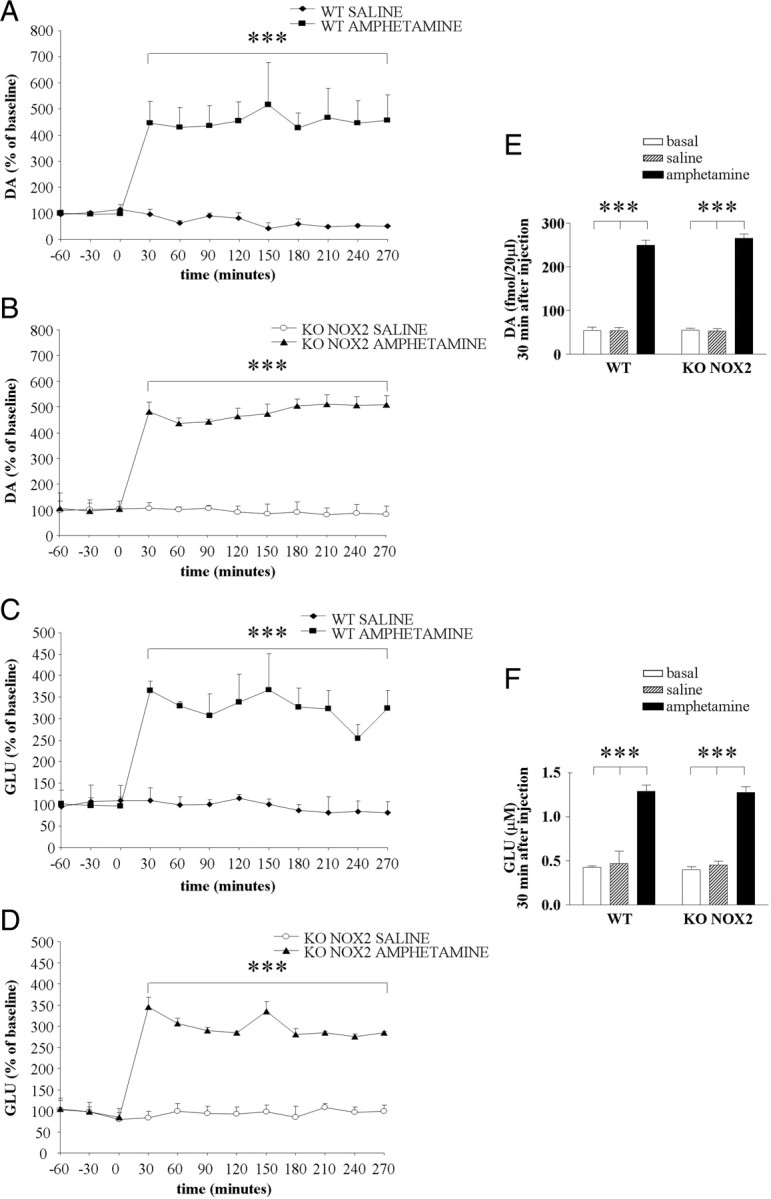
Neurotransmitter level elevation is similar in wild-type and NOX2-deficient mice after amphetamine exposure. A–D, Time-dependent effect of amphetamine or saline injection on extracellular dopamine (DA; A, B) and glutamate (GLU; C, D) levels was determined by microdialysis in the prefrontal cortex of WT (A, C) and KO NOX2 (B, D) mice. Data are expressed as the percentage of baseline (as described in Materials and Methods). E, F, Concentration of dopamine (E) and glutamate (F) in WT and KO NOX2 mice at basal level or 30 min after saline or amphetamine injection. ***p < 0.001 using two-way ANOVA for repeated measures followed by Tukey's post hoc test (n = 4 WT saline; n = 4 WT amphetamine; n = 4 KO NOX2 saline; n = 4 KO NOX2 amphetamine). For dopamine analysis: two-way ANOVA for repeated measures in KO NOX2 saline versus KO NOX2 amphetamine: Ftr(1,66) = 179.126, p < 0.001; Ft(11,66) = 62.712, p < 0.001; Ftr×t(11,66) = 63.847, p < 0.001; in WT amphetamine versus KO NOX2 amphetamine: Fgen(1,66) = 0.282, p = 0.615; Ft(11,66) = 62.241, p < 0.001; Ft×gen(11,66) = 0.413, p = 0.945; however, Tukey's post hoc test did not reveal any difference for time; in WT amphetamine versus KO NOX2 saline: Ftr(1,66) = 28.066, p = 0.002; Ft(11,66) = 18.176, p < 0.001; Ft×tr(11,66) = 18.651, p < 0.001; in WT saline versus KO NOX2 amphetamine: Ftr(1,66) = 198.908, p < 0.001; Ft(11,66) = 52.993, p < 0.001; Ftr×t(11,66) = 63.514, p < 0.001; in WT saline versus KO NOX2 saline: Fgen(1,66) = 0.239, p = 0.642; Ft(11,66) = 1.854, p = 0.062; Fgen×t(11,66) = 0.753, p = 0.684; in WT amphetamine versus WT saline: Ftr(1,66) = 30.049, p = 0.002; Ft(11,66) = 16.115, p < 0.001; Ftr×t(11,66) = 19.754, p < 0.001. For glutamate analysis: two-way ANOVA for repeated measures in KO NOX2 saline versus KO NOX2 amphetamine: Ftr(1,55) = 421.188, p ≤ 0.001; Ft(11,55) = 14.986, p ≤ 0.001; Ftr×t(11,55) = 14.751, p ≤ 0.001; in WT amphetamine versus KO NOX2 amphetamine: Fgen(1,55) = 0.490, p = 0.515; Ft(11,55) = 20.271, p < 0.001; Ft×gen(11,55) = 0.246, p < 0.993; however, Tukey's post hoc test did not reveal any difference for time; in WT amphetamine versus KO NOX2 saline: Ftr(1,66) = 42.943, p < 0.001; Ft(11,66) = 7.117, p < 0.001; Ft×tr(11,66) = 7.061, p < 0.001; in WT saline versus KO NOX2 amphetamine: Ftr(1,55) = 108.426, p < 0.001; Ft(11,55) = 9.661, p ≤ 0.001; Ftr×t(11,55) = 10.743, p ≤ 0.001; in WT saline versus KO NOX2 saline: Fgen(1,66) = 0.0686, p = 0.802; Ft(11,66) = 0.143, p = 0.999; Fgen×t(11,66) = 0.362, p = 0.966; in WT amphetamine versus WT saline: Ftr(1,66) = 35.877, p < 0.001; Ft(11,66) = 6.078, p < 0.001; Ftr×t(11,66) = 6.594, p < 0.001.
Figure 5.

Behavioral alterations are similar in wild-type and NOX2-deficient mice after amphetamine exposure. Thirty minutes after injection with amphetamine (1 mg/kg, i.p.) or saline, mice were placed in the arena for open-field test. A–D, Bar graphs represent the frequency of groomings (A), rearings (B), crossings (C), and sniffings (D) recorded during the 20 min of the test in WT and KO NOX2 mice. For groomings: Ftr×gen(1,11) = 2.299, p = 0.147; Ftr(1,11) = 415.537, p < 0.001; Fgen(1,11) = 1.123, p = 0.312. ***p < 0.001 WT saline versus WT amphetamine and KO NOX2 saline versus KO NOX2 amphetamine; NS: WT saline versus KO NOX2 saline, p = 0.837; WT amphetamine versus KO NOX2 amphetamine, p = 1.000. For rearings: Ftr×gen(1,11) = 0.363, p = 0.703; Ftr(1,11) = 21.464, p < 0.001; Fgen(1,11) = 0.0546, p = 0.819; **p < 0.01 WT saline versus WT amphetamine; *p < 0.05 KO NOX2 saline versus KO NOX2 amphetamine; NS: WT saline versus KO NOX2 saline, p = 0.485; WT amphetamine versus KO NOX2 amphetamine, p = 0.862. For crossings: Ftr×gen(1,11) = 0.00361, p = 0.996; Ftr(1,11) = 13.164, p = 0.001; Fgen(1,11) = 0.114, p < 0.741; **p < 0.01 WT saline versus WT amphetamine; *p < 0.05 KO NOX2 saline versus KO NOX2 amphetamine; NS: WT saline versus KO NOX2 saline, p = 0.796; WT amphetamine versus KO NOX2 amphetamine, p = 0.854. For sniffings: Ftr×gen(1,11) = 1.028, p = 0.327; Ftr(1,11) = 20.887, p < 0.001; Fgen(1,11) = 0.785, p = 0.390 NS: WT saline versus KO NOX2 saline, p = 0.927; KO NOX2 saline versus KO NOX2 ketamine, p = 0.211. ***p < 0.001; **p < 0.01; *p < 0.05 using two-way ANOVA followed by Tukey's post hoc test (n = 5 WT saline; n = 5 WT amphetamine; n = 4 KO NOX2 saline; n = 5 KO NOX2 amphetamine).
Downregulation of the NMDA receptor subunit 2A after repeated ketamine injections is prevented in KO NOX2 mice
Excessive glutamate increase after repetitive administration of ketamine can lead to the downregulation of postsynaptic receptors. Therefore, we have analyzed the presence of NMDAR-2A and NMDAR-2B in the prefrontal cortex and in the posterior cingulate cortex. Both of these brain regions are subject to alterations in response to prolonged ketamine exposure (Ellison, 1995; Olney et al., 1999; Tsai and Coyle, 2002). As shown in Figure 6, WT mice displayed reduced expression of the NMDAR-2A after repeated injections of ketamine compared with saline (Fig. 6, A vs B and E vs F). In contrast, KO NOX2 mice displayed similar immunostaining for NMDAR-2A after ketamine or saline injections (Fig. 6, C vs D and G vs H). The reduction of NMDAR-2A immunoreactivity was not associated with apparent morphological alterations in cortex layers or signs of cellular death, as indicated by DAPI staining (Fig. 6A–H). The expression of the NMDAR-2B was also evaluated, but its expression was not different in WT and KO NOX2 mice after ketamine injection in both the prefrontal cortex and the posterior cingulate cortex (Fig. 6I–P). Thus, repeated exposure to ketamine elicits alterations of the NMDA receptor, which is mediated by NOX2.
Figure 6.
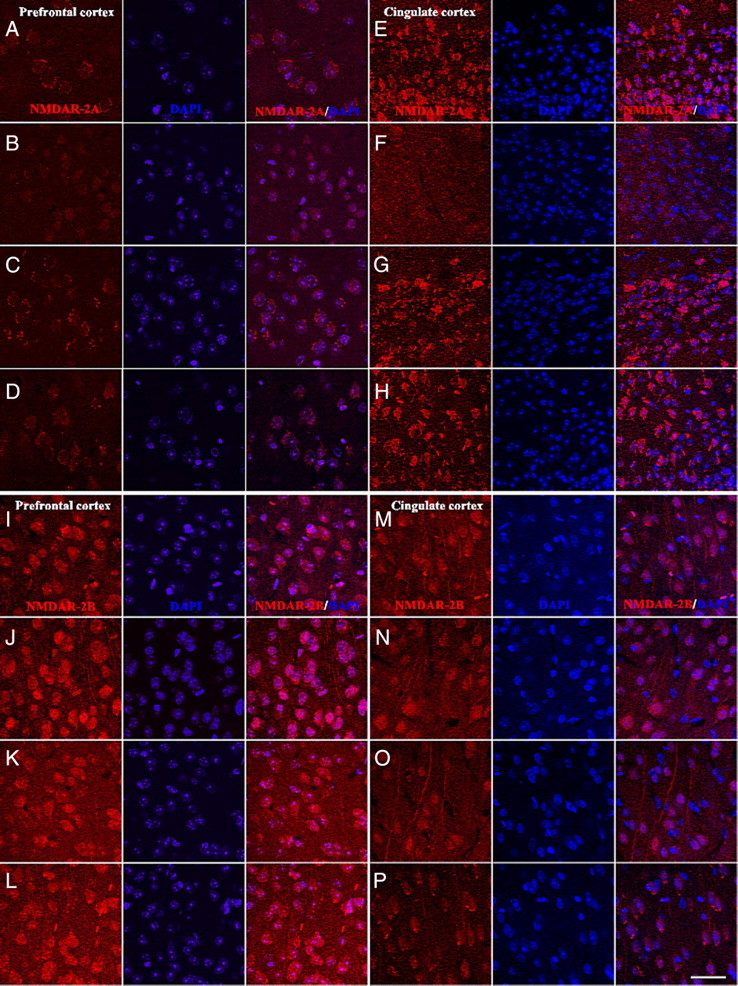
Expression of the NMDAR-2A and NMDAR-2B in wild-type and NOX2-deficient mice after repeated ketamine injections. Mice were treated with ketamine two times within an interval of 24 h and killed 18 h after the last injection. A–D, Representative images of immunohistochemistry for NMDAR-2A protein in the prefrontal cortex. NMDAR-2A staining in wild-type (A, B) and NOX2-deficient (C, D) mice treated with saline (A, C) or ketamine (B, D). n = 4 per group. E–H, Representative images of immunohistochemistry for NMDAR-2A protein in the posterior cingulate cortex. NMDAR-2A staining in wild-type (E, F) and NOX2-deficient (G, H) mice treated with saline (E, G) or ketamine (F, H). n = 4 per group. I–L, Representative images of immunohistochemistry for NMDAR-2B protein in the prefrontal cortex. NMDAR-2B staining in wild-type (I, J) and NOX2-deficient (K, L) mice treated with saline (I, K) or ketamine (J, L). n = 4 per group. M–P, Representative images of immunohistochemistry for NMDAR-2B protein in the posterior cingulate cortex. NMDAR-2B staining in wild-type (M, N) and NOX2-deficient (O, P) mice treated with saline (M, O) or ketamine (N, P). n = 4 per group. NMDAR-2A/DAPI, NMDAR-2B/DAPI, merged images for NMDAR-2A or NMDAR-2B immunoreactivity and DAPI staining. Scale bar, 40 μm.
Discussion
In this study, the involvement of NOX2 on acute behavioral and neurochemical effects elicited by ketamine administration was analyzed. Our results show that NOX2-deficient mice are completely protected from the behavioral alterations induced by ketamine. In addition, they do not display signs of oxidative stress in the brain and increased extracellular levels of glutamate and dopamine after ketamine administration. Thus, our data suggest a critical role of NOX2 activation in the development of psychotic symptoms after ketamine administration, through a direct effect on glutamate and dopamine elevation.
The presence of NOX2 in the brain has been widely documented. In particular, NOX2 is expressed in microglia, astrocytes, and neurons (Sorce and Krause, 2009). In neutrophils, NOX2 activity is tightly regulated and, during activation, requires the phosphorylation of specific cytosolic subunits and their translocation to the membrane to generate ROS (Bedard and Krause, 2007). The presence in the brain of all the cytosolic subunits required for NOX2 activation suggests that formation of the enzymatic complex can rapidly occur after ketamine exposure. Signs of oxidative stress were observed only in the brain of wild-type mice. Because behavioral alterations do not occur in NOX2-deficient mice, it appears that NOX2 can rapidly generate ROS in response to ketamine. Basically, the ketamine-induced NOX2 activation could be either a direct consequence of NMDA receptor inhibition or attributable to another pathway. The fact that two NMDA receptor antagonists, ketamine and dizocilpine [(+)-5-methyl-10,11-dihydro-5H-dibenzo [a,d] cyclohepten-5,10-imine maleate], both induce cerebral ROS generation during injection in rats (de Oliveira et al., 2009) and mice (Zuo et al., 2007) support the former possibility. However, given the lack of cellular studies specifically addressing this issue, it is, at this point, difficult to exclude that ketamine activates NOX2 through other mechanisms.
There are conflicting data in the literature concerning the duration of ketamine-induced behavioral alterations. Short-term (<2 h) behavioral changes in response to ketamine have been described previously (Hayase et al., 2006), whereas other observations suggest a markedly longer persistence (Uchihashi et al., 1994; Irifune et al., 1998). In our model, 4 h after ketamine injection, wild-type mice still displayed a marked enhancement of crossing, grooming, rearing, and sniffing. This is consistent with the prolonged elevations of neurotransmitters observed in our study.
ROS are important signaling molecules involved in physiological mechanisms in the brain, such as synaptic plasticity and memory (Kishida and Klann, 2007). In particular, ROS are able to modulate neurotransmission in cerebral circuitry. For example, the glutamate and GABA-controlled release of dopamine is dependent on hydrogen peroxide produced by mitochondria in the striatum (Avshalumov et al., 2003; Bao et al., 2009). Therefore, it is conceivable that one function of NOX2 in the brain is to produce ROS for the control of specific synaptic neurotransmission. Basal levels of glutamate and dopamine are similar in wild-type and NOX2-deficient mice, suggesting that absence of NOX2 does not impair normal synaptic activity. However, the fact that ketamine response is prevented in NOX2-deficient mice argues in favor of a NOX2-dependent ROS production that mediates neurochemical and behavioral alterations. This NOX2-dependent mechanism is specific for ketamine because the response to amphetamine in NOX2-deficient mice was similar to wild-type mice. Corroborating our results, Kishida et al. (2006) found an intact basal synaptic activity after pharmacological inhibition or genetic ablation of NOX2 but impairment in NMDA receptor-dependent long-term potentiation and memory formation, which require an increased release of glutamate (Kishida et al., 2006).
In addition to an early effect on the development of psychosis, it is tempting to speculate that NOX2-dependent elevation of glutamate levels induced by ketamine determines long-term alteration on neuronal connectivity. Indeed, the inhibition of GABAergic neurons by NMDA receptor antagonism eventually leads to their hyperstimulation because of the disinhibited glutamatergic inputs. This excessive stimulation of postsynaptic neurons is known to induce neurochemical and morphological changes, similar to those observed in schizophrenic patients (Farber, 2003). In this context, our data show that a repeated exposure to ketamine leads to a decrease in NMDA receptor 2A expression, which is prevented in NOX2-deficient mice. The downregulation of the NMDA receptor subunit 2A, but not 2B, is not specific for interneurons but likely occurs also in other neuronal subtypes. The downregulation of NMDA receptor subunit 2A is possibly attributable to sustained excessive glutamate concentration seen in wild-type mice. In adults, NMDA receptor 2A and 2B subunits are mainly expressed in cortex and hippocampus, in which they regulate numerous neuronal functions and, in particular, synaptic plasticity (Yashiro and Philpot, 2008). NMDA receptor subunit 2A is mainly localized in synaptic sites, whereas the 2B subunit is more expressed in extrasynaptic sites, and alterations of the NMDAR-2A/2B ratio influence neuronal activity and modify cortical functions (Yashiro and Philpot, 2008). The NMDA receptor subunit 2A is prone to adaptive responses in different models of glutamate and NMDA overstimulation (Wu et al., 2005; Gascón et al., 2008). Also, the NMDAR-2A subunit has relevant features for a role in ketamine-induced neurochemical alterations: (1) decrease of parvalbumin expression after prolonged ketamine exposure is specifically attributable to blockade of the NMDAR-2A subunit (Kinney et al., 2006), (2) the ratio of NMDAR-2A/NMDAR-2B subunits in GABAergic interneurons, specifically in parvalbumin-positive interneurons, is higher than in neighboring pyramidal neurons (Kinney et al., 2006; Xi et al., 2009), and (3) the NMDA receptors containing the 2A subunit contribute to the maintenance of parvalbumin and GAD67 phenotype in interneurons (Kinney et al., 2006). Thus, the observed adaptive reduction of NMDAR-2A could explain the loss of parvalbumin, observed after subchronic ketamine exposure (Behrens et al., 2008) or persistent social isolation rearing (Schiavone et al., 2009). In addition, lower density of NMDA receptor subunit 2A, but not 2B has been described in postmortem brain samples of patients with schizophrenia (Mechri et al., 2001; Beneyto and Meador-Woodruff, 2008).
The fact that NOX2 inhibition prevents both immediate and adaptive behavioral and neurochemical effects of ketamine raises the question whether a pharmacological inhibition of NOX2 represents a valuable therapeutic approach for schizophrenia. Indeed, decreasing the effect of NMDA receptor antagonists have been proposed to be a key approach for identifying such novel therapies (Krystal et al., 2003). Patients with genetic alteration of NOX2 present decreased levels of specific oxidative stress markers, such as isoprostanes (Violi et al., 2006, 2009). The same markers of oxidative stress are significantly increased in physiological fluids of schizophrenic patients (Dietrich-Muszalska and Olas, 2009). Even a single episode of psychosis induces such an increase (Mahadik et al., 1998; Khan et al., 2002; Zhang et al., 2009), indicating that oxidative damage occurs very early in the course of schizophrenia. The present study suggests that NOX2 is the source of this early generation of ROS, which is associated with the onset and the progression of schizophrenia. Therefore, targeting NOX2 in the CNS could be useful to prevent the development of the disease. A possible side effect of prolonged NOX2 inhibition would be reducing the ability of the neutrophils to kill some microorganisms and the development of local hyperinflammatory reactions (Schäppi et al., 2008). Indeed, the complete absence of NOX2 function results in chronic granulomatous disease (CGD), a hereditary disease characterized by the development of granulomas and by a susceptibility to certain fungal and bacterial infections (Schäppi et al., 2008). However, CGD patients in which only 5–10% of neutrophils generate ROS do not develop obvious symptoms (Kume and Dinauer, 2000). Thus, it appears that there is a good therapeutic window for such a putative therapy. However, although NOX enzymes are increasingly recognized as therapeutic targets, no specific NOX2 inhibitor is available so far for use in humans (Jaquet et al., 2009).
Although ketamine represents mostly a model of psychosis, there is also an interesting connection with depression. A single subanesthetic dose of ketamine has rapid and robust antidepressant effects in depressed patients, which might be attributable to ketamine-induced glutamate release (Hashimoto, 2009; Skolnick, 2009). Similar effects of ketamine have been observed in mouse models of depression (Maeng and Zarate, 2007; Maeng et al., 2008; Mathew et al., 2008). The role of NOX2 activation in the antidepressant effect of ketamine remains to be solved. Future research should also aim at understanding whether NOX2 controls glutamate release in physiological events, e.g., learning, and other pathological processes, such as neurodegenerative disorders (Sorce and Krause, 2009).
Footnotes
This work was funded by Swiss National Foundation Grant 125115 (Division Biologie et Médecine) (K.H.K.) and by grants from the Italian PRIN 2007 (L.T.). We thank Stéphanie Julien, Graziella Chakroun-Ciglie, and Olivier Plastre for excellent technical assistance.
References
- Avshalumov MV, Chen BT, Marshall SP, Peña DM, Rice ME. Glutamate-dependent inhibition of dopamine release in striatum is mediated by a new diffusible messenger, H2O2. J Neurosci. 2003;23:2744–2750. doi: 10.1523/JNEUROSCI.23-07-02744.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao L, Avshalumov MV, Patel JC, Lee CR, Miller EW, Chang CJ, Rice ME. Mitochondria are the source of hydrogen peroxide for dynamic brain-cell signaling. J Neurosci. 2009;29:9002–9010. doi: 10.1523/JNEUROSCI.1706-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bassareo V, Di Chiara G. Differential influence of associative and nonassociative learning mechanisms on the responsiveness of prefrontal and accumbal dopamine transmission to food stimuli in rats fed ad libitum. J Neurosci. 1997;17:851–861. doi: 10.1523/JNEUROSCI.17-02-00851.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bedard K, Krause KH. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol Rev. 2007;87:245–313. doi: 10.1152/physrev.00044.2005. [DOI] [PubMed] [Google Scholar]
- Behrens MM, Ali SS, Dao DN, Lucero J, Shekhtman G, Quick KL, Dugan LL. Ketamine-induced loss of phenotype of fast-spiking interneurons is mediated by NADPH-oxidase. Science. 2007;318:1645–1647. doi: 10.1126/science.1148045. [DOI] [PubMed] [Google Scholar]
- Behrens MM, Ali SS, Dugan LL. Interleukin-6 mediates the increase in NADPH-oxidase in the ketamine model of schizophrenia. J Neurosci. 2008;28:13957–13966. doi: 10.1523/JNEUROSCI.4457-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belforte JE, Zsiros V, Sklar ER, Jiang Z, Yu G, Li Y, Quinlan EM, Nakazawa K. Postnatal NMDA receptor ablation in corticolimbic interneurons confers schizophrenia-like phenotypes. Nat Neurosci. 2010;13:76–83. doi: 10.1038/nn.2447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beneyto M, Meador-Woodruff JH. Lamina-specific abnormalities of NMDA receptor-associated postsynaptic protein transcripts in the prefrontal cortex in schizophrenia and bipolar disorder. Neuropsychopharmacology. 2008;33:2175–2186. doi: 10.1038/sj.npp.1301604. [DOI] [PubMed] [Google Scholar]
- Bubeníková-Valesová V, Horácek J, Vrajová M, Höschl C. Models of schizophrenia in humans and animals based on inhibition of NMDA receptors. Neurosci Biobehav Rev. 2008;32:1014–1023. doi: 10.1016/j.neubiorev.2008.03.012. [DOI] [PubMed] [Google Scholar]
- Castane A, Artigas F, Bortolozzi A. The absence of 5-HT(1A) receptors has minor effects on dopamine but not serotonin release evoked by MK-801 in mice prefrontal cortex. Psychopharmacology (Berl) 2008;200:281–290. doi: 10.1007/s00213-008-1205-9. [DOI] [PubMed] [Google Scholar]
- Cochran SM, Kennedy M, McKerchar CE, Steward LJ, Pratt JA, Morris BJ. Induction of metabolic hypofunction and neurochemical deficits after chronic intermittent exposure to phencyclidine: differential modulation by antipsychotic drugs. Neuropsychopharmacology. 2003;28:265–275. doi: 10.1038/sj.npp.1300031. [DOI] [PubMed] [Google Scholar]
- Coitinho AS, Dietrich MO, Hoffmann A, Dall'Igna OP, Souza DO, Martins VR, Brentani RR, Izquierdo I, Lara DR. Decreased hyperlocomotion induced by MK-801, but not amphetamine and caffeine in mice lacking cellular prion protein (PrP(C)) Brain Res Mol Brain Res. 2002;107:190–194. doi: 10.1016/s0169-328x(02)00526-0. [DOI] [PubMed] [Google Scholar]
- de Oliveira L, Spiazzi CM, Bortolin T, Canever L, Petronilho F, Mina FG, Dal-Pizzol F, Quevedo J, Zugno AI. Different sub-anesthetic doses of ketamine increase oxidative stress in the brain of rats. Prog Neuropsychopharmacol Biol Psychiatry. 2009;33:1003–1008. doi: 10.1016/j.pnpbp.2009.05.010. [DOI] [PubMed] [Google Scholar]
- Dietrich-Muszalska A, Olas B. Isoprostenes as indicators of oxidative stress in schizophrenia. World J Biol Psychiatry. 2009;10:27–33. doi: 10.1080/15622970701361263. [DOI] [PubMed] [Google Scholar]
- Ellison G. The N-methyl-d-aspartate antagonists phencyclidine, ketamine and dizocilpine as both behavioral and anatomical models of the dementias. Brain Res Brain Res Rev. 1995;20:250–267. doi: 10.1016/0165-0173(94)00014-g. [DOI] [PubMed] [Google Scholar]
- Farber NB. The NMDA receptor hypofunction model of psychosis. Ann NY Acad Sci. 2003;1003:119–130. doi: 10.1196/annals.1300.008. [DOI] [PubMed] [Google Scholar]
- Gainetdinov RR, Mohn AR, Caron MG. Genetic animal models: focus on schizophrenia. Trends Neurosci. 2001;24:527–533. doi: 10.1016/s0166-2236(00)01886-5. [DOI] [PubMed] [Google Scholar]
- Gascón S, Sobrado M, Roda JM, Rodríguez-Peña A, Díaz-Guerra M. Excitotoxicity and focal cerebral ischemia induce truncation of the NR2A and NR2B subunits of the NMDA receptor and cleavage of the scaffolding protein PSD-95. Mol Psychiatry. 2008;13:99–114. doi: 10.1038/sj.mp.4002017. [DOI] [PubMed] [Google Scholar]
- Gunduz-Bruce H. The acute effects of NMDA antagonism: from the rodent to the human brain. Brain Res Rev. 2009;60:279–286. doi: 10.1016/j.brainresrev.2008.07.006. [DOI] [PubMed] [Google Scholar]
- Harte MK, Powell SB, Swerdlow NR, Geyer MA, Reynolds GP. Deficits in parvalbumin and calbindin immunoreactive cells in the hippocampus of isolation reared rats. J Neural Transm. 2007;114:893–898. doi: 10.1007/s00702-007-0627-6. [DOI] [PubMed] [Google Scholar]
- Hashimoto K. Emerging role of glutamate in the pathophysiology of major depressive disorder. Brain Res Rev. 2009;61:105–123. doi: 10.1016/j.brainresrev.2009.05.005. [DOI] [PubMed] [Google Scholar]
- Hayase T, Yamamoto Y, Yamamoto K. Behavioral effects of ketamine and toxic interactions with psychostimulants. BMC Neurosci. 2006;7:25. doi: 10.1186/1471-2202-7-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Homayoun H, Moghaddam B. NMDA receptor hypofunction produces opposite effects on prefrontal cortex interneurons and pyramidal neurons. J Neurosci. 2007;27:11496–11500. doi: 10.1523/JNEUROSCI.2213-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imre G, Fokkema DS, Den Boer JA, Ter Horst GJ. Dose-response characteristics of ketamine effect on locomotion, cognitive function and central neuronal activity. Brain Res Bull. 2006;69:338–345. doi: 10.1016/j.brainresbull.2006.01.010. [DOI] [PubMed] [Google Scholar]
- Irifune M, Shimizu T, Nomoto M. Ketamine-induced hyperlocomotion associated with alteration of presynaptic components of dopamine neurons in the nucleus accumbens of mice. Pharmacol Biochem Behav. 1991;40:399–407. doi: 10.1016/0091-3057(91)90571-i. [DOI] [PubMed] [Google Scholar]
- Irifune M, Sato T, Kamata Y, Nishikawa T, Nomoto M, Fukuda T, Kawahara M. Inhibition by diazepam of ketamine-induced hyperlocomotion and dopamine turnover in mice. Can J Anaesth. 1998;45:471–478. doi: 10.1007/BF03012584. [DOI] [PubMed] [Google Scholar]
- Jaquet V, Scapozza L, Clark R, Krause KH, Lambeth JD. Small molecule NOX inhibitors: ROS-generating NADPH oxidases as therapeutic targets. Antioxid Redox Signal. 2009;11:2535–2552. doi: 10.1089/ars.2009.2585. [DOI] [PubMed] [Google Scholar]
- Jentsch JD, Roth RH. The neuropsychopharmacology of phencyclidine: from NMDA receptor hypofunction to the dopamine hypothesis of schizophrenia. Neuropsychopharmacology. 1999;20:201–225. doi: 10.1016/S0893-133X(98)00060-8. [DOI] [PubMed] [Google Scholar]
- Khan MM, Evans DR, Gunna V, Scheffer RE, Parikh VV, Mahadik SP. Reduced erythrocyte membrane essential fatty acids and increased lipid peroxides in schizophrenia at the never-medicated first-episode of psychosis and after years of treatment with antipsychotics. Schizophr Res. 2002;58:1–10. doi: 10.1016/s0920-9964(01)00334-6. [DOI] [PubMed] [Google Scholar]
- Kinney JW, Davis CN, Tabarean I, Conti B, Bartfai T, Behrens MM. A specific role for NR2A-containing NMDA receptors in the maintenance of parvalbumin and GAD67 immunoreactivity in cultured interneurons. J Neurosci. 2006;26:1604–1615. doi: 10.1523/JNEUROSCI.4722-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kishida KT, Klann E. Sources and targets of reactive oxygen species in synaptic plasticity and memory. Antioxid Redox Signal. 2007;9:233–244. doi: 10.1089/ars.2007.9.ft-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kishida KT, Hoeffer CA, Hu D, Pao M, Holland SM, Klann E. Synaptic plasticity deficits and mild memory impairments in mouse models of chronic granulomatous disease. Mol Cell Biol. 2006;26:5908–5920. doi: 10.1128/MCB.00269-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krystal JH, D'Souza DC, Mathalon D, Perry E, Belger A, Hoffman R. NMDA receptor antagonist effects, cortical glutamatergic function, and schizophrenia: toward a paradigm shift in medication development. Psychopharmacology (Berl) 2003;169:215–233. doi: 10.1007/s00213-003-1582-z. [DOI] [PubMed] [Google Scholar]
- Kume A, Dinauer MC. Gene therapy for chronic granulomatous disease. J Lab Clin Med. 2000;135:122–128. doi: 10.1067/mlc.2000.104458. [DOI] [PubMed] [Google Scholar]
- Lewis DA, Hashimoto T, Volk DW. Cortical inhibitory neurons and schizophrenia. Nat Rev Neurosci. 2005;6:312–324. doi: 10.1038/nrn1648. [DOI] [PubMed] [Google Scholar]
- Maeng S, Zarate CA., Jr The role of glutamate in mood disorders: results from the ketamine in major depression study and the presumed cellular mechanism underlying its antidepressant effects. Curr Psychiatry Rep. 2007;9:467–474. doi: 10.1007/s11920-007-0063-1. [DOI] [PubMed] [Google Scholar]
- Maeng S, Zarate CA, Jr, Du J, Schloesser RJ, McCammon J, Chen G, Manji HK. Cellular mechanisms underlying the antidepressant effects of ketamine: role of alpha-amino-3-hydroxy-5-methylisoxazole-4-propionic acid receptors. Biol Psychiatry. 2008;63:349–352. doi: 10.1016/j.biopsych.2007.05.028. [DOI] [PubMed] [Google Scholar]
- Mahadik SP, Mukherjee S, Scheffer R, Correnti EE, Mahadik JS. Elevated plasma lipid peroxides at the onset of nonaffective psychosis. Biol Psychiatry. 1998;43:674–679. doi: 10.1016/s0006-3223(97)00282-5. [DOI] [PubMed] [Google Scholar]
- Mathew SJ, Manji HK, Charney DS. Novel drugs and therapeutic targets for severe mood disorders. Neuropsychopharmacology. 2008;33:2080–2092. doi: 10.1038/sj.npp.1301652. [DOI] [PubMed] [Google Scholar]
- Mechri A, Saoud M, Khiari G, d'Amato T, Dalery J, Gaha L. Glutaminergic hypothesis of schizophrenia: clinical research studies with ketamine (in French) Encephale. 2001;27:53–59. [PubMed] [Google Scholar]
- Morrone LA, Rombolà L, Pelle C, Corasaniti MT, Zappettini S, Paudice P, Bonanno G, Bagetta G. The essential oil of bergamot enhances the levels of amino acid neurotransmitters in the hippocampus of rat: implication of monoterpene hydrocarbons. Pharmacol Res. 2007;55:255–262. doi: 10.1016/j.phrs.2006.11.010. [DOI] [PubMed] [Google Scholar]
- Nanduri J, Yuan G, Kumar GK, Semenza GL, Prabhakar NR. Transcriptional responses to intermittent hypoxia. Respir Physiol Neurobiol. 2008;164:277–281. doi: 10.1016/j.resp.2008.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olney JW, Newcomer JW, Farber NB. NMDA receptor hypofunction model of schizophrenia. J Psychiatr Res. 1999;33:523–533. doi: 10.1016/s0022-3956(99)00029-1. [DOI] [PubMed] [Google Scholar]
- Sansone M. Influence of benzodiazepine tranquilizers on amphetamine-induced locomotor stimulation in mice. Psychopharmacology (Berl) 1980;71:63–65. doi: 10.1007/BF00433253. [DOI] [PubMed] [Google Scholar]
- Schäppi MG, Jaquet V, Belli DC, Krause KH. Hyperinflammation in chronic granulomatous disease and anti-inflammatory role of the phagocyte NADPH oxidase. Semin Immunopathol. 2008;30:255–271. doi: 10.1007/s00281-008-0119-2. [DOI] [PubMed] [Google Scholar]
- Schiavone S, Sorce S, Dubois-Dauphin M, Jaquet V, Colaianna M, Zotti M, Cuomo V, Trabace L, Krause KH. Involvement of NOX2 in the development of behavioral and pathologic alterations in isolated rats. Biol Psychiatry. 2009;66:384–392. doi: 10.1016/j.biopsych.2009.04.033. [DOI] [PubMed] [Google Scholar]
- Sesack SR, Carr DB, Omelchenko N, Pinto A. Anatomical substrates for glutamate-dopamine interactions: evidence for specificity of connections and extrasynaptic actions. Ann NY Acad Sci. 2003;1003:36–52. doi: 10.1196/annals.1300.066. [DOI] [PubMed] [Google Scholar]
- Skolnick P, Popik P, Trullas R. Glutamate-based antidepressants: 20 years on. Trends Pharmacol Sci. 2009;30:563–569. doi: 10.1016/j.tips.2009.09.002. [DOI] [PubMed] [Google Scholar]
- Sorce S, Krause KH. NOX enzymes in the central nervous system: from signaling to disease. Antioxid Redox Signal. 2009;11:2481–2504. doi: 10.1089/ars.2009.2578. [DOI] [PubMed] [Google Scholar]
- Spielewoy C, Biala G, Roubert C, Hamon M, Betancur C, Giros B. Hypolocomotor effects of acute and daily d-amphetamine in mice lacking the dopamine transporter. Psychopharmacology (Berl) 2001;159:2–9. doi: 10.1007/s002130100901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sulzer D, Sonders MS, Poulsen NW, Galli A. Mechanisms of neurotransmitter release by amphetamines: a review. Prog Neurobiol. 2005;75:406–433. doi: 10.1016/j.pneurobio.2005.04.003. [DOI] [PubMed] [Google Scholar]
- Trabace L, Kendrick KM, Castrignanò S, Colaianna M, De Giorgi A, Schiavone S, Lanni C, Cuomo V, Govoni S. Soluble amyloid beta1–42 reduces dopamine levels in rat prefrontal cortex: relationship to nitric oxide. Neuroscience. 2007;147:652–663. doi: 10.1016/j.neuroscience.2007.04.056. [DOI] [PubMed] [Google Scholar]
- Tsai G, Coyle JT. Glutamatergic mechanisms in schizophrenia. Annu Rev Pharmacol Toxicol. 2002;42:165–179. doi: 10.1146/annurev.pharmtox.42.082701.160735. [DOI] [PubMed] [Google Scholar]
- Uchihashi Y, Kuribara H, Isa Y, Morita T, Sato T. The disruptive effects of ketamine on passive avoidance learning in mice: involvement of dopaminergic mechanism. Psychopharmacology (Berl) 1994;116:40–44. doi: 10.1007/BF02244869. [DOI] [PubMed] [Google Scholar]
- Violi F, Sanguigni V, Loffredo L, Carnevale R, Buchetti B, Finocchi A, Tesauro M, Rossi P, Pignatelli P. Nox2 is determinant for ischemia-induced oxidative stress and arterial vasodilatation: a pilot study in patients with hereditary Nox2 deficiency. Arterioscler Thromb Vasc Biol. 2006;26:e131–e132. doi: 10.1161/01.ATV.0000229710.13054.2d. [DOI] [PubMed] [Google Scholar]
- Violi F, Sanguigni V, Carnevale R, Plebani A, Rossi P, Finocchi A, Pignata C, De Mattia D, Martire B, Pietrogrande MC, Martino S, Gambineri E, Soresina AR, Pignatelli P, Martino F, Basili S, Loffredo L. Hereditary deficiency of gp91(phox) is associated with enhanced arterial dilatation: results of a multicenter study. Circulation. 2009;120:1616–1622. doi: 10.1161/CIRCULATIONAHA.109.877191. [DOI] [PubMed] [Google Scholar]
- Wu HY, Yuen EY, Lu YF, Matsushita M, Matsui H, Yan Z, Tomizawa K. Regulation of N-methyl-d-aspartate receptors by calpain in cortical neurons. J Biol Chem. 2005;280:21588–21593. doi: 10.1074/jbc.M501603200. [DOI] [PubMed] [Google Scholar]
- Xi D, Keeler B, Zhang W, Houle JD, Gao WJ. NMDA receptor subunit expression in GABAergic interneurons in the prefrontal cortex: application of laser microdissection technique. J Neurosci Methods. 2009;176:172–181. doi: 10.1016/j.jneumeth.2008.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao JK, Reddy RD, van Kammen DP. Oxidative damage and schizophrenia: an overview of the evidence and its therapeutic implications. CNS Drugs. 2001;15:287–310. doi: 10.2165/00023210-200115040-00004. [DOI] [PubMed] [Google Scholar]
- Yashiro K, Philpot BD. Regulation of NMDA receptor subunit expression and its implications for LTD, LTP, and metaplasticity. Neuropharmacology. 2008;55:1081–1094. doi: 10.1016/j.neuropharm.2008.07.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang XY, Chen da C, Xiu MH, Wang F, Qi LY, Sun HQ, Chen S, He SC, Wu GY, Haile CN, Kosten TA, Lu L, Kosten TR. The novel oxidative stress marker thioredoxin is increased in first-episode schizophrenic patients. Schizophr Res. 2009;113:151–157. doi: 10.1016/j.schres.2009.05.016. [DOI] [PubMed] [Google Scholar]
- Zuo DY, Wu YL, Yao WX, Cao Y, Wu CF, Tanaka M. Effect of MK-801 and ketamine on hydroxyl radical generation in the posterior cingulate and retrosplenial cortex of free-moving mice, as determined by in vivo microdialysis. Pharmacol Biochem Behav. 2007;86:1–7. doi: 10.1016/j.pbb.2006.05.010. [DOI] [PubMed] [Google Scholar]