

Abstract
In the nervous system, the induction of plasticity is coded by patterns of synaptic activity. Glial cells are now recognized as dynamic partners in a wide variety of brain functions, including the induction and modulation of various forms of synaptic plasticity. However, it appears that glial cells are usually activated by stereotyped, sustained neuronal activity, and little attention has been given to more subtle changes in the patterns of synaptic activation. To this end, we used the mouse neuromuscular junction as a simple and useful model to study glial modulation of synaptic plasticity. We used two patterns of motor nerve stimulation that mimic endogenous motor-neuronal activity. A continuous stimulation induced a post-tetanic potentiation and a phasic Ca2+ response in perisynaptic Schwann cells (PSCs), glial cells at this synapse. A bursting pattern of activity induced a post-tetanic depression and oscillatory Ca2+ responses in PSCs. The different Ca2+ responses in PSCs indicate that they decode the pattern of synaptic activity. Furthermore, the chelation of glial Ca2+ impaired the production of the sustained plasticity events indicating that PSCs govern the outcome of synaptic plasticity. The mechanisms involved were studied using direct photo-activation of PSCs with caged Ca2+ that mimicked endogenous plasticity. Using specific pharmacology and transgenic knock-out animals for adenosine receptors, we showed that the sustained depression was mediated by A1 receptors while the sustained potentiation is mediated by A2A receptors. These results demonstrate that glial cells decode the pattern of synaptic activity and subsequently provide bidirectional feedback to synapses.
Introduction
Neuronal information processing in the brain is not only coded by the frequency of neuronal activity but also by the pattern of action potential firing in neurons. This frequency and pattern coding greatly influences synaptic plasticity of CNS synapses as well as the neuromuscular junction (NMJ) (Magleby and Zengel, 1976; Guyonneau et al., 2004; Harris, 2005; Nicoll and Schmitz, 2005).
In addition to the neuronal elements of the brain, it is now increasingly recognized that glial cells are dynamic partners in a wide variety of brain functions including the induction and modulation of various forms of synaptic plasticity (Pascual et al., 2005; Haydon and Carmignoto, 2006; Perea and Araque, 2007; Barres, 2008; Gordon et al., 2009; Henneberger et al., 2010). Glial cells respond to different neurotransmitters with Ca2+ release from internal stores. These Ca2+ elevations represent the excitability of glial cells and are dependent on the frequency of neuronal and synaptic activity; that is, the responses are more reliable and larger at higher frequencies of stimulation (Pasti et al., 1997). Also, the activation of glial cells is regulated by a frequency-dependent threshold, below which no Ca2+ response has been detected so far. In turn, glial cells modulate neuronal activity and synaptic plasticity by releasing neuroactive substances called gliotransmitters (Haydon and Carmignoto, 2006).
Although glial cell activation varies according to different frequencies of stimuli (Pasti et al., 1997), it is still unclear whether glial cells can decode different patterns of neuronal activity (e.g., differences in burst duration), a critical property that would allow them to fully integrate neuronal information and, in return, accordingly adjust their feedback modulation to neuronal and synaptic activity. Furthermore, it is unknown whether the outcome of glial cell regulation of neuronal activity is dependent upon the property of the glial activation. Therefore, we set out to investigate the impact of different patterns of neuronal activity on glial cells and their subsequent modulation of synaptic transmission.
To this end, we took advantage of the NMJ, which is a simple synapse, easily accessible, and covered by perisynaptic Schwann cells (PSCs; ∼5 cells per NMJ). These are nonmyelinating glial cells that surround the synapse and have similar roles to astrocytes in the CNS (Auld and Robitaille, 2003).
We found that endogenously evoked synaptic post-tetanic potentiation and depression relied on different glial calcium signals. Therefore, our results demonstrate that glial cells detect differences in ongoing synaptic activity and decode these differences to provide specific feedback to the synapse. These glial calcium signals result in the differential activation of potentiating A2A adenosine receptor and depressing A1 receptors presynaptically. This feedback provides a critical level of modulation to regulate a balance of potentiating and depressing influences on the synapse.
Materials and Methods
Animals and preparation.
All experiments were performed in accordance with the guidelines of the Canadian Council of Animal Care and the Animal Care Committee at the Université de Montréal. Juvenile [postnatal day (P) 21–P28] male CD-1 mice (Charles River) were killed by decapitation under deep anesthesia (0.1 ml/g midazolam and hypnorm dissolved in distilled water, administered intraperitoneally). Soleus muscles, with the tibial nerve intact, were removed and pinned in Rees saline solution (Rees, 1978) as follows (in mm): NaCl 110; KCl 5; MgCl2 1; NaHCO3 25; CaCl2 2; glucose 11; glutamate 0.3; glutamine 0.4; BES buffer, 5; cocarboxylase 0.4 μm; and choline chloride 36 μm and bubbled with 95% O2/5% CO2. Experiments were performed at 30–32°C under continuous perfusion of saline.
A1 and A2A knock-out mice. The A1 −/− mice were a kind gift from Bertil Fredholm (Karolinska Institute, Stockholm, Sweden) and Stephen Tilley (University of North Carolina, Chapel Hill, NC). These mice have previously been described (Giménez-Llort et al., 2002). A2A −/− mice have been characterized by Chen et al. (1999) and kindly provided by Jiang-Fan Chen (Boston University, Boston, MA) and Michael Schwarzschild (Massachusetts General Hospital, Boston, MA).
Electrophysiological recordings.
The tibial nerve was stimulated through a suction electrode filled with extracellular saline. Muscle contractions were prevented with partial blockade of the postsynaptic ACh receptors using d-tubocurarine chloride (2.9–4.4 μm, Sigma). Intracellular recordings of postsynaptic potentials (PSPs) were performed using glass microelectrodes (1.0 mm OD; WPI) pulled to 50–70 MΩ (filled with 3M KCl) with a Brown–Flaming micropipette puller (Sutter Instruments). Recordings were amplified (200×) using an AM Systems 1200 amplifier connected to a WPI external amplifier, digitized using a National Instruments BNC 2110 board and acquired using WinWCP software (John Dempster, Strathclyde University, Glasgow, UK). It is noteworthy that the choice of the nicotinic receptor antagonist has no impact on synaptic plasticity events studied at the NMJ (Robitaille, 1998; Silinsky, 2005; Bélair et al., 2010).
The continuous stimulation paradigm consisted of 1800 pulses delivered at 20 Hz. The bursting stimulation paradigm (Fig. 1 A) consisted of 30 repetitions of 20 pulses at 20 Hz repeated every 2 s. This was repeated three times with 20 s of rest between repetitions. Synaptic efficacy was monitored with test pulses delivered at a frequency of 0.2 Hz, a frequency known to have no effect on synaptic efficacy. Control baselines were generated by stimulation at 0.2 Hz for 20 min for comparison with treatments. For comparison with photolysis experiments, baseline controls were performed where a UV flash was given in the absence of caged compound and the 0.2 Hz stimulation was continued for 15 min after, as with treatments. PSP amplitude before, during high-frequency stimulation, and after were normalized to the PSP amplitude of test pulses obtained during the baseline period, before 20 Hz stimulation. In some experiments, stimulation was performed using a protocol of two stimuli at 10 ms interval elicited at 0.2 Hz to measure paired-pulse facilitation (PPF). Recordings were discarded when the holding potential changed by >5 mV. Throughout, n indicates the number of NMJs studied. Only one NMJ was studied per muscle.
Figure 1.
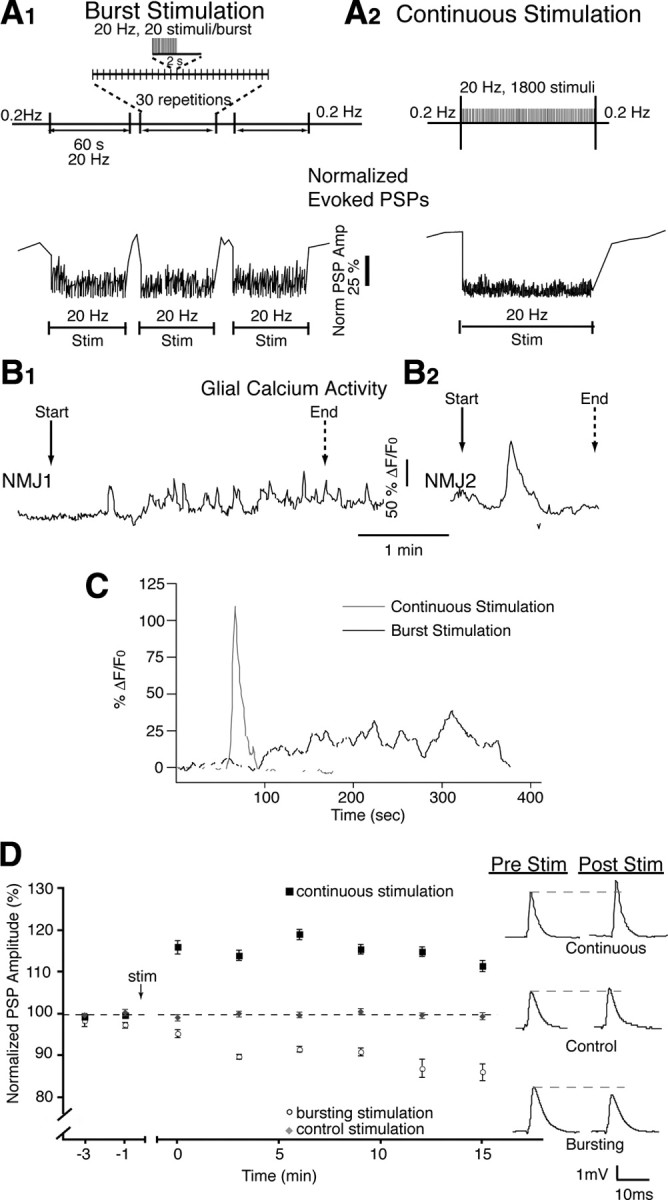
Different stimulation paradigms induce different glial calcium signals and post-tetanic plasticity. A1, Bursting stimulation protocol mimicking endogenous activity (top) induced repeated periods of synaptic depression (bottom), which recovered at the cessation of high-frequency activity. A2, Continuous stimulation protocol (top) induced a prolonged period of synaptic depression (bottom), which also recovered at the cessation of high-frequency activity. B1, B2, Multiple (B1) and single (B2) calcium elevations were correlated with bursting and continuous stimulation respectively. C, Average of calcium responses recorded in response to bursting and continuous stimulation. Responses were aligned on first rise. D, Normalized PSP amplitude over time showing that bursting stimulation caused post-tetanic depression, whereas continuous stimulation caused post-tetanic potentiation. PSPs are representative averages of 30 events taken from a single recording at the 15 min time point. Pre Stim, Before stimulation; Post Stim, after stimulation.
Calcium imaging and analysis in PSCs.
Dissected soleus muscles were incubated in 10 μm Fluo-4AM (Invitrogen) containing 0.02% pluronic acid (Invitrogen) for 1.5 h at room temperature. NMJs were located on the surface of muscle fibers using bright-field optics. Evoked calcium responses were obtained by stimulating the tibial nerve with one of the stimulation paradigms described above. Epifluorescent images were acquired on a Nikon E600N upright microscope fitted with a Princeton Instruments CCD-1300 camera. Digital acquisition was performed using MetaFluor software (Molecular Devices, a division of MDS Analytical Technologies) driving a Lamda 10-2 shutter wheel (Sutter Instruments). Images were acquired at a rate of one image per second with an integration time of 400 ms. Fluorescence was quantified by subtracting the background fluorescence from the neighboring muscle fiber and then performing the calculation (F − F0/F0)*100 to give the percentage ΔF/F0. Experiments were discarded when bleaching or focus drift occurred.
Photo-activation of caged molecules.
Caged compounds [diazo-2-AM, o-nitrophenyl (NP)-EGTA-AM; Invitrogen] were loaded with the following protocol. Muscles were incubated first with Fluo-4-AM (10 μm) alone for 30 min and then with both Fluo-4-AM and caged compound (20 μm) for 45 min followed by another period of 45 min with fresh solution. For direct injection of NP-EGTA in muscle fibers, Ringer solution was used as the intracellular solution containing NP-EGTA (5 mm). Iontophoretic injections of NP-EGTA were performed using current pulses (−10 nA, 200 ms) generated using a Grass S88 stimulator at a frequency of 2 Hz for 10 min.
Photolysis of NP-EGTA was performed using a Laser Science Nitrogen Pulsed UV laser (337 nm) delivering pulses of 4 ns duration. The UV pulses were aimed at the preparation using a fiberoptic probe with a diameter of ∼30 μm inserted into a pipette for guidance. The alignment of the fiberoptic probe was performed using visible red light emitted by an HeNe laser passing through the same optic fiber.
For multiple uncaging events mimicking Ca2+ response observed during bursting activity, 10 pulses generated at 60 Hz for 2 ms were used repeatedly. For a single large Ca2+ event observed during continuous stimulation, 15 pulses of 10 ms at 50 Hz were used. Two or more PSCs were targeted for photolysis during all experiments. Photolysis of NP-EGTA in the presynaptic terminal and muscle fiber as well as diazo-2 was induced using a regime of 15 pulses of 10 ms at 50 Hz. Calculated efficacy of the optic fiber is shown in supplemental Figure 1 (available at www.jneurosci.org as supplemental material).
A protocol was designed to specifically target PSCs taking advantage of the morphological organization of the NMJs at soleus muscles. In the first situation, we took advantage that PSCs were often ectopic, with their somata located off limit of the endplate area while their processes covered the nerve terminal. Also, NMJs were often found on the side of the muscle fiber (Fig. 2 A, side NMJs). Owing to these two characteristics, we could find side NMJs with ectopic PSC somata lying on the surface of the muscle fiber. This provided us with a morphological situation whereby the PSC somata could be selectively exposed to UV light without affecting the presynaptic terminal (for details, see Fig. 2). Second, we chose surface NMJs where the PSCs somata were clearly off limit from the endplate area and could be aimed selectively by the laser beam. This was possible because the alignment and positioning of the fiberoptic was performed using visible red light emitted by a red Diode laser and passed through the fiberoptic such that the cone of light covered at least two PSCs and not the endplate area.
Figure 2.
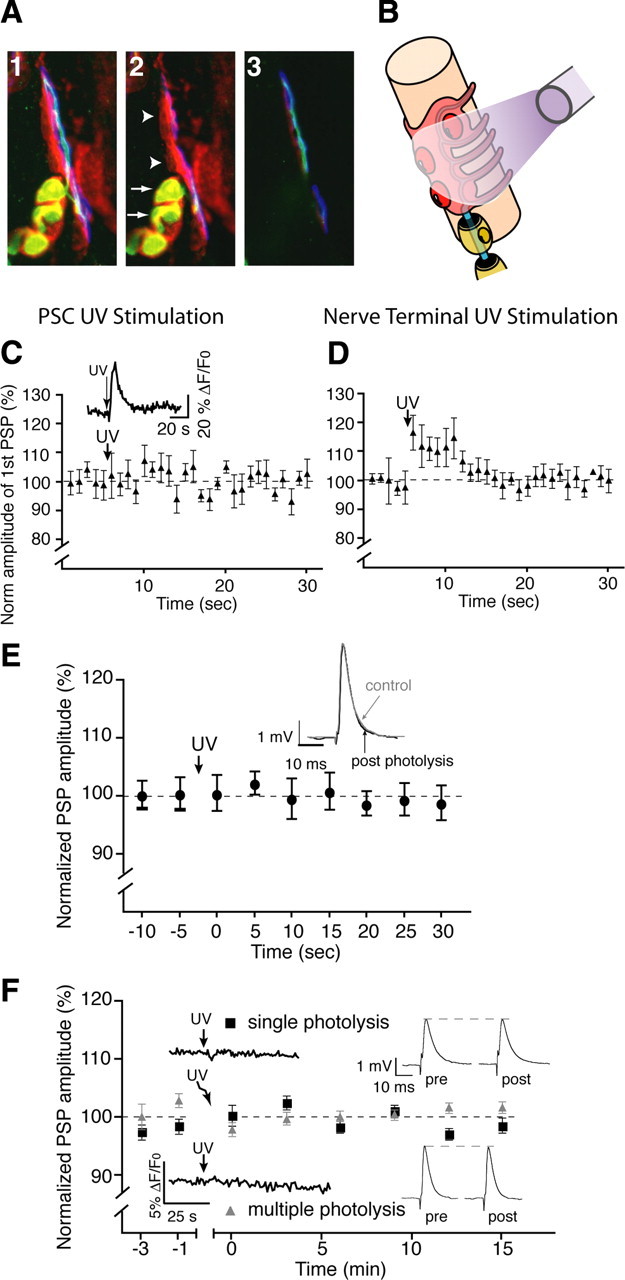
Selective photo-activation of PSCs. A1, A 9.6 μm Z-stack of immunofluorescent staining from a mouse NMJ with PSCs labeled red with anti-S100β, green anti-neurofilament in the presynaptic terminal, and blue α-bungarotoxin labeling postsynaptic nACh receptors. A2, A single optical section at the surface of the junction with the myelinating Schwann cells colocalized with the presynaptic terminal (arrows). PSCs are seen separated from the last myelinating segment overlying the presynaptic terminal (arrowheads). A3, The NMJ at 5.6 μm below the surface. PSC somata are located at the surface, while the presynaptic terminal is clearly visible at this depth. B, Arrangement of the fiberoptic used for photolysis and its position in relation to the synaptic components. C, Normalized average PSP amplitude showing that photo-activation of NP-EGTA in a PSC-targeted situation did not induce a rapid, transient increase in PSP amplitude indicative of a lack of direct effect on the presynaptic terminal. A PSC calcium elevation was detected (inset). D, Normalized average PSP amplitude showing that presynaptic photo-activation of NP-EGTA resulted in a rapid and transient increase in PSP amplitude. E, Changes of PSP amplitude before and after glial-targeted photo-activation of diazo-2. No rapid changes (up to 30 s after photolysis) were observed in PSP amplitude indicating that photolysis of diazo-2 had not direct presynaptic effect. F, Normalized changes in PSP amplitude showing that injection of cell impermeant NP-EGTA salt into muscle fibers had no effect on plasticity after photolysis. Insets show traces of calcium level measurements in PSCs (left) and PSP averages from representative individual experiments (left, n = 4).
Some uncaging experiments with diazo-2 were performed using an Olympus FV1000 (see Fig. 9 and supplemental Fig. 2, available at www.jneurosci.org as supplemental material). The 405 line of a UV laser was used to induce the photo-activation of diazo-2 with the tornado feature and the SIM scanner to simultaneously activate diazo-2 while imaging calcium. The power of the UV laser was set at 30%, which represents a final power of 8.3% since only 25% of the laser light is carried through the SIM scanner. An ROI was placed on top of at least two PSCs and scanned for up to 5 s to optimally activate diazo-2. Exposure to 405 laser light alone does not affect PSCs excitability as shown by their unaltered ability to respond to local agonists applications (data not shown).
Figure 9.

Role of adenosine receptors in post-tetanic plasticity induced by motor nerve stimulation. A, B, Changes in PSP amplitude before and after bursting stimulation in the presence of the A1 antagonists PSB-36 (A) and before and after continuous stimulation in the presence of the A2A antagonist SCH-58261 (B). Note that the A1 antagonist prevented the production of post-tetanic depression, whereas the A2A antagonist prevented the potentiation, as a depression was even observed. Insets represent representative PSPs before (Pre) and after (Post) the motor nerve stimulation. C, Changes in PSP amplitude before and after photo-activation of diazo-2 in PSCs (Flash) and the stimulation of the motor nerve using the continuous paradigm (Stim). Bath application of the A1 antagonist (PSB-36, black bar) was started 10 min after the end of the continuous stimulation period. Insets show a typical PSP before (Pre), after the diazo-2 photo-activation and motor nerve stimulation (Post) and during bath application of the A1 antagonist (PSB). Note that the A1 antagonist did not affect post-tetanic depression. D, Changes in PSP amplitude before and after photo-activation of diazo-2 in PSCs (Flash) and the stimulation of the motor nerve using the bursting paradigm (Stim). Bath application of the A2A antagonist (SCH-58261, black bar) was started 10 min after the end of the bursting stimulation paradigm. Insets indicate a typical PSP before (Pre), after the diazo-2 photo-activation and motor nerve stimulation (Post), and during bath application of the A2A antagonist (SCH). Note that the potentiation was not altered by the presence of the A2A antagonist.
Immunohistochemistry.
Dissected soleus muscles were pinned in a Sylgard-coated dish containing PBS and fixed for 10 min in 4% formaldehyde, at room temperature. Muscles were permeabilized in 100% cold methanol for 6 min, at −20°C. Nonspecific antibody labeling was prevented by incubating the muscles in 10% normal donkey serum (NDS) with 0.1% Triton X-100 for 20 min. Rabbit, anti-S100β (1:250, Dako) with 0.01% Triton X-100 and 2% NDS was incubated overnight at 4°C, then washed in normal serum muscles, to be then incubated in goat, anti-neurofilament (1:250; SC16143, Santa Cruz Biotechnology) for 90 min. Secondary antibodies Alexa 488 (α-goat) and 647 (α-rabbit, 1:500) were incubated together for 60 min at room temperature. After washout, muscles were incubated with α-bungarotoxin (Alexa 594, 0.75 μg/ml) for 30 min. After each step, soleus muscles were washed in PBS plus 0.01% Triton X-100 for 3 × 5 min. The preparations were then mounted in the Prolong Kit (Invitrogen) and observed using an Olympus FV1000 microscope. The three channels were observed simultaneously using the spectral detection feature of the confocal system. Pinholes were set to obtain an airy disk value of 1. Figures were not manipulated in any way after the acquisition.
Chemicals.
All chemicals were purchased from Sigma-Aldrich except for ARL 67156, CGS 21680, and PSB-36 (Tocris Bioscience).
Statistical analysis.
PSP values were compared with the control PSP amplitude with Student's t tests at the 15 min time point. When data were found not to conform to normality, Mann–Whitney U tests were used. Analyses were deemed significant at p ≤ 0.05.
Results
Glia decode the pattern of synaptic activity
We chose two physiological patterns of stimulation for the soleus muscle, similar to in vivo motoneuronal activity, which produced two forms of post-tetanic plasticity. This provided us with two different patterns of stimulation (bursting and continuous) at the same frequency (20 Hz) using the same number of pulses.
The first stimulation paradigm consisted of bursts of activity at 20 Hz with a total of 1800 pulses that induced three periods of synaptic depression each separated by a brief period of recovery (Fig. 1 A1), and closely replicates a form of endogenous activity (Hennig and Lømo, 1985). The second pattern used continuous delivery of 1800 pulses at 20 Hz, causing a sustained depression during stimulation (Fig. 1 A2). This is the frequency normally seen at the soleus muscle (Hennig and Lømo, 1985), and a pattern typical for studies of synapse–glia interactions at the NMJ (Rochon et al., 2001).
Transmitter release was evoked by stimulating the tibial motor nerve using different stimulation paradigms while we simultaneously recorded PSPs and monitored glial calcium responses using Fluo-4 AM (during and for 1 min after the 20 Hz stimulation). Interestingly, during burst stimulation, oscillatory calcium activity was observed in PSCs at each synapse where several small transient calcium responses (duration, 11.2 ± 1.6 s; amplitude, 47.2 ± 6.3% ΔF/F0) occurred in 27 of 29 cells. In addition, this stimulation induced an underlying increase in basal calcium that lasted on average 94.5 ± 6.6 s (Fig. 1 B1). Unlike bursting, continuous stimulation always elicited one to two calcium responses that lasted on average 27.3 ± 4.8 s with an average amplitude of 115.4 ± 28.7% ΔF/F0 (Fig. 1 B2). The individual responses were longer (p = 0.003, two-tailed t test) and had greater amplitude (p = 0.001, two-tailed t test) than the transient responses evoked with burst stimulation. However, as shown in Figure 1 C, on average, Ca2+ responses elicited by continuous stimulation had a faster rise time while the bursting motor nerve stimulation produced a slower and more sustained response.
In addition, the two patterns of motor nerve stimulation produced different types of post-tetanic plasticity. Indeed, as shown in Figure 1 D, the bursting stimulation induced post-tetanic depression of PSP amplitude that develops over a period of 10 min. Post-tetanic depression was 86.2 ± 2.0% of control at 15 min compared with control PSPs evoked using test pulses delivered at 0.2 Hz (Fig. 1 D, gray circles) (n = 5, p < 0.0001, two-tailed t test). However, rather than a post-tetanic depression, we found that continuous stimulation induced a post-tetanic potentiation of 111.5 ± 1.3% of control at 15 min compared with control stimulation (Fig. 1 D) (n = 8; p < 0.0001; two-tailed t test).
Hence, the two stimulation patterns induced different PSC activation and post-tetanic plasticity. More importantly, the difference in PSC calcium elevations induced by the two stimulation protocols indicates that they are differentially activated by these patterns, and thus, that glial cells decode the pattern of neuronal activity.
Selective PSC modulation with caged molecules
Next we wanted to directly manipulate glial cells to test whether the different calcium elevations induced by the two patterns determined the outcome of the two post-tetanic plasticities. However, owing to the complex structure of the mature mouse NMJ, we needed a protocol that would allow us to reliably manipulate multiple PSCs at the same time. To do this, we used loading of membrane-permeant, caged compounds combined with specific light activation of PSCs.
This technique allowed us to target PSCs overlying the presynaptic nerve terminal by using precise and specific placement of the fiberoptic probe, and by selecting NMJs with a favorable morphological conformation, as indicated in the Methods section (Fig. 2 A,B). Additionally, synapses were selected only when multiple PSCs were visible on the surface. The alignment of the fiberoptic probe was performed using visible red light emitted by an HeNe laser and passed through the optic fiber such that the cone of light covered at least two PSC somata and not the presynaptic terminal.
We controlled for possible direct presynaptic effects in each experiment by monitoring rapid changes (within seconds) (Kamiya and Zucker, 1994) in transmitter release and PPF. This approach is based on the rapid and readily reversible effects observed when manipulating presynaptic calcium levels using photolysis experiments. Finally, a brief calcium elevation in presynaptic terminals is known to rapidly and transiently increase transmitter release. However, UV exposure in conditions where PSCs were targeted had no rapid effect on transmitter release (Fig. 2 C) or on paired-pulse facilitation (PPF was 0.69 ± 0.06 in control and 0.72 ± 0.06 after uncaging, p > 0.05). This is consistent with the latency of the slower time course of the glial modulation compared with a direct presynaptic effect. In addition, no changes in PSP kinetics were observed (10–90% rise time, 2.41 ± 0.33 ms; before, 2.07 ± 0.17 ms; after; 10–90% decay 14.30 ± 3.25 ms; before, 14.97 ± 3.66 ms; after, significance at p < 0.05, n = 4). The photolysis was efficient since a Ca2+ elevation was observed in the PSCs (Fig. 2 C, inset). On the other hand, when photolysis was performed on an NMJ where the presynaptic terminal could be targeted, we observed a rapid and short-lived rise in transmitter release (Fig. 2 D) concomitant with a reduction in paired-pulse facilitation (0.89 ± 0.05 in control and 0.78 ± 0.04 after uncaging, p < 0.05).
Furthermore, the addition of a calcium chelator into the presynaptic terminal should effectively reduce transmitter release (Delaney et al., 1989, 1991; Adler et al., 1991; Robitaille and Charlton, 1992; Robitaille et al., 1993). This is known to be a sensitive assay for presynaptic effect. Importantly, the introduction of a calcium chelator will have only transient effects on transmitter release unless its concentration is continuously maintained (Adler et al., 1991). Indeed, a transient reduction of transmitter release was observed when the presynaptic terminal was targeted as indicated above (data not shown). However, such a transient reduction of transmitter release was never observed in our experiments when caged BAPTA (diazo-2) was photo-activated in a PSC-specific manner (Fig. 2 E). Hence, these control experiments indicate that the experimental strategy of choosing NMJs allowed us to selectively target PSCs without affecting the presynaptic terminal. Experiments were discarded when direct presynaptic effects were observed.
Finally, we controlled for possible postsynaptic effects by directly injecting the impermeant form of NP-EGTA in the muscle fiber. We found no evidence for a postsynaptic, muscle fiber contribution to synaptic transmission or plasticity when PSCs were exposed to our photolysis protocols (Fig. 2 F), as indicated by the lack of effect on PSP amplitude and the absence of Ca2+ elevation in PSCs.
Selective regulation PSC Ca2+
Using the approach described above for NMJ selection and specific PSC activation, we determined how to elicit different Ca2+ responses using different photo-activation protocols with NP-EGTA. Additionally, we determined how to block PSC activation by chelating Ca2+ using diazo-2, the caged BAPTA compound.
As shown in Figure 3 A, a calcium elevation in PSCs was induced by photo-activation of the caged calcium molecule NP-EGTA after its loading with the membrane permeant form (20 μm). These responses can be repeated on the same cells (Fig. 3 B). Additionally, we were able to control the properties of the calcium photolysis by adapting the pattern and duration of the UV exposure so that multiple, smaller calcium elevations in glial cells could be induced (Fig. 3 C). Conversely, activating caged BAPTA after loading the cells with membrane-permeant form (Diazo2-AM, Invitrogen) blocked Ca2+ responses elicited in PSCs. Indeed, as shown in Figure 3 D, ATP induced a first calcium response with average amplitude of 39.4 ± 8.5% ΔF/F0 (top), whereas the response was completely abolished after diazo-2 photo-activation (average change of 0.4 ± 0.2% ΔF/F0, bottom). Also, calcium responses in PSCs evoked by endogenous synaptic activity were no longer observed (Fig. 3 E). These results indicate that PSCs can be activated or blocked using photo-activation of caged molecules.
Figure 3.
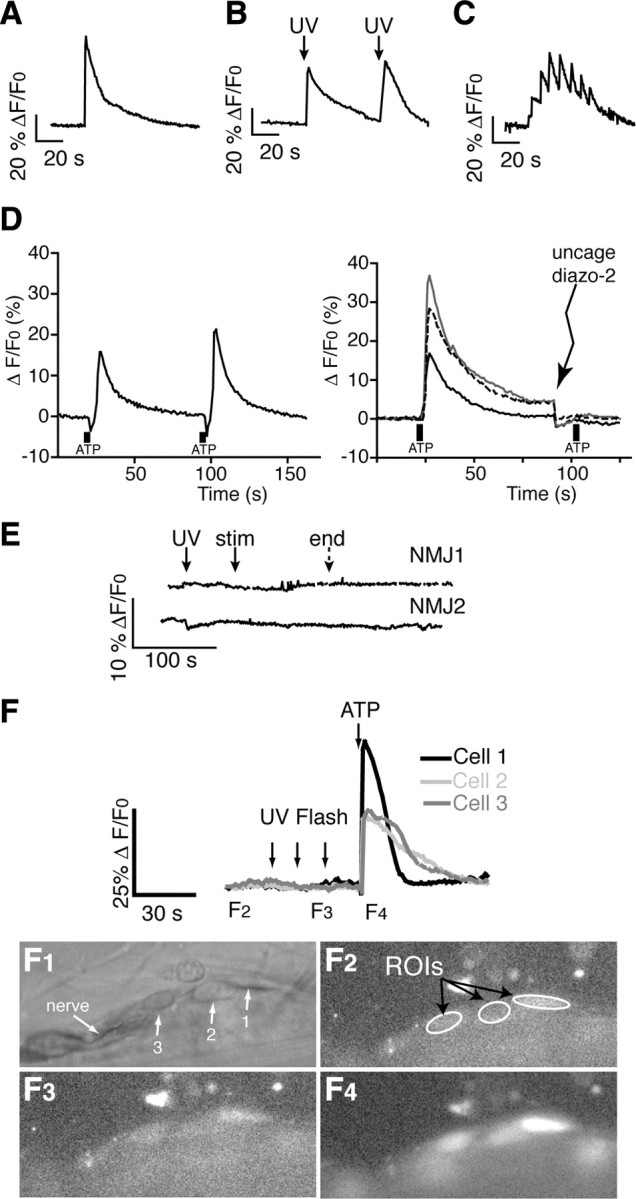
Manipulation of calcium in PSCs with caged compounds does not affect neuronal function. A, B, Single (A) or repeated (B) calcium elevations can be elicited in a single PSC using photo-activation of NP-EGTA. C, Multiple, consecutive calcium elevations can be evoked in the same PSC by modifying the protocol of photo-activation of NP-EGTA. D, Left, ATP-induced calcium responses in PSCs show little desensitization and are reproducible. Right, After a first ATP-induced calcium response with average amplitude of 39.4 ± 8.5% ΔF/F0, diazo-2 was photo-activated and ATP locally applied again on the same cells (shown are 3 responses from different NMJs), resulting in an average change of 0.4 ± 0.2% ΔF/F0. The second response was completely prevented when diazo-2 was uncaged before local application. E, Calcium elevations evoked with endogenous nerve stimulation were abolished by prior photolysis of diazo-2 in PSCs. F, Top, Large UV photolysis had no direct effect on the resting fluorescence of PSCs in the absence of any caged compound. Cell viability was confirmed by the ability of ATP to induce calcium responses that were similar to the ones elicited without prior exposure to UV flashes (n = 4). Bottom, Bright field and fluorescent images of the same neuromuscular junction as depicted graphically with cells 1, 2, and 3 indicated (F1). F2, Fluorescent image acquired during the baseline period before the UV flashes. Measured regions of interest (ROI) are indicated. F3, Fluorescent image acquired immediately after the second UV flash. F4, Fluorescent image acquired immediately after local application of ATP.
Importantly, direct exposure to UV light had no effect on PSCs or synaptic transmission. Indeed, in the absence of any caged compound, sustained and large UV photolysis had no direct effect on the resting fluorescence of PSCs (Fig. 3 F) (n = 4). Cell viability and excitability were confirmed by the ability of ATP to induce calcium responses similar to the ones elicited without prior exposure to UV flashes. In addition, PSPs evoked by paired-pulse stimulation in control were unaffected after UV photolysis in the absence of any caged compound (n = 4; PPF was 0.79 ± 0.06 in control and 0.77 ± 0.06 after uncaging, p > 0.05).
Endogenous glial calcium elevations determine the outcome of plasticity
After the validation of our technique, we next tested whether the pattern of glial activity caused the selective expression of short-term post-tetanic plasticity. If this is the case, we predict that preventing the glial Ca2+ rise should impair the outcome of the post-tetanic plasticity. We used the caged calcium chelator diazo-2 AM to rapidly buffer calcium upon photo-activation with UV light (Kamiya and Zucker, 1994). The UV fiberoptic probe was positioned to specifically target glial cells, as indicated above. The caged calcium chelator diazo-2 was then photo-activated in PSCs before nerve stimulation to effectively block PSC activity through inhibition of calcium elevations. As shown in Figure 4 A, chelation of glial cell calcium during bursting stimulation no longer produced post-tetanic depression, but rather resulted in a potentiation in PSP amplitude. PSP amplitude was 122.8 ± 1.8% at 15 min compared with control stimulation (n = 8; p < 0.0001; two-tailed t test). Conversely, photolysis of diazo-2 before continuous stimulation prevented post-tetanic potentiation and induced depression (Fig. 4 B) (94.9 ± 1.3%; n = 7; p < 0.0001; two-tailed t test).
Figure 4.
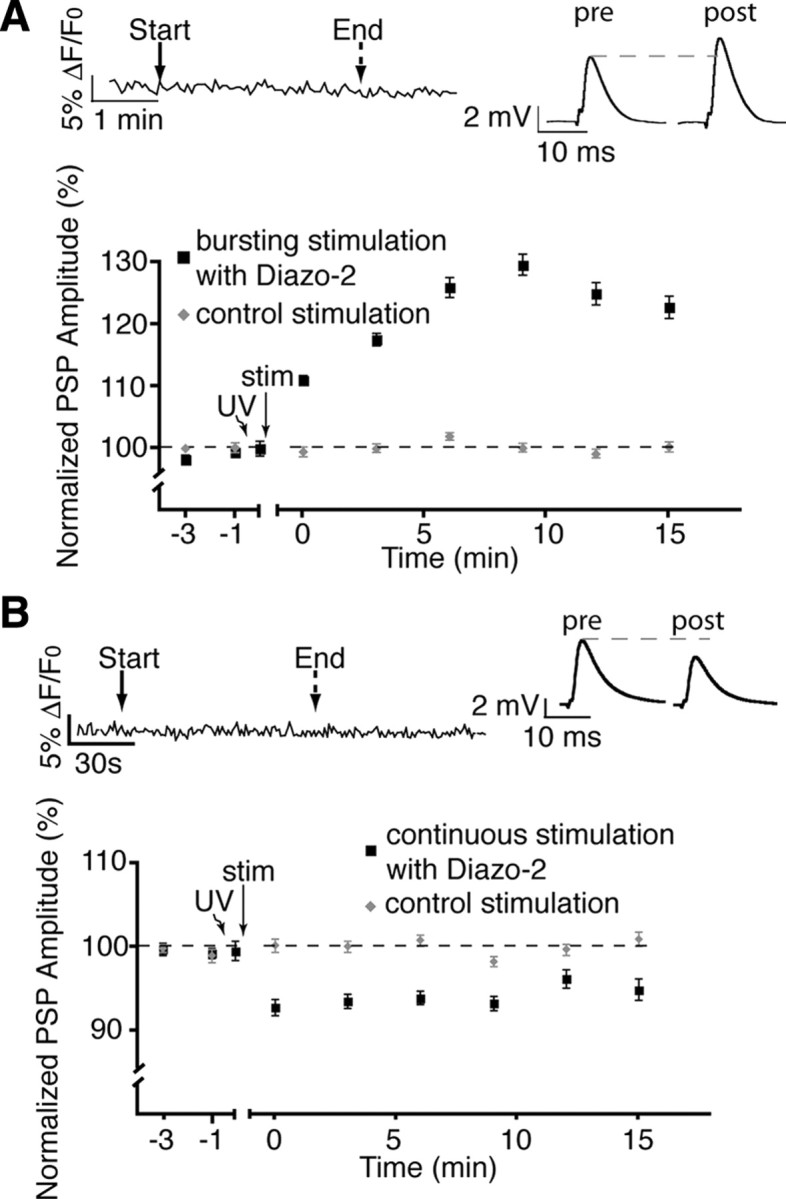
Inhibition of glial calcium elevations alters plasticity. A, Normalized PSP amplitude over time showing that post-tetanic depression was prevented by the photolysis of diazo-2 (caged BAPTA) in glial cells during burst stimulation revealing a potentiation. The top left inset shows the absence of a detectable Ca2+ rise in the PSCs. The top right inset shows representative PSPs (averages of 30 events) taken from a single recording at the 15 min time point. B, Photolysis of diazo-2 in glial cells during continuous stimulation blocked all post-tetanic potentiation. The top left inset shows that no detectable Ca2+ elevations were observed in glial cells after photolysis of diazo-2. The top right inset shows representative PSPs.
These results clearly indicate the importance of glial activation (via calcium) in the expression of appropriate endogenous synaptic plasticity. Notably, these data suggest that glial cell calcium elevations are not all-or-none events, but contain specific information concerning the ongoing activity of the synapse. Furthermore, these data indicate that glial cells govern the outcome of synaptic plasticity in a context-dependent manner. Hence, not only do glial cells decode neuronal activity, but their differential activation results in an adjusted feedback relevant to the current state of the synapse.
Glial calcium elevations are sufficient to induce plasticity in a pattern-dependent manner
To determine the importance of the distinct glial calcium signals observed during different patterns of synaptic activity, we designed protocols using photolysis of caged calcium compounds to mimic endogenously evoked calcium responses and, more importantly, the synaptic plasticity. Furthermore, direct glial activation would allow us to tease apart the underlying mechanisms, a task that would be difficult with endogenous activity alone since both the presynaptic terminal and PSCs bear many of the same types of receptors (e.g., A1 and muscarinic ACh receptors) (Hamilton and Smith, 1991; Correia-de-Sá et al., 1996; Grafe et al., 1999; Galkin et al., 2001; Rochon et al., 2001; Oliveira et al., 2002, 2004; De Lorenzo et al., 2004; Silinsky, 2004; Baxter et al., 2005; Moores et al., 2005; Todd and Robitaille, 2006; Dudel, 2007).
Based on the control experiments presented in Fig. 3, we established two different photolysis protocols to elicit either multiple calcium responses, thus mimicking the oscillatory responses seen with bursting stimulation, or a single calcium response, similar to that seen with continuous stimulation (Fig. 5 A, insets). Individual responses occurring during the burst activity elicited by the repeated photolysis protocol were not significantly different from endogenously evoked responses in amplitude or duration (p > 0.05, two-tailed t test). On average, each transient calcium response had an amplitude of 38.5 ± 5.1% ΔF/F0 and a duration of 8.2 ± 1.0 s (n = 25 events, from 8 PSCs and 8 NMJs). Similarly, calcium responses elicited by a single photo-activation were larger and longer (average amplitudes of 122.9 ± 14.2% ΔF/F0 and average duration of 51.9 ± 9.7 s, n = 11 PSCs from 11 NMJs) then the transient responses occurring during the bursting activity but were not different from the responses elicited by endogenous activity (p > 0.05, Student's t test) (Fig. 1 B2).
Figure 5.
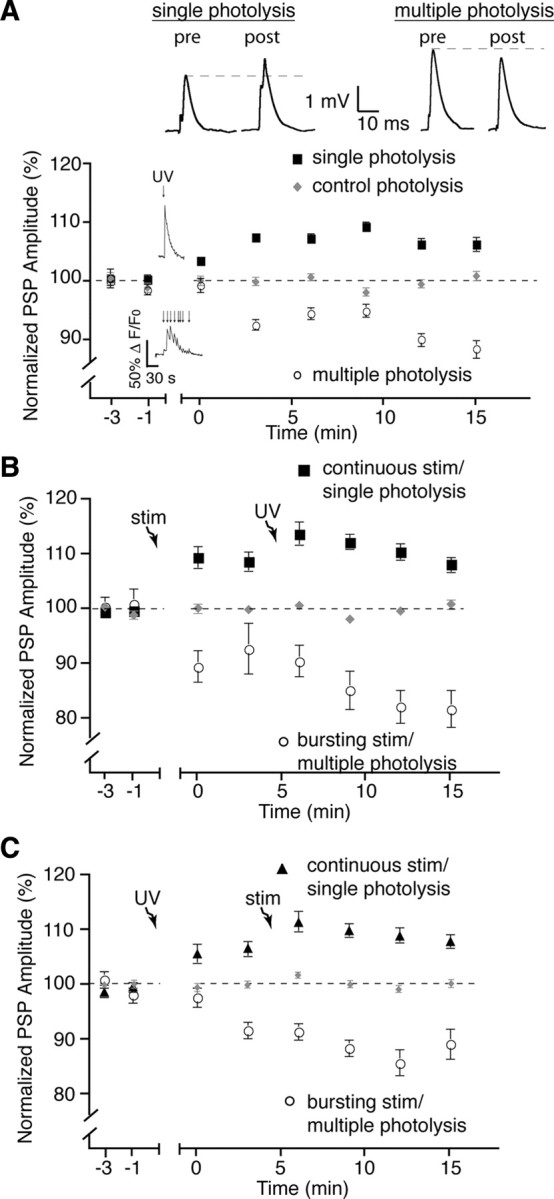
Direct induction of glial calcium elevations is sufficient for plasticity. A, Normalized PSP amplitude over time showing that multiple photolysis of NP-EGTA (caged calcium) in glial cells induced a prolonged elevation of glial calcium and post-tetanic depression similar to burst stimulation, whereas a single calcium elevation induced with photolysis of NP-EGTA in glia resulted in a post-tetanic potentiation similar to continuous stimulation. Insets in the graph illustrate a typical PSC Ca2+ responses elicited by photo-activation of NP-EGTA. Insets on the top show representative PSPs (averages of 30 events) taken from a single recording at the 15 min time point. B, Normalized PSP amplitude over time showing that photolysis of NP-EGTA performed after nerve stimulation did not induce further plasticity after that induced by the endogenous activity. C, Normalized PSP amplitude over time where photolysis of NP-EGTA was performed before nerve stimulation. No additional effect on post-tetanic plasticity was observed. Photolysis and stimulation-induced plasticity occluded each other. pre, Before; post, after; stim, stimulation.
Furthermore, the photo-activated responses in PSCs mimicked the endogenous synaptic plasticity events. Indeed, PSP amplitudes were depressed (88.6 ± 1.5%, p < 0.0003, two-tailed t test, n = 7) compared with controls after multiple photo-activations (Fig. 5 A), and were not statistically different from those evoked by endogenous 20 Hz burst stimulation (p > 0.05; Student's t test). In contrast, evoking a single calcium response induced a potentiation of PSP amplitude compared with controls (106.4 ± 1.2%, p = 0.0006, two-tailed t test, n = 8), similar to that seen after continuous stimulation (p > 0.05; Student's t test).
Importantly, not only did direct glial activation elicit plasticity events that were similar to the endogenous ones, but the two methods of glial activation occluded each other. Indeed, when nerve stimulation was followed by direct activation of glia, no further potentiation (108.2 ± 1.4% at 15 min, n = 6) or depression (81.9 ± 3.4% at 15 min, n = 6) was observed (Fig. 5 B). Similarly, photolysis-induced plasticity followed by stimulation did not result in greater plasticity (Fig. 5 C). Therefore, the two methods of glial activation and induction of synaptic plasticity (photolysis or endogenous) occlude each other indicating that photolysis-induced activation mimics endogenous activation of glial cells and the resulting changes in synaptic efficacy. More importantly, these results demonstrate that direct, differential activation of glial cells is sufficient to selectively induce post-tetanic potentiation and depression.
Purine receptors regulate PSC-induced plasticity
Knowing that glial cells were responsible for controlling bidirectional plasticity, we next investigated the mechanisms involved. A number of receptor systems offered the possibility of an opposing regulation of synaptic efficacy, such as muscarinic and purine receptor systems (Correia-de-Sá et al., 1996; Dudel, 2007). Since ATP is a prominent gliotransmitter involved in a number of neuronal regulations by glial cells (Fields and Burnstock, 2006), we tested the involvement of purines in our model of glial regulation of synaptic activity. We began by using the 5′-ectonucleotidase inhibitor ARL 67156 (50 μm) (Rebola et al., 2008) that prevents the degradation of ATP into its metabolites. As shown in Figure 6 A, rather than a depression of PSP amplitude, we observed a potentiation of 113.5 ± 1.8% compared with controls (n = 4; p < 0.0001, two-tailed t test) in the presence of ARL 67156 after the multiple photolysis protocol. Conversely, when we performed a single photolysis of caged calcium in the presence of ARL (Fig. 6 B), which normally produced potentiation of PSP amplitude, we observed no post-tetanic plasticity (102.0 ± 1.2%; n = 4; p > 0.05). These changes confirm that indeed a purine-dependent system is necessary for the glial-dependent modulation. However, it is unlikely that these results can be explained by the accumulation of ATP acting directly on P2 receptors since the presence of the nonselective P2 receptor antagonist suramin (100 μm) (Cunha et al., 1998) did not affect the outcome of the photolysis induced potentiation (105.9 ± 3.5%, p = 0.87) or depression (92.9 ± 1.8%, p = 0.13) (Fig. 6 C). The apparent lack of ATP receptor involvement is further supported by these results, considering that this antagonist is notorious for its weak specificity and is therefore likely to have inhibited any form of ATP receptor present. Hence, this suggests that the endogenous modulation of synaptic plasticity is due to the degradation of glial ATP into adenosine.
Figure 6.
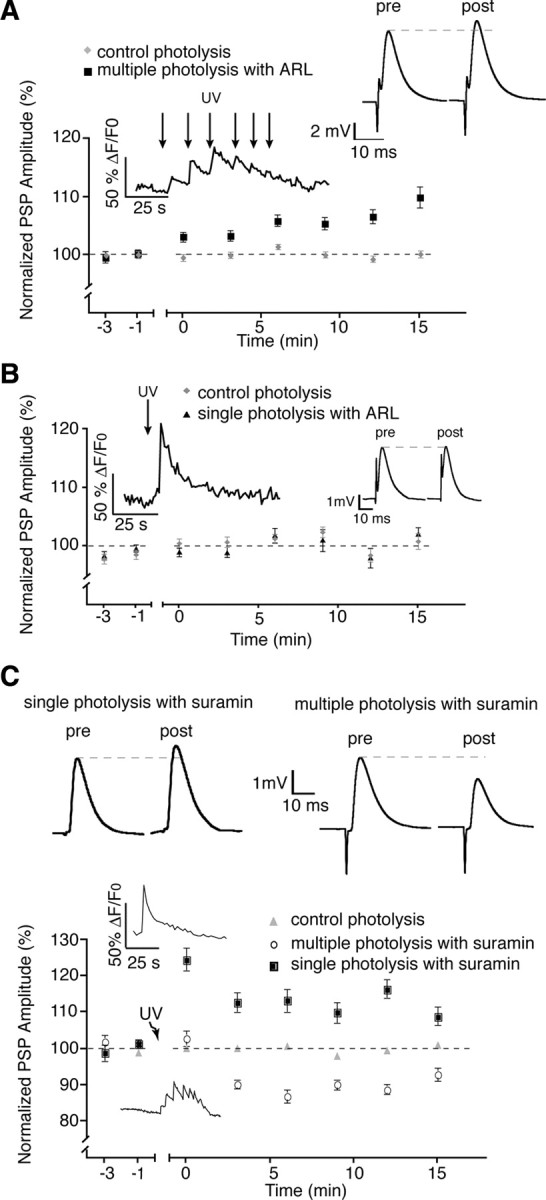
Purines but not ATP are implicated in plasticity. A, Blockade of ATP hydrolysis with ARL-67156 prevented post-tetanic depression normally observed when activating PSCs with the multiple photolysis protocol. B, The presence of ARL-67156 prevented post-tetanic potentiation normally induced by single photolysis of NP-EGTA. C, Addition of 100 μm suramin had no effect on potentiation or depression induced by the two photo-activation protocols. Insets in the graphs illustrate typical PSC Ca2+ responses elicited by photo-activation of NP-EGTA. Insets on the right show representative PSPs (averages of 30 events) taken from a single recording at the 15 min time point. pre, Before; post, after.
Adenosine A1 receptor mediate post-tetanic depression
We next tested the possibility that the blockade of ATP hydrolysis resulted in absence of adenosine, and an imbalance in A1 and A2A receptor regulation. Throughout the nervous system, A1 and A2A adenosine receptors mediate depression and potentiation, respectively (Correia-de-Sá et al., 1996; Lopes et al., 2002; Pousinha et al., 2010). Hence, one would predict that blocking A1 receptors would alter PSC-mediated depression, whereas blocking A2A receptors would alter PSC-mediated potentiation. Consistent with this possibility, bath application of an A1 receptor agonist (CPA, 30 nm) (O'Neill et al., 2007; Serpa et al., 2009) reduced PSP amplitude to 81.8 ± 5.7%% of control (p < 0.05), whereas an A2A agonist (CGS21680, 7.5 nm; Loram et al., 2009; D'Alimonte et al., 2009) increased PSP amplitude to 108.2 ± 2.2% relative to control (p < 0.05) (supplemental Fig. 2, available at www.jneurosci.org as supplemental material).
Consistent with the known role of A1 receptors at the neuromuscular synapse (Redman and Silinsky, 1994), addition of the A1 adenosine receptor antagonist PSB-36 (5 nm) (Weyler et al., 2006) did not affect base line synaptic transmission but when applied before photolysis-mediated activation of PSCs resulted in a post-photolysis potentiation of PSP amplitude rather than a post-tetanic depression (105.8 ± 1.3%, n = 8; p = 0.002, two-tailed t test) (Fig. 7 A). The involvement of A1 receptors in post-tetanic depression was further confirmed through the use of A1 receptor knock-out mice. We observed no depression in A1 −/− (98.7 ± 1.7%, n = 8) NMJ preparations (Fig. 7 B), whereas depression was normal in +/+ controls (88.6 ± 1.6%) after the multiple photolysis protocol (p < 0.0001). Furthermore, the PSC-induced depression was occluded by prior activation of A1 receptors with the selective (Lohse et al., 1988) A1 agonist CCPA (0.1 μm; 104.2 ± 1.5%; p = 0.05; two-tailed t test; n = 6) (Fig. 7 C).
Figure 7.
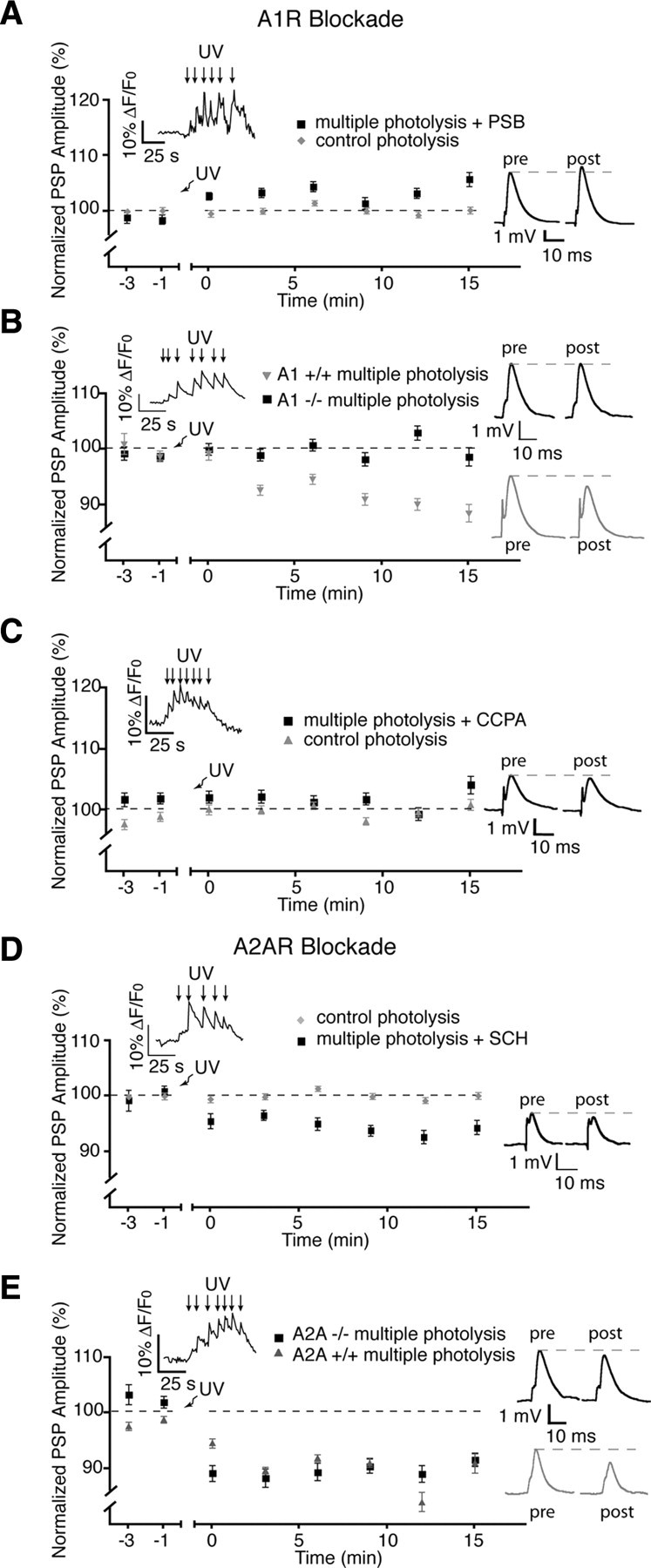
Post-tetanic depression is regulated by A1 adenosine receptors. A, Post-tetanic depression induced by multiple photolysis of NP-EGTA in glial cells was blocked by the A1 receptor antagonist PSB-36. B, No post-tetanic depression was elicited by multiple photolysis at NMJs of A1 −/− animals. C, Post-tetanic depression was occluded by prior addition of the A1 agonist CCPA. D, Presence of the A2A receptor antagonist SCH has no effect on post-tetanic depression. E, Post-tetanic depression was evoked by the multiple photolysis protocol at NMJs from A2A −/− mice. Insets in the graphs illustrate typical PSC Ca2+ responses elicited by photo-activation of NP-EGTA. Insets on the right show representative PSPs (averages of 30 events) taken from a single recording at the 15 min time point. pre, Before; post, after.
Finally, depression was not significantly altered when performing the multiple photolysis protocol in the presence of SCH-58261 (50 nm) (Rebola et al., 2008), an A2A receptor antagonist (depression of 94.4 ± 1.3%, from control) (Fig. 7 D) (p < 0.0001, n = 6) or when tested at NMJs from A2A −/− mice compared with +/+ controls (Fig. 7 E) (−/−, 91.6 ± 1.2%; +/+, 91.0 ± 1.7%; p > 0.05, n = 4). As a whole, these experiments indicate that the endogenous pattern of activity leading to glial-mediated post-tetanic depression involves A1 receptor activation after ATP degradation.
Adenosine A2 receptors mediate post-tetanic potentiation
Since our hypothesis is based on the different forms of short-term plasticity generated by a balanced A1 and A2A receptor activation, we next tested the involvement of A2A receptor activation in the modulation of glial-mediated potentiation. We used the single photolysis protocol to test for the involvement of A2A receptors in the glial-mediated post-tetanic potentiation. The presence of an A2A antagonist SCH-58261 (50 nm) had no effect on basal synaptic transmission but blocked the expression of the glial-induced post-tetanic potentiation, and resulted in depression (Fig. 8 A) (93.0 ± 1.5%; n = 5; p < 0.0001, two-tailed t test, n = 7). In addition, post-tetanic potentiation was absent at NMJs of A2A −/− mice (99.6 ± 1.3%) (Fig. 8 B) but not in the +/+ littermates (118.6 ± 3.6%; p < 0.0001, two-tailed t test, n = 4). Prior application of CGS-21680 (30 nm) (Rebola et al., 2008), an A2A agonist, occluded the PSC-induced potentiation and resulted in a depression (91.2 ± 1.2%) that was significantly reduced from controls (p < 0.0001, two-tailed t test, n = 6), further confirming the involvement of A2A receptors in potentiation (Fig. 8 C). Finally, use of the single photolysis protocol in the presence of the A1 receptor antagonist PSB-36 (5 nm) resulted in potentiation of PSP amplitude (109.1 ± 1.1%; p < 0.0001; n = 6) (Fig. 8 D), as did the use NMJs from A1 −/− mice (see Fig. 10 E) (115.6 ± 2.4%, n = 6), which was greater than +/+ littermates (106.3 ± 1.2%; p = 0.0001, n = 6). Together, the results strongly implicate a glial-regulated activation of A1 and A2A receptors in the generation of pattern-dependent induction of depression and potentiation at the NMJ.
Figure 8.
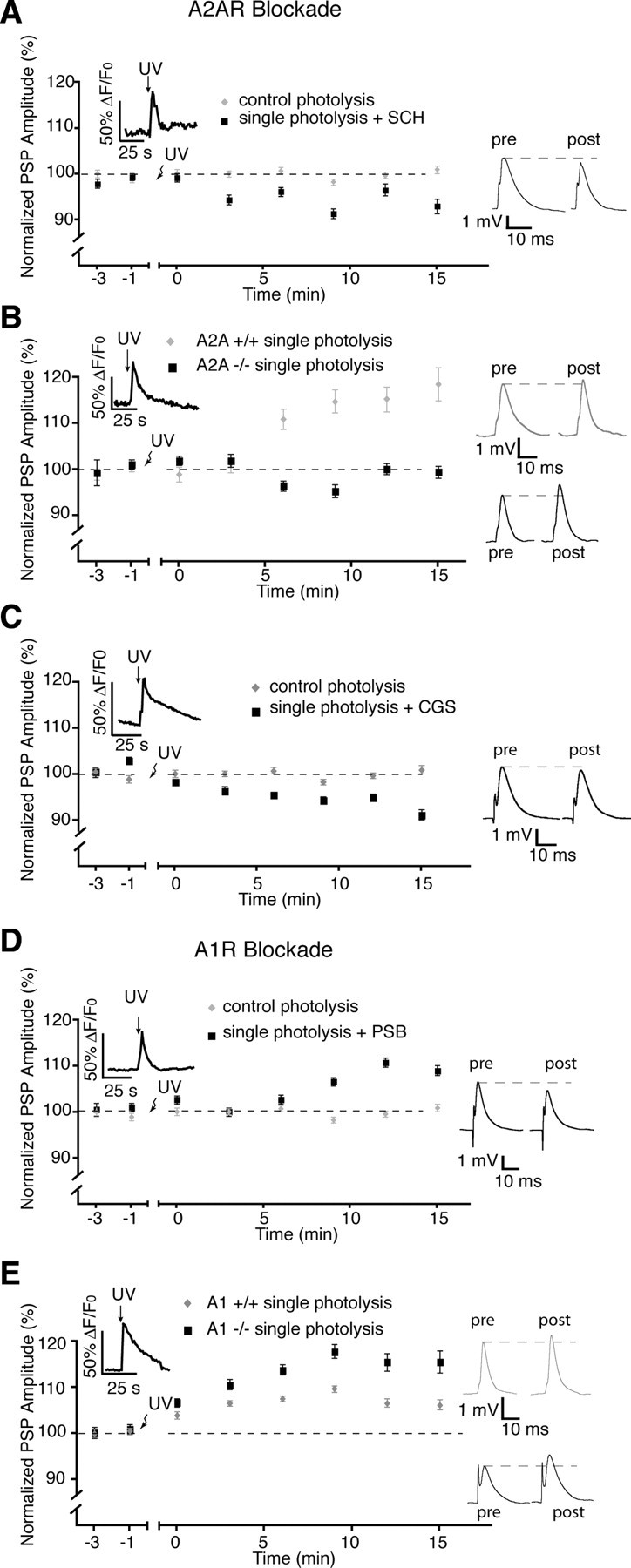
A2A receptors regulate post-tetanic potentiation. A, The presence of the A2A receptor antagonist, SCH, prevented post-tetanic potentiation induced by single photolysis of NP-EGTA in PSCs. B, No post-tetanic potentiation was elicited with the single photolysis protocol at NMJs of A2A −/− animals. C, Post-tetanic potentiation was occluded by prior addition of the A2A agonist CGS21680. D, Addition of the A1 receptor antagonist PSB-36 had no effect on post-tetanic potentiation. E, Post-tetanic potentiation was induced by the single photolysis protocol at NMJs from A1 −/− mice. pre, Before; post, after.
Figure 10.
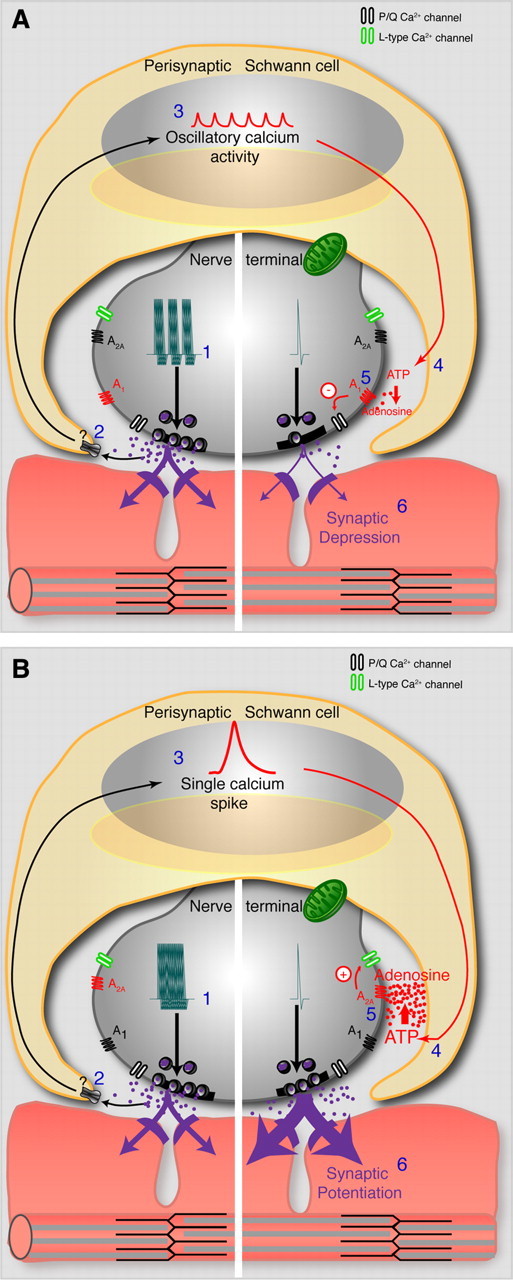
Model of glial-mediated bidirectional modulation of synaptic plasticity. A, Bursts of activity (1) induce the release of neurotransmitter to activate perisynaptic Schwann cells (2) and the postsynaptic terminal. Receptor activation on PSCs leads to oscillatory calcium elevations (3) and the release of a lesser amount of glial-derived ATP (4) that is degraded to adenosine. Relatively low levels of synaptic adenosine lead to synaptic depression through activation of A1 receptors and a decrease in presynaptic calcium entry through P/Q-type calcium channels (5 and 6). B, Continuous presynaptic activity (1) induces the release of neurotransmitter to activate perisynaptic Schwann cells (2) and the postsynaptic terminal. Receptor activation on PSCs leads to a single calcium elevation (3) and the release of a larger amount of glial-derived ATP (4) that is degraded to adenosine. Relatively high levels of synaptic adenosine lead to activation of A2A receptors, activation of L-type calcium channels (5) and synaptic potentiation (6).
Adenosine receptors mediate the plasticity induced by motor nerve stimulation
Knowing that the glial mechanisms regulating the sustained potentiation and depression involved a balance of antagonistic adenosine effects, we next tested whether these receptors were involved in the plasticity events induced by motor nerve stimulation and endogenous release of neurotransmitters. As shown in Figure 9, A and B, bath application of the A1 antagonist PSB-36 (5 nm) had no effect on basal level of neurotransmission but completely prevented the expression of the synaptic depression that is normally observed when using the bursting protocol, and a small potentiation was even observed (103% ± 1.4%, p < 0.05). Conversely, rather than the potentiation, a small depression (92.1 ± 1.9%, p < 0.05) was observed in the presence of the A2A receptor antagonist (SCH-58261, 50 nm) when stimulating the motor nerve with the continuous paradigm. Hence, these data suggest that the endogenous plasticity events are also mediated by a balanced A1/A2A regulation.
Adenosine receptors do not mediate the sustained plasticity after blockade of PSC activity
The observations that the polarity of post-tetanic plasticity events was reversed after the blockade of PSCs suggest that different mechanisms are recruited when glial regulation is perturbed. A possibility might be that the reversal is due to the activation of the antagonistic adenosine receptor, A1 rather than A2A and vice versa. This possibility would be consistent with the involvement of these receptors in the endogenous plasticity events elicited by motor nerve stimulation (Fig. 9 A,B). This possibility was examined by testing the effects of A1 and A2A receptor antagonists on the reversal of the plasticity events observed after Ca2+ chelation in PSCs.
As shown above, blockade of PSC activity by photo-activation of diazo-2 resulted in a depression of 65 ± 11% with continuous motor nerve stimulation (rather than potentiation) and a potentiation of 103% ± 1.1% with the bursting protocol (rather than depression) (Fig. 9 C,D). However, a depression was still observed in the presence of the A1 antagonist (PSB-36, 5 nm; 60 ± 11%) while PSP amplitude remained potentiated even when the A2A antagonist (SCH-58261, 50 nm) was present (106.1 ± 2.4%). Hence, the reversal observed in the post-tetanic plasticity cannot be explained by a switch in the adenosine receptor involved.
Discussion
Glial cells display finely tuned responsiveness to neuronal activity. They detect subtle changes in the frequency of activity (Pasti et al., 1997) and discriminate between different synaptic inputs (Perea and Araque, 2005). Here, we show that glia differentiate patterns of synaptic activity, an important element regulating synaptic plasticity. They integrate and decode the pattern of neuronal and synaptic activity to, in turn, influence synaptic transmission. Our data indicate that this regulation likely occurs by processing incoming information that alters and tunes subsequent feedback to a given synapse.
Neuronal communication depends on interplay between glial cells and neurons
Our data show that PSCs were differentially activated by different patterns of neuronal activity whereas blockade of their Ca2+-dependent activity perturbed the outcome of synaptic plasticity. This indicates that neurons are no longer the only cells that decode information from patterns of neuronal activity and process it to influence the outcome of synaptic communication. Owing to this decoding capability, glial cell regulation of short-term and long-term synaptic plasticity should no longer be seen as an all-or-none event, but rather as an adaptable regulation dependent on the context of previous neuronal activity. This additional level of detection would allow glial cells to mediate and regulate a large array of synaptic events. Also, our results are consistent with the notion presented earlier (Robitaille, 1998; Castonguay and Robitaille, 2001) that glial cells may not produce plasticity events themselves, but rather modulate and control presynaptic and postsynaptic mechanisms. Indeed, the persistence of some forms of plasticity after Ca2+ chelation in PSCs argues in favor of this, whereas the lack of purinergic sensitivity of the residual plasticity suggests that other receptor systems also regulate synaptic efficacy at the NMJ. Hence, glial cells add another level of regulation to neuronal synaptic plasticity.
Impacts on CNS synapses
The data obtained here using a PNS synapse are likely applicable to synapses throughout the nervous system. Indeed, it has been demonstrated previously that direct activation of retinal glial cells can induce both positive and negative changes in neuronal activity (Newman and Zahs, 1998), suggesting similar roles to those described here. Furthermore, Panatier et al. (2006) recently described a phenomenon similar to ours, occurring in the hypothalamus. They showed that glial cells determined the outcome of synaptic plasticity (potentiation or depression) as a result of changes in the glial synaptic coverage and release of d-serine. These are only some examples while more are continually added, suggesting the involvement of glial cells at synapses throughout the nervous system (Serrano et al., 2006; Perea and Araque, 2007; Fellin et al., 2009; Gordon et al., 2009; Henneberger et al., 2010). Here, we have built on the current knowledge of glial involvement in synaptic function by providing evidence that glial cells, acutely responding to neuronal activity, can supply context-dependent feedback to synapses by decoding the pattern of neuronal activity. Hence, our data indicate that glial regulation of neuronal plasticity is not only a matter of slow long-term modification to the glial environment, but that it occurs over a matter of minutes. Morphological synaptic changes are an interesting avenue to explore since they can occur within a similar time window (Matsuzaki et al., 2004).
Unlike the work discussed above, Agulhon et al. (2010) reported that Ca2+ elevation in astrocytes did not alter short-term and long-term plasticity. Interestingly, they evoked sustained and massive agonist-induced activation of a foreign receptor overexpressed in astrocytes, whereas Henneberger et al. (2010) and ourselves used more subtle glial regulation and stimulation paradigms. An interesting possibility might be that intense stimulation of glial cells puts them in a less responsive mode that depresses their normal Ca2+-dependent mechanisms. However, experiments directly addressing this issue need to be performed to solve this conundrum.
Glial cells decode neuronal signaling through calcium dynamics
We found that PSCs decode the pattern of activity by producing selective signature of global Ca2+ elevation. Notwithstanding spatial and temporal limitations inherent to the Ca2+ imaging technique, our data suggest that specific glial responses have direct consequences on the output of synaptic plasticity. Indeed, small and brief responses repeated over a longer period induced depression, whereas responses with larger amplitude and duration caused potentiation. Interestingly, this situation is reminiscent of the calcium hypothesis of the switch between long-term potentiation and long-term depression production in hippocampus where a rapid and short-lasting Ca2+ elevation leads to a potentiation, whereas smaller and repeated elevations lead to depression (Yang et al., 1999).
This observation suggests that the excitability of neurons and glial cells share similar biochemical mechanisms since both amplitude and duration of an intracellular chemical signal such as Ca2+ determine the outcome of the biological process. Also, considering Ca2+-related regulation, these data suggest that glial cells and neurons regulate neuronal communication within the same time domain. This strengthens the idea that glial and neuronal elements need to be in tune with each other for proper neuronal communication to occur (Serrano et al., 2006).
Glial regulation occurs through a balanced A1/A2A receptor activation
Activation of A1 and A2A receptors results in inhibition and activation of different types of presynaptic calcium channels to regulate transmitter release at synapses in general and at the mammalian NMJ in particular (Correia-de-Sá et al., 1996; De Lorenzo et al., 2004; Oliveira et al., 2004; Silinsky, 2005; Cunha, 2008). Moreover, an adenosine-dependent long-term depression has been reported (Redman and Silinsky, 1994) that involves an inhibition of presynaptic calcium currents (Silinsky, 2004, 2005). Our data indicate that the balanced A1-A2A regulation can be controlled, not only by the presynaptic pool itself in an autocrine manner, but also by perisynaptic glial cells. In addition, adenosine-dependent glial regulation appears both necessary and sufficient, as indicated by the blockade of the potentiation and depression induced by direct glial activation or by motor nerve stimulation.
Our data are consistent with the general perspective in the field of purinergic research where the synaptic regulation by adenosine depends on a balanced activation of inhibitory A1 and facilitatory A2A receptors. Importantly, this balanced regulation depends not only on the level of extracellular adenosine but also on direct interactions between A1 and A2A receptors through cross-regulatory actions (Cunha, 2001). This tight auto-regulated interaction is believed to be the basis for the unbalanced effects that are observed when one receptor type is specifically blocked. Indeed, similar to what we observed in this work, blocking a receptor type does not necessarily results in a simple blockade but rather to a reversed effect. Hence, the governance by adenosine receptors cannot be interpreted on the basis of individual receptor regulation but on combined and interactive functions of all receptors.
Model of bidirectional glial regulation
We propose a model (Fig. 10) based on the properties of A1-A2A receptor interactions as discussed above (see also Johansson et al., 2001), the ability of glial cells to release ATP (Fields and Burnstock, 2006), and the balanced regulation of cell activity during spatio-temporal changes in intracellular Ca2+ levels. Smaller but sustained accumulation of glial calcium elicited by bursting synaptic activity would induce the release of a smaller amount of ATP at any given time, leading to a lower concentration of adenosine in the synaptic cleft, the activation of A1 receptors, and post-tetanic depression. Conversely, post-tetanic potentiation would be induced by larger and more transient glial calcium responses elicited by sustained synaptic activity causing the release of a greater quantity of ATP, therefore leading to more adenosine and the activation of A2A receptors. Furthermore, our data and model are consistent with adenosine receptor pharmacology and knock-out manipulations reported previously (Fredholm et al., 2005) where the outcome could not be accounted for simply by the regulation of a single type of receptor. Importantly, possible involvement of other adenosine receptors such as A2B and A3 receptors cannot be ruled out completely.
Potential involvement in diseases
Our data show that PSC regulation leads to changes in synaptic potency that would facilitate or reduce motoneuronal control of the NMJ, which is based on the pattern of neuronal activity itself. Interestingly, the PSC-mediated adenosine regulation could be a target for possible treatments of muscular diseases (e.g., myasthenia gravis) or conditions that lead to weakened synapses (e.g., aging) whereby patterns of activity could be made to favor PSC-dependent potentiating pathway, thereby strengthening the efficacy of the NMJ. Albeit small, these changes in plasticity could influence the level of excitability when repeated over the course of the daily neuromuscular activity.
In addition, these principles of neuron–glial interactions may apply to the basic function of CNS synapses, and their perturbation may contribute to a number of malfunctions. For instance, there is evidence suggesting that inadequate interactions between A2A and D2 receptors may be in part responsible for certain aspects of the pathophysiology observed in Parkinson's disease and certain forms of drug addictions (Ferré et al., 2008). These interactions may not be solely neuronal in nature but may involve glial cells owing to their role in the regulation of adenosine receptors and their functions.
We demonstrate that glial cells can govern the outcome of synaptic plasticity based on their ability to decode the patterns of neuronal communication. These results indicate that glial cells can act as pattern detectors, a role that could influence many CNS functions.
Footnotes
R.R. was supported by the Canadian Institutes of Health Research (CIHR) and by an infrastructure grant to the Groupe de Recherche sur le Système Nerveux Central from the Fonds de la Recherche en Santé du Québec (FRSQ). K.J.T. was supported by the Natural Science and Engineering Research Council of Canada, Le Fonds Québécois de la Recherche sur la Nature et les Technologies, and currently by the FRSQ. R.R. held a Scientist scholar award from CIHR and is now a Chercheur-National of FRSQ. We thank Eve-Lyne Bélair and Audrée Pinard along with the rest of the Robitaille laboratory as well as Drs. M. Charlton, S. Oliet, A. Panatier, B. Fredholm, J.-F. Chen, and C. Zagami for helpful discussion and valuable critique of the manuscript. We also thank Aude Panatier for her help in preparing Figure 10, and Ozzy Murmut and Luc Langevin at Institut National d'Optique, Quebec, Canada, for their help in establishing the characteristics of the UV fiberoptic probe. Jiang-Fan Chen and Michael Schwarzschild kindly provided the A2A KO mice, while Bertil Fredholm and Stephen Tilley generously provided the A1 receptor knock-out mice.
References
- Adler EM, Augustine GJ, Duffy SN, Charlton MP. Alien intracellular calcium chelators attenuate neurotransmitter release at the squid giant synapse. J Neurosci. 1991;11:1496–1507. doi: 10.1523/JNEUROSCI.11-06-01496.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agulhon C, Fiacco TA, McCarthy KD. Hippocampal short- and long-term plasticity are not modulated by astrocyte Ca2+ signaling. Science. 2010;327:1250–1254. doi: 10.1126/science.1184821. [DOI] [PubMed] [Google Scholar]
- Auld DS, Robitaille R. Glial cells and neurotransmission: an inclusive view of synaptic function. Neuron. 2003;40:389–400. doi: 10.1016/s0896-6273(03)00607-x. [DOI] [PubMed] [Google Scholar]
- Barres BA. The mystery and magic of glia: a perspective on their roles in health and disease. Neuron. 2008;60:430–440. doi: 10.1016/j.neuron.2008.10.013. [DOI] [PubMed] [Google Scholar]
- Baxter RL, Vega-Riveroll LJ, Deuchars J, Parson SH. A2A adenosine receptors are located on presynaptic motor nerve terminals in the mouse. Synapse. 2005;57:229–234. doi: 10.1002/syn.20173. [DOI] [PubMed] [Google Scholar]
- Bélair EL, Vallée J, Robitaille R. Bidirectional plasticity of glial cells induced by chronic treatments in vivo. J Physiol 588. 2010;7:1039–1056. doi: 10.1113/jphysiol.2009.178988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castonguay A, Robitaille R. Differential regulation of transmitter release by presynaptic and glial Ca2+ internal stores at the neuromuscular synapse. J Neurosci. 2001;21:1911–1922. doi: 10.1523/JNEUROSCI.21-06-01911.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen JF, Huang Z, Ma J, Zhu J, Moratalla R, Standaert D, Moskowitz MA, Fink JS, Schwarzschild MA. A(2A) adenosine receptor deficiency attenuates brain injury induced by transient focal ischemia in mice. J Neurosci. 1999;19:9192–9200. doi: 10.1523/JNEUROSCI.19-21-09192.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Correia-de-Sá P, Timóteo MA, Ribeiro JA. Presynaptic A1 inhibitory/A2A facilitatory adenosine receptor activation balance depends on motor nerve stimulation paradigm at the rat hemidiaphragm. J Neurophysiol. 1996;76:3910–3919. doi: 10.1152/jn.1996.76.6.3910. [DOI] [PubMed] [Google Scholar]
- Cunha RA. Adenosine as a neuromodulator and as a homeostatic regulator in the nervous system: different roles, different sources and different receptors. Neurochem Int. 2001;38:107–125. doi: 10.1016/s0197-0186(00)00034-6. [DOI] [PubMed] [Google Scholar]
- Cunha RA. Different cellular sources and different roles of adenosine: A(1) receptor-mediated inhibition through astrocytic-driven volume transmission and synapse-restricted A(2A) receptor-mediated facilitation of plasticity. Neurochem Int. 2008;52:65–72. doi: 10.1016/j.neuint.2007.06.026. [DOI] [PubMed] [Google Scholar]
- Cunha RA, Sebastião AM, Ribeiro JA. Inhibition by ATP of hippocampal synaptic transmission requires localized extracellular catabolism by ecto-nucleotidases into adenosine and channeling to adenosine A1 receptors. J Neurosci. 1998;18:1987–1995. doi: 10.1523/JNEUROSCI.18-06-01987.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Alimonte I, D'Auro M, Citraro R, Biagioni F, Jiang S, Nargi E, Buccella S, Di Iorio P, Giuliani P, Ballerini P, Caciagli F, Russo E, De Sarro G, Ciccarelli R. Altered distribution and function of A2A adenosine receptors in the brain of WAG/Rij rats with genetic absence epilepsy, before and after appearance of the disease. Eur J Neurosci. 2009;30:1023–1035. doi: 10.1111/j.1460-9568.2009.06897.x. [DOI] [PubMed] [Google Scholar]
- Delaney KR, Zucker RS, Tank DW. Calcium in motor nerve terminals associated with posttetanic potentiation. J Neurosci. 1989;9:3558–3567. doi: 10.1523/JNEUROSCI.09-10-03558.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delaney K, Tank DW, Zucker RS. Presynaptic calcium and serotonin-mediated enhancement of transmitter release at crayfish neuromuscular junction. J Neurosci. 1991;11:2631–2643. doi: 10.1523/JNEUROSCI.11-09-02631.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Lorenzo S, Veggetti M, Muchnik S, Losavio A. Presynaptic inhibition of spontaneous acetylcholine release induced by adenosine at the mouse neuromuscular junction. Br J Pharmacol. 2004;142:113–124. doi: 10.1038/sj.bjp.0705656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudel J. The time course of transmitter release in mouse motor nerve terminals is differentially affected by activation of muscarinic M1 or M2 receptors. Eur J Neurosci. 2007;26:2160–2168. doi: 10.1111/j.1460-9568.2007.05770.x. [DOI] [PubMed] [Google Scholar]
- Fellin T, Halassa MM, Terunuma M, Succol F, Takano H, Frank M, Moss SJ, Haydon PG. Endogenous nonneuronal modulators of synaptic transmission control cortical slow oscillations in vivo. Proc Natl Acad Sci U S A. 2009;106:15037–15042. doi: 10.1073/pnas.0906419106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferré S, Quiroz C, Woods AS, Cunha R, Popoli P, Ciruela F, Lluis C, Franco R, Azdad K, Schiffmann SN. An update on adenosine A2A-dopamine D2 receptor interactions: implications for the function of G protein-coupled receptors. Curr Pharm Des. 2008;14:1468–1474. doi: 10.2174/138161208784480108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fields RD, Burnstock G. Purinergic signalling in neuron-glia interactions. Nat Rev Neurosci. 2006;7:423–436. doi: 10.1038/nrn1928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fredholm BB, Chen JF, Masino SA, Vaugeois JM. Actions of adenosine at its receptors in the CNS: insights from knockouts and drugs. Annu Rev Pharmacol Toxicol. 2005;45:385–412. doi: 10.1146/annurev.pharmtox.45.120403.095731. [DOI] [PubMed] [Google Scholar]
- Galkin AV, Giniatullin RA, Mukhtarov MR, Svandová I, Grishin SN, Vyskocil F. ATP but not adenosine inhibits nonquantal acetylcholine release at the mouse neuromuscular junction. Eur J Neurosci. 2001;13:2047–2053. doi: 10.1046/j.0953-816x.2001.01582.x. [DOI] [PubMed] [Google Scholar]
- Giménez-Llort L, Fernández-Teruel A, Escorihuela RM, Fredholm BB, Tobeña A, Pekny M, Johansson B. Mice lacking the adenosine A1 receptor are anxious and aggressive, but are normal learners with reduced muscle strength and survival rate. Eur J Neurosci. 2002;16:547–550. doi: 10.1046/j.1460-9568.2002.02122.x. [DOI] [PubMed] [Google Scholar]
- Gordon GR, Iremonger KJ, Kantevari S, Ellis-Davies GC, MacVicar BA, Bains JS. Astrocyte-mediated distributed plasticity at hypothalamic glutamate synapses. Neuron. 2009;64:391–403. doi: 10.1016/j.neuron.2009.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grafe P, Mayer C, Takigawa T, Kamleiter M, Sanchez-Brandelik R. Confocal calcium imaging reveals an ionotropic P2 nucleotide receptor in the paranodal membrane of rat Schwann cells. J Physiol. 1999;515:377–383. doi: 10.1111/j.1469-7793.1999.377ac.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guyonneau R, Vanrullen R, Thorpe SJ. Temporal codes and sparse representations: a key to understanding rapid processing in the visual system. J Physiol Paris. 2004;98:487–497. doi: 10.1016/j.jphysparis.2005.09.004. [DOI] [PubMed] [Google Scholar]
- Hamilton BR, Smith DO. Autoreceptor-mediated purinergic and cholinergic inhibition of motor nerve terminal calcium currents in the rat. J Physiol. 1991;432:327–341. doi: 10.1113/jphysiol.1991.sp018387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris KD. Neural signatures of cell assembly organization. Nat Rev Neurosci. 2005;6:399–407. doi: 10.1038/nrn1669. [DOI] [PubMed] [Google Scholar]
- Haydon PG, Carmignoto G. Astrocyte control of synaptic transmission and neurovascular coupling. Physiol Rev. 2006;86:1009–1031. doi: 10.1152/physrev.00049.2005. [DOI] [PubMed] [Google Scholar]
- Henneberger C, Papouin T, Oliet SH, Rusakov DA. Long-term potentiation depends on release of D-serine from astrocytes. Nature. 2010;463:232–236. doi: 10.1038/nature08673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hennig R, Lømo T. Firing patterns of motor units in normal rats. Nature. 1985;314:164–166. doi: 10.1038/314164a0. [DOI] [PubMed] [Google Scholar]
- Johansson B, Halldner L, Dunwiddie TV, Masino SA, Poelchen W, Giménez-Llort L, Escorihuela RM, Fernández-Teruel A, Wiesenfeld-Hallin Z, Xu XJ, Hårdemark A, Betsholtz C, Herlenius E, Fredholm BB. Hyperalgesia, anxiety, and decreased hypoxic neuroprotection in mice lacking the adenosine A1 receptor. Proc Natl Acad Sci U S A. 2001;98:9407–9412. doi: 10.1073/pnas.161292398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamiya H, Zucker RS. Residual Ca2+ and short-term synaptic plasticity. Nature. 1994;371:603–606. doi: 10.1038/371603a0. [DOI] [PubMed] [Google Scholar]
- Lohse MJ, Klotz KN, Schwabe U, Cristalli G, Vittori S, Grifantini M. 2-Chloro-N6-cyclopentyladenosine: a highly selective agonist at A1 adenosine receptors. Naunyn Schmiedebergs Arch Pharmacol. 1988;337:687–689. doi: 10.1007/BF00175797. [DOI] [PubMed] [Google Scholar]
- Lopes LV, Cunha RA, Kull B, Fredholm BB, Ribeiro JA. Adenosine A(2A) receptor facilitation of hippocampal synaptic transmission is dependent on tonic A(1) receptor inhibition. Neuroscience. 2002;112:319–329. doi: 10.1016/s0306-4522(02)00080-5. [DOI] [PubMed] [Google Scholar]
- Loram LC, Harrison JA, Sloane EM, Hutchinson MR, Sholar P, Taylor FR, Berkelhammer D, Coats BD, Poole S, Milligan ED, Maier SF, Rieger J, Watkins LR. Enduring reversal of neuropathic pain by a single intrathecal injection of adenosine 2A receptor agonists: a novel therapy for neuropathic pain. J Neurosci. 2009;29:14015–14025. doi: 10.1523/JNEUROSCI.3447-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magleby DL, Zengel JE. Stimulation-induced factors which affect augmentation and potentiation of transmitter release at the neuromuscular junction. J Physiol. 1976;260:687–717. doi: 10.1113/jphysiol.1976.sp011539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuzaki M, Honkura N, Ellis-Davies GC, Kasai H. Structural basis of long-term potentiation in single dendritic spines. Nature. 2004;429:761–766. doi: 10.1038/nature02617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moores TS, Hasdemir B, Vega-Riveroll L, Deuchars J, Parson SH. Properties of presynaptic P2X7-like receptors at the neuromuscular junction. Brain Res. 2005;1034:40–50. doi: 10.1016/j.brainres.2004.12.001. [DOI] [PubMed] [Google Scholar]
- Newman EA, Zahs KR. Modulation of neuronal activity by glial cells in the retina. J Neurosci. 1998;18:4022–4028. doi: 10.1523/JNEUROSCI.18-11-04022.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicoll RA, Schmitz D. Synaptic plasticity at hippocampal mossy fibre synapses. Nat Rev Neurosci. 2005;6:863–876. doi: 10.1038/nrn1786. [DOI] [PubMed] [Google Scholar]
- Oliveira L, Timóteo MA, Correia-de-Sá P. Modulation by adenosine of both muscarinic M1-facilitation and M2-inhibition of [3H]-acetylcholine release from the rat motor nerve terminals. Eur J Neurosci. 2002;15:1728–1736. doi: 10.1046/j.1460-9568.2002.02020.x. [DOI] [PubMed] [Google Scholar]
- Oliveira L, Timóteo MA, Correia-de-Sá P. Tetanic depression is overcome by tonic adenosine A(2A) receptor facilitation of L-type Ca(2+) influx into rat motor nerve terminals. J Physiol. 2004;560:157–168. doi: 10.1113/jphysiol.2004.067595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Neill C, Nolan BJ, Macari A, O'Boyle KM, O'Connor JJ. Adenosine A1 receptor-mediated inhibition of dopamine release from rat striatal slices is modulated by D1 dopamine receptors. Eur J Neurosci. 2007;26:3421–3428. doi: 10.1111/j.1460-9568.2007.05953.x. [DOI] [PubMed] [Google Scholar]
- Panatier A, Theodosis DT, Mothet JP, Touquet B, Pollegioni L, Poulain DA, Oliet SH. Glia-derived D-serine controls NMDA receptor activity and synaptic memory. Cell. 2006;125:775–784. doi: 10.1016/j.cell.2006.02.051. [DOI] [PubMed] [Google Scholar]
- Pascual O, Casper KB, Kubera C, Zhang J, Revilla-Sanchez R, Sul JY, Takano H, Moss SJ, McCarthy K, Haydon PG. Astrocytic purinergic signaling coordinates synaptic networks. Science. 2005;310:113–116. doi: 10.1126/science.1116916. [DOI] [PubMed] [Google Scholar]
- Pasti L, Volterra A, Pozzan T, Carmignoto G. Intracellular calcium oscillations in astrocytes: a highly plastic, bidirectional form of communication between neurons and astrocytes in situ. J Neurosci. 1997;17:7817–7830. doi: 10.1523/JNEUROSCI.17-20-07817.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perea G, Araque A. Properties of synaptically evoked astrocyte calcium signal reveal synaptic information processing by astrocytes. J Neurosci. 2005;25:2192–2203. doi: 10.1523/JNEUROSCI.3965-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perea G, Araque A. Astrocytes Potentiate Transmitter Release at Single Hippocampal Synapses. Science. 2007;317:1083–1086. doi: 10.1126/science.1144640. [DOI] [PubMed] [Google Scholar]
- Pousinha PA, Correia AM, Sebastião AM, Ribeiro JA. Predominance of adenosine excitatory over inhibitory effects on transmission at the neuromuscular junction of infant rats. J Pharmacol Exp Ther. 2010;332:153–163. doi: 10.1124/jpet.109.157255. [DOI] [PubMed] [Google Scholar]
- Rebola N, Lujan R, Cunha RA, Mulle C. Adenosine A(2A) receptors are essential for long-term potentiation of NMDA-EPSCs at hippocampal mossy fiber synapses. Neuron. 2008;57:121–134. doi: 10.1016/j.neuron.2007.11.023. [DOI] [PubMed] [Google Scholar]
- Redman RS, Silinsky EM. ATP released together with acetylcholine as the mediator of neuromuscular depression at frog motor nerve endings. J Physiol. 1994;477:117–127. doi: 10.1113/jphysiol.1994.sp020176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rees D. A non-phosphate-buffered physiological saline for in vitro electrophysiological studies on the mammalian neuromuscular junction [proceedings] J Physiol. 1978;278:8P–9P. [PubMed] [Google Scholar]
- Robitaille R. Modulation of synaptic efficacy and synaptic depression by glial cells at the frog neuromuscular junction. Neuron. 1998;21:847–855. doi: 10.1016/s0896-6273(00)80600-5. [DOI] [PubMed] [Google Scholar]
- Robitaille R, Charlton MP. Presynaptic calcium signals and transmitter release are modulated by calcium-activated potassium channels. J Neurosci. 1992;12:297–305. doi: 10.1523/JNEUROSCI.12-01-00297.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robitaille R, Garcia ML, Kaczorowski GJ, Charlton MP. Functional colocalization of calcium and calcium-gated potassium channels in control of transmitter release. Neuron. 1993;11:645–655. doi: 10.1016/0896-6273(93)90076-4. [DOI] [PubMed] [Google Scholar]
- Rochon D, Rousse I, Robitaille R. Synapse-glia interactions at the mammalian neuromuscular junction. J Neurosci. 2001;21:3819–3829. doi: 10.1523/JNEUROSCI.21-11-03819.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serpa A, Ribeiro JA, Sebastião AM. Cannabinoid CB(1) and adenosine A(1) receptors independently inhibit hippocampal synaptic transmission. Eur J Pharmacol. 2009;623:41–46. doi: 10.1016/j.ejphar.2009.09.020. [DOI] [PubMed] [Google Scholar]
- Serrano A, Haddjeri N, Lacaille JC, Robitaille R. GABAergic network activation of glial cells underlies hippocampal heterosynaptic depression. J Neurosci. 2006;26:5370–5382. doi: 10.1523/JNEUROSCI.5255-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silinsky EM. Adenosine decreases both presynaptic calcium currents and neurotransmitter release at the mouse neuromuscular junction. J Physiol. 2004;558:389–401. doi: 10.1113/jphysiol.2004.061457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silinsky EM. Modulation of calcium currents is eliminated after cleavage of a strategic component of the mammalian secretory apparatus. J Physiol. 2005;566:681–688. doi: 10.1113/jphysiol.2005.090647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Todd KJ, Robitaille R. Neuron-glia interactions at the neuromuscular synapse. Novartis Found Symp. 2006;276:222–229. discussion 229–237, 275–281. [PubMed] [Google Scholar]
- Weyler S, Fülle F, Diekmann M, Schumacher B, Hinz S, Klotz KN, Müller CE. Improving potency, selectivity, and water solubility of adenosine A1 receptor antagonists: xanthines modified at position 3 and related pyrimido[1,2,3-cd]purinediones. ChemMedChem. 2006;1:891–902. doi: 10.1002/cmdc.200600066. [DOI] [PubMed] [Google Scholar]
- Yang SN, Tang YG, Zucker RS. Selective induction of LTP and LTD by postsynaptic [Ca2+]i elevation. J Neurophysiol. 1999;81:781–787. doi: 10.1152/jn.1999.81.2.781. [DOI] [PubMed] [Google Scholar]